 Jiao Cheng
Jiao Cheng Liwen Zhang
Liwen Zhang Min Hui1,2,3
Min Hui1,2,3 Yuan Li
Yuan Li Zhongli Sha
Zhongli Sha- 1Department of Marine Organism Taxonomy and Phylogeny, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3Shandong Province Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 4College of Marine Science, University of Chinese Academy of Sciences, Qingdao, China
- 5Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
The heterogeneous seascapes in the northwestern Pacific (NWP) can be important selective forces driving adaptive divergence of marine coastal species distributed along the gradients. Here, we tested this hypothesis in Japanese mantis shrimp (Oratosquilla oratoria) with a wide distribution in the NWP and a significant north-south population structure. To this end, the full-length (FL) transcriptomes of northern and southern O. oratoria were firstly sequenced using PacBio single molecule real-time sequencing technology. Based on the FL transcriptome data, we captured large-scale FL transcripts of O. oratoria and predicted the FL transcriptome structure, including coding region, transcription factor and long noncoding RNA. To reveal the divergence between northern and southern O. oratoria, we identified 2,182 pairs of orthologous genes and inferred their sequence divergences. The average differences in coding, 5’ untranslated and 3’ untranslated region were 1.44%, 2.79% and 1.46%, respectively, providing additional support to previous proposition that northern and southern O. oratoria are two species. We provided further evolutionary context to our analysis by identifying positive selected genes (PSGs) between northern and southern O. oratoria. In total, 98 orthologs were found evolving under positive selection and involved several environmentally responsive genes associated with stress response, immunity and cytoskeletal organization, etc. Furthermore, we found PSGs also diverged in gene expression response of northern and southern O. oratoria to heat stress. These findings not only highlight the importance of genetic variation in these genes in adapting to environmental changes in O. oratoria, but also suggest that natural selection may act on the plasticity of gene expression to facilitate O. oratoria adaptation to environmental gradients. Overall, our work contributes to understanding how marine coastal species has evolved to adapt to heterogeneous seascapes in the NWP.
1 Introduction
In marine ecosystems, oceanographic heterogeneity (e.g., current interfaces, habitat transition zones, ecological gradients) can impose spatially varying selective pressures on populations or species distributed along the gradients. Such heterogeneous seascapes in coastal waters have been demonstrated to strongly influence adaptive divergence, particularly for species with large population sizes where selection is expected to be highly efficient (Sandoval-Castillo et al., 2018; Coscia et al., 2020; Pratt et al., 2022). The Japanese mantis shrimp Oratosquilla oratoria (De Haan, 1844) is benthic, neritic and burrowing shrimp with a wide distribution in the Northwestern Pacific (NWP) (Manning, 1971). Due to its high productivity as well as excellent meat quality, O. oratoria is commercially exploited in coastal waters throughout Japan, Korea and China (Kodama et al., 2006; Ahyong, 2012). This species occurs within a highly heterogeneous seascape that spans coastal bioregions with strong gradients in sea surface temperature (SST) and salinity (Figure 1). More specifically, SST is strongly influenced by the complex current systems with different temperature characteristics, including the cold Oyashio Current (OC), China Costal Current (CCC), a low-temperature Yellow Sea cold water mass and the warm Kuroshio Current as well as its branches (Figure 1A, Liu, 2013). A biogeographic boundary lies in line with the Yangtze River Estuary where the annual SST is above 20°C on the southern side of the estuary, and rapidly decreases to the value less than 15°C on the northern side (Figure 1B, Johnson and Boyer, 2015). Taxa with narrow thermal tolerance ranges are restricted to one side of the boundary as a result of this steep temperature gradient across the Yangtze River Estuary (Liu, 2013). In addition, a strong salinity gradient is generated by the collision of the Changjiang diluted water (CDW) with several coastal currents (e.g., CCC and Subei Coastal Current) (Figure 1C, Su and Yuan, 2005). Therefore, the wide distribution range across heterogeneous seascapes makes O. oratoria an ideal system to study how ecologically based natural selection has driven adaptive divergence of marine coastal species.
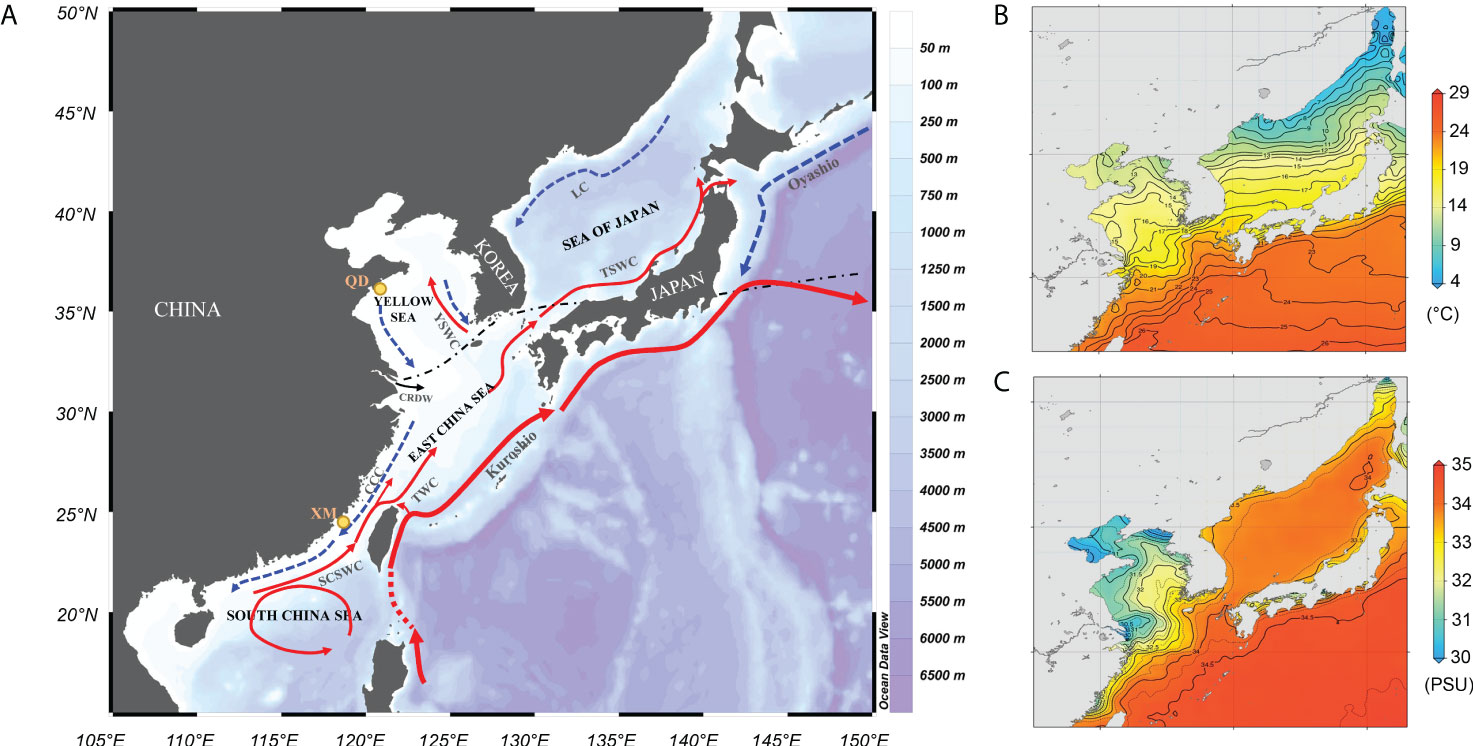
Figure 1 Map of northwestern Pacific showing the major surface currents in the winter (A) and spatial heterogeneity in sea surface temperature (B) and salinity (C). SCSWC, South China Sea Warm Current; TWC, Taiwan Warm Current; CCC, China Coastal Current; CRDW, Changjiang River Diluted Water; YSWC, Yellow Sea Warm Current; TSWC, Tsushima Warm Current; LC, Liman Current. Annual temperature (°C) and annual salinity (PSU) at the surface (ten-degree grid) were based on Johnson and Boyer, 2015. The dash/dotted line separates the distribution range of northern and southern O. oratoria. Two sampling locations in this study are marked in orange dots.
Several recent researches have revealed a remarkable pattern of genetic differentiation in Japanese mantis shrimp despite high dispersal potential during its planktonic larval stage (4–6 weeks, Hamano and Matsuura, 1987). Our previous analyses have revealed two highly divergent lineages in O. oratoria with clear allopatric distribution by integrating mitochondrial and nuclear evidence (Cheng and Sha, 2017; Cheng et al., 2020). The distribution range of the two O. oratoria lineages exhibits a remarkable latitudinal cline: the northern lineage is restricted to cold waters with temperate affinities, while the southern lineage inhabits the subtropical and tropical regions influenced by the Kuroshio Current and its branches. The deep genetic divergence, which is so far unlinked to any apparent morphological variation, substantiates the existence of cryptic speciation in Japanese mantis shrimp (Cheng and Sha, 2017). In the coast of China, the sharp genetic break of O. oratoria is in line with the Yangtze River Estuary (Du et al., 2016; Cheng and Sha, 2017; Cheng et al., 2020). Nevertheless, these studies are constrained by small numbers of markers that precludes an accurate description of genome-wide DNA sequence divergence. Meanwhile, we know little about how seascape heterogeneity in the NWP influences adaptative evolution of O. oratoria as well as the roles of genetic variation and plasticity in local adaptation to environmental gradients. The polygenic basis of traits governing physiological tolerance puts forward challenges for linking genetic variation to ecological differences among populations adapting to different environments (Storz and Wheat, 2010; Rockman, 2012). Thus, the genetic underpinnings of diversification and adaptation in Japanese mantis shrimp remain unclear due to the lack of genomic information.
Transcriptome represents a sample of the spatiotemporally expressed genome that can be generated in a short time at a reduced cost compared to whole genome sequencing, and can be used as an alternative to genomic approaches for non-model organisms (Morozova and Marra, 2008). Beyond uncovering the molecular bases of physiological responses to a particular environmental challenge (Gracey and Cossins, 2003), transcriptomics has also been used in a comparative framework to reveal numerous aspects of ecological speciation and adaptation (e.g., Zhang et al., 2015a; Cheng et al., 2019). As a low-cost next-generation sequencing technology, RNA sequencing (RNA-seq) has become a versatile tool for studying transcriptomics. However, RNA-seq is incapable of providing full-length (FL) transcripts due to its inherent length limitations, which presents major challenges for further structural and functional genomic studies (Steijger et al., 2013; Tilgner et al., 2013). Another limitation is that the presence of different isoforms and differences in transcript abundance has greatly hampered transcriptome assembly from short reads. The third-generation sequencing technology seems to offer an opportunity to overcome the shortcomings of short-read sequences by enabling the generation of kilobase-sized sequencing reads without assembly (Sharon et al., 2013). To date, an increasing number of FL transcriptome analyses have been conducted in a variety of marine organisms (e.g., Yi et al., 2018; Pootakham et al., 2020; Wang et al., 2022), which greatly facilitate the transcriptome research in the ocean life.
