 Wensi Wu1
Wensi Wu1 Lixing Huang
Lixing Huang Yingxue Qin
Yingxue Qin Qingpi Yan
Qingpi Yan- 1Fisheries College, Jimei University, Xiamen, China
- 2Key Laboratory of Special Aquatic Feed for Fujian, Fujian Tianma Technology Company Limited, Fuzhou, China
Pseudomonas plecoglossicida is responsible for visceral white spot disease in economically valuable marine fish such as Larimichthys crocea and Epinephelus coioides. Based on RNA sequencing, we previously showed that P. plecoglossicida fliS gene expression is significantly up-regulated in E. coioides spleens during infection. Here, to explore the role of this gene in pathogenicity, RNA interference (RNAi) was performed to silence fliS in P. plecoglossicida, and the mutant with the best silencing efficiency (89%) was chosen for further studies. Results showed that fliS silencing significantly attenuated motility, chemotaxis, adhesion, and biofilm formation of P. plecoglossicida. Furthermore, E. coioides infected with the fliS-RNAi strain recorded no deaths and showed fewer pathogens in the spleen and fewer white spots on the spleen surface compared to those fish infected with the wild type P. plecoglossicida strain. RNAi of fliS significantly affected the spleen transcriptome and metabolome in infected E. coioides. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that the cytokine-cytokine receptor interaction pathway was the most enriched immune-related pathway, and the arginine biosynthesis pathway was the most enriched metabolism-related pathway. These findings suggest that fliS is a virulence gene of P. plecoglossicida and is involved in the regulation of motility, chemotaxis, adhesion, and biofilm formation, as well as the inflammatory and immune responses of E. coioides to P. plecoglossicida infection.
Introduction
The continuous emergence of various diseases remains a considerable challenge for the aquaculture industry (Borrego et al., 2017; Mugimba et al., 2021). Pseudomonas plecoglossicida is an opportunistic pathogen widely distributed in seawater and was originally isolated from cultured ayu (Plecoglossus altivelis) presenting with bacterial hemorrhagic ascites (Nishimori et al., 2000). More recently, it has been reported that P. plecoglossicida can infect Larimichthys crocea and Epinephelus coioides under low-temperature conditions, resulting in economically costly visceral white spot disease with high infection and mortality rates (Tang et al., 2020; Li et al., 2020). Pathogenicity is regulated by different genes, many of which are closely related to virulence regulation in aquatic pathogens, e.g., toxR in Vibrio alginolyticus (Cai et al., 2017), sodA and sodB in Aeromonas hydrophila (Zhang et al., 2020), and exbB and fliG in P. plecoglossicida (Jiao et al., 2021; Tang et al., 2022). However, despite extensive research on P. plecoglossicida, our understanding of its virulence genes remains poor. Previous dual RNA-seq data of E. coioides spleens infected with P. plecoglossicida (NCBI, SRP114910 and SRP115064) in our laboratory found that the P. plecoglossicida fliS gene was highly expressed in the host during infection (Luo et al., 2020), suggesting that this gene might be involved in the regulation of P. plecoglossicida virulence.
Bacterial flagella are complex motility organs composed of multiple protein subunits, including molecular motors, hook-basal-body complexes, and filaments (Lowenthal et al., 2009; Lertsethtakarn et al., 2011). Flagella are responsible for bacterial movement and are one of the factors that affect pathogenicity (Duan et al., 2013). The flagellum-specific export system is a specialized type III secretion system (T3SS) that exists in pathogenic bacteria for toxin export (Condry and Nilles, 2017; Miao et al., 2010), facilitated by special proteins called T3SS chaperones. The FliS protein, located in the flagellar hook, is a T3SS chaperone (Gu et al., 2017) that binds to the C-terminal region of flagellin to facilitate its export (Ozin et al., 2003; Muskotal et al., 2006). The fliS gene, which encodes FliS, is closely related to flagellum formation, motility, and bacterial toxicity (Yokoseki et al., 1995; Yokoseki et al., 1996). In Yersinia pseudotuberculosis, the fliS mutant strain has shorter flagella and weaker mobility. After complementation, the flagellum length of strain restored to wild type state (Xu et al., 2014). In addition, deletion of fliS in Campylobacter jejuni was reported to result in loss of motility and shorter flagella (Radomska et al., 2017). To date, however, the role of fliS in P. plecoglossicida pathogenicity has not yet been reported.
Given the considerable threat of P. plecoglossicida to cultured fish and the potential impact of fliS on bacterial virulence, we used RNA interference (RNAi) to silence the fliS gene in the P. plecoglossicida NZBD9 strain. We then explored differences in virulence between the wild type and fliS-RNAi strains as well as differences in the immune response of E. coioides to different strain infection using transcriptomic and metabolomic sequencing and analysis.
Materials and methods
Bacterial strains and culture conditions
The pathogenic P. plecoglossicida strain (NZBD9) was isolated from the spleen of naturally infected L. crocea with white spot disease and stored at -80°C (Luo et al., 2019). The P. plecoglossicida strain was routinely grown in Luria Bertani (LB) broth at 18 or 28°C with shaking at 220 rpm. Escherichia coli DH5α was obtained from TransGen Biotech (Beijing, China) and grown in LB broth at 37°C with shaking at 220 rpm.
Construction of P. plecoglossicida RNAi strain
The RNAi strain was constructed based on previous study (Luo et al., 2020). The sequences of five pairs of short hairpin RNA (shRNA) were designed according to the sequences of the fliS gene using RNAi Designer (http://rnaidesigner.thermofisher.com) and are provided in Supplementary Table 1. The five pairs of shRNAs were annealed and ligated to the linearized pCM130/tac vector. The recombined pCM130/tac vector was transformed into competent E. coli DH5α by heat shock, and competent P. plecoglossicida was eventually electrotransformed. The tetracycline resistance marker (10 μg/mL; final concentration) was used for screening stably silenced clones. The mRNA levels of fliS in the five mutants were determined by quantitative real-time polymerase chain reaction (qRT-PCR).
Growth rate assay
Overnight culture of P. plecoglossicida in LB broth was adjusted to an optical density at 600 nm (OD600 nm) of 0.1 and diluted 10 000-fold with fresh LB broth. Aliquots (200 μL) of the diluted bacterial culture were added to 10 wells of a microtiter plate, then incubated at 28°C. The OD600 nm of each well was automatically detected each hour for 48 h.
Soft agar plate motility assay
Swarming motility was determined following previously described methods (Zuo et al., 2019). After overnight cultivation in LB medium, OD600 nm was adjusted to 0.2. The bacterial suspension (1 μL) was inoculated on semisolid agar plates. The plates were incubated at 28°C for 10–16 h to determine colony diameter. Three replicates were performed per group.
Biofilm formation assay
Biofilm formation was explored following previous research (Jiao et al., 2021). P. plecoglossicida in the exponential growth period was adjusted to OD600nm = 0.2 with fresh LB broth. Then, 100 μL of diluted bacterial suspension was added into each well of a 96-well plate and incubated at 28°C for 24 h. Subsequently, each well was washed twice with 200 μL of sterile phosphate-buffered saline (PBS), stained with 125 μL of crystal violet (0.1%) for 15 min, washed three times with sterile PBS, and dried under sterile conditions. Finally, 200 μL of 33% acetic acid was added to dissolve the dyed biofilm. The OD590nm of each well was measured, and each strain was repeated 10 times independently.
Chemotaxis assay
Overnight culture of P. plecoglossicida was adjusted with sterile PBS to OD600nm = 1.0, and the bacterial suspension (0.25 mL) was then aspirated into a sterile syringe (1 mL). The open end of a capillary tube (inner diameter 0.1 mm, sealed at one end) was filled with E. coioides skin mucus (3 μL), horizontally dipped into the bacterial suspension in the syringe, then incubated at 28°C for 1 h. Finally, the number of bacteria in the mucus was determined by plate counting (Mao et al., 2020). Sterile PBS in a capillary tube was used as the control. Three independent biological replicates were performed per group.
In vitro adhesion assay
In vitro adhesion was determined following previous study (He et al., 2021). First, sterile mucus of E. coioides (20 μL) was evenly coated onto a slide and fixed with 200 μL of 4% methanol for 30 min. Then, 200 μL of bacterial suspension (OD600nm = 0.3) was evenly coated on the area with mucus. After the slides were incubated at 28°C for 2 h, they were washed three times with PBS to remove adherent cells. The bacterial cells adhered to the glass slide were fixed with 200 μL of 4% methanol for 30 min and stained with 0.1% crystal violet for 3 min, after which the unstained crystal violet was washed with PBS. Five slides were performed per group, and the adhered bacteria in 20 randomly selected fields were counted under a microscope (×1 000) (Leica DM4000 B LED, Leica, Germany).
Fish infection and sampling
Healthy size-matched E. coioides (14.7 ± 0.7 cm in length) were purchased from Zhangzhou city (Fujian, China) and adaptively maintained at 18°C for one week in laboratory seawater (water volume 320 L; pH = 7.8–8.2; continuous oxygen). The seawater temperature remained at 18 ± 1°C throughout the experiment.
For survival rate assay, 60 fish were randomly divided into three groups: i.e., fliS-RNAi strain group, wild type strain group, and PBS group. Each fish in the infection groups was intraperitoneally injected with the corresponding strain at a dose of 5 × 104 colony-forming units (CFU)/fish. For the negative control, 20 fish were injected with 200 μL of PBS. Survival was observed and recorded every 12 h after injection to 10 days post infection (dpi).
