 Hao Kang1,2†
Hao Kang1,2† Yongxiang Yu2,3†
Yongxiang Yu2,3† Meijie Liao2,3*
Meijie Liao2,3* Yingeng Wang2,3*
Yingeng Wang2,3* Guanpin Yang1
Guanpin Yang1 Zheng Zhang2,3Bin Li2,3Xiaojun Rong2,3Chunyuan Wang2,3
Zheng Zhang2,3Bin Li2,3Xiaojun Rong2,3Chunyuan Wang2,3- 1College of Marine Life Sciences, Ocean University of China (OUC), Qingdao, China
- 2Key Laboratory of Maricultural Organism Disease Control, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 3Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
Vibrio bacteria, particularly members of the Harveyi clade, are the most important pathogens of aquatic organisms that cause significant economic losses in the world. It is difficult to provide specific data on taxa of the Harveyi clade for biological research and prevention strategies. Therefore, we conducted an extensive phenotypic and antibiotic resistance study, as well as phylogenetic and molecular typing of 192 isolates of the Harveyi clade collection from 2000 to 2020 with a typical interannual difference from a coastal area in China. The isolates had a significant interspecific genetic and antibiotic resistance diversity. Based on the multilocus sequence analysis (MLSA) of housekeeping genes (gyrB, pyrH, recA, and atpA), 192 Harveyi clade isolates were rapidly and accurately classified into 10 species. The population of these isolates was composed of 95 sequence types (STs), of which 92 STs were newly identified, indicating a high degree of genetic diversity. ST327 ranked first, accounting for 11.5% of the total number of isolates (22 out of 192), followed by ST215 with 6.25%, while 63 STs included single isolates. At the metabolic level, the physiological and biochemical experiments revealed that all the Harveyi clade isolates were positive for oxidase and negative for melibiose. The isolates showed a varied tolerance to 11 antibiotics. No isolates were resistant to neomycin. The percentages of sulfadimidine-resistant strains (61 out of 192), sulfadiazine (44 out of 192), sulfamonomethoxine (44 out of 192), sulfamethoxazole (33 out of 192), thiamphenicol (34 out of 192), ciprofloxacin (52 out of 192), and enrofloxacin (31 out of 192) were 31.77%, 22.92%, 22.92%, 17.19%, 17.71%, 27.08%, and 16.15%, respectively. A proportion of 61.8% of the isolates presented a multiple antibiotic resistance index (MARI) lower than 0.1, indicating that the risk of antibiotic resistance transmission of most of the Harveyi clade is low in mariculture systems in China. These results provide substantial data to support further studies on the identification and genetic and metabolic diversity of Harveyi clade isolates in mariculture systems in China.
Introduction
Vibrio spp. are curved rods of Gram-negative bacteria composed of halophile species with significant biodiversity and present naturally in marine, estuarine, and freshwater systems worldwide (Thompson et al., 2004; Baker-Austin et al., 2017; Baker-Austin et al., 2018; Hackbusch et al., 2020). A recent evolutionary event within the genus Vibrio occurred 39 million years ago (Sawabe et al., 2007). The genus Vibrio has over 100 species that have been classified into 14 clades (Ruwandeepika et al., 2012; Romalde et al., 2014). The colloquially “Harveyi clade” is considered as the most severely pathogenic Vibrio cluster of aquatic organisms, capable of causing more than 50 different types of aquatic animal diseases and potentially disrupting the aquaculture system due to its high mortality (Del Gigia-Aguirre et al., 2017; Wang et al., 2021). Multiple Harveyi clade species have been implicated in the aquatic diseases from different countries, causing mortalities up to 100% of some common economic aquatic animals such as Litopenaeus vannamei and tilapia, which are widely farmed around the world (Tran et al., 2013; Prithvisagar et al., 2021). Harveyi clade species were thought to be the major pathogens threatening the health development of the aquaculture industry globally.
The Harveyi clade consists of V. harveyi and 11 related species, i.e., V. alginolyticus, V. parahaemolyticus, V. campbellii, V. rotiferianus, V. natriegens, V. azureus, V. mytili, V. owensii, V. jasicida, V. diabolicus, and V. sagamiensis (Lin et al., 2010; Cano-Gomez et al., 2011; Ruwandeepika et al., 2012; Goudenège et al., 2014). These species share high phenotypic and genotypic homology, with 16S rDNA gene sequence similarities greater than 97% and DNA–DNA re-association values close to 70%, making it challenging to differentiate them (Sawabe et al., 2007; Cano-Gomez et al., 2009). Furthermore, because the maritime environment is complex and changing through time and space, marine microorganisms that live in it have adapted to this environment, and hence present a diverse range of species, metabolic types, functional gene composition, and ecological functions (Fraser et al., 2007). Traditional biochemical tests and single-gene sequencing led often to the misidentification of different species of the Harveyi clade (Gomez-Gil et al., 2004; Cano-Gomez et al., 2009).
The development in bacterial identification technology has been promoted by molecular identification techniques such as multilocus sequence analysis (MLSA), core genome tree, average nucleotide identity (ANI), DNA–DNA hybridization (DDH), and genomic characteristic dissimilarity (Richter and Rosselló-Móra (2009); Thompson et al., 2009; Fu et al., 2015). Meanwhile, bacterial typing systems are used to distinguish genera, species, and strains according to their phenotypic and genetic characteristics, e.g., restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), pulsed-field gel electrophoresis (PFGE), multilocus sequence typing (MLST), core genome MLST (cgMLST), and enterobacterial repetitive intergenic consensus PCR (ERIC-PCR) (Botella et al., 2002; Yang et al., 2017; Alikhan et al., 2018; Yan et al., 2021). MLSA is rapid and robust in classifying all prokaryotes using a universal set of genes, and successful in establishing species-level taxonomy within the Harveyi clade based on different levels of genes (Gevers et al., 2005; Cano-Gomez et al., 2009). Pascual et al. (2010) reported that the concatenated sequences of rpoD, rctB, and toxR can be used to identify species of Vibrio strains appropriately. Coincidentally, the gene combination topA-mreB has also provided a practical yet accurate approach for routine identification of V. harveyi-related species (Cano-Gomez et al., 2011). Furthermore, MLST outperformed the other typing techniques in studies of evolutionary, phylogenetic, and population genetics, as it provided unambiguous data using a large international database (https://pubmlst.org/) (Maiden et al., 1998; Aanensen and Spratt, 2005; Harun et al., 2021). Typing can also be performed by directly identifying the nucleotide sequence and variation of multiple housekeeping genes of strains, which has been applied to numerous prokaryotes as well eukaryotic organisms, including the Harveyi clade species V. parahaemolyticus and the general database of Vibrio spp. (Han et al., 2015; Jelocnik et al., 2019).
Antibiotic resistance of Harveyi clade isolates is an important indicator of metabolic and phenotypic diversity. As the most important pathogen clade in aquatic organisms, Harveyi clade strains caused more than 50 kinds of aquatic animal diseases and were considered to be a major economic threat to the aquaculture industry (Lin et al., 2010; Goudenège et al., 2014; Wang et al., 2021). Among them, Vibrio harveyi is one of the most serious Vibrio pathogenic, which can infect the most diverse range of aquatic animals in the world (Santhyia et al., 2015). Therefore, analyzing the antimicrobial resistance of pathogens in a particular area is crucial to the development of effective preventive measures.
China is the world’s largest aquaculture producer. The total content of aquatic products exceeded 65 million tons in 2020, which guaranteed the national demand for high-quality protein (China Fisheries Yearbook, 2021). However, pathogens have become a restrictive factor for the development of aquaculture in China, resulting in annual losses of over 20 billion RMB, with Vibrio spp. proved as the most important pathogen of marine organisms (China Fisheries Yearbook, 2021). Moreover, the Chinese mariculture sector is characterized by rich and diversified breeding. In different aquaculture systems, a single bacterial species exhibits diverse metabolic phenotypes and ecological functions. During the past two decades, various species of the Harveyi clade have been isolated from infected animal in mariculture in China with dominance higher than 60% in our laboratory. In the present study, the genetic populations and evolutionary relationship of Harveyi clade strains isolated from coastal areas of a mariculture system in China were investigated based on different gene classes and levels. Following that, the prevalence, antibiotic resistance, and genetic diversity of all isolates were analyzed using phenotypic and molecular typing methods. The results of this study provide a theoretical foundation for understanding the differences in multivariate epigenetic mechanisms among species of the Harveyi clade from China with typical interannual variations, as well as basic data for prevention and treatment of aquatic animal diseases.
Materials and methods
Strains and culture conditions
We chose 192 isolates identified as Harveyi clade with typical temporal differences, isolated from animal parts of marine and mariculture environments infected with bacteria with prevalence and dominance of more than 60% in coastal areas of China from 2000 to 2020. Furthermore, 12 type strains served as process control, namely, V. harveyi ATCC 14348, V. campbellii ATCC 25920, V. owensii DSM 23055, V. alginolyticus ATCC 17749, V. natriegens ATCC 14048, V. mytili LMG 19157, V. parahaemolyticus ATCC 17802, V. jasicida DSM 21061, V. rotiferianus DSM 17186, V. azureus NBRC 104587, V. sagamiensis NBRC 104859, and V. diabolicus HE800.
All the strains were preserved in our laboratory in cryo-vials with 20% (v/v) glycerol at −80°C. Data on the isolates are depicted in Supplementary Table 1. The isolates were cultivated on trypticase soy broth (TSB) plates (TSB supplemented with 1.5% agar) at 28°C for 24 h until use.
DNA extraction
Genomic DNA was extracted from the cultivated isolates following the protocol described by Rahman et al. (2014) with minor modifications. A single colony was collected from a fresh culture and resuspended in 100 μl of nuclease-free water, vortexed for 5 s, and incubated at 99°C for 10 min. After that, the suspension was vortexed again and centrifuged at 12,000 relative centrifugal force (RCF) for 5 min. The supernatant was then transferred to a fresh tube and stored at −20°C. The concentration and purity of the extracted DNA were assessed using NanoDrop 2000. Solutions with a 260/280 ratio of 1.7 to 2.0 were used for PCR assays.
PCR amplification and sequencing
PCR amplification and sequencing of the 16S rDNA or 16S rRNA gene were performed according to Weisburg et al. (1991) using the primers 27F and 1492R. The four housekeeping genes gyrB, pyrH, recA, and atpA were amplified and sequenced using the primers and the amplification conditions described by Rahman et al. (2014). The complete list of genes analyzed in this study and all primers used for PCR amplification and sequencing are listed in Table 1. A PCR template of approximately 100 ng of DNA was used for amplification. PCR products were visualized on a 1.5% agarose gel. The qualified rate of the products was determined according to the molecular weight standard. Sequencing and purification of PCR products were performed by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China). The DNA sequences were analyzed using the BLAST tool of GenBank and optimized for highly similar sequences (Mega BLAST) (Kumari et al., 2020).
