
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 26 April 2023
Sec. Marine Evolutionary Biology, Biogeography and Species Diversity
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1087507
This article is part of the Research TopicEcological and Genetic Insights into Seaweeds’ Diversity and AdaptationView all 7 articles
 Mi Yeon Yang1
Mi Yeon Yang1 Myung Sook Kim1,2*
Myung Sook Kim1,2*An increasing number of studies have demonstrated that genetic differentiation and cryptic diversity in the sea occur over considerably smaller spatial scales than previously comprehended, considering the wide distribution range of many morphologically defined macroalgal species. However, knowledge of the turf-forming red alga Caulacanthus is incomplete regarding its species diversity, as well as genetic differentiation within the genus. We analyzed Caulacanthus specimens from the NW Pacific, NE Pacific, Central Pacific, SW Pacific, SE Indian, NE Atlantic, and SE Atlantic Ocean using mitochondrial cytochrome oxidase subunit I (COI-5P), plastid ribulose-1,5-bisphosphate carboxylase/oxygenase (rbcL), and Rubisco spacer (rbcL-S). The objectives of this study were to 1) determine the number of species that exhibit the morphology of C. ustulatus, 2) investigate the present distribution pattern of Caulacanthus species, and 3) estimate the degree of genetic connectivity between the populations of Caulacanthus species from different regions. Our results revealed molecular evidence that the genus Caulacanthus comprises of at least seven species with deep genetic divergence, which is indicative of not only a strong geographical subdivision but also a relatively long temporal discontinuity. Most species exhibited limited geographic distribution, showing considerable genetic divergence in the populations isolated by distance. Our study provides evidence of a greater evolutionary independence of Caulacanthus populations, which have undergone a series of allopatric diversification events.
The existence of widespread or cosmopolitan species has been widely accepted in marine organisms because of their high dispersal potential and the continuity of the marine environment (Avis, 1998). However, recent studies using molecular data have revealed that many supposedly widespread macroalgal species consist of cryptic species complexes (Díaz-Tapia et al., 2018; Nauer et al., 2019; Vieira et al., 2019), raising questions concerning the real distribution of such cosmopolitan species. Widespread species have frequently been found to be highly genetically differentiated across regional boundaries (Díaz-Tapia et al., 2018; Yang et al., 2020) and sometimes exhibit fine-scale endemism (Leliaert et al., 2018; Yang and Kim, 2018). Generally, regional endemism is considered to have evolved in regions that have experienced a stable environment over a long period (Myers and Giller, 1988).
With the discovery of cryptic speciation, molecular analyses have been developed to identify and delimit species (Díaz-Tapia et al., 2020; Lagourgue et al., 2022). Among the widespread macroalgal species, molecular data have clarified the real distribution of traditionally identified species solely based on morphological characteristics (Yang et al., 2015; Boo et al., 2019; Kang et al., 2021), with deep genetic divergence seen between regional populations owing to their limited natural dispersal ability (Nauer et al., 2019; Yang et al., 2021b). Other widely distributed species showed genetic affinities among populations despite separation by large oceanic distances, suggesting other dispersal mechanisms for the present distribution pattern (Fraser et al., 2013; Sherwood et al., 2014). Such long dispersal mechanisms can be explained by the artificial distribution of non-buoyant macroalgae (Fraser et al., 2013). Although the hidden algal diversity is increasingly being discovered, our knowledge on the global diversity and distribution of red macroalgae is still incomplete.
The turf-forming red algal genus, Caulacanthus Kützing, is a component of intertidal ecosystems and is commonly found in temperate and tropical marine waters (Guiry and Guiry, 2023). The genus Caulacanthus includes three currently accepted species, Caulacanthus okamurae Yamada, Caulacanthus salifugus A. B. Cribb, and Caulacanthus ustulatus (Turner) Kützing (Guiry and Guiry, 2023). The type species of the genus, C. ustulatus, was described from Cádiz, Spain, and subsequently reported in various regions, including the Western Pacific Ocean, western North America, Mediterranean Sea, Africa, Australia, and New Zealand (Guiry and Guiry, 2023). Whether the actual distribution of this species belong to the widespread species, Caulacanthus was investigated by Zuccarello et al. (2002), who conducted molecular analyses using Rubisco and cox2‐3 spacers to confirm the phylogenetic relationships between collections from the Northwest (NW) Pacific (including Korea and China), tropical Western (W) Pacific (including the Philippines), Southwest (SW) Pacific (including Australia), Northeast (NE) Pacific (including the USA), and the NE Atlantic (including France, Spain, and Portugal). They found that C. ustulatus included two distinct phylogenetic groups (Pacific and Atlantic) and concluded that the Pacific lineage indicated C. okamurae, which has long been regarded as a synonym of C. ustulatus (Searles, 1968; West and Calumpong, 1990; Rueness, 1997). C. okamurae was described based on the morphological features of vegetative plants (Yamada, 1933). Since then, taxonomic position of these species has been under debate (Searles, 1968; West and Calumpong, 1990); however, the distinctness of C. okamurae has been accepted based on different cystocarpic development and gemination patterns of tetraspores (Kamura, 1963), as well as molecular data (Rueness and Rueness 2000; Zuccarello et al., 2002). This suggests the need to confirm the actual distribution range of C. ustulatus in different regions, comparing with specimens from type locality through molecular analyses.
C. okamurae is native to the NW Pacific, including Japan, Korea, China, and Taiwan, and has recently been reported in many regions, including France (Zuccarello et al., 2002; Burel et al., 2019), Washington (Zuccarello et al., 2002), California (Miller, 2012; Hartnell College Genomics Group et al., 2020), Gulf of California (Norris et al., 2017), Italy (Petrocelli et al., 2020), and Spain (Bárbara et al., 2019). Recently, after molecular examinations, the name of C. ustulatus has been changed to C. okamurae in several region (Zuccarello et al., 2002; Petrocelli et al., 2020). C. okamurae forms a dense turf on substrates, thereby providing shelter for other macroalgae and marine invertebrates (Petrocelli et al., 2020). Another taxonomically accepted species, C. salifugus, was described based on sterile material growing in a semi-marine cavern in Queensland, Australia (Cribb, 1965), and has not been recorded since then. Other species, such as C. spinellus (Hooker f. Harvey) Kützing from New Zealand, C. indicus Weber-van Bosse from Indonesia, and C. rigidus Kützing from Senegal, were previously accepted within the genus. However, they have been regarded as synonyms of C. ustulatus because of their similar morphologies (Searles, 1968; West and Calumpong, 1990; Adams, 1994).
Mitochondrial DNA sequences generally constitute a powerful tool for species delimitation, particularly in red algal groups that are difficult to resolve using morphological data (Yang et al., 2021a; Muangmai et al., 2022; Preuss et al., 2022). The advent of species delimitation methods based on DNA sequences has provided a useful method for validating previously described species and identifying new evolutionary units (Muangmai et al., 2022; Boo et al., 2022). The present study involves Caulacanthus samples from 14 countries and addresses species diversity within the genus. We collected the specimens from Cádiz in Spain, which is type locality of C. ustulatus, to determine the regional distribution to which C. ustulatus was distributed.
In this study, using mitochondrial COI-5P and rbcL data with a previously determined rbcL‐S spacer, we addressed several questions concerning taxa regarded as being C. ustulatus and C. okamurae. Specifically, we aimed to 1) determine the number of species that exhibit the morphology of C. ustulatus, 2) investigate the present distribution pattern of the genus Caulacanthus, and 3) estimate the degree of genetic connectivity between the populations of Caulacanthus species from different regions.
Samples identified as Caulacanthus based on morphological criteria were collected during low tide from Korea, Japan, China, Taiwan, New Zealand, and Spain. The COI-5P sequences from Canada and Australia were provided by Prof. Gary W. Saunders of the University of New Brunswick. Additionally, we obtained sequences from France, the USA, the Philippines, Italy, Spain, Portugal, and Namibia from the GenBank to construct the phylogenetic trees for mitochondrial COI-5P and plastid rbcL and rbcL‐S spacer dataset. Details of the specimens and sampling details are listed in Table S1. For molecular analyses, specimens were air-dried and preserved in silica gel, and epibionts were then carefully removed under a dissecting microscope in a laboratory prior to DNA analysis. For anatomical observations, specimens were preserved in 5% formalin, and voucher specimens were deposited in the herbarium of Jeju National University (JNUB), Korea.
The generitype, C. ustulatus, from type locality was used to analyze the detailed morphology. Specimens were sectioned using a freezing microtome (MFS no 222; Nipon Optical Works, Tokyo, Japan). Sections were stained with 1% aniline blue acidified with 1% HCl and mounted in 35% corn syrup solution. Photomicrographs were taken using a BX43 microscope (Olympus, Tokyo, Japan) using an EOS 600D digital camera (Canon, Tokyo, Japan). Digitized images were edited to produce a plate using Adobe Photoshop software v. 6.1 (Adobe System Inc., San Jose, CA, USA).
