 Busu Li
Busu Li Huan Wang
Huan Wang Ang Li1,2
Ang Li1,2 Ling Zhu
Ling Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 16 June 2022
Sec. Aquatic Physiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.916373
Skeletal muscles of teleost are mainly composed of slow-twitch muscles (SM) and fast-twitch muscles (FM) differed in contractile properties, metabolic capacities, and regeneration rate. The transcriptional regulatory mechanisms that control different muscle types have been elucidated in teleost according to transcriptome between SM and FM. However, the differences between SM and FM were affected not only by genotype but also by complicated epigenetic effects, including DNA methylation, which usually regulates genes in transcription level. To determine the essential role of DNA methylation in the regulation of different muscle types, we analyzed whole-genome methylation profiles of pelagic migratory fish Pseudocaranx dentex with abundant and well-separated SM and integrated DNA methylation profiles with the previously obtained transcriptome data. A total of 4,217 differentially methylated genes (DMGs) were identified, of which 3,582 were located in the gene body and 635 in the promoter. These DMGs mainly participated in muscle metabolite and cell junction. Enriched cell junction pathway reflected different capillary distribution between SM and FM. Through comprehensive analysis of methylome and transcriptome, 84 differentially expressed genes (DEGs) showed significant methylation variation in promoters between SM and FM, indicating that their expression was regulated by DNA methylation. Hypomethylated and highly expressed oxygen storage protein Myoglobin (myg) in SM indicated demethylation of myg promoter could upregulate its expression, thus increasing O2 supplying and meeting oxygen demands of SM. Hypermethylated and lowly expressed tnn (Troponin) and rlc (myosin regulatory light chain) in SM may be associated with low mobility of myosin cross bridges, which lead to slower and less frequent muscle contraction in SM than in FM. In addition, hypomethylated and highly expressed lbx1 (Ladybird homeobox protein homolog 1) and epo (erythropoietin) may be related to increased satellite cell numbers, and Semaphorin/Plexin genes may be related to higher rate of neuromuscular connection reconstruction, which further promote high muscle regeneration efficiency in SM. Our study elucidated the potential DNA methylation mechanisms that regulate physiological characteristics differences between SM and FM, which could facilitate our understanding of skeletal muscle adaptation in pelagic migratory fishes and further enrich the theoretical basis for the study of physiological characteristics and adaptive evolution in teleost fishes.
The skeletal muscles of fish are critical tissues involved in swimming activity, which is of great significance for the survival of fish, supporting various physiological activities such as feeding, reproduction, clustering, migration, and escape from enemies. In most teleost, skeletal muscles are mainly composed of slow-twitch muscles (SM) and fast-twitch muscles (FM), which are differed in contractile properties, metabolic capacities, and regeneration efficiency (Bassaglia and Gautron, 1995; Jayaraman et al., 2013). SM contain higher level of mitochondria, capillaries, and myoglobin than FM. The energy metabolism of SM is aerobic supporting sustaining swimming, whereas FM rely mainly on glycogen for anaerobic metabolism for fast movements.
The proportion of SM and FM varies among different locomotion type fishes. Fishes with a more active mode of life have a higher proportion of SM (Greek-Walker and Pull, 1975; Teulier et al., 2019), making them an excellent model system for skeletal muscle difference study (Gibb, 2002). Oceanic migratory fishes such as Trachurus trachurus and Sardina pilchardus, which swim constantly in schools covering great distances have a higher proportion of SM than benthopelagic fishes such as Sparus aurata and Callionymus lyra (Greek-Walker and Pull, 1975; Gibb, 2002; Teulier et al., 2019). Pseudocaranx dentex, as a pelagic migratory fish with high nutritional value, is the candidate species for far-reaching marine aquaculture in China. The aquaculture conditions of P. dentex should meet their long-distance swimming requirement, but the underlying regulation mechanism is still unclear, which has become the limitation for the large-scale aquaculture of P. dentex. To meet their high energy requirement during oceanic migratory, P. dentex developed higher proportion of SM with more mitochondria and capillaries, which could provide energy and oxygen for sustaining swimming. Understanding the molecular differences between different muscle types could help us reveal the adaptation mechanism of the high proportion SM of pelagic migratory fishes to the long-distance swimming. The transcriptional regulatory mechanisms that control the different muscle types have been elucidated according to the transcriptome between SM and FM in several species (Mareco et al., 2015; Gao et al., 2017) including P. dentex (Wang et al., 2022).
However, studies over the last several decades have demonstrated that not only genetic but also epigenetic events especially DNA methylation might be involved in the skeletal muscle development and differences (Fang et al., 2017; Fan et al., 2020). DNA methylation refers to modification in gene expression without changing the DNA sequence itself and mediates numerous biological processes such as growth, development, and genomic imprinting (Razin and Cedar, 1991; Jones and Takai, 2001; Bird, 2007). In teleost, whole-genome DNA methylation analysis has been widely applied for exploration of physiological regulation mechanism. Several researchers have revealed the epigenetic effects on sex differences in Takifugu rubripes (Zhou et al., 2019), growth and gonad of large yellow croaker (Larimichthys crocea) (Zhang et al., 2019b; Zhou et al., 2019), and skin color in Crucian carp (Carassius carassius L.) (Zhang et al., 2017). Whole-genome analysis of DNA methylation has become an effective approach for researching physiological regulation in teleost. To reveal the epigenetic regulation process of physiological characteristics differences between SM and FM, we generated the whole-genome single-base DNA methylation profiles and integrated the methylome with the transcriptome data obtained from the previous study between SM and FM of P. dentex. We identified critical genes whose expression was regulated by promoter methylation, which may be related to phenotypic differences between SM and FM, and constructed an overview of essential genes and related physiological function regulated by DNA methylation between SM and FM of P. dentex. The results will provide a new perspective for understanding the long-distance swimming adaptability of pelagic migratory fish skeletal muscle and further enrich the theoretical basis for the study of physiological characteristics and adaptive evolution in teleost fish.
SM and FM from three adult P. dentex individuals (body length: 36.83 ± 0.67 cm; body weight: 1567.63 ± 147.05 g) collected in 2020 from Dalian Tianzheng Industrial Co., Ltd. (Dalian, Liaoning province, China) were used for whole-genome bisulfite sequencing (WGBS) with the Illumina HiSeq/NovaSeq platform (Illumina, CA, USA). High-quality, double-stranded genomic DNA was isolated from muscle tissues using a DNA extraction kit (TIANGEN, Beijing, China), following the manufacturer’s recommended instructions. Genomic DNA extraction quality was validated by agarose gels and NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). DNA concentration was quantified using the Qubit® DNA Assay Kit on a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA).
A total amount of 100 ng of genomic DNA spiked with 0.5 ng of lambda DNA was fragmented to 200–300 bp by sonication with Covaris S220. Bisulfite treatment of these fragments was performed using the EZ DNA Methylation-GoldTM Kit (Zymo Research) followed with PCR amplification to generate sequencing library. The libraries constructed by Novogene Corporation (Beijing, China) were sequenced using the Illumina Novaseq platform (Illumina, CA, USA). The quality of the libraries was assessed on the Agilent Bioanalyzer 2100 system. After image analysis and base calling performed with Illumina CASAVA pipeline, 150-bp paired-end reads were generated.
Thereafter, reads constructed by the Illumina pipeline were preprocessed through Trimmomatic software with the following parameters: SLIDINGWINDOW, 4:15; LEADING, 3; TRAILING, 3; mismatch: 2; ILLUMINACLIP: adapter. fa, 2:30:7. Then, reads unpaired or shorter than 36 nt after trimming were removed. Remaining reads passed all these filtering steps were counted as clean reads and used to perform all subsequent analysis. The quality of raw reads and clean reads were assessed by FastQC (fastqc_v0.11.5). Q30 and GC content were also calculated to evaluate data quality.
Bisulfite-treated reads were aligned to the P. dentex reference genome (PRJNA731999) using Bismark software with following parameters: (score_min L, 0, -0.2, -X 700). In brief, the P. dentex reference genome and clean reads were transformed into bisulfite-converted version (C-to-T and G-to-A converted), and clean reads were aligned to reference genome in a directional manner. Alignment strategy of Bismark was exhibited as a schematic diagram (Supplementary Figure 1) and the methylation state of all cytosine positions was inferred. The sequencing depth and coverage were calculated using deduplicated reads, which were reads that aligned to the same genome regions. The binomial distribution test for each C site was used to confirm C-site methylation by screening conditions for coverage ≥5× and FDR-corrected p-value < 0.05. The percentage of cytosine sequenced at cytosine reference positions in the lambda genome was also evaluated as the bisulfite non-conversation rate.
