 Jianhu Pang1Caiyuan Zhao2Hualong Su2Zhongcheng Liu1Qiang Lu1Xinyi He1Shaoping Weng1,3,4
Jianhu Pang1Caiyuan Zhao2Hualong Su2Zhongcheng Liu1Qiang Lu1Xinyi He1Shaoping Weng1,3,4 Jianguo He1,2,3,4*
Jianguo He1,2,3,4*- 1State Key Laboratory for Biocontrol, School of Life Sciences, Sun Yat-sen University, Guangzhou, China
- 2School of Marine Sciences, Sun Yat-sen University, Zhuhai, China
- 3Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
- 4Guangdong Province Key Laboratory of Aquatic Economic Animals, Sun Yat-sen University, Guangzhou, China
Enterocytozoon hepatopenaei (EHP) is a high-impact pathogen in shrimp farming, causing huge economic losses to the global shrimp farming industry every year. However, current EHP detection methods are primarily based on the development of polymerase chain reaction (PCR) techniques that rely on sophisticated and expensive instruments. Consequently, a rapid, practical, and sensitive protocol for the detection of EHP is necessary. Recombinase polymerase amplification combined with a lateral flow dipstick (LFD-RPA) assay was developed using a pair of primers and nfo-probe targeting the conserved region of the spore wall protein gene. Under optimized reaction conditions, the LFD-RPA assay can detect 10 copies/μL of standard plasmid within 20 min at 40°C. Furthermore, the specificity of the LFD-RPA was also verified with other common pathogens of shrimp. Thirty-nine samples of Litopenaeus vannamei were collected in shrimp farms and detected using LFD-RPA and nested PCR. Thirty-two positive samples were detected by LFD-RPA. Compared with those of nested PCR, the diagnostic sensitivity and specificity of LFD-RPA were 100% and 100%, respectively. These results indicated the great application potential of the newly developed LFD-RPA assay for point-of-care diagnosis, epidemic surveillance, and epidemiological investigation of EHP.
Introduction
Hepatopancreatic microsporidiosis, which displays a typical symptom of retarded growth in shrimp, has a great influence on the shrimp culture industry because of a new microsporidian species, Enterocytozoon hepatopenaei (EHP). EHP has a wide range of hosts, such as Litopenaeus vannamei, Penaeus monodon, and P. stylirostris (Tang et al., 2015; Chaijarasphong et al., 2021). The incidence of EHP has been reported to be high in major shrimp farming species such as L. vannamei and P. monodon in Asian countries, including China, Indonesia, Malaysia, Vietnam, Thailand, and India (Kim et al., 2022), causing huge economic losses to the shrimp farming industry every year (Rajendran et al., 2016; Shen et al., 2019; Aranguren Caro et al., 2020; Patil et al., 2021). Therefore, a variety of effective measures must be implemented to address the current critical situation.
Rapid and effective detection methods can be used to screen specific pathogen-free broodstocks in a timely and accurate manner during the selection of shrimp larvae. During the culture process, regular testing enables us to keep up with the infection of EHP, providing us with effective references for later decision and avoiding greater economic losses (Pang et al., 2022). Thus, a rapid, accurate, and sensitive detection technique is necessary to detect EHP in shrimp culture and control the spread of EHP.
However, current mainstream diagnosis methods of EHP are based on a molecular biological approach (such as PCR, qPCR, and nested PCR). Several polymerase chain reaction (PCR)-based methods have been developed using the EHP small subunit ribosomal RNA (SSU rRNA) gene sequence (Liu et al., 2018a; Mai et al., 2020; Tang et al., 2015). However, when PCR-based assays targeted SSU rRNA, false-positive results were reported (Jaroenlak et al., 2016a). The nested PCR detection method based on cell wall protein (SWP) gene of EHP established by Jaroenlak et al. is currently recommended by Office International Des Epizooties (OIE) for EHP detection, which is sensitive but laborious and time-consuming (Jaroenlak et al., 2016a). Isothermal nucleic acid amplification methods, such as loop-mediated isothermal amplification (LAMP) targeting conserved regions of the SSU rRNA gene, have also been developed for the specific detection of EHP. However, the design of primers (six primers) is complicated (Ma et al., 2019a).
Recombinase polymerase amplification (RPA) is a novel isothermal nucleic acid amplification method developed by TwistDx (Cambridge, UK), which has simple instrumentation (Piepenburg et al., 2006). RPA amplifies detectable amounts of DNA within 20 min. In RPA, a recombinase and polymerase are used to anneal oligonucleotide primers to the template DNA for extension and amplification at an isothermal temperature (35-42°C) (Figure 1) (Pang et al., 2019). To date, RPA methods have been developed for the rapid detection of several important pathogens, including viruses, bacteria, and parasite (Abd et al., 2013; Yang et al., 2015; Yehia et al., 2015; Pang et al., 2019; Ma et al., 2021a). Pang et al. also established detection method of EHP based RPA, but the device (such as fluorescence acquisition devices and electrophoresis facilities) dependence of these methods limits their application in remote areas (Li et al., 2021; Ma et al., 2021a; Pang et al., 2022; Zhou et al., 2020).
