 Zhibin Gan
Zhibin Gan Diana S. Jones
Diana S. Jones Xinming Liu4
Xinming Liu4 Dong Dong
Dong Dong Xinzheng Li
Xinzheng Li- 1Department of Marine Organism Taxonomy and Phylogeny, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 3Collections and Research, Western Australian Museum, Welshpool DC, WA, Australia
- 4Institute of Marine Drugs, Guangxi University of Chinese Medicine, Nanning, China
- 5College of Marine Science, University of Chinese Academy of Sciences, Beijing, China
- 6Laboratory for Marine Biology and Biotechnology, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, China
Thoracican barnacles represent a unique group that has evolved in parallel identical somatotype s (sessile, stalked and asymmetric) in both normal and chemosynthetic environments. Hydrothermal vents and methane seeps are typical extreme deep-sea chemosynthetic habitats for marine macrobenthos. Characterizing the evolutionary history and adaptive strategy of barnacles is fundamentally important for understanding their origin, speciation, and diversification. Herein, we performed a series of phylogenetic analyses focusing on the mitochondrial genomes of the main extant barnacle lineages. Phylogenetic inferences and topology tests contradict the view of the sister relationship between verrucomorphs and balanomorphs, instead revealing that pollicipedids, calanticids and balanomorphs share common ancestor. Selective pressure analyses indicate that the two barnacle lineages of chemosynthetic ecosystems exhibit similar patterns in their evolution of adaptive characters, but have diverse and specific positive substitution sites of mitogenomes. Divergence times suggest that chemosynthetic barnacles originated in the Cenozoic, coinciding with the origins of other metazoan animals in chemosynthetic habitats as well as the Paleogene mass extinction and oceanic anoxic events. It is reasonable to suppose that ecological niche vacancy, sitotaxis, gene specificity in adaptive stress responses, and the subdivision of the ecological niche contributed to the origin and diversification of barnacles in chemosynthetic ecosystems.
Introduction
The deep-sea ecosystem is generally characterized by high hydrostatic pressures, darkness, hypoxic, low temperatures, and limited food resources (Danovaro et al., 2014). Typical chemosynthetic habitats, such as deep-sea hydrothermal vents and cold seeps, sustain highly productive biocoenoses (Grassle, 1985; Zhao et al., 2020), despite these environments having higher levels of hydrogen, sulfide, methane, anoxia, heavy metals and other suffused chemicals than the surrounding deep-sea environments (Le Bris et al., 2017). Mounting evidence indicates that some modern metazoan species in chemosynthetic ecosystems radiated during the Cenozoic; examples include mytilid mussels (Lorion et al., 2013), alvinocarid shrimp (Yang et al., 2013), and siboglinid tubeworms (Chevaldonné et al., 2002). These findings challenge the traditional hypothesis that chemosynthetic species represent relic taxa (Little and Vrijenhoek, 2003). Nevertheless, the factors and mechanisms shaping their speciation, diversification and evolution remain unclear. Barnacles (Cirripedia: Thoracica) are not only organisms that are distributed throughout normal and chemosynthetic environments (e.g., neolepadoids, probathylepadids, and Eochionelasmus are found in chemosynthetic environments (Herrera et al., 2015; Ren & Sha, 2015)), but also represent a distinct group that have convergently evolved into various somatotypes (i.e., sessile, stalked, and asymmetric somatotypes) in both photosynthesis-supporting and chemosynthesis-supporting biotopes. Barnacles represent excellent models for evolutionary and ecological studies in the oceans, as most of their adults are almost permanently attached to the substrate, and confined in relatively narrow marine zones. Nonetheless, no studies to date have investigated the molecular adaptive characters of these sessile taxa surviving in the severe deep-sea chemosynthetic habitats, although a variety of potential mechanisms were detected in studies of organisms that thrive in harsh deep-sea habitats (Lan et al., 2017; Wang et al., 2019; Gan et al., 2020a).
Since at least Darwin time, the phylogenetic history of thoracican barnacles has been recognized as both fascinating and controversial, owing to their unique configuration, morphological diversity, convergent evolution, and ubiquity in ocean habitats (Newman & Ross, 1976; Jones, 2012; Gan et al., 2020b). However, the origin, diversification and other evolutionary process of barnacles have remained poorly understood, especially at higher taxonomic levels (Newman, 1987; Newman & Yamaguchi, 1995; Buckeridge, 1996; Gale, 2015). The main issues concern the evolution of the peduncle, asymmetry, and the origin of the chemosynthetic group. Several different evolutionary scenarios have been proposed on the basis of resemblant morphological features, ontogeny, and the fossil record. The most influential hypotheses have been those of the polyphyletic evolution (Pilsbry, 1916; Utinomi, 1968; Ghiselin & Jaffe, 1973; Newman & Ross, 1976; Newman, 1982) and monophyletic evolution (Newman & Yamaguchi, 1995; Buckeridge, 1996) of sessile and asymmetric barnacles. The latter was deemed more plausible on the basis of the barnacles (Neoverruca and Neobrachylepas) found in the hydrothermal vents; nonetheless, this hypothesis was soon rejected through the establishment of modern molecular phylogeny (Pérez-Losada et al., 2008; Linse et al., 2013; Rees et al., 2014; Herrera et al., 2015; Lin et al., 2015; Kim et al., 2022) as well as paleontological evidence (Gale, 2014a; Gale, 2014b; Gale & Sørensen, 2014). Building on these molecular results, as well as morphological and paleontological arguments, Chan et al. (2021) recently documented the thoracican phylogenetic tree (see Figure 10 in Chan et al., 2021), and proposed a comprehensive taxonomic framework for barnacles. This was the first attempt to integrate, update, and revise the taxonomy of barnacles since the work of Martin & Davis (2001). Nonetheless, some evolutionary issues have remained unresolved. As mentioned above, the findings of previous molecular researches (Spears et al., 1994; Harris et al., 2000; Pérez-Losada et al., 2004; Pérez-Losada et al., 2008; Pérez-Losada et al., 2012; Linse et al., 2013; Pérez-Losada et al., 2014; Rees et al., 2014; Herrera et al., 2015; Lin et al., 2015) significantly challenged the classical view of barnacle evolution, but some relationships for higher taxa remain unresolved due to their low confidence values, or inconsistent topology (Chan et al., 2021). This is possibly because all these analyses employed few, identical, or similar molecular markers (i.e., mostly 18S, 28S, H3 and COI). The use of mitogenomes has great advantages, owing to their universal homology, low recombination and various genome-level characters, in evolutionary analyses of organisms (Larget et al., 2002; James et al., 2016; Tsang et al., 2017; Kong et al., 2020; Bartáková et al., 2021).
Due to next-generation sequencing technology, the complete mitochondrial genome can now be more easily and precisely captured. In this study, we first sequenced and annotated the complete mitogenomes of 13 barnacle species from various families and habitats. Next, by integrating the data with publicly available mitogenome data on GenBank (http://www.ncbi.nlm.nih.gov/genbank), we performed a comprehensive mitogenomic analysis of barnacles. The main objectives were to explore the evolutionary history of major taxa within the barnacle lineage, to characterize the genetic adaptive features of chemosynthetic barnacles living in harsh marine environments, and to understand how the origin and diversification of chemosynthetic barnacles were associated with historical ecological events and other factors.
Materials and methods
Sample collection, DNA extraction and sequencing
Individual barnacles were collected in the Indian Ocean and the West Pacific from habitats such as mid-oceanic ridges, basins, trenches, hydrothermal vents, seamounts, shelf seas, and tidal zones. Of these, the deep-sea samples were collected during multiple expeditions led by the Institute of Oceanology Chinese Academy of Sciences, the Second Institute of Oceanography Ministry of Natural Resources, the Institute of Deep-sea Science and Engineering Chinese Academy of Sciences and Tongji University. The samples were obtained using a variety of remotely operated vehicles and manned submersibles. All samples were preserved in either 75% ethanol or at -80°C, and deposited in the Marine Biological Museum Chinese Academy of Sciences, Qingdao, China, and the Sample Repository of the Second Institute of Oceanography Ministry of Natural Resources, Hangzhou, China (Table 1).

Table 1 Collection and mitogenome information for the newly sequenced specimens used in this study.
Total genomic DNA was extracted from pedunculate or abdominal muscle using a modified cetyltrimethylammonium bromide method (Allen et al., 2006), and a 500-bp paired-end library was constructed using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, Ipswich, United States). Next-generation sequencing was performed on the Illumina NovaSeq 6000 platform (BIOZERON Co., Ltd, Shanghai, China).
Mitogenome assembly and annotation
The raw data, 150 bp paired-end reads, were filtered by removing the following: reads with adaptors, reads with a quality score of below 20 (Q<20), reads containing a percentage of uncalled bases (“N” characters) that was equal to or greater than 10%, and duplicated sequences. A combination of de novo and reference-guided assemblies was used to reconstruct the mitochondrial genome. The filtered reads were first assembled into contigs using GetOrganelle v1.6.4 (Jin et al., 2020). Next, the contigs were aligned with mitogenomes of closely related barnacle species using BLAST searching, and were also optimized using scaffolds from SPAdes 3.13.0 (Bankevich et al., 2012). Finally, the assembled sequence was reordered and oriented according to the reference mitochondrial genomes, thus generating the final assembled mitochondrion genomic sequence.
The assembled mitogenomes were annotated using the online MITOS tool (http://mitos.bioinf.unileipzig.de). The default parameters were used to predict protein coding genes (PCGs), transfer RNA (tRNA) genes, and ribosome RNA (rRNA) genes. These genes were further verified using the Open Reading Frame Finder on NCBI (https://www.ncbi.nlm.nih.gov/orffinder), tRNAscan-SE 2.0 (Chan & Lowe, 2019) and rRNAmmer 1.2 (Lagesen et al., 2007), respectively. Manual corrections of genes for start/stop codons were performed in SnapGene Viewer (https://www.snapgene.com) by referencing the reference mitochondrial genome. The circular mitochondrial genome map was drawn using the CGview Server (Grant & Stothard, 2008).