The methylation level of an individual C-sites was calculated with the following formula: ML(C) = reads(mC)/[reads(mC) + reads(umC)], where mC is the reads number of methylated C-site counts, and umC is the unmethylated. The methylome data had been deposited in the Sequence Read Archive database with the accession number PRJNA796775.
Differentially methylated regions (DMRs) were identified by DSS software with the following parameters: smoothing = TRUE, smoothing.span = 200, delta = 0, p.threshold = 1e-05, minlen = 50, minCG = 3, dis.merge = 100, pct.sig = 0.5. By definition, DMRs have at least three methylation sites in the region, Q-value ≤ 0.01, and the absolute mean methylation difference greater than 50%. Subsequently, DMR-related genes (DMGs) were defined as genes whose gene body region [from Transcriptional Start Site (TSS) to Transcriptional End Site (TES)] or promoter region (2 kb upstream from the TSS) have 1 bp overlap with the DMRs.
Gene Ontology (GO) is an international standardized gene function classification system. We implemented GO enrichment analysis of DMGs using the GOseq R package (Young et al., 2010) with gene length bias correction. Significantly enriched signal transduction pathways represented by DMGs were determined using pathway enrichment analysis. KOBAS software (Mao et al., 2005) was used to test the statistical enrichment of DMR-related genes in KEGG pathways.
We matched the promoter regions of WGBS data with the RNA-seq data produced by identical materials from present study which are publicly available at the Sequence Read Archive database with the accession number PRJNA733284 (Wang et al., 2022). By integrated analysis of DMGs and differentially expressed genes (DEGs), we obtained a set of genes related to muscle difference and performed functional analysis of these genes. The STRING database was used to protein-protein analyze interaction networks of candidate DMGs (http://string-db.org/) (Franceschini et al., 2013).
Genome-wide DNA methylation analysis of the skeletal muscles in P. dentex was performed by WGBS with >99.9% conversion efficiency. In present study, 24.12-GB and 24.45-GB raw bases were generated on average for SM and FM, respectively. After quality control, approximately 80 million clean reads were generated for each group, with the Q30 of clean, full-length reads ranging from 92.76% to 93.58%. The mapped reads were used for subsequent analysis, as the rates ranged from 83.57% to 84.71% (Table 1). These results indicated a reliable sequencing outcome.
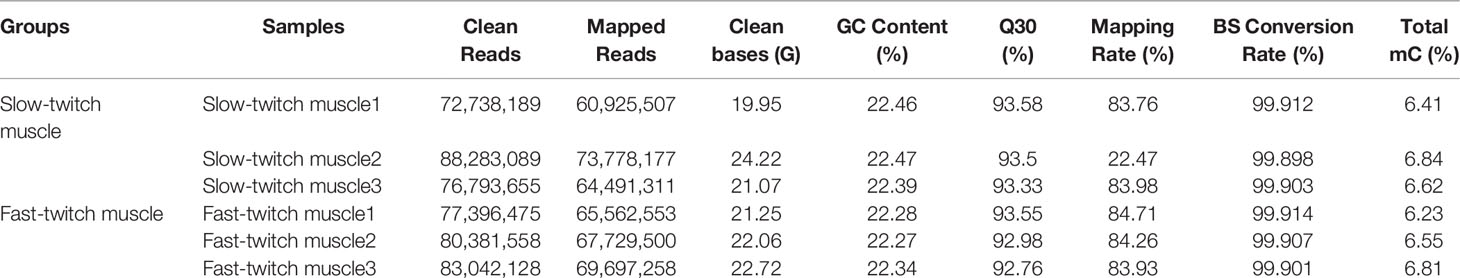
Table 1 Summary of whole-genome bisulfite sequencing (WGBS) sequencing data in P. dentex skeletal muscles.
Approximately 2.5 billion methylated cytosines (mCs) were detected throughout the whole genome, accounting for 6.5% of C sites in muscle samples of P. dentex. DNA methylation occurs at three different sequence sites, the cytosine-phosphate-guanosine (CG) dinucleotides, CHG and CHH sites (where H is A, C, or T). The classification of mCs showed a similar proportion in SM and FM genome. In the SM, 98.28% of the mCs were mCG type, whereas about 0.37% and 1.35% were mCHG and mCHH types (Figure 1A), and, in FM, the proportion of mCG, mCHG and, mCHH were 97.56%, 0.44%, and 2.00% (Figure 1B), respectively (Supplementary Table 1). Methylation in CG motifs was further analyzed in subsequent results. Furthermore, we investigated the DNA methylation on a chromosomal level. The highest mCG was on chromosome 1 with 77% and the lowest mCG was on chromosome 21 with 70% in both SM and FM (Figure 1C and Supplementary Table 2).
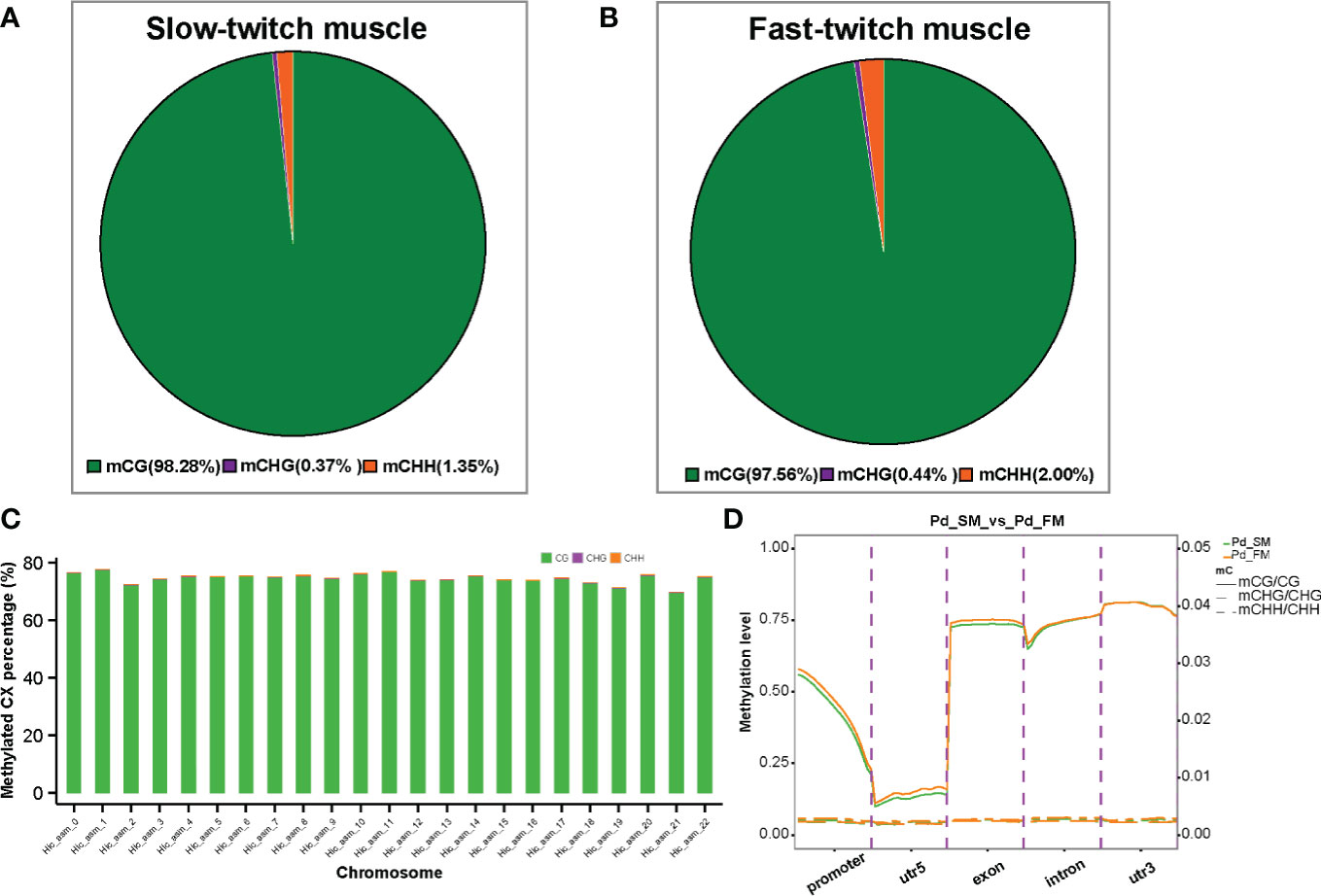
Figure 1 The DNA methylation characteristics of slow-twitch muscles and fast-twitch muscles in P. dentex. The average ratio of DNA methylation types in slow- (A) and fast-twitch muscles (B). The green, purple, and orange colors represent methylated CG, CHG, and CHH, respectively. (C) Representative image of methylation level distribution on each chromosome in P. dentex. The methylation level data of mCG/mCHH/mCHG on each chromosome in slow- and fast-twitch muscles could be got in Supplementary Table 2. (D) DNA methylation levels in functional regions of the genome.