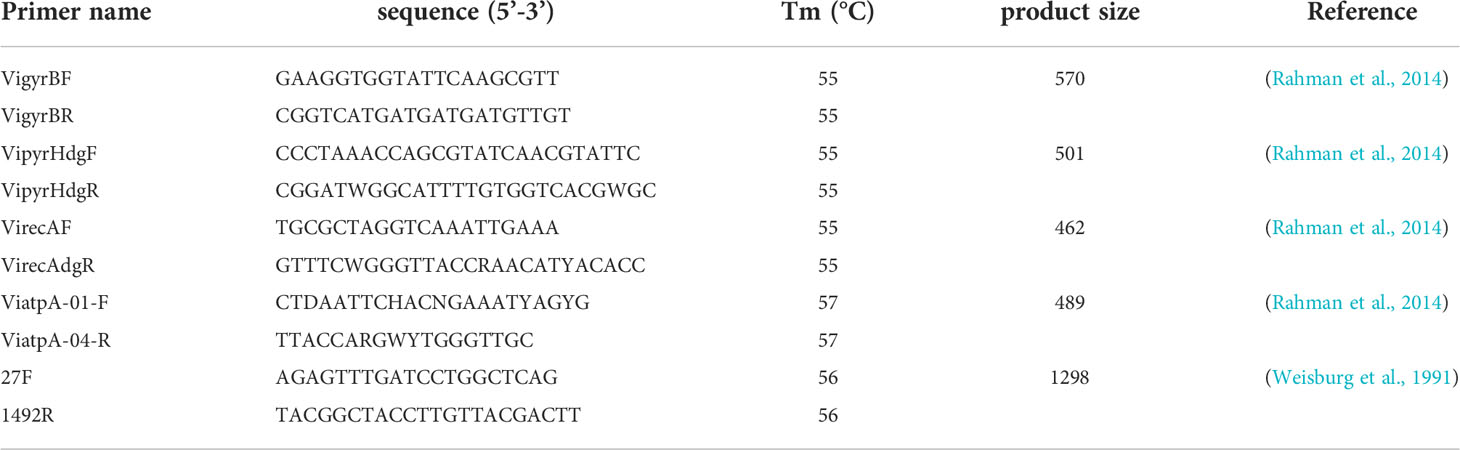
Table 1 Primers used for amplification and sequencing of Harveyi clade isolates.
Phylogenetic analysis and genetic diversity
Sequences of the genes 16S rDNA, gyrB, pyrH, recA, and atpA were aligned using ClustalX (Tamura et al., 2013). Phylogenetic analysis based on the genes 16S rDNA (1298 bp), gyrB, pyrH, recA, and atpA, as well as various concatenations of four protein-coding loci (gyrB, pyrH, recA, and atpA), were conducted using the neighbor-joining (NJ) approach. Bootstrap (BT) support for individual nodes was calculated with the Kimura 2-parameter model using 1000 BT replications. The trees were constructed using MEGA 6.0 software. A fully resolved and highly harmonious tree topology was obtained, depicting individual strain relationships.
Morphological, physiological, and biochemical analyses
Single colonies of the Harveyi clade isolates were picked up and inoculated on thiosulfate-citrate–bile salts–sucrose (TCBS) agar, TSB, and HB7011-5 Vibrio chromogenic medium (hopebio Biotechnology Co., Ltd, China) plates and incubated at 28°C for 24 h, after which colony morphology was observed. Twenty-three biochemical tests were evaluated by the following tests: gram staining, o-Nitrophenyl β-D-galactopyranoside (ONPG), urease, xylose, sucrose, Simmons citrate agar, lysine decarboxylase, gelatin, amygdalin, melibiose, α-L-rhamnopyranose monohydrate, hydrogen sulfide (H2S), malonic acid disodium salt, mannitol, D-glucose, methyl red and Voges-Proskauer tests (MR-VP), inositol, ornithine decarboxylase, DL-arabinose, sorbitol, lactose, arginine double hydrolase, tryptone water, and glucose oxidase. All the selected biochemical tests were determined with the Bacterial biochemical identification kit (hopebio Biotechnology Co., Ltd, China), according to the manufacturer’s instructions.
Antimicrobial susceptibility testing
The antibiotic susceptibility of the Harveyi clade isolates was tested on TSB following the Kirby–Bauer disc diffusion method (K-B method) and the Clinical and Laboratory Standards Institute [CLSI] (2017) guidelines. Eleven antimicrobial agents were tested, namely, aminoinositols: florfenicol (FFC, 30 μg/disk) and thiamphenicol (THI, 30 μg/disk); tetracycline: neomycin (NEO, 30 μg/disk) and doxycycline (DOX, 30 μg/disk); quinolones: ciprofloxacin (CIP, 5 μg/disk), enrofloxacin (ENR, 10 μg/disk), and flumequine (FLU, 30 μg/disk); and sulfonamides: trimethoprim/sulfamethoxazole (SMZ, 1.25 μg/disk and 25 μg/disk, respectively), sulfamonomethoxine (SMM, 250 μg/disk), sulfadiazine (SDI, 250 μg/disk), and sulfadimidine (SDM, 250 μg/disk). The isolates were inoculated in TSB with 1.5% NaCl solution and adjusted to an optical density (OD) 0.5 McFarland standard after a 24-h incubation period at 28°C. The antibacterial discs were then applied after 100 μl of the bacterial solution was equally spread over the agar plates. The inhibition zones were measured using the SCAN 4000 automatic image analysis colony counter (Interscience, France) after 20 h of incubation at 28°C. The reference strain Escherichia coli ATCC 25922 was used for quality control. All the experiments were performed in triplicates. Each strain was classified as resistant, intermediate, or susceptible, according to Clinical and Laboratory Standards Institute [CLSI] (2017). The multiple antibiotic resistance index (MARI) was calculated based on the formula described by Krumperman (1983).
MLST and UPGMA analysis
PCR primers, amplification conditions, and housekeeping gene sequencing methods were carried out following the methods of Rahman et al., (2014). The Vibrio spp. Pubmlst database (http://pubmlst.org/Vibrio.spp) was used to obtain the allele number and define the sequence type (ST). If STs or alleles other than the database are identified, submit the new allele to the database administrator to obtain a serial number of the newly identified allele or STs (Jiang et al., 2019). Using goeBURST (http://goeBURST.phyloviz.net) and the MLST classification data, we created a minimal spanning tree based on PHYLOViZ 2.0 (Ribeiro-Gonçalves et al., 2016) and assigned STs to clonal complexes (CC). The nucleotide diversity was determined using the software DNA Sequence Polymorphism DnaSP version 6.12.03 (Rozas et al., 2017). The number of polymorphic sites, GC content, and the ratio of nonsynonymous to synonymous substitutions (dN/dS) were calculated by START v 2.0 (Jolley et al., 2001).
Results
Epidemiological investigation of the Harveyi clade isolates
A total number of 192 isolates were identified, of which 101 isolates were isolated from fish (52.7%), 60 in flatfish (31.3%), 22 in grouper (11.6%), 19 in other species (9.8%), 64 in shrimp (L. vannamei) (33.3%), 23 in sea cucumber (12%), and 4 in crab (2%) (Figure 1). In addition, 63 of the total strains (33.2%) were from 2010 and earlier, while 129 strains (66.8%) were collected from 2011 to 2020. Based on the BLASTn search of the 16S rDNA gene, V. harveyi (79 isolates), V. alginolyticus (63 isolates), V. owensii (19 isolates), V. rotiferianus (11 isolates), V. natriegens (9 isolates), V. azureus (3 isolates), V. parahaemolyticus (2 isolates), V. campbellii (2 isolates), V. jasicida (2 isolates), and V. sagamiensis (2 isolates) were identified.
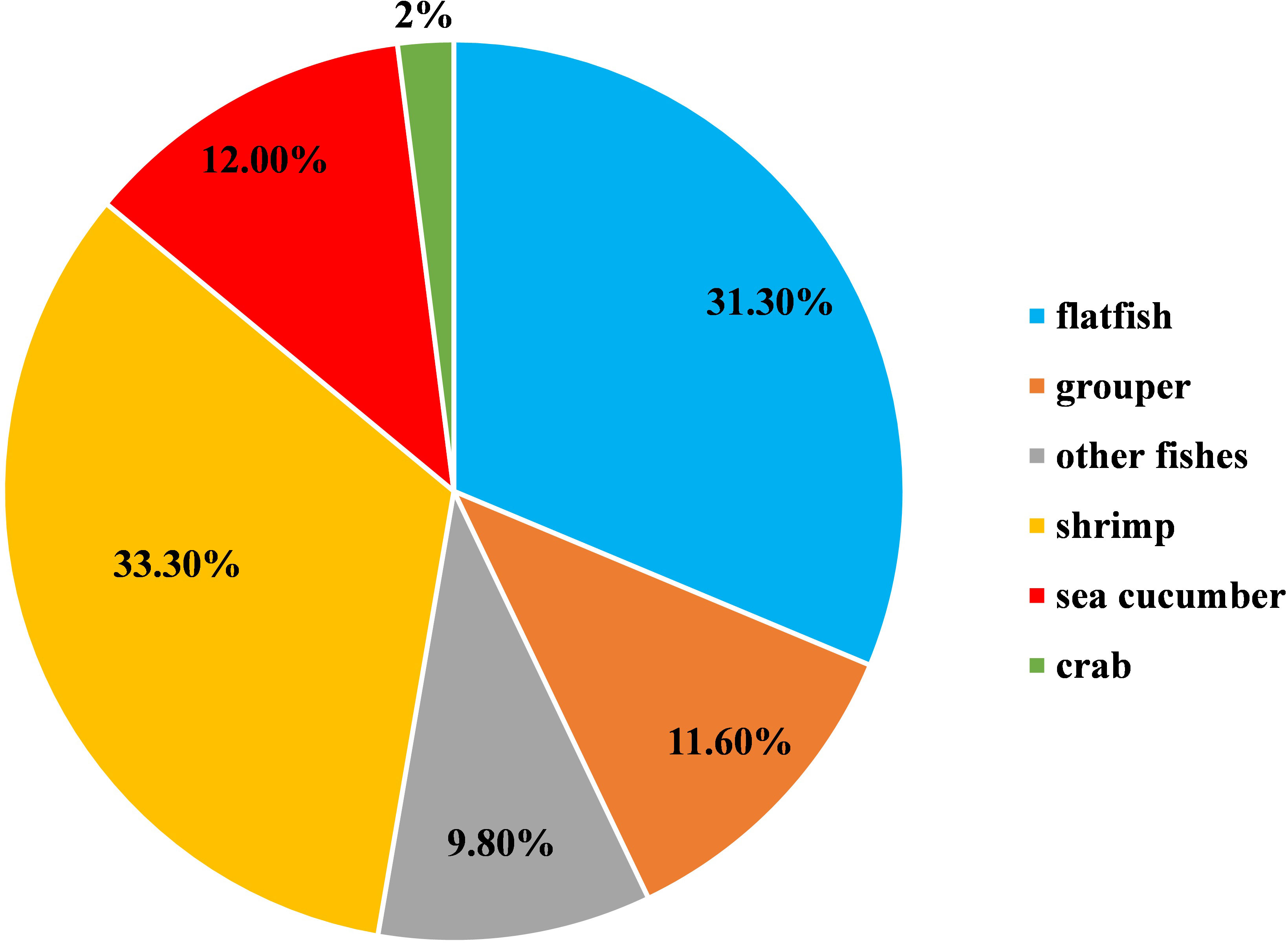
Figure 1 Host composition of the 192 Harveyi clade isolates (100%).
