
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 16 June 2022
Sec. Marine Molecular Biology and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.914095
This article is part of the Research TopicUnderstanding the Effects of Climate Change on Marine Biodiversity through Genomics and TranscriptomicsView all 6 articles
 Svenja J. Müller1,2*
Svenja J. Müller1,2* Katharina Michael1,2
Katharina Michael1,2 Ilenia Urso3
Ilenia Urso3 Gabriele Sales3
Gabriele Sales3 Cristiano De Pittà3
Cristiano De Pittà3 Lavinia Suberg2
Lavinia Suberg2 Wiebke Wessels1
Wiebke Wessels1 Evgeny A. Pakhomov4,5,6
Evgeny A. Pakhomov4,5,6 Bettina Meyer1,2,7*
Bettina Meyer1,2,7*The pelagic tunicate Salpa thompsoni is recognized as a major metazoan grazer in the Southern Ocean. Long term observations show an increase in this species’ biomass and a southward shift in its distribution both of which are positively correlated with ocean warming and winter sea ice decline around the Antarctic Peninsula. However, our understanding on how salps adapt their life cycle to the extreme seasonality of the Southern Ocean and the putative differences between its two reproductive forms (aggregates, solitaries) is rudimentary. In particular, our current knowledge of whether and how S. thompsoni overwinter is limited, largely due to winter sampling constraints. In this study, we investigated the form-specific gene expression profiles of Salpa thompsoni during the austral autumn and winter. Between the seasons, genes related to translation showed the biggest difference in gene expression. We found more genes were upregulated in solitaries compared to aggregates, indicating a potentially form-specific overwintering strategy. Our data provide first insights into the seasonal and form-specific physiology of salps by considering their complex life cycle, thereby contributing to a more comprehensive understanding of the response of salps to seasonal changes in their environment and to anthropogenic induced global climate change.
The pelagic tunicate Salpa thompsoni, along with Antarctic krill and copepods, are the dominant metazoan grazers in the Southern Ocean (SO) (Voronina, 1998). Unlike copepods and krill, salps were long considered to be a trophic dead end due to their low nutritional value. During the last decade, however, their trophic importance has been reconsidered because of their increasing abundance in the SO, high potential for carbon sequestration, and ability to outcompete other grazers due to their efficient filtering capacity. The habitat of S. thompsoni was historically located in the low Antarctic (45-55°S), but long-term observations have shown an ongoing southward expansion of their distribution range, which may be a direct consequence of ongoing ocean warming and sea ice decline (Loeb et al., 1997; Pakhomov et al., 2006; Pakhomov et al., 2011; Atkinson et al., 2017; Henschke et al., 2018).
Salps can grow rapidly under favorable environmental conditions due to the short generation times of their chain-forming sexual (aggregate) and asexual (solitary) life forms, and are therefore able to form large blooms (Foxton, 1966; Andersen, 1998). While aggregates are known to be protogynous hermaphrodites, in which the ovary develops first and the testes later, solitaries reproduce asexually by budding and form “chains” of aggregates (Figures 1A, B). Salps also exhibit a seasonal life cycle (Figure 1C). At the onset of spring, solitaries usually release massive numbers of aggregates, which subsequently dominate the upper water column from summer until early autumn. At the end of the productive season, the aggregates give birth to solitaries which consequently dominate the salp community during winter (Foxton, 1966; Loeb and Santora, 2012). These first insights were further complemented with studies considering seasonal patterns of abundance and distribution (Chiba et al., 1999; Pakhomov et al., 2011), body composition (Dubischar et al., 2012) and feeding dynamics (von Harbou et al., 2011).
