
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 08 September 2022
Sec. Deep-Sea Environments and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.957762
This article is part of the Research TopicDeep-sea Chemosynthetic Ecosystems: Living in Extreme EnvironmentsView all 13 articles
 Xinyi Zhai1†
Xinyi Zhai1† Xiaochong Shi1,2,3†
Xiaochong Shi1,2,3† Haojin Cheng1
Haojin Cheng1 Peng Yao2,4Bin Zhao2,4Lin Chen2,4
Peng Yao2,4Bin Zhao2,4Lin Chen2,4 Jiwen Liu1,2,3Lei Cao5Minxiao Wang5Lulu Fu5
Jiwen Liu1,2,3Lei Cao5Minxiao Wang5Lulu Fu5 Xiao-Hua Zhang1,2,3
Xiao-Hua Zhang1,2,3 Min Yu1,2,3*
Min Yu1,2,3*Site F is the most vigorous cold seep known on the continental slope of the northern South China Sea. Up to now, the microbial community structures in sediments of Site F based on the high-throughput sequencing of the 16S rRNA genes have been studied extensively. However, few studies investigated the microbial community structures at fine vertical scales of Site F and control stations outside Site F. In this study, a comprehensive investigation of microbial communities in sediments of Site F along the depths varying from 0 to 24 cm below sea floor (cmbsf) of four sampling sites—SRS (Southern Reduced Sediment), NRS (Northern Reduced Sediment), Control 1 (close to Site F), and Control 2 (far from Site F)—was carried out. The high relative abundances of anaerobic methanotrophic archaea (ANME), Desulfobacterota [sulfate-reducing bacteria (SRB)], and Campylobacteria [sulfur-oxidizing bacteria (SOB)] in SRS and NRS indicated that these two sites were newborn cold seep sites compared with non-seep sites, Control 1, and Control 2. A positive correlation between ANME-1b, ANME-2, and SEEP-SRB and an enrichment of Sulfurovum and Methlomonadaceae were found in the surface sediments of both SRS and NRS, indicating that the processes of anaerobic oxidation of methane (AOM), sulfur oxidation, and sulfate reduction might occur in seep sites. SRS was enriched with ANME-1b and SEEP-SRB2 with a proposed sulfate-methane transition zone (SMTZ) approximately located at 8 cmbsf. The high abundance of ANME in SRS may due to the high concentration of methane. NRS was enriched with ANME-2, Desulfatiglans, Sulfurovum, and Methanosarcinaceae with a proposed SMTZ at about 10 cmbsf. According to the analyses of microbial community structure and environmental factors, NRS could be described as a notable cold seep reduced sediment site with low sulfate and high H2S that nourished abundant SEEP-SRB1, ANME-2, Methanosarcinales, and Sulfurovum, which showed similar distribution pattern. Our study expands the current knowledge on the differences of microbial communities in cold seep sites and non-seep sites and sheds light on the horizontal and vertical heterogeneity of sediment microbial community in Site F.
Deep-sea cold seeps usually refer to the ecosystems located at continental slopes, where dissolved and gaseous methane and reductive fluids emitted from oceanic subsurface (Boetius and Wenzhöfer, 2013; Li et al., 2021). The fluids enriched with hydrocarbons (primarily methane) and hydrogen sulfide sustain distinct microbiomes involved in various reactions in marine sediments, making the cold seep an oasis enriched with key functional groups including methanotrophs, methanogens, hydrocarbon degraders, and sulfate-reducing bacteria (SRB) (Jørgensen and Boetius, 2007; Boetius and Wenzhöfer, 2013; Li et al., 2021). Microorganism-mediated anaerobic oxidation of methane (AOM) is a dominant process at cold seep ecosystems and a major global sink of methane (Knittel et al., 2005; Sun et al., 2020). AOM is often associated with the reduction of sulfate, and the total reaction is , which is usually performed by a consortium of anaerobic methanotrophic archaea (ANME) and SRB (Hoehler et al., 1994; Cui et al., 2019). ANME, which could be classified into ANME-1, ANME-2, and ANME-3, are found widely distributed in cold seep sediments (Knittel et al., 2005; Vigneron et al., 2013; McKay et al., 2016; Niu et al., 2017; Wu et al., 2018; Cui et al., 2019; Zhang et al., 2020). ANME-1 are divided into ANME-1a and ANME-1b subgroups, whereas ANME-2 are most widely distributed and divided into ANME-2a, ANME-2b, ANME-2c, and ANME-2d subgroups (Orphan et al., 2002; Mills et al., 2003; Knittel et al., 2005).
In general, the syntrophic partners of ANME-1 and ANME-2 are SRB affiliated with Desulfosarcina/Desulfococcus (DSS; within the Desulfobacteraceae) or Desulfobulbus (DBB; within the Desulfobulbaceae) (Orphan et al., 2001a; Orphan et al., 2001b; Orphan et al., 2002; Boetius et al., 2000; Jørgensen and Boetius, 2007; Zhang et al., 2020). It was found that ANME-1 were associated to SEEP-SRB2 (within the Desulfuribacteraceae) (Ruff et al., 2016); ANME-2 were associated to SEEP-SRB1 (members of DSS), SEEP-SRB2, and SEEP-DBB (members of DBB) (Orphan et al., 2001a; Pernthaler et al., 2008; Kleindienst et al., 2012; Green-Saxena et al., 2014; Ruff et al., 2016); and ANME-3 were associated with DBB and SEEP-SRB1 (Niemann et al., 2006; Lösekann et al., 2007; Schreiber et al., 2010). Furthermore, ANME-1 may perform AOM process via reversal of the methanogenic pathway without a bacterial partner (Orphan et al., 2002; Knittel et al., 2005; Stokke et al., 2012; Cho et al., 2017). Sulfur-oxidizing bacteria (SOB) also inhabit widely in cold seep sediments, many of which are affiliated with Campylobacterota and Gammaproteobacteria (Feng et al., 2018; Sun et al., 2020; Li et al., 2021). SOB play an important role in sulfide oxidation, which occurs in the near-surface sediment and supplies sulfate to the layers below (Li et al., 2021). Sulfate diffuses from above and methane rises from below, resulting in a region where sulfate and methane coexist called the sulfate-methane transition zone (SMTZ) (Borowski et al., 1996). The microbial consortia of ANME and SRB involved in AOM and sulfate reduction are often found in SMTZ (Boetius et al., 2000; Orphan et al., 2002).
Since the first cold seep discovered in the South China Sea (SCS) in 2004, more than 40 cold seeps were found on the continental margins of the SCS (Chen et al., 2005; Feng et al., 2018). Among them, Site F (22°06.922N; 119°17.130E) is the first intensely vigorous cold seep known on the continental slope of the northern SCS (Feng and Chen, 2015; Wang et al., 2021). Site F is found extremely active and characterized by an authigenic carbonate mound, which interspersed with cold seeps and benthic communities as well as surrounded by shell bed features and reduced sediments (Wang et al., 2021). In Site F, sufficient cold seep fluids are migrating along and escaping at the bottom of the authigenic carbonate mound, resulting in presence of the newly formed reduced sediment patches (Wang et al., 2021).
Up to now, a series of studies focused on the microbial communities in sediments of Site F have been carried out with high-throughput sequencing based on the 16S rRNA genes. It was found that Sulfurovum was the dominated microorganisms in the surface sediments, whereas Deltaproteobacteria, Methanomicrobia, and Dehalococcoidia were abundant in the subsurface (Sun et al., 2020). In addition, Nitrosopumilus was also found dominant and type I methanotrophs was found conducted aerobic methane oxidization in Site F sediments (Jing et al., 2020). In the SMTZ, the high relative abundance of ANME-1b may perform AOM in collaboration with ANME-2c and Desulfobacteraceae (Cui et al., 2019). Studies on Jiaolong cold seep closed to Site F revealed that Sulfurimonas, Sulfurovum, and ANME-1 dominated the surface sediment layers, whereas SEEP-SRB1, ANME-2, ANME-1, and ANME-3 dominated the deeper sediment layers (Wu et al., 2018; Li et al., 2020; Li et al., 2021). It was found that ANME, especially ANME-2a-2b, as well as SOB and SRB were dominant microorganisms in the SMTZ of Jiaolong (Li et al., 2021). However, the conclusions varied among studies because of the inconsistency of sampling locations and depths. Many studies were limited by the number of sampling stations and few studies investigated stations apart from the cold seeps as control. In addition, the microbial community structures and metabolic potentials along the fine vertical scales of these seeps are still unknown (Jing et al., 2020).
In this study, we investigated the microbial communities of sediment samples among four sites around Site F by high-throughput sequencing based on the 16S rRNA gene amplicons. Our study aimed the following: to (i) determine and compare the difference of microbial communities in the cold seep sites and non-seep control sites along the horizontal and vertical directions at fine vertical scales and to infer the SMTZ depths of cold seep sites; (ii) to investigate the relationship of SRB, SOB, ANME, and other methane-metabolizing microbes to depict the ecological integrity of cold seep sites; and (iii) to improve the understanding of microbial biogeochemical processes in cold seeps of SCS.