Phylogenetic analyses
The analyses were achieved using the mitogenomes of the 13 species obtained in the present study, as well as the open-access mitogenome datasets of other barnacle species, including an iblomorph, three asymmetrical barnacle species (including one chemosynthetic species), 17 stalked barnacle species (including three chemosynthetic species), and 31 acorn barnacle species (including two chemosynthetic species). A first-pass phylogenetic inference based on all the functional genes, (i.e., concatenated 13 PCGs as well as 22 tRNA and two rRNA sequences) was conducted using the Flowchart analyses of PhyloSuites v1.2.2 (Zhang et al., 2020) with the maximum-likelihood optimality criterion. However, this produced a phylogenetic tree that was poorly supported, possibly owing to noise from gene heterogeneity and mutational saturation. Next, we retained only the 13 PCGs. An inspection of the substitution status of these 13 PCGs using DAMBE v5.3 (Xia & Xie, 2001) indicated that all had mutational saturation at the third codon. Thus, we excluded the third codons in subsequent analyses on the concatenated nucleotide data. Bayesian inference (BI) and maximum likelihood (ML) methods were used to analyze the data from both the concatenated nucleotide and the putative amino acid sequences of the 13 PCGs.
Nucleotide and amino acid sequences of each 13 PCGs were aligned using MAFFT v7 (Katoh & Standley, 2013) with default parameters, and the nucleotide sequences were aligned in codon manner. MACSE v2 (Ranwez et al., 2018) and Gblocks (Talavera & Castresana, 2007) were used to optimize alignments and to eliminate segments that were divergent and ambiguously aligned. Finally, the trimmed nucleotide and amino acid sequences were concatenated into independent dataset respectively. The best partitioning schemes and best-fit substitution models of each partition in the concatenated dataset were determined using ModelFinder (Kalyaanamoorthy et al., 2017) and PartitionFinder 2 (Lanfear et al., 2017) according to the Bayesian information criterion. All the above analyses were performed using the integrated and scalable desktop platform PhyloSuites v1.2.2. The ML tree was reconstructed using RaxML-NG (Kozlov et al., 2019), and branch support was assessed by transfer bootstrap expectation (TBE) with 1,000 replicates (Lemoine et al., 2018). The Bayesian analysis was conducted using a parallel version of Mrbayes v3.2.6 (Ronquist et al., 2012). Two independent runs were executed with four Markov Chain Monte Carlo (MCMC) for 10 million generations (beginning with a random tree, chains were sampled every 10,000 generations). Convergence of the analyses was ensured by monitoring the likelihood values and the standard deviation of partition frequencies (<0.01). The resultant outputs were further validated by using Tracer 1.7.1 (Rambaut et al., 2018) to diagnose the effective sample size (ESS) values of all sampled parameters. The first 25% of trees generated prior to the achievement of stationarity of the log-likelihood values were discarded as burn-in. The remaining trees were used to construct a 50% majority rule consensus tree and to estimate the posterior probabilities (PP) simultaneously. FigTree (http://tree.bio.ed.ac.uk/software/figtree/) was used to visualize the phylogenetic trees. Linearized mitochondrial gene arrangement patterns were superimposed on the phylogenetic trees using the iTOL webserver (Letunic & Bork, 2021).
To test the hypothesis that sessile barnacle species (i.e., verrucomorphs and balanomorphs) form a monophyletic group, we performed a likelihood-based approximately unbiased (AU) test in IQ-TREE v2.0 (Nguyen et al., 2015) with 1,000 replicates to assess the P-value of the test topology.
Divergence time estimation
The divergence times of barnacle lineages were estimated from the partitioned nucleotide dataset using BEAST v2.6.3 (Bouckaert et al., 2014). Variation in mutation rates amongst branches was allowed by assuming an uncorrelated relaxed lognormal molecular clock model. The birth-death model (Gernhard, 2008) was set as the tree’s priors. Unlinked site models were set for each partitioned dataset to allow independent substitution rates and base frequencies, and determined by bModelTest (Bouckaert & Drummond, 2017) implemented in the BEAST v2.6.3. Six fossil calibration points [C1: Praelepas jaworskii, 306.5–311.7 million years ago (MYA); C2: Calantica (Scillaelepas) ginginensis, 83.5–85.5 MYA; C4: Arcoscalpellum fossula, 70.6–89.3 MYA; C5: Pollicipes aboriginalis, 83.5–85.8 MYA; C9: Pachydiadema (Catophragmus) cretacea, 70.6–89.3 MYA; C11: Tetraclitella sp. cf. purpurascens, 20.4–23.0 MYA] were selected to calibrate the divergence time estimation; all points had been tested with high confidence in previous studies (Pérez-Losada et al., 2008; Linse et al., 2013; Herrera et al., 2015). Fossil ages were used as lower boundary constraints, and their prior exponential distribution were assumed with estimated mean values. The default prior distribution settings were assumed for all other parameters. The MCMC analysis was run for 10 million generations with a sampling frequency of 10,000 generations. Convergence diagnosis was examined by Tracer. After the first 25% of trees were discarded as burn-in, the maximum clade credibility tree with median nodal height was generated using TreeAnnotator 2.6.3 (Bouckaert et al., 2014). The time-scaled tree was mapped against geological time and visualized using the R packages phytools (Revell, 2012), PHYLOCH (http://www.christophheibl.de/Rpackages.html), strap (Bell & Lloyd, 2015), and CODA (Plummer et al., 2006).
Selective pressure analyses
Signatures of natural selection on the 13 PCGs were detected using the codon-based maximum likelihood (CodeML) implemented in the PAML 4 package (Yang, 2007), based on the tree that was reconstructed with the concatenated amino acid sequences. Both branch and branch-site models were used to identify the variation in selective pressures acting on the branch lineages and individual sites of PCGs. For the branch model, a one-ratio model and a two-ratio model were used to estimate the ω values across the whole lineage and foreground lineages (i.e., the chemosynthetic barnacles). The branch-site model assumed ω values to be alterable among sites in the proteins as well as across branches on the tree, and could detect any positive selection affecting a few sites along the foreground branches. Bayes Empirical Bayes (BEB) analysis was used to calculate the Bayesian posterior probability of the positively selected sites (ω>1). All the assumed models were compared against their corresponding null hypotheses using the Likelihood Ratio Test (LRT); the LRT P-values were then calculated using a Chi-square (χ2) analysis. Models with LRT P-values less than 0.05 and BEB larger than 95% were considered as the best-fit models for evolution and positive selection.
To achieve a more intuitive understanding of the significance of the sites under positive selection, we mapped the sites onto the three-dimensional (3D) structure of mitochondrial proteins. The SWISS-MODEL (Waterhouse et al., 2018) server and I-TASSER (Yang et al., 2015) server was used to model the 3D structure of the target proteins based on the sequence reconstructed using the PAML 4 package. Next, PyMOL (The PyMOL Molecular Graphics System, V2.5, Schrödinger, LLC.) was used for visualizing and depicting the positive selection sites in the proteins.
Results
The mitogenomes of 13 barnacle species (including three hydrothermal vent barnacles and two asymmetrical barnacle species) were successfully recovered. Of these, the mitogenome of Regioscalpellum regium was partially recovered with gaps in the putative control regions, and that of Octolasmis warwickii was partially recovered with gaps between the tRNA-P and tRNA-T genes. The lengths of complete mitogenomes were between 14,910 (Calantica studeri) and 20,032 bp (Ashinkailepas seepiophila). Nearly all mitogenomes contained the typical 13 PCGs, two rRNAs, and the 22 tRNAs, except for Glyptelasma gigas, which was missing a tRNA-Q gene. The annotated sequences of mitogenomes are stored in GenBank (Table 1), and the mitochondrial genome maps are shown in the Figure 1.
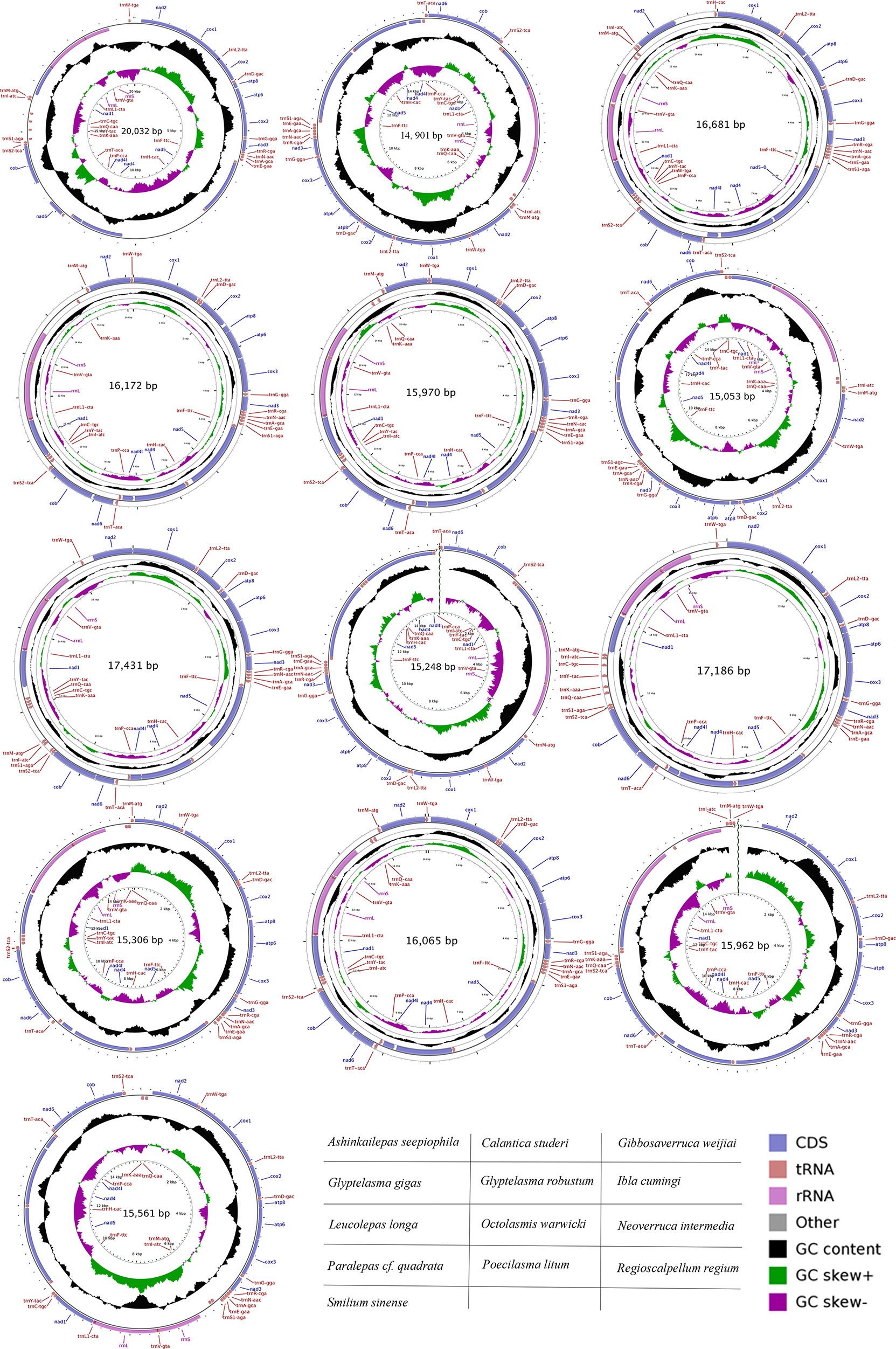
Figure 1 The organization of the mitogenomes of the newly sequenced specimens used in this study.