For spleen sampling, 240 fish were randomly divided into three groups: i.e., wild type strain infection group, fliS-RNAi strain infection group, and PBS injection group. The fish were treated as described above. Six spleens were randomly sampled from each group at 1, 2, and 3 dpi. Two spleens from each group were randomly mixed into one sample and subjected to bacterial load and gene expression assays. Thirty-six spleens were randomly sampled at 4 dpi, with six spleens from the same group randomly mixed into one sample and subjected to qRT-PCR, pathogen load assay, fliS gene expression determination, and metabolomic analysis. All spleen samples were frozen in liquid nitrogen and stored in a refrigerator at -80°C.
qRT-PCR
qRT-PCR was performed using a QuantStudio 6 Flex Real-Time PCR system (Life Technologies, Carlsbad, CA, USA). Primers were synthesized by Tsingke Biotechnology Co., Ltd., (China) as shown in Supplementary Table 2. The fliS gene expression levels of P. plecoglossicida were normalized using 16S rDNA. Pathogen load of P. plecoglossicida in the infected spleens was determined based on the copy number of the housekeeping gene gyrB (Xin et al., 2020). Relative gene expression in the different groups was calculated using the 2−△△CT method (Luo et al., 2020).
Transcriptomic analysis
An Illumina TruSeq™ RNA Sample Prep Kit was used for the RNA sequencing (RNA-seq) libraries. Total RNA was extracted from spleen samples, with RNA concentration and purity detected using a Nanodrop 2000 spectrophotometer. The RNA integrity number (RIN) was determined using an Agilent 2100 bioanalyzer. Oligo-dT was used to enrich mRNA, fragmentation buffer was added to fragment mRNA, cDNA was reverse synthesized, and adapters were ligated. Sequencing was performed using the Illumina NovaSeq 6000 sequencing platform by Majorbio Biotechnology Co., Ltd. (Shanghai, China).
Raw Illumina reads were trimmed and quality-controlled (QC) using fastp to obtain high-quality clean data to ensure the accuracy of subsequent analyses. The clean reads of each sample were compared with the reference sequences assembled by Trinity to obtain the mapping results of each sample and for subsequent unigene and transcript quantification of each sample.
To explore the related biological processes, BLAST2GO was used for Gene Ontology (GO) annotation (Diao et al., 2019) and molecular annotation of differentially expressed transcripts of samples. Differentially expressed genes (DEGs) of transcripts between the fliS-RNAi strain infection (experimental) and the wild type strain infection groups (control) were analyzed using edgeR (screening criteria: |log2FC| ≥ 1 and false discovery rate (FDR) < 0.05). GO enrichment analysis was performed on the obtained transcripts using GOATOOLS. Signaling pathway functional enrichment was analyzed by Kyoto Encyclopedia of Genes and Genomes (KEGG) (Zhao et al., 2018). KOBAS (http://kobas.cbi.pku.edu.cn/home.do) were used for KEGG pathway analysis of differential transcripts. Ten genes were randomly selected to verify the reliability of RNA-seq by qRT-PCR.
Metabolomic analysis
A certain amount of sample was accurately weighed and added to the extract obtained from the metabolite extraction process in a low-temperature environment. After centrifugation, the supernatant was collected for liquid chromatography-mass spectrometry (LC-MS) detection. Six replicates were performed per group.
LC-MS was performed using ultra-performance liquid chromatography tandem Fourier transform mass spectrometry (UPLC-MS/MS) with the UHPLC-Q Exactive HF-X system (Thermo Fisher). Chromatographic conditions were as follows: Acquity UPLC HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm); Waters, Milford, USA); mobile phase A 95% water + 5% acetonitrile (containing 0.1% formic acid) and mobile phase B 47.5% acetonitrile + 47.5% isopropanol + 5% water (containing 0.1% formic acid); injection volume 2 μL; column temperature 40°C. For mass spectrometry, the sample was subjected to electrospray ionization and mass spectrometry signals were collected in positive and negative ion scanning modes. Specific parameters are provided in Supplementary Table 3.
After UPLC-TOF/MS, the raw data were preprocessed, including original data missing value filtering, missing value filling, data normalization, QC verification, and data conversion. In addition, mass spectrometry information was exported and compared in the Human Metabolome database (HMDB) (http://www.hmdb.ca/) and Metlin database (https://metlin.scripps.edu/) to obtain specific information on metabolites.
The preprocessed data were uploaded to the MSG BioCloud platform (https://cloud.majorbio.com) for data analysis. The R package ropls (v1.6.2) was used for principal component analysis (PCA) and orthogonal least squares discriminant analysis (OPLS-DA). In addition, t-test and difference multiple analysis were performed. differentially expressed metabolites between the fliS-RNAi and wild type groups were screened (VIP > 1, FDR < 0. 05, |FC| ≥ 1), and the pathways involved in the differentially expressed metabolites were obtained using KEGG (https://www.kegg.jp/kegg/pathway.html). The Python software package SciPy was used for pathway enrichment analysis and Fisher’s exact test was used to obtain the most relevant biological pathways.
Combined analysis of transcriptomics and metabolomics
The VennDiagram package in R was used to analyze KEGG pathways involved in differentially expressed genes (DEGs) and differentially expressed metabolites. The top 10 KEGG pathways with the largest number of DEGs and differentially expressed metabolites were counted for visualization. The DEGs obtained by transcriptomics and differentially expressed metabolites obtained by metabolomics were integrated to analyze KEGG pathway data.
Statistical analyses
All data are expressed as means ± standard deviation (SD) from at least three sets of independent experiments. Data analysis was implemented using IBM SPSS Statistics v22.0 (New York, USA), with one-way analysis of variance (ANOVA) with Dunnett’s test used. P-values < 0.05 were considered statistically significant.
Data access
The RNA-seq data were deposited in the GenBank SRA database under accession numbers SRP 374606 (fliS-RNAi strain infection group), SRP 370956 (NZBD9 strain infection group), and SRP 370960 (PBS injection group). The metabolome data were deposited in the China National GeneBank Database under accession number CNP0003231.
Results
Silencing efficiency of RNAi strains
The expression level of fliS in five different RNAi strains was verified by qRT-PCR. As shown in Figure 1A, all five shRNAs significantly reduced fliS mRNA expression by 64.2%, 76.7%, 89.0%, 77.7%, and 84.8%, respectively. Thus, five stably silenced strains of P. plecoglossicida were successfully constructed. Among them, the fliS-RNAi-126 strain (hereinafter the fliS-RNAi strain) showed the best RNAi effect and was selected for subsequent experiments (Figure 1A). As shown in Figure 1B, there were no significant differences in the growth curves of the fliS-RNAi and wild type strains.
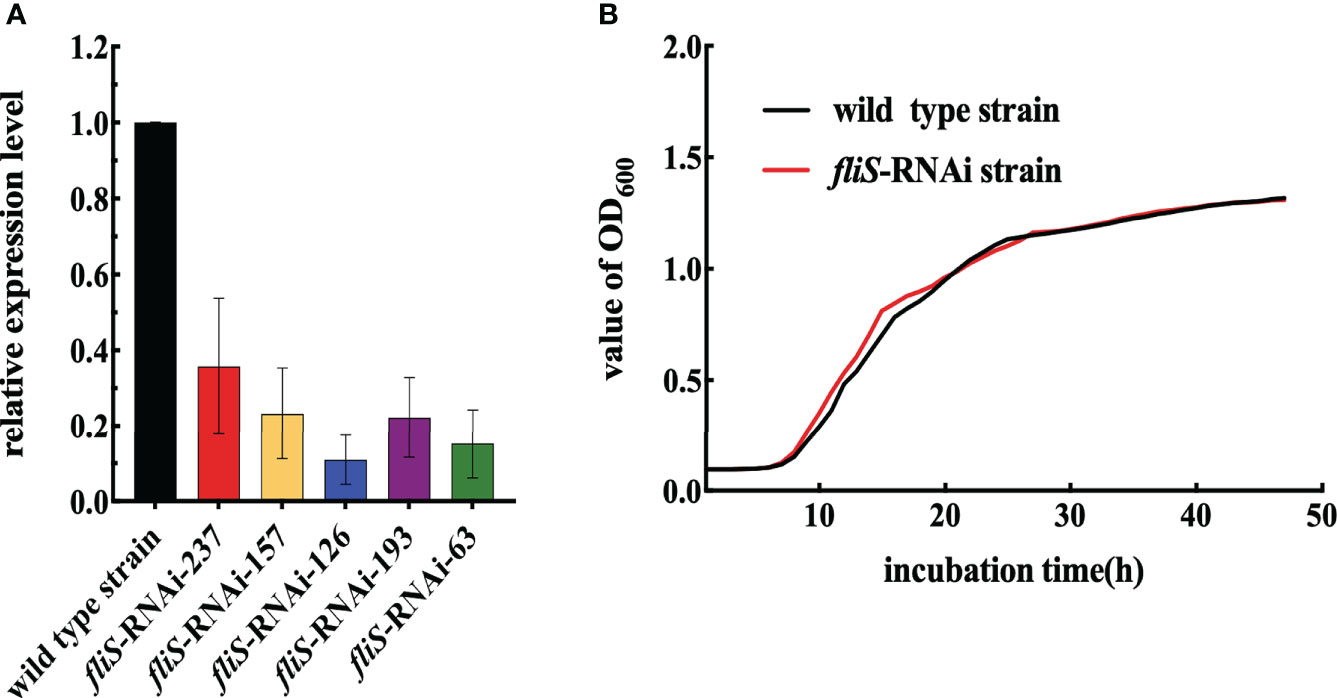
Figure 1 Construction and growth curve of P. plecoglossicida fliS-RNAi strain. (A) fliS mRNA levels of P. plecoglossicida strains. (B) Growth curve of wild type and fliS-RNAi strains.