Apical regions were separated from the thallus and extracted using the LaboPass Tissue Genomic DNA Isolation Kit (Cosmogenetech, Seoul, Korea), according to the manufacturer’s instructions. The extracted DNA was stored at ‐20°C and underwent polymerases chain reaction (PCR) amplification using AccuPower PCR Premix (Bioneer, Daejeon, Korea) in final volume of 20 μL. Targeted sequences of COI-5P and rbcL gene were amplified and sequenced using the primers GazF2 and GazR1 for COI-5P (Saunders, 2005; Lane et al., 2007), and F7–R898 and F762–R1442 for rbcL (Gavio and Fredericq, 2002; Kim et al., 2010). The PCR conditions were as described in Yang et al. (2021a). The PCR products were visually checked on a 1% agarose gel, purified using ExoSAP-IT (USB, Cleveland, Ohio, USA), and then commercially sequenced (Macrogen Inc., Seoul, Korea).
All successful amplifications were sequenced in both directions. Assembly and manual editing were conducted using Geneious v.9 (http://www.geneious.com). All edited sequences were combined with the available GenBank sequences. Multiple sequence alignment was performed using the MUSCLE algorithm in Geneious v.9. Catenella caespitosa was used as an outgroup.
Phylogenetic trees of the COI-5P gene were constructed using maximum likelihood (ML) and Bayesian inference (BI). ML analyses were performed using RAxML with 1,000 bootstrap replicates, and BI was performed with MrBayes (v.3.2.1) (Ronquist et al., 2012) using the Metropolis-coupled Markov Chain Monte Carlo (MCMC) with the models. Four million generations of two independent runs were performed with four chains and sampling trees for every 100 generations. The burn-in period was graphically identified by tracking the likelihoods in each generation to determine whether whey reached a plateau. Further, 25% of the saved trees was removed, and the remaining trees were used to calculate the Bayesian posterior probabilities (BPPs). For comparison with sequences from GenBank, we generated the combined phylogenetic tree using the sequences of rbcL and rbcL‐S spacer including the representative samples of each lineage on the COI-5P based phylogeny.
To estimate the genetic diversity of Caulacanthus species, we calculated the number of polymorphic sites (S), number of haplotypes (H), haplotype diversity (Hd) and nucleotide diversity (π) for COI-5P data using Arlequin v.3.5 (Excoffier and Lischer, 2010). Haplotype networks were built using the TCS method (Clement et al., 2000) in Arlequin. Genetic distances based on COI-5P were estimated using MEGA X v. 10.2.6 (Kumar et al., 2018). Pairwise fixation indices (FST) were calculated using Arlequin, to measure genetic differentiation among biogeographic regions within the lineage to assign the species as C. okamurae.
Species delimitation in the genus Caulacanthus was studied via the single and multiple-threshold generalized mixed Yule coalescent methods (GMYC; Pons et al., 2006; Fujisawa and Barraclough, 2013), automatic barcode gap discovery analysis (ABGD; Puillandre et al., 2012), and a Bayesian implementation of the Poisson tree processes (bPTP; Zhang et al., 2013). The GMYC method requires an ultrametric tree, which was obtained by Bayesian analyses in BEAST v.1.10.4 (Suchard et al., 2018), with divergence times estimated under an uncorrelated lognormal relaxed molecular clock model (Drummond et al., 2012) and the Yule-process as the prior tree. The Bayesian Markov chain Monte Carlo (MCMC) was run for 20 million generations, with trees and parameters were sampled every 100 generations. The output was checked for convergence using Tracer v.1.7.1 (Rambaut et al., 2018). After removing 25% of the trees as burn-in, the remaining trees were used to generate a single summarized tree in TreeAnnotator v.1.10.4 (part of the BEAST v.1.10.4 package) as an input file for GMYC analyses. GMYC analyses with a single threshold model were performed in R software (R Development Core Team, http://www.R-project.org) under the “splits” package using the “gmyc” function (R-Forge, http://r-forge.r-project.org/projects/splits/). The ABGD method was conducted via a webserver (ABGD web, https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html) under the default parameters, except for the related gap width (X) of 1.3, using the Kimura-2-parameter (K2P) distance matrix as the input file, generated in MEGA7. The bPTP analysis was performed using a bifurcated phylogenetic input tree, as implemented in the bPTP web server (https://species.h-its.org). The parameters were set as follows: 500,000 MCMC generations, thinning by a factor of 100, and a 10% burn-in.
The analysis of the mitochondrial COI-5P gene (552 bp) across 94 Caulacanthus sequences revealed 171 polymorphic sites, of which 139 were parsimony-informative sites. In total, 26 haplotypes were identified, of which 14 were by at least two samples and 12 were singletons. The average nucleotide compositions were as follows: A, 27.2%; C, 15.4%; G, 25.2%; and T, 32.2%. The total genetic diversity within a genus was 0.8518 ± 0.0296 for haplotype diversity and 0.05962 ± 0.0290 for nucleotide diversity.
Both ML and BI analyses for COI-5P data produced an identical phylogenetic topology (Figure 1) that was divided into six distinct lineages (Lineages 1‐6), which were strongly supported. Three different species delimitation methods suggested the existence of multiple cryptic Caulacanthus species, and these results were highly congruent with phylogenetic analyses (Figure 1). Both the GMYC and ABGD models defined six operational taxonomic units (OTUs), whereas bPTP estimated eight OTUs within the genus Caulacanthus (Figure 1). We recognized C. okamurae for Lineage 1 according to the phylogenetic lineage, which included samples from its native region, Japan, Korea, and China. The name C. ustulatus was assigned to the Lineage 2 based on the inclusion of the specimens from Cádiz in Spain, which is the type locality.
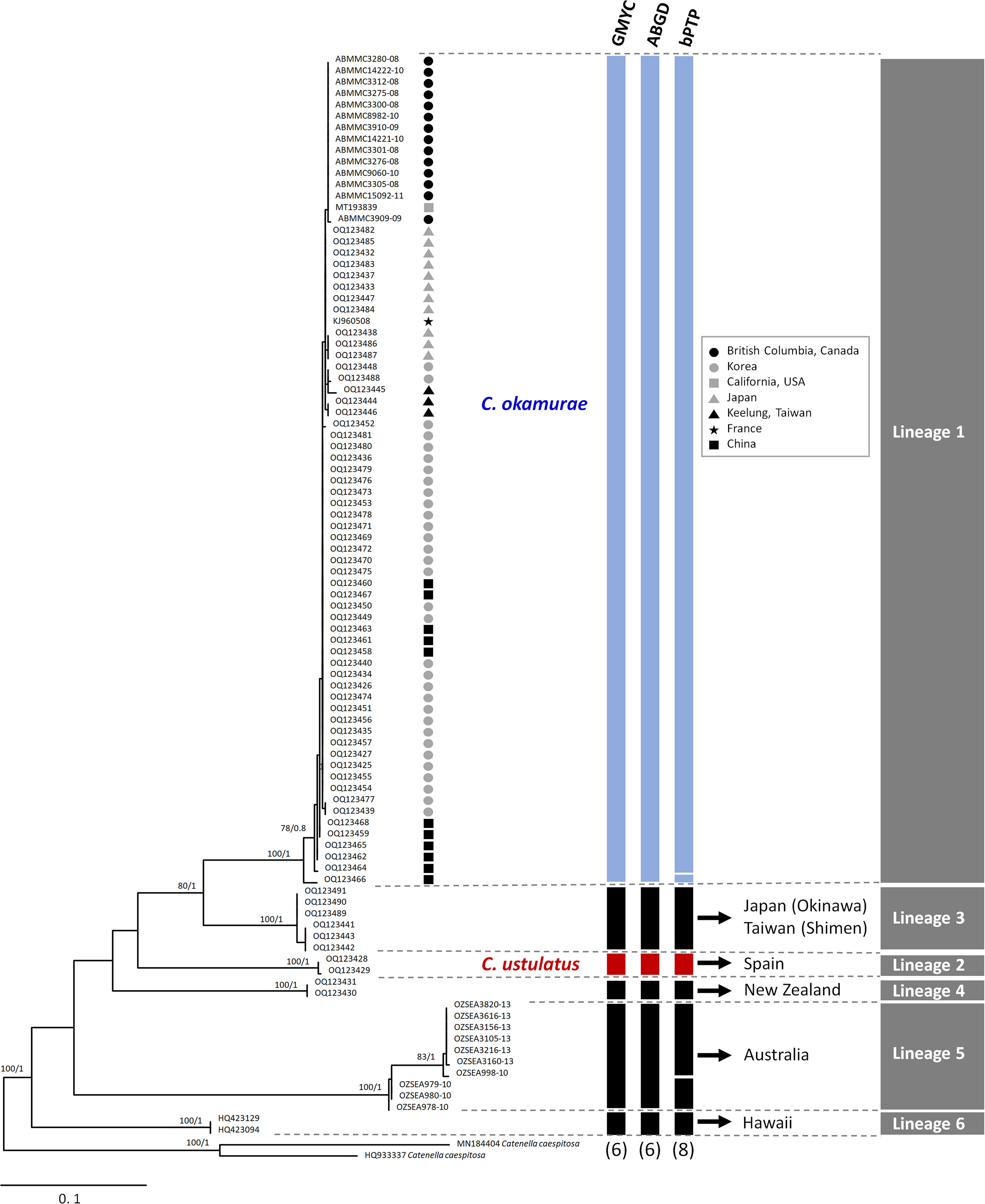
Figure 1 Maximum likelihood phylogeny of the genus Caulacanthus based on the COI-5P gene. ML bootstrap values (left) and Bayesian posterior probabilities (right) are indicated at the nodes. Values of <50% ML bootstrap and <0.8 posterior probabilities are not shown. The results of three species delimitation methods (GMYC, ABGD, and bPTP) for COI-5P gene are shown in the columns at the edge of the tree, with the number of putative species depicted at the bottom. Blue and red columns indicate Caulacanthus okamurae and C. ustulatus, respectively. Black columns indicate other lineages of Caulacanthus. Gray columns on right edge summarize the results for the tested species delimitation methods.