Phylogenetic analyses
The final aligned datasets for nucleotide and amino acid sequences consisted of 7,178 bp and 3,563 characters, respectively. The best partitioning schemes and best-fit substitution models as determined by ModelFinder and PartitionFinder are listed in the Supporting Information, Supplementary Table 1. Tree topologies resulting from the BI and ML analyses were identical for the same dataset, and only a few weakly supported internal nodes of Balanidae and Calanticidae were different between the two datasets. Furthermore, the topology that was revealed from the amino acid dataset (Figure 2) received higher nodal support than that from the nucleotide dataset (Supplementary Figure 1). But consistently, all orders received monophyletic support, and all families formed a monophyletic group except Balanidae in the phylogenetic analyses. Fortuitously, a conflict between the topologies of the nucleotide and amino acid datasets was found in the position of Smilium sinense. This species was recovered as a sister group of verrucids in the nucleotide topologies, which contrasted its coordinal species Calantica studeri in the amino acid topologies as well as in the topology generated by all the functional genes. Furthermore, the nodal support for S. sinense in nucleotide topologies was very poor (PP=0.55, BP=59) (Supplementary Figure 1). Comparisons across different barnacle species revealed that they have a highly conserved gene order that corresponded to their families, except for Chthamalidae and Calanticidae, which displayed conspicuous translocations or inversions of some gene blocks (Figure 3).
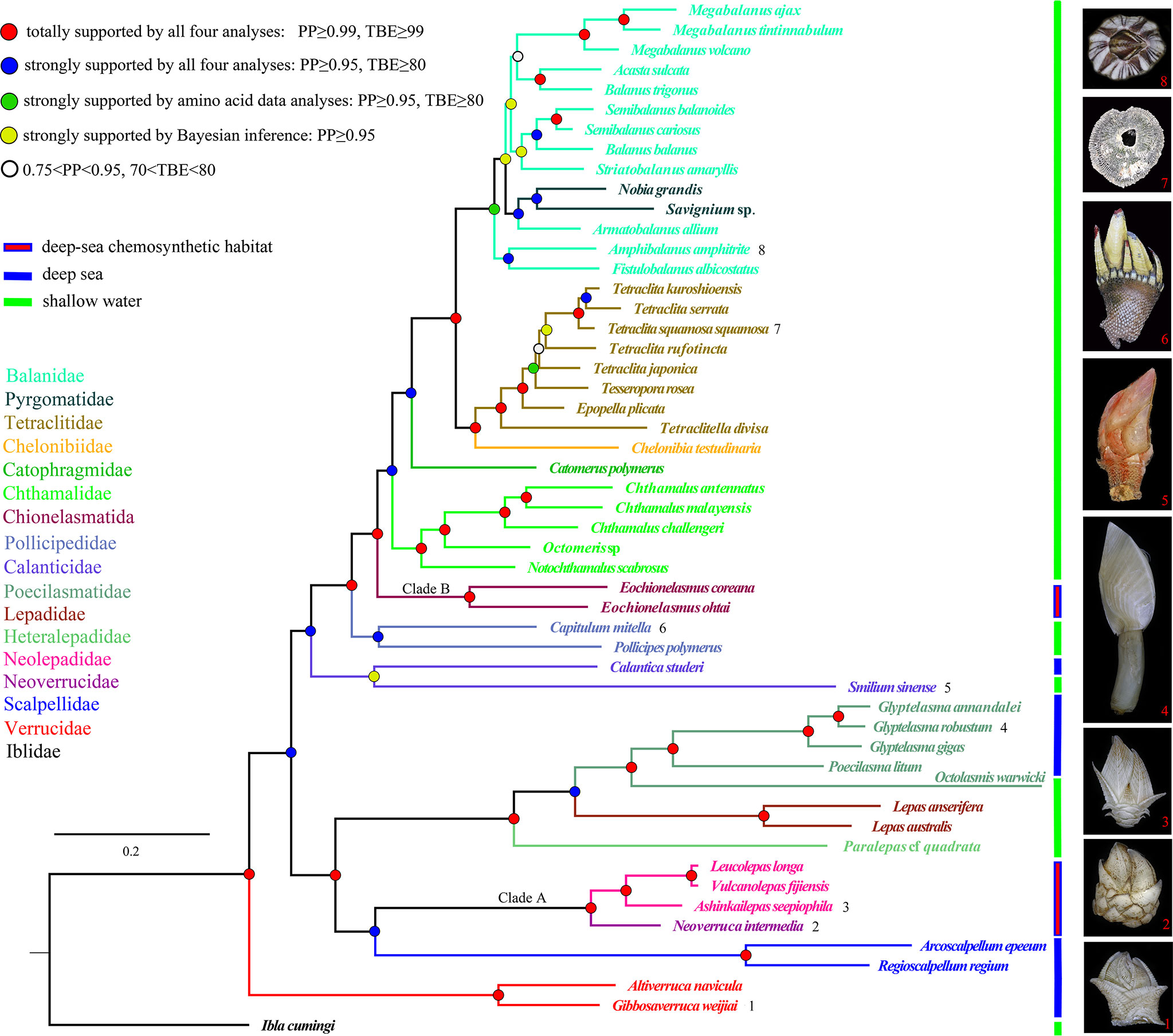
Figure 2 Phylogenetic topology inferred from the combined amino acid sequences of the complete mitogenomes of barnacles. Clades A and B indicate the two chemosynthetic barnacle lineages. The color of the species name and branch denotes the family classification of the taxon. The digits after the species name correspond to sample photos. Posterior probabilities (PP) and transfer bootstrap expectation (TBE) values from both the nucleotide and amino acid data are annotated using different colored dots.
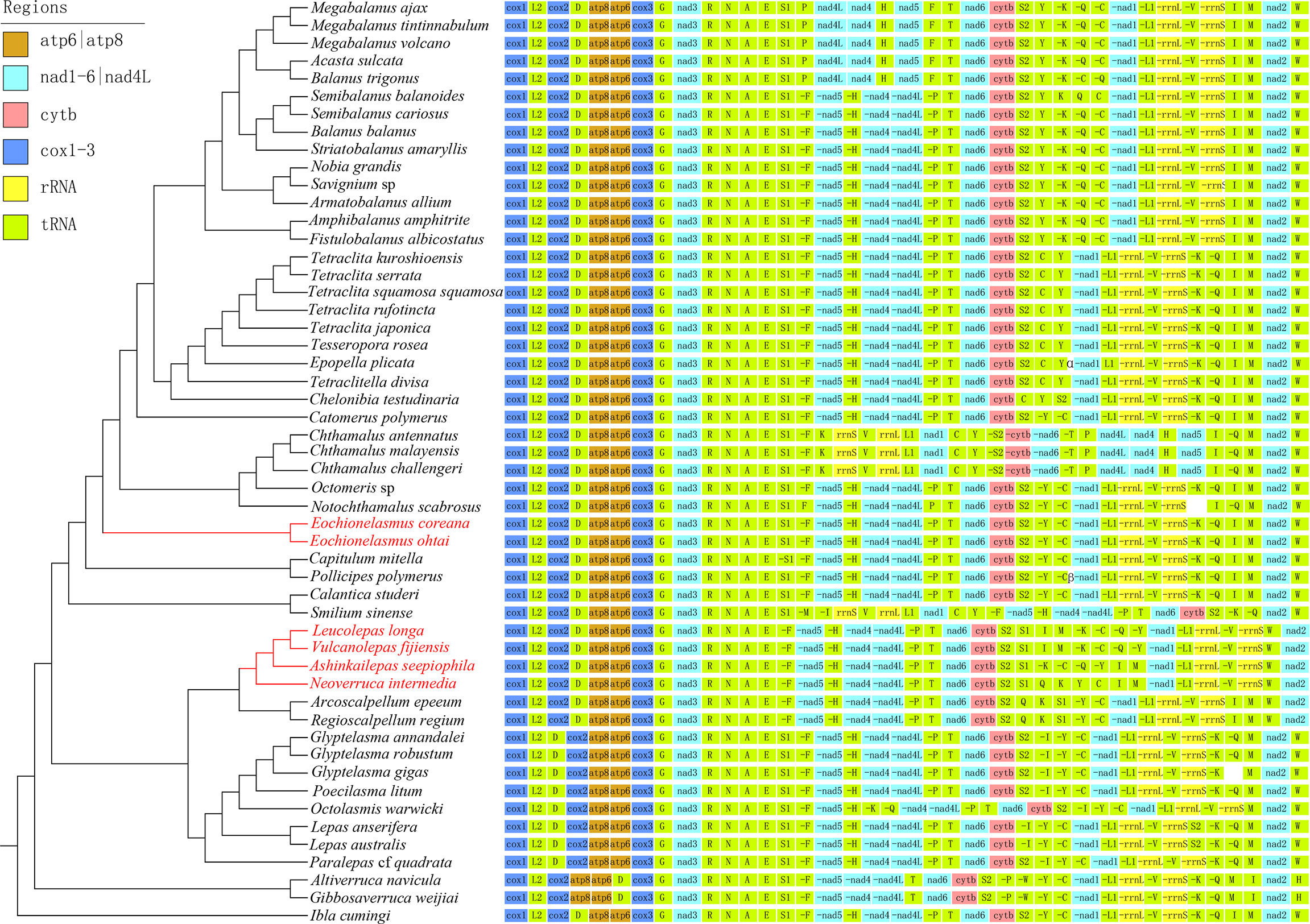
Figure 3 Mitogenomic gene arrangement patterns of barnacles superimposed on the phylogenetic tree. Genes encoded by the light-strand are initiated by a “-”. The symbol “α” represents the heavy-strand gene block “S2–C–Y”, while “β” represent the light-strand gene block “C–C”.