Taxonomic evolution of the Harveyi clade based on MLSA
As shown in Figure 2, the phylogenetic tree cluster analysis based on the concatenated sequences of the genes gyrB, pyrH, recA, and atpA divided the 192 isolates together with 12 reference strains into 10 distinct clades (representing 10 gene species) and three outgroups. The numbers of the 10 gene species were distributed as follows: 77 V. harveyi, 41 V. alginolyticus, 19 V. parahaemolyticus, 17 V. owensii, 17 V. natriegens, 12 V. rotiferianus, 3 V. campbellii, 3 V. diabolicus, 2 V. jasicida, and 1 V. mytili. Two subgroups emerged from all the strains of V. alginolyticus and V. diabolicus. When we compared the phylogenetic tree constructed using only 16S rDNA sequencing, we could not identify the strains of V. harveyi, V. campbellii, V. rotiferianus, V. parahaemolyticus, and V. owensii within the Harveyi clade (99% to 100% sequence identities) (Supplementary Figure 1).
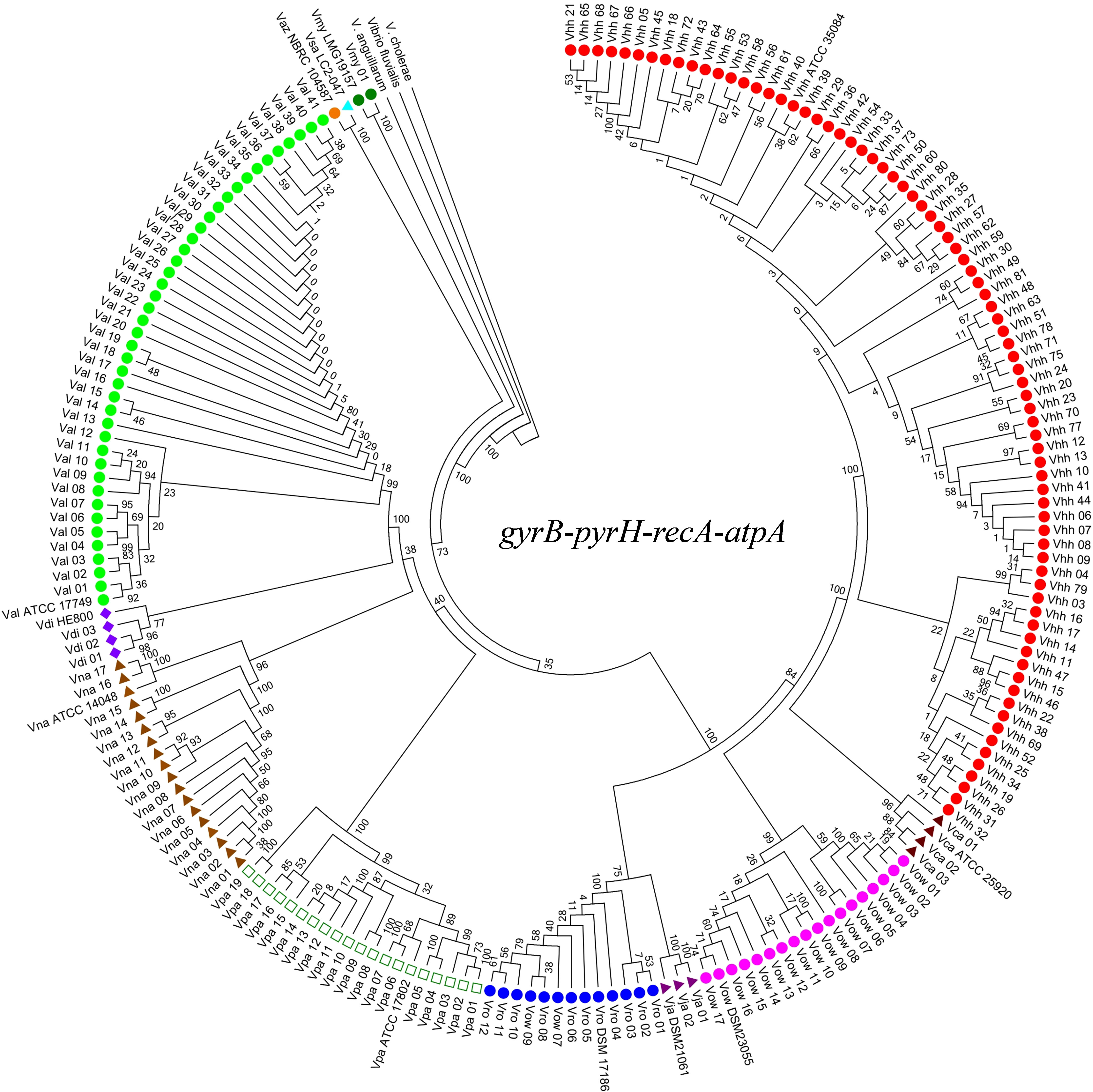
Figure 2 Neighbor-joining phylogenetic analysis based on concatenated gene sequences of four protein-coding loci (i.e., gyrB-pyrH-recA-atpA). Vhh, V. harveyi; Val, V. alginolyticus; Vro, V. rotiferianus; Vca, V. campbellii; Vpa, V. parahaemolyticus; Vow, V. owensii; Vna, V. natriegens; Vdi, V. diabolicus; Vsa, V. sagamiensis; Vaz, V. azureus; Vja, V. jasicida; Vmy, V. mytili. Bootstrap values based on 1,000 resamplings are shown as percentages at the branch nodes.
Furthermore, the phylogenetic analysis based on different gene combination levels indicated that it was possible to accurately classify Vibrio species within the Harveyi clade based on the phylogenetic tree constructed using the concatenated sequences of the four genes. After analyzing all the concatenated genes, the three-locus phylogeny (gyrB, pyrH, and atpA) was consistent with the four-locus phylogeny and the Harveyi clade was identified (Supplementary Figure 2). As for the two-locus concatenated sequences, the genes gyrB and atpA identified V. owensii, V. parahaemolyticus, and V. rotiferianus, while the genes gyrB and pyrH identified V. owensii (Supplementary Figure 3). A slightly more complicated grouping emerged from all the single-gene trees of the nucleotide sequences (Supplementary Figure 4). Nonetheless, atpA was the only gene with the highest discriminatory power. The aforementioned results indicate that the MLSA scheme based on the concatenated sequences of four protein-coding genes can offer a robust phylogenetic reconstruction to resolve unitary relationships of the Harveyi clade isolates. Meanwhile, the three-gene scheme based on gyrB, pyrH, and atpA is suitable for Harveyi clade identification to some extent.
The 16S rDNA gene, gyrB, pyrH, atpA, and recA sequences determined in this study have been deposited in the GenBank of NCBI under the accession numbers ON437347-ON437538, ON469581-ON469772, ON778048-ON778239, ON778240-ON778431, and ON4491819-ON492010, respectively.
Phenotypic and physiological characterization of Harveyi clade bacteria
The phenotypic analysis of the Harveyi clade isolates in this study was in accordance with Bergey’s manual of determinative bacteriology (Holt et al., 1994). All the Harveyi clade isolates growing on TCBS agar were yellowish or greenish colonies, Gram-negative, oxidase-positive, and glucose-fermenting bacteria. Macroscopic observation showed that the upper surface of the colonies of the Harveyi clade isolates on the solid medium was raised, smooth, and wet (Supplementary Figure 5). These characteristics can be first attributed to the genus Vibrio. The biochemical features indicated that the Harveyi clade isolates had a positive oxidase and were unable to ferment melibiose, rhamnose, and amygdalin. In addition, 153 isolates were able to ferment sucrose and showed negative MR-VP and positive inositol reactions, 85 isolates had a positive ONPG reaction, 106 isolates produced a positive urease reaction, 175 isolates showed a positive lysine decarboxylase test, 55 isolates had a positive gelatin test, and 107 isolates showed a positive arginine double hydrolase test and were unable to decompose hydrogen sulfide (Table 2). On the other hand, all the isolates of V. harveyi, V. alginolyticus, V. campbellii, V. owensii, and V. diabolicus tested positive for lysine decarboxylase. V. rotiferianus, V. campbellii, and V. diabolicus were all citrate and urease positive (Table 2). The results indicated that these biochemical markers may be employed for the early screening of these Harveyi clade species.
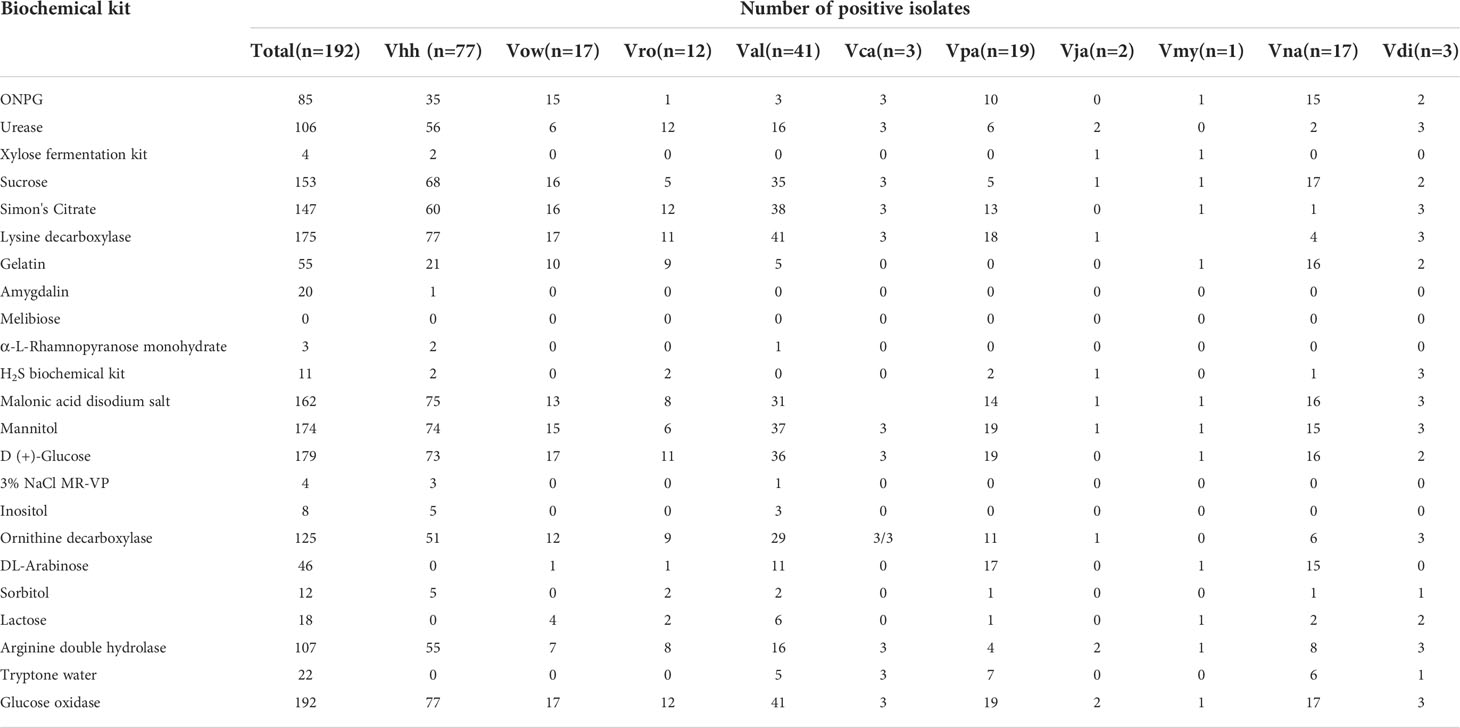
Table 2 Results of Harveyi clade isolates based on biochemical tests.