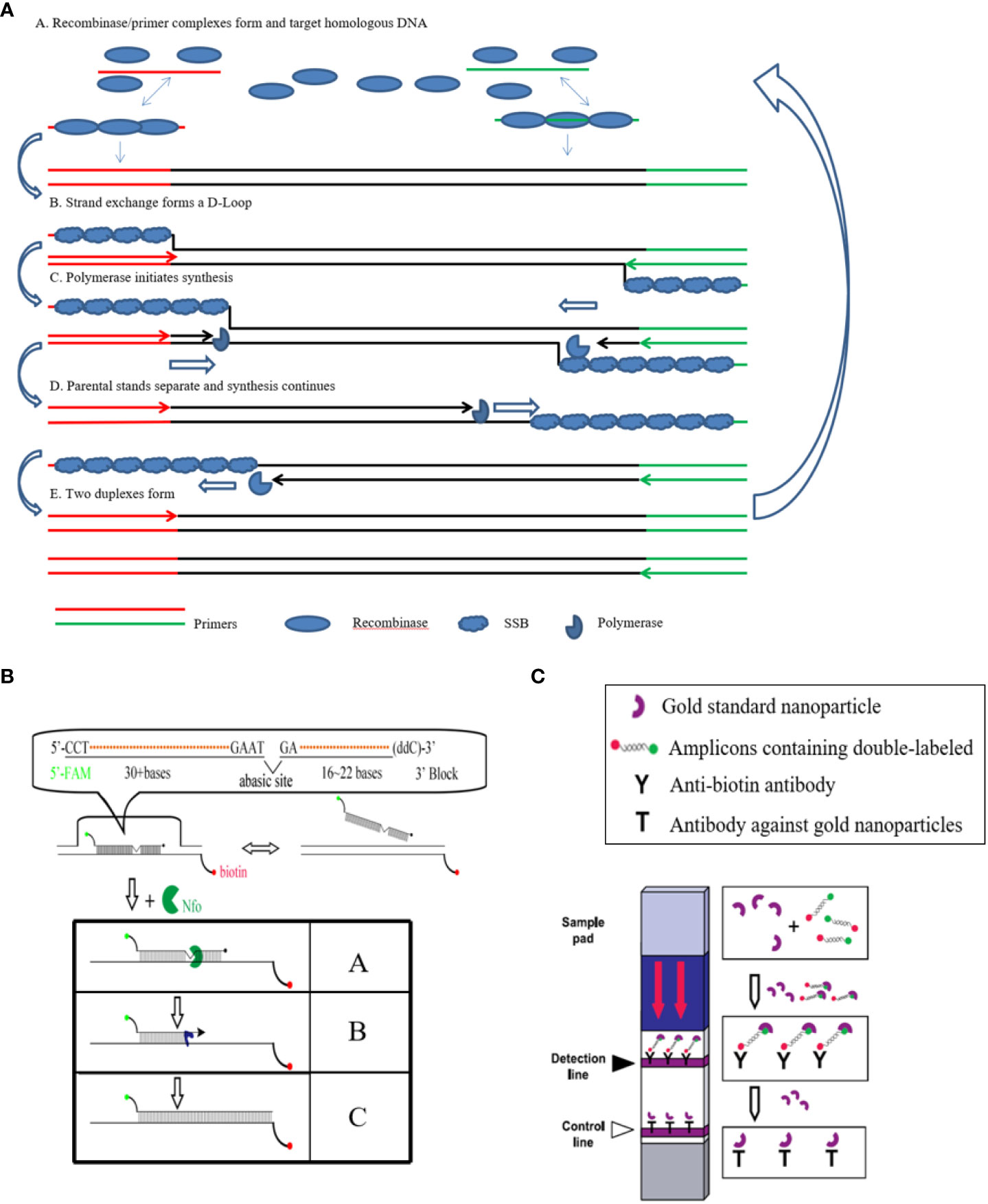
Figure 1 Principle of the LFD-RPA reaction. (A): A Recombinase/primer complexes form and target homologous DNA; B. strand exchange forms a D-Loop; C. polymerase initiates synthesis; D. parental stands separate and synthesis continues; E. two duplexes form; (B): A. Nfo enzyme recognizes the THF site in the amplified double-stranded sequence containing a 5’-FAM tag, an internal THF and a 3’ protective group, and shears it to form a polymerase Bsu-catalyzed extended 3’ hydroxyl substrate. B. The remaining part of the probe extends to fix its binding to the corresponding chain of the biotin-containing tag. C. Finally completes the double-labeled amplicon containing biotin and FAM; (C): In the sample loading area of the LFD test strip (Sample loading pad) containing gold standard nanoparticles with a specific antibody with FAM, can be combined with the product containing the corresponding antigen label, through the test strip on the two indicator lines of the antibody, one is the detection line, which is coated with the antibody of the product biotin, can intercept the double antigen label product that binds to the gold standard nanoparticle, thus showing red, indicating that the product has a double antigen label; the other is the control line, it is coated with an antibody with a FAT antibody on a gold standard nanoparticle, which can intercept the gold standard nanoparticle, thus showing a red color, indicating the effectiveness of the test strip.
In this study, we developed a rapid detection method for EHP based on RPA coupled with lateral flow dipstick (LFD) technology (LFD-RPA), which does not require complex and expensive equipment. In addition, we designed and optimized the LFD-RPA primers and probe and examined the analytical performance of this method. Finally, we tested the newly developed LFD-RPA assay on naturally infected samples to explore the application potential of this technology in point-of-care testing.
Materials and methods
Sample collection and DNA extraction
In November 2021, a sample of naturally infected EHP shrimp was collected from a shrimp pond in Guangzhou, Guangdong, China. The sample was confirmed to be EHP positive by sequencing. Fifty milligrams of hepatopancreatic tissue from each shrimp sample was collected to extract DNA using a tissue DNA kit (Omega, Norcross, GA, USA) following the manufacturer’s instructions. In brief, samples were homogenized and digested for 60 min at 55°C in 250 μL of lysis buffer containing 5 μg/mL of proteinase K. The mixture was centrifuged at 12000 x g (Eppendorf 5427 R, Germany) for 1 min at 4°C. After centrifugation, the supernatant was collected carefully. The genomic DNA of other common shrimp pathogens, such as white spot syndrome virus (WSSV), Decapod iridescent virus 1 (DIV1), Taura syndrome virus (TSV), Vibrio parahaemolyticus isolated from acute hepatopancreas necrosis, and infectious hypodermal and hematopoietic necrosis virus (IHHNV), were preserved by our laboratory. The quantity and quality of extracted DNA were determined by measuring the absorbance at 260 nm (A260) and the A260/A280 ratio, respectively, using a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA USA). The A260/A280 ratios for all genomic DNA samples were 1.8–2.0. All of the DNA samples were stored at −20°C.