Most of the phylogenetic relationships among species recovered from the present analyses were consistent with the results of previous studies based on multilocus molecular markers (Pérez-Losada et al., 2008; Linse et al., 2013; Rees et al., 2014; Herrera et al., 2015; Lin et al., 2015). Barnacles from deep-sea chemosynthetic ecosystems can be divided into two strongly supported clades (PP=1, BP=100). Clade A, containing Ashinkailepas seepiophila, Leucolepas longa, Neoverruca intermedia and Vulcanolepas fijiensis, was well supported as the sister group to the scalpellids clade. Clade B, containing Eochionelasmus coreana and E. ohtai, was strongly supported as the basal clade of the balanomorphs (Figure 2, Supplementary Figure 1). This result was highly congruent with the findings of Herrera et al., (2015), who suggested that deep-sea chemosynthetic barnacles have colonized hydrothermal vents and/or cold seeps at least twice in the history of thoracican evolution. The verrucomorph clade occupied the basal position in the topologies of all the trees recovered (Figure 2; Supplementary Figure 1), and the hypothesis that sessile barnacles (verrucomorph and balanomorph) form a monophyletic group was clearly rejected by the AU tests (P-value=3.73e-07 in the amino acid dataset, P-value=8.67e-06 in the nucleotide dataset).
Divergence time
The topology of the tree reconstructed from the divergence time estimation by BEAST2 was nearly identical to the topologies of trees recovered from the BI and ML analyses based on the nucleotide and amino acid data. Minor differences also appeared in a few shallow nodes of the Balanidae and Tetraclitidae. Divergence times of the barnacle lineages estimated under the prior settings are shown in Figure 4. Nearly all the node times lay between the corresponding values estimated by Linse et al. (2013) and Herrera et al., (2015). The occurrence time of the most recent common ancestor (MRCA) of Clade A was estimated at 46.8 million years ago, with the 95% highest posterior density interval (HPD) occurring at 25.5–74.1 MYA. This clade diverged from its sister clade at 168.4 MYA (95% HPD: 126.3–220.4). Within Clade A, the origin of the Neolepadidae was estimated to have occurred 29.3 MYA (95% HPD: 13.7–49.2). The MRCA of Clade B was older than that of Clade A, had occurred 65.3 MYA (95% HPD: 34.6–101.7), where it split from other balanomorphs at 124.6 MYA (95% HPD: 91.2–158.9). In general, the extant deep-sea chemosynthetic barnacles most probably originated from the early Paleogene or upper Cretaceous, and speciated during the subsequent periods.
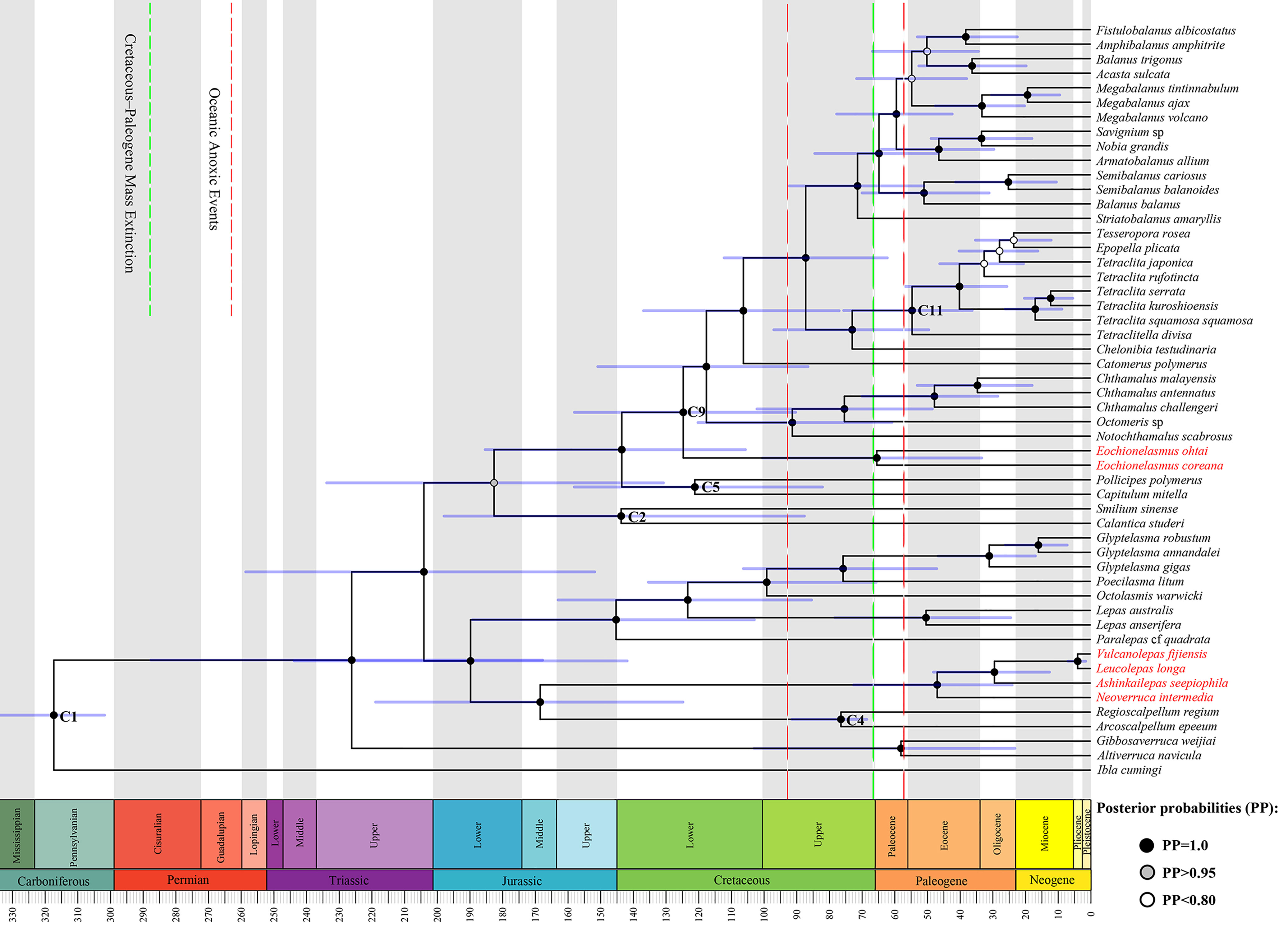
Figure 4 Maximum clade credibility time-scaled tree generated under the birth-death model based on the partitioned protein coding genes of barnacle mitogenomes. Fossil calibration points (C1, 2, 4, 5, 9, and 11) follow those used by Pérez-Losada et al. (2008) and Herrera et al., (2015). Species highlighted in red belong to the two chemosynthetic barnacle lineages.
Positive selection
To test if any selective pressure occurred specifically in chemosynthetic barnacle lineages, we compared the alternative model (a two-ratios model, with different lineages assigned different ω values) and the null model (a one-ratio model, with all lineages assigned the same ω value) by labelling the foreground branches in two different ways. The first involved labelling the entire clades of Clade A and Clade B respectively; the second involved labelling the individual branches of Clade A and Clade B. Both approaches produced very significant results (P<10-8) (Table 2). The results also showed that chemosynthetic barnacle species had a higher ω value than other barnacle species (Table 2, Figure 5), suggesting they had accumulated more nonsynonymous substitutions throughout their evolution and their adaptation to extreme environments. Notably, the ω values of the common ancestor to Clade A and Clade B were distinctly high. This could indicate that the ancestors of chemosynthetic barnacles had undergone relatively high selective pressures when they colonized the chemosynthetic biotope during their emergence.
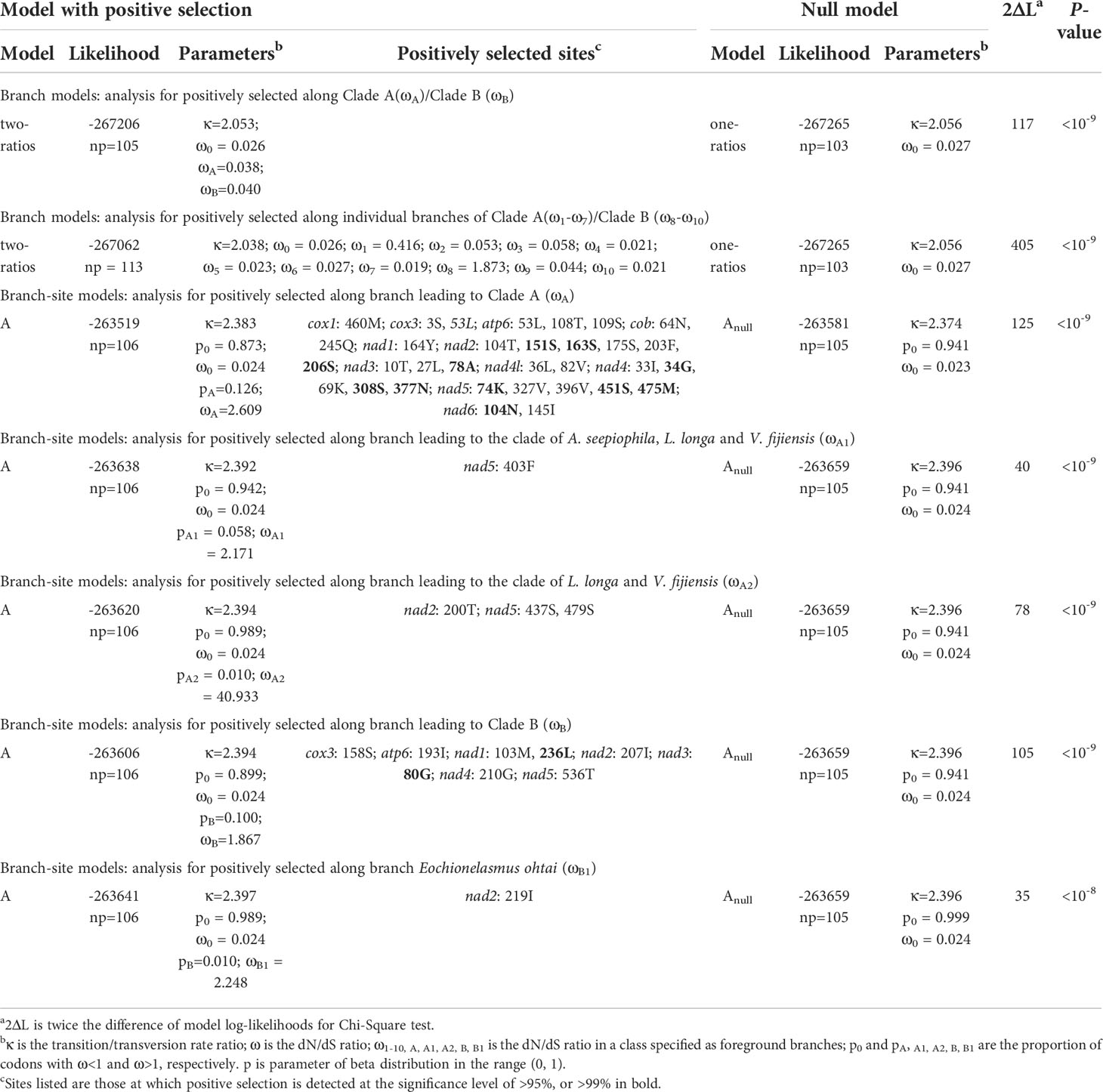
Table 2 Results of models of selective pressure acting on deep-sea chemosynthetic barnacles.