In addition to MLSA, all members of the V. campbellii, V. jasicida, and V. parahaemolyticus group showed green colonies on TCBS agar, indicating that they were unable to ferment sucrose. Nevertheless, V. diabolicus and V. owellii were sucrose positive. The other groups showed yellow and green colonies on TCBS, and thus were either sucrose positive or negative. The colonies of V. harveyi, V. campbellii, V. sagamiensis, and V. aliginolyticus appeared translucence on TSB, while the colonies of the other strains were transparent. On the modified Vibrio chromogenic medium, V. parahaemolyticus and V. jasicida showed large colonies of blue-green color and the colonies of V. alginolyticus were pale yellow or beige, while the other species were inhibited.
Diversity and clustering analysis of sequence types
The nucleotide sequence variations of the four gene fragments are summarized in Table 3. The GC contents of every locus were similar and varied per locus from 47.31% (atpA) to 48.53% (pyrH). The number of alleles of each locus in the 192 Harveyi clade isolates was distributed as follows: 53 pyrH, 56 atpA, 79 gyrB, and recA. The number of polymorphic sites ranged per locus from 137 (atpA) to 170 (recA). The nucleotide diversity varied from 0. 0.02643 (atpA) to 0.08482 (pyrH), indicating that allele loci had a low mutation rate. The value of dN/dS of each locus was lower than 0.25, suggesting a purifying selection of the four housekeeping genes for the 192 Harveyi clade isolates.
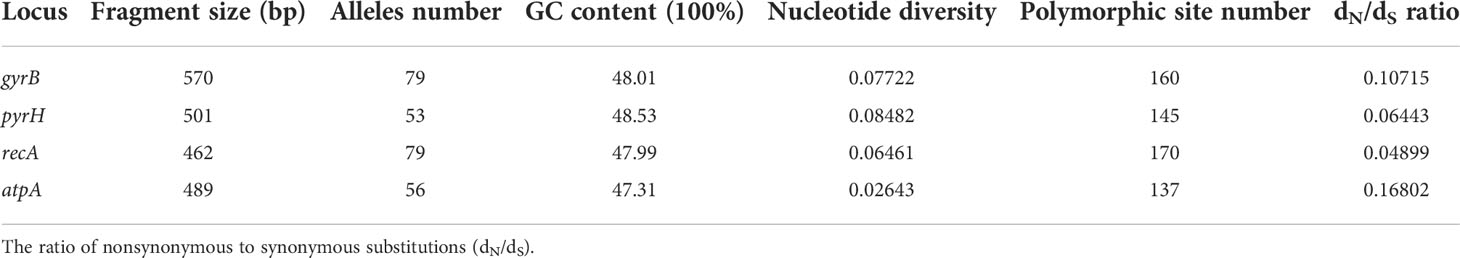
Table 3 Nucleotide sequence variations of each MLST locus for 192 Harveyi clade isolates.
A total of 95 STs were identified, of which 92 were newly identified STs (Supplementary Table 1). The MLST analysis revealed high molecular diversity among the Harveyi clade isolates, with most strains forming unique sequence types in the Chinese mariculture system of this study. ST327 was the most common among the newly identified STs, accounting for 11.5% of the total number of isolates (22 out of 192), with all of them being V. alginolyticus. ST215, represented by V. harveyi, was the second most common STs and accounted for 6.25% of the total number of isolates (12 out of 192). Sixty-three STs had single isolates. Twenty-seven were identified in 77 isolates of V. harveyi, with each ST comprising 1 to 12 isolates. Fifteen STs were found in 41 isolates of V. alginolyticus, with each ST having 1 to 22 isolates. Eleven STs were identified in 19 V. parahaemolyticus isolates, with each ST containing 1 to 5 isolates, followed by V. natriegens (15 out of 17), V. owensii (11 out of 17), V. rotiferianus (9 out of 12), V. campbellii (3 out of 3), V. diabolicus (3 out of 3), V. jasicida (2 out of 2), and V. mytili (1 out of 1). Furthermore, the statistical classification of the ST strains according to their host origin showed that 42 STs were present in 101 isolates from fish, 26 STs in 64 isolates from shrimp, 4 STs in 4 isolates from crab, and 23 STs in 23 isolates from sea cucumber.
The identification of the CC clustering patterns indicated that 95 STs were separated into 7 CCs (CC0, CC1, CC2, CC3, CC4, CC5, and CC6) and 4 doublets (D1–D5) (Figure 3), while the remaining 36 STs were singletons. The most common CC was CC0, which included 78 isolates with 5 STs, all of which were identified as V. harveyi. Based on UPGMA analysis, the STs that belong to the same CCs and doublets were also clustered together in the UPGMA tree (Figure 4). CC0 was composed of V. harveyi and represented the core group, which was divided into two major branches. One branch included V. campbellii, V. rotiferianus, V. owensii, V. mytili, and V. jasicida, while the other branch included V. parahaemolyticus, V. diabolicus, V. alginolyticus, and V. natriegens. Among them, V. parahaemolyticus and V. campbellii had the closest genetic distance to V. harveyi. V. jasicida (composed of D2) and V. natriegens (composed of CC1 and CC5) were the most genetically distant species from CC0.
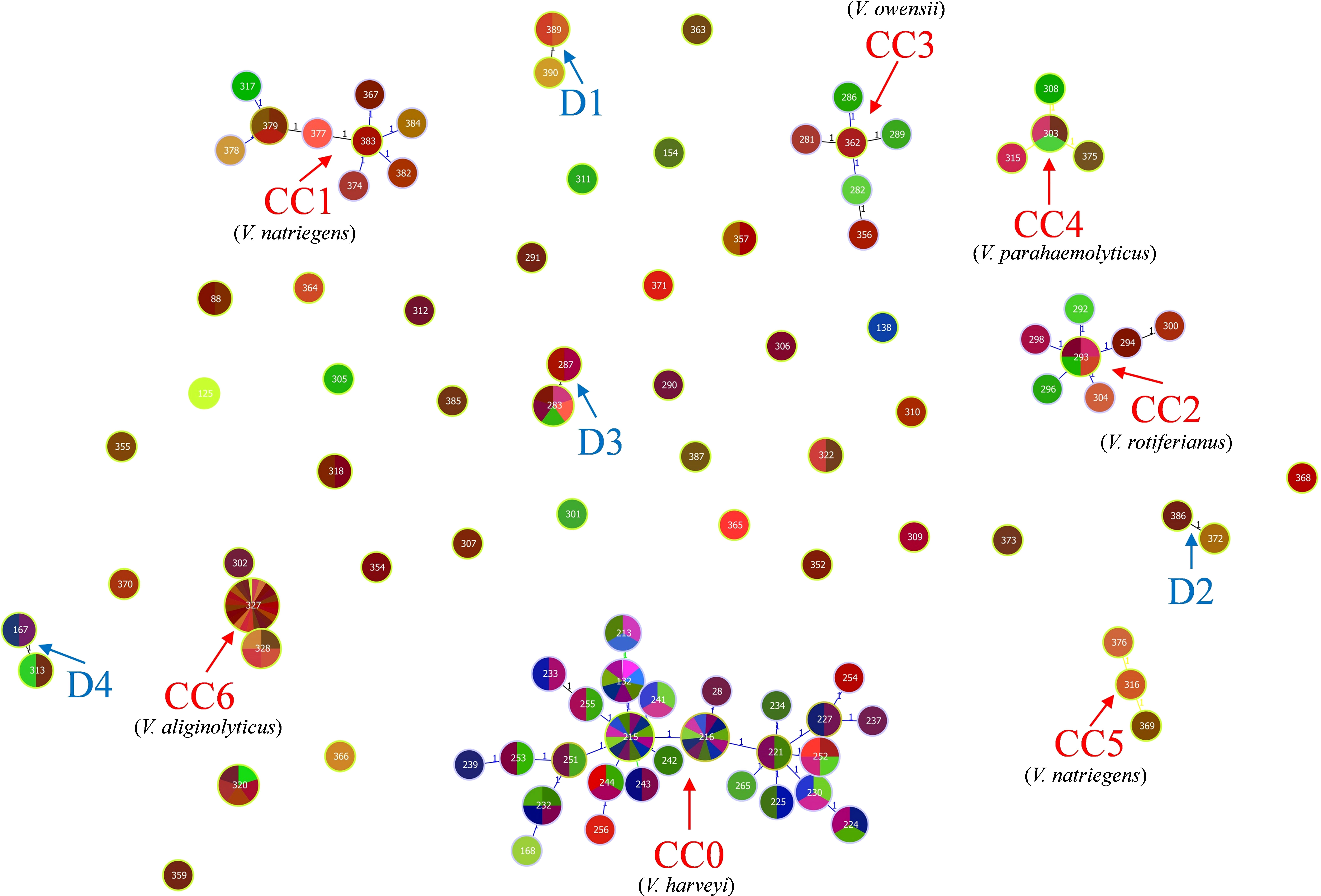
Figure 3 goeBURST minimum spanning tree for all the 95 sequence types obtained from the combination of all allele types of the four MLST loci gyrB, pyrH, recA, and atpA using the PHYLOViZ 2.0 analysis software. The genetic relationships between all the analyzed Chinese Harveyi clade isolates are indicated. Note: The different numbers in the circles represent different STs.