In the present study, we deep-sequenced the FL transcriptomes of northern and southern O. oratoria by using PacBio single molecule real-time (SMRT) sequencing technology. Based on the obtained FL transcriptomes, we first performed transcription factor prediction, long noncoding RNA prediction and transcript functional annotation. A comparative transcriptomic analysis was then carried out to identify orthologous genes and to evaluate the transcriptome-wide genetic divergence between northern and southern O. oratoria. With these datasets, we aimed to trace signatures of positive selection in O. oratoria during adaptation to heterogeneous environments and identify environmentally responsive genes that ecologically based natural selection may have acted on. Finally, we incorporated expression profiles of O. oratoria under heat stress to investigate the role of evolving plasticity in local adaptation to environmental gradients. As such, this study provides abundant transcriptome resources and new insights into the divergence and adaptation of Japanese mantis shrimp, and also provides ample evidence for adaptive evolution of marine coastal species in response to heterogeneous seascapes.
2 Materials and methods
2.1 Sample collection and RNA preparation
The adult O. oratoria individuals were collected from the coastal waters of Qingdao (QD) and Xiamen (XM), China, which correspond to northern and southern O. oratoria, respectively (Figure 1A). All individuals were acclimated over 24 h at 20°C. One individual at each sampling site was randomly selected and the gill, hepatopancreas, digestive tract, nervous chain, and abdominal muscle were immediately dissected out, frozen and stored in liquid nitrogen. Subsequently, all ten samples were subjected to RNA extraction using the TRIzol kit (Invitrogen, USA) according to the manufacturer’s instructions, and RNA degradation and contamination were detected with 1% agarose gels. The integrity and purity of RNA were assessed using NanoDrop 2000 (Thermo Scientific, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Only qualified RNA samples were used for cDNA library constructions.
2.2 Library construction and sequencing
RNA samples of five different tissues from each individual were mixed in equal amount to construct the PacBio sequencing library. Specifically, mRNA was purified from mixed total RNA using poly (T) oligo-attached magnetic beads and reverse-transcribed into FL cDNA using the SMARTer PCR cDNA Synthesis Kit (Clontech, USA). After PCR amplification, quality control and purification, the BluePippin Size Selection system (Sage Science, USA) was used for size selection of the FL cDNA and for producing libraries of differently sized cDNA. The screened cDNA was re-amplified by PCR, end repaired, connected to the SMRT dumbbell-type connector, and exonuclease digested. The cDNA products were then subjected to construction of SMRTbell Template libraries using SMRTBell Template Prep Kit. The concentration and quality of the libraries was assessed using Agilent 2100 Bioanalyzer and Qubit 2.0 (Life Technologies, USA). Finally, qualified libraries were sequenced on PacBio Sequel platform (Pacific Biosciences, USA).
The remaining RNA of each tissue was used for Illumina sequencing library construction. Five separate Illumina libraries were constructed for northern and southern O. oratoria using the protocol of NEBNext UltraTM RNA Library Prep Kit (NEB, USA), respectively. Briefly, poly (A) mRNA was purified from total RNA using Oligo (dT) magnetic beads and then broken into short fragments to synthesize first strand cDNA using random hexamer primer. Second-strand cDNA was then synthesized using DNA polymerase I and RNaseH. The cDNA was subjected to end-repair, phosphorylation, 3’ adenylation, and ligation to sequencing adaptors. Afterwards, cDNA libraries were generated by PCR amplification, and the library preparations were paired-end sequenced at 150 bp on an Illumina HiSeq X Ten platform. Clean Illumina reads were produced after removing adaptor sequences, reads containing ploy-N (with the ratio of ‘N’ to be more than 10%) and low quality reads (with quality score less than 5), and then all clean reads from the same site were merged together for de novo assembled by using Trinity v2.5.1 (Grabherr et al., 2011) with default parameters.
2.3 Pacbio read error correction
The SMRT Link 5.1 pipeline was used for PacBio data processing according to the official protocol. Circular consensus sequences (CCSs) were extracted from raw reads with the following parameters: max_drop_fraction, 0.8; max_length, 18000; min_length, 200; min_predicted_accuracy, 0.8; min_passes, 1; min_zscore, -999. After discarding CCS reads with length shorter than 50 bp, the remaining CCS reads were classified into full length non-chimeric (FLNC) or non-full-length (NFL) transcripts according to whether 5’/3’ cDNA primers and poly(A) tail were simultaneously observed. The iterative clustering for error correction (ICE) algorithm was used to obtain consensus isoforms by clustering FLNC sequences, and Arrow software (https://github.com/PacificBiosciences/GenomicConsensus) was used to refine consensus isoforms using the NFL reads to produce polished consensus sequences. All polished consensus sequences were corrected in aid of Illumina RNA-seq data using LoRDEC (Salmela and Eric, 2014). Furthermore, redundant sequences were removed from the transcriptome isoform sequences to obtain unigenes by using CD-HIT (Fu et al., 2012) with an amino-acid sequence identity threshold of 95%. Finally, the completeness of two FL transcriptomes was assessed by examining the coverage of the 1066 conserved core genes of Arthropoda (https://busco.ezlab.org/) using BUSCO 3 (Simão et al., 2015).
2.4 Coding sequences, transcription factor and long noncoding RNA prediction
All the isoforms were used to predict the coding sequences (CDS) and protein sequences using ANGEL software (Shimizu et al., 2006). We used the confidence protein sequences of O. oratoria or closely related species for ANGLE training, and then run the ANGLE predictions for given sequences. The 5’UTR and 3’UTR (untranslated regions) sequences were predicted based on the CDS, start and stop codons. Transcripts containing the 5’ and 3’UTRs and complete CDSs were defined as FL transcripts. Transcription factors (TFs) were predicted using AnimalTFDB v2.0 (Animal Transcription Factor Database) (Zhang et al., 2015b). Because O. oratoria was not included in the database, hmmsearch was used to identify TFs based on the Pfam search results of the TF family.
Long noncoding RNAs (lncRNAs) are defined as RNAs which are at least 200 nucleotides in length and lack protein-coding capacity (Rinn and Chang, 2012). On the basis of this, lncRNA of O. oratoria transcriptome was first predicted by screening the coding potential of transcripts using PLEK (Li et al., 2014), Coding-Non-Coding-Index (CNCI) (Sun et al., 2013) and Coding Potential Calculator (CPC) (Kong et al., 2007). The transcript sequences predicted using PLEK, CNCI and CPC tools were further used to search against the Pfam-A and Pfam-B databases using hmmscan. Transcripts with predicted coding potential according to the results of all four methods were filtered out, and those without coding potential constituted the candidate set of lncRNAs.
2.5 Gene function annotation
The non-redundant transcript sequences were mapped to public databases to obtain the annotation information. Transcripts were compared against NCBI NT (non-redundant nucleotide sequences) using BLAST v2.2.31 (Altschul et al., 1997) with cut-off E-value < 1E-5, NCBI NR (non-redundant protein sequences), Swiss-Prot (http://www.ebi.ac.uk/uniprot/), KOG (euKaryotic Ortholog Group), KEGG (Kyoto Encyclopedia of Genes and Genomes) using diamond v0.8.36 (Li et al., 2002) with cut-off E-value < 1E-5. According to the annotation results of the NR database, the Blast2GO v2.5 software (Conesa et al., 2005) was used to perform Gene Ontology (GO) annotation and classification. KEGG classification was performed using KASS (vr140224) (Moriya et al., 2007) and the KEGG Automatic Annotation Server. HMMER v3.1 (Nastou et al., 2016) was used to compare amino acid sequences of transcripts against the Protein family (Pfam) database for Pfam annotation.
2.6 Putative orthologs identification and sequence divergence analyses
Orthologous groups were constructed from the BLASTP results using OrthoMCL (Li et al., 2003) based on the Markov Cluster algorithm (mcl) with default settings. The 5’UTR, coding and 3’UTR regions were separately extracted from each pair of orthologs. The CDS and UTR regions were aligned separately to each other using MUSCLE (Edgar, 2004) with default settings. For the CDS region, pair-wise alignments were performed for the putative orthologous pairs based on protein sequences, and back-translated to DNA sequences for subsequent analysis. The phylogenetic tree was conducted on the basis of amino acid sequence alignment using the maximum likelihood method as implemented in PhyML v3.0 (Guindon et al., 2010). The node reliability was calculated from 1000 bootstrap replications. According to the method of Ye et al. (2014), sequence divergences between the homologous regions of each gene pair were calculated by dividing the number of substitutions by the number of base pairs compared.
2.7 Test for positive selection
The ratio of non-synonymous to synonymous nucleotide substitutions provides information about the evolutionary forces operating on a gene (Biswas and Akey, 2006). We estimated non-synonymous substitutions per nonsynonymous site (Ka) and synonymous substitutions per synonymous site (Ks) among putatively orthologous coding regions using a maximum-likelihood method implemented in the codeml program in the PAML package (Yang, 2007) with one-ratio model (model = 0). In this study, Ka/Ks > 1 was considered to indicate that genes were under positive selection and Ka/Ks < 0.1 was regarded as a signature of purifying selections. Positively selected genes (PSGs) with Ka/Ka > 1 were characterized with GO enrichment analysis implemented in the GOseq R packages (Young et al., 2010) based on the Wallenius noncentral hypergeometric distribution. Go terms with a P value < 0.05 were considered significantly enriched. The R bioconductor package topGO (Alexa and Rahnenführer, 2010) was used to present the directed acycline graph of the enriched GO category.
2.8 Expression of PSGs response to thermal stress
To infer the potential role of positive selection in O. oratoria adaptation, we explored the expression level of PSGs in two representative geographic populations (Qingdao in the Yellow Sea and Zhoushan in the East China Sea) in response to the same heat stress (20°C–28°C, Lou et al., 2019a). Taking advantage of range-wide sampling, our previous analyses of mitochondrial and nuclear sequences indicated genetic homogeneity among O. oratoria populations from the East and South China Seas (Cheng and Sha, 2017; Cheng et al., 2020). These two heat-stressed populations reported in Lou et al. (2019a) correspond to northern and southern O. oratoria in this study, respectively. The heating schemes of the two populations were similar, with detailed description in Lou et al. (2019a). Briefly, the control condition was kept at a water temperature of 20°C after acclimation, whereas water temperature for the heat stress condition was raised at 1°C per day to 28°C and then held constant for 24h. All raw reads in the FASTQ format (BioProject accession: PRJNA475657) were filtered by removing the reads with sequencing adaptors, unknown nucleotides (N ratio > 10%) and low quality (quality scores ≤ 5). Clean reads of each sample were then mapped to the orthologous assembly of each respective group by using RSEM (Li and Dewey, 2011). The expression level was determined by averaging the expression values of three biological replications for each experimental condition. The differentially expressed orthologous genes were identified using the DESeq R package v1.18.0 (Anders and Huber, 2010) with the filtering thresholds of |log2FC| ≥ 1 and P ≤ 0.05. Hierarchical clustering was performed using the heatmap.2 function in the gplots R package (R Development Core Team, 2022).