Samples were collected from Site F cold seep in the SCS during the scientific cruise of R/V “Kexue” organized by the Institute of Oceanology, Chinese Academy of Sciences in June, 2021. The four sampling sites are SRS, NRS, Control 1, and Control 2 (Figure 1A). SRS is the reduced sediment site at southeastern periphery of authigenic carbonate mound and about 50 m from the known seepage, and NRS is the reduced sediment site at northern periphery of the authigenic carbonate mound and about 120 m from the seepage. Control 1 is on the south near Site F and about 85 m from the seepage, whereas Control 2 is on the southwest far from Site F and about 320 m from the seepage (Wang et al., 2021). Sediment samples from these sites were collected by Discover manned submersible dives 261, 263, 267, 268, and 271, respectively, with pushcore equipment. The cores were washed with sterile water and kept closed before sampling. After sampling, the cores were closed again and taken on board. The core was opened on board, and the sediment inside was pushed up from the bottom of the core. When the sample emerged at the top end of the core, subsample was taken for every 2 cm with a stainless‐steel cutter, and considered as one sample. To analyze the concentration of methane in sediment methane, 3 ml of sample was transferred with a 5-ml cutoff syringe to a 22-ml serum vial containing 3 ml of NaOH (2 mol /L) (purged with 99.999% N2), crimp sealed, and stored at 4°C for subsequent next measurement (Zhuang et al., 2019). In addition, the pore waters were extracted with Rhizons samplers (The Netherlands) inserted into the pre-drilled holes in PVC tubes and attached to vacuum test tubes by 23G needles under N2 atmosphere (Zhao et al., 2017). Notably, site SRS was sampled twice by two dives and named as SRS 1 and SRS 2. All sampling depths ranged from 12 to 22 cm below sea floor (cmbsf). A total of 53 sediment samples were obtained, named as SRS 1_0-20, SRS 2_0-22, NRS_0-20, Control 1_0-12, and Control 2_0-22. All the samples were stored in sterile sampling bags and were immediately frozen at −80°C.
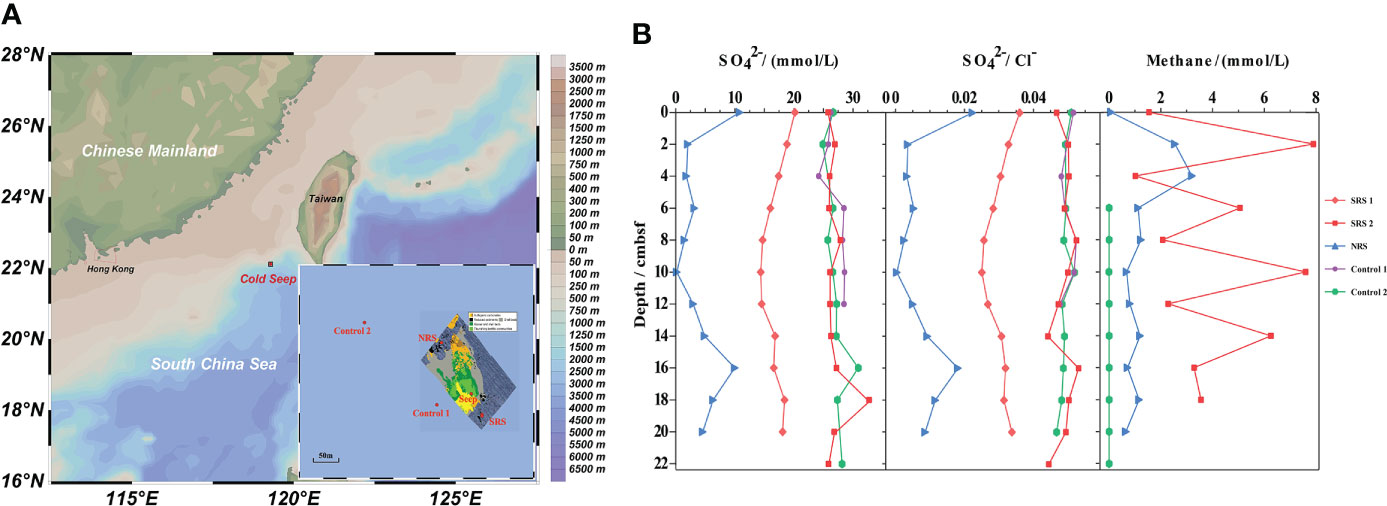
Figure 1 The locations and environmental characteristics of sampling sites. (A) The locations of sampling sites. Seabed features associated with cold seep activity at Site F referred to Wang et al. (2021). (B) The chemical properties of sediment samples.
Methane concentrations were measured using a gas chromatograph (GC; SRI Instruments) equipped with a HayeSep column and a flame ionization detector by injecting 500 μl of headspace gas, and the analytical precision was better than 1.5% (Zhuang et al., 2019). The sulfate concentration in pore waters was determined with a Dionex ICS-3000 ion chromatography (USA) on 1:200 diluted aliquots in Milli-Q water, and the relative standard deviation of duplicate analyses was less than 1% (Zhao et al., 2017).
Microbial community genomic DNA was extracted from sediment samples using the FastDNA® Spin Kit for Soil (MP Biomedicals) and a FastPrep‐24 cell disrupter (MP Biomedicals) according to manufacturer’s instructions. The extracted DNA was checked on 1% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). The hypervariable region V4 of prokaryotic (bacterial and archaeal) 16S rRNA genes was amplified with primer pairs 515FmodF (5′‐GTGYCAGCMGCCGCGGTAA‐3′) and 806RmodR (5′‐GGACTACNVGGGTWTCTAAT‐3′) by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA) (Walters et al., 2015). The PCR amplification of 16S rRNA gene was performed as follows: initial denaturation at 95°C for 3 min; 29 cycles of denaturing at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s; single extension at 72°C for 10 min and end at 10°C. The PCR mixtures contain 5× TransStart FastPfu buffer of 4 μl, 2.5 mM dNTPs of 2 μl, forward primer of (5 μM) 0.8 μl, reverse primer of (5 μM) 0.8 μl, TransStart FastPfu DNA Polymerase of 0.4 μl, bovine serum albumin (BSA) of 0.2 μl, template DNA of 10 ng, and finally ddH2O of up to 20 μl. PCR reactions were performed in triplicate. The PCR product was extracted from 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s instructions and quantified using Quantus™ Fluorometer (Promega, USA).
Purified amplicons were pooled in equimolar and paired-end sequenced on an Illumina NovaSeq PE250 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The raw reads have been deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: PRJNA827936).
The raw 16S rRNA gene sequencing reads were demultiplexed, quality-filtered by fastp version 0.20.0 (Chen et al., 2018), and merged by FLASH version 1.2.7 (Magoč and Salzberg, 2011) with the following criteria: (i) the 300 bp reads were truncated at any site receiving an average quality score of <20 over a 50-bp sliding window, and the truncated reads shorter than 50 bp and containing ambiguous characters were discarded; (ii) only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of overlap region is 20%, and reads that could not be assembled were discarded; (iii) samples were distinguished by barcoding (exact matches) and primers (allowing 2-nucleotide mismatches). Operational taxonomic units (OTUs) with 97% similarity cutoff (Edgar, 2013) were clustered using UPARSE version 7.1 (Edgar, 2013), and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.2 (Wang et al., 2007) against the 16S rRNA gene database (Silva v138) (http://www.arb-silva.de) using confidence threshold of 70% (Quast et al., 2013).
Quantitative PCR was performed to quantify the abundance of total bacterial and archaeal 16S rRNA gene with the specific primer sets 967R/1046R (Sogin et al., 2006) and 967F/1060R (Cadillo-Quiroz et al., 2006), respectively. A 20-µl reaction system contained 10 µl of SYBR Premix ExTaq II (2×), 0.4 µl of ROX Reference Dye II (50×) (TaKaRa, Tokyo, Japan), 0.4 µl (0.8 µl for archaea; 10 µM) of primers, and 2 µl of template. For bacterial 16S rRNA gene, the thermal cycling steps consisted of an initial denaturation at 95°C for 5 min, 40 cycles of 95°C for 30 s, 53°C for 1 min, and 72°C for 15 s and a final extension at 72°C for 10 min. For archaeal 16S rRNA gene, the thermal cycling steps contain an initial denaturation at 95°C for 30 s, 40 cycles of 95°C for 5 s, 50°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 5 min. All assays with negative controls were conducted in triplicate, using a QuantStudio 5 System (Thermo Fisher Scientific). Standard curves were generated by PCR amplification of a 10-fold serial dilution of plasmids containing target gene fragments. The amplification curves showed good linear relationships (R2 > 0.999) and the amplification efficiencies >0.90.
The analyses of microbial diversity were performed with the “vegan” package in R software. Chao1 and Shannon indices of α‐diversity based on similarities (ANOSIM) were chosen to display microbial community richness and community diversity, respectively. The distribution patterns of microbial communities were analyzed by non-metric multidimensional scaling (NMDS) and hierarchical clustering of β-diversity based on Bray–Curtis distances. The relationship between the community structures at the OTU level and the environmental variables was displayed by distance-based redundancy analysis (db-RDA) based on Bray–Curtis distances. The line charts showing sulfate and methane concentrations, bar graphs showing microbial community composition at various taxonomic levels, as well as ternary plot showing the distribution and relative abundance of the top 50 genera between sediment sites were performed by Originpro 2021 software (https://www.originlab.com).
Co-occurrence network analyses were conducted in R software with “igraph” and “Hmisc” packages. The co-occurrence patterns were generated with the top 50 abundant microbial genera, as well as the top 50 abundant bacterial and archaeal genera across all samples to reduce network complexity. Co-occurrence paired with a Spearman’s correlation coefficient >0.7 or<−0.7 and a P-value<0.01 (Benjamini and Hochberg adjusted) was considered as a valid relationship. Gephi 0.9.2 software (http://gephi.org) was used for network visualization (Jacomy et al., 2009).