To further compare the genome-wide distribution and the methylation levels of various functional genomic elements between SM and FM, we analyzed the methylation level of CGs in five genomic elements including promoters, 5′UTR (un-translated region), exons, introns, and 3′UTRs (Figure 1D). The highest level of mCG methylation was detected in the 3′UTR region, which was approximately 80% followed by the exon regions, which was about 75% (Supplementary Table 3). The lowest methylation level was detected around TSS. However, mCHG and mCHH methylation level were highest in the exon and intron, respectively (Supplementary Table 3).
We identified 50,070 differentially methylated CG regions, 541 differentially methylated CHH regions and 47 differentially methylated CHG regions. Among the DMRs, 29,779 (CG: 29,696 + CHH: 67 + CHG: 16) were hypermethylated and 20,879 (CG: 20,374 + CHH: 474 + CHG: 31) were hypomethylated (Figure 2A). In present study, methylation in CG motifs that were the most abundant methylated DMGs was further analyzed in subsequent results. The DMRs in CG motifs were mostly located at introns. More detailed information was listed in Figure 2B. We noticed that most DMRs in CG motifs were about length 250 bp, and the length follows a normally distributed model (Figure 2C).
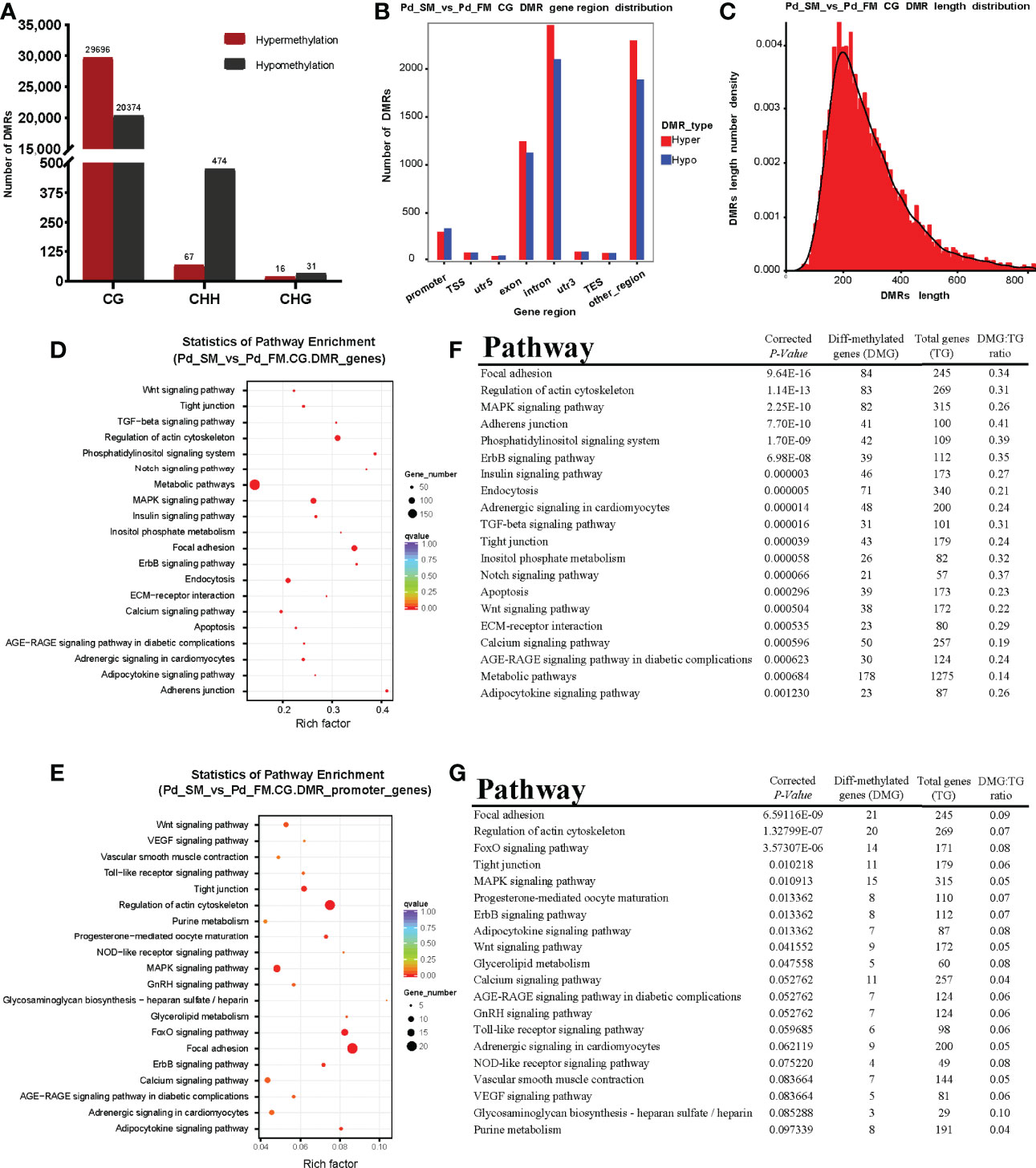
Figure 2 The characteristics of DMRs and function analysis of DMGs. (A) The number of differentially methylated regions among different methylation types. (B) The distribution numbers of hyper/hypo DMRs in different genomic elements. (C) DMR length distribution density of mCGs contexts. (D, E) Scatterplot of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched by the DMGs in gene body (D) and promoter region (E) between slow- and fast-twitch muscles. The vertical axis represents the name of the pathway, and the horizontal axis shows the enrichment factor. The size of the plot denotes the number of DMGs, and the color corresponds to the Qvalue. A deeper color represents a smaller Qvalue and indicates more significant enrichment of the pathway. (F, G) The top 20 pathways enriched by DMGs in gene body (F) and promoter region (G) between slow- and fast-twitch muscles.
To explore the methylation changes of genes in different muscles of P. dentex, the GO and KEGG databases were used to annotate 4,217 DMGs detected in the DMRs which contained 3,582 DMGs in gene body and 635 in promoter. We further performed functional characterization of DMGs in CG motifs. On the basis of the GO database, only DNA binding term was significantly enriched for DMGs between SM and FM in P. dentex (Supplementary Figure 2). According to the KEGG pathway analysis, DMGs of gene body were significantly enriched in focal adhesion, adherens junction, tight junction, MAPK, TGF-β, Notch, Wnt, and calcium signaling pathway. Thirty pathways were significantly enriched (P-value < 0.05) between SM and FM, the top 20 of which were shown in Figures 2D, F. In addition, DMGs of promoter were significantly enriched in “focal adhesion” pathway (Figures 2E, G).