The molecular phylogenetic analysis using the rbcL and rbcL‐S spacer included overlapping portion (60–82 bp) for 24 Caulacanthus sequences, and excluded the Lineages 5–6, which were with only COI-5P sequences. The plastid phylogenetic tree was resolved into five lineages (Figure 2), four of which corresponded with the previously determined lineages in the study using rbcL-S spacer (Zuccarello et al., 2002). C. okamurae (Lineage 1) included sequences from the NW Pacific (Korea, Japan, China, and Taiwan), NE Pacific (the USA) and NE Atlantic (France and Spain) oceans, and Mediterranean Sea (Italy). The sequences from Cádiz (OQ136657 and OQ136658) generated in this study, referred as C. ustulatus (Lineage 2), were grouped with sequences from Portugal, Spain, and Namibia. Additionally, we unexpectedly found that Lineage 3 matched the two sequences (AF453721 and AF453720) from Queensland, Australia. Lineage 4 was placed at the most basal position in this phylogenetic tree. The independent Lineage 7 from the Philippines was a sister group to the Lineage 1, C. okamurae.
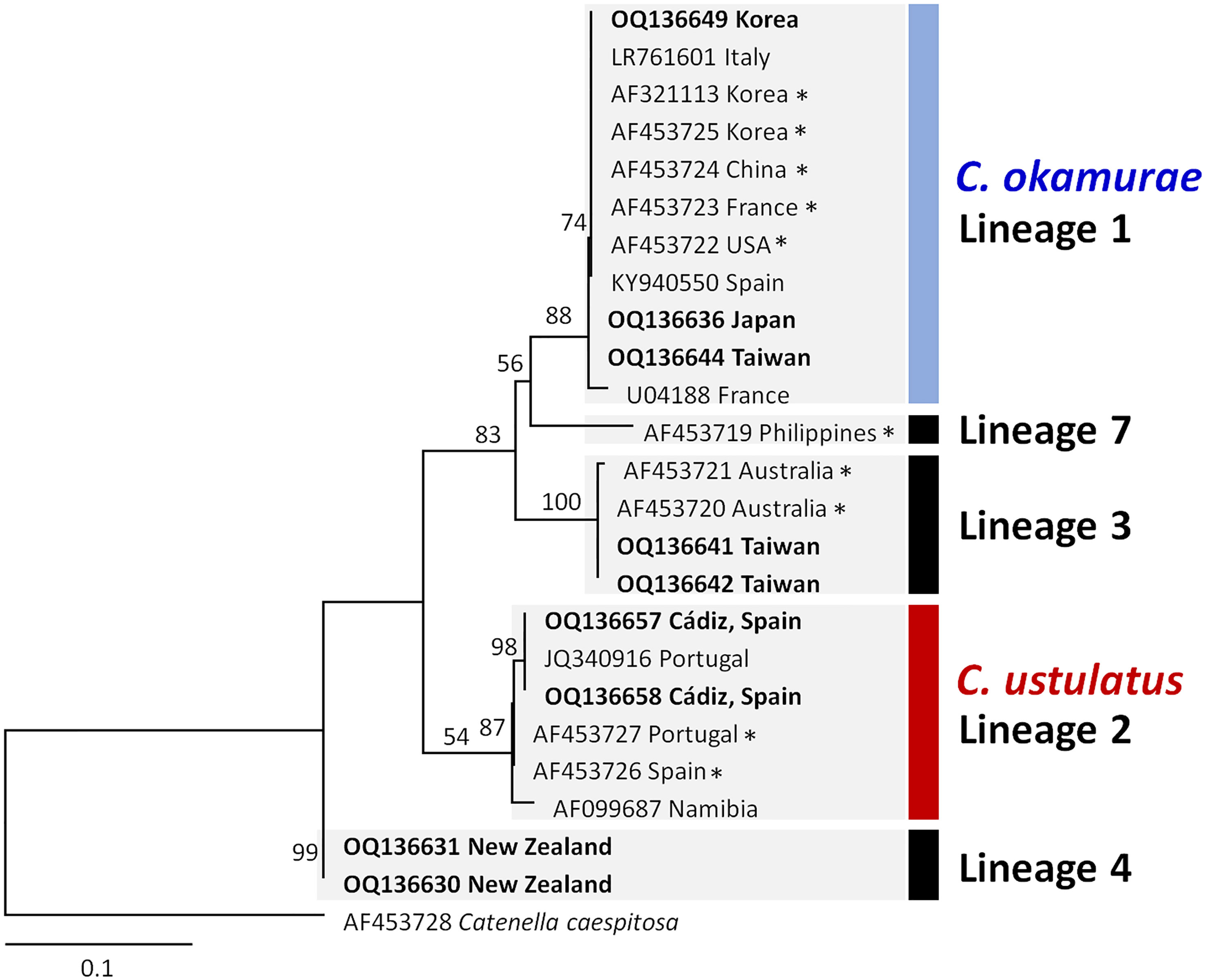
Figure 2 Maximum likelihood phylogeny of the genus Caulacanthus based on the combined sequences of rbcL and rbcL-S spacer. ML bootstrap values are indicated at the nodes. Numbering of lineages was applied as in Figure 1. Asterisk (*) behind the sequence name indicates the rbcL-S sequences downloaded from the GenBank. Bold type indicates the sequences generated in this study.
The TCS haplotype network analysis based on the COI-5P data (Figure 3A) revealed six major groups corresponding to the six COI-5P based phylogenetic lineages and the GMYC/ABGD based OTUs. Each lineage was separated from the others by 52–86 missing steps. Among them, C. okamurae (Lineage 1) showed the highest diversity (accounting for 78% of the total sample diversity), and relatively complex haplotype network with 15 haplotypes. Within C. okamurae, the most abundant haplotype H04 and its connected haplotypes, H05–H08, occurred in Korea and China. Haplotypes H01–H02 were only observed in the populations of the NE Pacific Ocean (British Columbia and California; Table S1). The central haplotype H03 and H13 comprised specimens from Japan and France. The singletons of H10–H11 and H14–H15 occurred in Korea and China, respectively, whereas H09 and H12 were observed only from Taiwan. C. ustulatus (Lineage 2) had two haplotypes from Cádiz and was connected to Lineage 3, which comprised two haplotypes that represented Japan (Okinawa) and Taiwan (Shimen) by 71 mutational steps. The other three lineages were confined to a single country, such as New Zealand (Lineage 4; one haplotype), Australia (Lineage 5; five haplotypes), and Hawaii (Lineage 6; one haplotype). A clear gap between the intra-lineage and inter-lineage p-distances was detected for the COI-5P gene (Figure 3B). The minimum inter-lineage divergence of 9.4% (between Lineages 1 and 3) was considerably higher than the maximum intra-lineage divergence of 3.62% (Lineage 5; Table 1), indicating that each lineage was genetically distinct.