The sulfate concentration in porewater of most sediment samples and the methane concentration of SRS 2, NRS, and Control 2 were determined (Figure 1B). The SO42−/Cl− value could show the exact sulfate concentration by eliminating the influence of salinity. Site NRS exhibited an obvious lower. SO42−/Cl− value than SRS and Control 2 at all depths. Meanwhile, SO42−/Cl− value in NRS decreased with depths and was almost 0 at 10 cmbsf, whereas that in Control 2 remained comparatively constant. As for the content of methane, Control 2 remained almost 0 at all depths, whereas in NRS, increased obviously from 0 to 4 cmbsf and then decreased to stabilization. Despite great variations with depths, SRS 2 exhibited a superiority of methane than NRS in general.
A total of 3,194,466 reads were generated after quality control, discard of singletons, and then rarefaction. The sequencing statistics of all samples were showed in Table S1. Each sample was rarefied and clustered into 26,046 OTUs at a 97% similarity level (22,802 bacterial OTUs and 3,162 archaeal OTUs) for further analyses. Chao1 and Shannon indices indicated the microbial community richness and diversity of different sampling sites. Figures 2A, B show that Chao1 and Shannon indices varied among sites, ranging from 3,786.8 to 4,224.9 and 5.06 to 6.95, respectively. In general, there was significant differences in richness and diversity among seep sites (SRS and NRS) and non-seep sites (Control 1 and Control 2). Sites SRS and NRS, especially NRS, had lower richness and diversity indices compared with Control 1 and Control 2. In addition, the Shannon indices of SRS 1, SRS 2, and NRS did not show significant difference.
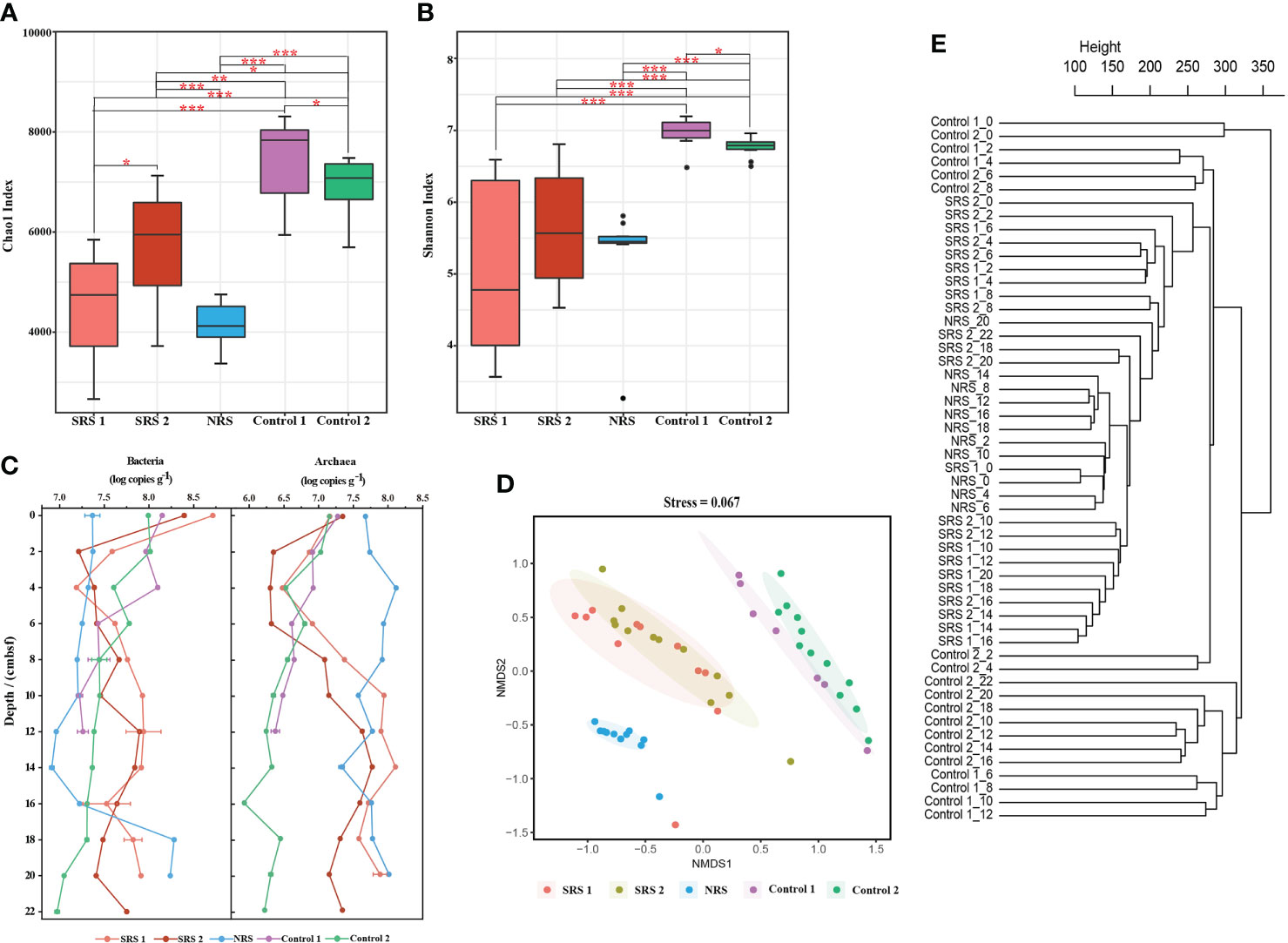
Figure 2 The α-diversity and β-diversity indices, as well as 16s rRNA gene abundance of all samples. (A) Horizontal Chao1 index at the OTU level. (B) Horizontal Shannon index at the OTU level. (C) Horizontal and vertical changes of bacterial and archaeal 16S rRNA gene abundance. (D) Non-metric multidimensional scaling ordination of microbial communities based on the Bray–Curtis dissimilarity. (E) Hierarchical clustering of microbial communities based on the Bray–Curtis distances. * 0.01 ≤ P value ≤ 0.05; ** 0.001 ≤ P value ≤ 0.01; *** P value ≤ 0.001.
Quantitative PCR showed that the abundances of bacterial 16S rRNA gene were about 1.0 order of magnitude higher than that of archaea in SRS and Controls; however, the opposite result was found in NRS (Figure 2C). The bacterial and archaeal 16S rRNA gene abundances decreased at 0–4 cmbsf, and a sharp increase of archaeal abundance was observed at 6–12 cmbsf in SRS 1 and SRS 2. In NRS, the abundance of bacteria increased below 14 cmbsf, and the archaeal abundances were relatively high at all depths. In general, the abundances of archaeal 16S rRNA gene showed more distinguishable among sites than that of bacteria.
NMDS and hierarchical clustering based on Bray–Curtis distances are performed to visualize the differences and relationship of the microbial communities in the 53 samples along the sites and depths (Figures 2D, E). Samples from each site were distinguished with each other at OTU level of the total prokaryotes (stress = 0.067), bacteria (stress = 0.067), and archaea (stress = 0.1018) (Figures 2D, E and Figures S1, S2). Samples from SRS 1 and SRS 2 clustered together and had similar microbial compositions. Moreover, bacterial and archaeal communities from sites Control 1 and Control 2 were clustered together and were apart from SRS and NRS. Hierarchical clustering also revealed that the sediment samples clustered by site other than depth. However, there were still slight vertical clusters observed within each site, and assemblages of sediments clustered with 10 cmbsf as the boundary (<10 and >10 cmbsf) in general.
The relative abundances of major microbial communities of the 53 samples along the sampling sites and vertical depths were analyzed at the levels of phylum, class, order, and family (Figure 3). The dominant groups varied significantly among sites. At the phylum level, 92 phyla were observed. Chloroflexi (0.5%–48.1%), Halobacterota (0.3%–42.6%), and Desulfobacterota (6.6%–20.9%) were dominant groups in SRS; Campylobacterota (7.6%–61.0%), Chloroflexi (8.6%–19.4%), and Desulfobacterota (7.5%–19.7%) were dominant groups in NRS; and Proteobacteria (6.7%–29.3% and 10.7%–33.7%, respectively), Planctomycetota (8.7%–20.2% and 8.2%–31.6%, respectively), and Chloroflexi (5.4%–25.2% and 5.0%–21.0%, respectively) were dominant groups in both Control 1 and Control 2. However, Control 1 contained more Desulfobacterota (7.5%–19.7%), whereas Control 2 contained more Actinobacteriota (3.1%–15.3%). In addition, 250 classes, 594 orders, and 944 families were observed. Within all sequences of each sample, Campylobacterales (0.7%–43.4%), Anaerolineales (composed of Anaerolineaceae, 0.3%–39.7%), and ANME (mainly composed of ANME-1b, 0.01%–39.4%) were dominant groups in SRS; Campylobacterales (mainly composed of Sulfurovaceae, 5.9%–57.5%), Anaerolineales (composed of Anaerolineaceae, 0.8%–15.0%), Methanosarciniales (0.7%–11.8%), and Desulfobulbales (4.4-9.7%) were dominant groups in NRS; and Gammaproteobacteria (2.9%–22.1% and 4.5%–20.6%, respectively), Anaerolineae (0.8%–16.0% and 0.5%–11.5%, respectively), and Alphaproteobacteria (3.0%–8.8% and 6.2%–15.1%, respectively) were dominant groups in both Control 1 and Control 2. However, proportions of microbial communities in Control 1 and Control 2 differed at the family level, for example, Control 2 contained more Pirellulaceae (3.1%–9.2%), Nitrosopumilaceae (0.5%–17.6%), and Scalinduaceae (0.03%–13.6%). Microbial composition of different sites also showed comparative differences at the same depth. In the shallow sediments above 6 cmbsf, SRS and NRS were mainly composed of Campylobacterales, whereas Controls were mainly composed of Nitrosopumilales. In the sediments deeper than 6 cmbsf, SRS were mainly composed of Anaerolieales and ANME-1b, and NRS were mainly composed of Campylobacterales, whereas Controls did not show a dominant group.