DNA methylation in promoter regions can inhibit gene expression (Wagner et al., 2014), whereas the association between DNA methylation within the gene body and gene expression is still poorly understood (Jones, 2012). To further identify the key differential genes involved in the regulation of skeletal muscle differences in teleost, we integrated the RNA-seq data that were obtained in previous study (Wang et al., 2022) and WGBS data of promoter regions to reveal methylated candidate genes between SM and FM in P. dentex. Our results showed that there were 84 DEGs with statistically significant methylation variation in promoter region, including 31 hypermethylated and downregulated genes, and 53 hypomethylated and upregulated genes in SM (Figures 3A, B). Functional characterization indicated that these genes were involved in process of neuron guidance and cell junction (Supplementary Table 4). Muscle-related genes also showed both methylation and expression difference between SM and FM, such as Myoglobin (myg), Troponin (tnn), and Myosin regulatory light chain (rlc) (Figure 3C). The promoter of myg was hypomethylated and its expression was enhanced in SM. The promoter of tnn and rlc were hypomethylated, which induced their mRNA expression in FM. As muscle is a major metabolic tissue and two types of skeletal muscles have different metabolic properties, it was not unexpected that metabolic genes, including ADP/ATP translocase (adt3), glyceraldehyde-3-phosphate dehydrogenase (gapdh), and insulin-like growth factor-binding protein (igfbp7) showed both methylation and expression difference (Supplementary Table 5). Satellite cells related transcription factor lbx1 (ladybird homeobox protein homolog 1) and epo (erythropoietin) genes were hypomethylated and upregulated in SM (Figure 3C and Supplementary Table 5). Semaphorin and Plexin that are axon guidance molecules were highly expressed and hypomethylated in SM compared with FM (Figure 3C). Furthermore, we performed an association analysis and constructed the interaction networks of 84 DEGs with methylation variation in promoter regions (Figure 4A). These results illustrated that DMGs related to muscle contraction including myg, tnn, and rlc (Figure 4B) and axon guidance (Figure 4C) including Semaphorin and Plexin were highly correlated with each other.
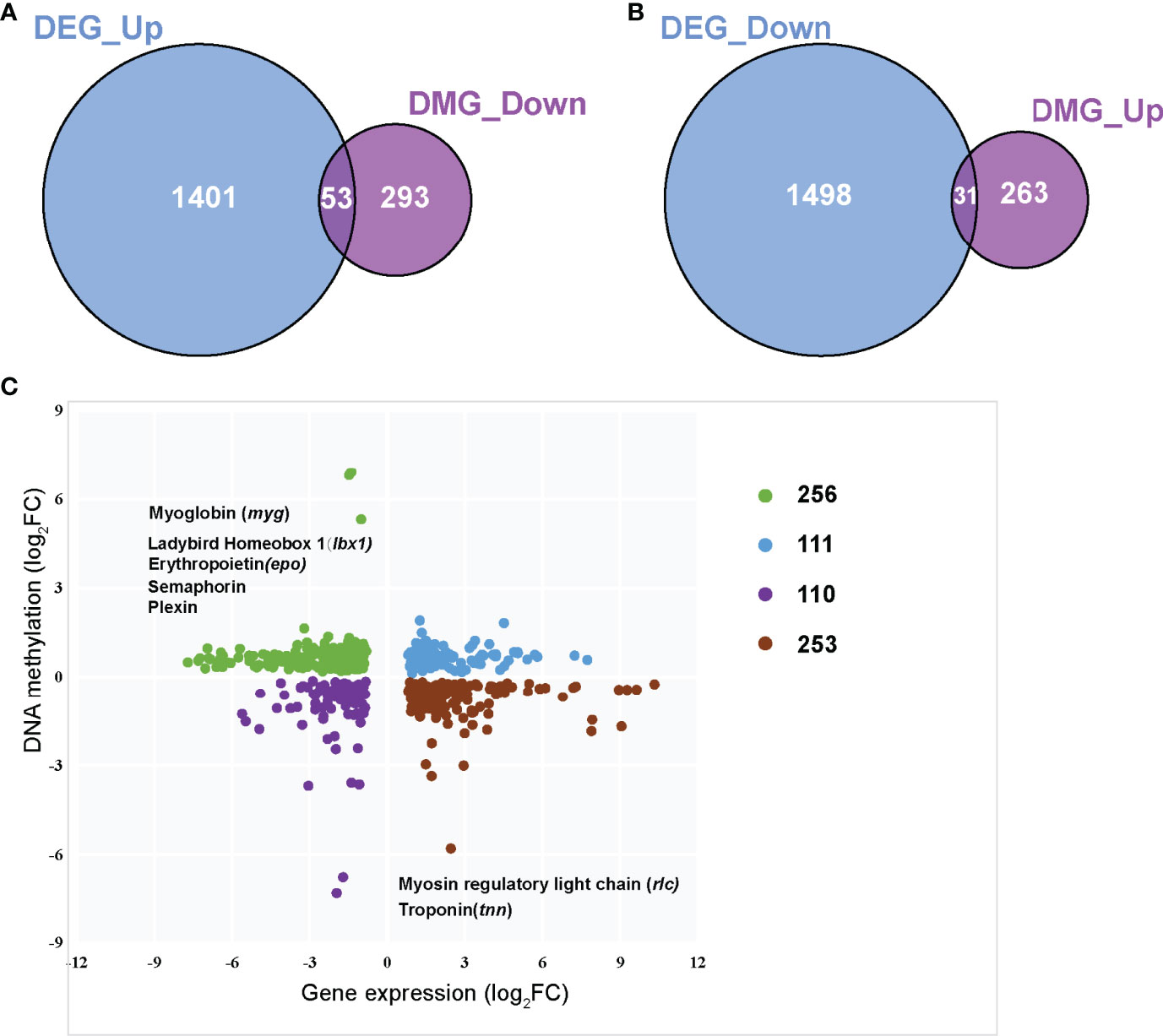
Figure 3 Integrated analysis of the genome-wide DNA methylation and gene expression profiles between slow- and fast-twitch muscles. (A) Venn diagram of overlapped hypo DMGs in promoters and upregulated DEGs in SM. (B) Venn diagram of overlapped hyper DMGs in promoters and downregulated DEGs in SM. (C). Integrated analysis of DNA methylation levels and gene expression levels. The vertical axis represents the DNA methylation level, and the horizontal axis shows the gene expression. FCs (fold changes) indicates the DNA methylation or gene expression of SM relative to that of FM.
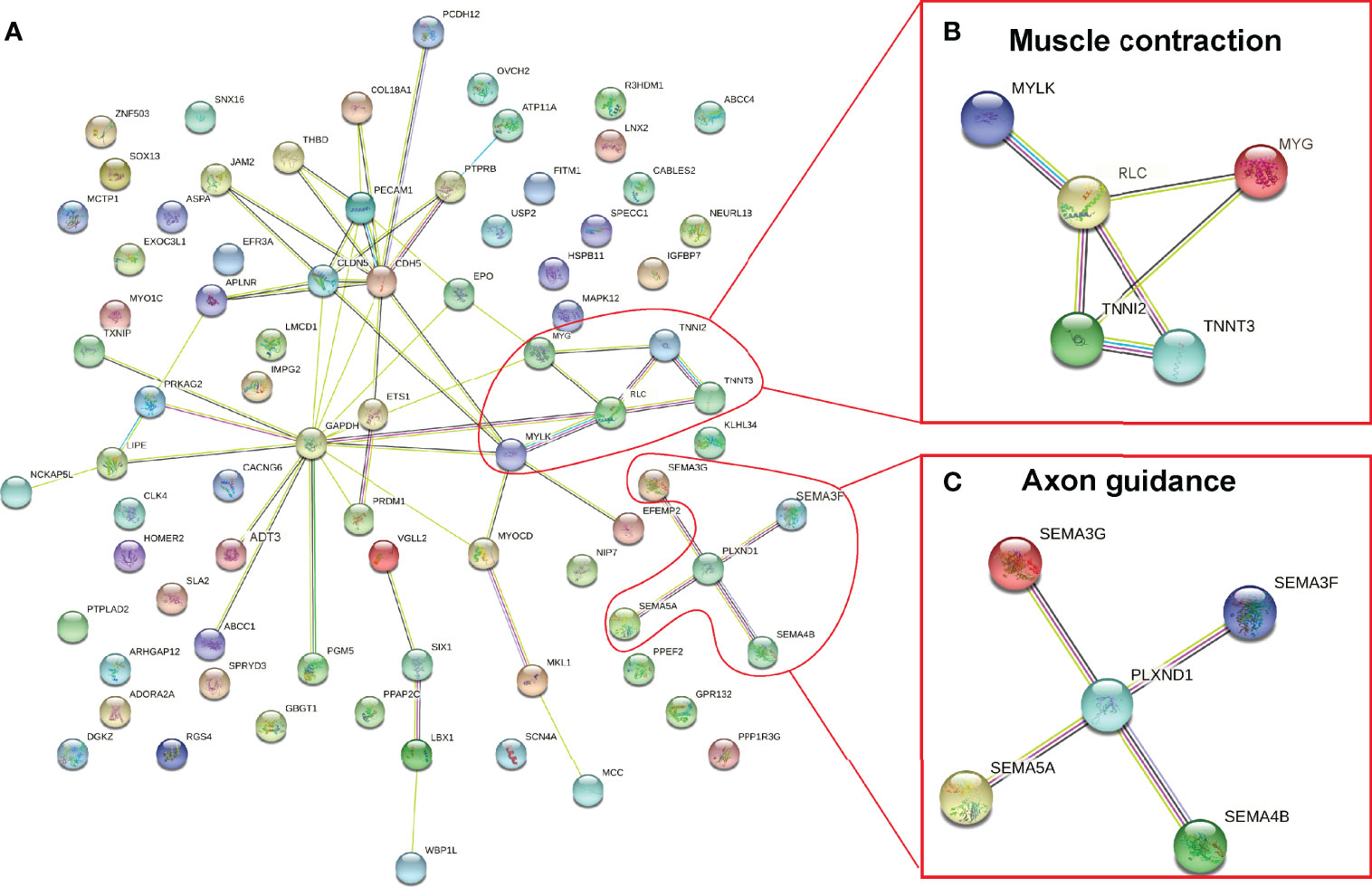
Figure 4 Co-regulation network of DEGs and DMGs between slow- and fast-twitch muscles of P. dentex (A). Enriched muscle contraction (B) and axon guidance (C) networks. The network nodes indicate proteins, and lines represent there are predicted associations between these two proteins.