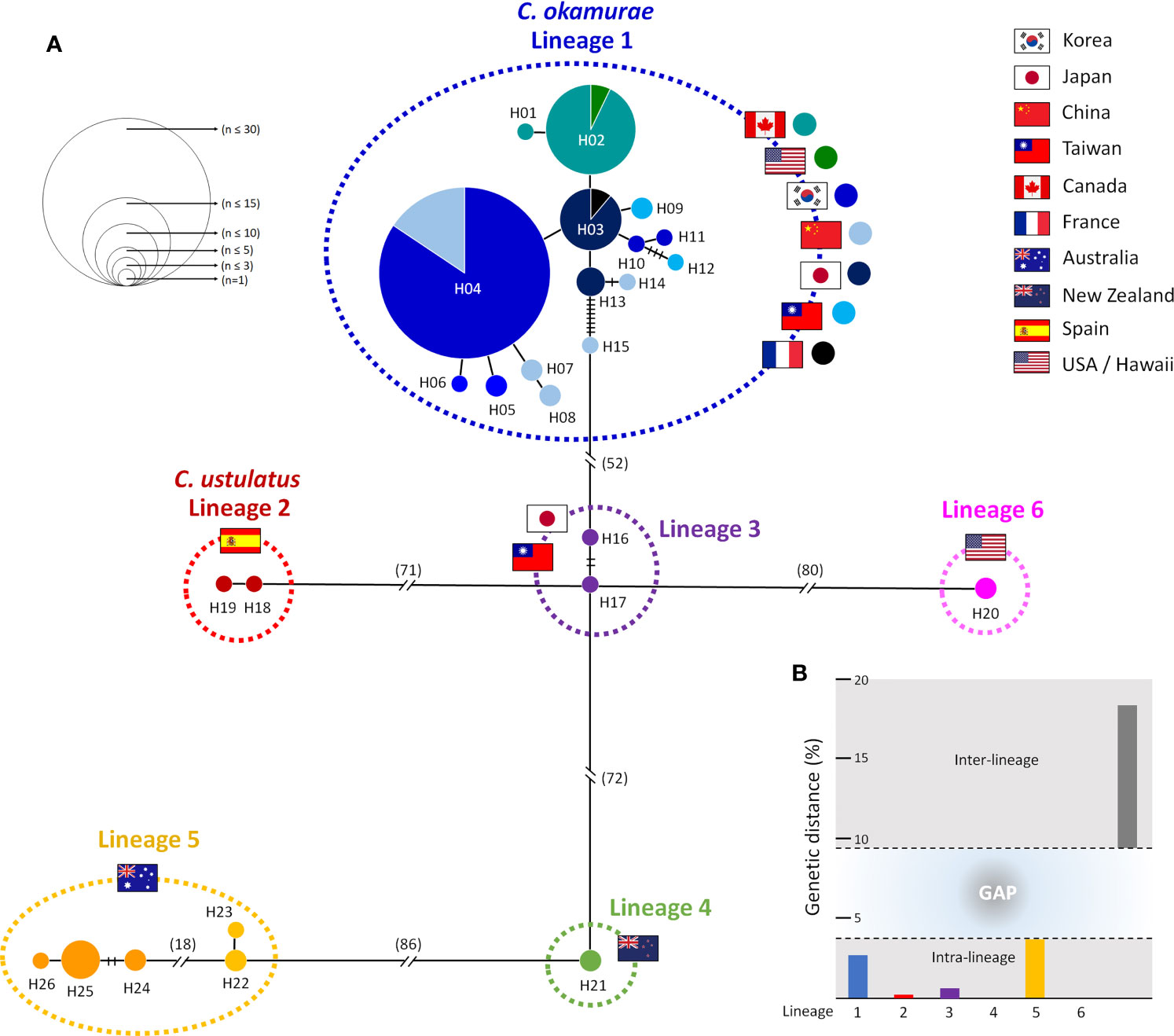
Figure 3 Haplotype network and genetic distance for six lineages of Caulacanthus using the COI-5P sequences. In the network (A), each circle denotes a single haplotype, and each connecting line indicates one mutation step between haplotypes. The size of the circles is proportional to haplotype frequency. The pie chart colors within Lineage 1 correspond to the matching colors next to the flag. The cross bars represent the number of mutational steps between two haplotypes when the mutational steps are >1. Flags indicate the country of origin of specimens for each lineage. Minimum and maximum inter-lineage (gray), and intra-lineage (colors refer to lineages defined in the haplotype network) genetic distances (p-distance) in percent are shown in (B).
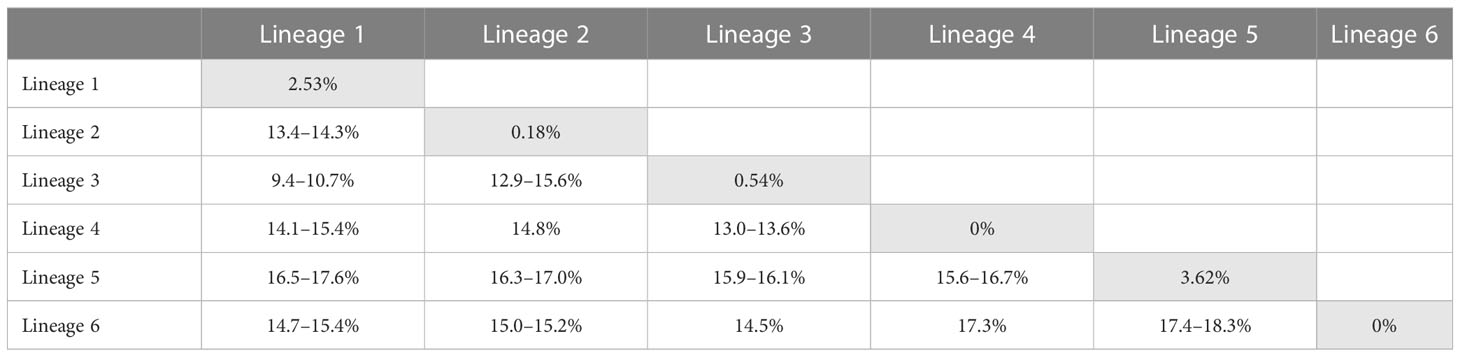
Table 1 Intra- (gray) and inter-lineage p-distances of Caulacanthus calculated using the COI-5P gene.
Pairwise FST values were calculated only for C. okamurae based on the COI-5P data (Table 2), as the other lineages had only up to ten samples collected from one or two locations. For C. okamurae, we divided into the following five populations, excluding the French sample, which contained only one specimen; (1) Pop1 (n = 16) included W Korea and Dailan, (2) Pop2 (n = 20) from southern (S) Korea and eastern (E) Korea, (3) Pop3 (n = 8) from the coasts from Nanji Island and Taiwan, (4) Pop4 (n = 11) from the coast of mainland Japan, and (5) Pop5 (n = 15) from the NE Pacific coast. Among the five populations, low FST value (FST range = 0.0349) was observed within the populations from Korea and Dailan (Pop1/Pop2) populations. The mainland Japan population (Pop4) showed genetic affinity (FST range = 0.1412) to those along the East China Sea (Pop3) rather than to the populations of Korea and Dailan (Pop1/Pop2; FST range = 0.6140–0.6887). Moderate to high FST values (FST range = 0.4655–0.8646) were detected between populations from the NE Pacific Ocean (Pop5) and others (Pop1/Pop2/Pop3/Pop4).
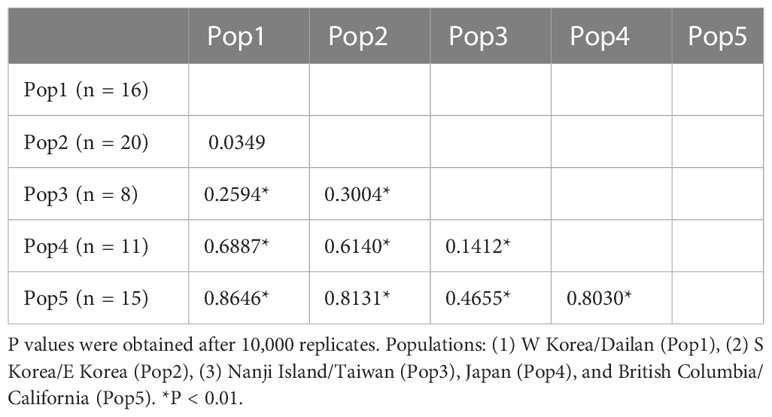
Table 2 Pairwise estimates of FST between five groups in C. okamurae based on COI-5P data.
In the global distribution of the genus Caulacanthus based on the combined result of mitochondrial and plastid data (Figure 4), C. okamurae was most widespread species, found in the NW Pacific (Korea, Japan, China, and Taiwan), NE Pacific (Canada and the USA), and NE Atlantic (Spain and France) oceans and Mediterranean Sea (Italy). In contrast to this result, C. ustulatus was distributed only in the NE Atlantic Ocean (Spain and Portugal) and Namibia. In the NW Pacific, two Caulacanthus species (C. okamurae and Lineage 3) co-occurred with partial geographic division, indicating that the Lineage 3 was distributed from subtropical to tropical region, including Okinawa, Keelung, Cooktown, and Magnetic Island (Figure 4). Another Lineages 4–7 were found only in a single region, whereas Lineage 5 occurred in the SE Indian and SW Pacific oceans. On examining the detailed distribution of C. okamurae within the NW Pacific Ocean based on the COI-5P haplotype (Figure 5), the haplotypes from mainland Japan were distinct to those from Korea/China; however, a part of haplotype from Nanji Island were connected to those from Korea.