Effects of fliS silencing on P. plecoglossicida virulence-related characteristics
The LB agar plate-cultured colonies of the fliS-RNAi and wild type strains of P. plecoglossicida are shown in Figure 2A. Results indicated that the fliS-RNAi strain colonies were significantly smaller (6.58 ± 0.11 mm, p < 0.01) than those of the wild type strain (8.37 ± 0.08 mm) (Figure 2B).
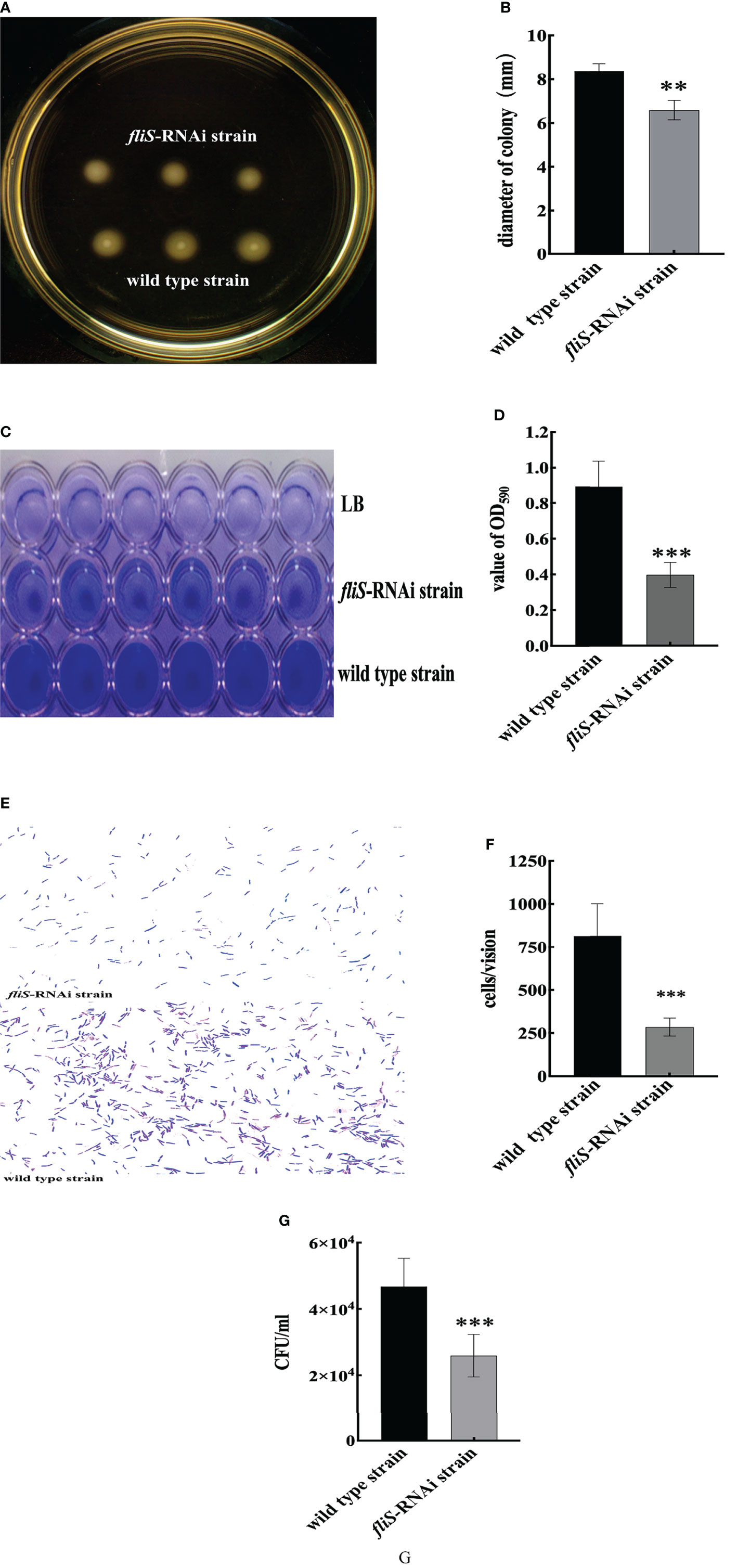
Figure 2 Characteristics of two P. plecoglossicida strains. (A) Colonies of wild type and fliS-RNAi strain swarms on soft agar plates. (B) Colony diameters of wild type and fliS-RNAi strains. (C) Biofilm of wild type and fliS-RNAi strains. (D) Biofilm formation ability of wild type and fliS-RNAi strains. (E) Adhering bacteria under the microscope. (F) Adhesion ability of wild type and fliS-RNAi strains. (G) Chemotaxis capacity of wild type and fliS-RNAi strains. Data are mean ± SD. *p < 0. 05, **p < 0. 01, ***p < 0. 001.
Biofilm formation of the fliS-RNAi and wild type strains of P. plecoglossicida is shown in Figure 2C. Compared to the wild type strain, the fliS-RNAi biofilm was lighter and deeper than that of LB. Results also showed that the OD590 of fliS-RNAi was significantly lower (0.39 ± 0.02 nm, p < 0.001) than that of the wild type strain (0.89 ± 0.04 nm) (Figure 2D).
Adhesion of the fliS-RNAi and wild type strains of P. plecoglossicida to the mucus surface of E. coioides is shown in Figure 2E. Results indicated that the adhesion ability of the fliS-RNAi strain was markedly weakened, with significantly fewer (286 ± 6.20 cells/vision, p < 0.001) adhered bacteria compared to the wild type strain (814 ± 22.20 cells/vision) (Figure 2F).
Chemotaxis capacity of the fliS-RNAi and wild type strains of P. plecoglossicida is shown in Figure 2G. Results indicated that fliS-RNAi strain chemotaxis was marked weakened, with significantly lower (2.6 × 104 CFU/mL, p < 0.001) chemotaxis compared to the wild type strain (4.7 × 104 CFU/mL).
Effects of fliS silencing on P. plecoglossicida pathogenicity
The E. coioides infected with the wild type strain of P. plecoglossicida began to die at 2 dpi, with 100% mortality reached at 6 dpi. In contrast, no E. coioides infected with the fliS-RNAi strain died by 10 dpi, as also found in the PBS-injected controls (Figure 3A). Furthermore, the apparent characteristics of E. coioides spleens at 4 dpi (Figure 3B) showed many white nodules on the spleen surface of E. coioides infected by the wild type strain, but almost no white spots on the spleen surface of fish infected by the fliS-RNAi strain or PBS. As shown in Figure 3C, although pathogen load in the fliS-RNAi strain-infected E. coioides spleens did not change significantly from 1 dpi to 4 dpi, it was consistently significantly lower than that of the wild type strain, and relative pathogen load (fliS-RNAi strain pathogen load/wild type strain pathogen load) decreased sharply with the duration of infection. The fliS gene of both strains was more highly expressed in the spleen than in vitro, and fliS expression in the fliS-RNAi strain was consistently lower than that in the wild type strain (Figure 3D).
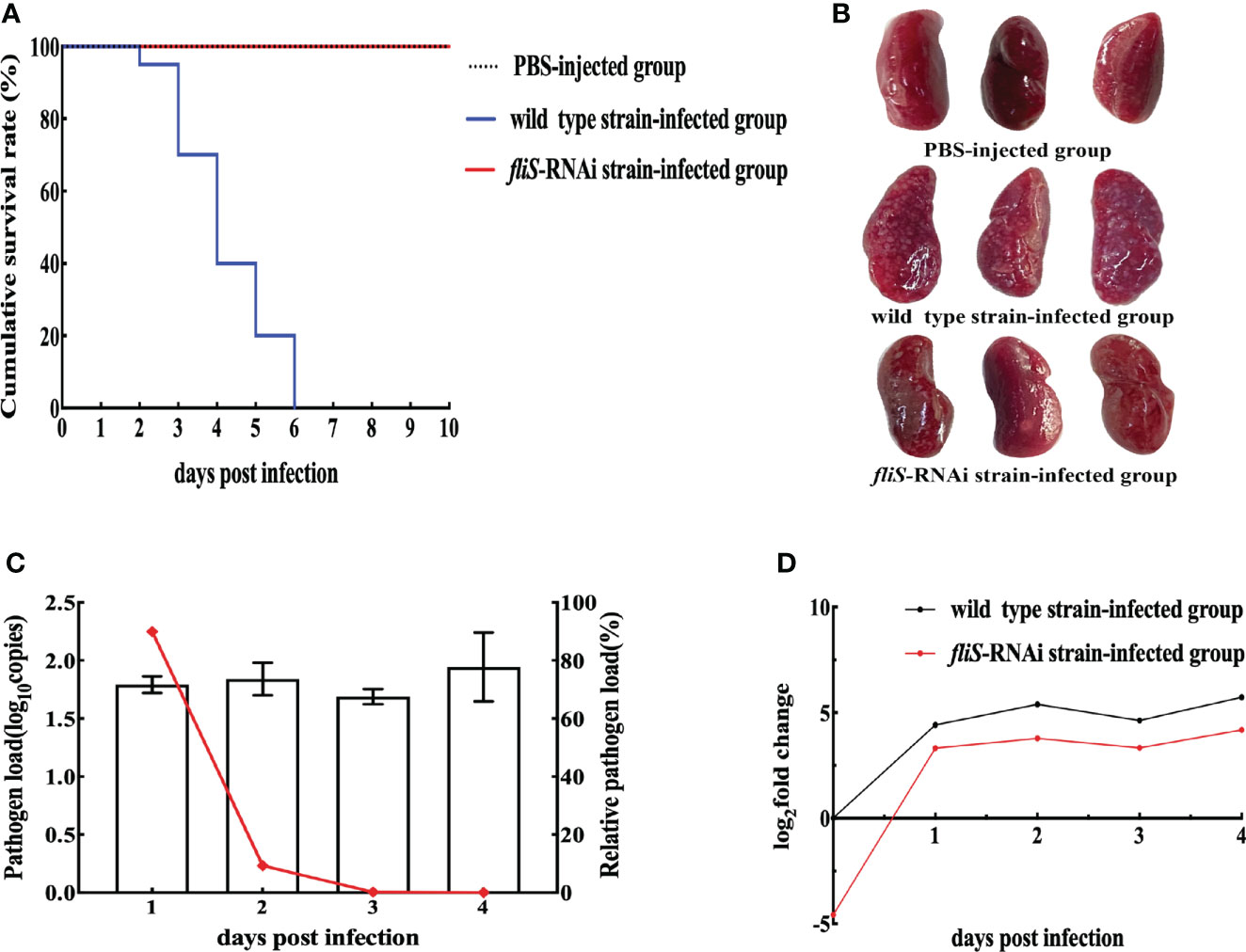
Figure 3 Pathogenicity of two P. plecoglossicida strains against E coioides. (A) Survival rate. (B) Symptoms in E coioides spleens at 4 dpi. (C) Pathogen load of fliS-RNAi strain in E coioides spleens during infection. Column represents pathogen load in spleen, represented by copy number of housekeeping gene gyrb. Line graph represents relative pathogen load value (i.e., copy number of gyrB gene of fliS-RNAi strain/copy number of gyrB gene of wild type strain). (D) mRNA expression level of fliS gene of P. plecoglossicida in E coioides spleens during infection.
