 Baoquan Gao
Baoquan Gao Jianjian Lv1,2
Jianjian Lv1,2 Xianliang Meng
Xianliang Meng Jitao Li
Jitao Li Ping Liu
Ping Liu- 1Key Laboratory of Sustainable Development of Marine Fisheries, Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 2Laboratory for Marine Fisheries and Aquaculture, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
Background
The blue crab Callinectes sapidus is native to the western Atlantic Ocean from Uruguay toNova Scotia (Millikin and Williams, 1984; Johnson, 2015) where it represents a commercially valuable shellfish product (Mancinelli et al., 2017). The blue crab is the target of several large recreational and commercial fisheries ($219 million annually in the U.S.) (National Marine Fisheries Service, 2016), and playing important roles in the ecologically environments they inhabit (Roegner and Watson, 2020). Considering their economic and ecological significance, several studies have been conducted to explore the mechanism of spawning, soft shell crab culture, and physiological processes in blue crab. For example, Bembe et al. studied the optimal temperature and photoperiod for the spawning of blue crabs (Bembe et al., 2017). Spitznagel et al. (2019) investigated the risk factors for mortality and reovirus infection in aquaculture production of soft-shell blue crabs. Further, Roegner and Watson (2020) reported de novo transcriptome assembly and functional annotation for adult blue crab Y-organs; they also performed Illumina sequencing for differential gene expression analysis between Y-organs of intermolt and premolt crabs. Yednock et al. (2015) used RNA-Seq to examine short-term transcriptomic responses in two tissues from juvenile blue crabs exposed to crude oil in a laboratory exposure experiment. The genome assembly at the chromosome level of blue crab has been completed, resulting in a 985Mb assembly with a scaffold N50 of 153kb, 88% (888/1013)of which were complete and single copies by arthropod BUSCO (Benchmarking Universal Single-Copy Orthologs) (Bachvaroff et al., 2021).
Single molecule real-time (SMRT) sequencing can generate kilobase-sized sequencing reads, facilitating the assembly of FL transcripts (Eid et al., 2009; Sharon et al., 2013). The FL transcriptome has a lot of advantages. First, FL transcript sequences can be directly obtained to provide detailed information pertaining to the transcriptome of sequenced species. Second, various alternative splicing events can be detected. Besides, new functional genes can be discovered, and perfectideal genome annotation is feasible.
Herein we used Pacific Biosciences (PacBio) SMRT sequencing to report, for the first time, the FL transcriptome of C. sapidus. Based on the obtained data, we conducted some important studies, including transcript functional annotation, coding sequence (CDS) prediction, lncRNA prediction, transcription factor (TF) prediction, and simple sequence repeat (SSR) analysis. FL transcriptome database can prove valuable for studying, for example, the genetic evolution, genetic breeding, and physiological mechanisms of C. sapidus.
Data Description
Sample Collection and RNA Sample Preparation
Six healthy adult C. sapidus (223.4 ± 18.4 g) were purchased from an aquatic product market. The crabs were reared for a week in an indoor closed seawater tank (water, 10,000-L; temperature, 19°C; salinity, 30 ppt; pH 8.0). Subsequently, the hemocyte, eyestalk, muscle, hepatopancreas, heart, stomach, gill, and thymus were extracted from three randomly chosen crabs, respectively. These samples were frozen in liquid nitrogen. Total RNA was extracted separately using TRIzol (Invitrogen, USA). RNA quality was assessed by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), and a mixed pool sample was used for single molecule FL transcriptome sequencing.
Library Preparation and SMRT Sequencing
The cDNA sequencing library was constructed using the aforementioned mixed pool sample, which was sequenced on a single PacBio SMRT cell. Briefly, first- and second-strand cDNA was generated from mRNA using the SMARTerTM PCR cDNA Synthesis Kit (Pacific Biosciences, USA), and >4-Kb size selection was performed using BluePippin® (Sage Science, USA). Subsequently, >4-Kb cDNA was mixed in equal amounts with non-size-selected cDNA. SMRTbellTM hairpin adapters were ligated after a round of PCR and end-repair. On exonuclease digestion, a cDNA library was obtained.
PacBio Long Read Processing
With minFullPass = 1 and minPredictedAccuracy = 0.80, subreads were processed into error-corrected reads of insert using the Iso-seq pipeline (Pacific Biosciences, Menlo Park, CA, USA). By searching for the polyA tail signal and 5’- and 3’-cDNA primers in reads of insert, FL, and FL non-chimeric (FLNC) reads were identified. Iterative clustering for error correction was used to obtain FLNC consensus isoforms. The LoRDEC software was employed to correct polished consensus isoforms using Illumina short-read RNA-seq data (Salmela and Rivals, 2014). The CD-HIT software was used to remove redundancy of high-quality transcripts (Fu et al., 2012). Gene function was annotated by BLAST v2.2.26 (Altschul et al., 1997) based on the databases of NR (Li et al., 2002), GO (Michael et al., 2000), NT, Pfam, KOG/COG (Tatusov et al., 2003), KEGG (Kanehisa et al., 2004), and Swiss-Prot (Bairoch and Apweiler, 2000).