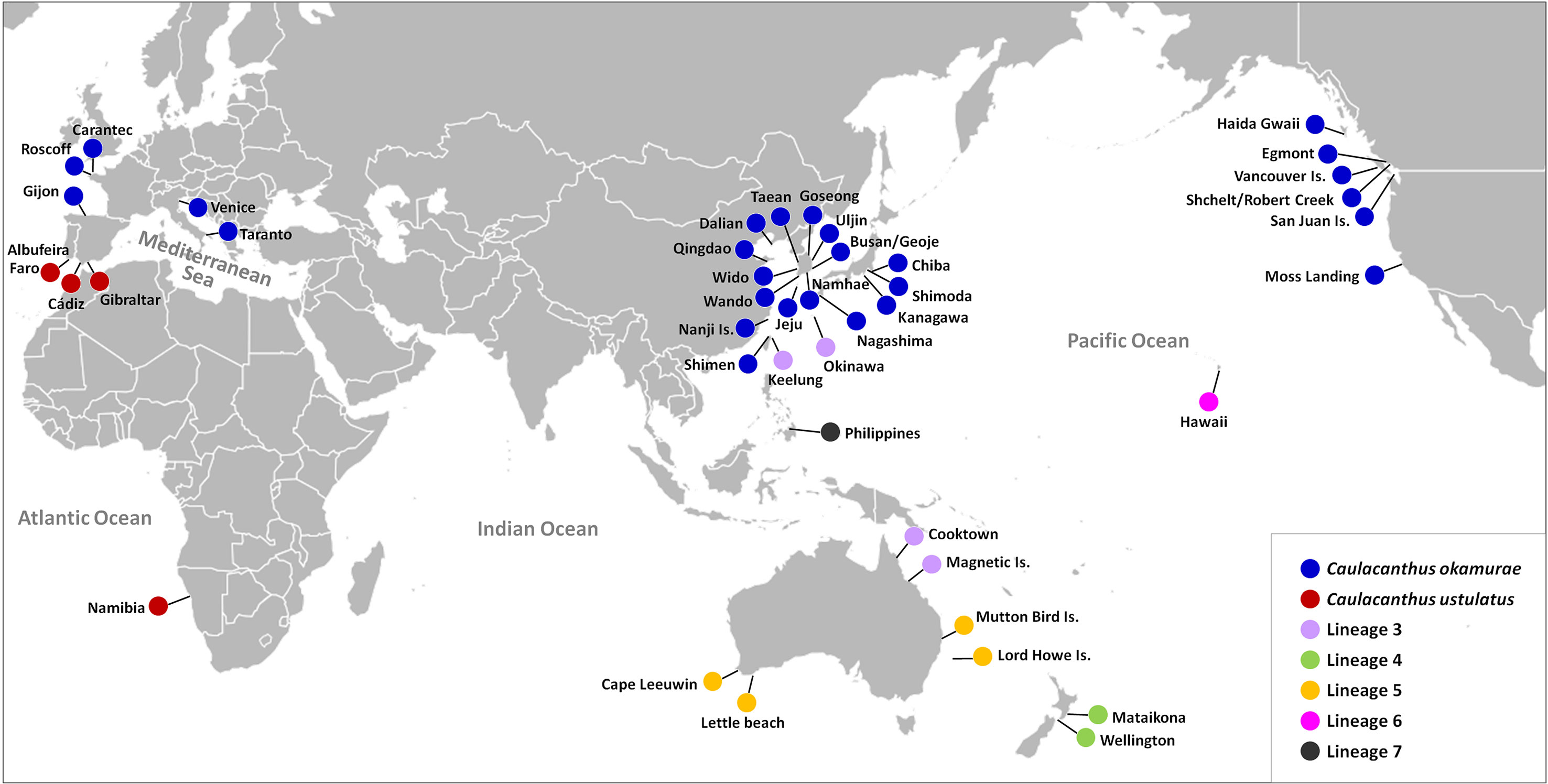
Figure 4 Map showing updated global distribution of seven Caulacanthus lineages in the world according to the combined result of mitochondrial and plastid sequence data.
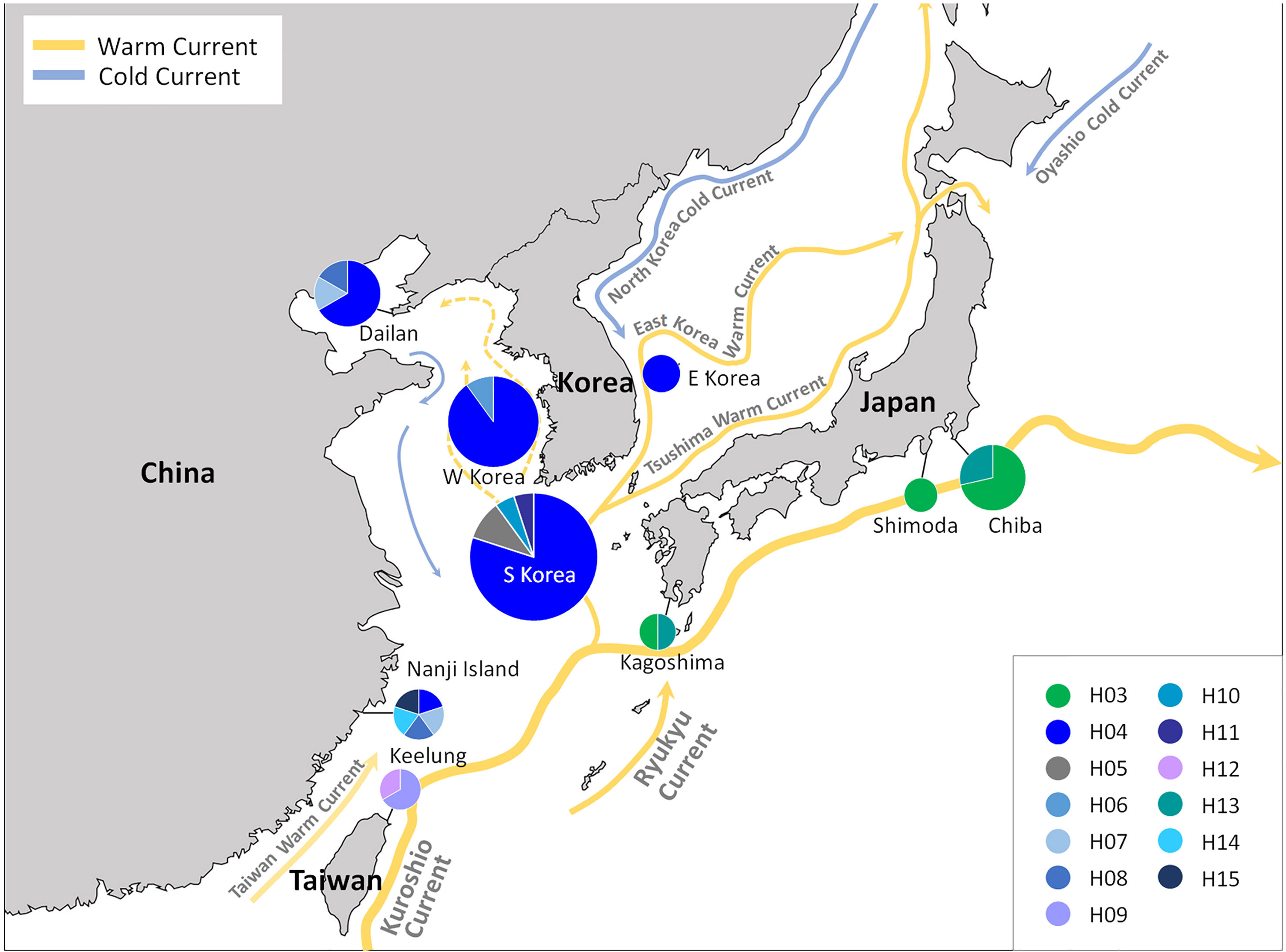
Figure 5 COI-5P haplotype distribution of Caulacanthus distributed in the NW Pacific Ocean. C. okamurae occurs in Korea, Japan, China, and Taiwan, and Lineage 3 is distributed only in Shimen and Okinawa in this region. Each pie chart represents the proportion of the haplotype presented within the site.