3 Results
3.1 Full-length transcriptome sequencing
Bases on the PacBio SMRT sequencing technology, a total of 116.92 Gb and 110.32 Gb subreads were obtained, with N50 of 3,465 bp and 2,350 bp for the northern and southern O. oratoria, respectively. As shown in Table 1 and Figure S1, these subreads yielded 1,192,616 CCSs with a mean length of 3,714 bp for the northern O. oratoria and 1,312,861 CCSs with a mean length of 2,390 bp for the southern O. oratoria. According to the presence or absence of 5’/3’ cDNA primers and poly(A) tail, 1,022,216 (85.71%) FLNC reads and 162,789 NFL reads were further identified from the CCS reads of the northern O. oratoria. For the southern O. oratoria, 988,172 (75.27%) FLNC reads and 311,339 NFL reads were generated from the CCS reads. After isoform-level clustering based on the ICE algorithm and polishing based on the Arrow algorithm, a total of 46,871 and 55,408 polished FL consensus isoforms with an average length of 3,439 bp and 2,275 bp were generated from the FLNC reads of northern and southern O. oratoria, respectively. After removing redundancy, the consensus isoforms were finally clustered into a total of 18,630 unigenes with N50 value of 3,852 bp for the northern O. oratoria, and 21,843 unigenes with N50 value of 3,061 bp for the southern O. oratoria (Table 1). Of the 1066 conserved core genes of Arthropoda, 682 genes (64.0%) and 840 genes (78.8%) were mapped to the FL transcriptome of northern and southern O. oratoria, respectively (Table S1), indicating the sequencing data in this study were relatively complete and suitable for downstream analyses.
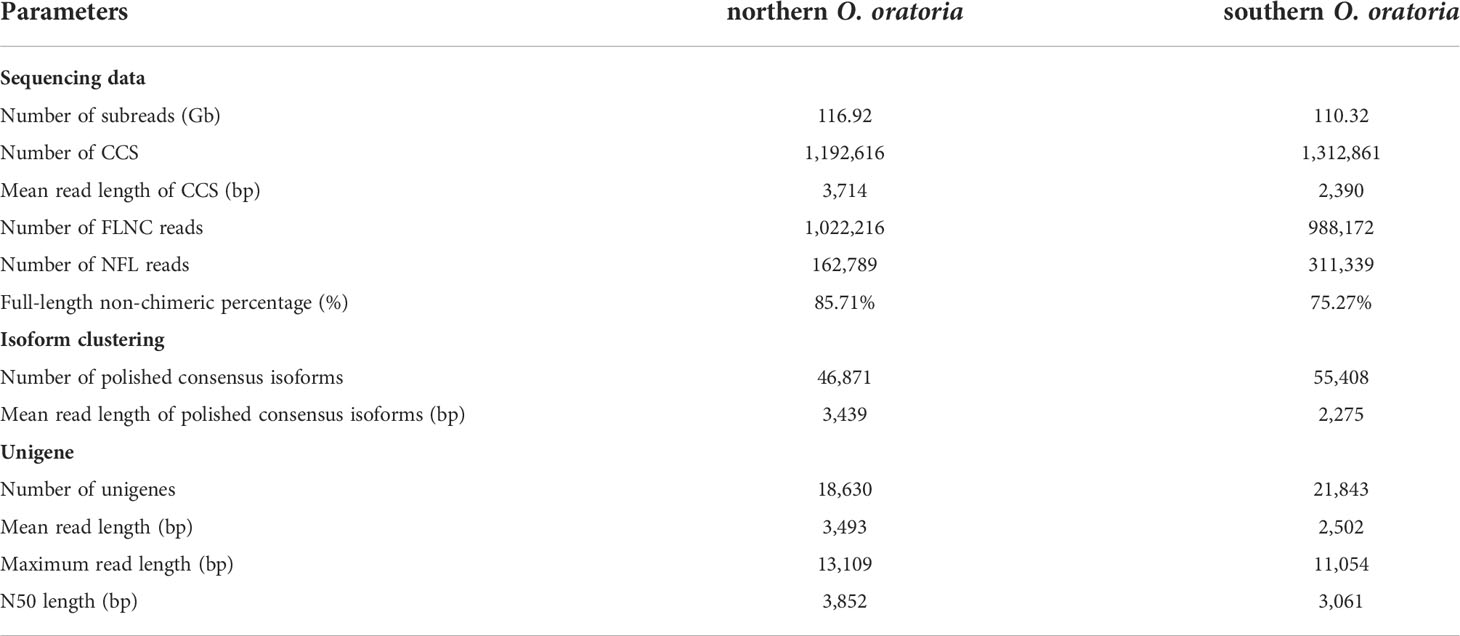
Table 1 Summary for the full-length transcriptome of northern and southern O. oratoria using PacBio sequencing.
3.2 Comparison between PacBio and Illumina unigenes
The transcriptomes of five tissues of northern and southern O. oratoria were separately sequenced using the Illumina platform (Table S2). After trimming and filtering, clean reads were assembled into 60,802 unigenes with an average length of 1,212 bp and N50 value of 2,140 bp for the northern O. oratoria, and 117,403 unigenes with an average length of 997 bp and N50 value of 1,563 bp for the southern O. oratoria, which were obviously shorter than unigenes in both FL transcriptomes. Most of PacBio unigenes of northern and southern O. oratoria had lengths > 2,000 bp, accounting for 87.1% and 61.0% of the total number, while most Illumina unigenes of northern and southern O. oratoria had lengths < 2,000 bp, accounting for 83.5% and 88.7%, respectively (Figure 2). The similarity between PacBio and Illumina unigenes were further conducted using BLAST v2.2.31 with the parameter set to 1E-5. The results showed that only 9.78% (5944) and 7.91% (9288) of Illumina unigenes had high similarity to 83.0% (15394) and 86.1% (18790) of PacBio unigenes in northern and southern O. oratoria, respectively.
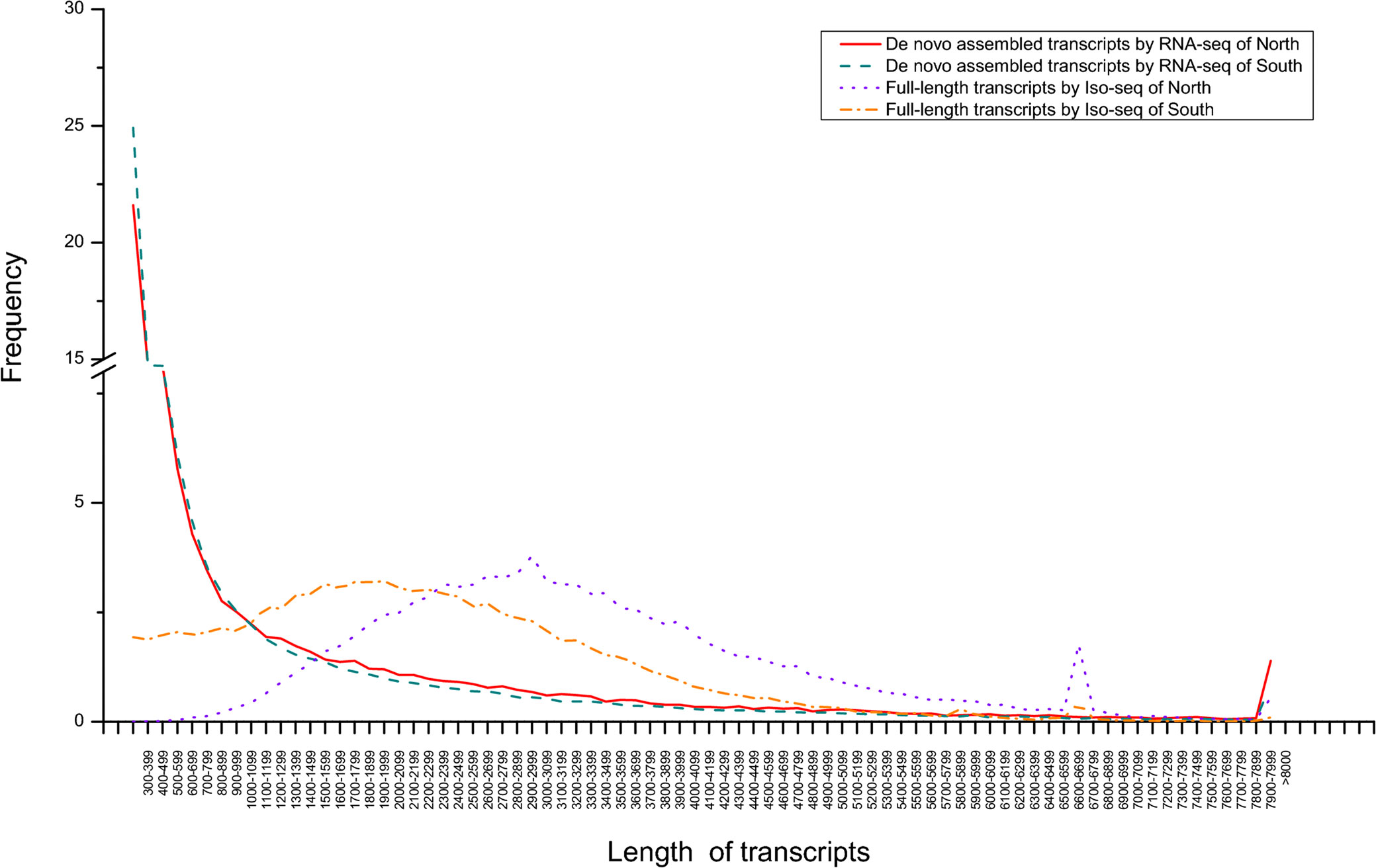
Figure 2 The comparison of unigene length distributions between PacBio sequencing and Illumina sequencing.
3.3 Full-length transcriptome structure
3.3.1 Coding sequences
Through the ANGEL predictions, a total of 18,716 CDSs were generated for the northern O. oratoria, of which 10,351 containing the start and stop codons were defined as complete open reading frames (ORFs). For southern O. oratoria, a total of 21,275 CDSs were predicted, including 10,286 complete ORFs. The length distributions of CDSs are shown in Figure S2. The CDS length of northern O. oratoria ranged from 49 bp to 3,590 bp with an average length of 477 bp, and the CDS length of southern O. oratoria ranged from 49 bp to 3,943 bp with an average length of 614 bp.
3.3.2 Transcription factor
In total, 1,549 putative TFs from 29 TF gene families were identified in the northern O. oratoria, and 944 putative TFs from 29 TF gene families were identified in the southern O. oratoria (Figure S3A). In both FL transcriptomes, Zinc finger C2H2 (zf-C2H2) was the most abundant TF gene family, followed by the family of BTB domain and Zinc Finger-containing (ZBTB) transcription factors. A GO enrichment analysis was further conducted to determine the potential functions of genes in which TFs have been determined. For both FL transcriptomes, binding (GO:0005488), metal ion binding (GO:0046872) and cation binding (GO:0043169) were the most enriched GO terms (Figure S3B).
3.3.3 Long noncoding RNA
Four prediction approaches were applied to identify lncRNAs, and a total of 6,579 and 9,048 were predicted in the FL transcripts of northern and southern O. oratoria, respectively. A Venn diagram was generated to visualize the respective contribution of different approaches to lncRNA prediction, and 565 and 1,624 were shared among the four approaches for northern and southern O. oratoria, respectively (Figure S4). The linkages between the identified lncRNAs and CDSs were further checked. The lncRNAs were only related to 6,415 (34.3%) CDSs in the northern O. oratoria and 8614 (40.5%) in the southern O. oratoria, suggesting that lncRNAs were not fully determined.