Effects of fliS silencing on infected E. coioides spleen transcriptome
RNA-seq was performed on the spleens of E. coioides infected with the fliS-RNAi or wild type strains of P. plecoglossicida. Quality control of the original sequencing data showed that the Q20 (percentage of bases with quality value ≥ 20) of each sample was above 97.41%, Q30 (percentage of bases with quality value ≥ 30) was above 93.94%, and GC content was about 50% (Supplementary Table 4). The sequencing error rate was less than 0.1%, and sequencing accuracy was high. Thus, the quality of the sequencing data met the requirements for subsequent data processing and analysis.
Through the correlation analysis between biological repeat samples, on the one hand to test whether the variation between biological repeats meets the expectations of experimental design, on the other hand to provide basic reference for DEG analysis. Pearson correlation analysis was used to assess the associations between the wild type strain-infected group and fliS-RNAi strain-infected groups. The minimum and maximum Pearson correlation coefficients between groups were 0.098 and 0.164, respectively, and within groups were 0.99 and 1, respectively (Supplementary Figure 1). Thus, biological repeatability was deemed good, with strong intra-group correlation and small inter-group correlation. The experimental data were therefore reliable and could be used for further analysis of DEGs.
EdgeR was used for differential transcript analysis of E. coioides. The statistical standard of mRNA expression level of transcriptome data was FDR < 0.05 and |log2FC| ≥ 1 as GO and KEGG pathway enrichment analysis of DEGs. Transcriptomic analysis of E. coioides spleens sampled at 4 dpi identified 52 006 DEGs between the fliS-RNAi and wild type strain-infected spleens, including 38 393 up-regulated DEGs and 13 613 down-regulated DEGs (Figure 4A). The top 50 most significantly up-regulated and down-regulated DEGs were selected for analysis (Figure 4B).
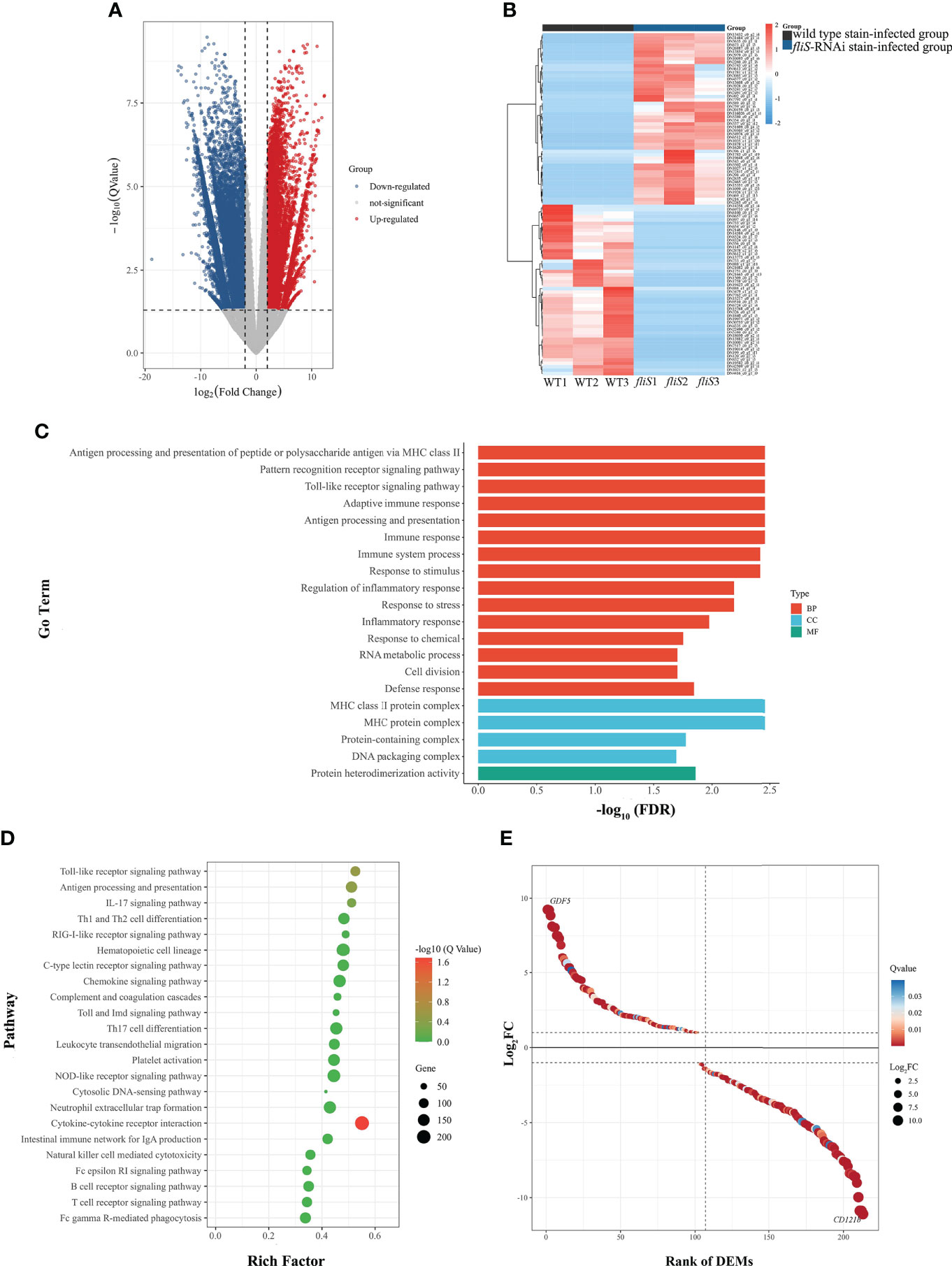
Figure 4 Comparative transcriptomic analysis of E coioides spleens infected by fliS-RNAi and wild type strains of P. plecoglossicida. (A) Volcano plot obtained from EdgeR analysis. (B) Heat map of top 50 up-regulated (red) and down-regulated DEGs (blue). Deeper color indicates higher expression difference. (C) GO enrichment analysis of DEGs. (D) KEGG pathway enrichment analysis of DEGs. (E) Expression level of DEGs in cytokine-cytokine receptor interaction pathway.
Figure 4C shows the top 20 GO enrichment results. Based on GO functional annotation, the DEGs were annotated into three categories, i.e., biological process (BP), cell composition (CC), and molecular function (MF), with significant enrichment in protein-containing complex and immune system process and response to stimulus.
Further enrichment analysis identified 23 KEGG pathways involved in the immune response (Figure 4D), with 23.8% of DEGs enriched in immune system pathways. Based on Fisher’s exact test, 213 DEGs were significantly enriched in the cytokine-cytokine receptor interaction pathway, including 102 significantly up-regulated DEGs and 111 significantly down-regulated DEGs (Figure 4E). Most genes involved in this pathway were chemokines, including class I helical cytokines, class II helical cytokines, IL1-like cytokines, IL17-like cytokines, TNF family, and TGF-β family. Notably, chemokines such as CXCL8, CXCR2, CCL11, and CXCL12 were up-regulated, while certain inflammatory genes such as IL1B, IL1R2, IL17, and IL17RA were down-regulated. Furthermore, TNF gene expression was down-regulated, while TGFB1 and TGFB2 gene expression levels were up-regulated. qRT-PCR verification was performed on 10 randomly selected DEGs from the RNA-seq database. Results showed that the expression trends of the selected DEGs were consistent with those of transcriptome sequencing (Supplementary Figure 2).
Effects of fliS silencing on infected E. coioides spleen metabolome
To determine the cumulative differences in spleen metabolites between the fliS-RNAi and wild type strain-infected groups, PCA was performed to obtain scores of the two principal components. In the total ion PCA model, PC1 = 60.30%, PC2 = 8.52% (Figure 5A), R2X = 0.603, and the cumulative difference explanatory value was greater than 0.5, so the established PCA model was stable and could be used for subsequent metabolic difference analysis. To display the metabolic differences between the two groups more accurately, the supervised OPLS-DA model was used for further analysis. The fliS-RNAi strain-infected samples were mainly distributed in the negative half of PC1, while the wild type strain-infected samples were mainly distributed in the positive half of PC1, so the model could effectively distinguish the two groups of samples (Figure 5B). The replacement test of OPLS-DA model showed that R2X and R2Y were 0.753 and 0.991, and Q2 was 0.986, which were close to 1 and had good reliability (Supplementary Table 5).