The detailed morphology of specimens collected from Cádiz, where is the type locality of the generitype, C. ustulatus, was observed. Thalli of C. ustulatus were erect or creeping, entangled, cartilaginous, 10–35 mm in height, and dark brown to dark red (Figures 6A–C). Branches were irregular to sub-dichotomous with short branchlets, divided at right angles or slightly curved toward the apex, and tapering to points (Figures 6B, C). Small projections were attached to the substratum or adjacent branches (Figure 6C). On a surface view, cortical cells of a thallus exhibited various from circular to polygonal (Figure 6D). The thallus was uniaxial (Figure 6E) with siphonous axis articulated and divided dichotomously toward the thallus surface (Figure 6F). The main axes were cylindrical and 350–390 μm in diameter (Figure 6G). In the cross-sectional view, the central core of axial filament was surrounded by 3–4 large and polygonal cells with 50–85 μm in diameter (Figures 6G, H). The cortex comprised mainly two layered ovoid cells (Figure 6H). Tetrasporangia formed in the basal to mid part of the branchlets (Figure 6I). Mature zonately divided tetrasporangia were 22–28 μm in diameter and 48–56 μm in length (Figure 6J).
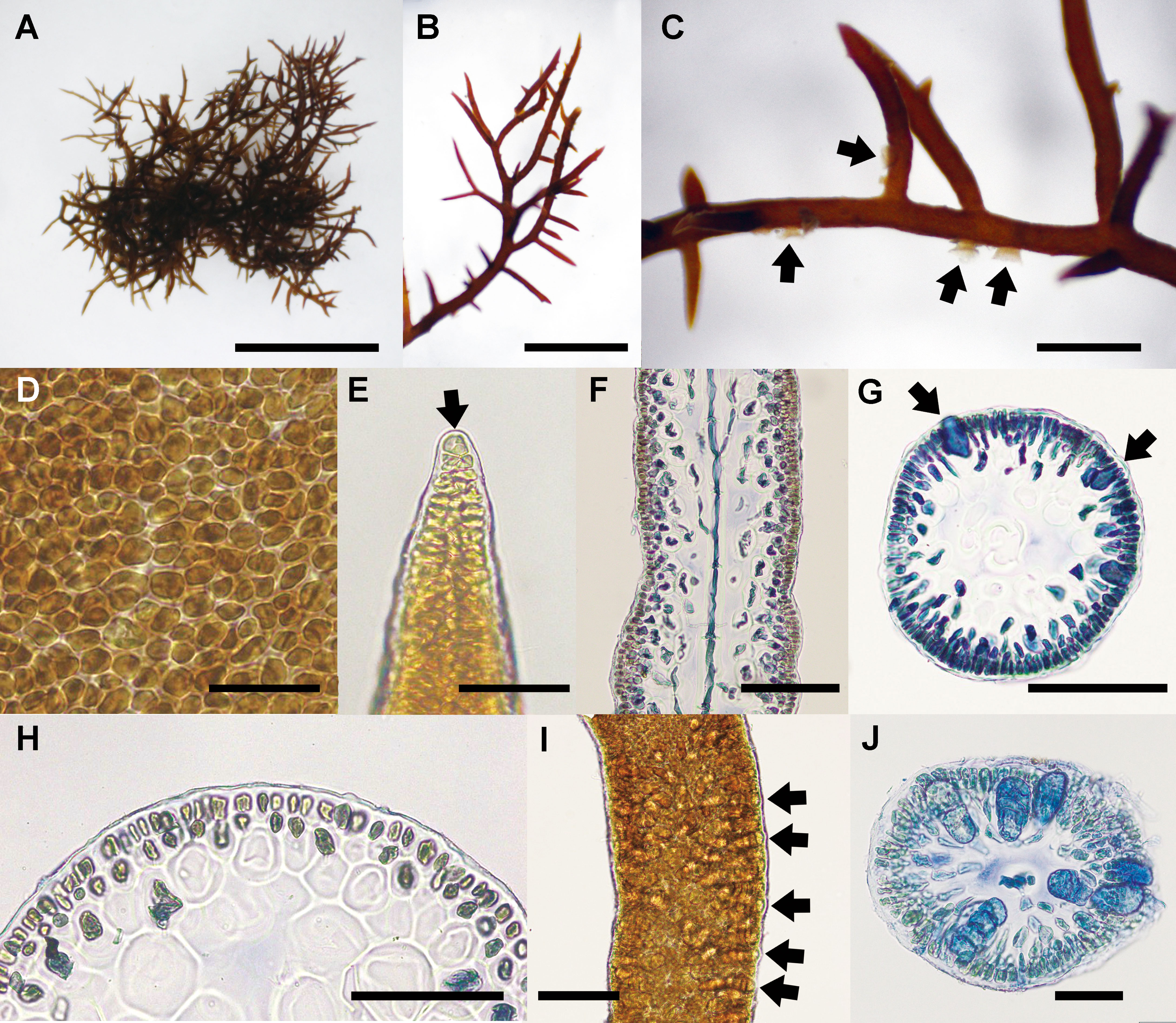
Figure 6 Caulacanthus ustulatus from Cádiz, Spain. (A) Habit of thallus collected at San Sebastian, Cádiz, on April 28, 2014. Scale bar = 300 μm. (B) Apical part of branches with numerous short branchlets. Scale bar = 200 μm. (C) Several projections (arrows) on a branch. Scale bar = 100 μm. (D) Surface view of a thallus showing circular to polygonal cortical cells. Scale bar = 30 μm. (E) Apical cell (arrow) of a thallus tip. Scale bar = 50 μm. (F) Longitudinal section of a thallus median part showing an axial filament. Scale bar = 100 μm. (G) Cross section of distal portion of tetrasporophytic thallus showing immature tetrasporangia (arrows). Scale bar = 100 μm. (H) Layer of ovoid cortical cells. Scale bar = 30 μm. (I) Tetrasporangia of a branchlet. Scale bar = 50 μm. (J) Zonately divided tetrasporangia (arrows) in the cortex. Scale bar = 50 μm.
We suggest at least seven OTUs within the genus Caulacanthus, through a combination of phylogenetic analyses using the mitochondrial and plastid sequence data, molecular species delimitation methods, and haplotype network, although bPTP resolved two additional OTUs within the Lineages 1 and 6 (Figures 1, 2). The finding of cryptic species diversity was similar to that for other red algal taxa, particularly turf algae, which has typically been overlooked because of their small size, structural simplicity, and densely entangled nature (Díaz-Tapia et al., 2020). Together with a detailed morphological description, our study provides molecular evidence that the generitype C. ustulatus is not a species with a widespread distribution but rather confined to the E Atlantic Ocean (Figures 5, 6). Another previously recognized species, C. okamurae, was found in the Northern Hemisphere, including nine countries. This finding was consistent in terms of previous studies that reported that C. okamurae was introduced from several countries (Petrocelli et al., 2020). The other five lineages were distributed in Japan/Taiwan/Australia (Lineage 3), New Zealand (Lineage 4), Australia (Lineage 5), Hawaii (Lineage 6), and the Philippines (Lineage 7) (Figure 4).
The COI-5P haplotype network of Caulacanthus clearly showed deep genetic divergence with considerable mutational steps between the lineages (Figure 3). The pairwise level of sequence divergence reported herein provided a clear gap between maximum intra-lineage divergence of 3.62% and minimum inter-lineage divergence of 9.4%, which provided the evidence for an independent genetic lineage. The high level of divergence within the Caulacanthus complex is indicative of not only a strong geographical subdivision but also a long temporal discontinuity (Fraser et al., 2010). Deep genetic divergences detected among the highly divergent lineage requires taxonomic validation and formal description through sufficient observations of regional specimens. In the case of Australia and New Zealand, some species names have previously been misattributed owing to morphological similarity within the region (Cribb, 1965; Searles, 1968; Rueness, 1997). C. salifugus was first reported in Paradise Cave in the SE Queensland (Cribb, 1965), in the middle of distribution between the Lineages 3 and 5, therefore, molecular evidence is acquired to confirm its accurate taxonomic position. C. spinellus was originally described in New Zealand, after it was reduced to synonymy with C. ustulatus. In addition, further studies are needed in other regions, such as Indonesia and Senegal, where synonyms of C. ustulatus have been reported (Searles, 1968; West and Calumpong, 1990; Rueness, 1997).
