
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 09 March 2023
Sec. Marine Biology
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1087339
This article is part of the Research Topic Echinoderms: Biology, Ecology and Exploitation View all 17 articles
 Fang Su1,2,3,4,5
Fang Su1,2,3,4,5 Shilin Liu1,2,3,4,5
Shilin Liu1,2,3,4,5 Lili Xing1,2,3,4,5
Lili Xing1,2,3,4,5 Da Huo1,2,3,4,5
Da Huo1,2,3,4,5 Hongsheng Yang1,2,3,4,5,6
Hongsheng Yang1,2,3,4,5,6 Lina Sun1,2,3,4,5*
Lina Sun1,2,3,4,5*Embryonic development, especially metamorphosis and settlement, has a major impact on the life history of marine invertebrates. Apostichopus japonicus is an economically important species of sea cucumber. In this study, we performed RNA sequencing on six key stages of A. japonicas development: fertilized eggs, blastula, gastrula, auricularia, doliolaria, and pentactula. A total of 32,353 genes were identified and annotated as a reference gene set for subsequent pairwise comparison analysis. After filtering out low-quality genes, the dynamic molecular responses to development were revealed by WGCNA. The results showed that of the 20 modules, genes in the blue, yellow, and darkslateblue modules were highly correlated with the gastrula, auricularia, and blastula stages, respectively. GO terms for “RNA” and “proteasome complex” were most significantly enriched in the blue module. In the darkslateblue and yellow module, receptors of signaling pathways and metabolic processes were significantly enriched, respectively. All DEGs were categorized into 34 terms, mainly associated with signal transduction and cellular immunity. The expression pattern of genes associated with adhesion, cell cycle, signal, transcription factor, extracellular matrix (ECM), and cytoskeleton was analyzed according to gene function. The results of this study facilitated a more comprehensive understanding of the molecular characteristics of sea cucumber embryonic development and will provide theoretical guidance for larva rearing in sea cucumber culture.
Apostichopus japonicus (Selenka) is distributed in shallow temperate and temperate-cold waters along the coasts of China, Japan, Korea, and Russia. This species is considered to be one of the most valuable sea foods, due to its extraordinary nutritional and medicinal properties (Yang et al., 2015). A. japonicus also displays some unique biological characteristics, such as evisceration, regeneration, aestivation, and autolysis (Yang et al., 2015). It has been extensively studied from many perspectives, including feeding behavior and movement patterns (Pan et al., 2015; Sun et al., 2015), reproduction and breeding (Kato et al., 2009; Ahmed et al., 2011; Soliman et al., 2013), responses to environmental changes (temperature, salt, light, and dissolved oxygen) (Dong et al., 2010; Meng et al., 2011; Xu et al., 2016; Huo et al., 2017), disease and immunity (Gu et al., 2010; Zhang et al., 2014), and physiology and behavior of regeneration or aestivation (Chen et al., 2015; Miao et al., 2017; Sun et al., 2017a; Sun et al., 2017b). In 2017, we published the A. japonicus genome, the first fine sea cucumber genome, making an important contribution to sea cucumber and echinoderm research (Zhang et al., 2017). A. japonicus, as a representative species for sea cucumbers, is becoming a good model species for research.
Development is the most important biological process for species. In recent years, there have been a number of studies on sea cucumber development, promoting our understanding of these species. Researchers have elucidated the development and growth morphogenesis patterns of different kinds of sea cucumbers, including Cucumaria frondose (Gianasi et al., 2018), Stichopus sp. (Curry fish) (Hu et al., 2010), Isostichopus fuscus (Hamel et al., 2003), and A. japonicus (Yang et al., 2015). A. japonicus has received the most attention from researchers due to its high economic value in China and Asia. The life cycle of A. japonicus can be divided into eight major developmental stages. Embryonic and larval development before the juvenile phase can be divided into six key stages as follows: fertilization of eggs, blastula, gastrula, auricularia, doliolaria, and pentactula (Yang et al., 2015). To date, there have been numerous studies on the effect of environmental factors (such as density, temperature, salinity, and pH) on the development of A. japonicus (Li and Li, 2009; Liu et al., 2010; Yuan et al., 2015), mainly focusing on survival rate and metamorphosis. However, there has been a lack of studies on genetic mechanisms and gene regulation associated with development.
Morphological changes in the development of fertilized eggs to juvenile have been reported in the literature. When the fertilization membrane is lifted, it indicates that the ovum is successfully fertilized. After multiple cell divisions, the number of cells increased and the cell volume became smaller. The blastula is a hollow sphere surrounded by a thin layer of cells, and the egg membrane is clearly visible. The embryo is detachable by rotation and then rotate freely with the oscillation of surface cilia in a finger ring shape. As development proceeds, the vegetal pole invades and forms mesenchymal cells. The digestive tract is not fully differentiated, and morphological distinctions are not obvious. The gastrula further develops into auricular larvae, and the digestive system begins to improve. The food particles enter the mouth with the water flow formed by the oscillation of the cilia. Calcareous ossicle begin to form and continue to elongate towards the posterior end. According to the development morphology of the water cavity, the auricular larvae are divided into three stages: early-auricularia larva, mid-auricularia larva, late-auricularia larva (Qiu, 2013). At the late-auricularia larva stage, five small vesicles grow outward the water cavity, resembling finger-like branches (Wang et al., 2019). The body shrinks to a cylindrical shape and the larvae metamorphose into doliolaria. Most of the cilia fall off, and the movement ability and swimming ability of the larvae decrease. At the pentactula stage, the cilia rings degenerate and disappear completely. The larvae change from planktonic life to benthic life, with five tentacles protruding. The larvae sink to the attachment base and crawl by the first tube foot and tentacles. However, the global gene regulation patterns of A. japonicus development and key regulatory gene families are still not well understood.
With the development of high-throughput sequencing techniques, a number of A. japonicus transcriptomes have been successfully constructed, including transcriptomes of intestine regeneration (Sun et al., 2011), intestines, respiratory trees and coelomic fluid (Du et al., 2012), response to pathogen infection (Zhou et al., 2014; Gao et al., 2015), and albinism biogenesis (Xing et al., 2018). However, comparative transcriptomes of all key stages of A. japonicus development have not yet been constructed, as it is very hard to assemble 18 transcriptomes de novo with mega data. Fortunately, the fine A. japonicus genome provides a good reference, which makes this possible (Zhang et al., 2017). In the current study, we applied transcriptomic techniques to identify differentially expressed genes (DEGs) of six different developmental stages of A. japonicus. The results of this study provided a better understanding of the mechanisms of A. japonicus development. Furthermore, these data will increase the amount of genetic information for A. japonicus, providing a good foundation for investigating the molecular/genetic mechanisms of sea cucumbers.
Parent culture, spawning, and larva rearing were performed in our breeding base in Qingdao. The embryos and larvae of A. japonicus were cultivated in 2 m×5 m×1.5 m cement pools at 20 ± 1°C. Samples from six major developmental stages were collected: fertilized eggs (F, ~20 min after fertilization), blastula (B, ~10 h), gastrula (G, ~26 h), auricularia (A, ~ 8 d), doliolaria (D, ~ 10 d), and pentactula (P, ~ 12 d). The samples were monitored under a microscope to ensure developmental synchrony. Three samples at each developmental stage were collected as biological replicates. Each sample was washed with ddH2O, frozen, and stored in liquid nitrogen. Finally, a total of 18 samples were prepared for sequencing to obtain 18 transcriptomes.
Total RNA was extracted from 18 samples as mentioned above using the RNAeasy mini kit (Qiagen, USA) according to the manufacturer’s instructions. The quality and concentration of RNA was measured by NanoDrop 1000 (Thermo). A total of 18 RNA samples were used for isolation, fragmentation, and construction of the cDNA library. Extracted total RNA was treated with DNase I, Oligo (dT) and used for RNA isolation. A total of 3 μg RNA per sample was used as input material for the RNA sample reparations. Fragmentation was carried out using divalent cations under elevated temperatures in NEBNext First Strand Synthesis Reaction Buffer (5×). Then cDNA was synthesized using the mRNA fragments as templates. Short fragments were purified and resolved with EB buffer (10 mM Tris-HCl, pH 8.5) for end reparation and single nucleotide A (adenine) addition. Subsequently, the short fragments were connected with adapters. Suitable fragments were selected for PCR amplification. During the QC steps, an Agilent 2100 Bioanalyzer and an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) were used in the quantification and qualification of the sample library. Finally, sequencing was carried out using Illumina HiSeq 4000 by the Beijing Genome Institute (BGI) (Shenzhen, China). Table S1 (supplementary material) gives a brief overview of the condition of the samples and summarizes the key characteristics of the transcriptome sequencing.
The sequencing reads which were of low quality, adaptor-polluted, or had a high number of unknown base (N) reads, were removed prior to further analyses. Filtering was performed using internal software of BGI. All sequencing data and quality information can be found in Table S1 in the supplementary material. HISAT (v0.1.6-beta) (Kim et al., 2015) was used to map all clean reads to the sea cucumber genome (NCBI accession number: PRJNA354676) (Zhang et al., 2017). After genome mapping, we used GATK to call SNP and INDEL variants for each sample (McKenna et al., 2010). After that, StringTie (v1.0.4) was used to reconstruct novel transcripts (Pertea et al., 2015), cuffcompare (v2.2.1) was used to compare reconstructed transcripts to draft genome annotation, and CPC (v0.9-r2) was used to predict the coding potential of novel transcripts (Kong et al., 2007). After novel transcript detection, we merged novel coding transcripts with reference transcripts to get complete reference, then we mapped clean reads to it using Bowtie2 (V2.25) (Langmead and Salzberg, 2012), then calculated gene expression level for each sample with RSEM (V1.2.12) (Li and Dewey, 2011). The gene expression summary was shown in Table S1 in the supplementary material. With gene expression results, NOIseq based on a noise distribution model was chosen to detect DEGs (Parameters: Fold Change ≥ 2.00 and Probability ≤0.8).