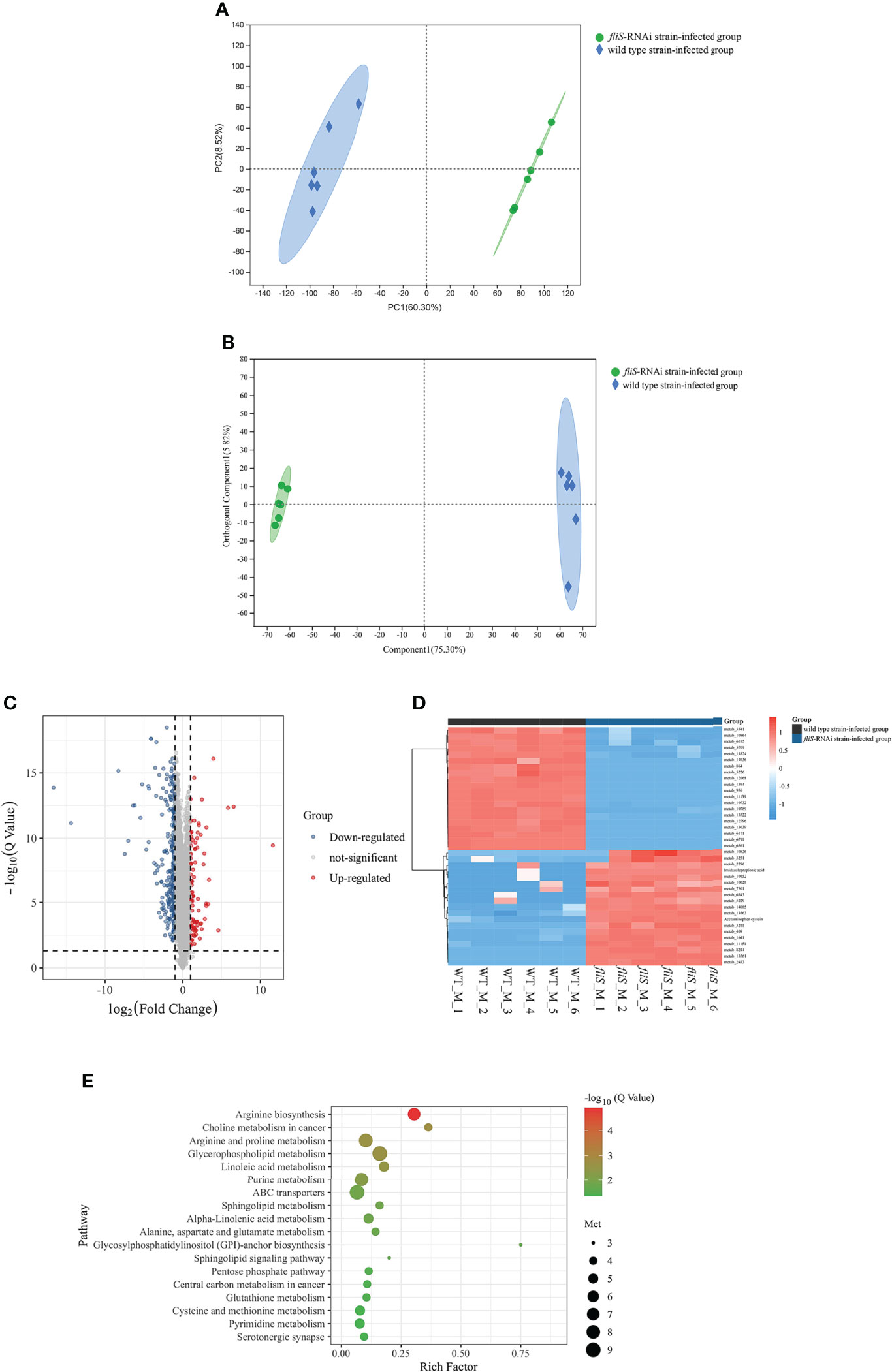
Figure 5 Comparative metabolomic analysis of E coioides spleens infected by fliS-RNAi and wild type strains of P. plecoglossicida. (A) PCA diagram. X-axis shows first principal component, Y-axis shows second principal component. (B) OPLS-DA score plot. Component 1 represents first predictive principal component interpretation and Orthogonal Component 1 represents first orthogonal component interpretation. (C) Volcano plot of differentially expressed metabolites. (D) Heat map comparison of differentially expressed metabolites. (E) KEGG enrichment of differentially expressed metabolites.
The differentially expressed metabolites between the two infection groups were analyzed. Based on screening criteria (FDR < 0. 05, VIP > 1, FC > 1, FC < 1), 326 differentially expressed metabolites were identified, mainly divided into organic acids, lipids, carbohydrates, nucleic acids, peptides, vitamins and coenzyme factors, steroids, hormones and transmission media, and antibiotics according to their biological function. Compared with the wild type strain-infected group, 107 and 219 differentially expressed metabolites in the fliS-RNAi strain-infected group were up-regulated (FC > 1) and down-regulated (FC < 1), respectively (Figure 5C). The top 20 up-regulated and down-regulated differentially expressed metabolites were selected for further analysis (Figure 5D).
Based on KEGG pathway enrichment analysis, 105 metabolic pathways were annotated, with 18 found to be significantly enriched (P < 0.05), including glycosylphosphatidylinositol (GPI)-anchor biosynthesis, glycerophospholipid metabolism, arginine biosynthesis, arginine and proline metabolism, ABC transporters, linoleic acid metabolism, alpha-linolenic acid metabolism, sphingolipid signaling pathway, and glutathione metabolism. Six metabolic pathways were highly significantly enriched (P < 0.01), including linoleic acid metabolism, arginine and proline metabolism, glycerophospholipid metabolism, choline metabolism in cancer, purine metabolism, and especially arginine biosynthesis pathway (Figure 5E).
Comprehensive transcriptomic and metabolomic analysis
The transcriptomic and metabolomic results were correlated. Venn analysis indicated that 101 KEGG pathways were shared between the transcriptomes and metabolomes, accounting for 94.2% of the total metabolome pathways (105) and 29.1% of the total transcriptome pathways (347) (Figure 6A). Figure 6B shows the top 10 KEGG pathways with the largest number of DEGs and differentially expressed metabolites. We found that arginine biosynthesis pathway was most involved (Figure 6B), and the correlation between metabolites and DEGs was the best (Figure 6C). So focused on this pathway to explore the relationship between them, further in-depth analysis was conducted.
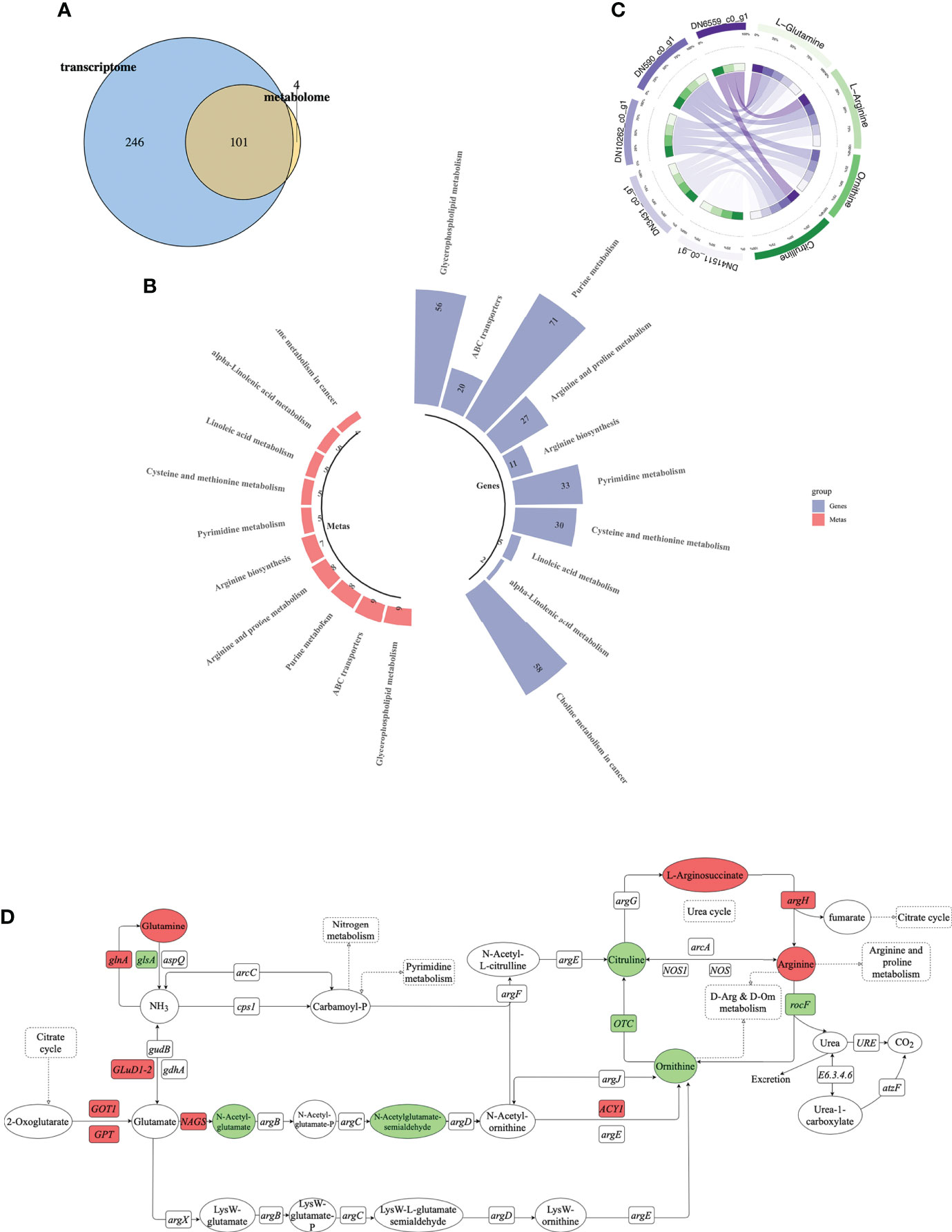
Figure 6 Combined transcriptomic and metabolomic analysis. (A) Venn diagram of DEM-enriched pathways and differential metabolite-enriched pathways (blue is transcriptome, yellow is metabolome, and round cross region represents number of shared KEGG pathways). (B) Top 10 pathways containing most DEGs and differentially expressed metabolites. Each column represents a KEGG pathway. Blue represents transcriptome, red represents metabolome. (C) Correlation analysis between differentially expressed metabolites and DEGs. Purple represents DEGs, green represents differentially expressed metabolites. (D) Schematic of arginine biosynthesis pathway. Green indicates down-regulation, red indicates up-regulation. Circle represents metabolites, rectangle represents genes.