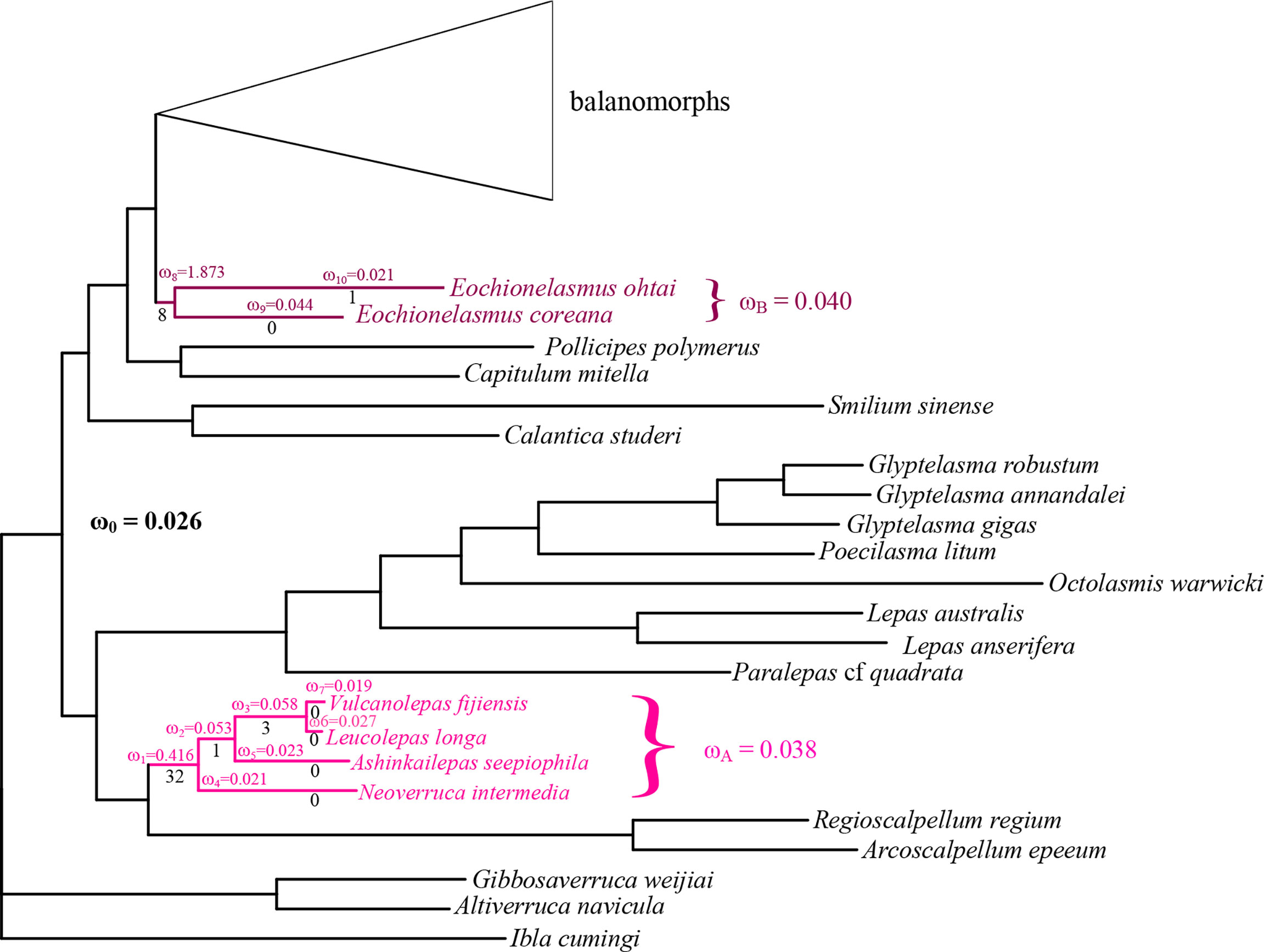
Figure 5 Barnacle tree (based on amino acid sequences) annotated with selective pressure. Different colors indicate different chemosynthetic barnacle lineages. ω is the ratio of non-synonymous to synonymous substitutions with a different branch or lineage. The number of adaptive substitutions (determined by PAML) is shown below each branch.
Studies suggest that positive selection always occurs episodically at a few sites or lineages in response to variation of the surrounding environment (Yang, 2007; Shen et al., 2010). Thus, we performed a series of branch-site model tests (ω>1 along the foreground branch vs. ω≤1 along the foreground and background branches) among individual branches of Clade A and Clade B. However, only the results of tests along branches leading to Clade A, Clade B, internal branches of Clade A and the Eochionelasmus ohtai branch received statistically significant support (P<0.05) (Table 2). This pattern reflected the pattern observed in the results of the branch model analysis. BEB analysis showed that positive selection sites occurred in all mitochondrial PCGs of Clade A, except in cox2 and atp8. In contrast, for Clade B, positive selection sites occur in cox3, atp6, nad1, nad2, nad3, nad4 and nad5. No common positive selection site between the two lineages was found (Table 2). The predicted 3D structure of the proteins revealed that these positive selection sites appeared as both conformation of α-helix structures and random coils (Supplementary Figure 2).
Discussion
Phylogeny and evolutionary history of barnacles
Our findings pertaining to barnacle evolutionary history largely corroborated those of previous studies, particularly with regards to the independent origins of the two chemosynthetic barnacle lineages. However, our findings also suggested alternative evolutionary scenarios for other thoracicans. Most apparent among these was the phylogenetic position of verrucomorph barnacles. Our analysis indicated that verrucomorphs were basal to all barnacles except iblomorphs (Figure 2; Supplementary Figure 1). This partly corresponded to the initial speculation that verrucomorphs and balanomorphs were of independent origins (Pilsbry, 1916; Newman & Ross, 1976). The sister relationship between verrucomorphs and balanomorphs was clearly rejected by the monophyletic test in our analysis. It had also not been confirmed in the mitochondrial genome analyses of Kim et al. (2018); Kim et al., (2019) and Lee et al. (2019), or the multiple gene analyses of Linse et al. (2013); Rees et al. (2014), Herrera et al., (2015), and Kim et al. (2022). Furthermore, the mitogenome arrangement of verrucomorph species, which displayed unique gene blocks (Figure 3), was distinctly different from that of the balanomorphs and other barnacles. The ontogeny of verrucomorph species (Verruca stroemia) also differed from that of balanomorph species (Semibalanus balanoides) owing to a lack of lateral plates in individual development (Newman, 1989). Although limited evidence of morphology and fossil records to fully support current phylogenetic result, there were some clues to speculate that verrucomorphs might originate from archaeolepadomorphs. Archaeolepadomorphs (such as Archaeolepas and Bosquetlepas) and verrucomorphs only possess six plated capitula (i.e., the paired scuta and terga, a carina, and a scutum) and lack any lateral plates, the arrangement thus provides the most parsimonious evolutionary interpretation. Moreover, our analysis indicates that the origin of verrucomorphs is in the upper Triassic (Figure 4), which corresponds with the divergence time of the archaeolepadomorphs indicated by Chan et al. (2021). However, considering that incongruent phylogenetic position of verrucomorphs was present in molecular phylogeny. The natural evolutionary history of verrucomorphs must be further verified with more evidence from added data analysis.
Another controversial issue in thoracican phylogeny concerns the calanticids and pollicipedids, which were traditionally placed in Scalpelliformes, until Chan et al. (2021) proposed the orders Calanticomorpha and Pollicipedomorpha for them, however, their phylogenetic status is uncertain owing to the incongruent results of separate molecular analyses. (Pérez-Losada et al., 2008; Linse et al., 2013; Rees et al., 2014; Lin et al., 2015; Herrera et al., 2015). Our mitogenome data indicate that the pollicipedids diverged immediately before the balanomorphs, and were followed closely by the calanticids (Figure 2, Supplementary Figure 1) with robust support. These findings coincide with the Bayesian results of Linse et al. (2013), as well as the relatively constant position occupied by the pollicipedids in all mitochondrial genome-based phylogenetic analyses (Song et al., 2017; Cai et al., 2018; Kim et al., 2018; Kim et al., 2019; Chen et al., 2019; Lee et al., 2019; Tian et al., 2020). The mitogenome arrangement of calanticids and pollicipedids more closely resembles the patterns of balanomorphs, except for Smilium sinense which shows remote inversion events of the gene blocks, C–Y–nad1–L1–rrnL–V–rrnS and I–M (Figure 3). Anderson (1983) demonstrated that the evolution of balanomorphs from calanticids was functionally feasible. Gale (2014b) indicated that Etcheslepas was morphologically intermediate between Capitulum and Pycnolepas, which suggested that pollicipedomorphs and brachylepadomorphs shared a close relationship. He further proposed a phylogenetic hypothesis for balanomorphs based on their morphology and paleontology, with calanticomorphs and pollicipedomorphs located at the basal position (Gale & Sørensen, 2014). In the morphometric analyses on antennule of cyprid larvae, Al-Yahya et al. (2016) showed similar structures among pollicipedomorphs and balanomorphs. Collectively, the evidence indicates that balanomorphs, pollicipedomorphs and calanticomorphs might share a common ancestor before radiating into the respective lineages observed today.