Recombinant plasmid construction
A recombinant plasmid was used as positive controls to assess analytical sensitivity of the EHP assay. Standard plasmids were constructed using purified DNA and primers (swp-514-F and swp-514-R) in published literature (Table 1) (Jaroenlak et al., 2016a). A 514 bp amplicon was cloned into a pGEM1-T Easy Vector by using the DNA Ligation Kit Ver.2.1 (Takara, Japan) and transformed into Escherichia coli DH5α. The recombinant plasmids were confirmed by PCR and sequencing. The plasmid was extracted from Escherichia coli DH5a and used as a standard plasmid for further experiments. The concentration of recombinant plasmids was converted to copy number using the following formula: Number of copies = (M×6.02×1023×10−9)/(n×660), where M is the amount of DNA in nanogram, and n is the length of the plasmid. Furthermore, the average weight of one base pair was assumed to be 660 Da (Ma et al., 2019a).

Table 1 Primers and probes used in this study.
LFD-RPA primer and probe design
According to the appendix of the TwistAmp® reaction kit manual (www.twistdx.co.uk), we manually designed four primer pairs and a probe. The sequences were screened for homology using the blastn algorithm to ensure specificity (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Four pairs of primers (Table 1) were designed on the basis of the conserved domains of spore wall protein (SWP) gene (Accession: KX258197). All of the primer pairs produced amplicons, ranging in size less than 500 bases as recommended. Then, we selected the best primer combination by using the TwistAmp Basic kit (TwistDx, Cambridge, UK). The reaction was performed in a total volume of 50 μL. Master mixes were made, which contained 2.4 μL of each primer(480nM), 11.2 μL of ddH2O, 29.5 μL of Twist amplification rehydration buffer, 2 μL of template (108 copies/μL of standards recombinant plasmids), or 2 μL of nuclease-free water, as the no template control (NTC). To start the reaction, 2.5 μL of magnesium acetate was added to each tube (to bring the total Mg2+ concentration to 280 mM) by pipetting into the tube lids and centrifuging immediately before inserting the tubes into a dry heat bath (Thermomixer Comfort 5355, Eppendorf, Hamburg, Germany) and incubating at 38°C for 4 min. After 4 min, the samples were removed from the incubator, inverted vigorously 8–10 times to mix, spun down, and returned to the incubator block. Incubation was continued for a total incubation time of 20 min. Each basic RPA product (5 μL) was subjected to electrophoresis in a 2% (w/v) agarose gel.
After the primer screening assay, the preferred reverse primer was modified with a biotin label at its 5′ end. In addition, a probe (LFD-probe) was designed (Table 1), which was comprised of three groups. The upstream region is a dT-FAM (carboxyfluorescein) antigenic label. There is a tetrahydrofuran (THF) abasic-site mimic and biotin in each of the inner and downstream regions, which serve as substrate sites and 3’ blocking groups for nucleases in the reaction, respectively. The THF abasic-site mimic was designed following a previously published principle (Boyle et al., 2014). All of the primers and probes in the study were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
Optimization of the LFD-RPA assay conditions
LFD-RPA reactions were performed in 50 μL volumes using the TwistAmp nfo kit (TwistDx, Cambridge, UK). Master mixes containing 420 nM of each primer (the best primer combination selected by the basic RPA), 120 nM of nfo probe, 11.2 μL of ddH2O, and 29.5μL of Twist amplification rehydration buffer were prepared in reaction tubes supplied with dried enzyme pellets. The LFD-RPA reaction process is described in LFD-RPA Primer and Probe Design section.
Different concentrations of probe (60, 90, 120, and 150 nM), reaction temperature (10°C, 20°C, 30°C, 35°C, 40°C, 45°C, and 50°C), and time (5, 10, 15, 20, 25, 30, 35, and 40 min)—all of which are critical parameters—were explored to determine the optimal reaction condition for amplification in the LFD-RPA assay. A commercially available LFD (Milenia® GenLine HybriDetect Kit, Milenia Biotec GmbH, Giessen, Germany) was used for the detection of RPA amplicons. Two microliters of the amplification products was mixed with 98 μL of PBST supplied in the kit. Then, 10 μL of the diluted solution was pipetted onto the LFD. The dipsticks were immerged in PBST running buffer to make the solution flow, and after approximately 10 min, the results were visually analyzed.
Determination of the sensitivity and specificity of the LFD-RPA assay
The sensitivity of the LFD-RPA assay was determined by using the TwistAmp® nfo kit via testing 10-fold serial dilutions of standard plasmids. The reactions were prepared by adding different concentrations of standard plasmids (100 copies, 101 copies, 102 copies, 103 copies, 104 copies, 105 copies, 106 copies, 107 copies, and 108 copies) in 2 μL of each template or 2 μL of nuclease-free water as NTC. The amplification products were comparatively analyzed by the LFD. The operation procedures and reaction conditions were in accordance with the results of Optimization of the LFD-RPA Assay Conditions section.
As previously described, we used nested PCR as recommended by the OIE as a reference method (Jaroenlak et al., 2016a). The PCR reaction mixture for the two steps (25 μL) contained 0.2 μM of each primer and 0.5 unit of Taq DNA polymerase (Accurate Biology, Changsha, China). For the first-step PCR, the template was added with different concentrations of the abovementioned standard plasmids. The first PCR reaction used the primers swp-514-F and swp-514-R (Table 1), and amplification conditions were as follows: 95°C for 5 min initial denaturation, followed by 30 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 58°C and extension for 45 s at 68°C with a final 5 min extension step at 68°C. The expected PCR product was 514 bp. The PCR protocol for the second PCR reaction used the primers swp-148-F and swp-148-R (Table 1), and the expected second PCR product was 148 bp. The amplification program was as follows: 95°C for 5 min, followed by 20 cycles of 95°C for 30 s, 64°C for 30 s, 68°C for 30 s, and a final extension step of 68°C for 5 min. The amplicons were analyzed by 2% agarose gel electrophoresis with ethidium bromide staining using a 2000 bp DNA ladder marker (TaKaRa, Japan).
Other shrimp pathogens (WSSV, DIV1, TSV, V. parahaemolyticus, and IHHNV) were used to investigate LFD-RPA specificity. Ten nanograms of DNA isolated from the abovementioned pathogens were subjected to LFD-RPA amplification. The positive control used standard plasmids (108 copies/μL). Furthermore, the NTC employed nuclease-free water.