Epigenetic effects were defined as processes that can modify gene expression without any DNA sequence change and were heritable to subsequent generations. DNA methylation was the most relevant epigenetic event in vertebrates (Varriale, 2014) that can regulate gene expression (Zhang et al., 2019a). Increased DNA methylation were usually associated with gene repression, whereas reduced DNA methylation could induce gene activation (Siegfried and Simon, 2010). In present study, we investigated the essential role of DNA methylation in the regulation of the different muscle types according to the analysis of whole-genome methylation profiles and integrated analysis of DNA methylation profiles and RNA-seq data between SM and FM of P. dentex. Pathways regulated skeletal muscle physiological characteristics and cell junctions were enriched according to the DMGs. Furthermore, combing methylome data with transcriptome data, we identified critical genes whose expression were regulated by promoter methylation, which may be related to phenotypic differences between SM and FM, and constructed an overview of essential genes and related physiological function regulated by DNA methylation between SM and FM of P. dentex.
Skeletal muscle development, also termed as myogenesis, was a complex-multistep process requiring a very precise, space- and time-controlled regulation that occurs both during embryonic development as well as regeneration process of the muscle (Lehka and Redowicz, 2020). In present study, KEGG analysis showed that the MAPK, TGF-β, Notch, Wnt, Calcium, and focal adhesion signaling pathways involved in myogenesis and skeletal muscle regeneration of teleost (Pan et al., 2021; Wang et al., 2021) were enriched according to DMGs between SM and FM of P. dentex. MAPK signaling pathway was reported negatively regulates skeletal muscle differentiation (Xie et al., 2018). TGF-β was reported able to inhibit myoblasts proliferation and specific inhibition of TGF-β signaling pathway could significantly improve capability of muscle regeneration (Delaney et al., 2017). Inhibition of Notch signaling that is a key player in skeletal muscle development and regeneration was shown to impair muscle regeneration, whereas enhancing Notch activation facilitated the repair process (Conboy et al., 2003; Buas and Kadesch, 2010). Wnt signaling played a role in fiber type determination during development but have not been precisely linked to adult myofiber typing maintenance in adulthood (Girardi and Le Grand, 2018). Although Wnt signaling was poorly activated in mature skeletal muscles (Kuroda et al., 2013), it was reported that timely regulation of Wnt signaling is essential for muscle regeneration (Rudolf et al., 2016). Focal adhesion was the signaling center of numerous intracellular pathways that regulate cell growth and differentiation, which played an important role in the development of skeletal muscles (Sastry and Burridge, 2000; Li et al., 2019). The calcium signaling pathway was the key pathway exerting allosteric regulation on many proteins, including through ion channel activation or by acting as a secondary messenger, which could directly affect skeletal muscle metabolism (Fang et al., 2017). Therefore, we speculated that MAPK, TGF-β, Notch, Wnt, and calcium signaling pathway played important role in epigenetic regulation of various skeletal muscle types differences.
According to our results, cell junction pathway such as tight junction and adherens junction showed significant methylation difference between two types of fibers in P. dentex, which is consist with the results in Siniperca chuatsi that DEGs between SM and FM were enriched in adherens junction and tight junction (Pan et al., 2021). Skeletal muscle, however, was rarely reported to have any cell–cell junctions. However, SM generally had higher capillary volume than mostly FM in meeting the demand for oxygen during sustained swimming (Sjogaard, 1982; Murakami et al., 2010; Buckley and Bossen, 2013). One of the main cellular constituents of capillary was endothelial cells, which express both adherens and tight junctions (Dejana, 2004) contribute to maintain vascular integrity (Le Guelte and Gavard, 2011). Multiple enriched cell junction pathways according to DMGs between SM and FM in P. dentex may explain the capillary distribution difference of these two types of muscles.
According to the integrated analysis of WGBS and RNA-seq data, we identified three critical genes (myg, tnn, and rlc) played important role in skeletal muscle differences and their gene expression was regulated by DNA methylation (Figures 5A, B). The promoter of myg was hypomethylated; thus, their expression was enhanced in SM. The promoter of tnn and rlc were hypermethylated, which reduced their mRNA expression in SM.
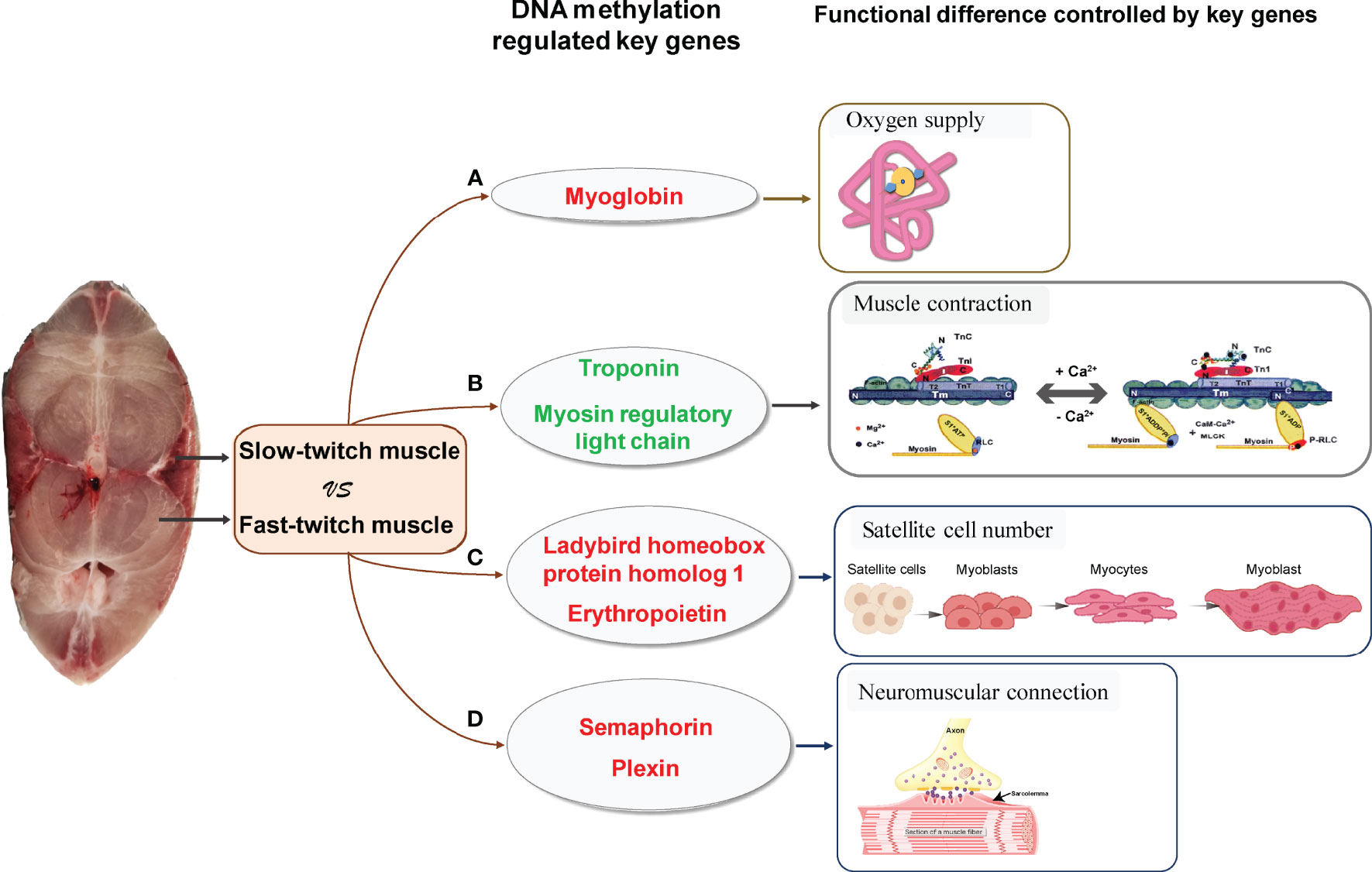
Figure 5 Critical methylated genes associated with phenotypic differences between slow- and fast-twitch muscles of P. dentex. (A) Hypomethylated and highly expressed myg in SM (red letter) indicated demethylation of the myg promoter could upregulate its expression, thus increasing O2 supplying and meeting the oxygen demands of SM. (B) Hypermethylated and lowly expressed tnn and rlc (green letter) in SM may be associated with low mobility of myosin cross bridges, which lead to slower and less frequent muscle contraction in SM than in FM. (C) Hypomethylated and highly expressed lbx1 and epo genes (red letter) may be related to increased satellite cell numbers, and Semaphorin/Plexin genes (red letter) (D) may be related to higher rate of neuromuscular connection reconstruction, which further promote high muscle regeneration efficiency in SM. The schematic diagram of gene physiological function mainly referred to Jahan (Jahan) and Szczesna (Szczesna, 2003) and was slightly modified.