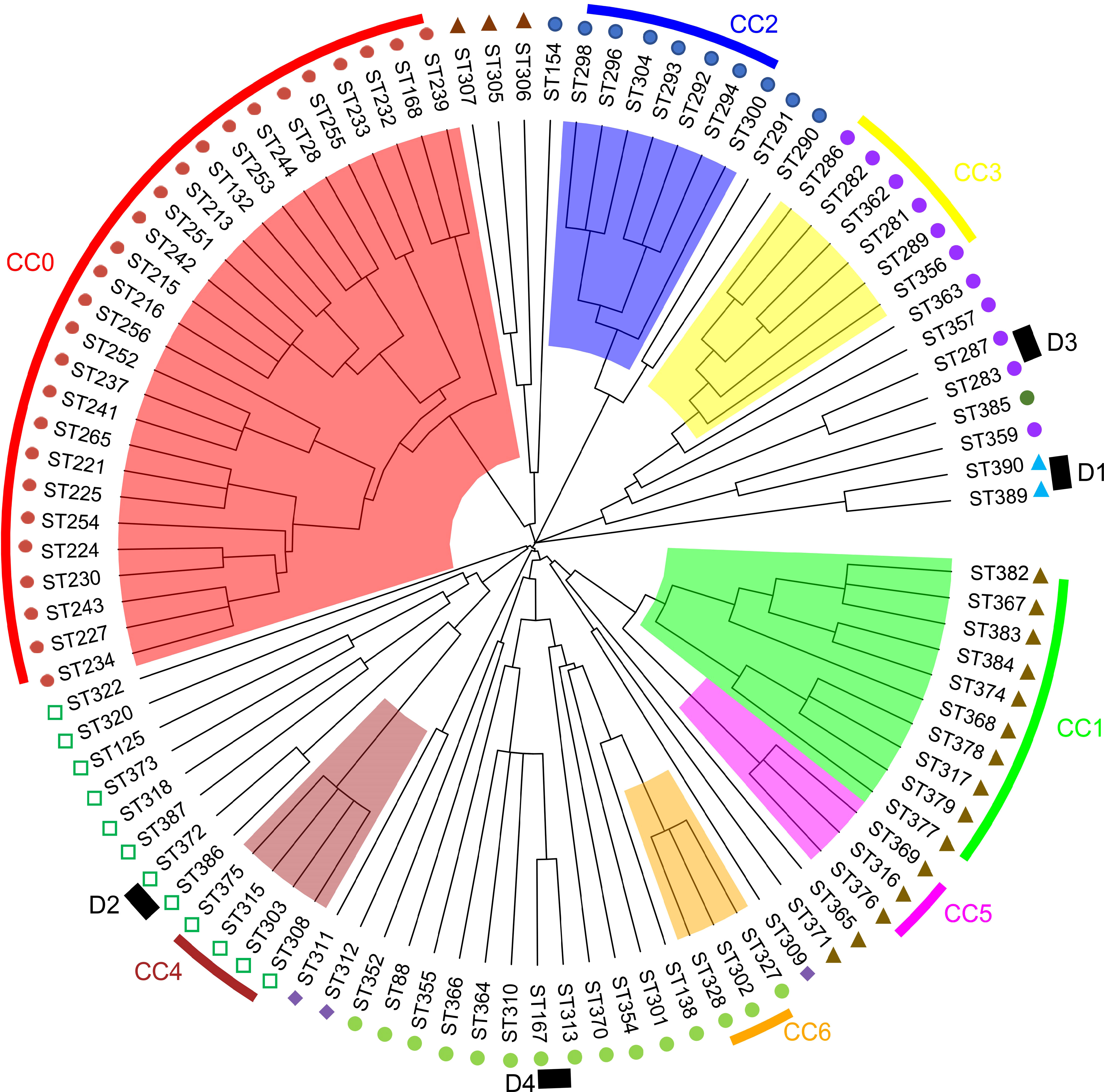
Figure 4 MLST hierarchical clustering phylogenetic map of the Harveyi clade.  : V. harveyi;
: V. harveyi;  : V. parahaemolyticus;
: V. parahaemolyticus;  : V. diabolicus;
: V. diabolicus;  : V. alginolyticus;
: V. alginolyticus;  : V. natriegens;
: V. natriegens;  : V. jasicida;
: V. jasicida;  : V. owensii;
: V. owensii;  V. mytili;
V. mytili;  : V. rotiferianus.
: V. rotiferianus.
Antimicrobial susceptibility characteristics of the Harveyi clade isolates
The antibiotic resistance profiles of each Harveyi clade isolate are illustrated in Figure 5. The results showed that 93.8% of the Harveyi clade isolates were resistant to NEO. SDM resistance was found in 31.9% of the isolates. SDI and SMM resistance were observed in 22.9% of the isolates. Resistance to SMZ, THI, and ENR was recorded in 17.3%, 14.6%, and 8.3% of the isolates, respectively. On the other hand, Harveyi clade isolates were highly sensitive to certain antibiotics. Susceptibility to FFC, THI, FLU, ENR, CIP, SMZ, SMM, and SDI was recorded in 92.8%, 82.3%, 89%, 84%, 73%, 78%, 72%, and 61% of the isolates, respectively (Figure 6).
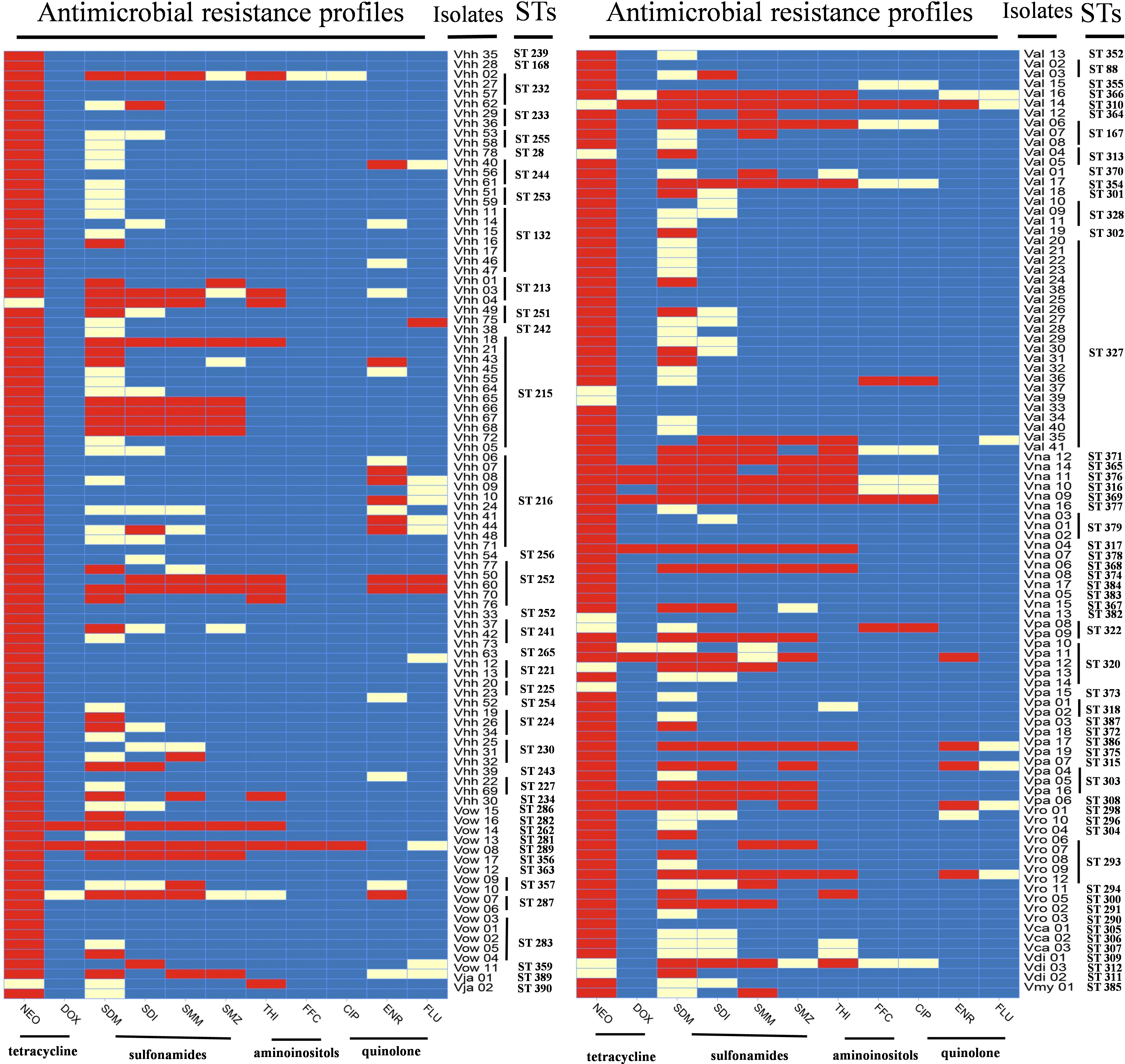
Figure 5 Levels of antibiotic resistance profiles of 11 tested antibiotics. The black bar, the gray bar, and the light gray bar represent the proportion of resistant strains, intermediate strains, and sensitive strains, respectively. The gray striped bar represents the proportion of the multiple antibiotic resistance index of the strains.
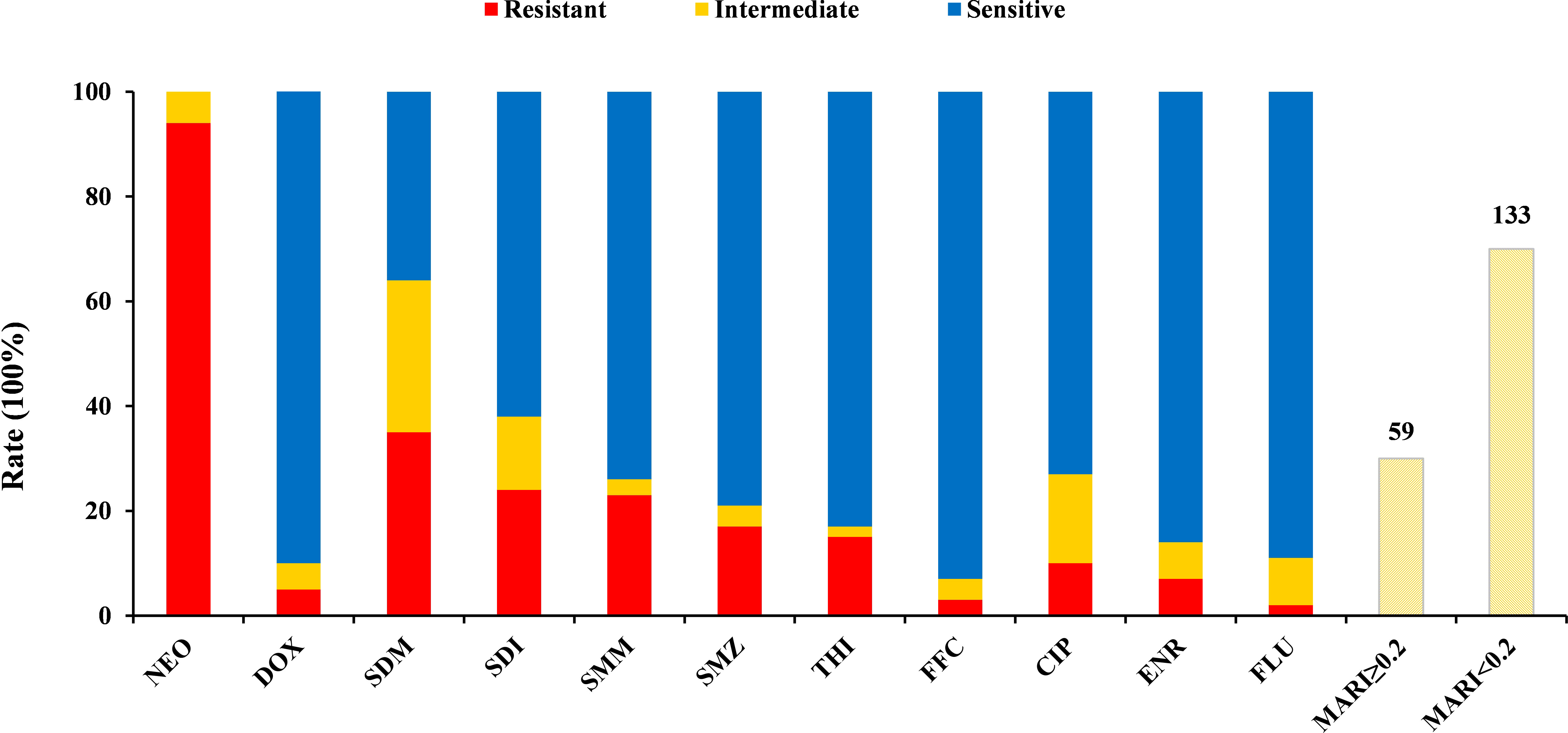
Figure 6 Antibiotic resistance profiles of the Harveyi clade isolates. The colors represent different levels of resistance.  : resistance,
: resistance,  : intermediate,
: intermediate,  : susceptibility. Note: MAR index is calculated by dividing the total numbers of the tested antibiotics into the numbers of antibiotics, to which the isolate was resistant.