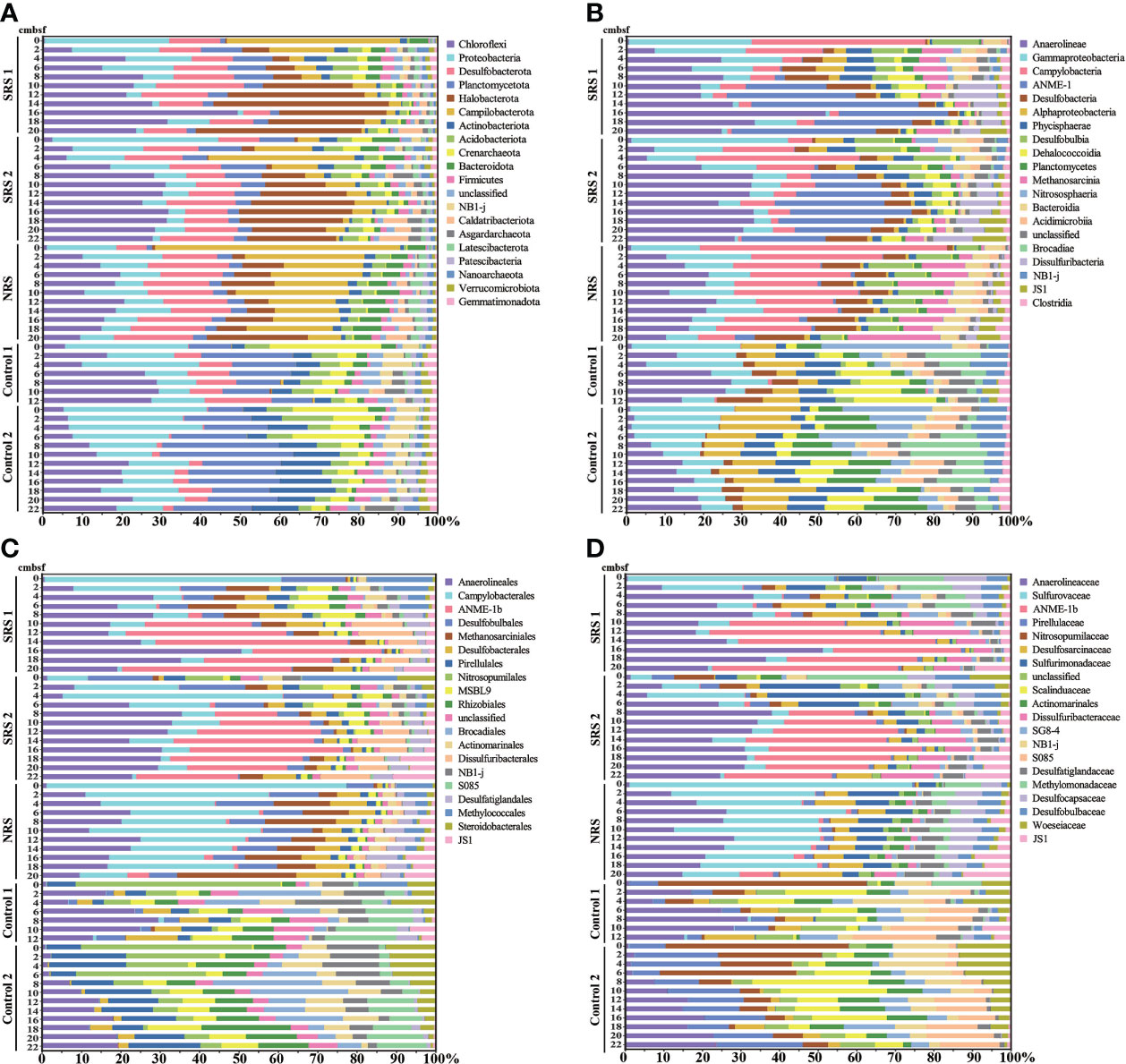
Figure 3 Microbial community compositions in all samples. (A) Microbial community compositions of the top 20 phyla. (B) Microbial community compositions of the top 20 classes. (C) Microbial community compositions of the top 20 orders. (D) Microbial community compositions of the top 20 families.
The abundance and distribution of microorganisms involved in methane and sulfur metabolisms were analyzed in detail (Figure 4). ANME were more abundant in SRS (0.04%–42.5%) and NRS (0.6%–11.7%) and almost not available in Control 1 (0%–0.006%) and Control 2 (0%–0.035%). ANME showed higher relative abundances in SRS (0.04%–42.5%) and were mainly composed of ANME-1b (0.01%–39.4%). Nevertheless, more ANME-2a-2b (0.4%–3.8%) and ANME-2c (0.2%–4.7%) compared with ANME-1 (0.01%–4.1%) were observed in NRS. As for methylotrophs and methanogens, they also showed higher sequence abundances in SRS and NRS compared with Control 1 and Control 2. Methylomonadaceae (0.01%–12% and 0.5%–7.4%, respectively) and Methanosarcinaceae (0.02%–5.4% and 0.1%–0.3%, respectively) were dominant methylotrophs and methanogens in SRS and NRS. Few methanogens were found in Controls, but methylotrophs were identified, such as Methloligellaceae (0.9%–2.7%) and Methylomirabilaceae (0.01%–0.9%) in Control 2 and Methloligellaceae (0.32%–1.07%) and Methylomonadaceae (0.12%–0.45%) in Control 1. The phylum Campylobacterota were abundant in SRS (0.7%–43.4%) and NRS (7.6%–61%) and not affluent in Control 1 (0.04%–0.36%) and Control 2 (0.003%–0.009%). At the genus level, Sulfurovum was the most abundant genus belonging to Campylobacterota in SRS and NRS, followed by Sulfurimonas. Similarly, the phylum Desulfobacterota was enriched in SRS (6.5%–20.7%) and NRS (6.8%–17.5%), but almost not available in Control 1 (0.09%–12.7%) and Control 2 (0.07%–7.5%). For SRB, SEEP-SRB2 was the most abundant genus in SRS, followed by SEEP-SRB1. Desulfatiglans and SEEP-SRB1 were abundant in NRS, whereas unclassified Syntrophobacterales were abundant in Control 1 and Control 2. Above all, the distribution and abundance of ANME, methylotrophs, methanogens, SOB, and SRB showed clear differences among sampling sites. It is suggested that a great series of active biogeochemical reactions might occur in SRS and NRS, including sulfur oxidation, sulfate reduction, anaerobic and aerobic oxidation of methane, and even methane production.
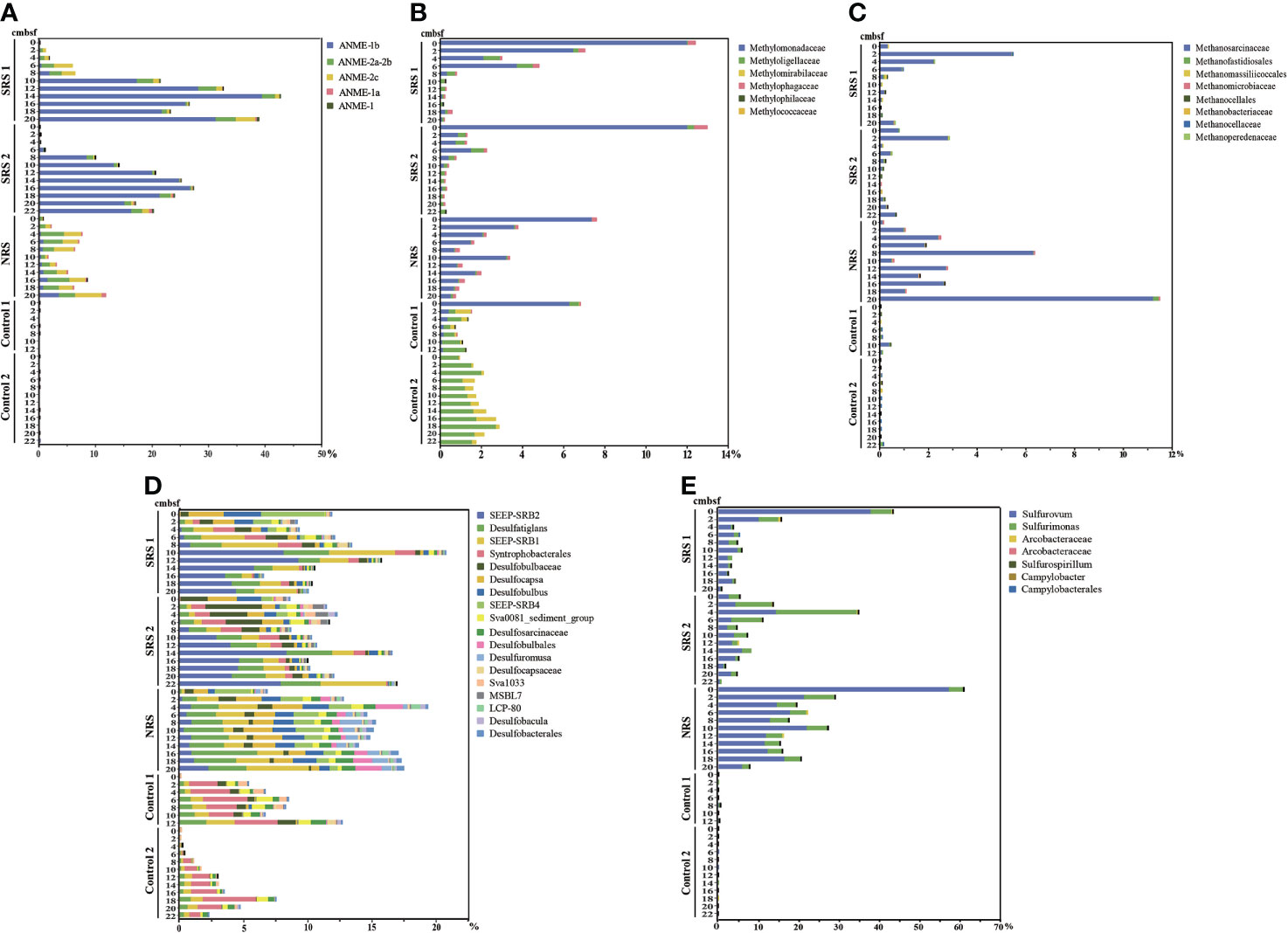
Figure 4 The abundances of main functional microbial groups in all samples. (A) Main genera of the ANME group. (B) Main genera of the methylotrophs group. (C) Main genera of the methanogens group. (D) Main genera of the SRB in the phylum Desulfobacterota. (E) Main genera of the SOB in the phylum Campylobacterota.