Myg is a cytoplasmic hemoprotein and functions as an oxygen storage protein in muscles (Ordway and Garry, 2004). It can bind one molecule of O2 per molecule of protein (Feher, 2017). At onset of muscle contraction, myoglobin immediately releases its bound O2 to the mitochondria (Takakura et al., 2015). SM contain higher levels of mitochondria than FM. Correspondingly, SM generate energy by oxidative metabolism of mitochondria for continuous contractions with less fatigue, whereas FM rely on anaerobic respiration to produce ATP for bursts of movement (Bassel-Duby and Olson, 2006). Moreover, it was reported that fishes with a more active mode of life have a higher proportion of SM (Webb, 1984; Dwyer et al., 2014; Teulier et al., 2019). Elevated myoglobin expression regulated by methylation in SM of P. dentex enables oxygen storage for aerobic metabolism maintenance during endurance swimming of migratory (Figure 5A).
It was reported that rlc in skeletal muscles modulates Ca2+-dependent tnn regulation of contraction (Kamm and Stull, 2011). Tnn binding with Ca2+ induced allosteric changes in the thin filament allowing the myosin head to form a strong myosin cross-bridge with F-actin to activate myosin ATPase (Leavis and Gergely, 1984). Rlc phosphorylation increased the mobility of myosin cross bridges such that they move away from the thick filament surface toward actin thin filaments in skeletal muscles (Stull et al., 2011). Therefore, Rlc phosphorylation and Ca2+- binding Tnn played an important modulatory role in striated muscle contraction (Szczesna, 2003). Hypermethylation and lower expression of tnn and rlc in SM may be associated with low mobility of myosin cross bridges, resulting in slower and less frequent muscle contraction compared with FM (Figure 5B). In conclusion, DNA methylation could regulate myg, tnn, and rlc expression variation, thus affecting oxygen supply and muscle contraction, which result in differences between SM and FM.
Skeletal muscles were reported retain a tremendous regenerative capacity, which was attributed to the presence of satellite cells (Dumont et al., 2015). Satellite cells were identified as multipotent stem cells of the skeletal muscle tissue and played a central role in the growth, maintenance, and repair of the muscles. Adult satellite cells were quiescent under resting conditions but can quickly re-enter the cell cycle following stimuli such as physical trauma or growth signals. Activated satellite cells will migrate extensively, proliferate, differentiate, and fuse to form regenerating myofibers (Schmalbruch, 1976; Verdijk et al., 2014). During muscle regeneration process, lbx1 (Ladybird homeobox protein homolog 1) was strongly expressed in satellite cells (Watanabe et al., 2007) and epo (erythropoietin) contributed to increasing satellite cell number, which were all critical in differentiation and maintenance of satellite cells (Jia et al., 2012).
The regeneration efficiency of SM and FM differs. SM were reported contain more satellite cells than FM (Collins et al., 2005; Ono et al., 2010) and the initial activation of satellite cells occurs more rapidly which resulted in high regeneration efficiency in SM (Rosenblatt et al., 1995; Lagord et al., 1998; Ono et al., 2010) to satisfy the regeneration demand of these highly active postural muscles. In present study, we found that the DNA methylation level of satellite cells related lbx1 and epo genes were downregulated in SM compared with FM, which was the opposite of that observed for their expression levels. Because lbx1 and epo genes play important roles in satellite cells maintenance as mentioned above, we hypothesized that methylation of lbx1 and epo genes may be the key functional regulators of high muscle regeneration efficiency in SM (Figure 5C).
In addition to the satellite cell numbers, successful neuromuscular connections were also critical for restoring skeletal muscle function and physiological properties during muscle regeneration process (Do et al., 2011; Anderson et al., 2017). Semaphorin/Plexin were axon guidance molecules identified as specific ligands and receptors in the neuromuscular connection system (Tanaka et al., 2007). Several species of experimental evidence supported the idea that dysregulation of Semaphorin/Plexin might trigger the denervation of neuromuscular junctions (Van Battum et al., 2015; Grice et al., 2018). Semaphorin and Plexin genes were hypomethylated and highly expressed in SM compared with FM, which indicated that higher regeneration efficiency in SM maybe related to the neuromuscular connection reconstruction speed (Figure 5D).
In conclusion, our study supplied DNA methylation atlas and identified methylation differences between SM and FM of P. dentex. According to the integrative analysis of methylome and transcriptome, we identified critical DNA methylation genes and pathways controlled phenotypic differences between SM and FM. Our results provide valuable data of genome epigenetic mechanism in skeletal muscle differences, which could provide new perspective for understanding the long-distance swimming adaptability of pelagic migratory fish skeletal muscle and further enrich the theoretical basis for the study of physiological characteristics and adaptive evolution in teleost fishes.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession numbers can be found at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA796775, PRJNA796775.
The animal study was reviewed and approved by Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences.