: susceptibility. Note: MAR index is calculated by dividing the total numbers of the tested antibiotics into the numbers of antibiotics, to which the isolate was resistant.
Multiple antibiotic resistance analysis indicated that the multiple antibiotic resistance index (MARI) of all the isolates ranged from 0 to 0.82 (Figure 7 and Supplementary Table S1), which revealed that 97.9% of the isolates (188 out of 192) were resistant to at least one antibiotic. A MARI value higher than 0.2 was observed in 30.7% of the resistant isolates (59 out of 192). These isolates have a transmission potential since the MARI was higher than 0.2, indicating a high antibiotic exposure risk. The isolates Vhh 50, Vhh 60, Val 14, and Vna 09 had the highest MARI value (0.82) and were resistant to nine antibiotics. The remaining 55 isolates showed multidrug resistance to at least three tested antibiotics (Figure 6).
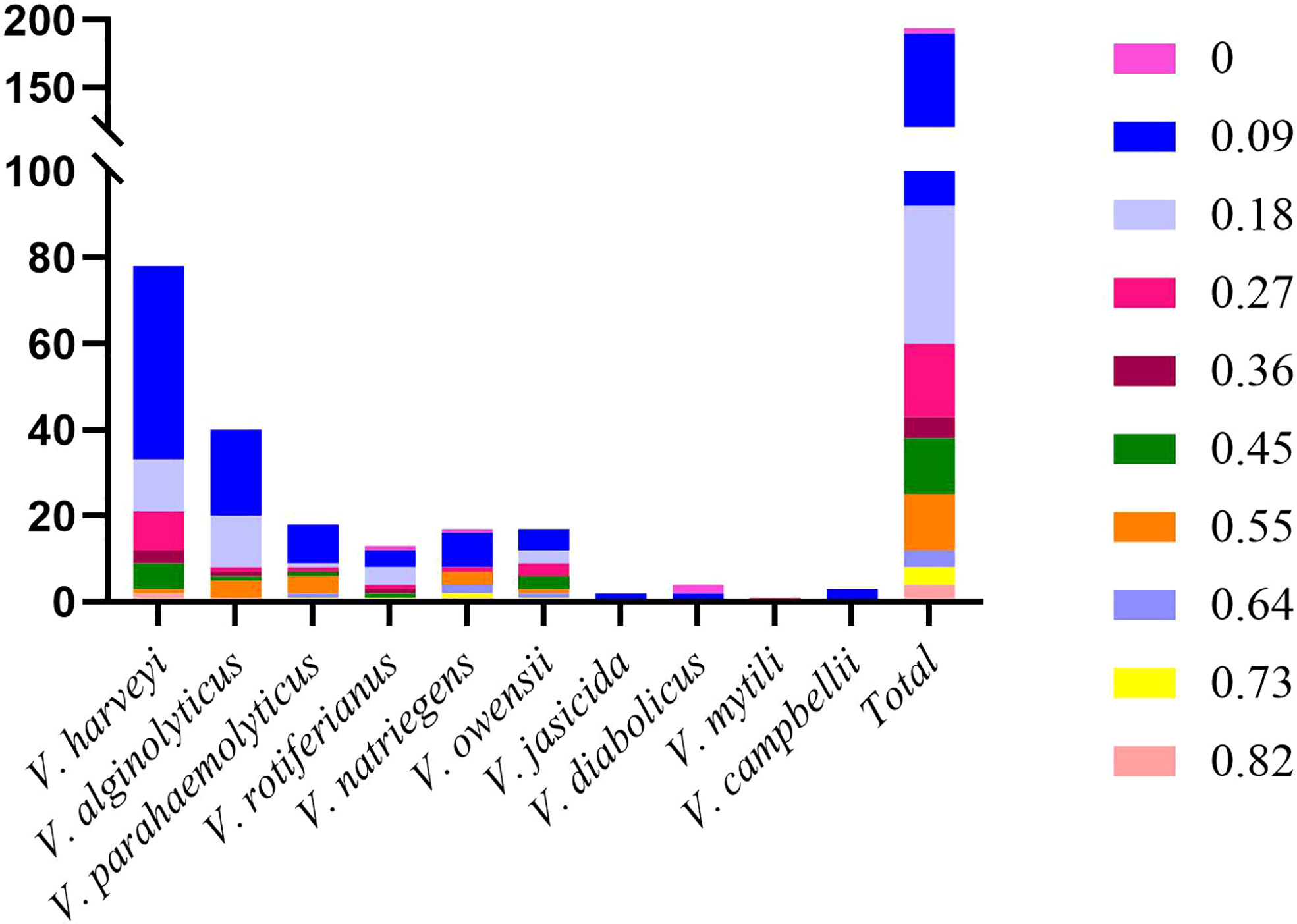
Figure 7 Distribution of multi-antibiotic resistance strains.
Association analysis of antimicrobial resistance with the genetic population of the isolates is shown in Figure 5. ST132 consisted of seven isolates of V. harveyi, all of which were resistant to neomycin, while the other STs had sensitive phenotypes. The resistance profiles of the other isolates of the same ST showed different profiles. Moreover, 59 strains were resistant to the three antibiotics mentioned above, including 40 non-repetitive STs scattered in different years. Due to the large number of various STs and the significantly varying ST numbers, the relationship between the different STs and antimicrobial resistance was difficult to determine.
Discussion
We analyzed a total of 192 isolates of the Harveyi clade from infected parts of animals from marine and mariculture environments in coastal areas of China from 2000 to 2020. The phenotypic, metabiotic, and genotypic properties of these isolates were investigated thoroughly in this study. Many researchers confirmed that the phenotypic traits do not affect the differentiation of Harveyi clade isolates (Gauger and Gómez-Chiarri, 2002; Thompson, 2003). Lukjancenko et al. (2012) showed that the traditional phenotypes were unable to distinguish sister species due to large conserved regions in the proteome prediction. Gomez-Gil et al. (2004) demonstrated that V. harveyi, V. campbellii, and V. rotiferianus had similar phenotypes and were misidentified. The results of this study also revealed that the phenotypic methods have a limited ability to discriminate Harveyi clade members. Although 16S rDNA gene sequencing is considered as a gold standard method for bacterial taxonomy, it does not have sufficient resolution to correctly identify Harveyi clade species (Gomez-Gil et al., 2004; Janda and Abbott, 2007; Chatterjee and Haldar, 2012). The low discriminating ability of the 16S rDNA gene in identifying the 192 isolates in the present research was consistent with previous studies. In recent years, studies have shown that MLSA can accurately identify Harveyi clade strains with high resolution and reproducibility (Thompson et al., 2005; Xie et al., 2020). Rahman et al. (2014) found that most Vibrio species could be easily identified using population and phylogenetic analyses based on the combination of the genes gyrB-pyrH-atpA-recA. Using five housekeeping genes (i.e., rpoA, pyrH, topA, ftsZ, and mreB), Cano-Gomez et al. (2011) identified 36 Vibrio harveyi-related isolates as V. harveyi, V. campbellii, V. rotiferianus, and V. owensii. Furthermore, Pascual et al. (2010) identified rapidly and accurately 44 Vibrio core groups as V. harveyi, V. campbellii, V. rotiferianus, and V. parahaemolyticus using seven concatenated genes (i.e., 16S rDNA, recA, pyrH, rpoD, gyrB, rctB, and toxR). Xie et al. (2020) have recently demonstrated that the two pathogenic bacteria HM-12 and HM-14 were confirmed as V. harveyi and V. alginolyticus, respectively, using the five housekeeping genes ftsZ, gapA, gyrB, mreB, and topA. Furthermore, the NJ tree analysis of the species V. alginolyticus and V. diabolicus revealed two subgroups, which supports the reports stating that the two species were originally one species (Rahman et al., 2014). In the present study, 192 Harveyi clade isolates were accurately identified to 10 species using the four concatenated genes gyrB, pyrH, atpA, and recA. Likewise, V. rotiferianus and V. jasicida revealed two subgroups, which led us to speculate that these two species may also have originated from one species. These studies clearly show that MLSA can effectively resolve phylogenetic relationships at the genus and species level; however, there are no general criteria for selecting the genes and their number for MLSA. Therefore, the use of MLSA needs to be improved to make its application more viable and ubiquitous.
Molecular subtyping is widely used for epidemiological and population genetic analysis of pathogenic bacteria. MLST was first introduced in the epidemiology of pathogenic Neisseria meningitidis strains (Maiden et al., 1998). MLST has been widely employed in recent years for V. parahaemolyticus. Han et al. (2015) found that 218 V. parahaemolyticus clinical isolates in China produced 137 STs, indicating that V. parahaemolyticus from China has a high genetic diversity. Using MLST, Jiang et al. (2019) found that 90 V. parahaemolyticus strains from Bohai and Yellow Seas of China were composed of 68 sequence types, displaying a high level of genetic diversity. Rahman et al. (2014) reported that 182 Vibrio strains obtained from the Venice Lagoon and a marine environment were classified into 162 STs and were processed as distinct species/taxa. We analyzed in the present study the extent of genetic diversity on nucleotide variations among the Harveyi clade isolates. Han et al. (2015) and Turner et al. (2013) reported the same phenomena in V. parahaemolyticus strains. The observed alleles, polymorphic site numbers, and nucleotide diversity reveal the richness and uniqueness of the Harveyi clade isolates from coastal areas of China.