3.4 Functional annotation of O. oratoria full-length transcriptome
To obtain more comprehensive genetic information of O. oratoria, we combined the FLNC reads of the two FL transcriptomes to produce 123,824 polished FL consensus isoforms with N50 of 3,250 bp, and 42,735 unigenes with N50 of 3,472 bp after removing redundant sequences (Table S3). A total of 33,741 (80.0%) unigenes had at least one significant hit in the Nr, Nt, SwissProt, KOG, GO, KEGG or Pfam database, and a total of 8,517 (19.9%) unigenes were annotated in all databases (Figure S5A). By querying the Nr database, we found that the largest number of unigenes (11,309, 33.5%) were aligned to Hyalella azteca, followed by Zootermopsis nevadensis (1,616, 4.8%) and Limulus polyphemus (1,077, 3.2%) (Figure S5B). In KOG analysis, the main function classifications were found to be ‘General function prediction only’, ‘Signal transduction mechanisms’, ‘Cytoskeleton’ and ‘Posttranslational modification, protein turnover, chaperones’ (Figure S5C). In addition, 31,883 unigenes were mapped to 357 KEGG pathways and clustered significantly in ‘signal transduction’ and ‘transport and catabolism’ (Figure S5D). We further used the GO classification system to functionally categorize the unigenes based on Nr annotations. In total, 24,028 (56.2%) unigenes were successful annotated to three GO categories, and the largest category was biological process (49,789, 43.6%) followed by cellular component (34,354, 30.1%) and molecular function (29,974, 26.3%) (Figure S5E). Notably, in the biological process category, we found that 2,866 genes were involved in response to stimulus, 212 genes were related to immune system process, and 464 genes were associated with reproduction. In the molecular function category, O. oratoria had 48 genes related to antioxidant activity. These annotation and classification will aid understanding of the gene function in O. oratoria.
3.5 Sequence divergence between northern and southern O. oratoria orthologous genes
A total of 2,182 orthologous gene pairs were identified between northern and southern O. oratoria, and the phylogenic tree was constructed with the other two arthropods Daphnia pulex and Penaeus vannamei (Figure 3A). The UTR regions of each unigene pair was identified based on the predicted coding region. Among the 2182 pairs of orthologs, 884 pairs contain 5’UTR, coding and 3’UTR regions. Differences between coding regions of northern and southern orthologous genes occur at 1.44% of the positions, while the overall difference of 5’UTR and 3’UTR between northern and southern O. oratoria is 2.79% and 1.46%, respectively (Table S4).
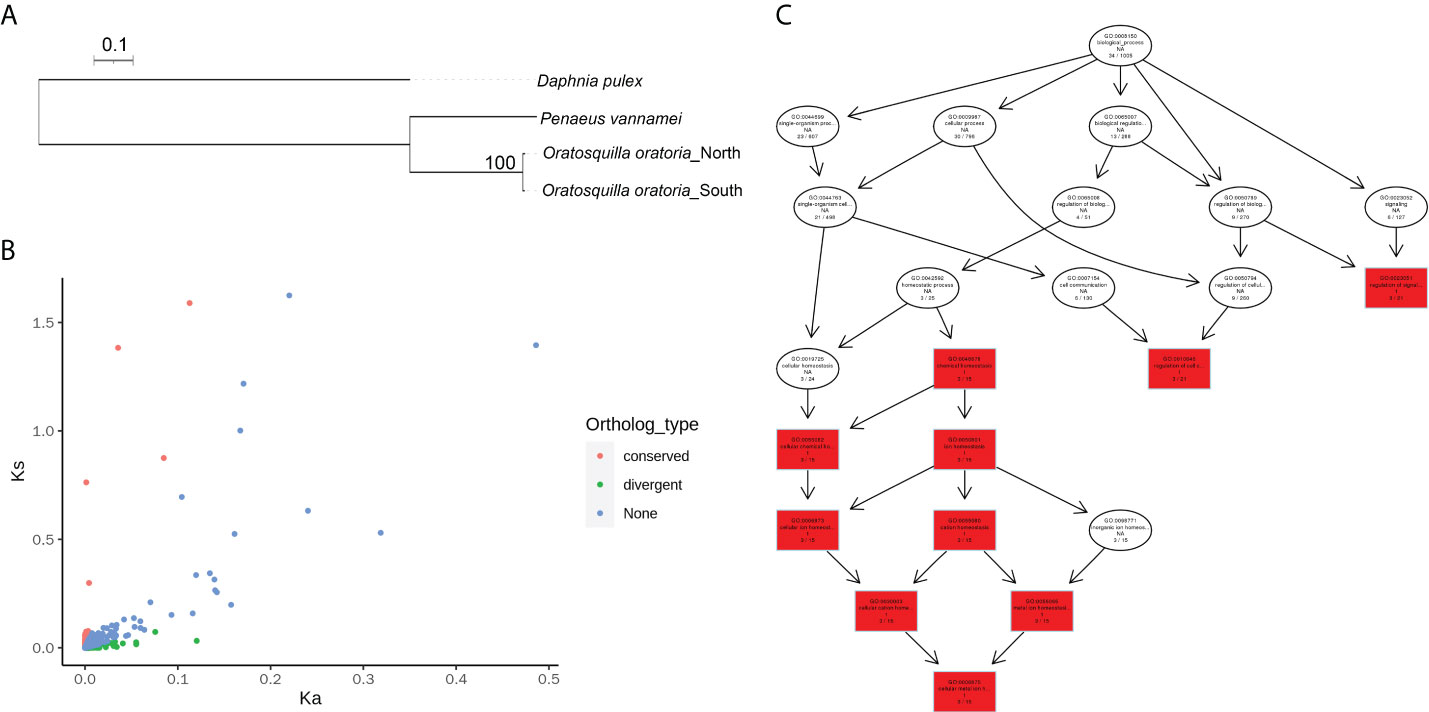
Figure 3 Orthologous genes analysis results. (A) Phylogenetic tree of three arthropods based on the orthologous genes. (B) Distribution of Ka/Ks for the orthologous genes. We define genes with Ka/Ks > 1 as divergent genes, and genes with Ka/Ks < 0.01 as conserved genes. (C) The directed acycline graph of the GO category in the biological process significantly enriched in 98 genes with Ka/Ks > 1 by topGO analysis. The degree of enrichment is represented by color intensities.
3.6 Identification of genes under positive selection
The ratio of Ka/Ks is an indicator of selection acting on a protein-coding gene. Among the 2,182 pairs of orthologs identified, both Ka and Ks could be calculated for 1,509 (69.2%) orthologs. For the remaining orthologs, we could only calculate either Ka (84 orthologs, 3.8%) or Ks (477 orthologs, 21.9%), or the orthologs were identical (112, 5.1%). For the orthologous pairs for which Ka/Ks ratio could be calculated, the mean values of Ka, Ks, and Ka/Ks were 0.452, 0.013, and 0.951, respectively. Among them, 98 orthologs (6.5%) had a Ka/Ks > 1, indicating these genes have evolved under positive selection (Figure 3B, Table S5). Except the orthologous pairs with high Ka/Ks, 361 pairs (24.0%) had a Ka/Ks < 0.1, suggesting that these genes have evolved under high selective constraint (Figure 3B, Table S5).
GO enrichment analysis of 98 genes under positive selection showed that 50 GO terms covering 23 genes were significantly over-presented (P < 0.05) (Table S6). The top enriched gene groups were mainly related to cellular ion homeostasis (GO:0048878, GO:0050801, GO:0055082) and regulation of cell communication (GO:0010646) (Figure 3C). Notably, GO terms (GO:0010628, GO:0045893) associated with transcription regulation were also significantly enriched. Although not enriched in the GO analysis, the following genes under positive selection may also play important roles in local adaptation in O. oratoria (Table 2): two gene involved in cytoskeletal organization (Rho guanine nucleotide exchange factor 7, ARHGEF7; cell adhesion molecule 2, CADM2), six genes involved in stress response (glutathione peroxidase 3, GPx3; ankyrin repeat and zinc finger domain-containing protein 1, ANKZF1; two copies of ferritin and poly [ADP-ribose] polymerase 12, PARP12), five genes involved in innate immune response (L-type lectin, LTL; fibrinogen-related protein 2, FREP2; prophenoloxidase activating factor, PPAF; two copies of antilipopolysaccharide factor, ALFs), five genes involved in genetic information processing (reverse transcriptase; ribonuclease Z; zinc finger protein 271, ZNF271; two copies of zinc finger protein 791, ZNF791).
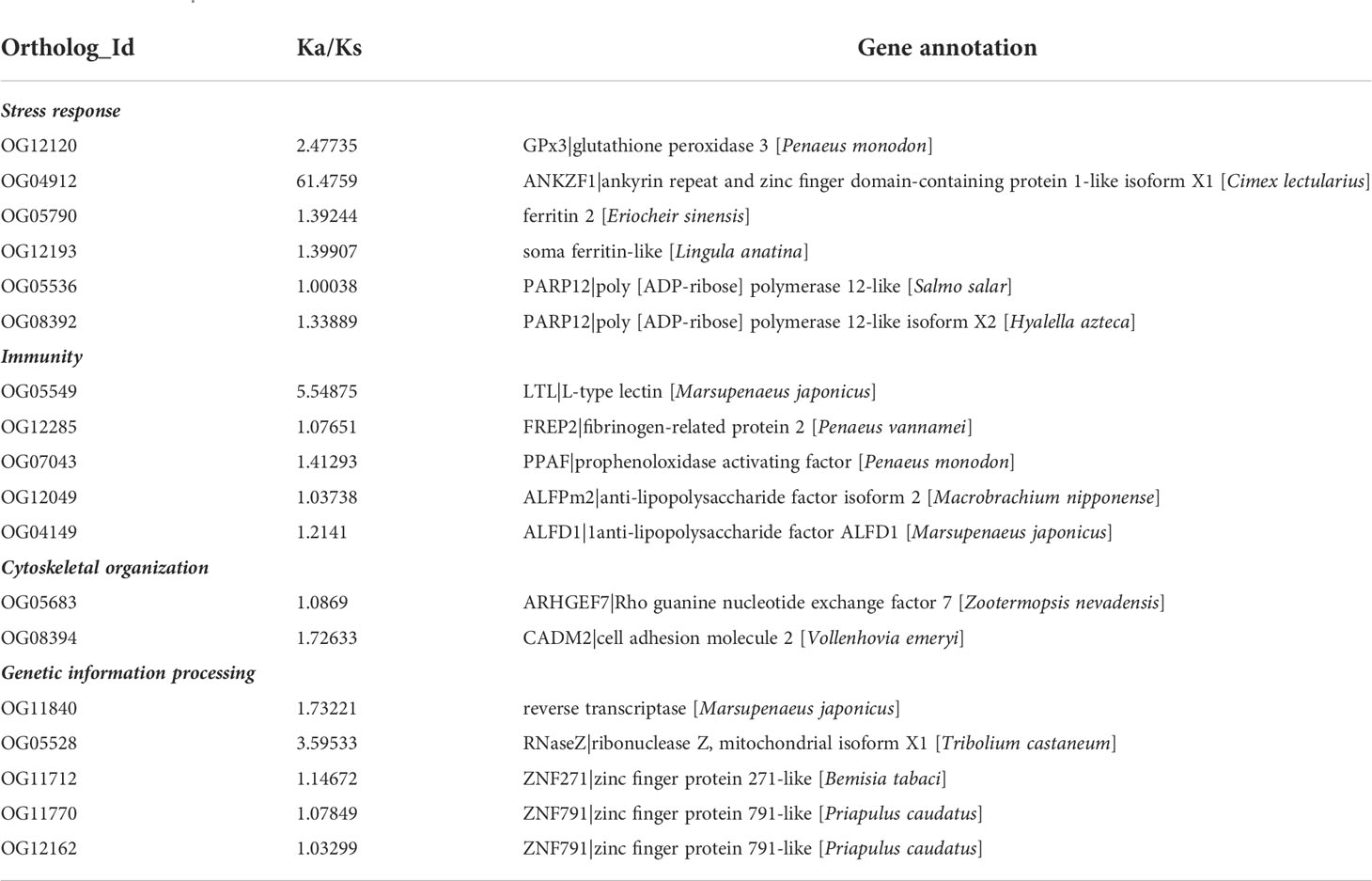
Table 2 PSGs related to stress response, immunity, cytoskeletal organization and genetic information processing between northern and southern O. oratoria.
