 Shun Zhou1
Shun Zhou1 Jiwen Liu1,2,3
Jiwen Liu1,2,3 Peng Yao4
Peng Yao4 Liang Fu5
Liang Fu5 Zuosheng Yang6Yulin Zhang1
Zuosheng Yang6Yulin Zhang1 Rui Du1Chao Jia1
Rui Du1Chao Jia1 Lin Chen4Jinchang Liang1
Lin Chen4Jinchang Liang1 Xiaolei Wang1
Xiaolei Wang1 Xiaochong Shi1,2,3
Xiaochong Shi1,2,3 Xiao-Hua Zhang1,2,3
Xiao-Hua Zhang1,2,3 Min Yu1,2,3*
Min Yu1,2,3*- 1Frontiers Science Center for Deep Ocean Multispheres and Earth System, and College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Laboratory for Marine Ecology and Environmental Science, Laoshan Laboratory, Qingdao, China
- 3Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China
- 4Key Laboratory of Marine Chemistry Theory and Technology, Ministry of Education, Ocean University of China, Qingdao, China
- 5Sansha Track Ocean Coral Reef Conservation Research Institute, Sansha, China
- 6College of Marine Geosciences, Ocean University of China, Qingdao, China
Deep ocean blue holes possess steep physicochemical gradients, especially low dissolved oxygen concentration, which shape the extraordinary microbial communities. However, the environmental responses of microorganisms with different lifestyles and knowledge of culturable microorganisms in the blue holes are still unknown. Here, we investigated the bacterial community structure with different lifestyles of the world’s deepest blue hole - the Yongle Blue Hole (YBH) in the South China Sea using both culture-dependent and -independent methods. YBH can be divided by oxygen content into an oxic zone, a suboxic zone and two anoxic zones. The abundance of bacteria, archaea, genes dsrB and soxB were all higher in the free-living (FL) lifestyle than in the particle-associated (PA) lifestyle, yet the diversity and richness of PA bacteria were higher than that of FL bacteria. More Gammaproteobacteria and less Alphaproteobacteria, Chloroflexi and Nitrospinae were observed within the FL fraction than within the PA fraction. The relative abundance of sulfur-oxidizing bacteria (SOB) was dominant between 100-140 m (anoxic zone I) in YBH, with a maximum of 90.0% (140 m FL fraction). The SOB in YBH were mainly colorless sulfur bacteria and purple non-sulfur bacteria, of which Thiomicrorhabdus and Sulfurimonas were the main representatives. In addition, a total of 294 bacterial strains were isolated on a variety of media and culture conditions, and 22.2% (18/81) of anaerobic strains were identified as potential novel species. Our study reveals a distinction between FL and PA bacteria in YBH. It contributes to further understanding of the bacterial community in deep ocean blue holes, and provides bacterial resources for subsequent studies on their adaptation to extreme marine environments.
Introduction
A marine blue hole is a submerged marine cave, generally considered to be an underwater carbonate karst system with a large horizontal scale, found in all the world’s ocean waters (Smart et al., 1988; Mylroie et al., 1995; Schwabe, 1999; Bishop et al., 2015). Most of the marine blue holes in the world are offshore and are the product of repeated global sea level rise and fall (Mylroie et al., 1995; Becking et al., 2011; Smith et al., 2011). The relatively isolated internal environment causes physical-chemical jumps in temperature, salinity, dissolved oxygen and hydrogen sulfide, and stable anoxic environments (Seymour et al., 2007; Iliffe and Kornicker, 2009; Pohlman, 2011). The blue hole is named for its dark blue color, which contrasts with the lighter colored waters surrounding it (Mylroie et al., 1995). The world’s proven ocean blue holes include Yongle Blue hole (300.89 m) (Xie et al., 2019), Bahamas Dean’s Blue Hole (202.08 m) (Wilson, 1994), Egypt Dahab Blue Hole (131.06 m) (Naumann et al., 2015) and Belize Great Blue Hole (124.05 m) (Schmitt et al., 2020).
The Yongle Blue Hole (YBH, also known as the Dragon Hole) is located between the reefs of Jinqing Island and Shiyu Island in Yongle Atoll, Xisha Islands, Sansha City. YBH is 300.89 m in depth and about 130 m in diameter (Xie et al., 2019), and its formation may date back to the Miocene (Li et al., 2018a; Han et al., 2020). Due to the barrier effect of the pycnocline, the vertical exchange of dissolved oxygen is blocked, which in turn leads to a sharp decrease in the dissolved oxygen content at about 80-100 m (Bi et al., 2018; Xie et al., 2019; Yao et al., 2020). Below the depth of 100 m, there is an anoxic state in YBH since it is semi-enclosed. In such an environment, the water body transforms from oxic to hypoxia and then to anoxic at a depth of nearly 200 m (Bi et al., 2018; Yao et al., 2020). Such unique and rare oxygen conditions provide an excellent natural laboratory for biogeochemical studies (Xie et al., 2019). The biological community structure and potential functions of YBH have been investigated, including the phytoplankton (Liu et al., 2019; Fu et al., 2020; Ge et al., 2020), zooplankton (Chen et al., 2018; Li et al., 2020b), archaeal (He et al., 2019) and bacterial components (He et al., 2019; He et al., 2020; Li et al., 2020a; Zhang et al., 2021), and most research focused on the diversity and function of bacteria.
Previous studies (Zhang et al., 2021) divided YBH into three zones according to the oxygen concentration, including the oxic zone (0-80 m), the hypoxic zone (80-100 m) and the anoxic zone (100-300 m). Although the water depth in YBH is only 300 m, several studies found multiple dramatic changes in the bacterial community structure from the surface to the bottom waters based on the high-throughput sequencing of 16S rRNA gene amplicon (He et al., 2019; He et al., 2020; Li et al., 2020a; Zhang et al., 2021). For the oxic zone, in addition to the overwhelmingly dominant Pseudomonadota (Proteobacteria), a relatively high abundance of Bacteroidetes was also found, with over 20% at 0 m and 20-50 m (Zhang et al., 2021). In the hypoxic zone, the abrupt increased in abundances of bacterial, archaeal and anammox 16S rRNA genes, and genes nirS and dsrB were found by qPCR, possibly due to the presence of more phytoplankton in this zone (He et al., 2019). Moreover, genes associated with carbon (methane for example), nitrogen and sulfur cycles appeared enriched in the hypoxic zone by metagenome sequencing (He et al., 2020). In the anoxic zone, bacteria belonging to Pseudomonadota were dominant, especially at depths of 130-170 m and 270 m where the relative abundance even reached 97.0% (Zhang et al., 2021). At the genus level, Thiomicrospira and Arcobacter contributed significantly to the microbial community in the anoxic zone of YBH, with abundances of up to 96.1% and 31.5% at 270 m and 130-150 m, respectively (Zhang et al., 2021). Dissolved oxygen (DO), temperature, salinity, pH, sulfur and nutrient concentrations were considered to be the main environmental factors affecting the microbial community within YBH (He et al., 2019; He et al., 2020). Current results indicated that microbial communities in YBH shifted along depths and the environment in YBH shaped the unique microbial communities. In addition, many studies reported that free-living (FL) and particle-associated (PA) communities showed different responses to various environmental factors, resulting in diverse microbial communities (DeLong et al., 1993; Tarn et al., 2016; Mestre et al., 2017; Liu et al., 2020b). However, little is known about the microbial communities of FL and PA lifestyles in YBH. Therefore, it is hypothesized that microbial communities of FL and PA lifestyles may show different distribution patterns in YBH, and there are still a lot of unclassified microorganisms in YBH that could be explored using various culture conditions.
In this study, we comprehensively revealed the structure of the bacterial community within YBH at a fine scale combining cultivation-independent methods with traditional cultivation-dependent methods. On the one hand, high-throughput sequencing and quantitative PCR were used to study the composition and abundance of the bacterial communities of FL and PA lifestyles. On the other hand, a variety of media and cultural conditions were used for isolating diverse bacteria including sulfur oxidizing bacteria and anaerobic bacteria.
Methods and materials
Sample collection
Water samples were collected in mid-March 2017 with YBH (Yao et al., 2020). Briefly, due to the shallow water around YBH, the research vessel (Qiong Qionghai 03138) had to be moored in the lagoon at Yongle Atoll during the survey, approximately 1.7 km from YBH. There was a buoyant platform built on the water surface of YBH, and a winch was placed with a Niskin water collector (8 L, Keruiou, Tianjin) mounted for water sample collection. All water samples were collected on the buoyant platform and immediately transferred to the research vessel for subsequent processing. Water samples were taken at a total of 13 depths, including 0, 10, 40, 70, 80, 85, 90, 100, 120, 140, 170, 190 and 230 m within YBH. About 800 of ml seawater was filtered through 3 µm and 0.22 µm polycarbonate membranes (Millipore, USA) to obtain PA and FL fractions. The membranes were immediately frozen in liquid nitrogen and then stored on board at -20°C before being transferred to -80°C in the laboratory. Environmental factors, such as temperature, salinity, concentrations of dissolved oxygen (DO), , , and H2S, were determined on site as described previously (Yao et al., 2020). The environmental factors involved in this paper are presented in Table S1. Unfiltered seawater was collected for bacterial culture experiments.
Based on previous studies (Yao et al., 2020) on the anoxic marine zones (AMZs) (Canfield et al., 2010; Garcia-Robledo et al., 2017; Zakem et al., 2020) and the oxygen minimum zones (OMZs) (Trouwborst et al., 2006; Moore et al., 2009), the water column within YBH in this study was divided according to the DO concentration, with 0-80 m being the oxic zone (DO≥3 mg/L), 80-100 m being the suboxic zone (0<DO<3 mg/L). Under 100 m was the anoxic zone, which can be further subdivided into two zones, that were 100-140 m being the anoxic zone I (DO below the lower detection limit of the sensor or assay but with markers of metabolic processes that can demonstrate the presence of oxygen, such as nitrate and small amounts of hydrogen sulfide) and below 140 m being the anoxic zone II (nitrate and nitrite were not available, but a significant amount of hydrogen sulfide can be detected, indicating that the DO was zero).
DNA extraction and quantitative PCR (qPCR)
The total genomic DNA was extracted from membranes following the methods described previously (Liu et al., 2015). The bacterial 16S rRNA gene, archaeal 16S rRNA gene, and genes encoding for dissimilatory sulfite reductase β subunit (dsrB) and SoxB, were amplified with Eub338F/Eub518R (Lane, 1991), 967F/1060R (Cadillo-Quiroz et al., 2006), DSRp2060F/DSR4R (Cabrera et al., 2006) and soxB1164F/soxB1446B (Krishnani et al., 2010), respectively. PCR reactions were carried out as follows: initial denaturation at 95°C for 3 min followed by 35 cycles of 95°C for 30 s, primer specific annealing temperature for 30 s and 72°C for 30 s. The melting curve was then performed as follows: denaturing at 95°C for 1 min, starting at annealing temperature in 0.5°C increments and collecting the signal. A standard curve was generated using a plasmid containing the target gene. In addition to template DNA, each reaction included serial dilutions of the plasmid and a negative control to ensure that the qPCR assay was free from contamination and stable. All samples were run on the StepOne™ real-time PCR system (Applied Biosystems, USA) and the data obtained were analyzed by the StepOne software (version 2.2).