BL, SL, and ZZ contributed to conception and design of the study. BL, HW, and SL collected the samples. BL, AL, CA, and, LZ organized the database. BL performed the data analysis. BL wrote the first draft of the manuscript. BL and SL supervised, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the major project of Shandong province of China for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2022QNLM050101-3), the National Natural Science Foundation of China (42076132 and 42106129), and China Agriculture Research System of MOF and MARA (CARS-47).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dalian Tianzheng Industry Co., Ltd. (Dalian, Liaoning, China) for providing specimens for this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.916373/full#supplementary-material
Anderson J. E., Do M. Q., Daneshvar N., Suzuki T., Dort J., Mizunoya W., et al. (2017). The Role of Semaphorin3a in Myogenic Regeneration and the Formation of Functional Neuromuscular Junctions on New Fibres. Biol. Rev. Camb Philos. Soc. 92 (3), 1389–1405. doi: 10.1111/brv.12286
Bassaglia Y., Gautron J. (1995). Fast and Slow Rat Muscles Degenerate and Regenerate Differently After Whole Crush Injury. J. Muscle Res. Cell Motil. 16 (4), 420–429. doi: 10.1007/BF00114507
Bassel-Duby R., Olson E. N. (2006). Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 75, 19–37. doi: 10.1146/annurev.biochem.75.103004.142622
Buas M. F., Kadesch T. (2010). Regulation of Skeletal Myogenesis by Notch. Exp. Cell Res. 316 (18), 3028–3033. doi: 10.1016/j.yexcr.2010.05.002
Buckley A. F., Bossen E. H. (2013). Skeletal Muscle Microvasculature in the Diagnosis of Neuromuscular Disease. J. Neuropathol. Exp. Neurol. 72 (10), 906–918. doi: 10.1097/NEN.0b013e3182a7f0b8
Collins C. A., Olsen I., Zammit P. S., Heslop L., Petrie A., Partridge T. A., et al. (2005). Stem Cell Function, Self-Renewal, and Behavioral Heterogeneity of Cells From the Adult Muscle Satellite Cell Niche. Cell 122 (2), 289–301. doi: 10.1016/j.cell.2005.05.010
Conboy I. M., Conboy M. J., Smythe G. M., Rando T. A. (2003). Notch-Mediated Restoration of Regenerative Potential to Aged Muscle. Science 302 (5650), 1575–1577. doi: 10.1126/science.1087573
Dejana E. (2004). Endothelial Cell-Cell Junctions: Happy Together. Nat. Rev. Mol. Cell Biol. 5 (4), 261–270. doi: 10.1038/nrm1357
Delaney K., Kasprzycka P., Ciemerych M. A., Zimowska M. (2017). The Role of TGF-Beta1 During Skeletal Muscle Regeneration. Cell Biol. Int. 41 (7), 706–715. doi: 10.1002/cbin.10725
Do M. K., Sato Y., Shimizu N., Suzuki T., Shono J., Mizunoya W., et al. (2011). Growth Factor Regulation of Neural Chemorepellent Sema3A Expression in Satellite Cell Cultures. Am. J. Physiol. Cell Physiol. 301 (5), C1270–C1279. doi: 10.1152/ajpcell.00257.2011
Dumont N. A., Bentzinger C. F., Sincennes M. C., Rudnicki M. A. (2015). Satellite Cells and Skeletal Muscle Regeneration. Compr Physiol 5(3), 1027–1059. doi: 10.1002/cphy.c140068
Dwyer G. K., Stoffels R. J., Pridmore P. A. (2014). Morphology, Metabolism and Behaviour: Responses of Three Fishes With Different Lifestyles to Acute Hypoxia. Freshwater Biol. 59 (4), 819–831. doi: 10.1111/fwb.12306
Fang X., Zhao Z., Yu H., Li G., Jiang P., Yang Y., et al. (2017). Comparative Genome-Wide Methylation Analysis of Longissimus Dorsi Muscles Between Japanese Black (Wagyu) and Chinese Red Steppes Cattle. PloS One 12 (8), e0182492. doi: 10.1371/journal.pone.0182492
Fan Y., Liang Y., Deng K., Zhang Z., Zhang G., Zhang Y., et al. (2020). Analysis of DNA Methylation Profiles During Sheep Skeletal Muscle Development Using Whole-Genome Bisulfite Sequencing. BMC Genomics 21 (1),327. doi: 10.1186/s12864-020-6751-5
Feher J. (2017). “6.4 - Oxygen and Carbon Dioxide Transport,” in Quantitative Human Physiology, 2nd ed. Ed. Feher J. (Boston: Academic Press), 656–664.
Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., et al. (2013). STRING V9.1: Protein-Protein Interaction Networks, With Increased Coverage and Integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Gao K., Wang Z., Zhou X., Wang H., Kong D., Jiang C., et al. (2017). Comparative Transcriptome Analysis of Fast Twitch Muscle and Slow Twitch Muscle in Takifugu Rubripes. Comp. Biochem. Physiol. Part D: Genomics Proteomics 24, 79–88. doi: 10.1016/j.cbd.2017.08.002
Gibb A. C. (2002). Functional Morphology and Biochemical Indices of Performance: Is There a Correlation Between Metabolic Enzyme Activity and Swimming Performance? Integr. Comp. Biol. 42 (2), 199–207. doi: 10.1093/icb/42.2.199
Girardi F., Le Grand F. (2018). Wnt Signaling in Skeletal Muscle Development and Regeneration. Prog. Mol. Biol. Transl. Sci. 153, 157–179. doi: 10.1016/bs.pmbts.2017.11.026
Greek-Walker M., Pull G. A. (1975). A Survey of Red and White Muscle in Marine Fish. J. Fish Biol. 7 (3), 295–300. doi: 10.1111/j.1095-8649.1975.tb04602.x
Grice S. J., Sleigh J. N., Cader M. Z. (2018). Plexin-Semaphorin Signaling Modifies Neuromuscular Defects in a Drosophila Model of Peripheral Neuropathy. Front. Mol. Neurosci. 11. doi: 10.3389/fnmol.2018.00055
Jahan I. Skeletal Muscle Damage? Satellite Cells to the Rescue!. Available at: https://www.stemside.co.uk/post/skeletal-muscle-damage-satellite-cells-to-the-rescue.
Jayaraman A., Liu M., Ye F., Walter G. A., Vandenborne K. (2013). Regenerative Responses in Slow- and Fast-Twitch Muscles Following Moderate Contusion Spinal Cord Injury and Locomotor Training. Eur. J. Appl. Physiol. 113 (1), 191–200. doi: 10.1007/s00421-012-2429-2
Jia Y., Suzuki N., Yamamoto M., Gassmann M., Noguchi C. T. (2012). Endogenous Erythropoietin Signaling Facilitates Skeletal Muscle Repair and Recovery Following Pharmacologically Induced Damage. FASEB J. 26 (7), 2847–2858. doi: 10.1096/fj.11-196618
Jones P. A. (2012). Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 13 (7), 484–492. doi: 10.1038/nrg3230
Jones P. A., Takai D. (2001). The Role of DNA Methylation in Mammalian Epigenetics. Science 293 (5532), 1068–1070. doi: 10.1126/science.1063852
Kamm K. E., Stull J. T. (2011). Signaling to Myosin Regulatory Light Chain in Sarcomeres. J. Biol. Chem. 286 (12), 9941–9947. doi: 10.1074/jbc.R110.198697
Kuroda K., Kuang S., Taketo M. M., Rudnicki M. A. (2013). Canonical Wnt Signaling Induces BMP-4 to Specify Slow Myofibrogenesis of Fetal Myoblasts. Skelet Muscle 3 (1), 5. doi: 10.1186/2044-5040-3-5
Lagord C., Soulet L., Bonavaud S., Bassaglia Y., Rey C., Barlovatz-Meimon G., et al. (1998). Differential Myogenicity of Satellite Cells Isolated From Extensor Digitorum Longus (EDL) and Soleus Rat Muscles Revealed In Vitro. Cell Tissue Res. 291 (3), 455–468. doi: 10.1007/s004410051015
Leavis P. C., Gergely J. (1984). Thin Filament Proteins and Thin Filament-Linked Regulation of Vertebrate Muscle Contraction. CRC Crit. Rev. Biochem. 16 (3), 235–305. doi: 10.3109/10409238409108717
Le Guelte A., Gavard J. (2011). Role of Endothelial Cell-Cell Junctions in Endothelial Permeability. Methods Mol. Biol. 763, 265–279. doi: 10.1007/978-1-61779-191-8_18
Lehka L., Redowicz M. J. (2020). Mechanisms Regulating Myoblast Fusion: A Multilevel Interplay. Semin. Cell Dev. Biol. 104, 81–92. doi: 10.1016/j.semcdb.2020.02.004
Li R., Zhang R., Yi J., Guo W., Cheng Q., Zhi L., et al. (2019). Characterization and Expression Profiles of Muscle Transcriptome in Schizothoracine Fish, Schizothorax Prenanti. Gene 685, 156–163. doi: 10.1016/j.gene.2018.10.070