The obtained genes were annotated according to the sea cucumber genome annotation (Zhang et al., 2017). The novel transcripts were annotated using BLAST programs with SwissProt, NCBI non-redundant (Nr) protein, and NCBI non-redundant nucleotide sequences (Nt) databases. The GO (http://www.geneontology.org/) and KEGG databases (http://www.genome.jp/kegg/) were used to classify and group the identified proteins. A hypergeometric test was used to define significantly enriched GO terms and pathways of differentially expressed genes. The terms which FDR not larger than 0.001 were defined as significant enriched.
Co-expression networks were constructed using the WGCNA (v1.47) package in R (Langfelder and Horvath, 2008) to identify specific modules associated with larva development at different stages. The fragments per kilobase of transcript per million mapped reads (FPKM) of each gene was calculated as the gene expression level. Genes were filtered out when FPKM < 2 in more than 70% of samples and outlier samples were deleted. The rest genes in the 17 libraries were imported into WGCNA to construct co-expression modules using the automatic network construction function block wise Modules with default settings. To find biologically or clinically significant modules, module eigengenes were used to calculate the correlation coefficient with samples. Intramodular connectivity (K.in) and module correlation degree (MM) of each gene was calculated with the R package WGCNA. Genes with high connectivity tended to be hub genes which might have important functions. To identify modules significantly associated with different developmental stages, correlation analysis was performed using the module eigengene with gene expression levels in different groups (F, B, G, A, D, P). Modules with high gene expression were considered as target modules. To further analyze the biological functions of genes in each target module, GO and KEGG pathway enrichment analysis were performed. GO terms and pathways with p-value corrected by FDR ≤ 0.05 were considered to be significantly enriched.
The same RNA as used for Illumina sequencing was used in this experiment. The first strand cDNA was synthesized in a 25-μL reaction system as follows: 1) 4 μL RNA and 1 μL oligodT18 were denatured at 70°C for 5 min; 2) 1 μL M-MLV reverse transcriptase (Promega), 5 μL M-MLV buffer (25 mM KCl, 10 mM Tris-HCl, 0.6 mM MgCl2, and 2 nM DTT, pH 8.3), 5 μL dNTP, 1 μL ribonuclease inhibitor, and 8 μL RNase-free water were added at 42°C for 1 h. The synthesized cDNA was diluted with RNase-free water and stored at -80°C for subsequent quantitative real-time PCR. All gene mRNA expression levels were determined using the SYBR Green® real-time PCR assay with an Eppendorf Mastercycler® ep realplex (Eppendorf, Hamburg, Germany). The primers were designed using Primer3 (v0.4.0; http://bioinfo.ut.ee/primer3-0.4.0/primer3/) and are listed in Table S2 in the supplementary material. The amplification volume was 25 μL, and it contained 12.5 μL SYBR Green Master Mix (Takara), 0.5 μL (each) forward and reverse primer (10 μM), 1 μL diluted cDNA, and 10.5 μL RNase-free water. Thermal cycling was as follows: 1) 95°C for 5 s and 2) 40 cycles at 95°C for 10 s, 60°C for 20 s, and 72°C for 30 s. Melting curve analysis of the amplification products was performed to demonstrate the specificity of the PCR products. NADH dehydrogenase was used as a housekeeping gene for internal standardization (Sun et al., 2011; Sun et al., 2017b). The 2-ΔΔCT method was used to analyze mRNA expression levels. All data are reported as means ± SE (N = 5), and the level of statistical significance was set at P < 0.05. Analysis was carried out using SPSS18 software.
In the current study, 8.25~8.98 Gb × 18 data from sea cucumbers at six key developmental stages were generated by high-throughput sequencing techniques, and have been submitted to NCBI (accession NO. PRJNA889469). After read filtering, 53.20~59.96 Mb× 18 clean reads were obtained to construct 81,146 transcripts with contig N50 of 3910bp, which were further grouped into 32,353 genes. Among all transcripts, 48,098 transcripts were novel transcripts by reconstruction after mapping sequenced reads to the reference genome. Of these, 24,275 were previously unknown splicing events for known genes, 5,494 were novel coding transcripts without any known features, and the remaining 18,329 were long noncoding RNAs. The mRNA expression levels of all genes can be found in Table S3 in the supplementary material.
Thanks to the development of high-throughput sequencing techniques, many transcriptomes have been constructed to understand global gene regulation or response mechanisms to different types of biological processes in sea cucumbers (Sun et al., 2011; Du et al., 2012; Zhang et al., 2013; Gao et al., 2015; Xing et al., 2018). However, the gene expression patterns of development, one of most important biological processes, are still not known. In 2012, Du et al. (Du et al., 2012). constructed the transcriptomes of embryos and larvae. However, these data could only provide relatively complete gene sets and did not reveal the gene regulation patterns of development of sea cucumbers, because the embryo and larva samples were pooled to construct two libraries and the sampling times did not coincide with five key developmental stages. In this study, we revealed the global gene expression patterns of six developmental stages, which detailed all gene regulation patterns during sea cucumber development.
To better understand the gene co-expression network during sea cucumber development, WGCNA analysis was carried out to obtain 20 modules based on hierarchical clustering of calculated dissimilarity after filtering low-quality samples and genes (Figure 1A; Supplementary Table S4). Genes with similar expression patterns are represented on the same branch. Each branch represents a co-expression module. According to the similarity of the module eigengene, the modules with similar expression patterns are merged to obtain the final merged dynamic modules. Genes that do not fit into any module are shown in gray. The numbers of genes in modules ranged from 56 to 2,925 (Supplementary Table S5), and a heat map of sample expression patterns based on module eigengene was generated to visualize the correlation matrix between modules and groups (Figure 1B; Supplementary Table S6). Some modules were found to relate to specific developmental stages. The genes in the greenyellow, lightcyan, yellow, and lavenderblush3 modules were highly expressed in the auricularia, doliolaria, and pentactula stages, but were expressed at low levels in fertilized eggs, blastula, and gastrula stages. However, the darkmagenta, darkorange2, floralwhite, indianred4, and lightpink4 modules showed a completely opposite correlation. Furthermore, the results showed that the blue and yellow modules were positively correlated with the gastrula and auricularia stages, respectively. Additionally, the darkslateblue module had the highest correlation coefficient with the blastula stage. Therefore, we treated the three modules that were highly associated with these developmental stages as the target modules.
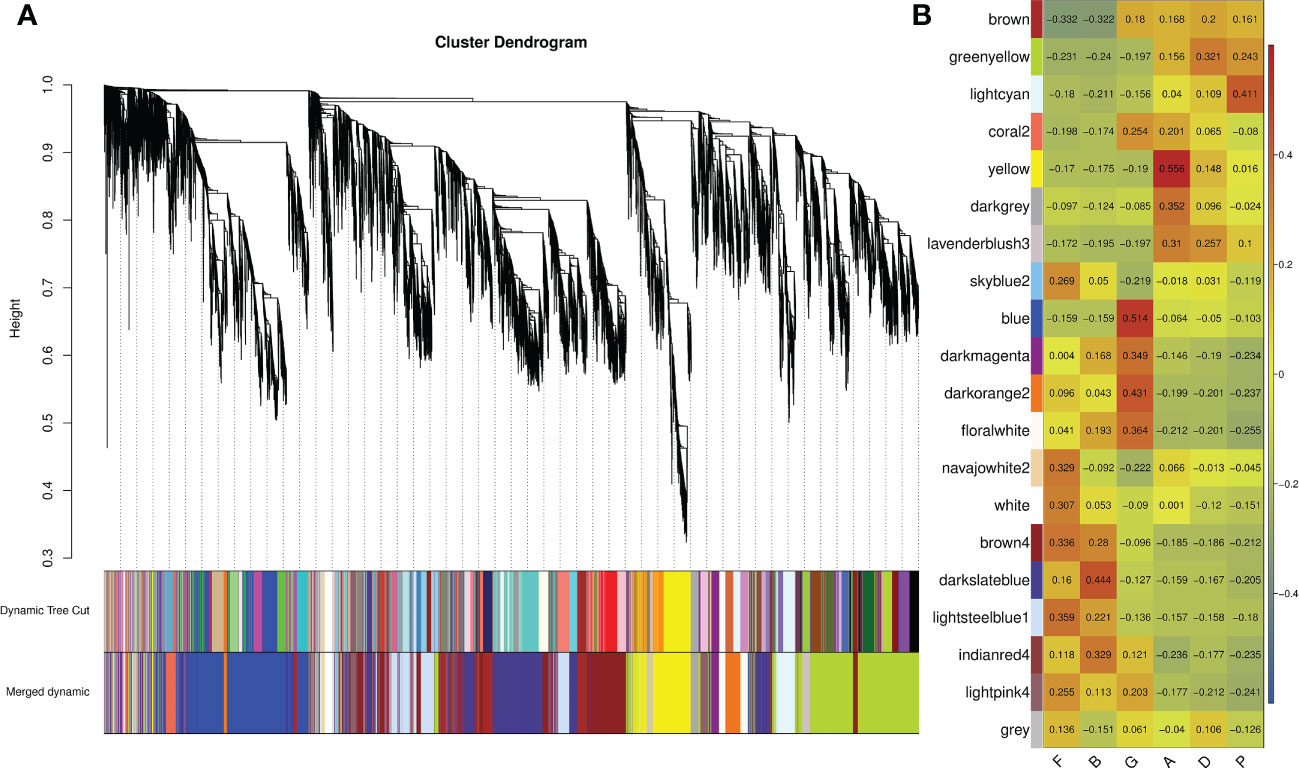
Figure 1 Weighted gene correlation network analysis (WGCNA) of all genes in different developmental stages. (A) Hierarchical cluster tree showing correlation modules identified by WGCNA. Each leaf in the tree represents one gene and each major tree branch represents one module, labelled with different colors. Modules with similar expression patterns are merged as merged dynamic. (B) Correlation matrix between modules and developmental stages. Red represents high expression and blue represents low expression.