High-throughput sequencing and analyses
Primers 338F (5’-ACTCCTACGGGAGGCAGCA-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) were used to amplify the V3-V4 region of the bacterial 16S rRNA gene. Amplification was performed in triplicate for each sample in a reaction system containing 2 µl of 10X polymerase chain reaction (PCR) buffer, 2 µl of 2.5 mM dNTPs, 0.5 µl of each primer (5 µM), 0.2 µl of rTaq polymerase, 0.2 µl of bovine serum albumin and 10 ng of template DNA. The PCR reaction was 95°C for 3 min, 25 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s and a final extension at 72°C for 5 min. After the amplification, the triplicate PCR products were mixed for the downstream analysis. The PCR amplicons were purified using the AxyPrepDNA Gel Extraction Kit (Axygen, China) and quantified using the Quant-iT PicoGreen Double Stranded DNA Detector (Invitrogen, USA). The purified amplicons were pooled together at equimolar concentrations and sequenced on the Illumina PE 250 platform at Majorbio Bio‐pharm Tech. Co., Ltd. (Shanghai, China). The raw data were merged and filtered for subsequent analyses. All screened reads were clustered into operational taxonomic units (OTUs) with a similarity cutoff of 97%. The taxonomy of each 16S rRNA gene sequence was analyzed by the RDP classifier against the SILVA v128 16S rRNA database with a 70% confidence level. Taxa assigned to sulfur-oxidizing bacteria (SOB) and sulfate-reducing bacteria (SRB) were based on the 16S rRNA gene sequencing data according to the taxonomy (Ghosh and Dam, 2009; Rosenberg et al., 2014). The raw data were deposited in the National Center for Biotechnology Information Short Read Archive database under the accession number SRP178367.
Enrichment cultures, isolation and identification of bacteria
Seawater from different depths were diluted in a serial dilution (10-1 to 10-5), and aliquots (100 μl) of each dilution were spread on marine agar 2216E (MA) (Patrick, 1978) or marine R2A (Gibbs and Hayes, 1988) plates, respectively. The inoculated plates were incubated at room temperature on board and at 20°C in the laboratory. The numbers of colonies (colony forming units, CFU) were counted after 7 days’ cultivation when most morphologically distinct colonies appeared on MA plates. Individual colonies were randomly selected and purified by streaking three times on MA or R2A plates at 20°C. The purified isolates were stored at -80°C in sterile 0.85% (w/v) NaCl supplemented with 15% (v/v) glycerol. The isolates were identified using 16S rRNA gene sequencing and the EzBioCloud server (Fu et al., 2018). For the enrichment of sulfur-oxidizing bacteria, seawater samples from 10, 110, 140 and 270 m were inoculated at 2% inoculum in serum bottles containing 50 ml liquid SPG medium (the components of media are available in Table S2) on board, respectively. Once the color of the culture changed from red to yellow or purplish-red, the subculture was performed using 2% inoculum for three times. After the enrichment, cultures were spread onto SPG plates for isolation and purification. For anaerobic bacteria, 1 ml water sample from 90, 130, 160 and 190 m was injected by syringe into an anaerobic flask containing 5 ml anaerobic marine broth 2216E (MB) on board, respectively. After incubation at 16°C for one month, or the enrichment becoming turbid, subsequent purification and identification were carried out (Fu et al., 2018).
Statistical analysis
Alpha diversity indices, including Chao 1 and Shannon, were performed by Mothur (Schloss, 2020) to estimate the community richness and diversity. For beta diversity analysis, non-metric multidimensional scaling (NMDS) was performed based on Bray-Curtis dissimilarity. Differences in community structure between the two lifestyles were tested for significance by analysis of similarities (ANOSIM). Both NMDS and ANOSIM analyses were conducted using the “vegan” package of R software (version 4.1.1) (Oksanen et al., 2013). Relationships between bacterial community structure and environmental factors were assessed by distance-based redundancy analysis (db-RDA), and were performed using Canoco software (version 5.0) (Šmilauer and Lepš, 2014) with a 9,999 Monte Carlo permutation test. Correlations between environmental factors and the abundance of functional genes were tested using Pearson correlation tests. The relative contributions of nutrition and environmental factors were estimated by Variance Partitioning Analysis (VPA) using the “vegan” package of R software (version 4.1.1) (Oksanen et al., 2013), with adjusted R2 coefficients based on RDA. The relationship between the two lifestyles and environmental factors was tested using the “ade4” package of R software (version 4.1.1) (Dray and Dufour, 2007).
Neutral community model (NCM) (Sloan et al., 2006; Zhou and Ning, 2017) was used to assess the potential contribution of stochastic processes to the assembly of bacterial communities, which can be expressed simply as the following equation.
N described the metacommunity size, and it was the total abundance of all OTUs in each sample here. m quantified the migration rate at the community level. Nm was the product of N and m, quantifying the estimate of dispersal between communities. The calculations were performed using the “Hmisc”, “minpack.lm” and “stats4” packages in R (version 4.1.1) (Chen et al., 2019). Statistical analyses were performed by SPSS version 24.0 (SPSS Inc., USA).
Results
Abundance, diversity and richness of bacteria
The abundances of bacterial and archaeal 16S rRNA genes (Figure 1A) in YBH were analyzed by qPCR. The trends in the abundance of 16S rRNA genes in FL (1.74×107-4.87×108 copies/L) and PA fractions (1.49×106-3.85×107 copies/L) were approximately the same. The abundance of bacteria remained essentially constant in the oxic zone, increased slightly in the suboxic zone, and reached the highest values in the anoxic zone I and decreased gradually in the anoxic zone II. The results for archaea were similar, with the difference that FL archaea had a substantial increase in the oxic zone, and the 16S rRNA gene abundance of FL and PA archaea were 8.36×103-2.09×106 copies/L and 9.65×102-1.76×104 copies/L, respectively.
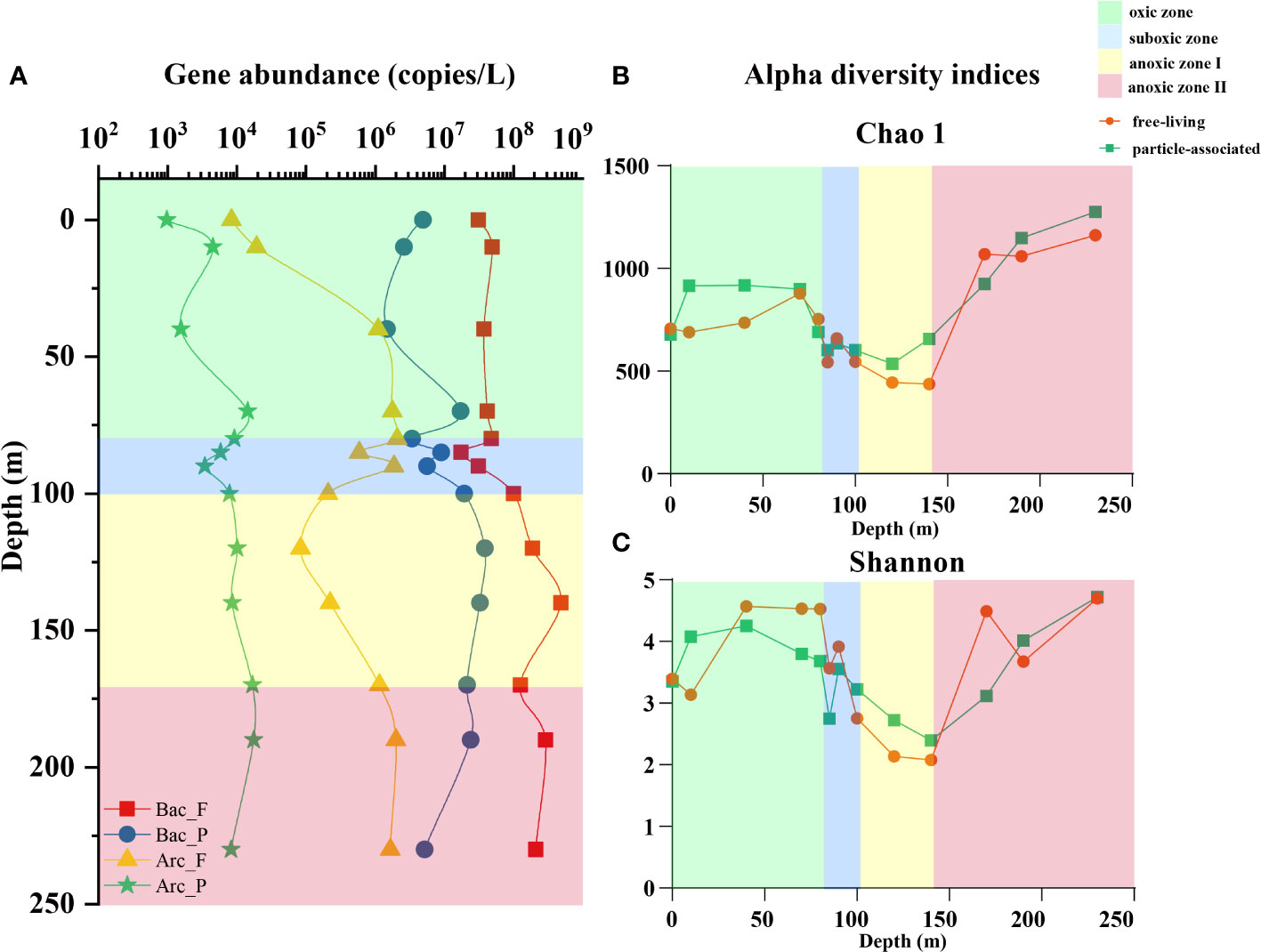
Figure 1 (A) The abundance of bacterial and archaeal 16S rRNA genes for free-living and particle-associated lifestyles. F, free-living lifestyle. P, particle-associated lifestyle. Alpha indices including (B) Chao 1 index and (C) Shannon index for bacteria in the two lifestyles. The orange dots represent free-living bacteria and the green squares represent particle-associated bacteria. Green background represents the oxic zone, the blue background represents the suboxic zone, the yellow background represents anoxic zone I, and the red background represents the anoxic zone II.
For the 26 samples from 13 water layers, a total of 947,543 sequences were obtained after high-throughput sequencing. After rarefaction, the number of sequences in each sample was 27,593, and the coverage of all samples was above 99%. Divided by a threshold of 97% similarity, a total of 1,988 OTUs were generated. The indices for alpha diversity analyses are shown in Figures 1B, C and Figure S1, including estimators for community richness (OTU numbers, Chao 1, Ace), community diversity (Shannon, Simpson) and coverage. Generally, these indices revealed that the diversity and richness of the prokaryotic community in the PA lifestyle were moderately higher than that in the FL lifestyle but with no significant difference (P>0.05). In the vertical direction, the community richness and diversity remained relatively stable in the oxic zone, then consistently decreased in the suboxic zone and anoxic zone I, and increased in the anoxic zone II.