Validation of LFD-RPA with clinical samples
Clinically suspected samples were collected in Maoming, Guangdong, China, in November 2021. Samples were refrigerated and returned to the laboratory immediately. DNA was extracted from each shrimp sample using an E.Z.N.A. Animal Tissue DNA Kit (Omega, Norcross, GA, USA). A total of 39 clinical samples were detected by LFD-RPA and nested PCR assays (Jaroenlak et al., 2016a).
Results
Establishment and optimization of the LFD-RPA assay
The optimal primer group should result in high amplification efficacy. Four pairs of primer candidates (Table 1) were screened through a basic-RPA reaction on 2% agarose to design the optimal primer set. After amplification, the products were also analyzed by agarose gel electrophoresis, and the results were visually determined (Figure 2). As shown in Figure 2, each reaction generated an amplicon at the expected size. Nevertheless, the F1/R1 primer set showed the best amplification efficiency. Therefore, the primer group F1/R1 was utilized in the later experiment (Table 1). After modification, the F1 and R1 primers were renamed to LFD-F1 and LFD-R1.
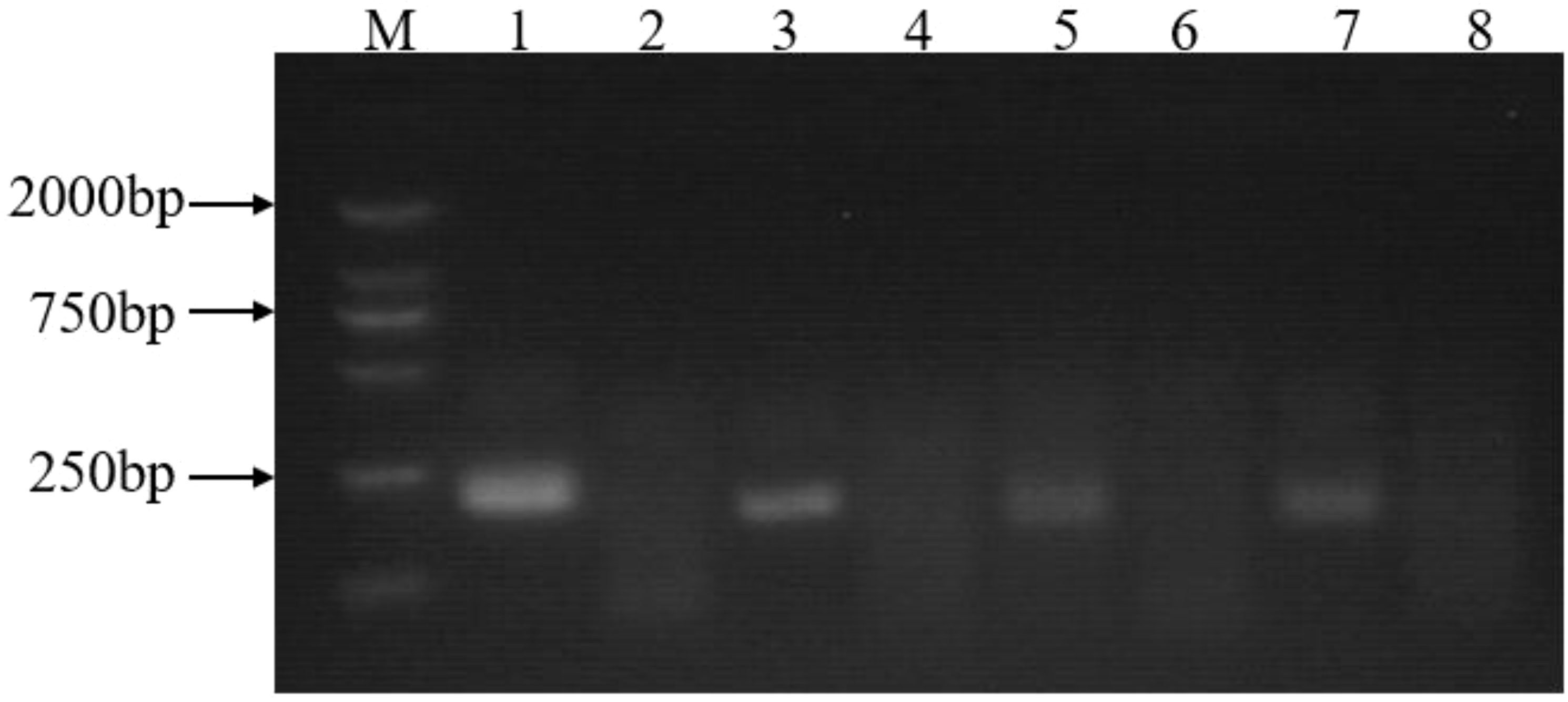
Figure 2 Basic-RPA primer screening. Lane1: F1/R1 primers with a 239-bp product; Lane2: no template control for F1/R1 primers; Lane3: F2/R2 primers with a 197-bp product; Lane4: no template control for F2/R2 primers; Lane5: F3/R3 primers, with a 184-bp product; Lane6: no template control for F3/R3 primers; Lane7: F4/R4 primers, with a 187-bp product; Lane8: no template control for F4/R4 primers; M: 2000 bp molecular marker.
Different concentrations of probe, incubation temperatures, and times were used to optimize the LFD-RPA assay. As shown in Figure 3A, test lines of the experimental strip developed a band with a deeper color when the concentration of probe was 120 nM, whereas test lines of the experimental strip in the control group developed a band with an indistinct color once the probe concentration reached 150 nM. The optimal concentration of the probe was 120 nM. In determining the optimal temperature for the LFD-RPA assay, seven different temperatures, including 10°C, 20°C, 30°C, 35°C, 40°C, 45°C, and 50°C, were used to perform the LFD-RPA reaction. The findings showed that the LFD-RPA assay presented a positive band over a temperature range of 10°C–50°C (Figure 3B). However, the test lines displayed the clearest band at 40°C. In addition, an amplification time of less than 20 min yielded an indistinct color on the strip, whereas 20 min yielded a band with the deepest color (Figure 3C). Considering high sensitivity, specificity, and swiftness, an amplification time of 20 min at 40°C with a probe concentration of 120 nM was selected for the optimized LFD-RPA detection method.