3.7 Expression analysis of PSGs
The publicly available RNA-seq data of two ecologically divergent O. oratoria populations (corresponding to northern and southern O. oratoria in this study) under thermal stress (Lou et al., 2019a) was used for expression analysis of PSGs. We firstly compared gene expression between genes under positive selection and those under neutral and purifying selection. Interestingly, relatively high levels of gene expression were consistently found in PSGs in comparison with genes under neutral and purifying selection regardless of experimental condition (Figure 4). Then, we focused on the expression profiles of PSGs in the northern and southern O. oratoria in response to the same heat stress (20°C–28°C). The expression heatmap showed remarkably different expression patterns of 98 PSGs (Figure 5A). We found specific clusters of highly expressed PSGs in the northern and southern O. oratoria exposed to heat stress. When considered separately for population-specific cluster, there was a significantly higher number of up-regulated PSGs under heat stress in the northern O. oratoria (28/98) than its southern counterpart (4/98). The highly expressed PSGs involved in “response to stimulus” (GO:0051716), “signaling” (GO:0035556) and “biological regulation” (GO:0008199) were only detected in heat-stressed northern O. oratoria (Figure 5B). However, only two proteins with known function were abundant in heat-stressed southern O. oratoria, a ribosome-binding protein and a CD63 antigen.
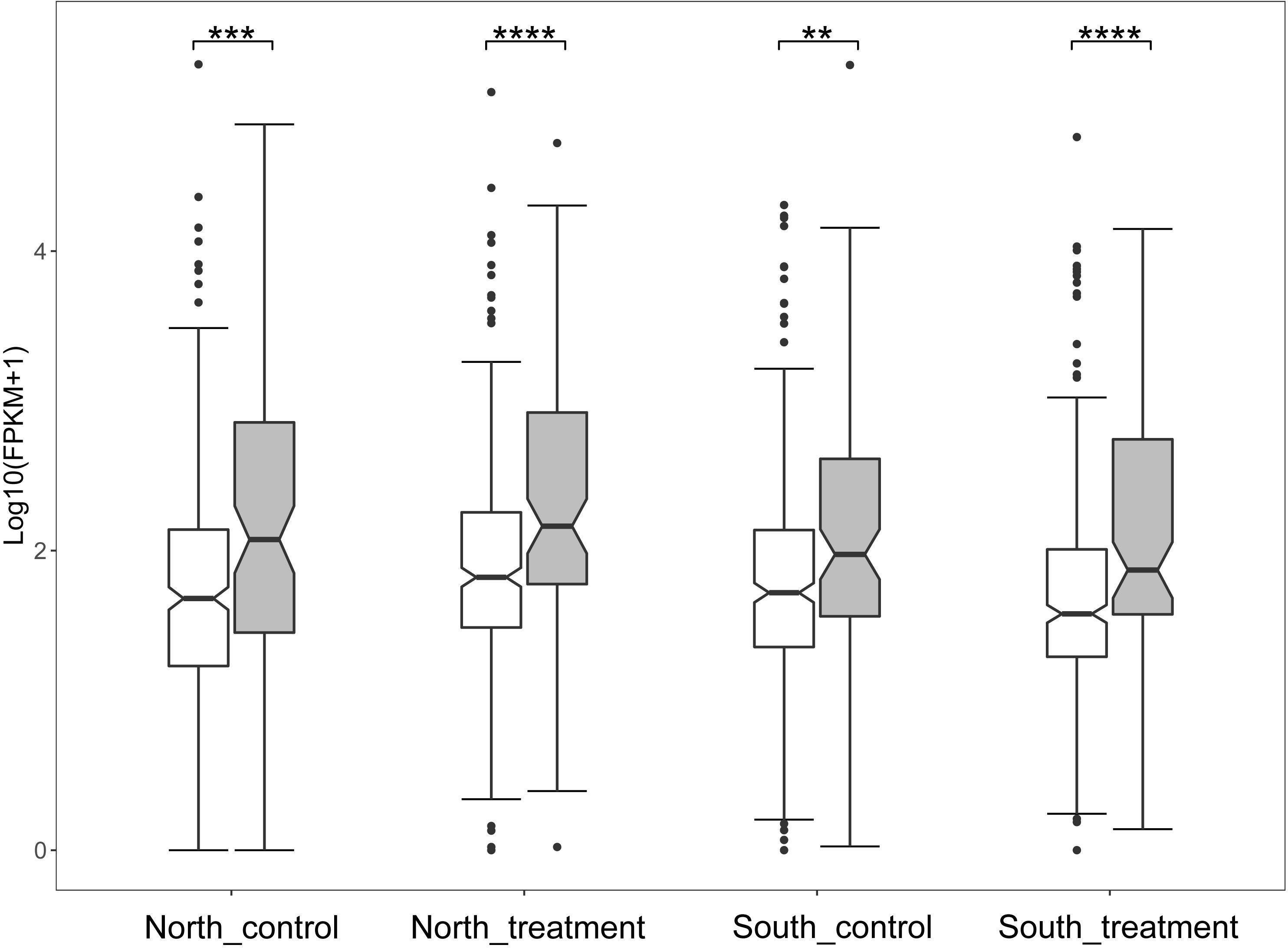
Figure 4 Boxplot of the expression level of genes under positive selection (gray), neutral and purifying selection (white). P-value was calculated by Mann–Whitney test. **P < 0.01; ***P < 0.001; ****P < 0.0001.
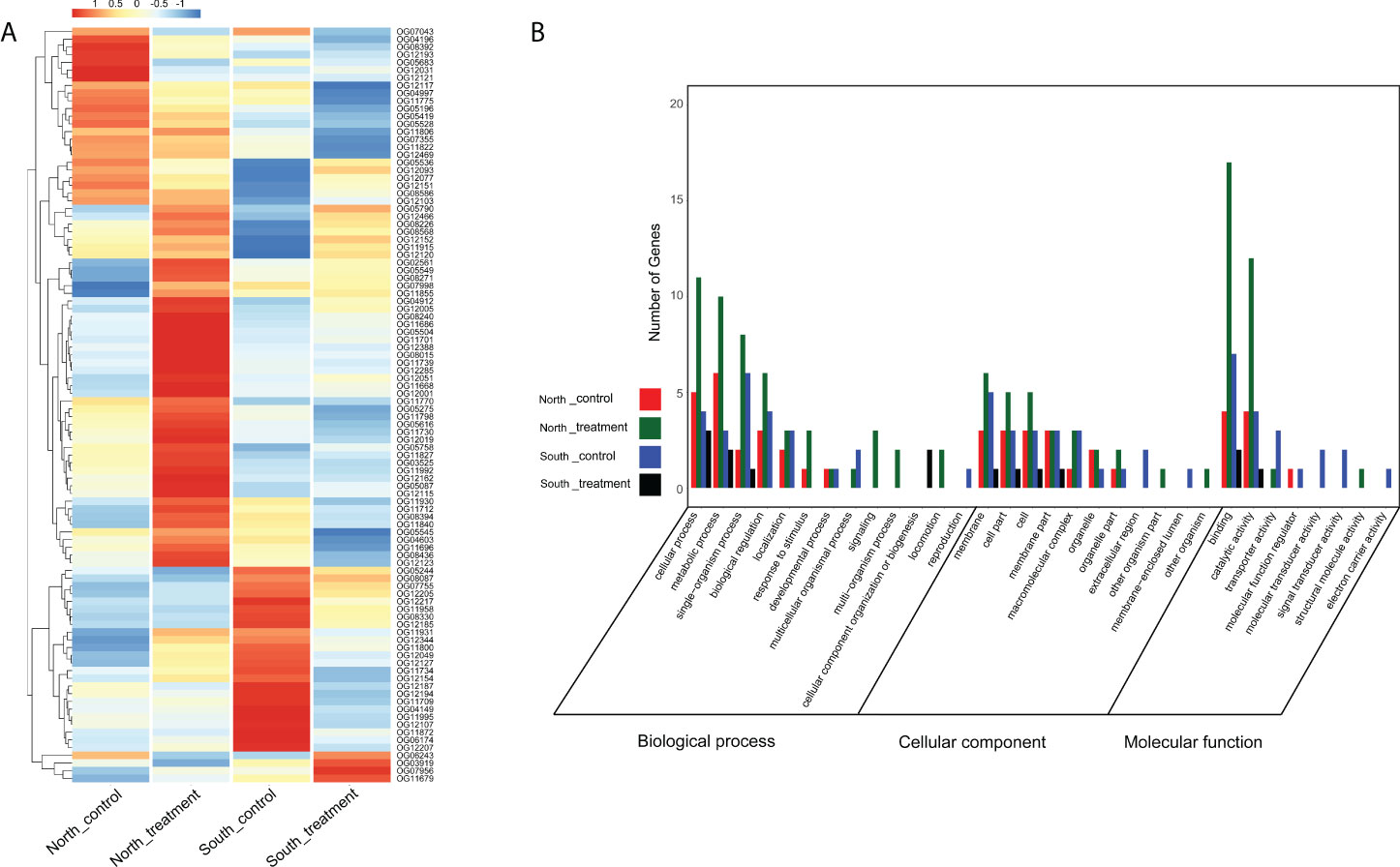
Figure 5 (A) Expression levels of all 98 PSGs and (B) GO classification of PSGs that highly expressed under different experimental condition in population-specific clusters. The expression level is represented by color intensities (red color indicates the higher expression, and blue color indicates the lower expression of the gene).