To further explore the function of module genes, we performed GO and KEGG enrichment analysis for each target module (Figure 2; Supplementary Tables S7, S8). GO terms for “RNA” and “proteasome complex”, such as “translation factor activity, RNA binding (GO:0008135)”, “RNA binding (GO:0003723)”, “RNA processing (GO:0006396)” and “proteasome complex (GO:0000502)”, were most significantly enriched in the blue module. In the darkslateblue module, receptors of signaling pathways were significantly enriched. The representatives were “immune response-activating cell surface receptor signaling pathway (GO:0002429)”, “immune response-regulating cell surface receptor signaling pathway (GO:0002768)” and “antigen receptor-mediated signaling pathway (GO:0050851)”. Additionally, genes grouped in the yellow module were significantly enriched in metabolic processes, such as “neurotransmitter metabolic process (GO:0042133)”, “single-organism carbohydrate catabolic process (GO:0044724)”, and “carbohydrate catabolic process (GO:0016052)”. According to the KEGG pathway enrichment results, “proteasome (ko03050)”, “spliceosome (ko03040)”, and “protein export (ko03060)” pathways related to gene information processes were enriched in the blue module. In the darkslate and yellow modules, metabolic pathways were significantly enriched. Overall, GO and KEGG enrichment analysis demonstrated the role of modular genes in the larval development of A. japonicus.
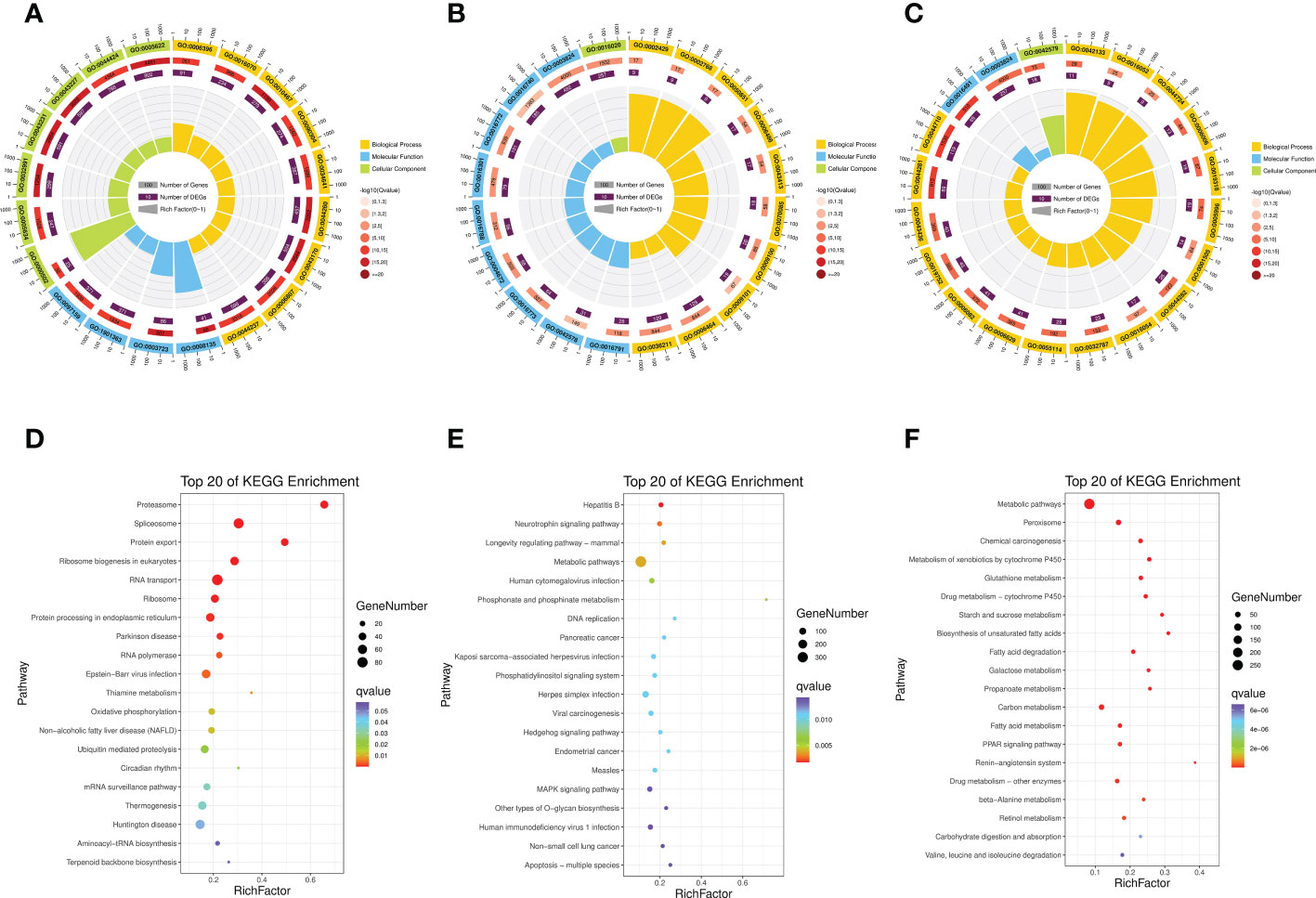
Figure 2 Gene Ontology (GO) enrichment analysis of genes in blue module (A), darkslateblue module (B) and yellow module (C). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of genes in blue module (D), darkslateblue module (E) and yellow module (F).
The correlation network of significantly enriched genes in the three target modules is illustrated in Figure 3. According to the target module size, the top five genes with the highest All.kWithin values were identified as hub genes (Table S9 in the supplementary material). In the correlation network of the blue module, protein mago nashi homolog (MAGOH), zinc finger C4H2 domain-containing protein-like isoform X1(AC4H2), N-acetyltransferase 10 (NAT10), nuclear pore complex protein Nup107 isoform X1 (Nup107 complex), and eukaryotic translation initiation factor 3 subunit F (eIF3f) were identified as hub genes (Figure 3A; Supplementary Table S9). MAGOH is one of the three core proteins of the exon junction complex (EJC), and is essential for the normal development of embryos and nerves (Asthana et al., 2022). AC4H2 plays a vital role in regulating nervous system development, and has already been identified in multiple species (May et al., 2015; Ma et al., 2017). NAT10 promotes cell proliferation and thereby participates in malignant transformation of various cancers. In addition, NAT10 is expressed in the central nervous system of the embryo (Guo et al., 2020). Nup107 complex is part of the Nuclear Pore Complex (NPC) structural scaffold and the role of the Nup107 complex in gene expression is not restricted to nucleo-cytoplasmic transport, but also participates in neural differentiation in vertebrates (Gonzalez-Aguilera and Askjaer, 2012). The first nerve cells were detected in the apical region of the epidermis in the late gastrula stage (Nakano et al., 2006). Thus, the blue module was closely related to neurological genesis. Genes for enzymes associated with digestion and immunity, such as lactase-phlorizin hydrolase-like, aminopeptidase N (APN), dipeptidase 1-like (DPEP1), mucin-17 isoform X2 (MUC17), and cAMP-regulated D2 protein-like (cAMP-D2) were identified as hub genes of the yellow module (Figure 3B, Table S9 in the supplementary material). During the auricularia stage, the digestive system begins to improve. For the darkslateblue module, tyrosine-protein kinase RYK isoform X4 (RYK), sodium-dependent multivitamin transporter (SMVT), lysosomal amino acid transporter 1 (LYAAT-1), cyclin-J, and ELAV-like protein 3 isoform X2 (elavl3) were identified as hub genes (Figure 3C, Table S9 in the supplementary material). RYK may be a co-receptor along with FZD8 of Wnt proteins, such as WNT1, WNT3, WNT3A and WNT5A. LYAAT-1 defines a subgroup of lysosomal transporters in the amino acid/auxin permease family (Sagne et al., 2001). These genes were involved in the transportation of substances and the cell cycle.