In the NMDS analysis at the OTU level, the bacterial communities in YBH could be clearly distinguished according to the two lifestyles, FL and PA (ANOSIM, R2 = 0.48, P<0.05; Figure 2A). As can be seen, along the first axis, the samples were well differentiated by DO concentration and could be clustered into oxic, suboxic and anoxic groups. At the same time, the two lifestyles within the same zone were separated. Furthermore, bacterial community structure varied significantly between lifestyles within each zone defined (ANOSIM, R2 = 0.51 for the oxic zone; R2 = 0.42 for the suboxic zone, R2 = 0.60 for anoxic zone I; R2 = 0.46 for anoxic zone II, respectively; P<0.05; Figure 2B).
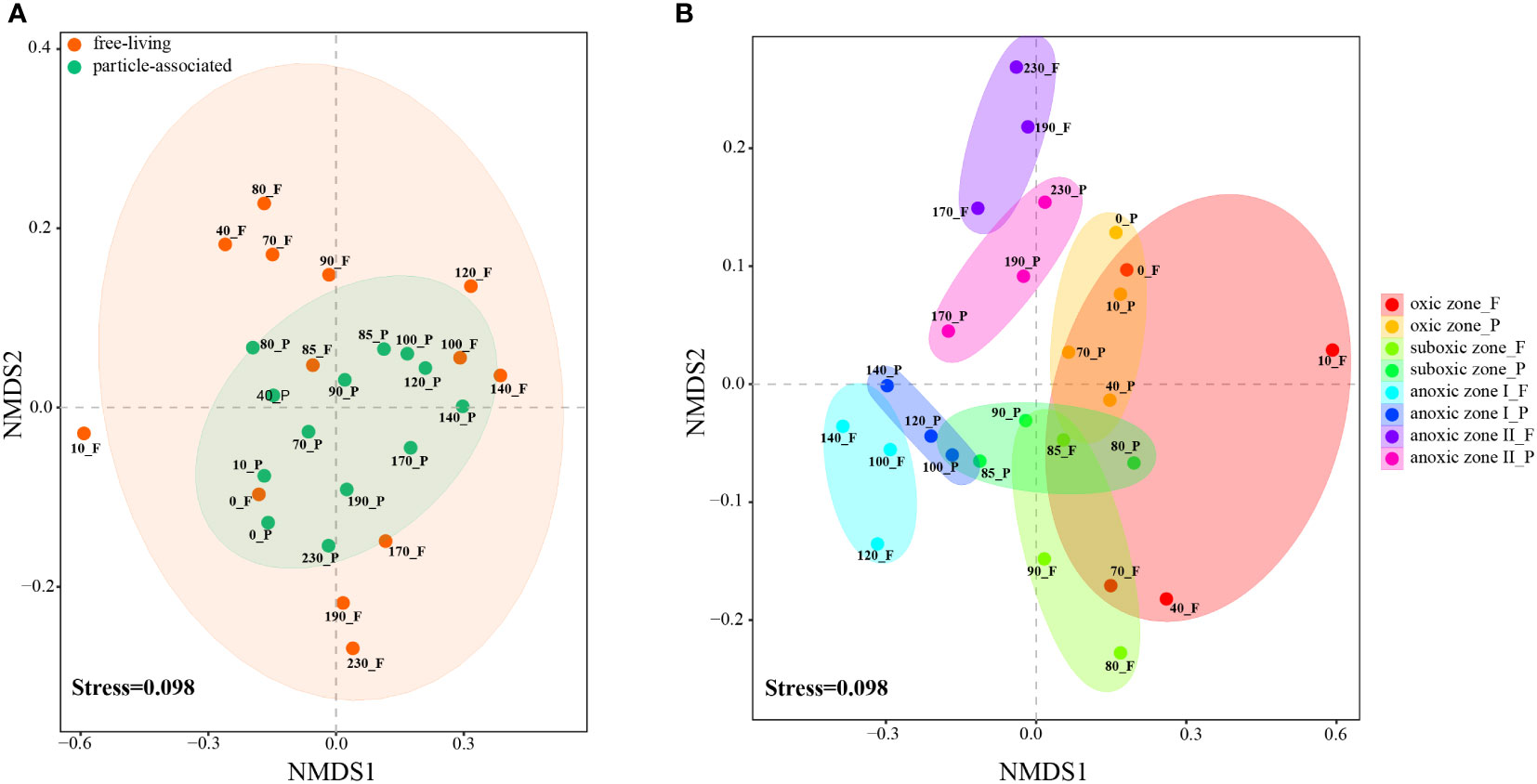
Figure 2 NMDS plots at OTU level show the clustering relationships between the two lifestyles of bacteria in all samples (A) and in the four zones (B). Samples in different groups are color-coded. F, free-living bacteria. P, particle-associated bacteria.
Bacterial community structure
Although the depth of YBH was only 300 m, the community structure of bacteria showed substantial variations among depths. At the phylum/class level (for bacteria within the phylum Pseudomonadota, the individual classes were used for analysis), the most dominant taxon was Gammaproteobacteria for both PA (41.17%-86.62%) and FL fractions (16.88%-78.31%) across all depths, with a significantly higher proportion (P<0.05) in the PA lifestyle (Figures 3A, B). The oxic zone was dominated by Gammaproteobacteria and Alphaproteobacteria, with FL fraction having more Alphaproteobacteria and less Gammaproteobacteria compared to PA fraction (P<0.01). Additionally, more Bacteroidetes, Chloroflexi, SAR406 clade and Actinobacteria were observed among the FL fraction (P<0.01). There were a number of Cyanobacteria in the 10 m and 40 m samples for both lifestyles in this zone. As for the suboxic zone, the main distinction from the oxic zone was the emergence of Campylobacterota. In the anoxic zone I, the abundance of Campylobacterota increased in both lifestyles, while Alphaproteobacteria almost disappeared, and Gammaproteobacteria changed little compared to the oxic and suboxic zones. In the anoxic zone II, the diversity of microbial communities was much higher, and Desulfobacterota, SAR406 clade and Parcubacteria were more abundant in the FL fraction. Meanwhile, there were more Alphaproteobacteria in the PA than in the FL lifestyle (all P<0.01).
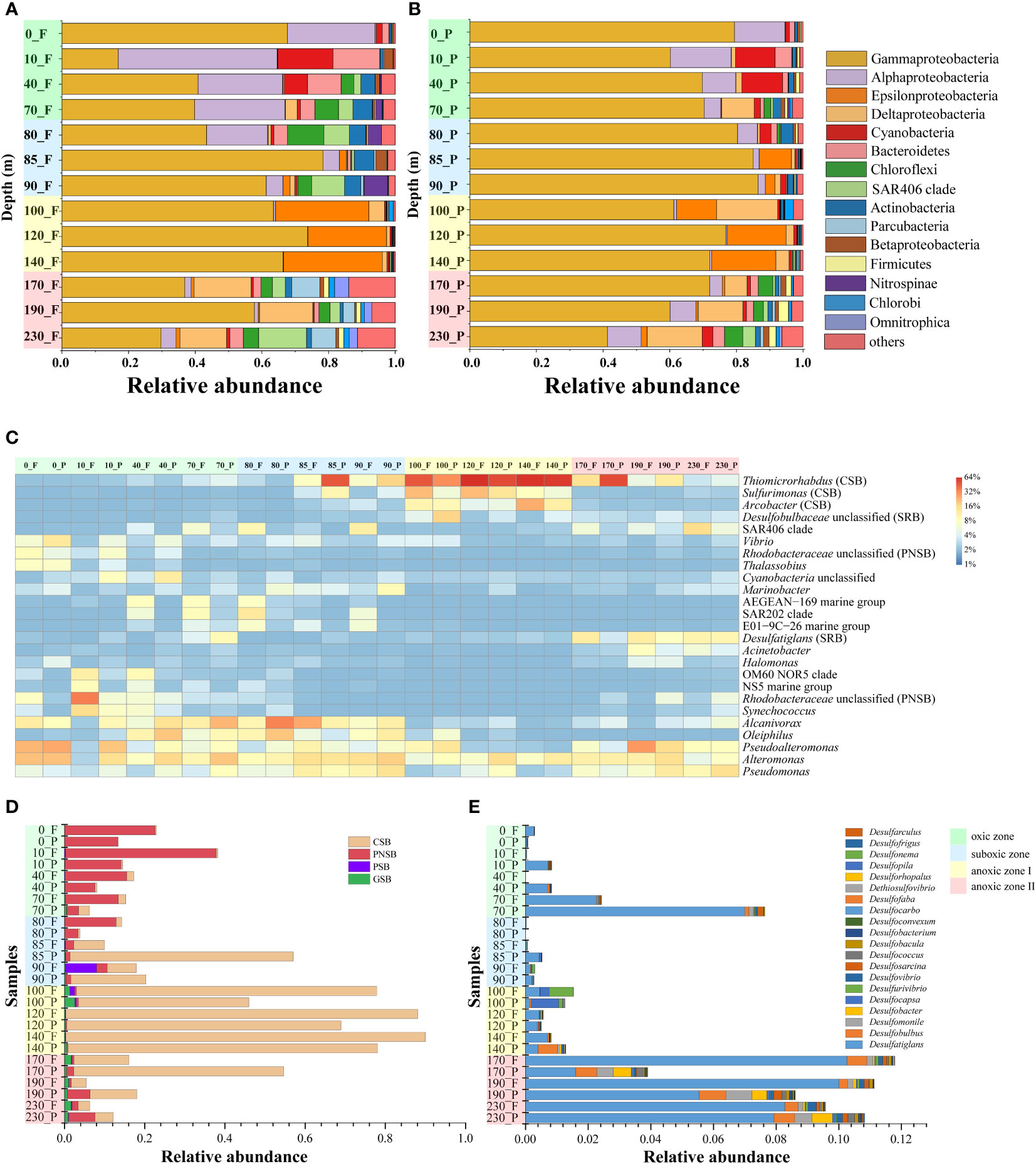
Figure 3 Bacterial communities at phylum level (for bacteria within the phylum Pseudomonadota, the individual classes under it were used for analysis) for free-living (A) and particle-associated bacteria (B). (C) Heatmap for TOP 25 genus within the bacterial communities. F, free-living bacteria. P, particle-associated bacteria. Green background represents the oxic zone, the blue background represents the suboxic zone, the yellow background represents anoxic zone I, and the red background represents the anoxic zone II. CSB, colorless sulfur bacteria. PNSB, purple non-sulfur bacteria. SRB, sulfate-reducing bacteria. The composition of sulfur-oxidizing bacteria (D) and sulfate-reducing bacteria (E). F, free-living bacteria. P, particle-associated bacteria. CSB, colorless sulfur bacteria. PNSB, purple non-sulfur bacteria. PSB, purple sulfur bacteria. GSB, green sulfur bacteria. Green background represents the oxic zone, the blue background represents the suboxic zone, the yellow background represents anoxic zone I, and the red background represents the anoxic zone II.