In this study, we identified 95 STs among 192 Harveyi clade isolates. In comparison to the pubMLST database, 92 STs were newly discovered, demonstrating that our research contributed substantially to the diversity of Vibrio spp. in the MLST database. ST28, ST88, and ST132 were found to be identical to the sequence types of isolates recovered from the Venice Lagoon (Rahman et al., 2014). When examining the three STs with the largest number, no significant relationship between the STs and the annual change was observed. The analysis of the STs and their origin (host and collection data) shows that the isolates with the same ST may have originated from the same host (e.g., ST327). Nevertheless, the majority of the STs consisted of isolates from different regions and sampling years (e.g., ST215 and ST216). However, cgMLST correlates with time and regions of bacterial isolates well (Alikhan et al., 2018). As a bacterial typing system, it has become an innovative tracing tool in recent years (Monte et al., 2021). Gonzalez-Escalona et al. (2017) demonstrated that cgMLST clearly showed higher resolution than traditional MLST for V. parahaemolytics. With the development of whole genome sequencing technology, we will use comparative genomics and cgMLST scheme for higher-resolution analysis of Harvey clade isolates.
Antibiotic resistance of Harveyi clade isolates is an important indicator of metabolic and phenotypic diversity. In the current investigation, antibiotic susceptibility tests showed that all the Harveyi clade isolates were resistant to neomycin, but showed low resistance to other antibiotics. Similarly, Obaidat et al. (2017) reported that all V. parahaemolyticus isolates in the Harveyi clade were neomycin resistant. In the Indian subcontinent, Vibrio spp. exhibited high neomycin resistance (Guardiola et al., 2012). Ciprofloxacin was the most active quinolone antibiotic against the Harveyi clade in this study, which is consistent with the results of Zanetti et al. (2001). The increased resistance to quinolones in Vibrio spp., with spatial and temporal differences, may be related to various antibiotic resistance mechanisms (Blair et al., 2015; Blanco et al., 2016; Deng et al., 2020). In this study area, compared with other areas, we found the uniqueness and diversity of Harveyi clade isolates between the coastal areas of China and other countries. For instance, the MARI values of the Harveyi clade isolates ranged from 0 to 0.82. The MARI reflects the degree of the environmental pollution caused by antibiotics that may be dangerous to human health (Tanil et al., 2005; Páll et al., 2021). A value higher than 0.2 indicates a high antibiotic exposure risk, while a value lower than 0.2 indicates a low antibiotic exposure risk (Mohamad et al., 2019). According to our findings, 61.8% of the Harveyi clade isolated from coastal areas in the past 20 years have a MARI value less than 0.1, indicating that the risk of antibiotic resistance transmission of most Harveyi clade is low in mariculture systems in China. However, the resistance to the 11 antibiotics was found in various combinations, showing that this resistance was not concentrated in a single ST. Thus, the Harveyi clade isolates analyzed by MLST are multidrug-resistant bacteria. New non-antibiotic bacteriostatic medications and alternatives should be developed to reduce the impact of the extensive use of antibiotics on the environment and ecological health (Tan et al., 2016). In recent years, antibiotic drugs have been made into dietary microspheres by scholars to achieve high availability, attractiveness and digestibility to fish, which will promote the source of antibiotics, reduce antibiotic contamination in fisheries, and ease the control of antibiotic resistance (Zhang et al., 2021). Therefore, analyzing the drug resistance of pathogens in different regions of the eastern coast of China in the past 20 years is of great significance for formulating antibacterial drug reduction policies and effective antibacterial drug use programs.
Conclusions
Our analyses showed that the strains have significant interspecific genetic and antibiotic resistance diversity. The Harveyi clade isolates were classified rapidly and robustly using the MLSA approach. The MLST analysis showed high genetic diversity and uniqueness of the Harveyi clade isolates from China with multiple sequence types. Furthermore, our study confirmed the presence of multiple antibiotic resistance in the Harveyi clade from the coastal areas of China collected in the last two decades. Although the MARI demonstrated that most Harveyi clade isolates from marine and mariculture environments in coastal areas of China had a low probability of antibiotic resistance transmission, they were still highly resistant to various antibiotics, emphasizing the need to increase the development of non-antibiotic drugs and antibiotic alternatives. These results will provide a basis for further studies on the genetic and metabolic diversity of Harveyi clade isolates in mariculture systems in China.
Data availability statement
The 16S rDNA gene, gyrB, pyrH, atpA, recA sequences determined in this study have been deposited in the GenBank of NCBI under the accession numbers: ON437347-ON437538, ON469581-ON469772, ON778048-ON778239, ON778240-ON778431, ON4491819-ON492010.
Author contributions
The research was conceived and designed by YW, ML, and YY. The experiments were executed by HK, YY, and CW. ZZ assisted in statistical analysis. YY and BL analyzed the data. HK and YY drafted the manuscript. ML, XR, and GY reviewed the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (2019YFD0900102), Central Public-interest Scientific Institution Basal Research Fund, CAFS (2022GH02), Basic Scientific Research Funds for Central Non-profit Institutes, Yellow Sea Fisheries Research Institutes (20603022021013), and Central Public-interest Scientific Institution Basal Research Fund, CAFS (2020TD40).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.932255/full#supplementary-material
Supplementary Table 1 | Allele profiles, sequence types, multiple antibiotic resistance index values (MARI), antimicrobial resistance profiles’ identification based on multilocus sequence analysis (MLSA), and sources of 192 Harveyi clade isolates.
Supplementary Figure 1 | Neighbor-joining (NJ) phylogenetic analysis based on 16S rDNA gene sequences of Harveyi clade isolates and reference strains used in this study. Numbers at nodes represent the bootstrap (BT) values based on 1000 resamplings.
Supplementary Figure 2 | NJ phylogenetic analysis based on all three-locus concatenated sequences of Harveyi clade isolates and reference strains used in this study. Numbers at nodes represent the BT values based on 1000 resamplings.
Supplementary Figure 3 | NJ phylogenetic analysis based on two-locus concatenated sequences of Harveyi clade isolates and reference strains used in this study. Numbers at nodes represent the BT values based on 1000 resamplings.
Supplementary Figure 4 | Phylogenetic reconstructions based on individual analyses of gyrB, pyrH, recA, and atpA using the NJ method. Numbers at nodes represent the BT values base on 1000 resamplings.
Supplementary Figure 5 | Morphology of Harveyi clade type strains. (A) growth of Harveyi clade type strains on TCBS agar; (B) microscopy Harveyi clade type strains on TCBS agar.
References
Aanensen D. M., Spratt B. G. (2005). The multilocus sequence typing network: mlst.net. Nucleic Acids Res. 33, 728–733. doi: 10.1093/nar/gki415
Alikhan N. F., Zhou Z., Sergeant M. J., Achtman M. (2018). A genomic overview of the population structure of salmonella. PloS Genet. 14, e1007261. doi: 10.1371/journal.pgen.1007261
Baker-Austin C., Oliver J. D., Alam M., Ali A., Waldor M. K., Qadri F., et al. (2018). Vibrio spp. infections. Nat. Rev. Dis. Primers. 4, 8. doi: 10.1038/s41572-018-0005-8
Baker-Austin C., Trinanes J., Gonzalez-Escalona N., Martinez-Urtaza J. (2017). Non-cholera Vibrios: The microbial barometer of climate change. Trends Microbiol. 25, 76–84. doi: 10.1016/j.tim.2016.09.008
Blair J. M., Webber M. A., Baylay A. J., Ogbolu D. O., Piddock L. J. (2015). Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51. doi: 10.1038/nrmicro3380
Blanco P., Hernando-Amado S., Reales-Calderon J. A., Corona F., Lira F., Alcalde-Rico M., et al. (2016). Bacterial multidrug efflux pumps: Much more than antibiotic resistance determinants. Microorganisms 4, 14. doi: 10.3390/microorganisms4010014
Botella S., Pujalte M. J., Macián M. C., Ferrús M. A., Hernández J., Garay E. (2002). Amplified fragment length polymorphism (AFLP) and biochemical typing of photobacterium damselae subsp. damselae. J. Appl. Microbiol. 93, 681–688. doi: 10.1046/j.1365-2672.2002.01748.x
Cano-Gomez A., Bourne D. G., Hall M. R., Owens L., Hoj L. (2009). Molecular identification, typing and tracking of vibrio harveyi in aquaculture systems: current methods and future prospects. Aquaculture 287, 1–10. doi: 10.1016/j.aquaculture.2008.10.058
Cano-Gomez A., Høj L., Owens L., Andreakis N. (2011). Multilocus sequence analysis provides basis for fast and reliable identification of Vibrio harveyi-related species and reveals previous misidentification of important marine pathogens. Syst. Appl. Microbiol. 34, 561–565. doi: 10.1016/j.syapm.2011.09.001
Chatterjee S., Haldar S. (2012). Vibrio related diseases in aquaculture and development of rapid and accurate identification methods. J. Mar. Sci. Res. Dev. 1, 1–7. doi: 10.4172/2155-9910.S1-002
China Fisheries Yearbook (2021). Fisheries department of agriculture ministry of China (Beijing: China Agriculture Press).
Clinical and Laboratory Standards Institute [CLSI] (2017). Performance standards for antimicrobial susceptibility testing. 27th Edn (Wayne, PA: Clinical and Laboratory Standards Institute).
Del Gigia-Aguirre L., Sánchez-Yebra-Romera W., García-Muñoz S., Rodríguez-Maresca M. (2017). First description of wound infection with Vibrio harveyi in Spain. New Microbes New Infect. 19, 15–16. doi: 10.1016/j.nmni.2017.05.004
Deng Y., Xu L., Chen H., Liu S., Guo Z., Cheng C., et al. (2020). Prevalence, virulence genes, and antimicrobial resistance of Vibrio species isolated from diseased marine fish in south China. Sci. Rep. 10, 14329. doi: 10.1038/s41598-020-71288-0
Fraser C., Hanage W. P., Spratt B. G. (2007). Recombination and the nature of bacterial speciation. Science 315, 476–480. doi: 10.1126/science.1127573
Fu S., Octavia S., Tanaka M. M., Sintchenko V., Lan R. (2015). Defining the core genome of salmonella enterica serovar typhimurium for genomic surveillance and epidemiological typing. J. Clin. Microbiol. 53, 2530–2538. doi: 10.1128/JCM.03407-14
Gauger E. J., Gómez-Chiarri M. (2002). 16S ribosomal DNA sequencing confirms the synonymy of Vibrio harveyi and Vibrio carchariae. Dis. Aquat Organ 52, 39–46. doi: 10.3354/dao052039
Gevers D., Cohan F. M., Lawrence J. G., Spratt B. G., Coenye T., Feil E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Gomez-Gil B., Soto-Rodríguez. S., García-Gasca A., Roque A., Swings J. (2004). Molecular identification of Vibrio harveyi-related isolates associated with diseased aquatic organisms. Microbiology 150, 1769–1777. doi: 10.1099/mic.0.26797-0
Gonzalez-Escalona N., Jolley K. A., Reed E., Martinez-Urtaza J. (2017). Defining a core genome multilocus sequence typing scheme for the global epidemiology of vibrio parahaemolyticus. J. Clin. Microbiol. 55, 1682–1697. doi: 10.1128/JCM.00227-17
Goudenège D., Boursicot V., Versigny T., Bonnetot S., Ratiskol J., Sinquin C., et al. (2014). Genome sequence of Vibrio diabolicus and identification of the exopolysaccharide HE800 biosynthesis locus. Appl. Microbiol. Biotechnol. 98, 10165–10176. doi: 10.1007/s00253-014-6202-9
Guardiola F. A., Cuesta A., Meseguer J., Esteban M. A. (2012). Risks of using antifouling biocides in aquaculture. Int. J. Mol. Sci. 13, 1541–1560. doi: 10.3390/ijms13021541
Hackbusch S., Wichels A., Gimenez L., Döpke H., Gerdts G. (2020). Potentially human pathogenic Vibrio spp. in a coastal transect: Occurrence and multiple virulence factors. Sci. Total Environ. 707, 136113. doi: 10.1016/j.scitotenv.2019.136113
Han D., Tang H., Ren. C., Wang G., Zhou L., Han. C. (2015). Prevalence and genetic diversity of clinical Vibrio parahaemolyticus isolates from China, revealed by multilocus sequence typing scheme. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00291
Harun A., Kan A., Schwabenbauer K., Gilgado F., Perdomo H., Firacative C., et al. (2021). Multilocus sequence typing reveals extensive genetic diversity of the emerging fungal pathogen Scedosporium aurantiacum. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.761596
Holt J. G., Krieg N. R., Sneath P. H. A., Staley J. T., Williams S. T. (1994). Bergey's manual of determinative bacteriology. 9th edn (Baltimore: Williams & Wilkins).