Microbial community compositions also varied along depths in each sampling site. In NRS, ANME-1, ANME-2, SEEP-SRB1, and SEEP-SRB2 all reached to the minimum at 10 cmbsf and started to increase with depths. Considering that the concentration of sulfate is almost 0 at 10 cmbsf, the SMTZ was proposed to locate at 10 cmbsf in NRS. Meanwhile, the abundances of Sulfurovum and Methlomonadaceae were peaked in surface sediment and decreased with depths in NRS, whereas Methanosarcinaceae showed the opposite result. In SRS, the abundances of ANME-1, SEEP-SRB1, and SEEP-SRB2 were low above 8 cmbsf and increased rapidly at 8–10 cmbsf, and the abundances of ANME-2a-2b also showed slight increase at 8 cmbsf. Interestingly, ANME-2a-2b, SEEP-SRB1, and SEEP-SRB2 all decreased below 12 cmbsf. There might be a positive correlation between ANME and SEEP-SRB, and the proposed SMTZ was approximately at 8 cmbsf in SRS. In addition, the abundances of Sulfurovum and Sulfurimonas, as well as methylotrophs especially Methylomonadaceae, were peaked in surface sediment and decreased with depths in SRS.
The distribution and relative abundance of microbial groups were different among sampling sites (Figures 5A, B). As shown in the ternary plot, Anaerolineaceae was the most abundant genus across all samples, whereas Sulfurovum and Sulfurimonas were enriched in NRS, and ANME-1b was enriched in SRS (Figure 5A). Few ANME was found in Control 1 and Control 2. In addition, all the top 15 genera were found to have significant differences (p< 0.01) between different sites in multi-group comparison stamp (Figure 5B). It could also be seen that Sulfurovum and Sulfurimonas, especially Sulfurovum, were enriched in NRS sediments, indicating sulfur oxidation in NRS. ANME-1b and SEEP-SRB2 were enriched in SRS, whereas SEEP-SRB1 and Desulfatiglans were enriched in both SRS and NRS. In general, the composition and distribution of ANME and SOB showed significant differences and might indicate different biogeochemical processes between seep sites (SRS and NRS) and non-seep sites (Control 1 and Control 2).
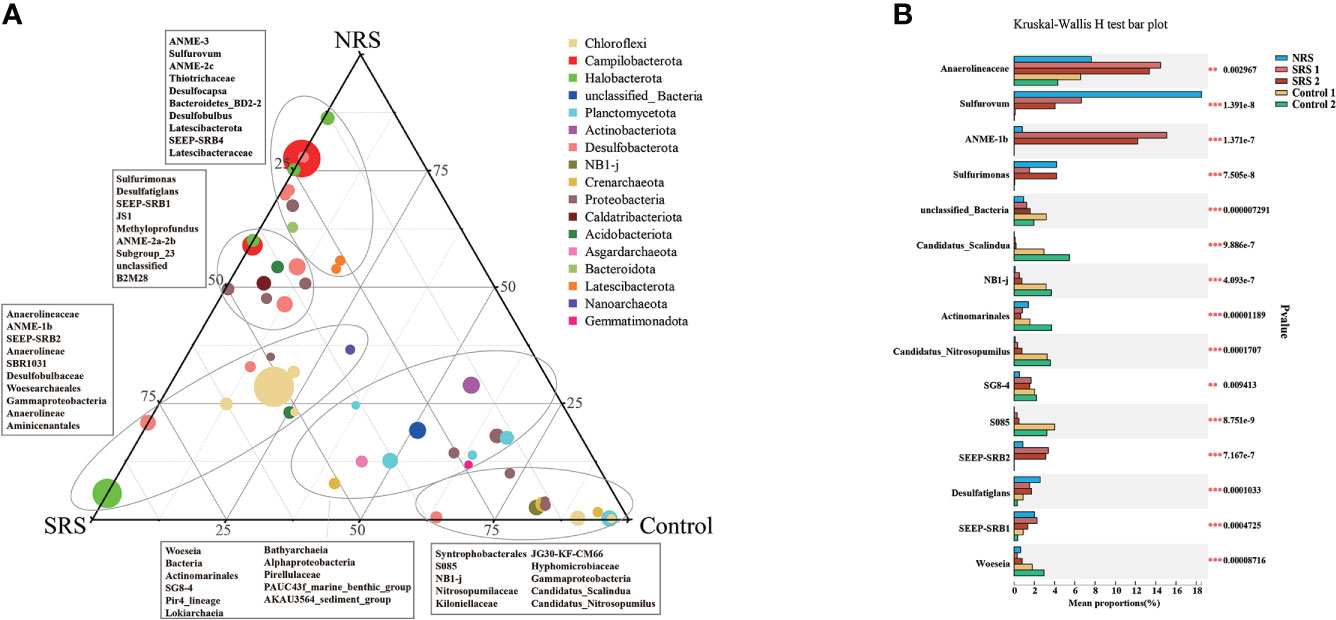
Figure 5 Distribution and relative sequence abundance of microbial groups between sampling sites. (A) Ternary plot showing the distribution and relative sequence abundance of the top 50 genera in all samples. Taxonomic affiliations were indicated by colors, whereas the size of the symbol indicated the relative sequence abundance of each genus. (B) Multi-group comparison stamp showing the relative sequence abundance and distribution differences of the top 15 genera in all samples. ** 0.001 ≤ P value ≤ 0.01; *** P value ≤ 0.001.
Co-occurrence networks were conducted to show the relationships of the top 50 microbial genera (Figure 6A), as well as the top 50 bacterial and the top 50 archaeal genera among all sediment samples (Figure S3). These three networks were all consisted of 49 nodes and 201 edges. ANME-1b was mainly associated with ANME-3, SEEP-SRB2, and Methyloprofundus; ANME-2a-2b was mainly associated with Nitrosopumilaceae and B2M28; ANME-2c was mainly associated with ANME-1b, ANME-2a-2b, ANME-3, Methyloprofundus, and Methanogenium. SEEP-SRB1 and SEEP-SRB2 were mainly associated with Desulfatiglans and JS1; SEEP-SRB2 was mainly associated with ANME-1b, ANME-2c, and Methyloprofundus; SEEP-SRB4 and Desulfobulbus were mainly associated with Candidatus Nitrosopumilus and Candidatus Scalindua. Sulfurovum and Sulfurimonas were mainly associated with Desulfobulbus, Desulfocapsa, and Thiotrichaceae. Our results found that ANME displayed a close relationship with SRB, especially ANME-1b with SEEP-SRB2.
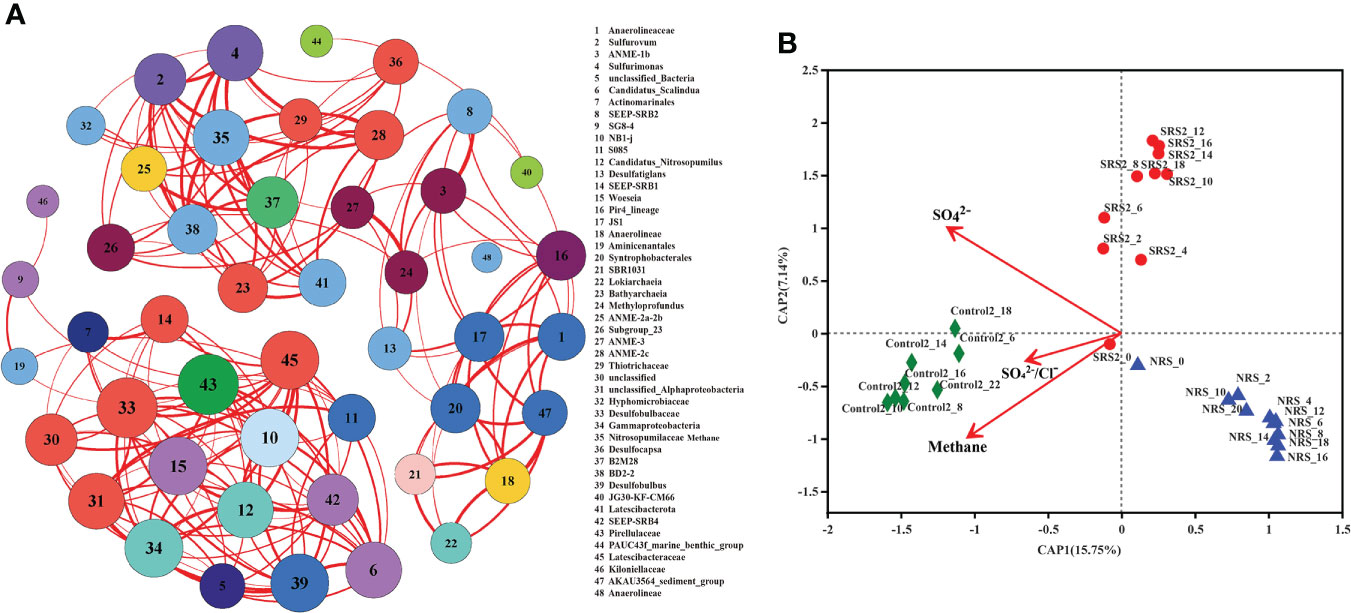
Figure 6 The relationship and environmental variables of microbial groups between sampling sites. (A) Co-occurrence networks of the top 50 genera in all samples with a Spearman’s correlation coefficient >0.7 or<−0.7 and a P-value<0.01. The size of each node indicates degree of connectivity and the color indicates the phylum that the genus affiliated to. The thickness of each line indicates strength of correlation. (B) Db-RDA the relationship of sulfate and methane changes and microbial community at the OTU level based on Bray–Curtis distances.