The detailed results of the community structure at the genus level are presented in Figure 3C. In the oxic zone, the main taxa were Alteromonas, Pseudoalteromonas and Alcanivorax. Notably, Rhodobacteraceae (not assigned to the genus, 35.08%) and Synechococcus (14.27%) showed a high abundance in the FL fraction of 10 m layer and Vibrio (11.36%) in the PA fraction of 0 m layer, respectively. In the suboxic zone, it was mainly composed of Alteromonas, Pseudoalteromonas, Alcanivorax and Oleiphilus, while the abundance of the two genera Thiomicrorhabdus and Sulfurimonas belonging to SOB also began to increase. It was remarkable that the two SOB genera were significantly more abundant in PA fraction in this zone (P<0.01). However, in the anoxic zone I, there was a massive difference from the oxic and suboxic zones, which was dominant by two SOBs, Thiomicrorhabdus (56.90% for FL fraction and 46.30% for PA fraction) and Sulfurimonas (15.38% for FL fraction and 6.84% for PA fraction), and both taxa were more abundant in FL lifestyles (P<0.01). In the anoxic zone II, the abundance of these two SOB genera, Thiomicrorhabdus and Sulfurimonas decreased, while Prosthecochloris (SOB, ranked 33rd in relative abundance) and Desulfatiglans (SRB) showed an increasing trend. In addition, Thiomicrorhabdus was more abundant in PA lifestyle (P<0.01) and Prosthecochloris was more abundant in FL fraction (P<0.05).
The above analyses found that several taxa were associated with the sulfur cycle in YBH, and the abundance and composition of bacteria involved in sulfur metabolism were further analyzed. Among them, bacteria identified as SOB could be clustered into the four main categories, which were green sulfur bacteria (GSB), purple sulfur bacteria (PSB), purple non-sulfur bacteria (PNSB) and colorless sulfur bacteria (CSB). The results are shown in Figure 3D. The relative abundance of SOB varied dramatically among all 26 samples (3.83% in the 80 m PA sample to 89.99% in the 140 m FL sample), with the taxon distribution shifting considerably with depth. The SOB in YBH were dominated by CSB and PNSB. For PNSB, it prevailed in the oxic zone (mainly in the FL lifestyle) and anoxic zone II (mainly in the PA lifestyle). Whereas, CSB was abundant in the suboxic zone and anoxic zone I. The relative abundances of both GSB and PSB were low, and GSB was concentrated at 100 m and in the anoxic zone II, while PSB was presented at 90 and 100 m. For SRB, they were primarily occurred at 70 m and in the anoxic zone II, with Desulfatiglans as the predominant genus (Figure 3E). For the two genes involved in sulfur metabolism, the range of variation was 9.53×102-9.35×105 copies/L for dsrB and 3.19×102-1.52×105 copies/L for soxB, respectively, with a dramatic and very rapid increase in the suboxic zone (Figure S2).
Correlation between bacterial composition and environmental factors
Environmental factors that determined the bacterial structure at each depth were analyzed by db-RDA (Figure 4A). The RDA1 and RDA2 axes explained 28.63% and 15.61% of the variation in bacterial composition, respectively. All the samples were clustered along the first axis into oxic, suboxic and anoxic groups. The 15 environmental factors included in the analysis had significant roles in shaping the community structure of bacteria within YBH, and the top 4 environmental factors in order of influence were silicate (F=6.5, P=0.002), phosphate (F=6.3, P=0.003), pH (F=6.2, P=0.012) and temperature (F=6.2, P=0.022). The VPA analysis showed that nutrition and physicochemical properties could explain more for the PA fraction than for the FL fraction (Figure S3, 0.4951 Vs. 0.3119). At the same time, both lifestyles were mainly influenced by a combination of nutrition and physicochemical properties with an explanation degree of 0.3691 and 0.3690 for FL and PA fraction, respectively. The mental test revealed that the components of the two lifestyles were mainly controlled by temperature, salinity, pH, DO and silicate. In addition, the effects of dissolved inorganic nitrogen (DIN), N/P and Si/N varied between the two lifestyles (Figure S4).
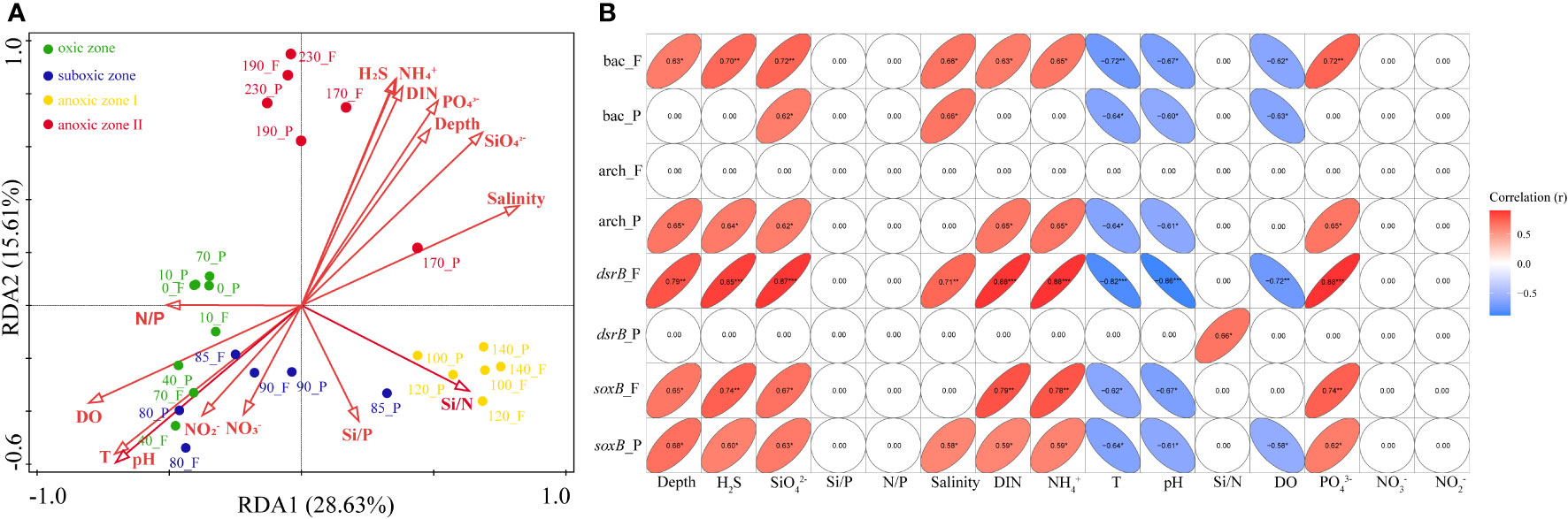
Figure 4 (A) Redundancy analysis (RDA) diagram illustrating the relationship between samples and environmental factors. Environmental factors are indicated by red arrows, and dots in different color represent samples from different zones. (B) Correlations between gene abundance and environmental factors. *0.01<P < 0.05; **=0.001<P ≤ 0.01; ***=P ≤ 0.001. bac, bacterial 16S rRNA gene. arch, archaeal 16S rRNA gene. F, free-living bacteria. P, particle-associated bacteria.
The abundance of bacteria in the PA fraction was mainly negatively regulated by temperature, DO and pH, and positively regulated by silicate as well as salinity, while the abundance of bacteria in the FL fraction was significantly positively regulated by ammonium, phosphate, DIN, hydrogen sulfide, depth, silicate and salinity. As for archaea, in the PA lifestyle were negatively regulated by temperature and pH, and positively regulated by ammonium, silicate, DIN, hydrogen sulfide, phosphate and depth. No environmental factors were significantly correlated with the abundance of FL archaea. Meanwhile, of the 15 environmental factors included in the study, the abundance of the dsrB gene in the FL fraction was significantly correlated with 10 factors, while the dsrB gene in the PA fraction was only positively correlated with Si/N. For soxB in both lifestyles of bacteria, it was primarily influenced by temperature, pH, ammonium, phosphate, silicate, DIN, hydrogen sulfide and depth. Furthermore, the abundance of soxB in the PA fraction was also correlated with salinity and DO. The detailed values of the correlation results are shown in Figure 4B.
The neutral community model at the OTU level explained more of the microbial community variation for the PA fraction (R2 = 0.595) than that for the FL fraction (R2 = 0.285) in YBH. The Nm-value (metacommunity size times immigration) was higher for the PA fraction (Nm=3,903) than the FL fraction (Nm=2,188). Since the number of sequences in each sample was 27,593, the m (migration rate) value was estimated to be 0.141 in PA and 0.079 in FL fraction, respectively (Figures 5A, B).
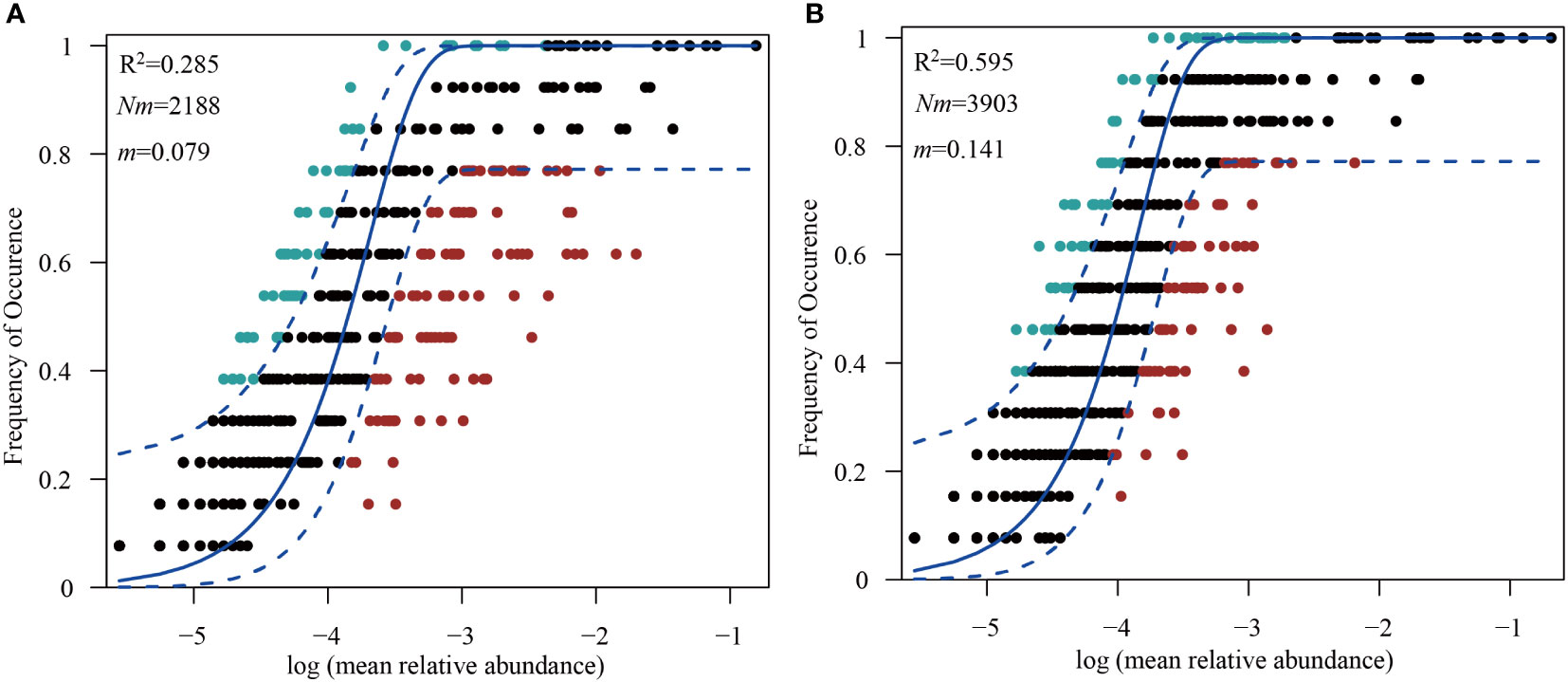
Figure 5 Fitting of a neutral community model (NCM) to the community assemblage at OTU level. The frequencies of occurrence of free-living (A) and particle-associated bacteria (B) are predicted respectively.