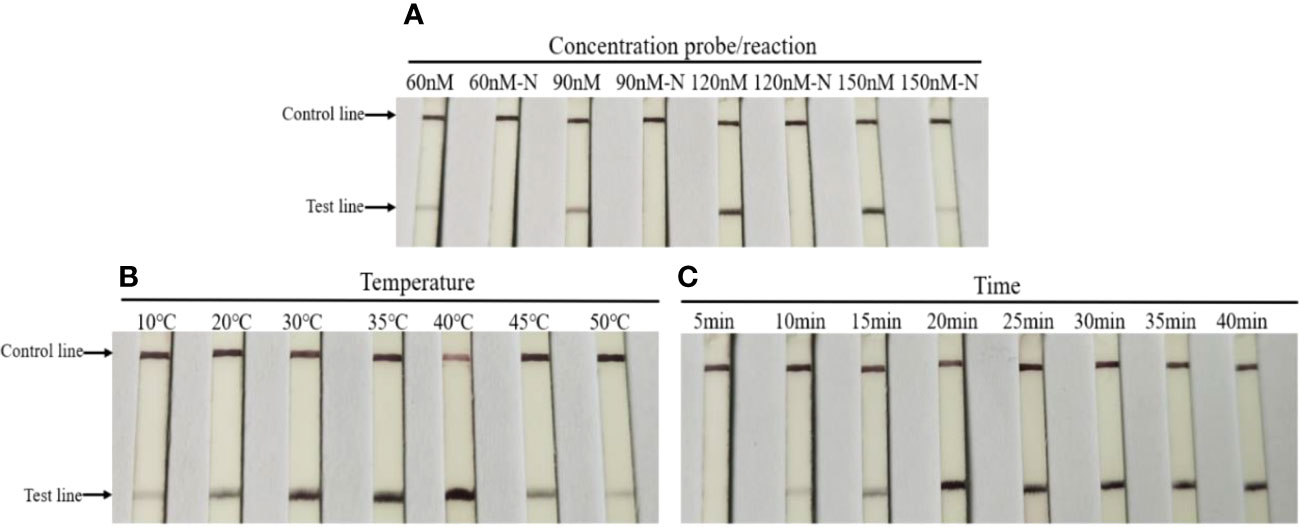
Figure 3 Optimization of the LFD-RPA detection assay. (A) Various concentrations of probe (60, 90, 120, and 150 nM) and no template control (60, 90, 120, and 150 nM-N) were evaluated using LFD-RPA. (B) Various reaction temperatures (10°C, 20°C, 30°C, 35°C, 40°C, 45°C, and 50°C) were used to select the optimal reaction temperature. (C) Various reaction times (10, 15, 20, 25, 30, 35, and 40 min) were used to select the optimal reaction time.
Sensitivity and specificity of the LFD-RPA assay
The sensitivity of the LFD-RPA detection method in detecting standard plasmids was evaluated. As shown in Figure 4A, 100 copies/μL to 108 copies/μL of standard plasmid DNA was used as the template in the assay. Nine dilutions were tested using the established LFD-RPA method and nested PCR. The LFD-RPA assay successfully detected standard plasmid DNA at a concentration of 10 copies of standard plasmid/50 μL (Figure 4A). By contrast, the nested PCR products were also detected by 2% agarose gel electrophoresis. The result indicated that the first step and the second step of nested PCR assay had a detection limit of 104 copies of standard plasmids (Figure 4B) and 10 copies (Figure 4C) of standard plasmids, respectively.
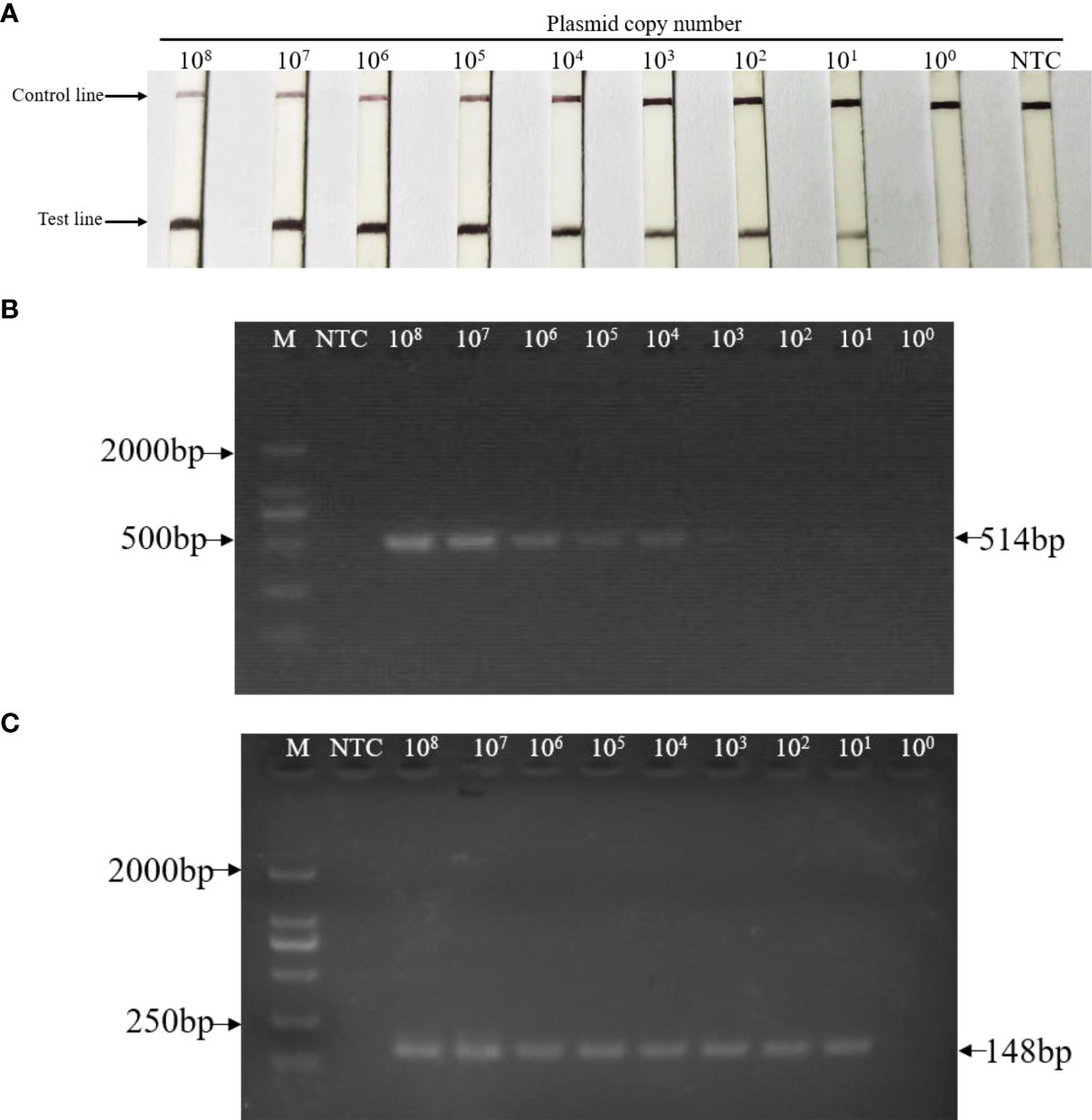
Figure 4 Sensitivity of LFD-RPA and nested PCR assay for the detection of EHP. Tenfold serial dilutions of standard plasmid DNA (108 copies to 100 copies) were tested by (A) LFD-RPA, the first-step PCR reactions (B), and the nested-step PCR (C). (A) The sensitivity of the LFD-RPA was ≥ 10 copies of template DNA per reaction. (B) Positive first-step PCR reaction products (514 bp) were detected on a stained agarose gel (2%) when the template DNA was ≥ 104 copies per reaction. (C) Positive nested-step PCR reaction products (148 bp) were detected on a stained agarose gel (2%) when the template DNA was ≥ 10 copies per reaction. NTC: no template controls contained water. M: DL2000TM DNA marker.
When analyzing the specificity of the LFD-RPA, genomic DNA from other shrimp pathogens and EHP were amplified in each reaction. The results showed that the reaction was only positive with the EHP, and none of the others were amplified (Figure 5). These data indicated that the LFD-RPA assay was a reliable method for the detection of EHP, and the assembly of the LFD-F1, LFD-R1, and LFD-probe was specific in detecting EHP (Table 1).

Figure 5 Specificity testing of the LFD-RPA assay. EHP and other shrimp pathogens were tested by LFD-RPA. Standard plasmid DNA (108 copies/μL) and no template controls containing water (NTC) were used as positive and negative control, respectively. Only the positive control group and EHP showed bands on the test line.
Assay performance of LFD-RPA and nested PCR with clinical samples
Thirty-nine clinically suspected samples were evaluated by LFD-RPA and nested PCR to evaluate the applicability and accuracy of our newly developed LFD-RPA assay. As shown in Table 2, among the 39 samples, 32 were positive and seven were negative by LFD-RPA and nested PCR, respectively. The sensitivity and specificity of LFD-RPA were consistent with the reference method nested PCR (Table S1).