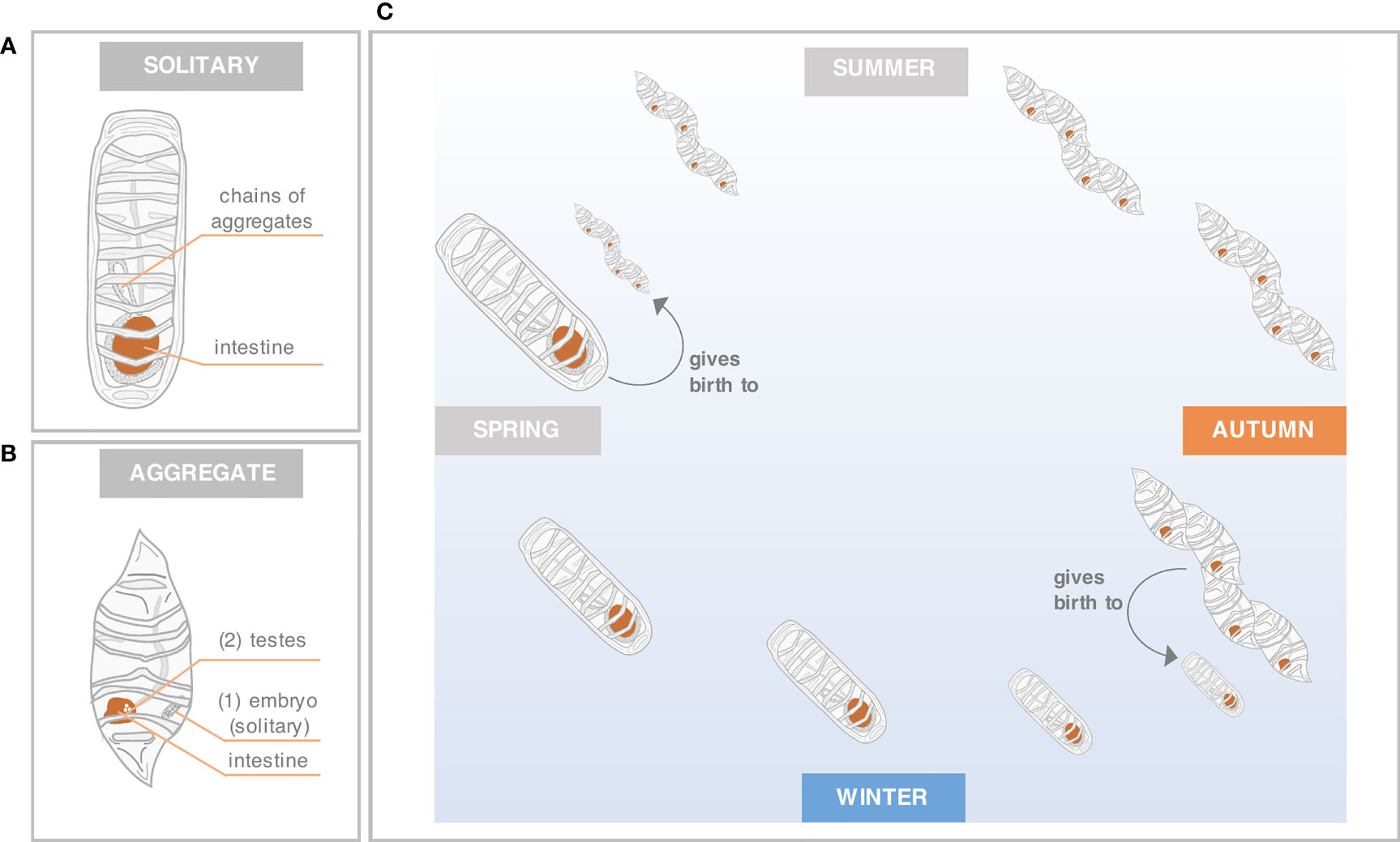
Figure 1 Forms and seasonal life cycle of S. thompsoni. Basic anatomy and reproductive features are shown for solitary (A) and aggregate (B) forms. Aggregates are characterized by their protogynous hermaphroditic nature, first they develop the ovary/embryo (1) and later the testes (2). The seasonal life cycle [C; modified from Loeb and Santora (2012)] reflects simplified seasonal migration, development and reproduction based on Foxton (1966) and Casareto and Nemoto (1986).
However, our understanding of biological and physiological seasonality of salps is rudimentary. There are still knowledge gaps in understanding how salps adapt their life cycle to the extreme seasonal environmental changes in the SO as well as in the putative differences between the reproductive forms. Jue et al. (2016) identified fast evolving genes involved in metabolic and immune processes in S. thompsoni, underscoring its potential as a model organism for studying the adaptive capacity of gelatinous zooplankton to changing polar environments. Batta-Lona et al. (2017) provided the first seasonal transcriptome study of S. thompsoni from different areas of the Western Antarctic Peninsula region, highlighting differentially expressed genes associated with reproduction and environmental stress between spring and summer. However, the study by Batta-Lona et al., (2017) was limited to the sexual reproducing generation (aggregates) and did not investigate both forms. Regarding the complex form-specific seasonal life cycle of salps, it is important to consider the responses of both reproductive generations.
Recent studies have demonstrated that a shift within the macrozooplankton community towards a higher abundance of salps in the high Antarctic regions could have profound implications for the biological carbon pump in the SO as well as possible pelagic food web consequences (Böckmann et al., 2021; Pauli et al., 2021a; Pauli et al., 2021b). These findings highlight the importance of understanding seasonal differences in the biology and physiology of salps in order to assess their resilience to environmental changes and how the population might develop under the predicted climate change scenarios.
In this study, we characterized the physiological response to seasonal environmental changes on transcriptome level, considering both reproductive generations of S. thompsoni. Transcriptomic studies have proven to be a powerful and established tool to study adaptation to extreme environmental changes in Antarctic krill and fish (Windisch et al., 2011; Windisch et al., 2014; Meyer et al., 2015; Höring et al., 2020). However, the ability of the Southern Ocean salp S. thompsoni to adapt to seasonal changes, particularly winter, and the underlying molecular mechanisms are still poorly understood.
By constructing a comprehensive de novo transcriptome of S. thompsoni and conducting a form-specific differential gene expression analysis of individuals obtained during autumn and winter we aimed to (i) identify differentially expressed genes underlying effective mechanisms involved in the life cycle of aggregates between seasons, and (ii) examine differences in gene expression patterns between both reproductive forms in winter in order to identify potential form-specific overwintering strategies. These findings will contribute to a better understanding of how each reproductive generation of S. thompsoni responds to seasonal changes of the environment and how it might adapt to anthropogenic induced global climate change. In turn, more knowledge of the seasonal life cycle may also help inform physiological models simulating salp life cycle and biomass dynamics in the future.