4 Discussion
4.1 Long read reference reconstruction of the full-length transcripts
This is the first FL transcriptome study on the mantis shrimp. Deep PacBio sequencing is currently one of the best methods for retrieving nucleic acid sequences from species with ultra-large and complex genomes. In this study, the FL transcriptome of O. oratoria has been deep-sequenced using the PacBio Sequel platform with a pooled RNA sample from different tissues to capture as many FL transcript isoforms as possible. Compared with the de novo assembled unigenes obtained from RNA-seq, the length is greatly longer in the non-assembled unigenes generated by PacBio SMRT sequencing. The annotation rate (80.0%) of PacBio unigenes is much higher than that reported in previous next-generation transcriptome studies (20.4%-33.7%, Yan et al., 2018; Lou et al., 2019b; Lou et al., 2019c). Although fewer unigenes were obtained by PacBio sequencing than by RNA-seq, a higher proportion of PacBio unigenes have been annotated than Illumina unigenes, indicating high-efficiency of PacBio SMRT sequencing in recovering FL transcripts. Thus, the FL transcripts produced in this study would serve as important basis for gene discovery, genome assembly and annotation of O. oratoria, and contribute towards better understanding of adaptive evolution in this species.
TFs play key roles in the transcriptional regulation of functional genes by sequence-specific binding to regulatory regions throughout the genome (Fulton et al., 2009). In this study, we have predicted 1,549 TFs in the northern O. oratoria and 944 TFs in the southern O. oratoria, laying a foundation for the next step in studying the potential regulatory roles of TFs in O. oratoria. Among them, zf-C2H2 and ZBTB are the most abundant TF families. C2H2 zinc-finger proteins are one of the most widespread transcription factor families in eukaryotes (Kubo et al., 1998), and participate in a wide range of biological processes, such as growth, development, and stress response (Liu et al., 2015). ZBTB TFs are generally considered transcriptional repressors, involving in a variety of developmental and cellular processes (Ren et al., 2019). This TF family has been shown to involve in developmental regulation of regenerative potential in Drosophila (Narbonne-Reveau and Maurange, 2019).
LncRNAs are a group of RNA molecules with the structure similar to mRNA and the length longer than 200 nucleotides (Rinn and Chang, 2012). Despite the lack of protein-coding capacity, LncRNAs can exert powerful biological functions by regulating gene expression at epigenetic, transcriptional and post-transcriptional levels (Kapranov et al., 2007; Cao, 2014). To date, the LncRNAs of O. oratoria has not been reported yet. We have identified 565 and 1,624 LncRNAs for northern and southern O. oratoria, respectively. Considering the important roles of lncRNAs in various biological processes regulation, our data provide a reference resource for further investigation on the underlying mechanism of lncRNA-related regulation in O. oratoria.
4.2 Environmental heterogeneity driving adaptive divergence in O. oratoria
For the O. oratoria complex, two cryptic species have been delineated based on a number of phylogenetic analysis (Cheng and Sha, 2017; Cheng et al., 2020). However, the genetic factors driving the evolution of O. oratoria are almost unknown due to the fact that few molecular data are available for inferring the evolutionary history and genetic divergence of this species. In this study, we have identified 2,182 orthologous gene pairs between northern and southern O. oratoria and determined the average sequence divergence to be 1.44% for the coding regions. This is comparable to the average divergence of two invasive whitefly species Asia II 3 and MEAM1 transcriptomes (1.73% in Wang et al., 2012), and much higher than the level of sequence divergence that was found between human and chimpanzee (0.45% in Hellmann et al., 2003) as well as between the two other whitefly cryptic species MEAM1 and MED (0.83% in Wang et al., 2011). When it comes to non-coding regions, the genetic divergence is more obvious for 5’UTR (2.79%) and 3’UTR (1.46%) regions, for which both ratios are higher than the reported mean 1.12% divergence of 5’UTR and 0.86% divergence of 3’UTR regions between human and chimpanzee (Hellmann et al., 2003). The relatively higher level of similarity at the coding regions compared to non-coding regions could be attributed to the presence of functional elements that are subject to purifying selection (Wang et al., 2011). Collectively, the substantial sequence divergence at the transcriptomic level in combination with previous phylogenetic results concur in suggesting that northern and southern O. oratoria constitute different species.
The heterogeneous seascapes in the NWP, such as temperature gradient governed by the oceanic currents system and salinity gradient surrounding the Yangtze River mouth, can impose spatially varying selective pressures on local populations distributed along the gradients. As expected, our analyses have identified a total of 98 orthologs under positive selection (Ka/Ks > 1), suggesting that those genes might play important roles in the speciation and adaptive evolution of O. oratoria. The candidate genes have been found to be involved in many biological processes, including stress response, immunity, and cytoskeletal organization. It should be noted that compared to a battery of thermally responsive genes, the relatively low number of salinity-tolerance associated genes identified here may be due to limited genomic information provided by transcriptome sequencing or relatively weak selective force generated by salinity gradient, or a combination of these. As discussed below, the functional inferences based on candidate genes suggest that seascape heterogeneity in the NWP, in particular differences in temperature, has facilitated adaptive evolution of O. oratoria.
It has been clearly established that any intense stress is usually accompanied by enhanced reactive oxygen species (ROS) generation, causing oxidative damage (Lushchak, 2011). The role of changes in temperature and salinity in induction of oxidative stress is highlighted in aquatic organisms (Liu et al., 2007; Madeira et al., 2013). In response to the production of ROS, these organisms have developed the cellular scavenging system to remove these radicals involving various enzymes. GPx3 is a secreted plasma protein that scavenges ROS in the extracellular compartment, and thereby protects cells against oxidative damage (Jin et al., 2011). ANKZF1 involves in the cellular response to hydrogen peroxide and plays a role in the maintenance of mitochondrial integrity under conditions of cellular stress (van Haaften-Visser et al., 2017). Ferritin is a highly conserved iron storage protein that can concentrate cellular iron to prevent the harmful ROS generation and reduce oxidative damage (Harrison and Arosio, 1996; McCord, 1996). Previous findings have suggested the roles of ferritin in salinity stress adaptation in the swimming crab Portunus trituberculatus (Huang and Xu, 2016). A ROS signature in cells can also trigger the activation of DNA repair (Mittler et al., 2011). RAPA proteins are involved in the detection of DNA damage and initiation of DNA repair by binding to damaged parts of DNA (Druzhyna et al., 2000). Therefore, selection on GPx3, ANKZF1, ferritin and RAPA12 may be associated with adaptation of O. oratoria in response to oxidative damage produced by temperature and salinity variation.
Water temperature is probably the most important environmental variable because it directly affects growth, metabolism and survival of marine organisms (Chen et al., 1995; Hennig and Andreatta, 1998). In the crab Carcinus maenas, Chisholm and Smith (1994) found the impact of the seasonal changes in water temperature on the antibacterial activity of haemocytes. Notably, representative genes involved in innate immune were found to be positively selected in warm- and cold-tolerant O. oratoria. Enzymes encoded by these genes include LTL that functions as pattern recognition receptors in shrimp immunity (Xu et al., 2014), FREP2 which is also known as fibrinogen-like domain immunolectins and plays a role in defense and protection against infection (Hanington and Zhang, 2011), PPAF involving in the prophenoloxidase activation pathway of innate immune system in invertebrates (Wang et al., 2015), and ALF which is a type of antimicrobial peptides with a vital role in crustacean antimicrobial defense (Li and Li, 2020). Previous common-garden experiments also suggested that differentially expressed genes after heat treatment in O. oratoria were enriched in pathways associated with immune (Lou et al., 2019a). Consequently, we hypothesize that these functional genes associated with immunity seem to evolve rapidly in O. oratoria to increase its capacity to manage thermal stresses. Cytoskeletal reorganization has been reported to undergo pronounced transformation under thermal stress (Richter et al., 2010). Herein, evolutionary forces acting on cytoskeletal organization-related genes (ARHGEF7 and CADM2) provided additional evidence reflective of thermal selection in O. oratoria. ARHGEF7 can regulate Rho GTPase activity and plays a role in regulating cytoskeletal and cell adhesion dynamics (Cheng et al., 2021), while CADM2 is a cell adhesion molecule that functions in cell recognition, adhesion, migration and differentiation (Edelman, 1983). Selection on genes involved in cytoskeletal organization has also been implicated in the warm-adapted marine mussel Mytilus galloprovincialis compared with its cold-adapted congeners (Popovic and Riginos, 2020). Collectively, these findings suggest that adaptation was driving the evolution of key genes associate with immune and cytoskeletal organization, potentially indicating their importance for O. oratoria to tolerant habitat temperature changes. Further validation of these candidates is necessary to determine their functional association with local thermal adaptation of O. oratoria.
4.3 Selection towards plasticity facilitating thermal adaptation of O. oratoria
Genetic variation and phenotypic plasticity both play important roles in adaptive evolution (Davis et al., 2005; Kelly, 2019). In particular, plasticity in gene expression is favored by organisms under dynamic environments (Li et al., 2018). Closely related species and genetically differentiated populations may exhibit differences in the patterns and extents of plastic responses, indicating there is genetic variation for plastic responses (Gao et al., 2008). This means that phenotypic plasticity can evolve in response to natural selection, which in turn suggests the existence of evolving plasticity (Pigliucci, 2005). As observed in the polar diatom Fragilariopsis cylindrus (Mock et al., 2017), we found PSGs in O. oratoria had significantly higher expression levels than those genes under neutral or purifying selection, indicative of a role of natural selection in driving gene expression.
Comparing gene expression responses to environmental conditions among locally adapted populations offers an opportunity to test for among-population variation in plasticity (DeBiasse and Kelly, 2016). The northern and southern O. oratoria are naturally distributed in different climate regions in the NWP and may vary in their sensitivity to thermal stress. As expected, they exhibit differences in the patterns of expression plastic response to the same heat stress (Figure 5). The northern O. oratoria that adapts to cold climate had greater differential gene expression in PSGs between control and heat-stressed samples than its southern counterpart, suggesting more sensitive of northern O. oratoria to heat stress. The higher expression plasticity in the heat-sensitive populations has also been observed in copepod Tigriopus californicus (Schoville et al., 2012), coral Porites astreoides (Kenkel et al., 2013) and snail Chlorostoma funebralis (Gleason and Burton, 2015). The observation of population-specific clusters of differentially expressed PSGs implies that natural selection may act on transcriptional plasticity to facilitate the evolution of lineage specific tolerance to heat stress in O. oratoria. Similarly, selection towards expression plasticity has been demonstrated in other aquatic animals, such as oyster populations from different environmental gradients (Li et al., 2018; Li et al., 2021). Along with previous studies, our results point to the potential importance of evolving plasticity in adaptation of marine organisms to heterogeneous seascapes.
5 Conclusion
In this study, we have demonstrated the advantage of PacBio sequencing to rapidly reconstruct FL transcripts compared to RNA-seq. By comparing transcriptome sequence divergence, our results support previous proposition that northern and southern O. oratoria constitute different species. The abundant PSGs related to environmental responsiveness are indicative of a strong selective force that O. oratoria might undergo during the adaptation process to heterogeneous seascapes in the NWP. In addition, we found genes underwent positive selection also exhibit divergence in expression plastic response to heat stress, suggesting that natural selection may act on the expression plasticity to facilitate O. oratoria adaptation. As such, our study strengthens the view that genetic variation and plasticity should be taken into account when attempting to understand the evolution of marine species distributed along the environmentally heterogeneous coast. In light of the important role of evolving plasticity in species’ responses to climate change, mechanistic studies are warranted to investigate the underlying processes that may mediate different adaptive potential, which is critical for O. oratoria in the context of a changing climate.