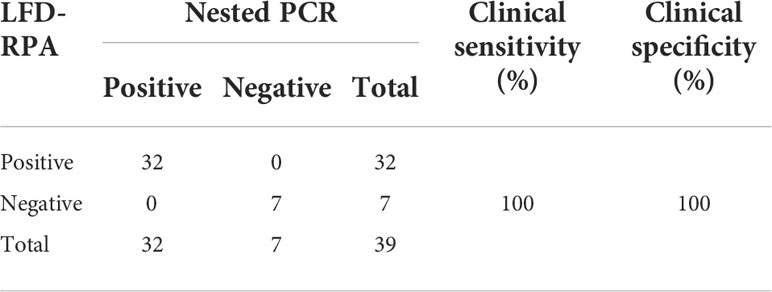
Table 2 The detection results of the clinical samples by the nested PCR and LFD-RPA (Jaroenram & Owens, 2014).
Discussion
EHP has caused several epidemics and infection in farmed shrimp. In this study, a specific method for the detection of EHP was developed by designing specific RPA primer and probe in combination with LFD.
At present, the commonly used EHP detection method is based on the PCR detection of the ribosomal 18S gene as the target gene, but these methods have certain non-specific amplification problems because of the high conservation of the 18S gene (Tang et al., 2015; Tang et al., 2016; Liu et al., 2018a). The SWP gene encodes a highly specific SWP, which is universally distributed in a variety of microsporidia. Spore walls of microsporidia participate in host–pathogen interaction through a species-specific SWP gene (Southern et al., 2007; Stentiford et al., 2013). Thus, the SWP gene has been regarded as an appropriate target gene for the identification of microsporidia, and OIE recommends it for the detection of EHP (Jaroenlak et al., 2016a).
Suitable primers and probes are key to the success of the RPA assay. A unique basic RPA assay was performed to screen the best primers from four primer sets in this study (Figure 2). Based on previous reports, the amplification efficiency of RPA changes with different conditions (Lillis et al., 2014; Crannell et al., 2015; Yang et al., 2017). Thus, in this study, incubation temperature, time, and probe concentration were optimized to explore the optimal conditions of LFD-RPA. Given its short amplicon and isothermal amplification, the incubation time for our LFD-RPA assay was shorter (20 min in LFD-RPA compared with 90 min in PCR). For most RPA assays, body temperature or a simple heating device can be used to achieve the detection temperature, which indicates its application potential for point-of-care diagnosis (Lillis et al., 2014).
Another molecular method for the detection of EHP was developed on the basis of PCR and LAMP techniques, which can obtain results within 90 and 60 min, respectively. The PCR developed by Hou et al. indicated that the assay had a detection limit of 20 copies of EHP plasmid DNA (Hou et al., 2021). TaqMan PCR was used for detecting EHP with sensitivity of 40 copies of purified EHP plasmid DNA (Liu et al., 2018a). One assay, particularly LAMP, was used to detect EHP. The LAMP system developed by Sathish et al. could detect EHP at a level of 10 copies within 45 min under isothermal conditions at 65°C (T et al., 2018). However, their selected target gene, namely, SSU rRNA gene, has been reported to have false positive, and their study did not use other related microsporidia to verify its specificity. Therefore, the specificity of their method must be further confirmed (Jaroenlak et al., 2016a).