The predictor variables of sulfate, SO42−/Cl−, and methane in SRS 2, NRS, and Control 2 were analyzed for db-RDA based on Bray–Curtis distances to understand the influence of environmental factors on microbial communities (Figure 6B). The results showed that sulfate, SO42−/Cl−, and methane explained 22.89% (15.75% by first axis and 7.14% by second axis) of the variation in community distribution. It seemed that sulfate and methane affected microbial community structure at the OTU level to the similar extent. Sulfate concentration showed obvious negative correlations with NRS samples compared with SRS 2 and Control 2 samples. In addition, methane concentration showed positive correlations with Control 2 samples and negative correlations with SRS 2 and NRS samples.
The microbial community compositions of the 53 samples exhibited an obvious discrepancy along four sites. Microbial community compositions and multi-group comparison showed that phyla Halobacterota, Campylobacterota, and Desulfobacterota were enriched in SRS and NRS. Halobacterota were mainly composed of ANME-1b, whereas Campylobacterota and Desulfobacterota represented the groups of SOB and SRB, respectively. Cold seeps supported an enormous biomass of microbial life, which was nourished by oxidation of methane, higher hydrocarbons, and sulfide (Jørgensen and Boetius, 2007). Many studies have showed that deep-sea cold seep sediments contained abundant ANME, SRB, SOB, and methane-oxidizing bacteria (MOB) (Sun et al., 2020). In our study, these microorganisms could be the indicators that differed SRS and NRS from Control 1 and Control 2 as cold seep sites. In Control 1 and Control 2, Alphaproteobacteria, Dehalococcoidia, Phycisphaerae, and Planctomycetes were abundant, all of which were found to show high abundances in SCS deep‐sea sediments (Zhang et al., 2021). Especially, our findings of abundant Dehalococcoidia and sulfate concentration were consistent with previous results that Dehalococcoidia mediating reductive dehalogenation and potential sulfate reduction were commonly observed in sulfate-replete and deep anoxic sediments in the SCS (Graw et al., 2018; Jochum et al., 2018). In addition, abundant Syntrophobacterales in SRB also hinted Control 1 and Control 2 contain adequate sulfate even as non-seep sites.
Bacteria and archaea in the four sites showed obvious site-tendentious distribution pattern. Except for the difference between cold seep sites and non-seep sites, interestingly, cold seep sites SRS and NRS showed distinct microbial compositions. SRS was well marked by high abundance of ANME-1b of Halobacterota, whereas NRS was well marked by that of Sulfurovum of Campylobacterota (Figure 5A). The dominant microbial communities in cold seep Site F sediments varied between previous studies. For example, Sun et al. (2020) found that Campylobacterota and Deltaproteobacteria were enriched in cold seep sediment, whereas Cui et al. (2019) found that ANME-1b were the dominant microbial communities (Cui et al., 2019; Sun et al., 2020). It could be seen that the microbial compositions varied with sampling sites in Site F, which could be attributed to different maturity of the sediments influenced by distances to the seeps and the availability of organic carbon (Jing et al., 2020). In addition, it was found that the presence of reduced sediment patches resulted from cold seep fluids migrating along the bottom of the authigenic carbonate mound and escaping at its boundary (Wang et al., 2021). The reduced sediment areas were newly formed because of the in situ measured upward advection of methane-rich fluids, the ongoing AOM processes producing sulfide or pyrite, and non-existent animal aggregations, which needed more time to settle down (Wang et al., 2021). Our study of microbial compositions in the reduced sediments SRS and NRS showed potential AOM processes because of the enriched methane-metabolizing microbes. As a result, we could infer that SRS and NRS were dissimilar newborn cold seep sites with distinct microbial communities.
In NRS, ANME-2 were the dominant microorganisms, and the abundances of ANME (especially ANME-2) and SRB showed consistent tendency along depths, suggesting the positive correlation between ANME-2 and SEEP-SRB. In addition, it was found that ANME-2a-2b dominated in the high-sulfate sediments and upper layers of estimated SMTZ and was replaced by ANME-1b in the bottom layers of estimated SMTZ (Niu et al., 2017). Our result showed that the abundance of ANME-2 was positively related to the sulfate concentration, especially at 10 cmbsf, where the sulfate concentration is almost 0. Therefore, the SMTZ in NRS was inferred to locate at 10 cmbsf. In addition, the sulfate concentration showed an increased trend below 10 cmbsf and reached maximum at about 16 cmbsf. This phenomenon was also found in Site F before (Sun et al., 2022), and the increased sulfate might be attributed to the reoxidation of H2S in the deeper layers of the sediment. Correspondingly, SOB showed a higher abundance at about 18 cmbsf, which may be responsible for the reoxidation of H2S.
In SRS, the high sequence abundance of ANME could be explained by pretty high level of methane (Figure 1B). ANME in SRS were mainly composed of ANME-1b, which was consistent with many studies (Knittel et al., 2005; Ruff et al., 2016; Niu et al., 2017; Wu et al., 2018; Cui et al., 2019). Our results of ANME-1b were consistent with previous studies that ANME-1 were dominated in the deeper, anoxic, sulfide-rich sediments, and the methanogenic zone even showed high relative abundance in SMTZ (especially bottom layers) as well (Ruff et al., 2016; Niu et al., 2017). Considering ANME-1b, SEEP-SRB1, and SEEP-SRB2, the proposed SMTZ in SRS was approximately at 8 cmbsf. In addition, the strong association between ANME-1b and SEEP-SRB2 showed by co-occurrence network was in correspondence with the finding, and ANME-1 was associated with SEEP-SRB2 in Pomonte methane seep, Italy (Ruff et al., 2016). In SMTZ, ANME-1 was found to perform AOM with ANME-2, SEEP-SRB1, SEEP-SRB2, and even Desulfobacteraceae as syntrophic partners (Wu et al., 2018; Cui et al., 2019; Dong et al., 2020). Our results proved the positive correlation between ANME and SEEP-SRB. However, the correlation between ANME and Desulfobacteraceae was not found because of the low abundance of Desulfobacteraceae in SRS. In addition, fluorescence in situ hybridization and other research have found cell numbers of ANME-1 increased with sediment depths and frequently existed as single cells, indicating that ANME-1 can also perform AOM process without an archaeal or bacterial partner (Orphan et al., 2002; Knittel et al., 2005; Stokke et al., 2012; Maignien et al., 2013). In deep-sea cold seep sediments with high concentrations of hydrocarbon gases, ANME-1 might participate in AOM process via reversal of the methanogenic pathway alone and utilize electron acceptors other than sulfate (Harrison et al., 2009; Lloyd et al., 2011; Cho et al., 2017; Bowles et al., 2019; Vigneron et al., 2019). In SRS, the abundant ANME-1b was not ruled out to perform AOM alone.
Methylotrophs Methylomonadaceae, belonging to the order Methylococcales, is type I aerobic methanotrophs and was found to mainly conduct aerobic methane oxidization in Site F sediments (Broman et al., 2020; Jing et al., 2020). This might account for the high abundance of Methylomonadaceae in the surface sediment of SRS and NRS. The exact opposite vertical distribution pattern of ANME and methylotrophs in SRS and NRS proved that methane was produced in anoxic sediments and diffused upward and was oxidized successively by ANME and aerobic methylotrophs in the surface sediment. As for SOB in SRS and NRS, the distributions of Sulfurovum and Sulfurimonas were consistent with previous reports that Campylobacterota, including Sulfurovum, were microaerobic and abundant in the surface sediments of cold seeps (Wu et al., 2018; Sun et al., 2020).
Compared with SRS, the low abundance of total ANME in NRS might be attributed to the low concentration of methane. ANME-2 was enriched in NRS, and it had distinct methane-oxidizing and electron-transporting pathways from ANME-1 (Wang et al., 2014). It was reported that ANME-2a shared similarities with Methanosarcinales and performed a reversal of H2-independent methanogenesis (Wang et al., 2014). Meanwhile, complete metabolic pathway converting methane to acetate identified in ANME-2a suggested that acetate produced by ANME-2a support the heterotrophic communities as a carbon source in cold seep ecosystem (Yang et al., 2020). The Methanosarcinales are tolerant to low levels of oxygen and usually predominant in environments with high acetate concentrations (Conklin et al., 2006). It was found that Methanosarcinales constituted a major component of methanogenic archaea in sediments of Haima cold seep, including plenty of methylotrophic Methanosarcinales distributed along all depths and a few acetogenotrophic Methanosarcinales within the methanogenic zone (Niu et al., 2017). In addition, methylotrophic and acetogenotrophic Methanosarcinales were also found predominant in sediments of Site F along all depths (Jing et al., 2020). Therefore, it can be speculated that ANME-2 and Methanosarcinales enriched in NRS cooperated to implement the active conversion between methane and acetate, which was also supported by the consistent vertical distribution tendency of them. In addition, methanogens were generally unable to assimilate sulfate and able to use sulfide as the sole sulfur source. For example, Methanococcales, usually inhabited anaerobic environments with high levels of sulfide (Liu et al., 2012). This could explain the consistency between high abundance of Methanosarcinales and low sulfate concentration in NRS. Moreover, we smelt smelly H2S in NRS sediment samples, suggesting high concentration of sulfide in NRS although we did not measure.