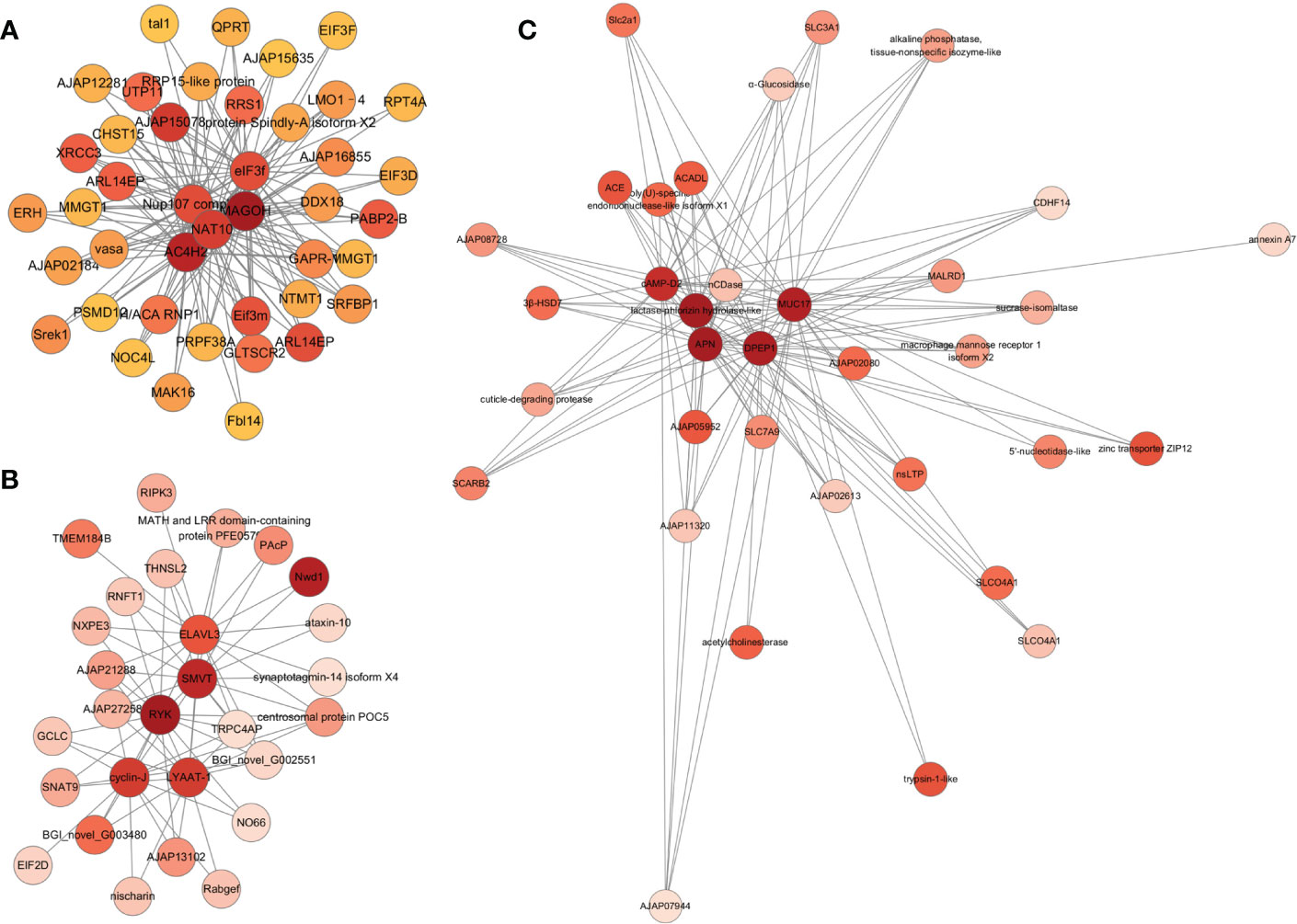
Figure 3 Correlation network visualization of the interactions between the top 5 genes with the highest connectivity in three target modules. Each node labelled with the abbreviation of gene name. Node colors represent connectivity, and darker nodes represent higher connectivity. (A–C) represent blue, darkslateblue and yellow module, respectively.
Using 32,353 genes as a reference, five comparison groups: F Vs B, B Vs G, G Vs A, A Vs D, and D Vs P, were used to analyze the differential expression of genes during A. japonicus development using NOIseq arithmetic (fold-change≥2.0 and probability≥0.8). The number of up-regulated genes and down-regulated genes was 20 and 112, 1,781 and 1,535, 535 and 97, 98 and 272, and 12 and 29, respectively (Figure 4). The highest number of genes showing differential expression were between the blastula to gastrula stage (B Vs G). All DEGs are listed in Supplementary Table S10.
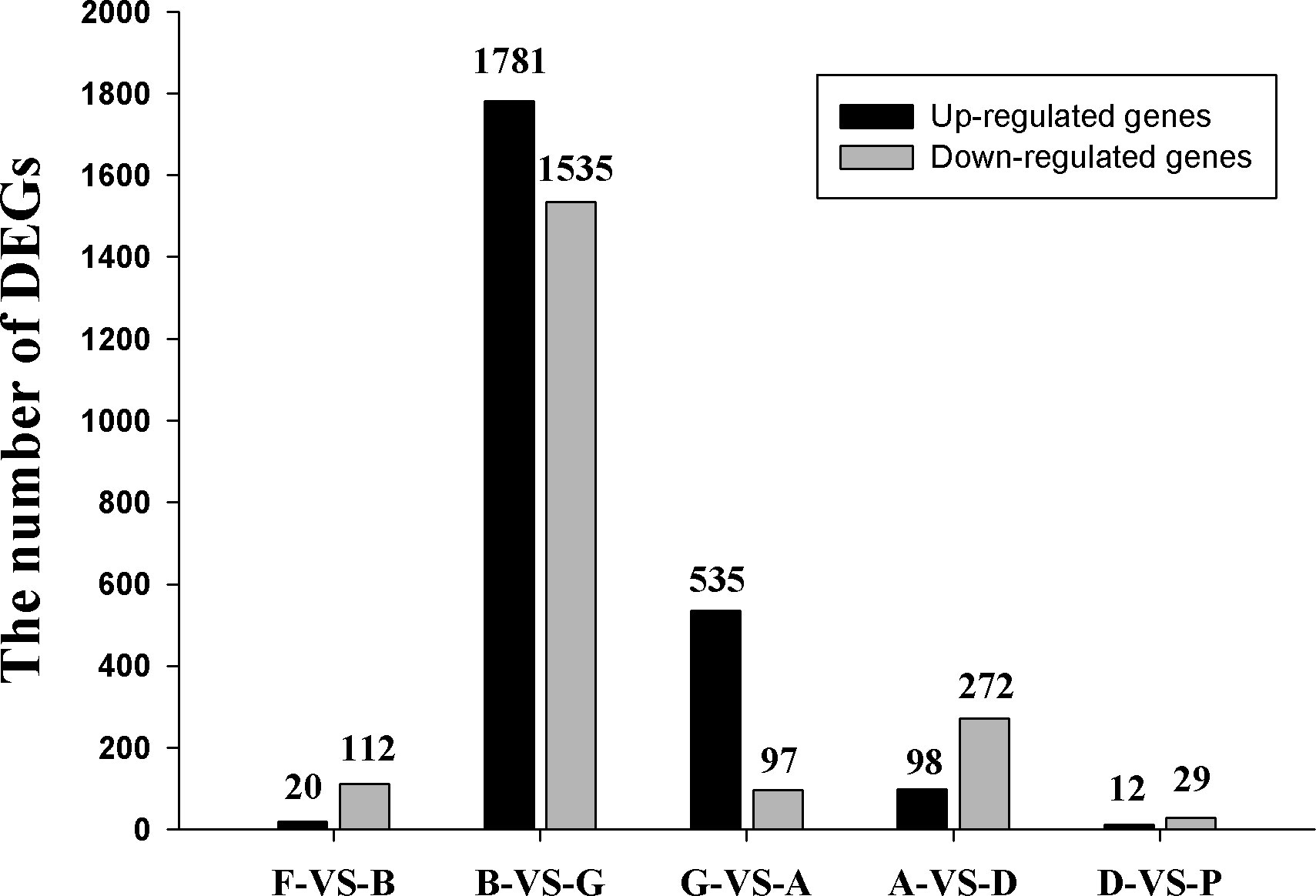
Figure 4 The number of differentially expressed genes (DEGs) in five comparison groups.
For validation purposes, RT-PCR analysis in four genes including cyclinB, ferritin, SoxB1, and cubilin-like protein was conducted in the six development stages (Figure 5). The results showed that the overall trend and variation of RT-PCR based expression patterns among these selected genes was coincident with those obtained by RNA-Seq based detection.

Figure 5 Validation of cyclin B (A), SoxB1 (B), ferritin (C) and cubilin-like (D) using RT-PCR. Data are shown as the mean ± SD. The lowercase letters indicate statistically significant differences (p < 0.05).
All DEGs could be categorized into 34 terms. “Signal transduction”, “Global and overview maps”, “Cancers”, “Endocrine system”, “Transport and catabolism”, “Infectious diseases: Viral”, “cellular community”, “Translation”, “Digestive system”, and “Signaling molecules and interaction” were the 10 most enriched terms during the entire development process of sea cucumbers (Supplementary Figure S1). Among almost all terms, the number of DEGs in the B Vs G comparison group was the largest, followed by the G Vs A comparison group (Figure 6), thus, there was strong gene expression regulation from blastula to auricularia. Compared with other pathways, the AVsD comparison group had the largest proportion in “cell growth and death”.