The LFD-RPA assay developed in this study was sensitive and specific. It can be used to detect EHP plasmid DNA with an analytical sensitivity of 10 copies. The results from the PCR assay were more sensitive than the results obtained by the LAMP assay reported by Sathish et al. (T et al., 2018; Hou et al., 2021) and were consistent with the detection limits of our LFD-RPA assay targeting the SWP gene. However, the complex primer design (six primers) inhibits the widespread application of LAMP (Savan et al., 2004). By contrast, our RPA assay required only 20 min incubation time, utilized two primers, and implemented a probe system to increase the assay specificity. The results of the specificity testing were demonstrated by distinguishing EHP from other common aquaculture shrimp pathogens (Figure 5). Furthermore, the result of our LFD-RPA assay relies on color changes in the LFD, which can be recognized by the naked eye.
Conclusion
In this study, a rapid, sensitive, specific, and visual LFD-RPA method for the detection of EHP was developed. This method could provide a useful alternative tool for the detection of EHP in practical applications and may serve as an important tool in the future surveillance of EHP.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Laboratory Animal Welfare and Ethics Committee CALAS.
Author contributions
JP and JH conceived this study, and SW, CZ, HS, ZL, QL and XH participated in its design. JP and ZL carried out the experiments, and CZ, HS participated in data analysis. JP drafted the manuscript, SW and JH revised the manuscript. All authors read and approved the final manuscript before submission. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Key Research and Development Program of China (2018YFD0900501), the China Agriculture Research System (CARS-48), and the Marine Life Processes and Utilization of Biological Resources (42000-02920001).
Acknowledgments
We thank LetPub for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.985314/full#supplementary-material
References
Abd E. W. A., El-Deeb A., El-Tholoth M., Abd E. K. H., Ahmed A., Hassan S., et al. (2013). A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PloS One 8 (8), e71642. doi: 10.1371/journal.pone.0071642
Aranguren Caro L. F., Mai H. N., Pichardo O., Cruz-Flores R., Hanggono B., Dhar A. K. (2020). Evidences supporting enterocytozoon hepatopenaei association with white feces syndrome in farmed penaeus vannamei in Venezuela and Indonesia. Dis. Aquat. Organisms 141, 71–78. doi: 10.3354/dao03522
Boyle D. S., McNerney R., Teng L. H., Leader B. T., Perez-Osorio A. C., Meyer J. C., et al. (2014). Rapid detection of mycobacterium tuberculosis by recombinase polymerase amplification. PloS One 9 (8), e103091. doi: 10.1371/journal.pone.0103091
Chaijarasphong T., Munkongwongsiri N., Stentiford G. D., Aldama-Cano D. J., Thansa K., Flegel T. W., et al. (2021). The shrimp microsporidian enterocytozoon hepatopenaei (EHP): Biology, pathology, diagnostics and control. J. Invertebrate Pathol. 186, 107458. doi: 10.1016/j.jip.2020.107458
Crannell Z. A., Cabada M. M., Castellanos-Gonzalez A., Irani A., White A. C., Richards-Kortum R. (2015). Recombinase polymerase amplification-based assay to diagnose giardia in stool samples. Am. J. Trop. Med. Hygiene 92 (3), 583–587. doi: 10.4269/ajtmh.14-0593
Hou Z., Yu J., Wang J., Li T., Chang L., Fang Y., et al. (2021). Development of a PCR assay for the effective detection of enterocytozoon hepatopenaei (EHP) and investigation of EHP prevalence in Shandong province, China. J. Invertebrate Pathol. 184, 107653. doi: 10.1016/j.jip.2021.107653
Jaroenlak P., Sanguanrut P., Williams B. A. P., Stentiford G. D., Flegel T. W., Sritunyalucksana K., et al. (2016a). A nested PCR assay to avoid false positive detection of the microsporidian enterocytozoon hepatopenaei (EHP) in environmental samples in shrimp farms. PloS One 11 (11), e166320. doi: 10.1371/journal.pone.0166320
Jaroenram W., Owens L. (2014). Recombinase polymerase amplification combined with a lateral flow dipstick for discriminating between infectious penaeus stylirostris densovirus and virus-related sequences in shrimp genome. J. Virological Methods 208, 144–151. doi: 10.1016/j.jviromet.2014.08.006
Kim J. H., Lee C., Jeon H. J., Kim B. K., Lee N., Choi S., et al. (2022). First report on enterocytozoon hepatopenaei (EHP) infection in pacific white shrimp (Penaeus vannamei) cultured in Korea. Aquaculture 547, 737525. doi: 10.1016/j.aquaculture.2021.737525
Li G., Cong F., Cai W., Li J., He D. (2021). Development of a recombinase polymerase amplification (RPA) fluorescence assay for the detection of enterocytozoon hepatopenaei (EHP). Aquacult. Rep. 19, 100584. doi: 10.1016/j.aqrep.2020.100584
Lillis L., Lehman D., Singhal M. C., Cantera J., Singleton J., Labarre P., et al. (2014). Non-instrumented incubation of a recombinase polymerase amplification assay for the rapid and sensitive detection of proviral HIV-1 DNA. PloS One 9 (9), e108189. doi: 10.1371/journal.pone.0108189
Liu Y., Qiu L., Sheng A., Wan X., Cheng D., Huang J. (2018a). Quantitative detection method of enterocytozoon hepatopenaei using TaqMan probe real-time PCR. J. Invertebrate Pathol. 151, 191–196. doi: 10.1016/j.jip.2017.12.006