Our result of co-occurrence network showed that ANME-1b was associated strongly with SEEP-SRB2, which was consistent with the finding conducted in Haima cold seep (Zhang et al., 2020). In addition, there were proportional abundances of SEEP-SRB1, ANME-2, and Methanosarcinales in NRS. It was reported that Methanosarcinales favored the growth of SRB by avoiding competition for the same substrate, and ANME-2a could produce acetate that might be a candidate electron shuttle between ANME-2 and SRB in AOM (Jing et al., 2020; Stams and Valentine, 2000; Plugge and Reeburgh, 2009; Yang et al., 2020). Above findings could explain the coincident distribution of Methanosarcinales, ANME-2, and SEEP-SRB1, which might coexist without competition and metabolize methane through acetate production and utilization in the cold seep environment. Compared with SRS, there was more Desulfatiglans in NRS. Desulfatiglans could oxidize acetate and pass the reducing equivalents to methanogens (Jochum et al., 2018), and it might be the reason that Desulfatiglans and Methanosarcinaceae were both abundant in NRS. Meanwhile, Desulfatiglans could produce acetate, which might be an important metabolic strategy for the microorganisms in sulfate-limited subsurface sediments and could be proved by the low sulfate concentration in NRS (Jochum et al., 2018).
SRB could produce acetate and hydrogen sulfide, which transferred upward for flourish of SOB in surface layers (Li et al., 2021). SOB played a role in sulfide oxidation, acetate assimilation, and carbon fixation in cold seep sediments (Li et al., 2021). Compared with SRS, NRS contained more SOB, especially Sulfurovum along the depths. Sulfurovum was microaerobic, and its distribution might imply differences of oxygen concentration among sites. In addition, the enrichment of SOB in NRS might be due to the inferred high concentration of sulfide, especially H2S. H2S might be released by methane oxidization and sulfate reduction (Sun et al., 2020). These might indicate comparative sulfate reduction in NRS.
In this study, a comprehensive investigation of sediment microbial communities of different sampling sites and depths in cold seep Site F was carried out. Both bacteria and archaea exhibited an obvious site-tendentious distribution pattern. The microbial community compositions and abundances of AMNE, Desulfobacterota (SRB), and Campylobacteria (SOB) indicate that SRS and NRS are newborn cold seep sites compared with non-seep sites Control 1 and Control 2. The close relationship between ANME-1b and SEEP-SRB2 was found in both SRS and NRS; however, the two cold seep sites have distinct microbial compositions. NRS can be described as notable cold seep reduced sediment with low sulfate and high H2S, nourishing abundant SEEP-SRB1, ANME-2, Methanosarcinales, and Sulfurovum, which showed similar distribution. The variations and relationships of microorganisms involved in methane and sulfur metabolisms depicted the ecological integrity of NRS commendably. Our study sheds light on the heterogeneity of different cold seep sites from the aspect of microbiology and expands current knowledge on the potential roles of microorganisms in cold seeps.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MY and X-HZ designed the experiments and analyzed the data. XZ performed most of the experiments, analyzed the data, prepared the figures and tables, and wrote the manuscript. XS attended the cruise, collected the samples and analyzed the data. HC performed the quantitative PCR analysis. PY, BZ, LiC, and LF analyzed the sulfate concentration. LeC and MW analyzed the methane concentration. MY, X-HZ, PY, and JL revised the manuscript. All the authors edited and approved the manuscript.
This work was supported by the Fundamental Research Funds for the Central Universities (202172002 and 202072003) and the National Natural Science Foundation of China (41730530, 91751202, and 41976137).
We acknowledge the Senior User Project of the R/V KEXUE (KEXUE2021GH01) for this cruise and all of the scientists and crews from Institute of Oceanology, Chinese Academy of Sciences, for their assistance with sampling during the cruise.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.957762/full#supplementary-material
Boetius A., Ravenschlag K., Schubert C. J., Rickert D., Widdel F., Gieseke A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nat. 407, 623–626. doi: 10.1038/35036572
Boetius A., Wenzhöfer F. (2013). Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6, 725–734. doi: 10.1038/ngeo1926
Borowski W. S., Paull C. K., Ussler W. (1996). Marine pore-water sulfate profiles indicate in situ methane flux from underlying gas hydrate. Geology. 24, 655–658. doi: 10.1130/0091-7613(1996)024<0655:MPWSPI>2.3.CO;2
Bowles M. W., Samarkin V. A., Hunter K. S., Finke N., Teske A. P., Girguis P. R., et al. (2019). Remarkable capacity for anaerobic oxidation of methane at high methane concentration. Geophys Res. Lett. 46, 12192–12201. doi: 10.1029/2019GL084375
Broman E., Sun X., Stranne C., Salgado M. G., Bonaglia S., Geibel M., et al. (2020). Low abundance of methanotrophs in sediments of shallow boreal coastal zones with high water methane concentrations. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01536
Cadillo-Quiroz H., Brauer S., Yashiro E., Sun C., Yavitt J., Zinder S. (2006). Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central new York state, USA. Environ. Microbiol. 8, 1428–1440. doi: 10.1111/j.1462-2920.2006.01036.x
Chen D. F., Huang Y. Y., Yuan X. L., Iii L. (2005). Seep carbonates and preserved methane oxidizing archaea and sulfate reducing bacteria fossils suggest recent gas venting on the seafloor in the northeastern south China Sea. Mar. Pet Geol. 22, 613–621. doi: 10.1016/j.marpetgeo.2005.05.002
Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics. 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cho H., Hyun J. H., You O. R., Kim M., Kim S. H., Choi D. L., et al. (2017). Microbial community structure associated with biogeochemical processes in the sulfatemethane transition zone (SMTZ) of gas-hydrate-bearing sediment of the ulleung basin, East Sea. Geomicrobiol J. 34, 207–219. doi: 10.1080/01490451.2016.1159767
Conklin A., Stensel H., Ferguson D., et al. (2006). Growth kinetics and competition between methanosarcina and Methanosaeta in mesophilic anaerobic digestion. Water Environ Res 78, 486–496. doi: 10.2175/106143006X95393
Cui H., Su X., Chen F., Holland M., Yang S., Liang J., et al. (2019). Microbial diversity of two cold seep systems in gas hydrate-bearing sediments in the south China Sea. Mar. Environ. Res. 144, 230–239. doi: 10.1016/j.marenvres.2019.01.009
Dong X., Rattray J. E., Campbell D. C., Webb J., Chakraborty A., Adebayo O., et al. (2020). Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a scotian basin cold seep. Nat. Commun. 11, 5825–5825. doi: 10.1038/s41467-020-19648-2
Edgar R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Feng D., Chen D. (2015). Authigenic carbonates from an active cold seep of the northern south China Sea: New insights into fluid sources and past seepage activity. Deep Sea Res. Part II: Topical Stud. Oceanography. 122, 74–83. doi: 10.1016/j.dsr2.2015.02.003
Feng D., Qiu J. W., Hu Y., Peckmann J., Guan H., Tong H., et al. (2018). Cold seep systems in the south China Sea: An overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Graw M. F., D'Angelo G., Borchers M., Thurber A. R., Johnson J. E., Zhang C., et al. (2018). Energy gradients structure microbial communities across sediment horizons in deep marine sediments of the south China Sea. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00729
Green-Saxena A., Dekas A. E., Dalleska N. F., Orphan V. J. (2014). Nitrate-based niche differentiation by distinct sulfate-reducing bacteria involved in the anaerobic oxidation of methane. ISME J. 8, 150–163. doi: 10.1038/ismej.2013.147
Harrison B. K., Zhang H., Berelson W., Orphan V. J. (2009). Variations in archaeal and bacterial diversity associated with the sulfate-methane transition zone in continental margin sediments (Santa Barbara basin, California). Appl. Environ. Microbiol. 75, 1487–1499. doi: 10.1128/aem.01812-08
Hoehler T. M., Alperin M. J., Albert D. B., Martens C. S. (1994). Field and laboratory studies of methane oxidation in an anoxic marine sediment: Evidence for a methanogen-sulfate reducer consortium. Global Biogeochem Cycles. 8, 451–463. doi: 10.1029/94GB01800
Jørgensen B. B., Boetius A. (2007). Feast and famine-microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/nrmicro1745
Jacomy M., Bastian M., Heymann S. (2009) Gephi: An open source software for exploring and manipulating networks. Available at: https://gephi.org/.