We identified 11 DEGs and seven differentially expressed metabolites involved in the arginine biosynthesis pathway (Figure 6D). Among them, glnA and argH were up-regulated and glsA and rocF were down-regulated, while glutamine and arginine were up-regulated and ornithine and citrulline were down-regulated. We found that glnA gene and argH gene in the transcriptome involved in arginine biosynthesis pathway, as well as glutamine and arginine in the metabolic group were significantly up-regulated. Among them, glnA gene was positively regulated with glutamine, and argH gene was positively regulated with arginine.
Discussion
RNAi technology is a highly efficient tool for investigating gene function (Dai et al., 2005; Fellmann et al., 2011; Liu et al., 2020). In this study, the five shRNAs used showed different fliS silencing efficiency, indicating the versatility of RNAi and differences at different sites. Stable and efficient silencing is essential for gene function studies. Here, the best fliS silencing efficiency obtained was 89%, and the effect persisted at all sampling times, thus laying the foundation for this research.
Motility mediated by the bacterial flagellar system increases the probability of contact, adhesion, and colonization of host cells, thus highlighting the critical role of flagella in the infection process (Houry et al., 2010; Chaban et al., 2015). Flagella are also involved in many other processes, including chemotaxis, biofilm formation, virulence factor secretion, and eukaryotic immune system regulation (Duan et al., 2013; Gallego-Hernandez et al., 2020). Here, fliS gene silencing significantly reduced P. plecoglossicida motility, biofilm formation, and adhesion ability, which are factors closely related to bacterial pathogenicity (Song et al., 2019; Zhang et al., 2020). We previously found that the P. plecoglossicida fliM gene was involved in flagellar assembly (Sun et al., 2019) and the fliG gene was involved in regulation of flagellar movement to regulate toxicity (Jiao et al., 2021).
Our results showed that E. coioides injected with the wild type strain of P. plecoglossicida began to die at 2 dpi and reached 100% mortality at 6 dpi, while the E. coioides fish infected by the fliS-RNAi strain were still alive at 10 dpi. Thus, fliS gene silencing resulted in a marked reduction in P. plecoglossicida pathogenicity, further suggesting that fliS is a virulence gene of P. plecoglossicida, as confirmed by the spleen symptoms and pathogen load. Silencing of fliS did not affect P. plecoglossicida growth, suggesting that the decrease in P. plecoglossicida virulence was not due to a decrease in the growth rate. However, whether P. plecoglossicida virulence is associated with motility, biofilm formation, and adhesion remains to be investigated.
Compared with the wild type-infected E. coioides spleen, we identified 52 006 DEGs in the fliS-RNAi-infected E. coioides spleen, including 38 393 up-regulated DEGs and 13 613 down-regulated DEGs. These findings indicated that fliS gene silencing significantly affected transcription in infected E. coioides.
Based on GO and KEGG pathway enrichment analysis of the 52006 DEGs, 23 immune-related pathways were enriched, especially the cytokine-cytokine receptor interaction pathway. Thus, fliS gene silencing significantly affected the immune response of P. plecoglossicida-infected E. coioides.
Cytokines are small molecular polypeptides or glycoproteins synthesized and secreted by a variety of tissue cells (mainly immune cells) (Wang et al., 2020; Chauhan et al., 2021), which regulate cell growth, differentiation, and immune response (Curfs et al., 1997). In this study, compared with the control group, IL-1B gene expression was significantly down-regulated in the spleens of E. coioides infected with the fliS-RNAi strain, with down-regulation also observed for the receptor gene IL1R2 and downstream gene TNF. The IL-1B gene encodes IL-1β, which can activate the release of other pro-inflammatory cytokines, such as TNF, in response to viral and bacterial stimulation (Eder, 2009; Fathy et al., 2019; Ren and Torres, 2009; Zhang et al., 2022). IL-1β also plays a key role in inflammation and immune response, with important implications in disease pathogenesis (Lukens et al., 2012). When fish are stimulated by bacteria, viruses, endotoxins, and other pathogenic factors, the transcription of IL-1β is significantly up-regulated in immune organs, skin, gills, intestines, and other tissues (Yang et al., 2017; Dong et al., 2021; Tian et al., 2021). In this study, the significant down-regulation of IL-1B in the fliS-RNAi strain-infected E. coioides spleens indicated a milder inflammatory response compared to those fish infected with the wild type strain, consistent with the lower pathogen load in the fliS-RNAi-infected spleens.
Our results also showed that TGFB1 and TGFB2 were up-regulated in the fliS-RNAi strain-infected E. coioides spleens compared with the wild type-infected group. TGFB1 and TGFB2 encode TGF-β, a multifunctional cytokine involved in immune response regulation (Haque and Morris, 2017), and play an important role in cell growth, proliferation, differentiation, adhesion, migration, polarization of Th17 reaction, generation, and apoptosis of reactive oxygen species (Morikawa et al., 2016; Seoane and Gomis, 2017; Wang et al., 2019). In the present study, up-regulation of TGFB1 and TGFB2 indicated that E. coioides infected with the fliS-RNAi strain showed a stronger TGF-β-based immune response than E. coioides infected with the wild type strain of P. plecoglossicida. Furthermore, E. coioides exhibited different inflammatory and TGF-β-based immune responses to infection by the wild type and fliS-RNAi strains of P. plecoglossicida, indicating complexity of the immune response.
Metabolomic analysis revealed significantly different patterns in the E. coioides infected with the wild type and fliS-RNAi strains. These results suggest that fliS gene silencing can significantly alter the metabolic pattern of E. coioides under P. plecoglossicida infection, consistent with the changes in the transcription of E. coioides infected with the two strains of P. plecoglossicida.
Combined with metabolomic analysis, our results showed that the differentially expressed metabolites were mainly concentrated in the arginine and proline metabolism, arginine biosynthesis, glutathione metabolism, and other metabolic pathways. Arginine biosynthesis was the most significantly enriched KEGG pathway. Seven differentially expressed metabolites were enriched in this pathway, including L-arginine and L-glutamine, which were significantly up-regulated. Amino acid metabolism plays an important role in the immune system to fight pathogen infection (Synne et al., 2016). The arginine biosynthesis pathway, including arginine, glutamine, and other metabolites, is involved in oxidative stress, biofilm formation, immune response, and inflammatory response (Wang et al., 2021; Huang et al., 2022; Long et al., 2022). In addition, arginine biosynthesis is a key metabolic pathway for the antitumor effects of drugs, with the arginine metabolite playing an important role (Chen et al., 2021). Previous research has shown that the survival rate of Aeromonas hydrophila-infected Jian carp (Cyprinus carpio var. Jian) significantly increases after arginine feeding, and arginine can enhance immunity and disease resistance (Chen et al., 2015). In addition, arginine regulates mRNA expression of the TOR pathway, as well as inflammatory cytokines and antioxidant-related signaling molecules (Habte-Tsion, 2020). Inhibition of the TOR pathway significantly hinders the arginine enzyme metabolism and Arg-NO metabolism pathways and improves humoral and cellular immunity (Habte-Tsion, 2020). In this study, we found that L-arginine was up-regulated in the spleens of E. coioides infected with the fliS-RNAi strain compared to the wild type strain, suggesting that virulence of the silenced strain was weakened, while the L-arginine metabolic pathway was strengthened, resulting in an enhanced immune response.
Similarly, studies have shown that dietary probiotics can improve L-glutamine content in Litopenaeus vannamei, thereby regulating the immune response (Chien et al., 2020). In Oreochromis niloticus, glutamine supplementation can enhance the bactericidal ability of Streptococcus through macrophages (Carvalho et al., 2018). In Scophthalmus maximus, glutamine can reduce the negative effects of soybean meal on intestinal immune function by increasing intestinal immune components and reducing intestinal inflammation (Gu et al., 2017). In this study, we found that glutamine levels were up-regulated in the spleens of E. coioides infected with the fliS-RNAi strain compared to those infected with the wild type strain. This may be due to stimulation of P. plecoglossicida, L-glutamine needs to be consumed by many immune cells, thereby increasing its synthesis to facilitate further immune response.
We speculate that infection with the fliS-RNAi strain of P. plecoglossicida may induce arginine biosynthesis pathway expression in E. coioides, which may help prevent pathogen infection, thereby weakening P. plecoglossicida damage to the host.