Data availability statement
Raw sequencing data analyzed for this study have been submitted to NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA853060.
Author contributions
JC and ZS conceived and designed the project. JC and YL collected the samples. JC conducted laboratory work. JC and LZ performed bioinformatics and statistical analyses. JC authored drafts of the manuscript. MH and LZ reviewed drafts of the paper. All authors contributed to the manuscript and approved it for submission and publication.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31872569), the Open Fund of CAS Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences (No. KF2021N002), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB42000000) and the National Key R&D Program of China (No. 2018YFD0900804).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.975686/full#supplementary-material
Supplementary Figure 1 | Length distributions of full-length transcriptomes of the northern and southern O. oratoria. (A) Distribution of the number and length of CCS reads. (B) Distribution of the number and length of FLNC sequences. (C) Distribution of the number and length of consensus isoforms.
Supplementary Figure 2 | Length distribution of CDSs of the northern (A) and southern (B) O. oratoria. Length of predicted CDS was plotted along the x-axis, while number of CDS transcripts was plotted along the left y-axis. The yellow line that represents the percentage of CDS length was plotted along the right y-axis.
Supplementary Figure 3 | Transcription factors identified in the northern and southern O. oratoria. (A) Classification of the detected TF families. Different types of transcript family were plotted along the x-axis, while the number of transcription factors were plotted along the y-axis. (B) GO enrichment reveals the functions of genes that have been determined to be TFs.
Supplementary Figure 4 | Venn diagram of the number of lncRNAs predicted in the northern (A) and southern (B) O. oratoria.
Supplementary Figure 5 | Functional annotation of O. oratoria full-length transcriptome. (A) Classification of unigene annotation in all databases, including Nr, SwissProt, KEGG, KOG, GO, Nt and Pfam. (B) Species distribution of highest scoring blastp match in Nr database. (C) KOG function classification of all O. oratoria unigenes. (D) KEGG pathway assignment of O. oratoria unigenes. (E) Distribution of GO terms for all annotated unigenes in biological process, cellular component and molecular function categories.
References
Ahyong S. T. (2012). The marine fauna of New Zealand: Mantis shrimps (Crustacea: Stomatopoda) (Packaging Ltd, Wellington: Graphic Press).
Alexa A., Rahnenführer J. (2010). topGO: Enrichment analysis for gene ontology. r package version 2.10.0.
Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106
Biswas S., Akey J. M. (2006). Genomic insights into positive selection. Trends Genet. 22, 437–446. doi: 10.1016/j.tig.2006.06.005
Cao J. (2014). The functional role of long non-coding RNAs and epigenetics. Biol. Proced. 16, 42. doi: 10.1186/1480-9222-16-11
Cheng J., Hui M., Sha Z. L. (2019). Transcriptomic analysis reveals insights into deep-sea adaptations of the dominant species, Shinkaia crosnieri (Crustacea: Decapoda: Anomura), inhabiting both hydrothermal vents and cold seeps. BMC Genomics 20, 388. doi: 10.1186/s12864-019-5753-7
Cheng K., Larabee S. M., Tolaymat M., Hanscom M., Shang A. C., Schledwitz A., et al. (2021). Targeted intestinal deletion of rho guanine nucleotide exchange factor 7, βPIX, impairs enterocyte proliferation, villus maturation, and mucosal defenses in mice. Am. J. Physiol. Gastrointest Liver Physiol. 320, G627–G643. doi: 10.1152/ajpgi.00415.2020
Cheng J., Sha Z. (2017). Cryptic diversity in the Japanese mantis shrimp Oratosquilla oratoria (Crustacea: Squillidae): Allopatric diversification, secondary contact and hybridization. Sci. Rep. 7, 1972. doi: 10.1038/s41598-017-02059-7
Cheng J., Zhang N., Sha Z. (2020). Nuclear microsatellites reveal population genetic structuring and fine-scale pattern of hybridization in the Japanese mantis shrimp Oratosquilla oratoria. PeerJ 8, e1027. doi: 10.7717/peerj.10270
Chen J. C., Lin M. N., Ting Y. Y., Chen J. C., Lin M. N., Ting Y. Y., et al. (1995). Survival, haemolymph osmolality and tissue water of Penaeus chinensis juveniles acclimated to different salinities and temperature levels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 110, 253–258. doi: 10.1016/0300-9629(94)00164-O
Chisholm J. R. S., Smith V. J. (1994). Variation of antibacterial activity in the haemocytes of the shore crab, Carcinus maenas, with temperature. J. Mar. Biol. Assoc. U K 74, 979–982. doi: 10.1017/S0025315400090238
Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M., et al. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Coscia I., Wilmes S. B., Ironside J. E., Goward-Brown A., O'Dea E., Malham S. K., et al. (2020). Fine-scale seascape genomics of an exploited marine species, the common cockle Cerastoderma edule, using a multimodelling approach. Evol. Appl. 13 (8), 1854–1867. doi: 10.1111/eva.12932
Davis M. B., Shaw R. G., Etterson J. R. (2005). Evolutionary responses to changing climate. Ecology 86 (7), 1704–1714. doi: 10.1890/03-0788
DeBiasse M. B., Kelly M. W. (2016). Plastic and evolved responses to global change: what can we learn from comparative transcriptomics? J. Hered. 107 (1), 71–81. doi: 10.1093/jhered/esv073
Development Core Team R. (2022). R: A language and environment for statistical computing (R Foundation for Statistical Computing). Available at: http://www.R-project.org.
Druzhyna N., Smulson M. E., LeDoux S. P., Wilson G. L. (2000). Poly(ADP-ribose) polymerase facilitates the repair of n-methylpurines in mitochondrial DNA. Diabetes 49 (11), 1849–1855. doi: 10.2337/diabetes.49.11.1849
Du X., Cai S., Yu C., Jiang X., Lin L., Gao T., et al. (2016). Population genetic structure of mantis shrimps Oratosquilla oratoria: Testing the barrier effect of the Yangtze river outflow. Biochem. Syst. Ecol. 66, 12–18. doi: 10.1016/j.bse.2016.02.033
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fulton D. L., Sundararajan S., Badis G., Hughes T. R., Wasserman W. W., Roach J. C., et al. (2009). TFCat: the curated catalog of mouse and human transcription factors. Genome Biol. 10 (3), R29. doi: 10.1186/gb-2009-10-3-r29
Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao L., Chen J., Yang J. (2008). Phenotypic plasticity: Eco-devo and evolution. J. Syst. Evol. 46 (4), 441–451. doi: 10.3724/SP.J.1002.2008.07170
Gleason L. U., Burton R. S. (2015). RNA-Seq reveals regional differences in transcriptome response to heat stress in the marine snail Chlorostoma funebralis. Mol. Ecol. 24, 610–627. doi: 10.1111/mec.13047
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gracey A. Y., Cossins A. R. (2003). Application of microarray technology in environmental and comparative physiology. Annu. Rev. Physiol. 65, 231–259. doi: 10.1146/annurev.physiol.65.092101.142716
Guindon S., Dufayard J., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hamano T., Matsuura S. (1987). Egg size, duration of incubation, and larval development of the Japanese mantis shrimp in the laboratory. Nippon Suisan Gakkaishi 53, 23–39. doi: 10.2331/suisan.53.23
Hanington P. C., Zhang S. M. (2011). The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation. J. Innate Immun. 3 (1), 17–27. doi: 10.1159/000321882
Harrison P. M., Arosio P. (1996). The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203. doi: 10.1016/0005-2728(96)00022-9
Hellmann I., Zollner S., Enard W., Ebersberger I., Nickel B., Paabo S. (2003). Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res. 13, 831–837. doi: 10.1101/gr.944903
Hennig O. L., Andreatta E. R. (1998). Effect of temperature in an intensive nursery system for Penaeus paulensis (Pérez farfante, 1967). Aquaculture 164, 167–172. doi: 10.1016/S0044-8486(98)00184-7
Huang S., Xu Q. (2016). Ferritin gene from the swimming crab (Portunus trituberculatus) involved in salinity stress adaptation. Turkish J. Fish. Aquat. Sci. 16, 141–153. doi: 10.4194/1303-2712-v16_1_15
Jin R. C., Mahoney C. E., Anderson L., Ottaviano F., Croce K., Leopold J. A., et al. (2011). Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 18, 1963–1973. doi: 10.1161/CIRCULATIONAHA.110.000034
Johnson D. R., Boyer T. P. (2015). East Asian Seas regional climatology (Version 2) (USA: National Centers for Environmental Information, NOAA). Available at: https://www.nodc.noaa.gov/OC5/regional_climate/EASclimatology/.