Janda J. M., Abbott S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Jelocnik M., Polkinghorne A., Pannekoek Y. (2019). Multilocus sequence typing (MLST) of chlamydiales. Methods Mol. Biol. 2042, 69–86. doi: 10.1007/978-1-4939-9694-0-7
Jiang Y., Chu Y., Xie G., Li F., Wang L., Huang J., et al. (2019). Antimicrobial resistance, virulence and genetic relationship of Vibrio parahaemolyticus in seafood from coasts of bohai Sea and yellow Sea, China. Int. J. Food Microbiol. 290, 116–124. doi: 10.1016/j.ijfoodmicro.2018.10.005
Jolley K. A., Feil E. J., Chan M. S., Maiden M. C. (2001). Sequence type analysis and recombinational tests (START). Bioinformatics 17, 1230–1231. doi: 10.1093/bioinformatics/17.12.1230
Krumperman P. H. (1983). Multiple antibiotic indexing of Escherichia coli to identify high-risk sources of fecal contamination of foods. Appl. Environ. Microbiol. 46, 165–170. doi: 10.1128/AEM.46.1.165-170.1983
Kumari P., Poddar A., Das S. K. (2020). Characterization of multidrug resistance in Vibrio species isolated from marine invertebrates from Andaman Sea. 3 Biotech. 10, 456. doi: 10.1007/s13205-020-02445-5
Lin B., Wang Z., Malanoski A. P., O'Grady E. A., Wimpee C. F., Vuddhakul V., et al. (2010). Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ. Microbiol. Rep. 2, 81–89. doi: 10.1111/j.1758-2229.2009.00100.x
Lukjancenko O., Ussery D. W., Wassenaar T. M. (2012). Comparative genomics of bifidobacterium, lactobacillus and related probiotic genera. Microb. Ecol. 63, 651–673. doi: 10.1007/s00248-011-9948-y
Maiden M. C., Bygraves J. A., Feil E., Morelli G., Russel J. E., Urwin R., et al. (1998). Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Mohamad N., Amal M., Saad M. Z., Yasin I., Zulkiply N. A., Mustafa M., et al. (2019). Virulence-associated genes and antibiotic resistance patterns of vibrio spp. isolated from cultured marine fishes in Malaysia. BMC Vet. Res. 15, 176. doi: 10.1186/s12917-019-1907-8
Monte D. F. M., Nethery M. A., Barrangou R., Landgraf M., Fedorka-Cray P. J. (2021). Whole-genome sequencing analysis and CRISPR genotyping of rare antibiotic-resistant salmonella enterica serovars isolated from food and related sources. Food Microbiol. 93, 103601. doi: 10.1016/j.fm.2020.103601
Obaidat M. M., Salman A. E. B., Roess A. A. (2017). Virulence and antibiotic resistance of vibrio parahaemolyticus isolates from seafood from three developing countries and of worldwide environmental, seafood, and clinical isolates from 2000 to 2017. J. Food Prot. 80, 2060–2067. doi: 10.4315/0362-028X.JFP-17-156
Páll E., Niculae M., Brudașcă G. F., Ravilov R. K., Șandru C. D., Cerbu C., et al. (2021). Assessment and antibiotic resistance profiling in Vibrio species isolated from wild birds captured in Danube delta biosphere reserve, Romania. Antibiot (Basel) 10, 333. doi: 10.3390/antibiotics10030333
Pascual J., Macian M. C., Arahal D. R., Garay E., Pujalte M. J. (2010). Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int. J. Syst. Evol. Microbiol. 60, 154–165. doi: 10.1099/ijs.0.010702-0
Prithvisagar K. S., Krishna Kumar B., Kodama T., Rai P., Iida T., Karunasagar I., et al. (2021). Whole genome analysis unveils genetic diversity and potential virulence determinants in Vibrio parahaemolyticus associated with disease outbreak among cultured Litopenaeus vannamei (Pacific white shrimp) in India. Virulence 12, 1936–1949. doi: 10.1080/21505594.2021.1947448
Rahman M. S., Martino M. E., Cardazzo B., Facco P., Bordin P., Mioni R., et al. (2014). Vibrio trends in the ecology of the Venice lagoon. Appl. Environ. Microbiol. 80, 2372–2380. doi: 10.1128/AEM.04133-13
Ribeiro-Gonçalves B., Francisco A. P., Vaz C., Ramirez M., Carriço J. A. (2016). PHYLOViZ online: web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 44, 246–251. doi: 10.1093/nar/gkw359
Richter M., Rosselló-Móra R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Romalde J. L., Dieguez A. L., Lasa A., Balboa S. (2014). New Vibrio species associated to molluscan microbiota: a review. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00413
Rozas J., Ferrer-Mata A., Saínchez-DelBarrio J. C., Guirao-Rico S., Librado P., Ramos-Onsins S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of Large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruwandeepika H., Jayaweera T., Bhowmick P. P., Karunasagar I., Bossier P., Defoirdt T. (2012). Pathogenesis, virulence factors and virulence regulation of Vibrios belonging to the harveyi clade. Rev. Aquacult. 4, 59–74. doi: 10.1111/j.1753-5131.2012.01061.x
Santhyia A. V., Mulloorpeedikayil R. G., Kollanoor R. J., Jeyaseelan P. M. (2015). Molecular variations in vibrio alginolyticus and v. harveyi in shrimp-farming systems upon stress. Braz. J. Microbiol. 46, 1001–1008. doi: 10.1590/S1517-838246420140410
Sawabe T., Kita-Tsukamoto K., Thompson F. L. (2007). Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tan L. T., Chan K. G., Lee L. H., Goh B. H. (2016). Streptomyces bacteria as potential probiotics in aquaculture. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00079
Tanil G. B., Radu S., Nishibuchi M., Rahim R. A., Napis S., Maurice L., et al. (2005). Characterization of Vibrio parahaemolyticus isolated from coastal seawater in peninsular Malaysia. Southeast Asian J. Trop. Med. Public Health 36, 940–945. doi: 10.1109/43.31533
Thompson F. L. (2003). Improved taxonomy of the family vibrionaceae. ph. d. thesis (Ghent, Belgium: Ghent University).
Thompson F. L., Gevers D., Thompson C. C., Dawyndt P., Naser S., Hoste B., et al. (2005). Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Appl. Environ. Microbiol. 71, 5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005
Thompson F. L., Iida T., Swings J. (2004). Biodiversity of Vibrios. Mol. Biol. Rev. 68, 403–431. doi: 10.1128/MMBR.68.3.403-431.2004
Thompson C. C., Vicente A. C., Souza R. C., Vasconcelos A. T., Vesth T., Alves N. Jr., et al. (2009). Genomic taxonomy of vibrios. BMC Evol. Biol. 9, 258. doi: 10.1186/1471-2148-9-258
Tran L., Nunan L., Redman R. M., Mohney L. L., Pantoja C. R., Fitzsimmons K., et al. (2013). Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis. Aquat Organ 105, 45–55. doi: 10.3354/dao02621
Turner J. W., Paranjpye R. N., Landis E. D., Biryukov S. V., Gonzalez-Escalona N., Nilsson W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the pacific northwest coast of the united states. PloS One 8, e55726. doi: 10.1371/journal.pone.0055726
Wang W., Liu J., Guo S., Liu L., Yuan Q., Guo L., et al. (2021). Identification of Vibrio parahaemolyticus and Vibrio spp. specific outer membrane proteins by reverse vaccinology and surface proteome. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.625315
Weisburg W. G., Barns S. M., Pelletier D. A., Lane D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Xie J., Bu L., Jin S., Wang X., Xu Y. (2020). Outbreak of vibriosis caused by Vibrio harveyi and Vibrio alginolyticus in farmed seahorse hippocampus kuda in China. Aquaculture 523, 735168. doi: 10.1016/j.aquaculture.2020.735168
Yang Y., Xie J., Li H., Tan S., Chen Y., Yu H. (2017). Prevalence, antibiotic susceptibility and diversity of Vibrio parahaemolyticus isolates in seafood from south China. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02566
Yan S., Zhang W., Li C., Liu X., Zhu L., Chen L., et al. (2021). Serotyping, MLST, and core genome MLST analysis of Salmonella enterica from different sources in China during 2004-2019. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.688614
Zanetti S., Spanu T., Deriu A., Romano L., Sechi L. A., Fadda G. (2001). In vitro susceptibility of Vibrio spp. isolated from the environment. Int. J. Antimicrob. Agents 17, 407–409. doi: 10.1016/s0924-8579(01)00307-7
Keywords: vibrio, harveyi clade, phenotype, multi-locus sequence analysis (MLSA), multi-locus sequence typing (MLST), antibiotic resistance
Citation: Kang H, Yu Y, Liao M, Wang Y, Yang G, Zhang Z, Li B, Rong X and Wang C (2022) Physiology, metabolism, antibiotic resistance, and genetic diversity of Harveyi clade bacteria isolated from coastal mariculture system in China in the last two decades. Front. Mar. Sci. 9:932255. doi: 10.3389/fmars.2022.932255
Received: 29 April 2022; Accepted: 28 July 2022;
Published: 22 August 2022.
Edited by:
Qingchao Wang, Huazhong Agricultural University, ChinaReviewed by:
Dao-Feng Zhang, Hohai University, ChinaQian Yang, Faculty of Bioscience Engineering, Ghent University, Belgium
Yiqin Deng, South China Sea Fisheries Research Institute (CAFS), China
Copyright © 2022 Kang, Yu, Liao, Wang, Yang, Zhang, Li, Rong and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meijie Liao, bGlhb21qQHlzZnJpLmFjLmNu; Yingeng Wang, d2FuZ3lnQCB5c2ZyaS5hYy5jbg==
†These authors have contributed equally to this work