Predicting Gene Structure of Isoforms
To identify protein CDSs from cDNAs, we applied the ANGEL pipeline (Shimizu et al., 2006). Animal TFDB 2.0 was used to analyze TFs (Zhang et al., 2015). CNCI (Sun et al., 2013), CPC (Kong et al., 2007), Pfam (Finn et al., 2016), and PLEK (Li et al., 2014) were used for lncRNA prediction. SSRs of the transcriptome were identified using MISA (http://pgrc.ipk-gatersleben.de/misa.html).
Results
Data Summary
In total, we obtained 17.19 Gb of subreads, including 6,986,978 subreads with the average length of 2,461 bp and the N50 length of 3,079 bp. Further, 368,306 circular consensus sequence reads were extracted, including 279,032 FLNC reads with the mean length of 3,010 bp. In addition, 142,291 consensus reads were obtained with their mean length of 3,072 bp. Polished transcripts were corrected by Illumina short reads; 142,291 high-quality transcripts with mean length of 3,087 bp were obtained for subsequent analyses. On subjecting high-quality transcripts to redundancy removal, we obtained 66,780 genes. The length of 1,147 (1.72%), 951 (1.42%), 7,580 (11.35%), 22,709 (34.00%), and 34,393 (51.50%) genes was 1–500 bp, 500–1000 bp, 1–2 kbp, 2–3 kbp, and >3 kbp, respectively (Table 1). In total, 13,626 genes have at least two transcripts (Supplementary Table 1).
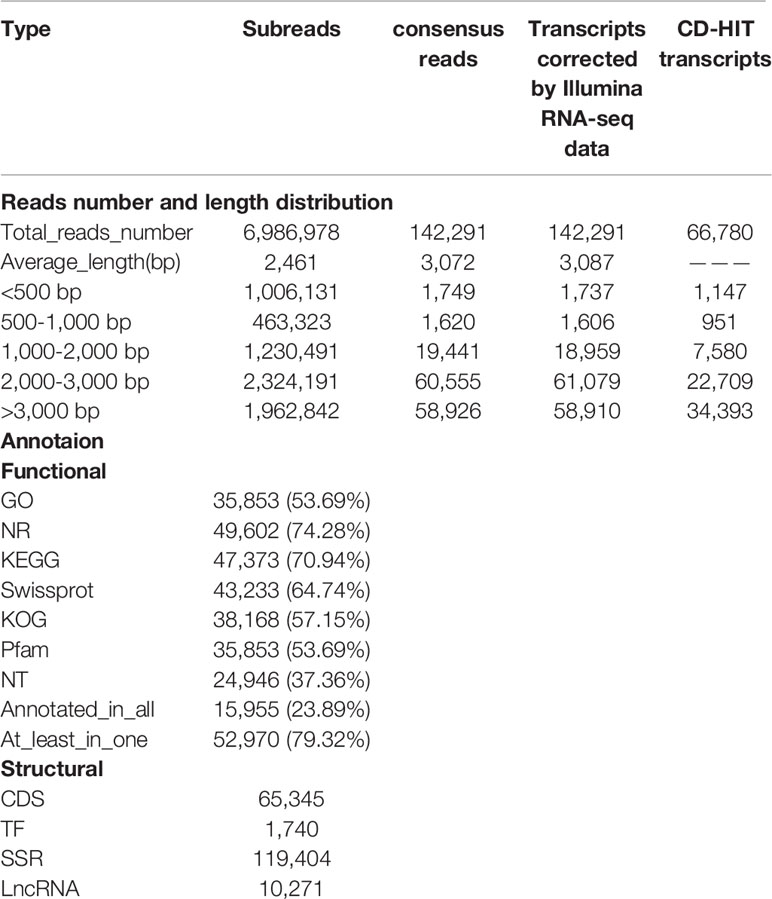
Table 1 Reads and annotation statistics for the ISO-seq transcripts.
Gene Annotations
We observed that 52,970 (79.32%) genes were successfully annotated by aligning to one or more of the seven databases, and 15,955 (23.89%) genes were annotated in all the seven aforementioned databases. Furthermore, 35,853 (53.69%) genes were annotated in GO, 49,602 (74.28%) in NR, 47,373 (70.94%) in KEGG, 43,233 (64.74%) in Swiss-Prot, 38,168 (57.15%) in KOG, 35,853 (53.69%) in Pfam, and 24,946 (37.36%) in NT (Table 1), respectively.