Diversity of culturable bacteria
The numbers of culturable bacteria on MA plates were found highest at the depths of 40 m (7,488 CFU/ml), 95 m (3,874 CFU/ml) and 0 m (2,232 CFU/ml). A total of 125 bacterial strains were isolated from MA and R2A plates. At the phylum/class level, 99 strains of Gammaproteobacteria, 19 strains of Alphaproteobacteria, 5 strains of Actinobacteria and 2 strains of Firmicutes were isolated. At the genus level (Figure 6A), the dominant taxa were Alcanivorax (34), Alteromonas (28), Vibrio (11), Pseudomonas (10), Erythrobacter (7), Marinobacter (7), and Pseudovibrio (6).
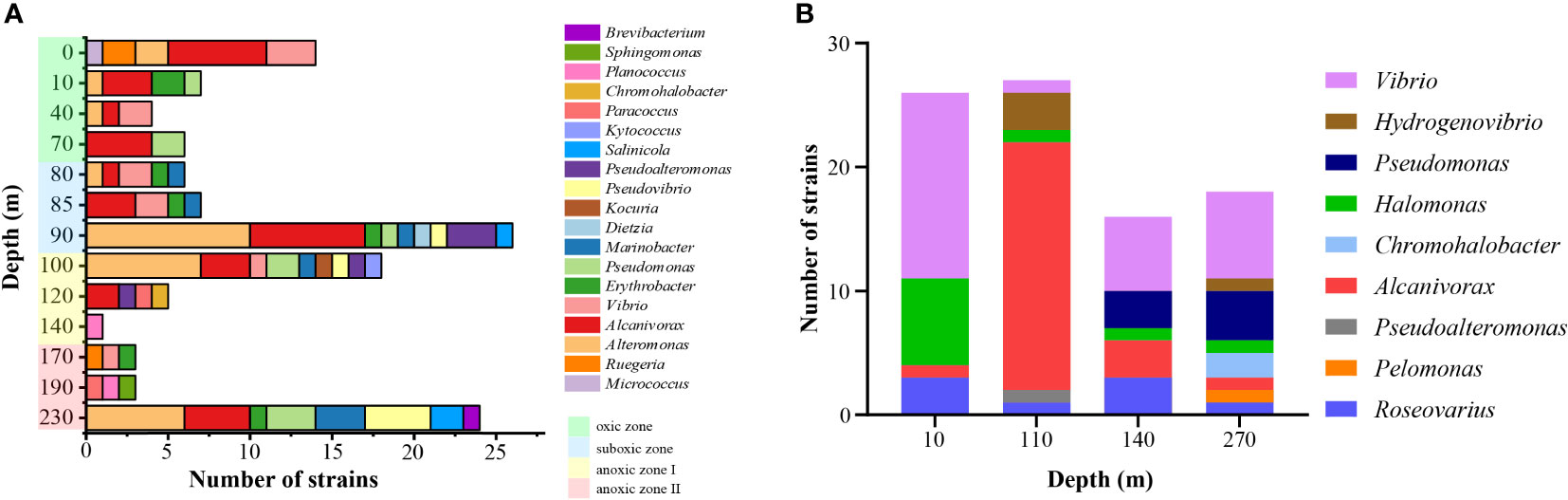
Figure 6 The number of culturable bacteria at the genus level in YBH. (A) Numbers of strains isolated on MA and R2A plates. (B) Numbers of sulfur-oxidizing bacteria enriched and isolated from SPG liquid medium. Green background represents the oxic zone, the blue background represents the suboxic zone, the yellow background represents anoxic zone I, and the red background represents the anoxic zone II.
For the enrichment of SOB, a total of 88 bacterial strains were obtained by enrichment and isolation of seawater samples from the four water layers (Figure 6B). All these strains belong to Pseudomonadota, including 77 strains of Gammaproteobacteria, 10 strains of Alphaproteobacteria and 1 strain of Betaproteobacteria. For samples from different depths, Vibrio was abundant at 10 m, 140 m and 270 m. Besides, more Halomonas were isolated at 10 m, whereas Alcanivorax at 110 m, Pseudomonas, Alcanivorax and Roseovarius at 140 m, and Pseudomonas at 270 m, respectively.
For the cultivation of anaerobic bacteria, 81 strains of anaerobic bacteria were isolated and identified from samples of 90, 130, 160 and 190 m. At the phylum level, Bacteroidetes had the highest abundance (21%, 17/81). At the genus level, the dominant genera were Saccharicrinis and Halodesulfovibrio, both in equal proportions (14%, 11/81 of each). Eighteen bacterial strains in 10 genera showed the 16S rRNA gene sequence similarity lower than 98.65% (90.24%-97.90%) with their closely related bacteria, which could be considered novel bacteria and need further analyses.
Discussion
As a specific habitat, studies on the abundance and community composition of bacteria in blue holes mainly focused on the Bahamas Blue Hole and YBH. However, current research on microorganisms in YBH was mainly based on the whole microbial communities, and there was a lack of understanding of the culturability of microorganisms in blue holes. Our results primarily showed differences in community structure, gene abundance and response to the environmental factors between bacteria with FL and PA lifestyles within YBH. We also used culture-dependent methods as a complementary to expand the knowledge of the diversity of culturable bacteria living in this specific habitat. The results indicated the presence of a large number of novel species in YBH.
Different lifestyles lead to separate bacterial composition
The two modes of bacterial lifestyles, FL and PA, have been reported in many studies, and there was a clear difference between the bacteria in these two lifestyles, both in terms of community structure and biogeochemical processes (DeLong et al., 1993; Tarn et al., 2016; Mestre et al., 2017; Liu et al., 2020b). Our previous studies on Vibrio in YBH also demonstrated that Vibrio preferred to FL lifestyle and had a different dominant composition in the two lifestyles (Li et al., 2020a). In this study, the bacteria from the two lifestyles showed significant differences in several aspects, such as gene abundance, diversity and richness. Both bacteria and archaea favored a FL lifestyle, which was also consistent with the results of previous studies (Li et al., 2015; Zhang et al., 2020; Zheng et al., 2020). The differences in abundance and diversity between the two lifestyles might be due to the suspended matters as well as the lower water fluidity in YBH. Specifically, the particulate matters in YBH consisted mainly of biogenous debris, terrestrial debris, authigenic particles, organic membrane-marine snow and debris from modern artificial materials (Sun et al., 2018). The suspended particles could provide bacteria with nutrients and were enriched in particular bacterial groups, which may exhibit less abundance and diversity than the FL fraction. Further, the abundance of PA bacteria might be less influenced by environmental factors compared with that of FL bacteria because of the particulate matters (Figure 5B). In addition, the bacterial community structure of the two lifestyles was distinctive. For example, the relative abundance of SOB in FL fraction was higher than in PA fraction in anoxic zone I, whereas the opposite was true in anoxic zone II. Meanwhile, in the oxic zone and suboxic zone, PNSB was more abundant in FL fraction. The reason was likely to be the different substrates oxidized by the different types of SOB and their different sulfur-oxidation pathways (Ghosh and Dam, 2009), combined with the interaction with the specific physicochemical properties in YBH. Certainly, these inferences needed to be supported by more detailed physicochemical parameters, as well as more in-depth analyses. Furthermore, the results for the effects of both nutrition and physicochemical properties, as well as the corresponding differences for each specific environmental factor, reflected the different distribution patterns of microorganisms in YBH for the two lifestyles.
Moreover, the differences brought about by the two lifestyles in YBH can be better revealed from the perspective of assembly process, which reflected the impact of deterministic or stochastic processes on the two lifestyles. The NCM model was a tool commonly used to explore this issue (Sloan et al., 2006; Zhou and Ning, 2017). Our results showed that the NCM model (Figures 5A, B) successfully estimated most of the relationship between the frequency of occurrence of OTUs and their relative abundance variation in YBH. The higher degree of explanation implied that the assembly of the PA fraction was more affected by stochastic processes, while that of the FL fraction was more affected by deterministic processes. This was because FL bacteria may have greater motility capability in YBH and were influenced more by environmental factors including temperature and salinity, which were deterministic processes. The stochastic processes played a greater influence on PA bacteria, and this was in accordance with previous findings in a subtropical river-bay system (Wang et al., 2020), but was quite different from previous studies in the marginal sea (Yuan et al., 2021). It was suggested that the bacterial assembly processes in different environments showed different results. The structure of bacterial composition with different lifestyles in YBH may be affected by various environmental factors because of the distinctive physicochemical factors in YBH from other marine environments.
Extremely specific and dynamic sulfur cycle
Current studies on the microbial constituents of the blue hole all found that bacteria involved in the sulfur cycle, particularly SOB, were dominated in the suboxic and anoxic zones, where rapid oxygen depletion and hydrogen sulfide production occurred (Gascoyne et al., 1979; Bottrell et al., 1991; Gonzalez et al., 2011; Haas et al., 2018; He et al., 2019; Gordon, 2020; He et al., 2020; Zhang et al., 2021). Bacteria belonging to Thiomicrospira was reported to be the most abundant SOB in YBH. Bacteria in the family Piscirickettsiaceae were reclassified in 2017, and a portion of species in the former genus Thiomicrospira was reclassified as the genera Hydrogenovibrio, Thioalkalimicrobium, and the newly named Thiomicrorhabdus (Boden et al., 2017; Kojima and Fukui, 2019; Liu et al., 2020a; Liu et al., 2021; Updegraff et al., 2022). In our study, based on the SILVA v128 database, six OTUs were annotated as Thiomicrospira (Figure S5), and the phylogenetic tree confirmed that five OTUs were affiliated with Thiomicrorhabdus and one was affiliated with Hydrogenovibrio (Figure S6). Our results confirmed that the relative abundance of SOB, represented by Thiomicrorhabdus, Sulfurimonas, Sulfurovum, Arcobacter and Rhodobacteraceae, reached almost 90% at 120 m water depth in YBH, agreed with previous studies (He et al., 2019; He et al., 2020; Zhang et al., 2021).