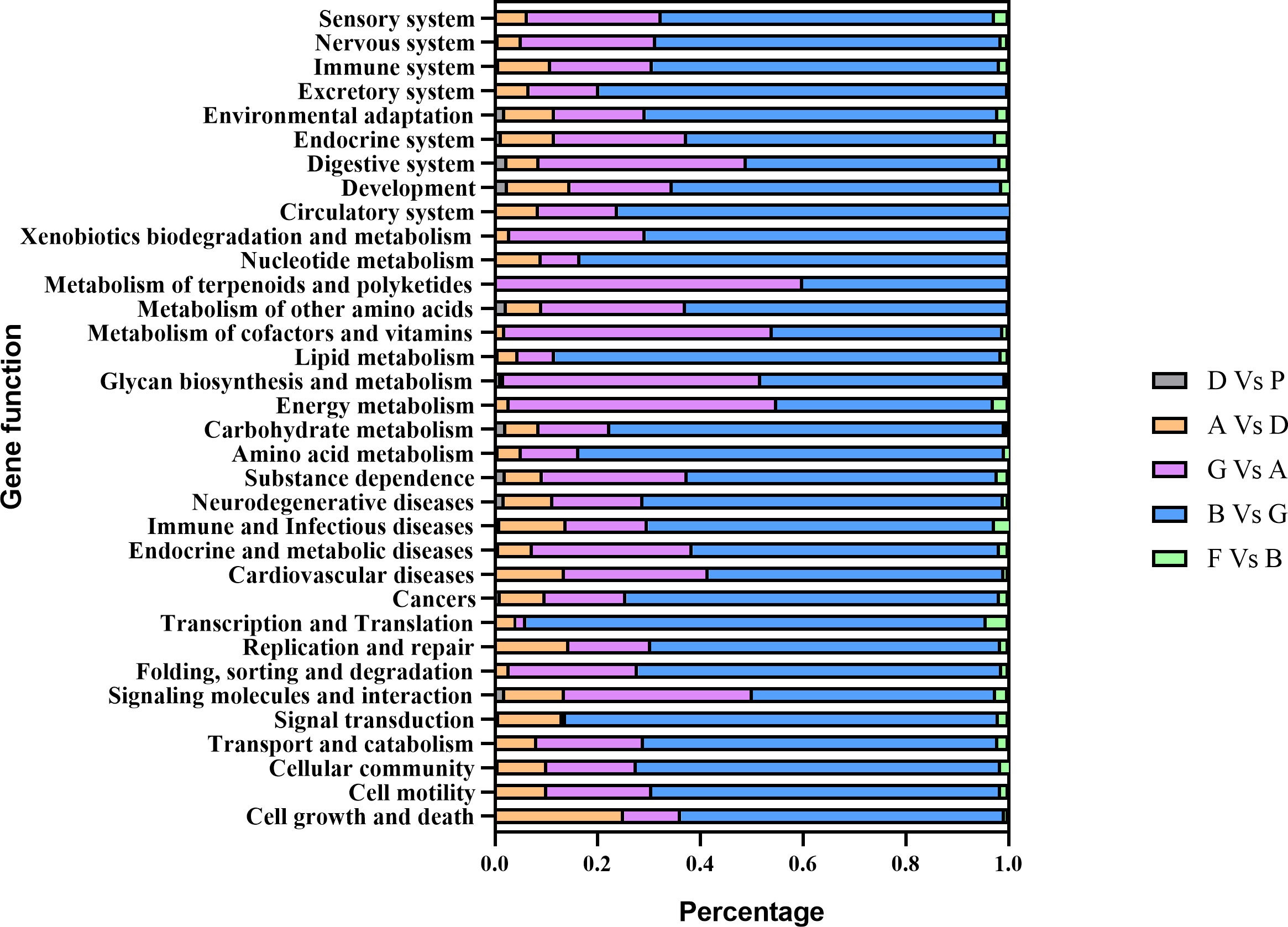
Figure 6 The percentage of DEGs enriched in each term at different developmental stages.
GO enrichment analysis showed significant enrichment of DEGs in three categories: molecular function (MF), cellular component (CC), and biological process (BP) (Table S11 in the supplementary material). In the MF category, 1, 14, 2, and 1 terms were enriched in the F Vs B, B Vs G, G Vs A, and D Vs P groups, respectively. In the CC category, 3, 23, 1, and 5 terms were enriched in the F Vs B, B Vs G, G Vs A, and A Vs D groups, respectively. In the BP category, 12, 17, 1, and 1 terms were enriched in the F Vs B, B Vs G, G Vs A, and A Vs D groups, respectively.
KEGG pathway enrichment analysis showed two pathways were enriched in F Vs B, including Ribosome and Vibrio cholera infection. Three pathways were enriched in B Vs G, including Ribosome, Protein export and Proteasome, while 16 pathways were enriched in G Vs A, including Protein digestion and absorption, Renin-angiotensin system as well as Pancreatic secretion. Six pathways were enriched in A Vs D, including cell cycle, oocyte meiosis, and p53 signaling pathway. Two pathways were enriched in D Vs P (Figure S2; Supplementary Table S12).
The transition from fertilized eggs to blastula is the first stage in sea cucumber development. It involves the expulsion of polar bodies, rapid cell division, and the expansion and break down of the fertilization envelope (Yang et al., 2015). About 1 hour after fertilization, the fertilized egg enters the cleavage stage. The cells gradually decrease in size after division and enter the blastocyst stage at about 7 to 9 h. The whole blastocyst stage lasts for about 13 h and is divided into a stationary stage and a rotational stage. Compared with fertilized eggs, blastocysts are multicellular embryos. We analyzed DEGs and found 20 up-regulated genes and 112 down-regulated genes during this stage. To evaluate the function of DEGs, we conducted GO and KEGG enrichment analysis. The significant GO cellular component terms were related to ribosome components, such as “ribosomal subunit” and “ribosome”. The biological process ontology includes terms associated with viral reproduction (Supplementary Table S11). The KEGG enrichment analysis results showed that DEGs are enriched in “Ribosome”, “Vibrio cholerae infection” and “Amoebiasis” (Figure S2). “Vibrio cholerae infection” and “Amoebiasis” are the common pathways during the comparative transcriptomic analysis of the early development in pacific white shrimp Litopenaeus vannamei (Wei et al., 2014). This suggests that the two pathways are involved in the embryonic development process. The ribosome as a complicated organelle mainly catalyzed the amino acids into peptide chains (translation). “Ribosome” is a basic cellular biology pathway involved in cell division and other cellular activities. Among the DEGs, lamin B receptor (LBR) was the most significantly upregulated gene. LBR is an integral membrane protein of the interphase nuclear envelope involved in mitotic nuclear envelope breakdown (Margalit et al., 2005; Olins et al., 2010), which called open mitosis. Thus, we speculated that LBR promoted NE breakdown to regulate open mitosis of sea cucumber cells. The results implied that two major events in this developmental process were DNA rapid replication for cell proliferation and immune response for protecting organisms from environmental stress.
The highest number of genes (up-regulated genes: 1,781; down-regulated genes: 1,535) showed differential expression during development from the blastula to gastrula stage (B Vs G), suggesting that this is a key period during sea cucumber development. It is worth noting that of the top 30 known up-regulated DEGs listed in Table S10 according to probability values and mRNA expression, a total of seven DEGs were associated with metalloproteinase, including three genes coding matrix metalloproteinase and four genes coding zinc metalloproteinase. In addition, many transcription factors, key genes in conservative signaling pathways as well as cell cycle regulation genes were significantly up-regulated during this stage, such as FoxQ1 (forkhead box protein Q1-like), FoxA, Wnt-8a, and frizzled 9-10. These results are consistent with the fact that during gastrulation the germ layers of the embryo are formed and the body plan of the organism is established (Tam and Loebel, 2007).
Most DEGs were categorized into “Immune and infectious diseases” (519 DEGs), “Cancers” (434 DEGs), and “Signal transduction” (408 DEGs) (Figure 6). The most enriched pathways in this process were “Ribosome”, “Protein export”, and “Proteasome”, which were responsible for translation and protein folding, sorting, and degradation (Figure S2B).
In this process, the digestive tract of sea cucumbers gradually starts to open and they start to feed. A total of 535 up-regulated genes and 97 down-regulated genes were screened during this stage. Among them, Proteinase T/proprotein convertase subtilisin/kexin type 9 preproprotein was up-regulated over 2,000-fold from the gastrula stage to the auricularia stage in sea cucumbers. This gene was the most highly expressed gene in the intestine of healthy A. japonicus, However, it was not highly expressed in the body wall or respiratory tree (Yang et al., 2009), indicating that the expression of this gene was very advantageous for intestine digestion.
At this stage, the majority of DEGs were categorized into “Immune and Infectious diseases” (122 DEGs), “Digestive system” (102 DEGs), and “Endocrine system” (94 DEGs) (Figure 6). The most enriched pathways were “Protein digestion and absorption”, “Renin-angiotensin system”, “Pancreatic secretion”, “Carbohydrate digestion and absorption”, and “Fat digestion and absorption”, which were responsible for both the digestive endocrine systems (Figure S2C). In light of these findings, we speculated that the formation of the digestive tract and the start of feeding were the key events in this process.
During the development of auricularia to doliolaria, the planktotrophic larva, the auricularia, undergoes a dramatic morphological transformation at the onset of metamorphosis (Lacalli and West, 2000). At this stage, 98 up-regulated genes and 272 down-regulated genes were found. The majority of DEGs were categorized into “Immune and Infectious diseases” and “Signal transduction”, accounting for 99 and 60 DEGs, respectively (Figure 6). MAP kinase-interacting serine/threonine-protein kinase 1 isoform X1 and PTSP-like peptide neurotransmitter precursor were most significantly up-regulated during the development from auricularia to doliolaria, and have been found to play important roles in the metamorphosis process. As mentioned above, nerve cells begin to form in the late gastrula stage and the adult nervous system develops during the doliolaria stage (Nakano et al., 2006). This indicates that metamorphosis is a multigenic synergistically involved process regulated by endogenous neurotransmitters (Matsuura et al., 2009). Among all the down-regulated genes, the two genes associated with DNA replication, zinc finger RAD18 domain-containing protein C1orf124 homolog (C1orf124) and spalt-like transcription factor (SALL), were the most significant. C1orf124 is a regulator of translesion synthesis and participates in polymerase switching (Ghosal et al., 2012). SALLs are evolutionarily conserved proteins that participate in embryonic development. In oncology studies, SALLs are involved in cellular apoptosis, angiogenesis, invasion, and metastasis (Ma et al., 2021). This is consistent with the most significantly enriched signaling pathway being the ‘cell cycle’, suggesting active cellular events during metamorphosis (Figure S2D).