Based on molecular evidence, C. okamurae showed broad trans-oceanic distribution in the Northern Hemisphere (including Korea, Japan, China, Taiwan, France, Spain, Italy, Canada, and the USA) (Figure 4). C. okamurae has been typically misidentified as C. ustulatus because of its resemblance to the gross morphology (Petrocelli et al., 2020). As the distribution of two different species, C. ustulatus and C. okamurae, has been identified in Europe, molecular screening of Caulacanthus specimens is required to assess their detail distribution. Moreover, our study provides the first molecular evidence of C. okamurae in British Columbia, Canada, where two haplotypes (H01–H02) were found (Figure 2). Populations from the NE Pacific Ocean with two haplotypes could serve as evidence that this population had been introduced from the NW Pacific Ocean earlier than the population from the NE Atlantic Ocean. The lack of shared haplotypes between the NW and NE Pacific oceans also supported this hypothesis. Non-native species have been increasingly reported in many regions, facilitated by both natural and anthropogenic stressors (Nelson et al., 2021). Climate change can be predicted to affect species range by encouraging the settlement of temperate non-native species (Miller et al., 2011). Drastic temperature shifts, including the series of El Niño/La Niña-Southern Oscillation events, along with increased sea temperature could have favored the settlement and spreading of Caulacanthus in several areas (Miller et al., 2011). Moreover, the spread of non-native seaweed C. okamurae could have facilitated increased abundance and diversity of other seaweeds and invertebrates by forming novel turf in intertidal zones (Smith et al., 2014; Petrocelli et al., 2020).
In the NW Pacific, two Caulacanthus species were found, namely C. okamurae and Lineage 3, of which Lineage 3 was found from tropical to subtropical regions, including Shimen and Okinawa, Cooktown, and Magnetic Island (Figure 4). Populations in the NW Pacific Ocean might be geographically separated by restricted gene flow. For example, the haplotypes from Korea/China were not shared with that from the Pacific coast of mainland Japan affected by Kuroshio Current, and this genetic division was supported by high FST values (Table 2). The distribution of the NW Pacific haplotypes is associated with ocean currents, which might mediate gene flow between populations. The gene flow by ocean currents was also shown in the distribution of haplotype H04, which occurred from Nanji Island to the coasts of Korea and Dailan (Figure 5). The occurrence of H04 in the entire coast of Korea may be the result of high gene flow between the Korean populations due to complex oceanic currents, which has also been observed in red algal species of the genus Chondrus Stackhouse (Yang and Kim, 2022). The influence of ocean currents affects population distribution may be confirmed in future studies including populations along the coasts where the Tsushima Warm Current passes.
The distribution of the seven Caulacanthus species provides information about the processes that have shaped the evolutionary history of this group, which were previously obscured by treating it as a single species. All lineages included a well-supported group with a deep split from each other, indicating that speciation occurred on an ocean basin scale. Therefore, our study provides evidence of a greater evolutionary independence among the populations of Caulacanthus, as measured by mtDNA divergence. Based on our phylogenetic reconstructions, we interpreted that the Caulacanthus species complex have undergone a series of allopatric diversification events (Derycke et al., 2008), showing considerable genetic divergence in the resulting population isolated by distance (Díaz-Tapia et al., 2020). The case of two subclades with genetic divergence of 3.62% within Lineage 5, that is, populations between the western and east coast of Australia, establishes genetic divergence by distance.
Accurately recognizing species boundaries is key to biodiversity research and conservation, as species are operational units for biodiversity quantification and management (Boo et al., 2019; Yang et al., 2020; Preuss et al., 2022). Our results revealed seven Caulacanthus lineages, most of which had limited geographic distribution. The lack of gene flow among lineages suggests that they have attained geographic isolation and may deserve to be assigned as species (Fraser et al., 2010). Five lineages would require validation and formal description. Additional studies should involve an integrative approach that considers other characteristics, such as morphology and ecology. Although the sampling in this study may not be sufficiently large to conclusively rule out the presence of lineages in an area, which was not sampled, the pattern emerging from our data showed seven lineages with regional distributions rather than a single cosmopolitan species. Further studies on the evolutionary relationships and genetic variation of regional populations, along with population collection from other countries, are essential to understand and effectively conserve and manage species of the genus Caulacanthus.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MYY and MSK conceived and designed the study and performed the collections. MYY performed laboratory work and the data analyses. MYY and MSK participated in the interpretation of the data and writing the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Basic Science Research Program (2019R1A6A1A10072987 and 2020R1I1A2069706) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education of Korea.
We are sincerely grateful to Dr. Gary W. Saunders and Dr. Zhong-Min Sun for collecting samples from Canada, Australia, and China, and Dr, Jeong Chan Kang and Dr. Hyung Woo Lee of the molecular phylogeny team of the Marine Algae Laboratory at Jeju National University. We also thank Dr. Wendy A. Nelson and Dr. Ricardo Bermejo Lacida for their assistance during samplings in New Zealand and Spain.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1087507/full#supplementary-material
Adams N. M. (1994). Seaweeds of New Zealand. An Illustrated guide. (Christchurch: Canterbury University Press).
Avis J. C. (1998). Conservation genetics in the marine realm. J. Hered. 89, 377–382. doi: 10.1093/jhered/89.5.377
Bárbara I., García-Redondo V., Díaz Tapia P., García-Fernández A., Piñeiro-Corbeira C., Peña V., et al. (2019). Adiciones y correcciones a la flora bentónica marina del atlántico ibérico norte. Acta Bot. Malacit. 44, 51–60. doi: 10.24310/abm.v44i0.5750
Boo G. H., Leliaert F., Le Gall L., Coppejans E., De Clerck O., Nguyen T. V., et al. (2022). Ancient tethyan vicariance and long-distance dispersal drive global diversification and cryptic speciation in the red seaweed Pterocladiella. Front. Plant Sci. 13, 849476. doi: 10.3389/fpls.2022.849476
Boo G. H., Qiu Y.-X., Kin J. Y., Bosch S., De Clerck O., He P., et al. (2019). Contrasting patterns of genetic structure and phylogeography in the marine agarophytes Gelidiophycus divaricatus and G. freshwateri (Gelidiales, rhodophyta) from East Asia. J. Phycol. 55, 1319–1334. doi: 10.1111/jpy.12910
Burel T., Le Duff M., Ar Gall E. (2019). Updated check-list of the seaweeds of the French coasts, channel and Atlantic ocean. Aod Les Cahiers Naturalistes L’Observatoire Marin 7, 1–38.
Clement M., Posada D., Crandall K. A. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Cribb A. B. (1965). An ecological and taxonomic account of the algae of a semi-marine cavern, paradise cave, Queensland. Papers Dept. Bot. Univ. Queensland 4, 259–282.
Derycke S., Remerie T., Backeljau T., Vierstaraete A., Vanfleteren J., Vincs M., et al. (2008). Phylogeography of the Rhabditis (Pellioditis) marina species complex: evidence for long-distance dispersal, and for range expansions and restricted gene flow in the northeast Atlantic. Mol. Ecol. 17, 3306–3322. doi: 10.1111/j.1365-294X.2008.03846.x
Díaz-Tapia P., Ly M., Verbruggen H. (2020). Extensive cryptic diversity in the widely distributed Polysiphonia scopulorum (Rhodomelaceae, rhodophyta): Molecular species delimitation and morphometric analyses. Mol. Phylo Evol. 152, 106909. doi: 10.1016/j.ympev.2020.106909
Díaz-Tapia P., Maggs C. A., Macaya E. A., Verbruggen H. (2018). Widely distributed red algae often represent hidden introductions, complexes of cryptic species or species with strong phylogeographic structure. J. Phycol. 54, 829–839. doi: 10.1111/jpy.12778
Drummond A. J., Suchard M. A., Xie D., Rambaut A. (2012). Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Excoffier L., Lischer H. E. L. (2010). Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fujisawa T, Barraclough TG (2013). Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol 62:707–24.
Fraser C. I., Thiel M., Spencer H. G., Waters J. M. (2010). Contemporary habitat discontinuity and historic glacial ice drive genetic divergence in Chilean kelp. BMC Evol. Biol. 10, 203. doi: 10.1186/1471-2148-10-203
Fraser C. I., Zuccarello G. C., Spencer H. G., Salvatore L. C., Garcia G. R., Waters J. M. (2013). Genetic affinities between trans-oceanic populations of non-buoyant macroalgae in the high latitudes of the southern hemisphere. PloS One 8, e69138. doi: 10.1371/journal.pone.0069138
Gavio B, Fredericq S (2002). Grateloupia turuturu (Halymeniaceae, rhodophyta) is the correct name of the non-native species in the Atlantic known as Grateloupia doryphora. Eur J Phycol. 37:349–59.
Guiry M. D., Guiry G. M. (2023) Algaebase (Galway: World-wide electronic publication, National University of Ireland). Available at: https://www.algaebase.org (Accessed March 20, 2023).