Kapranov P., Cheng J., Dike S., Nix D. A., Duttagupta R., Willingham A. T., et al. (2007). RNA Maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. doi: 10.1126/science.1138341
Kelly M. (2019). Adaptation to climate change through genetic accommodation and assimilation of plastic phenotypes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374(1768), 1768. doi: 10.1098/rstb.2018.0176
Kenkel C. D., Meyer E., Matz M. V. (2013). Gene expression under chronic heat stress in populations of the mustard hill coral (Porites astreoides) from different thermal environments. Mol. Ecol. 22, 4322–4334. doi: 10.1111/mec.12390
Kodama K., Kume G., Shiraishi H., Morita M., Horiguchi T. (2006). Relationship between body length, processed-meat length and seasonal change in net processed-meat yield of Japanese mantis shrimp Oratosquilla oratoria in Tokyo bay. Fish. Sci. 72, 804–810. doi: 10.1111/j.1444-2906.2006.01221.x
Kong L., Zhang Y., Ye Z. Q., Liu X. Q., Zhao S. Q., Wei L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Kubo K., Sakamoto A., Kobayashi A., Rybka Z., Kanno Y., Nakagawa H., et al. (1998). Cys2/His2 zinc-finger protein family of petunia: evolution and general mechanism of target-sequence recognition. Nucleic Acids Res. 26, 608–615. doi: 10.1093/nar/26.2.608
Li B., Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 12, 323. doi: 10.1186/1471-2105-12-323
Li W., Jaroszewski L., Godzik A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. doi: 10.1093/bioinformatics/18.1.77
Li S., Li F. (2020). The anti-lipopolysaccharide factors in crustaceans. Subcell Biochem. 94, 63–80. doi: 10.1007/978-3-030-41769-7_3
Li L., Li A., Song K., Meng J., Guo X., Li S., et al. (2018). Divergence and plasticity shape adaptive potential of the pacific oyster. Nat. Ecol. Evol. 2 (11), 1751–1760. doi: 10.1038/s41559-018-0668-2
Li A., Li L., Zhang Z., Li S., Wang W., Guo X., et al. (2021). Noncoding variation and transcriptional plasticity promote thermal adaptation in oysters by altering energy metabolism. Mol. Biol. Evol. 38 (11), 5144–5155. doi: 10.1093/molbev/msab241
Li L., Stoeckert C. J., Roos D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liu J. Y. (2013). Status of marine biodiversity of the China seas. PloS One 8, e50719. doi: 10.1371/journal.pone.0050719
Liu Y., Wang W. N., Wang A. L., Wang J. M., Sun R. Y. (2007). Effects of dietary vitamin e supplementation on antioxidant enzyme activities in Litopenaeus vannamei (Boone, 1931) exposed to acute salinity changes. Aquaculture 265, 351–358. doi: 10.1016/j.aquaculture.2007.02.010
Liu Q., Wang Z., Xu X., Zhang H., Li C. (2015). Genome-wide analysis of C2H2 zinc-finger family transcription factors and their responses to abiotic stresses in poplar (Populus trichocarpa). PloS One 10 (8), e0134753. doi: 10.1145/2818302
Li A., Zhang J., Zhou Z. (2014). PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 15, 311. doi: 10.1186/1471-2105-15-311
Lou F., Gao T., Han Z. (2019b). Transcriptome analyses reveal alterations in muscle metabolism, immune responses and reproductive behavior of Japanese mantis shrimp (Oratosquilla oratoria) at different cold temperature. Comp. Biochem. Physiol. Part D Genomics Proteomics 32, 100615. doi: 10.1016/j.cbd.2019.100615
Lou F., Gao T., Han Z. (2019c). Effect of salinity fluctuation on the transcriptome of the Japanese mantis shrimp Oratosquilla oratoria. Int. J. Biol. Macromol. 140, 1202–1213. doi: 10.1016/j.ijbiomac.2019.08.223
Lou F., Han Z., Gao T. (2019a). Transcriptomic responses of two ecologically divergent populations of Japanese mantis shrimp (Oratosquilla oratoria) under thermal stress. Animals 9, 399. doi: 10.3390/ani9070399
Lushchak V. I. (2011). Environmentally induced oxidative stress in aquatic animals. Aquat. Toxicol. 101, 13–30. doi: 10.1016/j.aquatox.2010.10.006
Madeira D., Narciso L., Cabral H. N., Vinagre C., Diniz M. S. (2013). Influence of temperature in thermal and oxidative stress responses in estuarine fish. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 166, 237–243. doi: 10.1016/j.cbpa.2013.06.008
Manning R. B. (1971). Keys to the species of Oratosquilla (Crustacea, stomatopoda), with descriptions of two new species. Smithson. Contrib. Zool. 71, 1–16. doi: 10.5479/si.00810282.71
McCord J. M. (1996). Effects of positive iron status at a cellular level. Nutr. Rev. 54, 85–88. doi: 10.1111/j.1753-4887.1996.tb03876.x
Mittler R., Vanderauwera S., Suzuki N., Miller G., Tognetti V. B., Vandepoele K., et al. (2011). ROS signaling: the new wave? Trends Plant Sci. 16 (6), 300–309.
Mock T., Otillar R. P., Strauss J., McMullan M., Paajanen P., Schmutz J., et al. (2017). Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 541, 536–540. doi: 10.1038/nature20803
Moriya Y., Itoh M., Okuda S., Yoshizawa A. C., Kanehisa M. (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, 182–185. doi: 10.1093/nar/gkm321
Morozova O., Marra M. A. (2008). Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264. doi: 10.1016/j.ygeno.2008.07.001
Narbonne-Reveau K., Maurange C. (2019). Developmental regulation of regenerative potential in Drosophila by ecdysone through a bistable loop of ZBTB transcription factors. PloS Biol. 17 (2), e3000149. doi: 10.1371/journal.pbio.3000149
Nastou K. C., Tsaousis G. N., Papandreou N. C., Hamodrakas S. J. (2016). MBPpred: Proteome-wide detection of membrane lipid-binding proteins using profile hidden Markov models. Biochim. Biophys. Acta Proteins Proteom. 1864, 747–754. doi: 10.1016/j.bbapap.2016.03.015
Pigliucci M. (2005). Evolution of phenotypic plasticity: Where are we going now? Trends Ecol. Evol. 20 (9), 481–486. doi: 10.1016/j.tree.2005.06.001
Pootakham W., Uengwetwanit T., Sonthirod C., Sittikankaew K., Karoonuthaisiri N. (2020). A novel full-length transcriptome resource for black tiger shrimp (Penaeus monodon) developed using isoform sequencing (Iso-seq). Front. Mar. Sci. 7, 172. doi: 10.3389/fmars.2020.00172
Popovic I., Riginos C. (2020). Comparative genomics reveals divergent thermal selection in warm- and cold-tolerant marine mussels. Mol. Ecol. 29 (3), 519–535. doi: 10.1111/mec.15339
Pratt E. A. L., Beheregaray L. B., Bilgmann K., Zanardo N., Diaz-Aguirre F., Brauer C., et al. (2022). Seascape genomics of coastal bottlenose dolphins along strong gradients of temperature and salinity. Mol. Ecol. 31 (8), 2223–2241. doi: 10.1111/mec.16389
Ren R., Hardikar S., Horton J. R., Lu Y., Zeng Y., Singh A. K., et al. (2019). Structural basis of specific DNA binding by the transcription factor ZBTB24. Nucleic Acids Res. 47 (16), 8388–8398. doi: 10.1093/nar/gkz557
Richter K., Haslbeck M., Buchner J. (2010). The heat shock response: life on the verge of death. Mol. Cell 40, 253–266. doi: 10.1016/j.molcel.2010.10.006
Rinn J. L., Chang H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi: 10.1146/annurev-biochem-051410-092902
Rockman M. V. (2012). The QTN program and the alleles that matter for evolution: All that's gold does not glitter. Evolution 66 (1), 1–17. doi: 10.1111/j.1558-5646.2011.01486.x
Salmela L., Eric R. (2014). LoRDEC: accurate and efficient long read error correction. Bioinformatics 30 (24), 3506–3514. doi: 10.1093/bioinformatics/btu538
Sandoval-Castillo J., Robinson N. A., Hart A. M., Strain L. W. S., Beheregaray L. B. (2018). Seascape genomics reveals adaptive divergence in a connected and commercially important mollusc, the greenlip abalone (Haliotis laevigata), along a longitudinal environmental gradient. Mol. Ecol. 27 (7), 1603–1620. doi: 10.1111/mec.14526
Schoville S. D., Barreto F. S., Moy G. W., Wolff A., Burton R. S. (2012). Investigating the molecular basis of local adaptation to thermal stress: population differences in gene expression across the transcriptome of the copepod Tigriopus californicus. BMC Evol. Biol. 12, 170. doi: 10.1186/1471-2148-12-170
Sharon D., Tilgner H., Grubert F., Snyder M. (2013). A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 31, 1009–1014. doi: 10.1038/nbt.2705
Shimizu K., Adachi J., Muraoka Y. (2006). ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 4, 649–664. doi: 10.1142/S0219720006002260
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Steijger T., Abril J. F., Engström P. G., Kokocinski F., Consortium RGASP., Hubbard T.J., et al. (2013). Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 10, 1177–1184. doi: 10.1038/nmeth.2714
Storz J. F., Wheat C. W. (2010). Integrating evolutionary and functional approaches to infer adaptation at specific loci. Evolution 64 (9), 2489–2509. doi: 10.1111/j.1558-5646.2010.01044.x
Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166. doi: 10.1093/nar/gkt646
Tilgner H., Raha D., Habegger L., Mohiuddin M., Gerstein M., Snyder M., et al. (2013). Accurate identification and analysis of human mRNA isoforms using deep long read sequencing. G3 (Bethesda) 3, 387–397. doi: 10.1534/g3.112.004812
Van Haaften-Visser D. Y., Harakalova M., Mocholi E., van Montfrans J. M., Elkadri A., Rieter E., et al. (2017). Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J. Biol. Chem. 92 (19), 7904–7920. doi: 10.1074/jbc.M116.772038
Wang J., Jiang K. J., Zhang F. Y., Song W., Zhao M., Wei H. Q., et al. (2015). Characterization and expression analysis of the prophenoloxidase activating factor from the mud crab Scylla paramamosain. Genet. Mol. Res. 14 (3), 8847–8860. doi: 10.4238/2015.August.3.8
Wang X. W., Luan J. B., Li J. M., Su Y. L., Xia J., Liu S. S. (2011). Transcriptome analysis and comparison reveal divergence between two invasive whitefly cryptic species. BMC Genomics 12, 458. doi: 10.1186/1471-2164-12-458
Wang A., Sha Z., Hui M. (2022). Full-length transcriptome comparison provides novel insights into the molecular basis of adaptation to different ecological niches of the deep-sea hydrothermal vent in alvinocaridid shrimps. Diversity 14, 371. doi: 10.3390/d14050371
Wang X. W., Zhao Q. Y., Luan J. B., Wang Y. J., Yan G. H., Liu S. S., et al. (2012). Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genomics 13, 529. doi: 10.1186/1471-2164-13-529
Xu S., Wang L., Wang X. W., Zhao Y. R., Bi W. J., Zhao X. F., et al. (2014). L-type lectin from the kuruma shrimp Marsupenaeus japonicus promotes hemocyte phagocytosis. Dev. Comp. Immunol. 44 (2), 397–405. doi: 10.1016/j.dci.2014.01.016
Yan H., Cui X., Shen X., Wang L., Jiang L., Liu H., et al. (2018). De novo transcriptome analysis and differentially expressed genes in the ovary and testis of the Japanese mantis shrimp Oratosquilla oratoria by RNA-seq. Comp. Biochem. Physiol. Part D Genomics Proteomics 26, 69–78. doi: 10.1016/j.cbd.2018.04.001
Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi: 10.1093/molbev/msm088
Ye X. D., Su Y. L., Zhao Q. Y., Xia W. Q., Liu S. S., and Wang X. W., et al. (2014). Transcriptomic analyses reveal the adaptive features and biological differences of guts from two invasive whitefly species. BMC Genomics 15 (1), 370. doi: 10.1186/1471-2164-15-370
Yi S. K., Zhou X. Y., Li J., Zhang M., Luo S. (2018). Full-length transcriptome of Misgurnus anguillicaudatus provides insights into evolution of genus Misgurnus. Sci. Rep. 8(1), 11699. doi: 10.1038/s41598-018-29991-6
Young M. D., Wakefield M. J., Smyth G. K., Oshlack A. (2010). Method Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Zhang H. M., Liu T., Liu C. J., Song S., Zhang X., Liu W., et al. (2015a). AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 43, D76–D81. doi: 10.1093/nar/gku887
Keywords: marine crustacean, adaptive divergence, natural selection, expression plasticity, full-length transcriptome, seascape heterogeneity
Citation: Cheng J, Zhang L, Hui M, Li Y and Sha Z (2022) Insights into adaptive divergence of Japanese mantis shrimp Oratosquilla oratoria inferred from comparative analysis of full-length transcriptomes. Front. Mar. Sci. 9:975686. doi: 10.3389/fmars.2022.975686
Received: 22 June 2022; Accepted: 28 July 2022;
Published: 23 August 2022.
Edited by:
Jonathan Sandoval, Flinders University, AustraliaReviewed by:
Xianliang Meng, Yellow Sea Fisheries Research Institute (CAFS), ChinaZhenming Lv, Zhejiang Ocean University, China
Copyright © 2022 Cheng, Zhang, Hui, Li and Sha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongli Sha, c2hhemxAcWRpby5hYy5jbg==