Sea cucumber larva develop from doliolaria to pentactula, and use their tentacles to attach to the substrate at ~ 12 days post-fertilization, which was a key part of substrate detection/selection and settlement 1. By this stage, the morphological structure of most of organs has taken shape. Hence, relatively few genes showed differential expression during this process. Four up-regulated genes and 10 down-regulated genes were screened. “Salivary secretion” and “Prion diseases” were the most enriched pathways (Figure S2E).
Key genes related to sea cucumber development were classified into seven groups according to gene function: adhesion, cell cycle, signal, transcription factor, extracellular matrix (ECM), and cytoskeleton. The detailed information of gene expression can be seen in Supplementary Table S13.
The expression profiles of 9 cadherin genes and 14 protocadherin Fat genes, as well as 18 integrin genes, are summarized in Figure 7A. Genes from the same family showed different expression patterns. The expression levels of Cadherin Ap showed an increasing trend during the developmental process, especially from doliolaria to pentactula. The expression trend of the remaining 7 cadherin genes was opposite to Cadherin Ap. The expression levels of most protocadherin Fat genes were gradually down-regulated. The integrin genes exhibited low expression levels from doliolaria to pentactula.
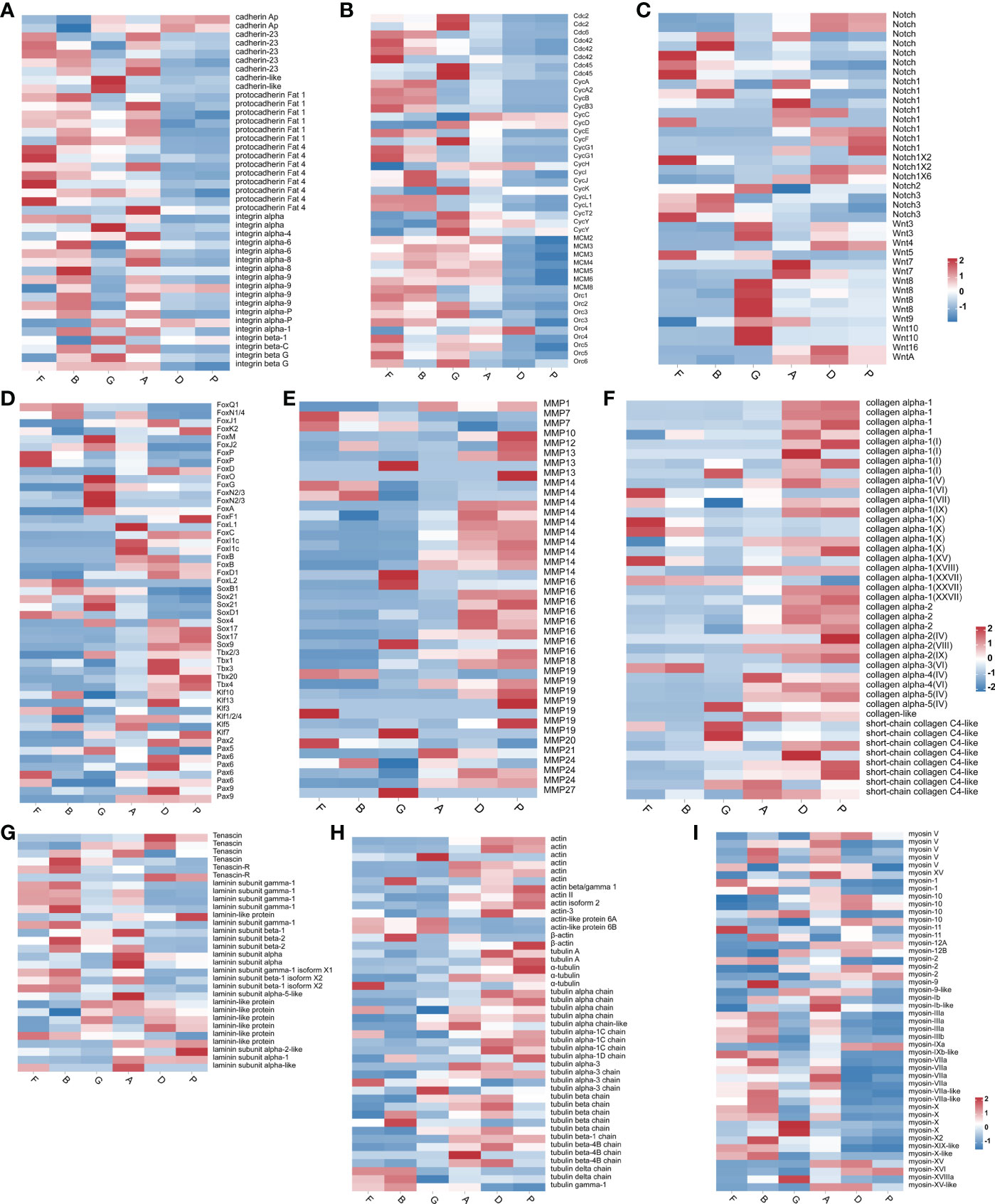
Figure 7 Heatmap diagram showing the expression levels of transcripts annotated as adhesion genes (A), cell cycle genes (B), signal genes (C), transcription factor (D), ECM genes (E–G) and cytoskeleton genes (H, I).
The expression profiles of 8 cell division control protein (Cdc) family genes, 19 cyclin family genes (Cyc), 7 DNA replication licensing factor genes (MCM), and 9 origin recognition complex genes (Orc) are summarized in Figure 7B. Cdc family genes exhibited low expression levels from auricularia to pentactula. Cdc6 is essential for the formation of pre-replication complexes at the origins of DNA replication (Davis et al., 2008). The ORC1 gene, which plays an important role in the initiation of DNA replication, was also highly expressed at the fertilized egg and blastula stages. Cyclin B accumulates and binds to Cdk1 at the G2-phase, playing a vital role in G2-to-M transition (Lemonnier et al., 2020). The expression level of MCM genes were up-regulated from fertilized egg to auricularia. During the development from fertilized eggs to blastula, cells proliferate through the mitotic cell cycle. Therefore, these genes regulate the cell division cycle.
Two family genes (Notch and Wnt) related to key signal pathways were screened (Figure 7C). There was no obvious pattern in Notch gene expression, and there were high expression genes in each stage. The expression levels of Wnt16 and WntA were up-regulated from auricularia to pentactula. Wnt4 and Wnt5 showed opposite expression levels.
A total of 23 forkhead transcription factor (Fox) genes were expressed during development. The expression patterns of eight Sox genes, five T-box transcription factors, six Krueppel-like factors, and eight paired box proteins are shown in Figure 7D. SoxB1, a multifunctional gene involved in maintaining stem cell multipotency, was highly expressed from blastula to auricularia stages. Tbx2/3 showed high expression during development from gastrula to pentactula. Interestingly, Pax5 was highly expressed from the fertilized egg to the gastrula stage and Pax9 was highly expressed from the auricularia to pentactula stages. The expression time of these two genes covers the entire larval development process.
Many genes associated with ECM were expressed during sea cucumber development, including 40 Matrix metalloproteinase (MMPs), 41 collagen, six tenascin, and 24 laminin (Figures 7E–G). The expression levels of most MMPs were up-regulated at doliolaria and pentactula stages. Most collagen genes were up-regulated from the auricularia to pentactula stage. However, laminin genes were highly expressed at the early stages of development.
During sea cucumber development, many cytoskeleton genes were expressed, including 14 actin, 46 myosin, and 30 tubulin (Figures 7H, I). The expression of many actin and tubulin genes were highly expressed from the auricularia to pentactula stages, while many myosin genes exhibited high expression levels at the early stages of development.
It has been clear for many years that matrix metalloproteinase hydrolyze ECM components, play a central role in embryogenesis, normal tissue remodeling, wound healing, and angiogenesis (Dolmatov et al., 2021). Wnt-8 and frizzled 9-10, the key genes of the Wnt signaling pathway, which controls the embryonic processes involved in body axis patterning, cell fate specification, cell proliferation, and cell migration (Clevers, 2006), were up-regulated over 200-fold at gastrula stage. The nervous system is developed from the ectoderm formed during gastrulation, so key genes, regulating neural development such as dorsal root ganglia, homeobox protein-like was significantly up-regulated. In addition, Cdc20-like expression was up-regulated to regulate cell division from the blastula to the gastrula stage (Yu, 2007), Cdc family genes were also up-regulated during blastula and gastrula stage. It is worth noting that TNF receptor-associated factor 6-like (TRAF6) was down-regulated over 250-fold, which may due to the fact that TRAF6 negatively regulates NF-κB.
In the present study, RNA-Seq analysis was conducted on six embryonic and larval stages of sea cucumber development as follows: fertilized eggs, blastula, gastrula, auricularia, doliolaria, and pentactula. A total of 53.20~59.96 Mb× 18 clean reads were obtained, and 32,353 genes were identified and annotated. After filtering out low-quality genes, the dynamic molecular responses to development were revealed by WGCNA. The blue module and the yellow module were positively correlated with the gastrula and auricularia stages, respectively. The darkslateblue module had the highest correlation coefficient with the blastula stage. GO terms for “RNA” and “proteasome complex” were most significantly enriched in the blue module. In the darkslateblue and yellow module, receptors of signaling pathways and metabolic processes were significantly enriched, respectively. Five comparison groups, F Vs B, B Vs G, G Vs A, A Vs D, and D Vs P, were used to analyze differential expression genes during the development of A. japonicus. All DEGs could be categorized into 34 terms, with “Immune and Infectious diseases” being the most enriched. There was strong gene expression regulation from blastula to Auricularia. Functional enrichment analysis was performed on DEGs from each comparison group. The behavioral events and molecular features are summarized in Figure 8. The expression pattern of genes associated with adhesion, cell cycle, signal, transcription factor, extracellular matrix (ECM) and cytoskeleton was analyzed according to gene function. These results suggested that development and metamorphosis relied on complex molecular mechanisms in which many genes were involved. The present study provided useful transcriptomic resources for further research on A. japonicus development.