The genes were assigned to 55 GO terms belonging to the following three main categories: cellular component, molecular function, and biological process. For biological process, most GO terms were enriched in cellular process (14,923, 41.62%), metabolic process (14,337, 39.99%), and single-organism process (10,618, 29.62%). For cellular component, the most abundant GO terms were cell (9,175, 25.59%), cell part (9,175, 25.59%), and organelle (6,197, 20.12%). Binding (21,792, 60.78%), catalytic activity (15,608, 43.53%), and transporter activity (2,522, 7.03%) represented the activity categories of molecular function (Figure 1A). Approximately 18,310 (36.91%) genes were aligned to Hyalella azteca, followed by Zootermopsis nevadensis (2,236, 4.51%) and Scylla paramamosain (1,426, 2.87%), by sequence alignment on the basis of the NR database (Figure 1B). With regard to KOG annotation, genes were divided into 26 subcategories, such as function R (general function prediction only), function T (signal transduction mechanisms), and function Z (cytoskeleton) (Figure 1C). In total, 357 pathways were derived from the KEGG database, and the pathway with the most genes was “metabolism” (116, 32.49%). The highest number of genes were involved in “signal transduction” (5,691, 12.01%), followed by “cardiovascular diseases” (3,140, 6.63%) (Figure 1D).

Figure 1 Characterization of functional annotation. (A) GO functional annotations. (B) NR functional annotations. (C) KOG functional annotations. (D) KEGG functional annotations.
CDS, TF, lncRNA Prediction and SSR Analyses
Using ANGEL, 65,345 CDSs were identified, including 33,461 complete CDSs (Supplementary Figure 1A). Besides, 142,291 high-quality transcripts were predicted to be TFs by searching the hidden Markov models of all the TFs in animal TFDB 2.0, which comprise 55 TF families. Supplementary Figure 1B shows the distribution of the top 30 TF families. In total, 10,271 lncRNA transcripts were predicted by all four computational approaches (Supplementary Figure 1C). Collectively, 88.04% lncRNAs were <4,000-nt long, and most were 2,000–3,000-nt long. Further, 142,291 high-quality transcripts were subjected to SSR analysis, including 119,404 SSRs. The numbers of mono-, di-, tri-, tetra-, penta-, and hexanucleotides were 26,035, 30,524, 22,474, 2,508, 433, and 115, respectively (Supplementary Figure 1D).
Reuse Potential
To the best of our knowledge, this is the first study to report the FL transcriptome of C. sapidus. The transcriptome data reported herein should support further studies on C. sapidus genetics and genomic information. Moreover, our data should be valuable to chromosome-level genome studies of C. sapidus and other related species.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/, CRA006442 https://figshare.com/articles/dataset/Full-length_Transcriptome_of_the_blue_crab_Callinectes_sapidus/19608261.
Ethics Statement
The relevant national and international guidelines were followed during the conductance of the animal experiments and the Yellow Sea Fisheries Research Institute approved the experiments. Endangered or protected species were not involved in this study.
Author Contributions
BG and JJL designed the experiment. XM raised Carb. JTL collected Carb tissue samples. YL uploaded data in CNCB-NGDC. BG drafted the manuscript. JL and PL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Marine Genetic Resource Center, Central Public-interest Scientific Institution Basal Research Fund, CAFS (NO. 2020TD46& NO.2021GH05).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.922188/full#supplementary-material
References
Altschul S. F., Madden T. L., Schaffer A. A., Zhang J. N. (1997). Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bachvaroff T. R., Mcdonald R. C., Plough L. V., Chung J. S. (2021). Chromosome-Level Genome Assembly of the Blue Crab, Callinectes Sapidus. G3 11 (9), jkab212. doi: 10.1093/g3journal/jkab212
Bairoch A., Apweiler R. (2000). The SWISS-PROT Protein Sequence Database and its Supplement TrEMBL in 2000. Nucleic Acids Res. 28 (1), 45–48. doi: 10.1093/nar/28.1.45
Bembe S., Liang D., Chung J. S. (2017). Optimal Temperature and Photoperiod for the Spawning of Blue Crab, Callinectes Sapidus, in Captivity. Aquaculture Res. 48 (11), 5498–5505. doi: 10.1111/are.13366
Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., et al. (2009). Real-Time DNA Sequencing From Single Polymerase Molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., et al. (2016). The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Research. Database Issue 44, 279–285. doi: 10.1093/nar/gkv1344
Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: Accelerated for Clustering the Next-Generation Sequencing Data. Bioinformatics 28 (23), 3150. doi: 10.1093/bioinformatics/bts565
Johnson D. S. (2015). The Savory Swimmer Swims North: A Northern Range Extension of the Blue Crab Callinectes Sapidus? J. Crust. Biol. 35, 105–110. doi: 10.1163/1937240X-00002293
Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. (2004). The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 32 (1), 277–280. doi: 10.1093/nar/gkh063
Kong L., Zhang Y., Ye Z. Q., Liu X. Q., Zhao S. Q., Wei L., et al. (2007). CPC: Assess the Protein-Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res. 36, 345–349. doi: 10.1093/nar/gkm391
Li W., Jaroszewski L., Godzik A. (2002). Tolerating Some Redundancy Significantly Speeds Up Clustering of Large Protein Databases. Bioinformatics 18 (1), 77–82. doi: 10.1093/bioinformatics/18.1.77
Li A. M., Zhang J. Y., Zhou J. Y. (2014). PLEK: A Tool for Predicting Long non-Coding RNAs and Messenger RNAs Based on an Improved K-Mer Scheme. BMC Bioinf. 15, 311. doi: 10.1186/1471-2105-15-311
Mancinelli G., Chainho P., Cilenti L., Falco S., Kapiris K., Katselis G., et al. (2017). On the Atlantic Blue Crab (Callinectes Sapidus Rathbun 1896) in Southern European Coastal Waters: Time to Turn a Threat Into a Resource? Fish. Res. 194, 1–8. doi: 10.1016/j.fishres.2017.05.002
Michael A., Catherine A. B., Judith A. B., David B., Butler H., Cherry J. M., et al. (2000). Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 25 (1), 25–29. doi: 10.1038/75556
Millikin M. R., Williams A. B. (1984) Synopsis of Biological Data on Blue Crab Callinectes Sapidus Rathbun. FAO Fisheries Synopsis 138, NOAA Technical Report, NMFS 1, 43 pp.
National Marine Fisheries Service. (2016) Fisheries of the United States 2015. U.S. Department of Commerce, NOAA Current Fishery Statistics No. 2015 (Silver Spring). Available at: https://www.st.nmfs.noaa.gov/commercial-fisheries/fus/fus15/index.
Roegner M. E., Watson R. D. (2020). De Novo Transcriptome Assembly and Functional Annotation for Y-Organs of the Blue Crab (Callinectes Sapidus), and Analysis of Differentially Expressed Genes During Pre-Molt. General and. Comp. Endocrinol. 298, 113567. doi: 10.1016/j.ygcen.2020.113567
Salmela L., Rivals E. (2014). LoRDEC: Accurate and Efficient Long Read Error Correction. Bioinformatics 30 (24), 3506–3514. doi: 10.1093/bioinformatics/btu538
Sharon D., Tilgner H., Grubert F., Snyder M. (2013). A Single-Molecule Long-Read Survey of the Human Transcriptome. Nat. Biotechnol. 31, 1009–1014. doi: 10.1038/nbt.2705
Shimizu K., Adachi J., Muraoka Y. (2006). ANGLE: A Sequencing Errors Resistant Program for Predicting Protein Coding Regions in Unfinished cDNA. J. Bioinf. Comput. Biol. 4 (3), 649–664. doi: 10.1142/S0219720006002260
Spitznagel M. I., Small H. J., Lively J. A., Shields J. D., Schott E. J. (2019). Investigating Risk Factors for Mortality and Reovirus Infection in Aquaculture Production of Soft-Shell Blue Crabs (Callinectes Sapidus). Aquaculture 502, 289–295. doi: 10.1016/j.aquaculture.2018.12.051
Sun L., Luo H. T., Bu D. C., Zhao G. G., Yu K. T., Zhang C. H., et al. (2013). Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-Coding Transcripts. Nucleic Acids Res. 41 (17), 166. doi: 10.1093/nar/gkt646
Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., et al. (2003). The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinf. 4 (1), 41. doi: 10.1186/1471-2105-4-41
Yednock B. K., Sullivan T. J., Neigel J. E. (2015). De Novo Assembly of a Transcriptome From Juvenile Blue Crabs (Callinectes Sapidus) Following Exposure to Surrogate Macondo Crude Oil. BMC Genomics 16, 521. doi: 10.1186/s12864-015-1739-2
Keywords: blue crab, Callinectes sapidus, full-length transcriptome, PacBio sequencing, ISO-seq
Citation: Gao B, Lv J, Meng X, Li J, Li Y, Liu P and Li J (2022) Full-Length Transcriptome Construction of the Blue Crab Callinectes sapidus. Front. Mar. Sci. 9:922188. doi: 10.3389/fmars.2022.922188
Received: 17 April 2022; Accepted: 09 May 2022;
Published: 13 June 2022.
Edited by:
Yangfang Ye, Ningbo University, ChinaReviewed by:
Hongbo Jiang, Shenyang Agricultural University, ChinaYuquan Li, Qingdao Agricultural University, China
Qi Liu, Dalian Ocean University, China
Copyright © 2022 Gao, Lv, Meng, Li, Li, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Liu, bGl1cGluZ0B5c2ZyaS5hYy5jbg==