Meanwhile, the above-mentioned taxa all belonged to CSB. They were found to obtain energy from the oxidation of reduced sulfur compounds and fix dissolved inorganic carbon (DIC) through the Calvin-Benson-Bassham (CBB) or reverse tricarboxylic acid cycles (Li et al., 2018b; Scott et al., 2018). could be oxidized under acidic and low O2 conditions through extracellular deposition of S0 by Thiomicrospira, and transient accumulation of sulfite and even polythionates could be detected as products of thiosulfate and S0 oxidation (Javor et al., 1990; Scott et al., 2006). It was suggested that the Thiomicrospira was able to oxidize reductive sulfur-containing compounds, especially at low pH and hypoxic conditions. Both oxygen (reduction from 5 mg/L to 0) and pH (reduction from 8.2 to 7.6) decreased rapidly in the anoxic zone I of YBH (Yao et al., 2018), providing an adequate material basis for the proliferation of the Thiomicrospira. In addition, Thiomicrorhabdus, Sulfurovum, Arcobacter and Thiothrix were also found to be important participants in the process of sulfur cycling. For example, Thiomicrorhabdus can form extracellular S0 spheres at low oxygen/pH and transient accumulation of or polysulfate during S0 spheres or oxidation (Liu et al., 2020a; Liu et al., 2021).
SOB were the most abundant bacteria in blue holes, but the dominant taxa of SOB varied from different habitats. Chlorobium (Gonzalez et al., 2011; Gordon, 2020) and Arcobacter (Iwanowicz et al., 2021) were the most abundant SOB in the Bahamian Blue Hole, while Thiomicrorhabdus was the dominant taxa in YBH. Chlorobium was a member of GSB, which was also a group of strictly anaerobic bacteria that can undergo photoautotrophy in very low light conditions (Imhoff, 2014; Nielsen et al., 2016). In YBH, the two photoautotrophic taxa, PSB and GSB, were much less abundant than CSB and PNSB (P<0.05). Furthermore, although Arcobacter was also found in YBH, its relative abundance (7.24% of SOB) was much lower than that in the Bahamas Blue Hole (over 50% of SOB). It was demonstrated that the composition of SOB and possible sulfur oxidation processes in YBH were very different from those in the Bahamas Blue Hole.
In addition, chemoautotrophic bacteria derived from the genera Thiomicrorhabdus, Sulfurovum, Arcobacter, Thiomicrospira, Hydrogenovibrio were also dominant SOBs in other sulfide-rich habitats, including OMZs, hydrothermal vents, cold spring, soda and salt lakes and marine sediments (Cao et al., 2014; Li et al., 2018b; Scott et al., 2019; Magnuson et al., 2021). For example, SUP05 was dominant in the OMZ and hydrothermal vent (Crowe et al., 2018; van Vliet et al., 2021), and it could account for up to 50% of the entire community in some samples, which would be a high level to be reported (Crowe et al., 2018; van Vliet et al., 2021). In the present study, the relative abundance of Thiomicrohabdus in YBH exceeded 50% in five samples (64.25% in 120 m FL, 59.78% in 140 m FL, 50.61% in 120 m PA, 57.87% in 140 m PA and 51.48% in 170 m PA), that was extremely rare in marine environments.
The Sox system was reported to be a prevalent mechanism of SOB (Friedrich et al., 2001; Friedrich et al., 2005; Ghosh and Dam, 2009), and the soxB gene was used as a biomarker of SOB in hydrothermal vents (Li et al., 2018b). The results of soxB gene abundance in our study were similar to previous studies in the same region (Luo et al., 2018). However, there were differences with the results from hydrothermal vents (Li et al., 2018b), where they found that SOB was positively correlated with soxB abundance. At the same time, we observed considerable amounts of Alcanivorax and Oleiphilus within YBH, suggesting that the sulfur oxidation process in YBH maybe also related to the carbon cycle. It was found that the sulfur oxidation processes in YBH were distinctive from other blue holes and the hydrothermal vents where sulfur oxidation is active. Further data, including but not limited to the transcriptome as well as the metagenome, will certainly be essential to fully reveal the extremely unique sulfur cycle within YBH.
A rich source of unknown culturable bacteria
The bacterial structure in blue holes was well studied using high-throughput sequencing, but for such a unique environment, there was a high probability that novel microorganisms could be obtained using different culture methods. While next-generation sequencing methods can provide a comprehensive and complete picture of the microbial community, in many studies, such as the physiological and biochemical characteristics of strains, the utilization of different substrates could only be carried out if the strain resources were available. At the genus level, Alcanivorax and Vibrio were found to be isolated at almost all depths (Figure 6A), indicating that both genera were well adapted to the environment in YBH, especially the absence of oxygen. This was consistent with previous studies of the Marianas Trench (Zhao et al., 2020), where both genera were also found in the water column from the surface to nearly 10,000 m. In addition, the numbers of bacteria isolated at 90, 100 and 230 m were much higher than other depths, most likely due to the rapid change in environmental factors represented by DO from the suboxic zone to the anoxic zone I and the accumulation of large amounts of dissolved inorganic carbon in the anoxic zone II (Yao et al., 2020).
Our anaerobic bacterial isolation and identification in YBH yielded a large number of potentially new species. Of the 81 anaerobic bacterial strains obtained, 18 strains had 16S rRNA similarities below the 98.65% criterion for new species determination. The relative abundance of novel bacteria in YBH was 22.2% (18/81), which was significantly higher than that in Marianas Trench waters (7.2%) (Zhao et al., 2020), Red Sea corals (10.0%) (ElAhwany et al., 2015), Antarctic soils (1.5%) (Ouyang et al., 2021) and South Korean Yellow Sea sediments (3.8%) (Velmurugan et al., 2011). In contrast, for the isolation of aerobic strains, only 2.4% of the strains were potentially new species. From the results of culturable anaerobic bacteria, it could be assumed that a considerable proportion (approximately 25%) of the anaerobes in YBH were unknown. At the same time, these anaerobic bacteria should play an important role in the unique biogeochemical cycle in YBH, as well as possess a special adaptation mechanism to the anaerobic environment in YBH. This suggested that the novelty and specificity of the culturable anaerobic bacteria in YBH provided a great opportunity to study their metabolic characteristics and further application.
As expected, there were differences in the microbial composition identified in the samples using culture-dependent methods compared to culture-independent methods (Demko et al., 2021). For example, Thiomicrorhabdus, which had the highest relative abundance in the high-throughput sequencing results, was not obtained under any of the various culture conditions used. However, four strains of Hydrogenovibrio, which belonged to the same family as Thiomicrorhabdus and had a similar phylogenetic relationship, was isolated in the SOB-enriched SPG medium (Figure 6B). Three of the strains were isolated from 110 m and one from 270 m. This was in agreement with the high-throughput sequencing result that Thiomicrorhabdus was predominantly distributed in the anoxic zone I. This demonstrated that, for specific habitats such as YBH, a culture-dependent approach that restores as much of the real-life condition as possible will help to obtain a larger, more representative number of strains. In addition to the differences, a number of strains that were abundant and important in high-throughput sequencing were also obtained, such as Alcanivorax, Vibrio, Alternomonas, Pseudomonas and Pseudoalteromonas. It was suggested that the results for culturable bacteria reflect to some extent the structure of the microbial community in YBH. Further, it also provided the strain resources for in-depth studies of the adaptation mechanism of bacteria to the unique environment of YBH.
Conclusion
In this article, the bacterial composition in YBH was studied by both culture-dependent and -independent methods. In YBH, there were many differences between bacteria with FL and PA lifestyles. FL bacteria show stronger variation internally compared to PA bacteria. Also, FL bacteria are relatively more influenced by deterministic processes. A large number of sulfur-oxidizing bacteria, represented by Thiomicrorhabdus, drive a very complex sulfur cycling process in YBH. In addition, a large number of new taxa have been identified among culturable anaerobic bacteria. Our study expanded current knowledge on microbial community structure from a lifestyle-based perspective in deep ocean blue holes, and provided a vast resource of bacterial strains for further studies on their adaptation to extreme marine environments.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
X-HZ and MY designed the experiments and analyzed the data. SZ conducted most of the experiments, analyzed the data, prepared graphs and tables, and wrote the manuscript. ZY, PY, LF, and LC attended the cruise, collected the samples and analyzed the data. YZ and JCL carried out the culture, isolation and analysis of aerobic bacteria. RD carried out the culture, isolation and analysis of sulfur-oxidizing bacteria. CJ and XS carried out the culture, isolation and analysis of anaerobic bacteria. XW performed the quantitative PCR analysis. MY, SZ, X-HZ, and JWL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (92251303, 41730530 and 41976137), the National Key Research and Development Program of China (2018YFE0124100), and the Fundamental Research Funds for the Central Universities (202172002).