Conclusions
Our study showed that the fliS gene is involved in the regulation of P. plecoglossicida motility, adhesion, and biofilm formation, and may be a virulence gene of P. plecoglossicida. Silencing the fliS gene significantly affected the transcriptome and metabolome of the E. coioides spleen following P. plecoglossicida infection. The cytokine-cytokine receptor interaction pathway was the most significantly enriched immune-related pathway in the transcriptome, while the arginine biosynthesis pathway was the most significantly enriched pathway in the metabolome. The transcriptome and metabolome changes indicated that the inflammatory response of E. coioides infected with the fliS-RNAi strain was milder than that infected with the wild type strain.
Data availability statement
The RNA-seq data were deposited in the GenBank SRA database under accession numbers SRP 374606 (fliS-RNAi strain infection group), SRP 370956 (NZBD9 strain infection group), and SRP 370960 (PBS injection group). The metabolome data were deposited in the China National GeneBank Database under accession number CNP0003231.
Ethics statement
The animal protocols were approved by the Animal Ethics Committee of Jimei University (Acceptance NO JMULAC201159).
Author contributions
Seven authors contributed to the article, QY conceived the experiments. WW and LZ conducted the experiments. All authors assisted in the collection and interpretation of data. WW and QY wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Fujian Province under contract No. 2021J01828, National Natural Science Foundation of China under contract No. 31972836, open fund of Fujian Province Key Laboratory of Special Aquatic Formula Feed under contract No. TMKJZ2101.
Conflict of interest
Authors JNZ and JLZ were employed by Fujian Tianma Technology Company Limited, Fuzhou, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.987825/full#supplementary-material
References
Borrego J., Valverde E., Labella A., Castro D.(2017). Lymphocystis disease virus: its importance in aquaculture. Rev. Aquac. 9 (2), 179–193. doi: 10.1111/raq.12131
Cai S., Cheng H., Pang H., Jian J. (2017). Role of the toxR gene from fish pathogen Vibiro alginolyticus in the physiology and virulence. Indian J. Microbiol. 57 (4), 477–484. doi: 10.1007/s12088-017-0685-x
Carvalho P., Yamamoto F., Barros M., Gatlin Iii D. (2018). L-glutamine in vitro supplementation enhances Nile tilapia Oreochromis niloticus (Linnaeus 1758) leukocyte function. Fish Shellfish Immunol. 80, 592–599. doi: 10.1016/j.fsi.2018.06.043
Chaban B., Hughes H. V., Beeby M. (2015). The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 46, 91–103. doi: 10.1016/j.semcdb.2015.10.032
Chauhan P., Nair A., Patidar A., Saha B. (2021). A primer on cytokines. Cytokine 145, 155458. doi: 10.1016/j.cyto.2021.155458
Chen G., Liu Y., Jiang J., Jiang W., Kuang S., Tang L., et al. (2015). Effect of dietary arginine on the immune response and gene expression in head kidney and spleen following infection of jian carp with Aeromonas hydrophila. Fish Shellfish Immunol. 44 (1), 195–202. doi: 10.1016/j.fsi.2015.02.027
Chen Y., Si L., Zhang J., Yu H., Liu X., Chen Y., et al. (2021). Uncovering the antitumor effects and mechanisms of shikonin against colon cancer on comprehensive analysis. Phytomedicine 82, 153460. doi: 10.1016/j.phymed.2021.153460
Chien C., Lin T. Y., Chi C., Liu C. (2020). Probiotic, Bacillus subtilis E20 alters the immunity of white shrimp, Litopenaeus vannamei via glutamine metabolism and hexosamine biosynthetic pathway. Fish Shellfish Immunol. 98, 176–185. doi: 10.1016/j.fsi.2020.01.014
Condry D., Nilles M. (2017). Introduction to type III secretion systems. Methods Mol. Biol. 1531, 1–10. doi: 10.1007/978-1-4939-6649-3_1
Curfs J., Meis J., Hoogkamp-korstanje J. (1997). A primer on cytokines: Sources, receptors, effects, and inducers. Clin. Microbiol. Rev. 10 (4), 742–780. doi: 10.1128/CMR.10.4.742
Dai F., Yusuf F., Farjah G., Brand-Saberi B. (2005). RNAi-induced targeted silencing of developmental control genes during chicken embryogenesis. Dev. Biol. 285 (1), 80–90. doi: 10.1016/j.ydbio.2005.06.005
Diao J., Liu H., Hu F., Li L., Wang X., Gai C., et al. (2019). Transcriptome analysis of immune response in fat greenling (Hexagrammos otakii) against Vibrio harveyi infection. Fish Shellfish Immunol. 84, 937–947. doi: 10.1016/j.fsi.2018.10.067
Dong W., Gao W., Cui J., Sun Y. (2021). microRNA-148 is involved in NF-κB signaling pathway regulation after LPS stimulation by targeting IL-1β in miiuy croaker. Fish Shellfish Immunol. 118, 66–71. doi: 10.1016/j.fsi.2021.08.028
Duan Q., Zhou M., Zhu L., Zhu G. (2013). Flagella and bacterial pathogenicity. J. Basic Microbiol. 53 (1), 1–8. doi: 10.1002/jobm.201100335
Eder C. (2009). Mechanisms of interleukin-1beta release. Immunobiology 214 (7), 543–553. doi: 10.1016/j.imbio.2008.11.007
Fathy S., Mohamed M., Ali M., Alattar A. (2019). Influence of IL-6, IL-10, IFN-γ and TNF-α genetic variants on susceptibility to diabetic kidney disease in type 2 diabetes mellitus patients. Biomarkers 24 (1), 43–55. doi: 10.1080/1354750X.2018.1501761
Fellmann C., Zuber J., McJunkin K., Chang K., Malone C., Dickins R., et al. (2011). Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell 41 (6), 733–746. doi: 10.1016/j.molcel.2011.02.008
Gallego-Hernandez A., DePas W., Park J., Teschler J., Hartmann R., Jeckel H., et al. (2020). Upregulation of virulence genes promotes Vibrio cholerae biofilm hyperinfectivity. Proc. Natl. Acad. Sci. U.S.A. 117 (20), 11010–11017. doi: 10.1073/pnas.1916571117
Gu H. (2017). Role of flagella in the pathogenesis of Helicobacter pylori. Curr. Microbiol. 74 (7), 863–869. doi: 10.1007/s00284-017-1256-4
Gu M., Bai N., Xu B., Zhang Z. (2017). Protective effect of glutamine and arginine against soybean meal-induced enteritis in the juvenile turbot (Scophthalmus maximus). Fish Shellfish Immunol. 70, 95–105. doi: 10.1016/j.fsi.2017.08.048
Habte-Tsion H. (2020). A review on fish immuno-nutritional response to indispensable amino acids in relation to TOR, NF-κB and Nrf2 signaling pathways: Trends and prospects. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 241, 110389. doi: 10.1016/j.cbpb.2019.110389
Haque S., Morris J. (2017). Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin Immunother. 13 (8), 1741–1750. doi: 10.1080/21645515.2017.1327107
He L., Wang L., Zhao L., Zhuang Z., Wang X., Huang H., et al. (2021). Integration of RNA-seq and RNAi reveals the contribution of znuA gene to the pathogenicity of Pseudomonas plecoglossicida and to the immune response of Epinephelus coioides. J. Fish Dis. 44, 1831–1841. doi: 10.1111/jfd.13502
Houry A., Briandet R., Aymerich S., Gohar M. (2010). Involvement of motility and flagella in Bacillus cereus biofilm formation. Microbiol. (Reading) 156 (Pt 4), 1009–1018. doi: 10.1099/mic.0.034827-0
Huang L., He X., Peng W., He X., Xu B., Xu H., et al. (2022). Hyperuricemia induces liver injury by upregulating HIF-1α and inhibiting arginine biosynthesis pathway in mouse liver and human L02 hepatocytes. Biochem. Biophys. Res. Commun. 617, 55–61. doi: 10.1016/j.bbrc.2022.05.096
Jiao J., Zhao L., Huang L., Qin Y., Su Y., Zheng W., et al. (2021). The contributions of fliG gene to the pathogenicity of Pseudomonas plecoglossicida and pathogen-host interactions with Epinephelus coioides. Fish Shellfish Immunol. 119, 238–248. doi: 10.1016/j.fsi.2021.09.03
Lertsethtakarn P., Ottemann K., Hendrixson D. (2011). Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 65, 389–410. doi: 10.1146/annurev-micro-090110-102908
Liu Z., Zhao L., Huang L., Qin Y., Zhang J., Zhang J., et al. (2020). Integration of RNA-seq and RNAi provides a novel insight into the immune responses of Epinephelus coioides to the impB gene of Pseudomonas plecoglossicida. Fish Shellfish Immunol. 105, 135–143. doi: 10.1016/j.fsi.2020.06.023
Li C., Wang S., Ren Q., He T., Chen X.. (2020). An outbreak of visceral white nodules disease caused by Pseudomonas plecoglossicida at a water temperature of 12 degrees c in cultured large yellow croaker (Larimichthys crocea) in China. J. Fish Dis. 43 (11), 1353–1361. doi: 10.1111/jfd.13206
Long S., Dong X., Liu H., Yan X., Tan B., Zhang S., et al. (2022). Effect of dietary oxidized fish oil on liver function in hybrid grouper (♀ Epinephelus fuscoguttatus × ♂ Epinephelus lanceolatus). Aquac. Rep. 22, 101000. doi: 10.1016/j.aqrep.2021.101000