Jing H., Wang R., Jiang Q., Zhang Y., Peng X. (2020). Anaerobic methane oxidation coupled to denitrification is an important potential methane sink in deep-sea cold seeps. Sci. Total Environ. 748, 142459. doi: 10.1016/j.scitotenv.2020.142459
Jochum L. M., Schreiber L., Marshall I. P. G., Jørgensen B. B., Schramm A., Kjeldsen K. U. (2018). Single-cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans-related Deltaproteobacteria widely distributed in marine sediment. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02038
Kleindienst S., Ramette A., Amann R., Knittel K. (2012). Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ. Microbiol. 14, 2689–2710. doi: 10.1111/j.1462-2920.2012.02832.x
Knittel K., Lösekann T., Boetius A., Kort R., Amann R. (2005). Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 71, 467–479. doi: 10.1128/AEM.71.1.467-479.2005
Li W. L., Dong X., Lu R., Zhou Y. L., Zheng P. F., Feng D., et al. (2021). Microbial ecology of sulfur cycling near the sulfate-methane transition of deep-sea cold seep sediments. Environ. Microbiol. 23, 6844–6858. doi: 10.1111/1462-2920.15796
Liu Y., Beer L. L., Whitman W. B. (2012). Methanogens: a window into ancient sulfur metabolism. Trends Microbiol. 20 (5), 251–258. doi: 10.1016/j.tim.2012.02.002
Li H., Yang Q., Zhou H. (2020). Niche differentiation of sulfate- and iron-dependent anaerobic methane oxidation and methylotrophic methanogenesis in deep sea methane seeps. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01409
Lloyd K. G., Alperin M. J., Teske A. (2011). Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ. Microbiol. 13, 2548–2564. doi: 10.1111/j.1462-2920.2011.02526.x
Lösekann T., Knittel K., Nadalig T., Fuchs B., Niemann H., Boetius A., et al. (2007). Diversity and abundance of aerobic and anaerobic methane oxidizers at the haakon mosby mud volcano, barents Sea. Appl. Environ. Microbiol. 73, 3348–3362. doi: 10.1128/aem.00016-07
Magoč T., Salzberg S. L. (2011). Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maignien L., Parkes R. J., Cragg B., Niemann H., Knittel K., Coulon S., et al. (2013). Anaerobic oxidation of methane in hypersaline cold seep sediments. FEMS Microbiol. Ecol. 83, 214–231. doi: 10.1111/j.1574-6941.2012.01466.x
McKay L., Klokman V. W., Mendlovitz H. P., LaRowe D. E., Hoer D. R., Albert D., et al. (2016). Thermal and geochemical influences on microbial biogeography in the hydrothermal sediments of guaymas basin, gulf of California. Environ. Microbiol. Rep. 8, 150–161. doi: 10.1111/1758-2229.12365
Mills H. J., Hodges C., Wilson K., Macdonald I. R., Sobecky P. A. (2003). Microbial diversity in sediments associated with surface-breaching gas hydrate mounds in the gulf of Mexico. FEMS Microbiol. Ecol. 46, 39–52. doi: 10.1016/s0168-6496(03)00191-0
Niemann H., Lösekann T., de Beer D., Elvert M., Nadalig T., Knittel K., et al. (2006). Novel microbial communities of the haakon mosby mud volcano and their role as a methane sink. Nat. 443, 854–858. doi: 10.1038/nature05227
Niu M., Fan X., Zhuang G., Liang Q., Wang F. (2017). Methane-metabolizing microbial communities in sediments of the haima cold seep area, northwest slope of the south China Sea. FEMS Microbiol. Ecol. 93, 10.1093. doi: 10.1093/femsec/fix101
Orphan V. J., Hinrichs K. U., Ussler W. 3rd, Paull C. K., Taylor L. T., Sylva S. P., et al. (2001a). Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl. Environ. Microbiol. 67, 1922–1934. doi: 10.1128/aem.67.4.1922-1934.2001
Orphan V. J., House C. H., Hinrichs K. U., McKeegan K. D., DeLong E. F. (2001b). Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science. 293, 484–487. doi: 10.1126/science.1061338
Orphan V. J., House C. H., Hinrichs K. U., McKeegan K. D., DeLong E. F. (2002). Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc. Natl. Acad. Sci. U. S. A. 99, 7663–7668. doi: 10.1073/pnas.072210299
Pernthaler A., Dekas A. E., Brown C. T., Goffredi S. K., Embaye T., Orphan V. J. (2008). Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics. Proc. Natl. Acad. Sci. U. S. A. 105, 7052–7057. doi: 10.1073/pnas.0711303105
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Ruff S. E., Kuhfuss H., Wegener G., Lott C., Ramette A., Wiedling J., et al. (2016). Methane seep in shallow-water permeable sediment harbors high diversity of anaerobic methanotrophic communities, Elba, Italy. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00374
Schreiber L., Holler T., Knittel K., Meyerdierks A., Amann R. (2010). Identification of the dominant sulfate-reducing bacterial partner of anaerobic methanotrophs of the ANME-2 clade. Environ. Microbiol. 12, 2327–2340. doi: 10.1111/j.1462-2920.2010.02275.x
Sogin M. L., Morrison H. G., Huber J. A., Mark Welch D., Huse S. M., PR N., et al. (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc. Natl. Acad. Sci. U. S. A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Stams A. J., Plugge C. M. (2009). Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 7, 568–577. doi: 10.1038/nrmicro2166
Stokke R., Roalkvam I., Lanzen A., Haflidason H., Steen I. H. (2012). Integrated metagenomic and metaproteomic analyses of an ANME-1-dominated community in marine cold seep sediments. Environ. Microbiol. 14, 1333–1346. doi: 10.1111/j.1462-2920.2012.02716.x
Sun Y., Niu M. Y., Liu Q., Zhuang G. C., Wang F. P. (2022). Diversity and distribution of microorganisms in the sediment of Formosa cold seep in south China Sea. Acta Microbiol. Sin. 62, 2001–2020. doi: 10.13343/j.cnki.wsxb.20220345
Sun Q. L., Zhang J., Wang M. X., Cao L., Du Z. F., Sun Y. Y., et al. (2020). High-throughput sequencing reveals a potentially novel sulfurovum species dominating the microbial communities of the seawater-sediment interface of a deep-sea cold seep in south China Sea. Microorganisms. 8, 687. doi: 10.3390/microorganisms8050687
Valentine D. L., Reeburgh W. S. (2000). New perspectives on anaerobic methane oxidation. Environ. Microbiol. 2, 477–484. doi: 10.1046/j.1462-2920.2000.00135.x
Vigneron A., Alsop E. B., Cruaud P., Philibert G., King B., Baksmaty L., et al. (2019). Contrasting pathways for anaerobic methane oxidation in gulf of Mexico cold seep sediments. mSystems. 4, e0009–18. doi: 10.1128/mSystems.00091-18
Vigneron A., Cruaud P., Pignet P., Caprais J. C., Cambon-Bonavita M. A., Godfroy A., et al. (2013). Archaeal and anaerobic methane oxidizer communities in the Sonora margin cold seeps, guaymas basin (Gulf of California). ISME J. 7, 1595–1608. doi: 10.1038/ismej.2013.18
Walters W., Hyde E. R., Berg-Lyons D., Ackermann G., Humphrey G., Parada A., et al. (2015). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 1 (1), e00009–e00015. doi: 10.1128/mSystems.00009-15
Wang B., Du Z., Luan Z., Zhang X., Yan J. (2021). Seabed features associated with cold seep activity at the Formosa ridge, south China Sea: Integrated application of high-resolution acoustic data and photomosaic images. Deep Sea Res. Part I Oceanographic Res. Papers. 177, 103622. doi: 10.1016/j.dsr.2021.103622
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Wang F.-P., Zhang Y., Chen Y., He Y., Qi J., Hinrichs K.-U., et al. (2014). Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J. 8, 1069–1078. doi: 10.1038/ismej.2013.212
Wu Y., Qiu J.-W., Qian P.-Y., Wang Y. (2018). The vertical distribution of prokaryotes in the surface sediment of jiaolong cold seep at the northern south China Sea. Extremophiles. 22, 499–510. doi: 10.1007/s00792-018-1012-0
Yang S., Lv Y., Liu X., Wang Y., Fan Q., Yang Z., et al. (2020). Genomic and enzymatic evidence of acetogenesis by anaerobic methanotrophic archaea. Nat. Commun. 11, 3941. doi: 10.1038/s41467-020-17860-8
Zhang T., Xiao X., Chen S., Zhao J., Chen Z., Feng J., et al. (2020). Active anaerobic archaeal methanotrophs in recently emerged cold seeps of northern south China Sea. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.612135
Zhang Y., Yao P., Sun C., Li S., Shi X., Zhang X. H., et al. (2021). Vertical diversity and association pattern of total, abundant and rare microbial communities in deep-sea sediments. Mol. Ecol. 30, 2800–2816. doi: 10.1111/mec.15937
Zhao B., Yao P., Bianchi T. S., Xu Y., Liu H., Mi T., et al. (2017). Early diagenesis and authigenic mineral formation in mobile muds of the changjiang estuary and adjacent shelf. J. Mar. Syst. 172, 64–74. doi: 10.1016/j.jmarsys.2017.03.001
Keywords: cold seep, South China Sea, reduced sediments, microbial communities, anaerobic methanotrophic archaea, sulfate-reducing bacteria, sulfur-oxidizing bacteria
Citation: Zhai X, Shi X, Cheng H, Yao P, Zhao B, Chen L, Liu J, Cao L, Wang M, Fu L, Zhang X-H and Yu M (2022) Horizontal and vertical heterogeneity of sediment microbial community in Site F cold seep, the South China Sea. Front. Mar. Sci. 9:957762. doi: 10.3389/fmars.2022.957762
Received: 31 May 2022; Accepted: 25 July 2022;
Published: 08 September 2022.
Edited by:
Hongmei Jing, Institute of Deep-Sea Science and Engineering (CAS), ChinaReviewed by:
Xiyang Dong, Third Institute of Oceanography of the Ministry of Natural Resources, ChinaCopyright © 2022 Zhai, Shi, Cheng, Yao, Zhao, Chen, Liu, Cao, Wang, Fu, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Yu, eXVtaW5Ab3VjLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.