Ma C., Fan S., Wang Y., Yang H., Qiao Y., Jiang G., et al. (2021a). Rapid detection of enterocytozoon hepatopenaei infection in shrimp with a real-time isothermal recombinase polymerase amplification assay. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.631960
Mai H. N., Cruz-Flores R., Aranguren Caro L. F., White B. N., Dhar A. K. (2020). A comparative study of enterocytozoon hepatopenaei (EHP) challenge methods in penaeus vannamei. J. Invertebrate Pathol. 171, 107336. doi: 10.1016/j.jip.2020.107336
Ma B., Yu H., Fang J., Sun C., Zhang M. (2019a). Employing DNA binding dye to improve detection of enterocytozoon hepatopenaei in real-time LAMP. Sci. Rep. 9 (1):15860. doi: 10.1038/s41598-019-52459-0
Pang J., He X., Wu Y., Weng S., He J. (2022). Rapid and sensitive detection of enterocytozoon hepatopenaei in real-time recombinase polymerase amplification assay. Aquaculture 554, 738103. doi: 10.1016/j.aquaculture.2022.738103
Pang J., Wang Q., Fei Y., Zhu P., Qiao L., Huang H., et al. (2019). A real-time recombinase polymerase amplification assay for the rapid detection of vibrio harveyi. Mol. Cell Probes 44, 8–13. doi: 10.1016/j.mcp.2019.01.001
Patil P. K., Geetha R., Ravisankar T., Avunje S., Solanki H. G., Abraham T. J., et al. (2021). Economic loss due to diseases in Indian shrimp farming with special reference to enterocytozoon hepatopenaei (EHP) and white spot syndrome virus (WSSV). Aquaculture 533, 736231. doi: 10.1016/j.aquaculture.2020.736231
Piepenburg O., Williams C. H., Stemple D. L., Armes N. A. (2006). DNA Detection using recombination proteins. PloS Biol. 4 (7), 1115–1121. doi: 10.1371/journal.pbio.0040204
Rajendran K. V., Shivam S., Ezhil Praveena P., Joseph Sahaya Rajan J., Sathish Kumar T., Avunje S., et al. (2016). Emergence of enterocytozoon hepatopenaei (EHP) in farmed penaeus (Litopenaeus) vannamei in India. Aquaculture 454, 272–280. doi: 10.1016/j.aquaculture.2015.12.034
Savan R., Igarashi A., Matsuoka S., Sakai M. (2004). Sensitive and rapid detection of edwardsiellosis in fish by a loop-mediated isothermal amplification method. Appl. Environ. Microbiol. 70 (1), 621–624. doi: 10.1128/AEM.70.1.621-624.2004
Shen H., Qiao Y., Wan X., Jiang G., Fan X., Li H., et al. (2019). Prevalence of shrimp microsporidian parasite enterocytozoon hepatopenaei in jiangsu province, China. Aquacult. Int. 27 (3), 675–683. doi: 10.1007/s10499-019-00358-6
Southern T. R., Jolly C. E., Lester M. E., Hayman J. R. (2007). EnP1, a microsporidian spore wall protein that enables spores to adhere to and infect host cells In vitro. Eukaryot. Cell 6 (8), 1354–1362. doi: 10.1128/EC.00113-07
Stentiford G. D., Feist S. W., Stone D. M., Bateman K. S., Dunn A. M. (2013). Microsporidia: diverse, dynamic, and emergent pathogens in aquatic systems. Trends Parasitol. 29 (11), 567–578. doi: 10.1016/j.pt.2013.08.005
T S. K., A N. K., J J. S. R., M M., K. P. ,. J., S. V. ,. A., et al. (2018). Visual loop-mediated isothermal amplification (LAMP) for the rapid diagnosis of enterocytozoon hepatopenaei (EHP) infection. Parasitol. Res. 117 (5), 1485–1493. doi: 10.1007/s00436-018-5828-4
Tang K. F. J., Han J. E., Aranguren L. F., White-Noble B., Schmidt M. M., Piamsomboon P., et al. (2016). Dense populations of the microsporidian enterocytozoon hepatopenaei (EHP) in feces of penaeus vannamei exhibiting white feces syndrome and pathways of their transmission to healthy shrimp. J. Invertebrate Pathol. 140, 1–7. doi: 10.1016/j.jip.2016.08.004
Tang K. F. J., Pantoja C. R., Redman R. M., Han J. E., Tran L. H., Lightner D. V. (2015). Development of in situ hybridization and PCR assays for the detection of enterocytozoon hepatopenaei (EHP), a microsporidian parasite infecting penaeid shrimp. J. Invertebrate Pathol. 130, 37–41. doi: 10.1016/j.jip.2015.06.009
Yang Y., Qin X., Wang G., Zhang Y., Shang Y., Zhang Z. (2015). Development of a fluorescent probe-based recombinase polymerase amplification assay for rapid detection of orf virus. Virol. J. 12, 206. doi: 10.1186/s12985-015-0440-z
Yang Y., Qin X., Zhang W., Li Z., Zhang S., Li Y., et al. (2017). Development of an isothermal recombinase polymerase amplification assay for rapid detection of pseudorabies virus. Mol. Cell Probes 33, 32–35. doi: 10.1016/j.mcp.2017.03.005
Yehia N., Arafa A. S., Abd E. W. A., El-Sanousi A. A., Weidmann M., Shalaby M. A. (2015). Development of reverse transcription recombinase polymerase amplification assay for avian influenza H5N1 HA gene detection. J. Virological Methods 223, 45–49. doi: 10.1016/j.jviromet.2015.07.011
Keywords: detection, EHP, RPA, LFD, shrimp
Citation: Pang J, Zhao C, Su H, Liu Z, Lu Q, He X, Weng S and He J (2022) Rapid visual detection of Enterocytozoon hepatopenaei by recombinase polymerase amplification combined with lateral flow dipstick. Front. Mar. Sci. 9:985314. doi: 10.3389/fmars.2022.985314
Received: 03 July 2022; Accepted: 29 August 2022;
Published: 16 September 2022.
Edited by:
Ji Hyung Kim, Gachon University, South KoreaReviewed by:
Farman Ali, Fujian Agriculture and Forestry University, ChinaLunbiao Cui, Jiangsu Provincial Center for Disease Control and Prevention, China
Copyright © 2022 Pang, Zhao, Su, Liu, Lu, He, Weng and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianguo He, bHNzaGpnQG1haWwuc3lzdS5lZHUuY24=