Acknowledgments
We thank all the scientists and crew members on the Qiong Qionghai 03138 during the expedition for their great efforts and help in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1086117/full#supplementary-material
References
Becking L. E., Renema W., Santodomingo N. K., Hoeksema B. W., Tuti Y., de Voogd N. J. (2011). Recently discovered landlocked basins in Indonesia reveal high habitat diversity in anchialine systems. Hydrobiologia 677 (1), 89–105. doi: 10.1007/s10750-011-0742-0
Bi N., Fu L., Chen H., Liu R., Chen L., Liu Q., et al. (2018). Hydrographic features of the yongle blue hole in the south China Sea and their influential factors. Sci. Bull. 21, 2184–2194. doi: 10.1360/N972017-01329
Bishop R. E., Humphreys W. F., Cukrov N., Žic V., Boxshall G. A., Cukrov M., et al. (2015). ‘Anchialine’ redefined as a subterranean estuary in a crevicular or cavernous geological setting. J. Crustacean Biol. 35 (4), 511–514. doi: 10.1007/BF03175407.10.1163/1937240X-00002335
Boden R., Scott K. M., Williams J., Russel S., Antonen K., Rae A. W., et al. (2017). An evaluation of Thiomicrospira, Hydrogenovibrio and Thioalkalimicrobium: reclassification of four species of Thiomicrospira to each Thiomicrorhabdus gen. nov. and Hydrogenovibrio, and reclassification of all four species of Thioalkalimicrobium to Thiomicrospira. Int. J. Syst. Evol. Micr. 67 (5), 1140–1151. doi: 10.1099/ijsem.0.001855
Bottrell S. H., Smart P. L., Whitaker F., Raiswell R. (1991). Geochemistry and isotope systematics of sulphur in the mixing zone of Bahamian blue holes. Appl. Geochem. 6 (1), 97–103. doi: 10.1016/0883-2927(91)90066-X
Cabrera G., Pérez R., Gomez J. M., Abalos A., Cantero D. (2006). Toxic effects of dissolved heavy metals on Desulfovibrio vulgaris and desulfovibrio sp. strains. J. Hazard Mater. 135 (1-3), 40–46. doi: 10.1016/j.jhazmat.2005.11.058
Cadillo-Quiroz H., Bräuer S., Yashiro E., Sun C., Yavitt J., Zinder S. (2006). Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central new York state, USA. Environ. Microbiol. 8 (8), 1428–1440. doi: 10.1111/j.1462-2920.2006.01036.x
Canfield D. E., Stewart F. J., Thamdrup B., De Brabandere L., Dalsgaard T., Delong E. F., et al. (2010). A cryptic sulfur cycle in oxygen-minimum–zone waters off the Chilean coast. Science 330 (6009), 1375–1378. doi: 10.1126/science.1196889
Cao H., Wang Y., Lee O. O., Zeng X., Shao Z., Qian P. Y. (2014). Microbial sulfur cycle in two hydrothermal chimneys on the southwest Indian ridge. mBio 5 (1), e00980–e00913. doi: 10.1128/mBio.00980-13
Chen C., Fu L., Bi N., Ge R., Liu G., Zhuang Y., et al. (2018). Zooplankton community composition and diel vertical distribution in the yongle blue hole, xisha islands, south China Sea. Oceanologia Limnologia Sinica. 3, 594–603. doi: 10.11693/hyhz20171200328
Chen W., Ren K., Isabwe A., Chen H., Liu M., Yang J. (2019). Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 7 (1), 1–16. doi: 10.1186/s40168-019-0749-8
Crowe S. A., Cox R. P., Jones C., Fowle D. A., Santibañez-Bustos J. F., Ulloa O., et al. (2018). Decrypting the sulfur cycle in oceanic oxygen minimum zones. ISME J. 12 (9), 2322–2329. doi: 10.1038/s41396-018-0149-2
DeLong E. F., Franks D. G., Alldredge A. L. (1993). Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol Oceanogr. 38 (5), 924–934. doi: 10.4319/lo.1993.38.5.0924
Demko A. M., Patin N. V., Jensen P. R. (2021). Microbial diversity in tropical marine sediments assessed using culture-dependent and culture-independent techniques. Environ. Microbiol. 23 (11), 6859–6875. doi: 10.1101/2021.02.27.433211
Dray S., Dufour A. B. (2007). The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw 22, 1–20. doi: 10.18637/jss.v022.i04
ElAhwany A. M., Ghozlan H. A., ElSharif H. A., Sabry S. A. (2015). Phylogenetic diversity and antimicrobial activity of marine bacteria associated with the soft coral Sarcophyton glaucum. J. Basic Microb. 55 (1), 2–10. doi: 10.1002/jobm.201300195
Friedrich C. G., Bardischewsky F., Rother D., Quentmeier A., Fischer J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8 (3), 253–259. doi: 10.1016/j.mib.2005.04.005
Friedrich C. G., Rother D., Bardischewsky F., Quentmeier A., Fischer J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microb. 67 (7), 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Fu T., Jia C., Fu L., Zhou S., Yao P., Du R., et al. (2018). Marinifilum breve sp. nov., a marine bacterium isolated from the yongle blue hole in the south China Sea and emended description of the genus Marinifilum. Int. J. Syst. Evol. Micr. 68 (11), 3540–3545. doi: 10.1099/ijsem.0.003027
Fu M., Sun P., Li Y., Pu X., Yuan C., Zhang X. (2020). Phytoplankton community structure in sansha yongle blue hole and its adjacent area of the south China Sea. Adv. Mar. Sci. 2, 304–316. doi: 10.3969/j.issn.1671-6647.2020.02.010
Garcia-Robledo E., Padilla C. C., Aldunate M., Stewart F. J., Ulloa O., Paulmier A., et al. (2017). Cryptic oxygen cycling in anoxic marine zones. P Natl. Acad. Sci. U.S.A. 114 (31), 8319–8324. doi: 10.1073/pnas.1619844114
Gascoyne M., Benjamin G. J., Schwarcz H. P., Ford D. C. (1979). Sea-Level lowering during the illinoian glaciation: evidence from a bahama “Blue hole”. Science 205 (4408), 806–808. doi: 10.1126/science.205.4408.806
Ge R., Fu L., Bi N., Chen C., Liu G., Zhuang Y., et al. (2020). Diel vertical distribution of phytoplanktonin yongle blue hole, xisha islands in spring. J. Ocean Univ China. 50 (1), 65–73. doi: 10.16441/j.cnki.hdxb.20180428
Ghosh W., Dam B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33 (6), 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Gibbs R. A., Hayes C. R. (1988). The use of R2A medium and the spread plate method for the enumeration of heterotrophic bacteria in drinking water. Lett. Appl. Microbiol. 6 (2), 19–21. doi: 10.1111/j.1472-765X.1988.tb01205.x
Gonzalez B. C., Iliffe T. M., Macalady J. L., Schaperdoth I., Kakuk B. (2011). Microbial hotspots in anchialine blue holes: initial discoveries from the Bahamas. Hydrobiologia 677 (1), 149–156. doi: 10.1007/s10750-011-0932-9
Gordon M. J. (2020). Microbial community structures in three Bahamian blue holes. [master’s thesis]. [Tampa (FL)]: University of South Florida.
Haas S., de Beer D., Klatt J. M., Fink A., Rench R. M., Hamilton T. L., et al. (2018). Low-light anoxygenic photosynthesis and fe-s-biogeochemistry in a microbial mat. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00858
Han J., He N., Li Q., Lv B., Xing L., Lu K. (2020). “A preliminary study of the genesis of sansha yongle long blue hole based on the mini-multichannel seismic,” in Big Data and Geophysics - 16th National Security Geophysics Symposium, Tongren, Guizhou, China, 180–185.
He H., Fu L., Liu Q., Fu L., Bi N., Yang Z., et al. (2019). Community structure, abundance and potential functions of bacteria and archaea in the sansha yongle blue hole, xisha, south China Sea. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02404
He P., Xie L., Zhang X., Li J., Lin X., Pu X., et al. (2020). Microbial diversity and metabolic potential in the stratified sansha yongle blue hole in the south China Sea. Sci. Rep. 10 (1), 1–17. doi: 10.1038/s41598-020-62411-2
Iliffe T. M., Kornicker L. S. (2009). Worldwide diving discoveries of living fossil animals from the depths of anchialine and marine caves. Smithsonian Contributions to Mar. Sci. 38:269-280.
Imhoff J. F. (2014). “The family chlorobiaceae,” in The prokaryotes. Eds. Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. (Berlin: Springer Berlin Heidelberg), 501–514. doi: 10.1007/978-3-642-38954-2_142
Iwanowicz D. D., Jonas R. B., Schill W. B., Marano-Briggs K. (2021). Novel microbiome dominated by arcobacter during anoxic excurrent flow from an ocean blue hole in andros island, the Bahamas. PloS One 16 (8), e0256305. doi: 10.1371/journal.pone.0256305
Javor B. J., Wilmot D. B., Vetter R. D. (1990). pH-dependent metabolism of thiosulfate and sulfur globules in the chemolithotrophic marine bacterium Thiomicrospira crunogena. Arch. Microbiol. 154 (3), 231–238. doi: 10.1007/BF00248960
Kojima H., Fukui M. (2019). Thiomicrorhabdus aquaedulcis sp. nov., a sulfur-oxidizing bacterium isolated from lake water. Int. J. Syst. Evol. Micr. 69 (9), 2849–2853. doi: 10.1099/ijsem.0.003567
Krishnani K. K., Kathiravan V., Natarajan M., Kailasam M., Pillai S. M. (2010). Diversity of sulfur-oxidizing bacteria in greenwater system of coastal aquaculture. Appl. Biochem. Biotech. 162 (5), 1225–1237. doi: 10.1007/s12010-009-8886-3
Lane D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic acid techniques in bacterial systematics. Eds. Stackebrandt E., Goodfellow M. (New York, NY: John Wiley and Sons), 115–175.
Li T., Feng A., Liu Y., Li Z., Guo K., Jiang W., et al. (2018a). Three-dimensional (3D) morphology of sansha yongle blue hole in the south China Sea revealed by underwater remotely operated vehicle. Sci. Rep. 8 (1), 1–9. doi: 10.1038/s41598-018-35220-x
Li Q., Lei Y., Morard R., Li T., Wang B. (2020b). Diversity hotspot and unique community structure of foraminifera in the world’s deepest marine blue hole–sansha yongle blue hole. Sci. Rep. 10 (1), 1–11. doi: 10.1038/s41598-020-67221-0
Li B., Liu J., Zhou S., Fu L., Yao P., Chen L., et al. (2020a). Vertical variation in vibrio community composition in sansha yongle blue hole and its ability to degrade macromolecules. Mar. Life Sci. Technol. 2 (1), 60–72. doi: 10.1007/s42995-019-00003-4
Li Y., Tang K., Zhang L., Zhao Z., Xie X., Chen C. T. A., et al. (2018b). Coupled carbon, sulfur, and nitrogen cycles mediated by microorganisms in the water column of a shallow-water hydrothermal ecosystem. Front. Microbiol. 9, 2718. doi: 10.3389/fmicb.2018.02718
Liu X., Chen B., Lai Q., Shao Z., Jiang L. (2021). Thiomicrorhabdus sediminis sp. nov. and thiomicrorhabdus xiamenensis sp. nov., novel sulfur-oxidizing bacteria isolated from coastal sediments and an emended description of the genus Thiomicrorhabdus. Int. J. Syst. Evol. Micr. 71 (2), 4660. doi: 10.1099/ijsem.0.004660
Liu J., Fu B., Yang H., Zhao M., He B., Zhang X. H. (2015). Phylogenetic shifts of bacterioplankton community composition along the pearl estuary: the potential impact of hypoxia and nutrients. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00064
Liu Y., He H., Fu L., Liu Q., Yang Z., Zhen Y. (2019). Environmental DNA sequencing reveals a highly complex eukaryote community in sansha yongle blue hole, xisha, south China Sea. Microorganisms 7 (12), 624. doi: 10.3390/microorganisms7120624
Liu X., Jiang L., Hu Q., Lyu J., Shao Z. (2020a). Thiomicrorhabdus indica sp. nov., an obligately chemolithoautotrophic, sulfur-oxidizing bacterium isolated from a deep-sea hydrothermal vent environment. Int. J. Syst. Evol. Micr. 70 (1), 234–239. doi: 10.1099/ijsem.0.003744
Liu Y., Lin Q., Feng J., Yang F., Du H., Hu Z., et al. (2020b). Differences in metabolic potential between particle-associated and free-living bacteria along pearl river estuary. Sci. Total Environ. 728, 138856. doi: 10.1016/j.scitotenv.2020.138856
Li J., Wei B., Wang J., Liu Y., Dasgupta S., Zhang L., et al. (2015). Variation in abundance and community structure of particle-attached and free-living bacteria in the south China Sea. Deep Sea Res. Pt II. 122, 64–73. doi: 10.1016/j.dsr2.2015.07.006
Luo J., Tan X., Liu K., Lin W. (2018). Survey of sulfur-oxidizing bacterial community in the pearl river water using soxB, sqr, and dsrA as molecular biomarkers. 3 Biotech. 8 (1), 1–12. doi: 10.1007/s13205-017-1077-y
Magnuson E., Mykytczuk N. C., Pellerin A., Goordial J., Twine S. M., Wing B., et al. (2021). Thiomicrorhabdus streamers and sulfur cycling in perennial hypersaline cold springs in the Canadian high Arctic. Environ. Microbiol. 23 (7), 3384–3400. doi: 10.1111/1462-2920.14916
Mestre M., Borrull E., Sala M. M., Gasol J. M. (2017). Patterns of bacterial diversity in the marine planktonic particulate matter continuum. ISME J. 11 (4), 999–1010. doi: 10.1038/ismej.2016.166
Moore T. S., Mullaugh K. M., Holyoke R. R., Madison A. S., Yücel M., Luther G. W. III (2009). Marine chemical technology and sensors for marine waters: potentials and limits. Annu. Rev. Mar. Sci. 1, 91–115. doi: 10.1146/annurev.marine.010908.163817
Mylroie J. E., Carew J. L., Moore A. I. (1995). Blue holes: definition and genesis. Carbonate Evaporite. 10 (2), 225–233. doi: 10.1007/BF03175407
Naumann M. S., Bednarz V. N., Ferse S. C., Niggl W., Wild C. (2015). Monitoring of coastal coral reefs near dahab (Gulf of aqaba, red Sea) indicates local eutrophication as potential cause for change in benthic communities. Environ. Monit Assess. 187 (2), 1–14. doi: 10.1007/s10661-014-4257-9
Nielsen J. T., Kulminskaya N. V., Bjerring M., Linnanto J. M., Rätsep M., Pedersen M.Ø., et al. (2016). In situ high-resolution structure of the baseplate antenna complex in Chlorobaculum tepidum. Nat. Commun. 7 (1), 1–12. doi: 10.1038/ncomms12454
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O’hara R., et al. (2013). “Package “vegan”,” in Community ecology package, version 2.