Hartnell College Genomics Group, Aguilar A., Ahumada T. J., Amezcua Moreno N., Bohn J., Bustamante D. E., et al. (2020). The complete mitochondrial and plastid genomes of the invasive marine red alga Caulacanthus okamurae (Caulacanthaceae, rhodophyta) from moss landing, California, USA. Mitochondrial DNA Part B. 5, 2067–2069. doi: 10.1080/23802359.2020.1763870
Kamura S. (1963). On the tetraspore-germination of Caulacanthus okamurae yamada. Bull. Arts. Sci. Div. Ryukyu Univ. 6, 73–79.
Kang J. C., Lin S.-M., Miller K. A., Kim M. S. (2021). Taxonomic revision of hook-forming Acrosorium (Delesseriaceae, rhodophyta) from the northwestern pacific based on morphology and molecular data. Plants 10, 2269. doi: 10.3390/plants10112269
Kim MS, Kim SY, Nelson W (2010). Symphyocladia lithophila sp. nov. (Rhodomelaceae, ceramiales), a new Korean red algal species based on morphology and rbcL sequences. Bot Mar 53:233–41.
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lagourgue L., Gobin S., Brisset M., Vandenberghe S., Benneville C., Jauffrais T., et al. (2022). Ten new species of Ulva (Ulvophyceae, chlorophyta) discovered in new Caledonia: genetic and morphological diversity, and bloom potential. Eur. J. Phycol. 57, 458–478. doi: 10.1080/09670262.2022.2027023
Lane C. E., Lindstrom S. C., Saunders G. W. (2007). A molecular assessment of northeast pacific Alaria species (Laminariales, phaeophyceae) with reference to the utility of DNA barcoding. Mol. Phylo Evol. 44, 634–648. doi: 10.1016/j.ympev.2007.03.016
Leliaert F., Payo D. A., Gurgel C. F. D., Schils T., Draisma S. G. A., Saunders G. W., et al. (2018). Patterns and drivers of species diversity in the indo-pacific red seaweed Portieria. J. Biogeogra. 45, 2299–2313. doi: 10.1111/jbi.13410
Miller K. A. (2012). Seaweeds of california. updates of California seaweed species list (Berkeley: University of California Jepson Herbarium), 1–59.
Miller K. A., Aguilar-Rosas L. E., Pedroche F. F. (2011). A review of non-native seaweeds from California, USA and Baja California, Mexico. Hydrobiologica 21, 365–379.
Muangmai N., Preuss M., West J. A., Zuccarello G. C. (2022). Cryptic diversity and phylogeographic patterns of the Bostrychia intricata species complex (Rhodomelaceae, rhodophyta) along the coast of southeastern Australia. Phycologia 61, 27–36. doi: 10.1080/00318884.2021.1994768
Nauer F., Gurgel C. F. D., Ayres-Ostrock L. M., Plastino E. M., Oliveira M. C. (2019). Phylogeography of the Hypnea musciformis species complex (Gigartinales, rhodophyta) with the recognition of cryptic species in the western Atlantic ocean. J. Phycol. 55, 767–687. doi: 10.1111/jpy.12848
Nelson W. A., D’Archino R., Neill K. F., Robinson N. M. (2021). Introduced marine macroalgae: new perspectives on species recognition and distribution in New Zealand. Bot. Mar. 64, 379–393. doi: 10.1515/bot-2021-0042
Norris J. N., Aguilar-Rosas L. E., Pedroche F. F. (2017). “Conspectus of the benthic marine algae of the gulf of California: Rhodophyta, phaeophyceae, and chlorophyta,” in Smithsonian Contributions to botany, no. 106 (Washington, D. C: Smithsonian Institution scholarly Press), 125.
Petrocelli A., Wolf M. A., Cecere E., Sciuto K., Sfriso A. (2020). Settlement and spreading of the introduced seaweed Caulacanthus okamurae (Rhodophyta) in the Mediterranean Sea. Diversity 12, 1–10. doi: 10.3390/d12040129
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, et al (2006). Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609.
Preuss M., Nelson W. A., D’Archino R. (2022). Cryptic diversity and phylogeographic patterns in the Asparagopsis armata species complex (Bonnemaisoniales, rhodophyta) from New Zealand. Phycologia 61, 89–96. doi: 10.1080/00318884.2021.2015223
Puillandre D., Lambert A., Brouillet S., Achaz G. (2012). ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x
Rambaut A., Drummond A. J., Xie D., Baele G., Suchard M. A. (2018). Posterior summarization in Bayesian phylogenetic using tracer 1.7. Sys. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rueness J. (1997). A culture study of Caulacanthus ustulatus (Caulacanthaceae, gigartinales, rhodophyta) from Europe and Asia. Cryptogamie Algol. 18, 175–185.
Rueness J., Rueness E. K. (2000). Caulacanthus ustulatus (Gigartinales, rhodophyta) from Brittany (France) is an introduction from the pacific ocean. Cryptogamie Algol. 21, 355–363. doi: 10.1016/S0181-1568(00)01042-4
Saunders G. W. (2005). Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future application. Phil. Trans. R. Soc B. Biol. Sci. 360, 1879–1888. doi: 10.1098/rstb.2005.1719
Searles R. B. (1968). Morphological studies of red algae of the order gigartinales. univ. Calif. Publ. Bot. 43, 1–86.
Sherwood A. R., Carlile A. L., Neumann J. M., Kociolek J. P., Johansen J. R., Lowe R. L., et al. (2014). The Hawaiian freshwater algae biodiversity survey, (2009-2014): systematic and biogeographic trends with an emphasis on the macroalgae. BMC Ecol. 14, 28. doi: 10.1186/s12898-014-0028-2
Smith J. R., Vogt S. C., Creedon F., Lucas B. J., Eernisse D. J. (2014). The non-native turf-forming alga Caulacanthus ustulatus displaces space-occupants but increases diversity. Biol. Invasions 16, 2195–2208. doi: 10.1007/s10530-014-0658-5
Suchard M. A., Lemey P., Baele G., Ayres D. L., Drummond A., Rambaut A. (2018). Bayesian Phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016. doi: 10.1093/ve/vey016
Vieira C., De Clerck O., Millet L., Payri C. E. (2019). Description of ten new Lobophora species from the Bismarck Sea (Papau new Guinea). Phycol. Res. 67, 228–238. doi: 10.1111/pre.12372
West J, Calumpong HP (1990). New records of marine algae from the Philippines. Micronesica 23:181–90.
Yamada Y. (1933). Notes on some Japanese algae V. J. Fac. Sci. Hokkaido Imp. Univ. Ser. V. Bot. 2, 277–285.
Yang M. Y., Fujita D., Kim M. S. (2021a). Phylogeography of Gloiopeltis furcata sensu lato (Gigartinales, rhodophyta) provides the evidence of glacial refugia in Korea and Japan. Algae 36, 13–24. doi: 10.4490/algae.2021.36.3.3
Yang M. Y., Kim M. S. (2018). Cryptic species diversity of ochtodenes-producing Portieria species (Gigartinales, rhodophyta) from the northwest pacific. Algae 33, 205–214. doi: 10.4490/algae.2018.33.7.30
Yang M. Y., Kim M. S. (2022). Phylogeography of the economic seaweeds Chondrus (Gigartinales, rhodophyta) in the northwest pacific based on rbcL and COI-5P genes. Algae 37, 135–147. doi: 10.4490/algae.2022.37.5.29
Yang M. Y., Kim S. Y., Kim M. S. (2021b). Population genetic structure and phylogeography of co-distributed Pachymenipsis species (Rhodophyta) along the coast of Korea and Japan. Diversity 13, 336. doi: 10.3390/d13080336
Yang M. Y., Macaya E. C., Kim M. S. (2015). Molecular evidence for verifying the distribution of Chondracanthus chamissoi and C. teedei (Gigartinales, rhodophyta). Bot. Mar. 58, 103–113. doi: 10.1515/bot-2015-0011
Yang M. Y., Yang E. C., Kim M. S. (2020). Genetic diversity hotspot of the amphi-pacific macroalga Gloiopeltis furcata sensu lato (Gigartinales, rhodophyta). J. Appl. Phycol. 32, 2515–2522. doi: 10.1007/s10811-019-02017-y
Zhang J. J., Kapli P., Pavlidis P., Stamatakis A. (2013). A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876. doi: 10.1093/bioinformatics/btt499
Keywords: Caulacanthus, distribution pattern, haplotype diversity, macroalgae, species delimitation
Citation: Yang MY and Kim MS (2023) Deep genetic divergences and geographic distribution of the red algal genus Caulacanthus (Gigartinales). Front. Mar. Sci. 10:1087507. doi: 10.3389/fmars.2023.1087507
Received: 02 November 2022; Accepted: 24 March 2023;
Published: 26 April 2023.
Edited by:
Zi-Min Hu, Yantai University, ChinaReviewed by:
Fabio Rindi, Polytechnic University of Marche, ItalyCopyright © 2023 Yang and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myung Sook Kim, bXl1bmdza2ltQGplanVudS5hYy5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.