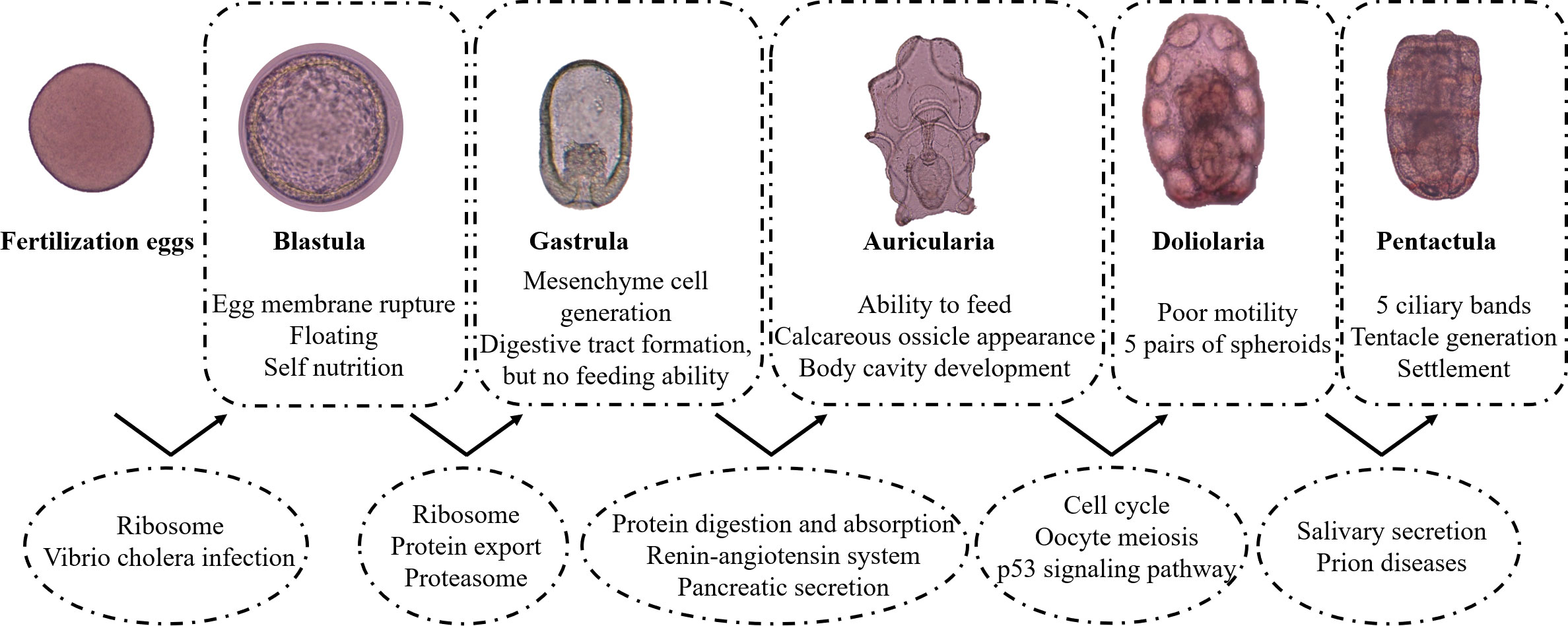
Figure 8 The major events during fertilization eggs to pentactula are shown in box. The significantly enriched signal pathways are shown in circle.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, PRJNA889469.
LS and HY conceived the study. FS carried out the field and laboratory work, participated in the data analysis, and drafted the manuscript. SL and LX collected the parent sea cucumbers. DH collected the experimental samples. LS, LX, and SL revised the manuscript. All authors approved the manuscript for publication. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (No. 42076093, 42276143), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA24030304), Youth Innovation Promotion Association CAS (No. 2019209).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1087339/full#supplementary-material
Supplementary Figure 1 | Diagram of the gene function in different comparison groups.
Supplementary Figure 2 | KEGG enrichment analysis of DEGs in F Vs B (A), B Vs G (B), G Vs A (C), A Vs D (D) and D Vs P (E).
Supplementary Table 4 | List of gene-module relationships.
Supplementary Table 6 | Sample expression pattern.
Supplementary Table 7 | GO enrichment analysis in three target modules.
Supplementary Table 8 | KEGG enrichment analysis in three target modules.
Supplementary Table 13 | The expression profile of key genes.
Ahmed H. O., Katow T., Katow H. (2011). Spatiotemporal expression pattern of gonad-stimulating substance-like peptide of the sea cucumber, Apostichopus japonicus. Dev. Growth Differ 53 (5), 639–652. doi: 10.1111/j.1440-169X.2011.01277.x
Asthana S., Martin H., Rupkey J., Patel S., Yoon J., Keegan A., et al. (2022). The physiological roles of the exon junction complex in development and diseases. Cells 11 (7). doi: 10.3390/cells11071192
Chen M., Zhu A., Storey K. B. (2015). Comparative phosphoproteomic analysis of intestinal phosphorylated proteins in active versus aestivating sea cucumbers. J. proteomics. 135, 141–150. doi: 10.1016/j.jprot.2015.09.016
Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127 (3), 469–480. doi: 10.1016/j.cell.2006.10.018
Davis A. J., Yan Z., Martinez B., Mumby M. C. (2008). Protein phosphatase 2A is targeted to cell division control protein 6 by a calcium-binding regulatory subunit. J. Biol. Chem. 283 (23), 16104–16114. doi: 10.1074/jbc.M710313200
Dolmatov I. Y., Nizhnichenko V. A., Dolmatova L. S. (2021). Matrix metalloproteinases and tissue inhibitors of metalloproteinases in echinoderms: Structure and possible functions. Cells 10 (9), 2331. doi: 10.3390/cells10092331
Dong G., Dong S., Wang F., Tian X. (2010). Effects of light intensity on daily activity rhythm of juvenile sea cucumber, Apostichopus japonicus (Selenka). Aquaculture Res. 41 (11), 1640–1647. doi: 10.1111/j.1365-2109.2010.02534.x
Du H., Bao Z., Hou R., Wang S., Su H., Yan J., et al. (2012). Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka 1867). PloS One 7 (3), e33311. doi: 10.1371/journal.pone.0033311
Gao Q., Liao M., Wang Y., Li B., Zhang Z., Rong X., et al. (2015). Transcriptome analysis and discovery of genes involved in immune pathways from coelomocytes of Sea cucumber (Apostichopus japonicus) after vibrio splendidus challenge. Int. J. Mol. Sci. 16 (7), 16347–16377. doi: 10.3390/ijms160716347
Ghosal G., Leung J. W., Nair B. C., Fong K. W., Chen J. (2012). Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis. J. Biol. Chem. 287 (41), 34225–34233. doi: 10.1074/jbc.M112.400135
Gianasi B. L., Hamel J.-F., Mercier A. (2018). Morphometric and behavioural changes in the early life stages of the sea cucumber Cucumaria frondosa. Aquaculture 490, 5–18. doi: 10.1016/j.aquaculture.2018.02.017
Gonzalez-Aguilera C., Askjaer P. (2012). Dissecting the NUP107 complex: Multiple components and even more functions. Nucleus 3 (4), 340–348. doi: 10.4161/nucl.21135
Gu M., Ma H., Mai K., Zhang W., Ai Q., Wang X., et al. (2010). Immune response of sea cucumber Apostichopus japonicus coelomocytes to several immunostimulants in vitro. Aquaculture 306 (1-4), 49–56. doi: 10.1016/j.aquaculture.2010.05.024
Guo X., Hao Y., Zhao Y., Shi H. (2020). Research advances on the function of n-acetyltransferase 10. Chem. Life 40 (11), 2014–2019.
Hamel J.-F., Hidalgo R. Y., Mercier A. (2003). Larval development and juvenile growth of the Galapagos sea cucumber. Isostichopus fuscus.