Origin and adaptive evolution to extreme deep-sea environments
Our result indicates the chemosynthetic barnacles diverged from the early Cenozoic (Figure 4). A similar divergence time has been proposed by Linse et al. (2013) and Herrera et al. (2015), and the divergence time of neolepadoids is also in accordance with the fossil records (Carriol et al., 2016; Gale et al., 2020). During the same period, other marine chemosynthetic taxa also emerged and radiated. For instance, mytilid mussels 45 MYA (Lorion et al., 2013), alvinocarid shrimp 48.4–55.9 MYA, bythograeid crabs 51.5–69.7 MYA (Yang et al., 2013) and siboglinid tubeworms 60 MYA (Chevaldonné et al., 2002). These divergence times occurred just posterior to the Cretaceous-Paleogene mass extinction (K-Pg extinction), which caused a near 50% decline in marine generic diversity (Sepkoski, 1996; D’Hondt, 2005) and over 50% of cirripede genera extinction (Gale & Sørensen, 2014). Jacobs & Lindberg (1998) have proposed that the oceanic anoxic/dysoxic events occurring at the Cenomanian/Turonian boundary as well as the latest Paleocene led to the elimination of much of the contemporary deep-sea benthos, as well as vent faunas; this argument has also been supported by Little & Vrijenhoek (2003). Accordingly, an abundance of empty ecological niches would have been created following these mass extinctions and anoxic/dysoxic events. Hsü et al. (1982); Zachos et al. (1989), and D’Hondt (2005) showed that the flux of organic matter to the deep seafloor declined tremendously during the K-Pg extinction. All these ecological events may have driven food scarcity among the contemporary deep-sea benthos. Hence, it is possible that the colonization of deep-sea chemosynthetic habitats by chemosynthetic animals was promoted by ecological niche vacancy and food scarcity. Furthermore, hydrothermal vents or cold seeps are widespread, energetic and highly productive ecosystems supported by chemoautotrophic microbes rather than by the sinking of photosynthesis-based organics (Grassle, 1987; Little & Vrijenhoek, 2003; Baker et al., 2016). Nonetheless, these extreme habitats are also characterized by hypoxic, chemical release, and more extreme temperature gradients in comparison with the surrounding deep-sea environments (Hourdez & Lallier, 2007; Le Bris et al., 2017). These conditions may have promoted the development of specialized adaptations in the genomes of chemosynthetic species.
Hypoxic, sulfide, methane and heavy metals suffused in the chemosynthetic ecosystem are toxic for the mitochondrial electron transfer chain, and can inhibit aerobic metabolism (Grieshaber & Völkel, 1998; Belyaeva et al., 2008). Mitogenome encode key enzymes involved in the process of aerobic respiration. Changes in survival environments will create selective pressures on genome to confront alternative environmental conditions, and fix nonsynonymous mutations as adaptive substitutions for species to cope with the new demands. Our results show that the mitogenomes of chemosynthetic barnacles encountered relatively high selective pressures, especially in ancestral taxa, which were the first to colonize chemosynthetic habitats (Figure 5, Table 2). Remarkably, the ω value of the ancestor of Clade B (Eochionelasmus) is higher than that of Clade A (neolepadoids). It is possible that the MRCA of neolepadoids was already a deep-sea dweller before it colonized deep-sea chemosynthetic habitats (Figure 2; Linse et al., 2013; Herrera et al., 2015). In contrast, Eochionelasmus is nested within the shallow-water clade (Figure 2; Pérez-Losada et al., 2014), indicating that its ancestor invaded into deep-sea chemosynthetic habitats directly from shallow waters. This means that the MRCA of Eochionelasmus likely encountered more selective constrains than that of the neolepadoids. This may also have accounted for the apparent differences in species diversity between the two chemosynthetic barnacle lineages (i.e., three species vs. over 15 species). Positive selection in specific sites of a functional protein is typically taken as evidence of an adaptive substitution responding to environmental changes (Yang & Nielsen, 2002; Rausher & Huang, 2016). The branch-site model analysis obtained a similar pattern as that from the branch model analysis, most of the adaptive substitutions occurred in their MRCAs of chemosynthetic barnacles (Figure 4, Table 2). The 3D structure shows that these adaptive substitutions are scattered throughout both α-helix and random coil conformations, and that some sites are located precisely in the transformation point of these two conformations, such as 245Q of the Cytb in the neolepadoid lineage, and 210G of the Nad4 of the Eochionelasmus lineage (Supplementary Figure 2). Meanwhile, some amino acids experienced radical substitutions in their physicochemical properties. For instance, the 104th site of the neolepadoids’ Nad2 arose as a substitution of a hydrophobic and nonpolar Valine with a hydrophilic and polar Threonine. These substitutions would have caused dramatic changes to protein conformation, in turn impacting their potency, just like the facts verified by physiological and biochemical experiments (Paulus et al., 2013; Laye et al., 2017). Interestingly, our findings also suggest that there is no identical positive selection site shared by neolepadoids and Eochionelasmus (Supplementary Figure 2), which may imply that genomic response to environmental change not only depends on external factors but also relates to themselves (i.e., lineage-specific). Studies have shown that diversifying selection on physiological process can reduce intraspecific gene flow and promote population divergence, ultimately incurring speciation (Ellegren & Sheldon, 2008; Scott et al., 2011; Morales et al., 2015).
No studies have specifically discussed the morphological or physiological adaptations of chemosynthetic barnacles for surviving in extreme environments, but Yorisue et al. (2013) showed potential strategies of larval development to settle down in proper habitats developed by chemosynthetic barnacles. In comparison with their deep-sea or shallow-water relatives, chemosynthetic barnacles have a relatively larger number of setae on the intermediate segments of the posterior three cirri (i.e., generally more than six pairs vs. less than six pairs), and a higher ratio of setal length to the width of the segment where the setae are rooted (generally 2.8–14.1 vs. mostly 2.0–3.5) (primary data summarized from Yamaguchi and Newman, 1990; Buckeridge, 1998; Newman, 2000; Southward & Jones, 2003; Buckeridge et al., 2013; Watanabe et al., 2021). We speculate that these features represent morphological adaptations of chemosynthetic barnacles, as the dense and long setae would enhance the volume of water currents flowing into the mantle cavity when their cirri are beating for respiration and feeding. Buckeridge et al. (2013) have shown that the powerful morphological plasticity of Vulcanolepas scotiaensis was triggered by microenvironmental variation in chemosynthetic habitats. Several chemosynthetic barnacle species, such as V. osheai, V. buckeridgei and V. verenae, had developed ectosymbioses with sulfur-oxidizing bacteria for feeding (Southward & Newman, 1998; Watanabe et al., 2021). Furthermore, all barnacle somatotypes (e.g, sessile, stalked and asymmetric) were developed in chemosynthetic habitats. Together, these phenomena suggest that a subdivision of the ecological niche may have also been conducive to the speciation and diversification of chemosynthetic barnacles.
Conclusions
Over all, the present study demonstrates a robustly supported tree for thoracican barnacles and outlines the evolutionary history of main barnacle lineages. A potentially integrated speciation and diversification mechanism, that is, one aroused by ecological events (e.g., niche vacancy and food scarcity), then progressed in specific responses of genome to environmental changes, and the subdivision of ecological niches, likely shaped the emergence and evolution of chemosynthetic barnacles. Such processes may also have been important in shaping the evolutionary trajectory of other modern chemosynthetic animals. Future studies involving more representative samples, nuclear genomes, and fossil investigations are needed to achieve a detailed understanding of these enigmatic organisms.
Data availability statement
The data presented in the study are deposited in the GenBank repository (https://www.ncbi.nlm.nih.gov), accession numbers OM009255—OM009267.
Author contributions
ZG, DD, and XZL conceived and designed the research. XZL harmonized the project. ZG, XML, and JS performed the experiments. ZG and DD analyzed the data. ZG wrote the manuscript. DJ, XZL, and DD revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Key R & D Program of China (No. 2018YFC0310800) and the Senior User Project of RV KEXUE (KEXUE2020GZ01).
Acknowledgments
We express our thanks to Dr Zhou D.Y. (Second Institute of Oceanography Ministry of Natural Resources) and Liu M.L. (North China Sea Marine Environment Monitoring Center, Guangxi) for their help with the sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.964114/full#supplementary-material
References
Allen G. C., Flores-Vergara M. A., Krasynanski S., Kumar S., Thompson W. F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325. doi: 10.1038/nprot.2006.384
Al-Yahya H., Chen H. S., Chan B. K. K., Kado R., Høeg J. T. (2016). Morphology of cyprid attachment organs compared across disparate barnacle taxa; does it relate to habitat? Biol. Bull. 231, 120–129. doi: 10.1086/690092
Anderson D. T. (1983). Catomerus polymerus and the evolution of the balanomorph form in barnacles (Cirripedia). Aust. Museum Memoir 18, 7–20.
Baker E. T., Resing J. A., Haymon R. M., Tunnicliffe V., Lavelle J. W., Martinez F., et al. (2016). How many vent fields? New estimates of vent field populations on ocean ridges from precise mapping of hydrothermal discharge locations. Planet Sci. Lett. 449, 186–196. doi: 10.1016/j.epsl.2016.05.031
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bartáková V., Bryjová A., Nicolas V., Lavrenchenko L. A., Bryja J. (2021). Mitogenomics of the endemic Ethiopian rats: Looking for footprints of adaptive evolution in sky islands. Mitochondrion 57, 182–191. doi: 10.1016/j.mito.2020.12.015
Bell M. A., Lloyd G. T. (2015). Strap: An r package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaentology 58, 379–389. doi: 10.1111/pala.12142
Belyaeva E. A., Dymkowska D., Więckowski M. R., Wojtczak L. (2008). Mitochondria as an important target in heavy metal toxicity in rat hepatoma AS-30D cells. Toxicol. Appl. Pharmacol. 231, 34–42. doi: 10.1016/j.taap.2008.03.017
Bouckaert R. R., Drummond A. J. (2017). bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 17, 1–11. doi: 10.1186/s12862-017-0890-6
Bouckaert R., Heled J., Kühnert D., Vaughan T., Wu C.-H., Xie D., et al. (2014). BEAST 2: A software platform for Bayesian evolutionary analysis. PloS Comput. Biol. 10, e1003537. doi: 10.1371/journal.pcbi.1003537
Buckeridge J. S. (1996). Phylogeny and biogeography of the primitive sessilia and a consideration of a tethyan origin for the group. Crustac Issues 10, 255–267.
Buckeridge J. S. (1998). A new coral inhabiting barnacle of the genus Chionelasmus (Cirripedia, balanomorpha) from new Caledonia, south-West pacific. Zoosystema 20, 167–176.
Buckeridge J. S., Linse K., Jackson J. A. (2013). Vulcanolepas scotiaensis sp. nov., a new deep-sea scalpelliform barnacle (Eolepadidae: Neolepadinae) from hydrothermal vents in the Scotia Sea, Antarctica. Zootaxa 3745, 551–568. doi: 10.11646/zootaxa.3745.5.4
Cai Y. F., Shen X., Zhou L., Chu K. H., Chan B. K. K. (2018). Mitochondrial genome of Tesseropora rosea: molecular evidence for non-monophyly of the genus Tetraclita. Mitochondrial DNA B Resour. 3, 92–94. doi: 10.1080/23802359.2017.1422412
Carriol R. P., Bonde N., Jakobsen S. L., Høeg J. T. (2016). New stalked and sessile cirripedes from the Eocene Mo clay, northwest Jutland (Denmark). Geodiversitas 38, 21–32. doi: 10.5252/g2016n1a2
Chan B. K. K., Dreyer N., Gale A. S., Glenner H., Ewers-Saucedo C., Pérez-Losada M., et al. (2021). The evolutionary diversity of barnacles, with an updated classification of fossil and living forms. Zool. J. Linn. Soc 193, 789–846. doi: 10.1093/zoolinnean/zlaa160
Chan P. P., Lowe T. M. (2019). “tRNAscan-SE: Searching for tRNA genes in genomic sequences,” in Gene prediction. Ed. Kollmar M. (New York: Humana), 1–14.