Ouyang Q., Yao B., Jiao Y., Jiang P., Li J. (2021). Diversity of culturable bacteria in soils from zhongshan and changcheng stations and marine sediments from Antarctica. Bio Resour. 2, 133–142. doi: 10.14188/j.ajsh.2021.02.005
Patrick F. M. (1978). The use of membrane filtration and marine agar 2216E to enumerate marine heterotrophic bacteria. Aquaculture 13 (4), 369–372. doi: 10.1016/0044-8486(78)90186-2
Pohlman J. W. (2011). The biogeochemistry of anchialine caves: progress and possibilities. Hydrobiologia 677 (1), 33–51. doi: 10.1007/s10750-011-0624-5
Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. (Eds.) (2014). The prokaryotes (Berlin: Springer).
Schloss P. D. (2020). Reintroducing mothur: 10 years later. Appl. Environ. Microb. 86 (2), e02343–e02319. doi: 10.1128/AEM.02343-19
Schmitt D., Gischler E., Birgel D., Peckmann J., Anselmetti F. S., Vogel H. (2020). Great blue hole (Lighthouse reef, belize): A continuous, annually-resolved record of common era sea surface temperature, Atlantic multidecadal oscillation and cyclone-controlled run-off. Quaternary Sci. Rev. 247, 106570. doi: 10.1016/j.quascirev.2020.106570
Schwabe S. J. (1999). Biogeochemical investigation of caves within Bahamian carbonate platforms. [doctoral dissertation]. Bristol: University of Bristol.
Scott K. M., Leonard J. M., Boden R., Chaput D., Dennison C., Haller E., et al. (2019). Diversity in CO2-concentrating mechanisms among chemolithoautotrophs from the genera Hydrogenovibrio, Thiomicrorhabdus, and Thiomicrospira, ubiquitous in sulfidic habitats worldwide. Appl. Environ. Microb. 85 (3), e02096–e02018. doi: 10.1128/AEM.02096-18
Scott K. M., Sievert S. M., Abril F. N., Ball L. A., Barrett C. J., Blake R. A., et al. (2006). The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PloS Biol. 4 (12), e383. doi: 10.1371/journal.pbio.0040383
Scott K. M., Williams J., Porter C. M., Russel S., Harmer T. L., Paul J. H., et al. (2018). Genomes of ubiquitous marine and hypersaline Hydrogenovibrio, Thiomicrorhabdus and thiomicrospira spp. encode a diversity of mechanisms to sustain chemolithoautotrophy in heterogeneous environments. Environ. Microbiol. 20 (8), 2686–2708. doi: 10.1111/1462-2920.14090
Seymour J. R., Humphreys W. F., Mitchell J. G. (2007). Stratification of the microbial community inhabiting an anchialine sinkhole. Aquat Microb. Ecol. 50 (1), 11–24. doi: 10.3354/ame01153
Sloan W. T., Lunn M., Woodcock S., Head I. M., Nee S., Curtis T. P. (2006). Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8 (4), 732–740. doi: 10.1111/j.1462-2920.2005.00956.x
Smart P. L., Dawans J. M., Whitaker F. (1988). Carbonate dissolution in a modern mixing zone. Nature 335 (6193), 811–813. doi: 10.1038/335811a0
Šmilauer P., Lepš J. (2014). Multivariate analysis of ecological data using CANOCO 5 (Cambridge, UK: Cambridge university pres). doi: 10.1017/CBO9780511615146
Smith D. E., Harrison S., Firth C. R., Jordan J. T. (2011). The early Holocene sea level rise. Quaternary Sci. Rev. 30 (15-16), 1846–1860. doi: 10.1016/j.quascirev.2011.04.019
Sun X., Fu L., Yang Z., Bi N., Fan D., Yao P., et al. (2018). Components and origin of suspended matter in the sansha yongle blue hole, south China Sea. Oceanologia Limnologia Sinica. 4, 779–792. doi: 10.11693/hyhz20171200332
Tarn J., Peoples L. M., Hardy K., Cameron J., Bartlett D. H. (2016). Identification of free-living and particle-associated microbial communities present in hadal regions of the Mariana trench. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00665
Trouwborst R. E., Clement B. G., Tebo B. M., Glazer B. T., Luther G. W. III (2006). Soluble Mn (III) in suboxic zones. Science 313 (5795), 1955–1957. doi: 10.1126/science.1132876
Updegraff T., Schiff-Clark G., Gossett H., Parsi S., Peterson R., Whittaker R., et al. (2022). Thiomicrorhabdus heinhorstiae sp. nov. and thiomicrorhabdus cannonii sp. nov.: novel sulphur-oxidizing chemolithoautotrophs isolated from the chemocline of hospital hole, an anchialine sinkhole in spring hill, Florida, USA. Int. J. Syst. Evol. Micr. 72 (3), 5233. doi: 10.1099/ijsem.0.005233
van Vliet D. M., von Meijenfeldt F. B., Dutilh B. E., Villanueva L., Sinninghe Damsté J. S., Stams A. J., et al. (2021). The bacterial sulfur cycle in expanding dysoxic and euxinic marine waters. Environ. Microbiol. 23 (6), 2834–2857. doi: 10.1111/1462-2920.15265
Velmurugan N., Kalpana D., Cho J. Y., Lee G. H., Park S. H., Lee Y. S. (2011). Phylogenetic analysis of culturable marine bacteria in sediments from south Korean yellow Sea. Microbiology 80 (2), 261–272. doi: 10.1134/S0026261711010188
Wang Y., Pan J., Yang J., Zhou Z., Pan Y., Li M. (2020). Patterns and processes of free-living and particle-associated bacterioplankton and archaeaplankton communities in a subtropical river-bay system in south China. Limnol Oceanogr. 65, S161–S179. doi: 10.1002/lno.11314
Wilson W. L. (1994). Morphometry and hydrology of dean's blue hole, long island. Bahamas J. Sci. 2 (1), 10–14.
Xie L., Wang B., Pu X., Xin M., He P., Li C., et al. (2019). Hydrochemical properties and chemocline of the sansha yongle blue hole in the south China Sea. Sci. Total Environ. 649, 1281–1292. doi: 10.1016/j.scitotenv.2018.08.333
Yao P., Chen L., Fu L., Yang Z., Bi N., Wang L., et al. (2018). Controls on vertical nutrient distributions in the sansha yongle blue hole, south China Sea. Sci. Bull. 63 (23), 2393–2402. doi: 10.1360/N972018-00155
Yao P., Wang X. C., Bianchi T. S., Yang Z. S., Fu L., Zhang X. H., et al. (2020). Carbon cycling in the world’s deepest blue hole. J. Geophys Res-Biogeo. 125 (2), e2019JG005307. doi: 10.1029/2019JG005307
Yuan H., Li T., Li H., Wang C., Li L., Lin X., et al. (2021). Diversity distribution, driving factors and assembly mechanisms of free-living and particle-associated bacterial communities at a subtropical marginal sea. Microorganisms 9 (12), 2445. doi: 10.3390/microorganisms9122445
Zakem E. J., Mahadevan A., Lauderdale J. M., Follows M. J. (2020). Stable aerobic and anaerobic coexistence in anoxic marine zones. ISME J. 14 (1), 288–301. doi: 10.1038/s41396-019-0523-8
Zhang Y., Jing H., Peng X. (2020). Vertical shifts of particle-attached and free-living prokaryotes in the water column above the cold seeps of the south China Sea. Mar. pollut. Bull. 156, 111230. doi: 10.1016/j.marpolbul.2020.111230
Zhang Y., Xu H., Wang L., Liu R., Fu L., Lin K. (2021). Unique bacterial communities and potential function along the vertical gradient in the deepest marine blue hole. Environ. Microbiol. Rep. 13 (6), 911–927. doi: 10.1111/1758-2229.13001
Zhao X., Liu J., Zhou S., Zheng Y., Wu Y., Kogure K., et al. (2020). Diversity of culturable heterotrophic bacteria from the Mariana trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2 (2), 181–193. doi: 10.1007/s42995-020-00027-1
Zheng Y., Wang J., Zhou S., Zhang Y., Liu J., Xue C. X., et al. (2020). Bacteria are important dimethylsulfoniopropionate producers in marine aphotic and high-pressure environments. Nat. Commun. 11 (1), 1–12. doi: 10.1038/s41467-020-18434-4
Keywords: Yongle Blue Hole, free-living, particle-associated, bacterial community, culture-dependent/independent methods
Citation: Zhou S, Liu J, Yao P, Fu L, Yang Z, Zhang Y, Du R, Jia C, Chen L, Liang J, Wang X, Shi X, Zhang X-H and Yu M (2023) Unique bacterial communities and lifestyles in deep ocean blue holes: Insights from the Yongle Blue Hole (South China Sea). Front. Mar. Sci. 10:1086117. doi: 10.3389/fmars.2023.1086117
Received: 01 November 2022; Accepted: 11 January 2023;
Published: 25 January 2023.
Edited by:
Cristiana Callieri, Department of Earth System Sciences and Technologies for the Environment (CNR), ItalyReviewed by:
Stefano Amalfitano, Department of Earth System Sciences and Technologies for the Environment (CNR), ItalyLuisa I. Falcon, National Autonomous University of Mexico, Mexico
Copyright © 2023 Zhou, Liu, Yao, Fu, Yang, Zhang, Du, Jia, Chen, Liang, Wang, Shi, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Yu, eXVtaW5Ab3VjLmVkdS5jbg==