Mao X., Cai T., Olyarchuk J. G., Wei L. (2005). Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 21 (19), 3787–3793. doi: 10.1093/bioinformatics/bti430
Mareco E. A., de la Serrana D. G., Johnston I. A., Dal-Pai-Silva M. (2015). Characterization of the Transcriptome of Fast and Slow Muscle Myotomal Fibres in the Pacu (Piaractus Mesopotamicus). BMC Genomics 16 (1), 182. doi: 10.1186/s12864-015-1423-6
Murakami S., Fujino H., Takeda I., Momota R., Kumagishi K., Ohtsuka A. (2010). Comparison of Capillary Architecture Between Slow and Fast Muscles in Rats Using a Confocal Laser Scanning Microscope. Acta Med. Okayama 64 (1), 11–18. doi: 10.18926/AMO/32859
Ono Y., Boldrin L., Knopp P., Morgan J. E., Zammit P. S. (2010). Muscle Satellite Cells are a Functionally Heterogeneous Population in Both Somite-Derived and Branchiomeric Muscles. Dev. Biol. 337 (1), 29–41. doi: 10.1016/j.ydbio.2009.10.005
Ordway G. A., Garry D. J. (2004). Myoglobin: An Essential Hemoprotein in Striated Muscle. J. Exp. Biol. 207 (Pt 20), 3441–3446. doi: 10.1242/jeb.01172
Pan Y., Chen L., Cheng J., Zhu X., Wu P., Bao L., et al. (2021). Genome-Wide DNA Methylation Profiles Provide Insight Into Epigenetic Regulation of Red and White Muscle Development in Chinese Perch Siniperca Chuatsi. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 256, 110647. doi: 10.1016/j.cbpb.2021.110647
Razin A., Cedar H. (1991). DNA Methylation and Gene Expression. Microbiol. Rev. 55 (3), 451–458. doi: 10.1128/mr.55.3.451-458.1991
Rosenblatt J. D., Lunt A. I., Parry D. J., Partridge T. A. (1995). Culturing Satellite Cells From Living Single Muscle Fiber Explants. In Vitro Cell Dev. Biol. Anim. 31 (10), 773–779. doi: 10.1007/BF02634119
Rudolf A., Schirwis E., Giordani L., Parisi A., Lepper C., Taketo M. M., et al. (2016). Beta-Catenin Activation in Muscle Progenitor Cells Regulates Tissue Repair. Cell Rep. 15 (6), 1277–1290. doi: 10.1016/j.celrep.2016.04.022
Sastry S. K., Burridge K. (2000). Focal Adhesions: A Nexus for Intracellular Signaling and Cytoskeletal Dynamics. Exp. Cell Res. 261 (1), 25–36. doi: 10.1006/excr.2000.5043
Schmalbruch H. (1976). The Morphology of Regeneration of Skeletal Muscles in the Rat. Tissue Cell 8 (4), 673–692. doi: 10.1016/0040-8166(76)90039-2
Siegfried Z., Simon I. (2010). DNA Methylation and Gene Expression. Wiley Interdiscip Rev. Syst. Biol. Med. 2 (3), 362–371. doi: 10.1002/wsbm.64
Sjogaard G. (1982). Capillary Supply and Cross-Sectional Area of Slow and Fast Twitch Muscle Fibres in Man. Histochemistry 76 (4), 547–555. doi: 10.1007/BF00489909
Stull J. T., Kamm K. E., Vandenboom R. (2011). Myosin Light Chain Kinase and the Role of Myosin Light Chain Phosphorylation in Skeletal Muscle. Arch. Biochem. Biophys. 510 (2), 120–128. doi: 10.1016/j.abb.2011.01.017
Szczesna D. (2003). Regulatory Light Chains of Striated Muscle Myosin. Structure, Function and Malfunction. Curr. Drug Targets Cardiovasc. Haematol Disord. 3 (2), 187–197. doi: 10.2174/1568006033481474
Takakura H., Furuichi Y., Yamada T., Jue T., Ojino M., Hashimoto T., et al. (2015). Endurance Training Facilitates Myoglobin Desaturation During Muscle Contraction in Rat Skeletal Muscle. Sci. Rep. 5, 9403. doi: 10.1038/srep09403
Tanaka H., Maeda R., Shoji W., Wada H., Masai I., Shiraki T., et al. (2007). Novel Mutations Affecting Axon Guidance in Zebrafish and a Role for Plexin Signalling in the Guidance of Trigeminal and Facial Nerve Axons. Development 134 (18), 3259–3269. doi: 10.1242/dev.004267
Teulier L., Thoral E., Queiros Q., McKenzie D. J., Roussel D., Dutto G., et al. (2019). Muscle Bioenergetics of Two Emblematic Mediterranean Fish Species: Sardina Pilchardus and Sparus Aurata. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 235, 174–179. doi: 10.1016/j.cbpa.2019.06.008
Van Battum E. Y., Brignani S., Pasterkamp R. J. (2015). Axon Guidance Proteins in Neurological Disorders. Lancet Neurol. 14 (5), 532–546. doi: 10.1016/s1474-4422(14)70257-1
Varriale A. (2014). DNA Methylation, Epigenetics, and Evolution in Vertebrates: Facts and Challenges. Int. J. Evol. Biol. 2014, 475981. doi: 10.1155/2014/475981
Verdijk L. B., Snijders T., Drost M., Delhaas T., Kadi F., van Loon L. J. (2014). Satellite Cells in Human Skeletal Muscle; From Birth to Old Age. Age (Dordr) 36 (2), 545–547. doi: 10.1007/s11357-013-9583-2
Wagner J. R., Busche S., Ge B., Kwan T., Pastinen T., Blanchette M. (2014). The Relationship Between DNA Methylation, Genetic and Expression Inter-Individual Variation in Untransformed Human Fibroblasts. Genome Biol. 15 (2), R37. doi: 10.1186/gb-2014-15-2-r37
Wang H., Cui J., Qiu X., Wang X. (2021). Differences in DNA Methylation Between Slow and Fast Muscle in Takifugu Rubripes. Gene 801, 145853. doi: 10.1016/j.gene.2021.145853
Wang H., Li B., Yang L., Jiang C., Zhang T., Liu S., et al. (2022). Expression Profiles and Transcript Properties of Fast-Twitch and Slow-Twitch Muscles in a Deep-Sea Highly Migratory Fish, Pseudocaranx Dentex. PeerJ 10, e12720. doi: 10.7717/peerj.12720
Watanabe S., Kondo S., Hayasaka M., Hanaoka K. (2007). Functional Analysis of Homeodomain-Containing Transcription Factor Lbx1 in Satellite Cells of Mouse Skeletal Muscle. J. Cell Sci. 120 (Pt 23), 4178–4187. doi: 10.1242/jcs.011668
Webb P. W. (1984). Body Form, Locomotion and Foraging in Aquatic Vertebrates. Am. Zoologist 24 (1), 107–120. doi: 10.1093/icb/24.1.107
Xie S. J., Li J. H., Chen H. F., Tan Y. Y., Liu S. R., Zhang Y., et al. (2018). Inhibition of the JNK/MAPK Signaling Pathway by Myogenesis-Associated miRNAs is Required for Skeletal Muscle Development. Cell Death Differ 25 (9), 1581–1597. doi: 10.1038/s41418-018-0063-1
Young M. D., Wakefield M. J., Smyth G. K., Oshlack A. (2010). Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 11 (2), R14. doi: 10.1186/gb-2010-11-2-r14
Zhang Y., Liu J., Fu W., Xu W., Zhang H., Chen S., et al. (2017). Comparative Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying Skin Color Variations in Crucian Carp (Carassius Carassius L.). BMC Genet. 18 (1), 95. doi: 10.1186/s12863-017-0564-9
Zhang Y., Shen W., Cao M., Li J., Zheng B., Lou Z., et al. (2019b). Dynamic Alterations in Methylation of Global DNA and Growth-Related Genes in Large Yellow Croaker (Larimichthys Crocea) in Response to Starvation Stress. Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 227, 98–105. doi: 10.1016/j.cbpb.2018.09.006
Zhang W., Zhang S., Xu Y., Ma Y., Zhang D., Li X., et al. (2019a). The DNA Methylation Status of Wnt and Tgfbeta Signals Is a Key Factor on Functional Regulation of Skeletal Muscle Satellite Cell Development. Front. Genet. 10. doi: 10.3389/fgene.2019.00220
Keywords: Pseudocaranx dentex, DNA methylation, slow-twitch muscles, fast-twitch muscles, satellite cells
Citation: Li B, Wang H, Li A, An C, Zhu L, Liu S and Zhuang Z (2022) The Landscape of DNA Methylation Generates Insight Into Epigenetic Regulation of Differences Between Slow-Twitch and Fast-Twitch Muscles in Pseudocaranx dentex. Front. Mar. Sci. 9:916373. doi: 10.3389/fmars.2022.916373
Received: 11 April 2022; Accepted: 03 May 2022;
Published: 16 June 2022.
Edited by:
Xiaotong Wang, Ludong University, ChinaReviewed by:
Zhiqiang Han, Zhejiang Ocean University, ChinaCopyright © 2022 Li, Wang, Li, An, Zhu, Liu and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shufang Liu, TGl1c2ZAeXNmcmkuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.