Chen P., Song J., Shen X., Cai Y., Chu K. H., Li Y., et al. (2019). Mitochondrial genome of Chthamalus challengeri (Crustacea: Sessilia): Gene order comparison within chthamalidae and phylogenetic consideration within balanomorpha. Acta Oceanol Sin. 38, 25–31. doi: 10.1007/s13131-019-1355-0
Chevaldonné P., Jollivet D., Desbruyeres D., Lutz R., Vrijenhoek R. (2002). Sister-species of eastern pacific hydrothermal vent worms (Ampharetidae, alvinellidae, vestimentifera) provide new mitochondrial COI clock calibration. Cah Biol. Mar. 43, 367–370.
Danovaro R., Snelgrove P. V., Tyler P. (2014). Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 29, 465–475. doi: 10.1016/j.tree.2014.06.002
D’Hondt S. (2005). Consequences of the Cretaceous/Paleogene mass extinction for marine ecosystems. Annu. Rev. Ecol. Syst. 36, 295–317. doi: 10.1146/annurev.ecolsys.35.021103.105715
Ellegren H., Sheldon B. C. (2008). Genetic basis of fitness differences in natural populations. Nature 452, 169–175. doi: 10.1038/nature06737
Gale A. S. (2014a). Origin and phylogeny of verrucomorph barnacles (Crustacea, cirripedia, thoracica). J. Syst. Palaeontol. 13, 753–789. doi: 10.1080/14772019.2014.954409
Gale A. S. (2014b). New thoracican cirripedes (Crustacea) from the Jurassic and Cretaceous of the UK. Proc. Geol. Assoc. 125, 406–418. doi: 10.1016/j.pgeola.2014.07.003
Gale A. S. (2015). Origin and phylogeny of the Cretaceous thoracican cirripede family stramentidae. J. Syst. Palaeontol. 14, 653–702. doi: 10.1080/14772019.2015.1091149
Gale A. S., Little C. T., Johnson J. E., Giosan L. (2020). A new neolepadid cirripede from a pleistocene cold seep, Krishna-godavari basin, offshore India. Acta Palaeontol. Pol. 65, 351–362. doi: 10.4202/app.00705.2019
Gale A. S., Sørensen A. M. (2014). Origin of the balanomorph barnacles (Crustacea, cirripedia, thoracica): New evidence from the late Cretaceous (Campanian) of Sweden. J. Syst. Palaeontol. 13, 791–824. doi: 10.1080/14772019.2014.954824
Gan Z., Xu P., Li X., Wang C. (2020a). Integrative taxonomy reveals two new species of stalked barnacle (Cirripedia, thoracica) from seamounts of the western pacific with a review of barnacles distributed in seamounts worldwide. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.582225
Gan Z., Yuan J., Liu X., Dong D., Li F., Li X. (2020b). Comparative transcriptomic analysis of deep- and shallow-water barnacle species (Cirripedia, poecilasmatidae) provides insights into deep-sea adaptation of sessile crustaceans. BMC Genomics 21, 240. doi: 10.1186/s12864-020-6642-9
Gernhard T. (2008). The conditioned reconstructed process. J. Theor. Biol. 253, 769–778. doi: 10.1016/j.jtbi.2008.04.005
Ghiselin M. T., Jaffe L. (1973). Phylogenetic classification in darwin's monograph on the sub-class cirripedia. Syst. Zool. 22, 132–140. doi: 10.1093/sysbio/22.2.132
Grant J. R., Stothard P. (2008). The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184. doi: 10.1093/nar/gkn179
Grassle J. F. (1985). Hydrothermal vent animals: Distribution and biology. Science 229, 713–717. doi: 10.1126/science.229.4715.713
Grassle J. F. (1987). The ecology of deep-sea hydrothermal vent communities. Adv. Mar. Biol. 23, 301–362. doi: 10.1016/S0065-2881(08)60110-8
Grieshaber M. K., Völkel S. (1998). Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu. Rev. Physiol. 60, 33–53.
Harris D. J., Maxson L., Braithwaite L. F., Crandall K. (2000). Phylogeny of the thoracican barnacles based on 18S rDNA sequences. J. Crust Biol. 20, 393–398. doi: 10.1111/mec.13054
Herrera S., Watanabe H., Shank T. M. (2015). Evolutionary and biogeographical patterns of barnacles from deep‐sea hydrothermal vents. Mol. Ecol. 24, 673–689. doi: 10.1111/mec.13054
Hourdez S., Lallier F. H. (2007). Adaptations to hypoxia in hydrothermal-vent and cold-seep invertebrates. Rev. Environ. Sci. Biotechnol. 6, 143–159. doi: 10.1007/s11157-006-9110-3
Hsü K. J., He Q., McKenzie J. A., Weissert H., Perch-Nielsen K., Oberhänsli H., et al. (1982). Mass mortality and its environmental and evolutionary consequences. Science 216, 249–256. doi: 10.1126/science.216.4543.249
Jacobs D. K., Lindberg D. R. (1998). Oxygen and evolutionary patterns in the sea: onshore/offshore trends and recent recruitment of deep-sea faunas. Nat.l Acad. Sci. U.S.A 95, 9396–9401. doi: 10.1073/pnas.95.16.9396
James J. E., Piganeau G., Eyre-Walker A. (2016). The rate of adaptive evolution in animal mitochondria. Mol. Ecol. 25, 67–78. doi: 10.1111/mec.13475
Jin J. J., Yu W. B., Yang J. B., Song Y., dePamphilis C. W., Yi T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Jones D. S. (2012). Australian Barnacles (Cirripedia: Thoracica), distributions and biogeographical affinities. Integr. Comp. Biol. 52, 366–387. doi: 10.1093/icb/ics100
Kalyaanamoorthy S., Minh B. Q., Wong T. K., Von Haeseler A., Jermiin L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim R. O., Chan B. K. K., Hou B. K., Ju S. J., Kim S. J. (2019). Complete mitochondrial genome of the deep-water epibiotic stalked barnacle, Glyptelasma annandalei (Cirripedia, lepadiformes, poecilasmatidae). Mitochondrial DNA B Resour. 4, 99–100. doi: 10.1080/23802359.2018.1536487
Kim S. J., Kang H. M., Corbari L., Chan B. K. K. (2018). First report on the complete mitochondrial genome of the deep-water scalpellid barnacle Arcoscalpellum epeeum (Cirripedia, thoracica, scalpellidae). Mitochondrial DNA B Resour. 3, 1288–1289. doi: 10.1080/23802359.2018.1532844
Kim S. J., Lee W. K., Ju S. J., Chan B. K. K. (2022). Phylogeny and shell form evolution of the hydrothermal vent asymmetrical barnacles (Cirripedia, thoracicalcarea, neoverrucidae). Mol. Phylogenet. Evol. 169, 107391. doi: 10.1016/j.ympev.2022.107391
Kong L., Li Y., Kocot K. M., Yang Y., Qi L., Li Q., et al. (2020). Mitogenomics reveals phylogenetic relationships of arcoida (Mollusca, bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Mol. Phylogenet. Evol. 150, 106857. doi: 10.1016/j.ympev.2020.106857
Kozlov A. M., Darriba D., Flouri T., Morel B., Stamatakis A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lagesen K., Hallin P., Rødland E. A., Stærfeldt H. H., Rognes T., Ussery D. W. (2007). RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lanfear R., Frandsen P. B., Wright A. M., Senfeld T., Calcott B. (2017). PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Lan Y., Sun J., Tian R., Bartlett D. H., Li R., Wong Y. H., et al. (2017). Molecular adaptation in the world's deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 26, 3732–3743. doi: 10.1111/mec.14149
Larget B., Simon D. L., Kadane J. B. (2002). Bayesian Phylogenetic inference from animal mitochondrial genome arrangements. J. R. Stat. Soc Ser. B Stat. Methodol. 64, 681–693. doi: 10.1111/1467-9868.00356
Laye V. J., Karan R., Kim J. M., Pecher W. T., DasSarma P., DasSarma S. (2017). Key amino acid residues conferring enhanced enzyme activity at cold temperatures in an Antarctic polyextremophilic β-galactosidase. Proc. Natl. Acad. Sci. U.S.A 114, 12530–12535. doi: 10.1073/pnas.1711542114
Le Bris N., Arnaud-Haond S., Beaulieu S., Cordes E., Hilario A., Rogers A., et al. (2017). “Hydrothermal vents and cold seeps,” in The first global integrated marine assessment: World ocean assessment (Cambridge: Cambridge University Press) 853–862. United Nations.
Lee W. K., Mi Kang H., Chan B. K. K., Ju S. J., Kim S. J. (2019). Complete mitochondrial genome of the hydrothermal vent stalked barnacle vulcanolepas fijiensis (Cirripedia, scalpelliforms, eolepadidae). Mitochondrial DNA B Resour. 4, 2725–2726. doi: 10.1080/23802359.2019.1644564
Lemoine F., Entfellner J. B. D., Wilkinson E., Correia D., Felipe M. D., De Oliveira T., et al. (2018). Renewing felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556, 452–456. doi: 10.1038/s41586-018-0043-0
Letunic I., Bork P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, 293–296. doi: 10.1093/nar/gkab301
Lin H. C., Høeg J. T., Yusa Y., Chan B. K. K. (2015). The origins and evolution of dwarf males and habitat use in thoracican barnacles. Mol. Phylogenet. Evol. 91, 1–11. doi: 10.1016/j.ympev.2015.04.026
Linse K., Jackson J. A., Fitzcharles E., Sands C. J., Buckeridge J. S. (2013). Phylogenetic position of Antarctic scalpelliformes (crustacea: cirripedia: thoracica). Deep Sea Res. 1 Oceanogr. Res. Pap 73, 99–116. doi: 10.1016/j.dsr.2012.11.006
Little C. T., Vrijenhoek R. C. (2003). Are hydrothermal vent animals living fossils? Trends Ecol. Evol. 18, 582–588. doi: 10.1016/j.tree.2003.08.009
Lorion J., Kiel S., Faure B., Kawato M., Ho S. Y., Marshall B., et al. (2013). Adaptive radiation of chemosymbiotic deep-sea mussels. Proc. R. Soc. B-Biol Sci. 280, 20131243. doi: 10.1098/rspb.2013.1243
Martin J. W., Davis G. E. (2001). An updated classification of the recent Crustacea (Los Angeles: Natural History Museum of Los Angeles County).