Lowenthal A., Hill M., Sycuro L., Mehmood K., Salama N., Ottemann K., et al. (2009). Functional analysis of the Helicobacter pylori flagellar switch proteins. J. Bacteriol. 191 (23), 7147–7156. doi: 10.1128/JB.00749-09
Lukens J., Gross J., Kanneganti T. (2012). IL-1 family cytokines trigger sterile inflammatory disease. Front. Immunol. 3, 315. doi: 10.3389/fimmu.2012.00315
Luo G., Sun Y., Huang L., Su Y., Zhao L., Qin Y., et al. (2020). Time-resolved dual RNA-seq of tissue uncovers Pseudomonas plecoglossicida key virulence genes in host-pathogen interaction with Epinephelus coioides. Environ. Microbiol. 22 (2), 677–693. doi: 10.1111/1462-2920.14884
Luo G., Zhao L., Xu X., Qin Y., Huang L., Su Y., et al. (2019). Integrated dual RNA-seq and dual iTRAQ of infected tissue reveals the functions of a diguanylate cyclase gene of Pseudomonas plecoglossicida in host-pathogen interactions with Epinephelus coioides. Fish shellfish Immunol. 95, 481–490. doi: 10.1016/j.fsi.2019.11.008
Mao L., Qin Y., Kang J., Wu B., Huang L., Wang S., et al. (2020). Role of LuxR-type regulators in fish pathogenic Aeromonas hydrophila. J. fish Dis. 43 (2), 215–225. doi: 10.1111/jfd.13114
Miao E., Mao D., Yudkovsky N., Bonneau R., Lorang C, Warren S, et al. (2010). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107 (7), 3076–3080. doi: 10.1073/pnas.0913087107
Morikawa M., Derynck R., Miyazono K. (2016). TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8 (5), a021873. doi: 10.1101/cshperspect.a021873
Mugimba K., Byarugaba D., Mutoloki S., Evensen O., Munang'andu H. (2021). Challenges and solutions to viral diseases of finfish in marine aquaculture. Pathogens 10 (6), 673. doi: 10.3390/pathogens10060673
Muskotal A., Kiraly R., Sebestyen A., Gugolya Z., Vegh B., Vonderviszt F., et al. (2006). Interaction of FliS flagellar chaperone with flagellin. FEBS Lett. 580 (16), 3916–3920. doi: 10.1016/j.febslet.2006.06.024
Nishimori E., Kita-Tsukamoto K., Wakabayashi H. (2000). Pseudomonas plecoglossicida sp. nov., the causative agent of bacterial haemorrhagic ascites of ayu, Plecoglossus altivelis. Int. J. Syst. Evol. Microbiol. 50 (1), 83–89. doi: 10.1099/00207713-50-1-83
Ozin A., Claret L., Auvray F., Hughes C. (2003). The FliS chaperone selectively binds the disordered flagellin c-terminal D0 domain central to polymerisation. FEMS Microbiol. Lett. 219 (2), 219–224. doi: 10.1016/s0378-1097(02)01208-9
Radomska K., Wosten M., Ordonez S., van Putten J. (2017). Importance of Campylobacter jejuni FliS and FliW in flagella biogenesis and flagellin secretion. Front. Microbiol. 8, 1060. doi: 10.3389/fmicb.2017.01060
Ren K., Torres R. (2009). Role of interleukin-1β during pain and inflammation. Brain Res. Rev. 60 (1), 57–64. doi: 10.1016/j.brainresrev.2008.12.020
Seoane J., Gomis R. (2017). TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 9 (12), a022277. doi: 10.1101/cshperspect.a022277
Song H., Kang Y., Zhang D., Chen L., Qian A., Shan X., et al. (2019). Great effect of porin(aha) in bacterial adhesion and virulence regulation in Aeromonas veronii. Microbial. Pathog. 126, 269–278. doi: 10.1016/j.micpath.2018.11.002
Sun Y., Zhuang Z., Wang X., Huang H., Fu Q., Yan Q., et al. (2019). Dual RNA-seq reveals the effect of the flgM gene of Pseudomonas plecoglossicida on the immune response of Epinephelus coioides. Fish Shellfish Immunol. 87, 515–523. doi: 10.1016/j.fsi.2019.01.041
Synne M., Rune W., Marit E. (2016). Functional amino acids in fish health and welfare. Front. Biosci. 8 (1), 143–169. doi: 10.2741/757
Tang Y., Jiao J., Zhao L., Zhuang Z., Wang X., Fu Q., et al. (2022). The contribution of exbB gene to pathogenicity of Pseudomonas plecoglossicida and its interactions with Epinephelus coioides. Fish Shellfish Immunol. 120, 610–619. doi: 10.1016/j.fsi.2021.12.026
Tang Y., Xin G., Zhao L., Huang L., Qin Y., Su Y., et al. (2020). Novel insights into host-pathogen interactions of large yellow croakers (Larimichthys crocea) and pathogenic bacterium Pseudomonas plecoglossicida using time-resolved dual RNA-seq of infected spleens. Zool. Res. 41 (3), 314–327. doi: 10.24272/j.issn.2095-8137.2020.035
Tian G., Xie M., Wang B., Gao W., Yuan H., Wei H., et al. (2021). Identification and expression of IL-1β in the endangered dabry’s sturgeon (Acipenser dabryanus). Aquac. Rep. 20, 100698. doi: 10.1016/j.aqrep.2021.100698
Wang N., Gao J., Yuan L., Jin Y., He G. (2021). Metabolomics profiling during biofilm development of Bacillus licheniformis isolated from milk powder. Int. J. Food Microbiol. 337, 108939. doi: 10.1016/j.ijfoodmicro.2020.108939
Wang L. Y., Liu Z. X., Zhao L. M., Huang L., Qin Y., Su Y., et al. (2020). Dual RNA-seq provides novel insight into the roles of dksA from Pseudomonas plecoglossicida in pathogen-host interactions with large yellow croakers (Larimichthys crocea). Zool. Res. 41 (4), 410–422. doi: 10.24272/j.issn.2095-8137.2020.048
Wang Y., Wang X., Ali F., Li Z., Fu Y., Yang X., et al. (2019). Comparative extracellular proteomics of Aeromonas hydrophila reveals iron-regulated secreted proteins as potential vaccine candidates. Front. Immunol. 10, 256. doi: 10.3389/fimmu.2019.00256
Xin G., Wang F., Zhao L., Qin Y., Huang L., Yan Q., et al. (2020). Integration of RNA-seq and RNAi provides a novel insight into the effect of pvdE gene to the pathogenic of Pseudomonas plecoglossicida and on the immune responses of orange-spotted grouper (Epinephelus coioides). Aquaculture 529, 735695. doi: 10.1016/j.aquaculture.2020.735695
Xu S., Peng Z., Cui B., Wang T., Song Y., Zhang L., et al. (2014). FliS modulates FlgM activity by acting as a non-canonical chaperone to control late flagellar gene expression, motility and biofilm formation in Yersinia pseudotuberculosis. Environ. Microbiol. 16 (4), 1090–1104. doi: 10.1111/1462-2920.12222
Yang Q., Chu Q., Zhao X., Xu T. (2017). Characterization of IL-1β and two types of IL-1 receptors in miiuy croaker and evolution analysis of IL-1 family. Fish Shellfish Immunol. 63, 165–172. doi: 10.1016/j.fsi.2017.02.005
Yokoseki T., Iino T., Kutsukake K. (1996). Negative regulation by FliD, FliS, and FliT of the export of the flagellum-specific anti-sigma factor, FlgM,in Salmonella typhimurium. J. Bacteriol. 178 (3), 899–901. doi: 10.1128/jb.178.3.899-901.1996
Yokoseki T., Kutsukake K., Ohnishi K., Lino T.. (1995). Functional analysis of the flagellar genes in the fliD operon of Salmonella typhimurium. Microbiology 141, 1715–1722. doi: 10.1099/13500872-141-7-1715
Zhang M., Qin Y., Huang L., Yan Q., Mao L., Xu X., et al. (2019). The role of sodA and sodB in Aeromonas hydrophila resisting oxidative damage to survive in fish macrophages and escape for further infection. Fish Shellfish Immunol. 88, 489–495. doi: 10.1016/j.fsi.2019.03.021
Zhang L., Song M., Yang N., Zhang X., Abbas Raza S., Jia K., et al. (2020). Nucleoside diphosphate kinases (ndk) reveals a key role in adhesion and virulence of Aeromonas veronii. Microb. Pathog. 149, 104577. doi: 10.1016/j.micpath.2020.104577
Zhang L., Yao Z., Tang H., Song Q., Song H., Yao J., et al. (2022). The lysine acetylation modification in the porin Aha1 of Aeromonas hydrophila regulates the uptake of multi-drug antibiotics. Mol. Cell. Proteomics 21 (9), 100248. doi: 10.1016/j.mcpro.2022.100248
Zhao L., Zhang X., Qiu Z., Huang Y.. (2018). De novo assembly and characterization of the Xenocatantops brachycerus transcriptome. Int. J. Mol. Sci. 19 (2), 520. doi: 10.3390/ijms19020520
Keywords: Epinephelus coioides, Pseudomonas plecoglossicida, fliS, pathogenicity, transcriptome, metabolome
Citation: Wu W, Zhao L, Huang L, Qin Y, Zhang J, Zhang J and Yan Q (2022) Transcriptomic and metabolomic insights into the role of fliS in the pathogenicity of Pseudomonas plecoglossicida against Epinephelus coioides. Front. Mar. Sci. 9:987825. doi: 10.3389/fmars.2022.987825
Received: 06 July 2022; Accepted: 22 August 2022;
Published: 13 September 2022.
Edited by:
Yunyun Zhuang, Ocean University of China, ChinaReviewed by:
Zhigang Zhou, Feed Research Institute (CAAS), ChinaJie Li, Yellow Sea Fisheries Research Institute (CAFS), China
Copyright © 2022 Wu, Zhao, Huang, Qin, Zhang, Zhang and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingpi Yan, eWFucXBAam11LmVkdS5jbg==