Hu C., Xu Y., Wen J., Zhang L., Fan S., Su T. (2010). Larval development and juvenile growth of the sea cucumber Stichopus sp. (Curry fish). Aquaculture 300 (1-4), 73–79. doi: 10.1016/j.aquaculture.2009.09.033
Huo D. L., Li X., Ru X., Liu S., Zhang L., et al. (2017). Differential expression of miRNAs in the respiratory tree of the Sea cucumber Apostichopus japonicus under hypoxia stress. G3 (Bethesda) 7 (11), 3681–3692. doi: 10.1534/g3.117.1129
Kato S., Tsurumaru S., Taga M., Yamane T., Shibata Y., Ohno K., et al. (2009). Neuronal peptides induce oocyte maturation and gamete spawning of sea cucumber, Apostichopus japonicus. Dev. Biol. 326 (1), 169–176. doi: 10.1016/j.ydbio.2008.11.003
Kim D., Langmead B., Salzberg S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi: 10.1038/nmeth.3317
Kong L., Zhang Y., Ye Z. Q., Liu X. Q., Zhao S. Q., Wei L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35 (Web Server issue), W345–W349. doi: 10.1093/nar/gkm391
Lacalli T. C., West J. E. (2000). The auricularia-to-doliolaria transformation in two aspidochirote holothurians, Holothuria mexicana and Stichopus californicus. Invertebrate Biol. 119 (4), 421–432. doi: 10.1111/j.1744-7410.2000.tb00112.x
Langfelder P., Horvath S. (2008). WGCNA: An r package for weighted correlation network analysis. BMC Bioinf. 9, 1–13. doi: 10.1186/1471-2105-9-559
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Lemonnier T., Dupre A., Jessus C. (2020). The G2-to-M transition from a phosphatase perspective: A new vision of the meiotic division. Cell Div 15, 9. doi: 10.1186/s13008-020-00065-2
Li B., Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinf. 12, 1–16. doi: 10.1186/1471-2105-12-323
Li L., Li Q. (2009). Effects of stocking density, temperature, and salinity on larval survival and growth of the red race of the sea cucumber Apostichopus japonicus (Selenka). Aquaculture Int. 18 (3), 447–460. doi: 10.1007/s10499-009-9256-4
Liu G., Yang H., Liu S. (2010). Effects of rearing temperature and density on growth, survival and development of sea cucumber larvae, Apostichopus japonicus (Selenka). Chin. J. Oceanology Limnology 28 (4), 842–848. doi: 10.1007/s00343-010-9092-4
Ma P., Ren B., Yang X., Sun B., Liu X., Kong Q., et al. (2017). ZC4H2 stabilizes smads to enhance BMP signalling, which is involved in neural development in xenopus. Open Biol. 7 (8), 170122. doi: 10.1098/rsob.170122
Ma T., Shi S., Jiang H., Chen X., Xu D., Ding X., et al. (2021). A pan-cancer study of spalt-like transcription factors 1/2/3/4 as therapeutic targets. Arch. Biochem. Biophys. 711, 109016. doi: 10.1016/j.abb.2021.109016
Margalit A., Vlcek S., Gruenbaum Y., Foisner R. (2005). Breaking and making of the nuclear envelope. J. Cell Biochem. 95 (3), 454–465. doi: 10.1002/jcb.20433
Matsuura H., Yazaki I., Okino T. (2009). Induction of larval metamorphosis in the sea cucumber Apostichopus japonicus by neurotransmitters. Fisheries Sci. 75 (3), 777–783. doi: 10.1007/s12562-009-0098-9
May M., Hwang K. S., Miles J., Williams C., Niranjan T., Kahler S. G., et al. (2015). ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum. Mol. Genet. 24 (17), 4848–4861. doi: 10.1093/hmg/ddv208
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi: 10.1101/gr.107524.110
Meng X.-L., Dong Y.-W., Dong S.-L., Yu S.-S., Zhou X. (2011). Mortality of the sea cucumber, apostichopus japonicus selenka, exposed to acute salinity decrease and related physiological responses: Osmoregulation and heat shock protein expression. Aquaculture 316 (1-4), 88–92. doi: 10.1016/j.aquaculture.2011.03.003
Miao T., Wan Z., Sun L., Li X., Xing L., Bai Y., et al. (2017). Extracellular matrix remodeling and matrix metalloproteinases (ajMMP-2 like and ajMMP-16 like) characterization during intestine regeneration of sea cucumber Apostichopus japonicus. Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 212, 12–23. doi: 10.1016/j.cbpb.2017.06.011
Nakano H., Murabe N., Amemiya S., Nakajima Y. (2006). Nervous system development of the sea cucumber Stichopus japonicus. Dev. Biol. 292 (1), 205–212. doi: 10.1016/j.ydbio.2005.12.038
Olins A. L., Rhodes G., Welch D. B., Zwerger M., Olins D. E. (2010). Lamin b receptor: Multi-tasking at the nuclear envelope. Nucleus 1 (1), 53–70. doi: 10.4161/nucl.1.1.10515
Pan Y., Zhang L., Lin C., Sun J., Kan R., Yang H. (2015). Influence of flow velocity on motor behavior of sea cucumber Apostichopus japonicus. Physiol. Behav. 144, 52–59. doi: 10.1016/j.physbeh.2015.02.046
Pertea M., Pertea G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi: 10.1038/nbt.3122
Qiu T. (2013). Research and application of key technical principles of sea cucumber (Apostichopus japonicus Selenka) ecological seed breeding (Doctoral dissertation, Doctoral Thesis. University of Chinese Academy of Sciences).
Sagne C., Agulhon C., Ravassard P., Darmon M., Hamon M., El Mestikawy S., et al. (2001). Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl. Acad. Sci. United States America 98 (13), 7206–7211. doi: 10.1073/pnas.121183498
Soliman T., Yamazaki Y., Niiyama H., Tsunoda K. (2013). Spontaneous captive breeding and larval development in the green and red variants of the Japanese sea cucumber Apostichopus japonicus(Selenka 1867). Aquaculture Res. 44 (5), 738–746. doi: 10.1111/j.1365-2109.2011.03078.x
Sun L., Chen M., Yang H., Wang T., Liu B., Shu C., et al. (2011). Large Scale gene expression profiling during intestine and body wall regeneration in the Sea cucumber Apostichopus japonicus. Comp. Biochem. Physiol. Part D: Genomics Proteomics 6), 195–205. doi: 10.1016/j.cbd.2011.03.002
Sun L., Sun J., Xu Q., Li X., Zhang L., Yang H. (2017a). Metabolic responses to intestine regeneration in sea cucumbers Apostichopus japonicus. Comp. Biochem. Physiol. Part D: Genomics Proteomics 22, 32–38. doi: 10.1016/j.cbd.2017.02.003
Sun L., Xu D., Xu Q., Sun J., Xing L., Zhang L., et al. (2017b). iTRAQ reveals proteomic changes during intestine regeneration in the sea cucumber Apostichopus japonicus. Comp. Biochem. Physiol. Part D: Genomics Proteomics 22, 39–49. doi: 10.1016/j.cbd.2017.02.004
Sun J., Zhang L., Pan Y., Lin C., Wang F., Kan R., et al. (2015). Feeding behavior and digestive physiology in sea cucumber Apostichopus japonicus. Physiol. Behav. 139, 336–343. doi: 10.1016/j.physbeh.2014.11.051
Tam P. P., Loebel D. A. (2007). Gene function in mouse embryogenesis: Get set for gastrulation. Nat. Rev. Genet. 8 (5), 368–381. doi: 10.1038/nrg2084
Wang C., Sun T., Guo J., Zhang K., Yin L. (2019). Studies on embryonic and larval development of yellow river delta Sea cucumber (Apostichopus japonicus). Fisheries Sci. Technol. Inf. 46 (04), 196–200+205. doi: 10.16446/j.cnki.1001-1994.2019.04.004
Wei J., Zhang X., Yu Y., Huang H., Li F., Xiang J. (2014). Comparative transcriptomic characterization of the early development in pacific white shrimp Litopenaeus vannamei. PloS One 9 (9), e106201. doi: 10.1371/journal.pone.0106201
Xing L., Sun L., Liu S., Li X., Zhang L., Yang H. (2018). Transcriptome analysis provides insights into the mechanism of albinism during different pigmentation stages of the albino sea cucumber Apostichopus japonicus. Aquaculture 486, 148–160. doi: 10.1016/j.aquaculture.2017.12.016
Xu D., Sun L., Liu S., Zhang L., Yang H. (2016). Understanding the heat shock response in the Sea cucumber Apostichopus japonicus, using iTRAQ-based proteomics. Int. J. Mol. Sci. 17 (2), 150–162. doi: 10.3390/ijms17020150
Yang H., Hamel J.-F., Mercier A. (Eds.). (2015). The sea cucumber Apostichopus japonicus: History, biology and aquaculture. Academic Press.
Yang A.-F., Zhou Z.-C., He C.-B., Hu J.-J., Chen Z., Gao X.-G., et al. (2009). Analysis of expressed sequence tags from body wall, intestine and respiratory tree of sea cucumber (Apostichopus japonicus). Aquaculture 296 (3-4), 193–199. doi: 10.1016/j.aquaculture.2009.08.016
Yu H. (2007). Cdc20: A WD40 activator for a cell cycle degradation machine. Mol. Cell 27 (1), 3–16. doi: 10.1016/j.molcel.2007.06.009
Yuan X., Shao S., Dupont S., Meng L., Liu Y., Wang L. (2015). Impact of CO2-driven acidification on the development of the sea cucumber Apostichopus japonicus (Selenka) (Echinodermata: Holothuroidea). Mar. pollut. Bull. 95 (1), 195–199. doi: 10.1016/j.marpolbul.2015.04.021
Zhang P., Li C., Zhang P., Jin C., Pan D., Bao Y. (2014). iTRAQ-based proteomics reveals novel members involved in pathogen challenge in sea cucumber Apostichopus japonicus. PloS One 9 (6), e100492. doi: 10.1371/journal.pone.0100492
Zhang P., Li C., Zhu L., Su X., Li Y., Jin C., et al. (2013). De novo assembly of the sea cucumber Apostichopus japonicus hemocytes transcriptome to identify miRNA targets associated with skin ulceration syndrome. PloS One 8 (9), e73506. doi: 10.1371/journal.pone.0073506
Zhang X., Sun L., Yuan J., Sun Y., Gao Y., Zhang L., et al. (2017). The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PloS Biol. 15 (10), e2003790. doi: 10.1371/journal.pbio.2003790
Keywords: Apostichopus japonicus, development, metamorphosis, RNA-Seq, WGCNA
Citation: Su F, Liu S, Xing L, Huo D, Yang H and Sun L (2023) Understanding gene regulation during the development of the sea cucumber Apostichopus japonicus using comparative transcriptomics. Front. Mar. Sci. 10:1087339. doi: 10.3389/fmars.2023.1087339
Received: 02 November 2022; Accepted: 22 February 2023;
Published: 09 March 2023.
Edited by:
Jinghui Fang, Chinese Academy of Fishery Sciences (CAFS), ChinaReviewed by:
Chunlin Wang, Ningbo University, ChinaCopyright © 2023 Su, Liu, Xing, Huo, Yang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lina Sun, c3VubGluYUBxZGlvLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.