Morales H. E., Pavlova A., Joseph L., Sunnucks P. (2015). Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol. Ecol. 24, 2820–2837. doi: 10.1111/mec.13203
Newman W. A. (1982). “Evolution within the Crustacea, part 3: Cirripedia,” in The biology of Crustacea. Ed. Abele L. (New York: Academic Press), 197–221.
Newman W. A. (1987). “Evolution of cirripedes and their major groups,” in Barnacle biology. Ed. Southward A. J. (Rotterdam: Balkema), 3–44.
Newman W. A. (1989). Juvenile ontogeny and metamorphosis in the most primitive living sessile barnacle, Neoverruca, from abyssal hydrothermal springs. Bull. Mar. Sci. 45, 467–477.
Newman W. A. (2000). A new genus and species of barnacle (Cirripedia, verrucomorpha) associated with vents of the lau. back-arc basin: Its gross morphology, inferred first juvenile stage and affinities. Zoosystema 22, 71–84.
Newman W. A., Ross A. (1976). Revision of the balanomorph barnacles, including a catalog of the species (San Diego: San Diego Society of Natural History).
Newman W. A., Yamaguchi T. (1995). A new sessile barnacle (Cirripedia, brachylepadomorpha) from the lau back-arc basin, tonga; first record of a living representative since the Miocene. Bull. Mus Natl. Hist Nat. 17, 221–244.
Nguyen L. T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Paulus J. K., Schlieper D., Groth G. (2013). Greater efficiency of photosynthetic carbon fixation due to single amino-acid substitution. Nat. Commun. 4, 1–7. doi: 10.1038/ncomms2504
Pérez-Losada M., Høeg J. T., Crandall K. A. (2004). Unraveling the evolutionary radiation of the thoracican barnacles using molecular and morphological evidence: A comparison of several divergence time estimation approaches. Syst. Biol. 53, 244–264. doi: 10.1080/10635150490423458
Pérez-Losada M., Høeg J. T., Crandall K. A., Achituv Y. (2012). Molecular phylogeny and character evolution of the chthamaloid barnacles (Cirripedia: Thoracica). Mol. Phylogenet. Evol. 65, 329–334. doi: 10.1016/j.ympev.2012.06.004
Pérez-Losada M., Høeg J. T., Simon-Blecher N., Achituv Y., Jones D., Crandall K. A. (2014). Molecular phylogeny, systematics and morphological evolution of the acorn barnacles (Thoracica: Sessilia: Balanomorpha). Mol. Phylogenet. Evol. 81, 147–158. doi: 10.1016/j.ympev.2014.09.013
Pérez-Losada M., Harp M., Høeg J. T., Achituv Y., Jones D., Watanabe H., et al. (2008). The tempo and mode of barnacle evolution. Mol. Phylogenet. Evol. 46, 328–346. doi: 10.1016/j.ympev.2007.10.004
Pilsbry H. A. (1916). The sessile barnacles (Cirripedia) contained in the collections of the U.S. national museum; including a monograph of the American species (Washington: U.S. Government Publishing Office).
Plummer M., Best N., Cowles K., Vines K. (2006). CODA: Convergence diagnosis and output analysis for MCMC. R News 6, 7–11.
Rambaut A., Drummond A. J., Xie D., Baele G., Suchard M. A. (2018). Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ranwez V., Douzery E. J., Cambon C., Chantret N., Delsuc F. (2018). MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35, 2582–2584. doi: 10.1093/molbev/msy159
Rausher M. D., Huang J. (2016). Prolonged adaptive evolution of a defensive gene in the solanaceae. Mol. Biol. Evol. 33, 143–151. doi: 10.1093/molbev/msv205
Rees D. J., Noever C., Høeg J. T., Ommundsen A., Glenner H. (2014). On the origin of a novel parasitic-feeding mode within suspension-feeding barnacles. Curr. Biol. 24, 1429–1434. doi: 10.1016/j.cub.2014.05.030
Ren X. Q., Sha Z. L. (2015). Probathylepadidae, a new family of scalpelliformes (Thoracica: Cirripedia: Crustacea), for Probathylepas faxian gen. nov., sp. nov., from a hydrothermal vent in the Okinawa trough. Zootaxa 4033, 144–150. doi: 10.11646/zootaxa.4033.1.9
Revell L. J. (2012). Phytools: An r package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Scott G. R., Schulte P. M., Egginton S., Scott A. L. M., Richards J. G., Milsom W. K. (2011). Molecular evolution of cytochrome c oxidase underlies highaltitude adaptation in the bar-headed goose. Mol. Biol. Evol. 28, 351–363. doi: 10.1093/molbev/msq205
Sepkoski J. J. (1996). “Patterns of phanerozoic extinction: a perspective from global data bases,” in Global events and event stratigraphy in the phanerozoic. Ed. Walliser O. H. (Heidelberg: Springer), 35–51.
Shen Y. Y., Liang L., Zhu Z. H., Zhou W. P., Irwin D. M., Zhang Y. P. (2010). Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. U.S.A 107, 8666–8671. doi: 10.1073/pnas.0912613107
Song J., Shen X., Chu K. H., Chan B. K. K. (2017). Mitochondrial genome of the acorn barnacle Tetraclita rufotincta pilsbr: highly conserved gene order in tetraclitidae. Mitochondrial DNA B Resour. 2, 936–937. doi: 10.1080/23802359.2017.1413305
Southward A. J., Jones D. S. (2003). A revision of stalked barnacles (Cirripedia: Thoracica: Scalpellomorpha: Eolepadidae: Neolepadinae) associated with hydrothermalism, including a description of a new genus and species from a volcanic seamount off Papua new Guinea. Senckenbergiana Maritima 32, 77–93. doi: 10.1007/BF03043086
Southward A. J., Newman W. A. (1998). Ectosymbiosis between filamentous sulphur bacteria and a stalked barnacle (Scalpellomorpha, neolepadinae) from the lau back arc basin, Tonga. Cah Biol. Mar. 39, 259–262.
Spears T., Abele L. G., Applegate M. A. (1994). Phylogenetic study of cirripedes and selected relatives (Thecostraca) based on 18S rDNA sequence analysis. J. Crust Biol. 14, 641–656. doi: 10.1080/10635150701472164
Talavera G., Castresana J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tian M., Chen P., Song J., He F., Shen X. (2020). The first mitochondrial genome of capitulum mitella (Crustacea: Cirripedia) from China: Revealed the phylogenetic relationship within thoracica. Mitochondrial DNA B Resour. 5, 2573–2575. doi: 10.1080/23802359.2020.1781564
Tsang L. M., Shen X., Cheang C. C., Chu K. H., Chan B. K. K. (2017). Gene rearrangement and sequence analysis of mitogenomes suggest polyphyly of archaeobalanid and balanid barnacles (Cirripedia: Balanomorpha). Zool. Scr 46, 729–739. doi: 10.1111/zsc.12246
Utinomi H. (1968). A revision of the deep-sea barnacles Pachylasma and Hexelasma from Japan, with a proposal of new classification of the Chthamalidae (Cirripedia, Thoracica). Publications of the Seto Marine Biological Laboratory, 16(1):21–396.
Wang K., Shen Y., Yang Y., Gan X., Liu G., Hu K., et al. (2019). Morphology and genome of a snailfish from the Mariana trench provide insights into deep-sea adaptation. Nat. Ecol. Evol. 3, 823–833. doi: 10.1038/s41559-019-0864-8
Watanabe H. K., Chen C., Chan B. K. K. (2021). A new deep-sea hot vent stalked barnacle from the Mariana trough with notes on the feeding ecology of Vulcanolepas. Mar. Biodivers. 51, 1–12. doi: 10.1007/s12526-020-01144-x
Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, 296–303. doi: 10.1093/nar/gky427
Xia X., Xie Z. (2001). DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373. doi: 10.1093/jhered/92.4.371
Yamaguchi T., Newman W. A. (1990). A new and primitive barnacle (Cirripedia: Balanomorpha) from the north Fiji basin abyssal hydrothermal field, and its evolutionary implications. Pac Sci. 44 (2), 135–155.
Yang Z. (2007). PAML 4: A program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591.
Yang J. S., Lu B., Chen D. F., Yu Y. Q., Yang F., Nagasawa H., et al. (2013). When did decapods invade hydrothermal vents? clues from the Western pacific and Indian oceans. Mol. Biol. Evol. 30, 305–309. doi: 10.1093/molbev/mss224
Yang Z., Nielsen R. (2002). Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. doi: 10.1093/oxfordjournals.molbev.a004148
Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. (2015). The I-TASSER suite: Protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yorisue T., Kado R., Watanabe H., Høeg J. T., Inoue K., Shigeaki K., et al. (2013). Influence of water temperature on the larval development of neoverruca sp. and Ashinkailepas seepiophila–implications for larval dispersal and settlement in the vent and seep environments. Deep Sea Res. 1 Oceanogr Res. Pap 71, 33–37. doi: 10.1016/j.dsr.2012.10.007
Zachos J. C., Arthur M. A., Dean W. E. (1989). Geochemical evidence for suppression of pelagic marine productivity at the Cretaceous/Tertiary boundary. Nature 337, 61–64. doi: 10.1038/337061a0
Zhang D., Gao F., Jakovlić I., Zou H., Zhang J., Li W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: barnacle, adaptive evolution, mitogenome, phylogeny, origin, chemosynthetic habitat
Citation: Gan Z, Jones DS, Liu X, Sui J, Dong D and Li X (2022) Phylogeny and adaptative evolution to chemosynthetic habitat in barnacle (Cirripedia: Thoracica) revealed by mitogenomes. Front. Mar. Sci. 9:964114. doi: 10.3389/fmars.2022.964114
Received: 08 June 2022; Accepted: 23 August 2022;
Published: 15 September 2022.
Edited by:
Benny K. K. Chan, Academia Sinica, TaiwanReviewed by:
Luisa Fernanda Dueñas, National University of Colombia, ColombiaRyota Hayashi, Nippon Koei Co., Ltd., Japan
Yoichi Yusa, Nara Women’s University, Japan
Copyright © 2022 Gan, Jones, Liu, Sui, Dong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Dong, ZG9uZ2RAcWRpby5hYy5jbg==; Xinzheng Li, bGl4emhAcWRpby5hYy5jbg==