For the construction of a seasonal de novo transcriptome of S. thompsoni, we used salps collected from different sampling campaigns and seasons (Table 1):
1. In winter onboard R.V. Nathaniel B. Palmer (August 2016) as part of the US Antarctic Marine Living Resources (AMLR) program
2. In autumn onboard R.V. Polarstern PS112 (April 2018)
3. In autumn onboard R.V. Polarstern PS112 (April 2018) and subsequent incubation in circular aquaria tanks at different temperatures for 48 - 72 hours
4. In summer onboard R.V. Polarstern ANT XXVIII/3 (January 2012)
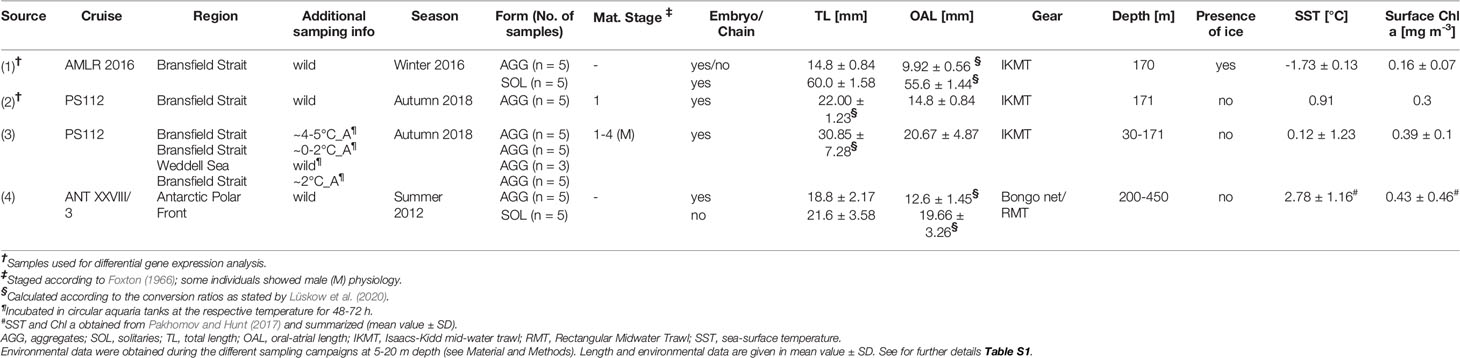
Table 1 Overview of samples used for the construction of the de novo transcriptome.
S. thompsoni were caught with an Isaacs-Kidd mid-water trawl (IKMT) net towed from ~ 30/170 m to the surface in the Bransfield Strait [(1)-(3)] and with a Bongo net/Multiple Rectangular Midwater Trawl (RMT) from 200-450 m depth in the Antarctic Polar Front (APF) region (4). Freshly caught salps were checked for the presence of an embryo/stolon and measured for oral-atrial length (OAL)/total length (TL) before they were shock-frozen in liquid nitrogen and stored at –80°C until further processing. For comparison between the different sampling campaigns, missing OAL and TL were calculated according to the conversion ratios as stated by Lüskow et al. (2020). Maturity stages (according to Foxton, 1966) were only determined during the R.V. Polarstern expedition PS112 in autumn 2018 [(2), (3)]. Further details are given in Tables 1 and S1.
Each sampling campaign (1-4) also evaluated environmental parameters [temperature, salinity and chlorophyll a (Chl a)] from the sea surface (5-20 m) at the time of zooplankton sampling by the following:
(1) The US AMLR program conducted CTD casts at all stations to 750 m depth or 10 m from the bottom if depth < 750 m using a Sea-Bird Scientific instrument (SBE 911; Sea- Bird Electronics). The CTD was attached to a rosette with 24 x 10 L Niskin bottles used to collect water for various biogeochemical parameters including Chl a. See Walsh and Reiss (2020) for further at- sea sampling information including sea ice analysis.
(2) + (3) PS112 employed a CTD rosette equipped with Sea-Bird Scientific SBE 911plus and Carousel Water Sampler SBE 32 with a chlorophyll sensor (Fluoro WetlabECO AFL FL).
(4) During research cruise ANT XXVIII/3 data were collected using a Sea- Bird Scientific SBE 911plus CTD equipped with a chlorophyll-sensitive fluorometer (WET Labs ECO FL). Environmental data used in the current study were obtained from literature (Pakhomov and Hunt, 2017). See Strass et al. (2017) for further details of the physical environment.
Frozen salps were transferred into RNAlater ICE (Thermo Fisher Scientific, USA) for 24 hours at -20°C prior to the extraction of total RNA. The stomach and embryo/stolon were dissected from each salp and the remaining tissue was transferred into 900-1350 µL lysis solution TR (STRATEC Molecular GmbH, Berlin, Germany) containing ß-mercaptoethanol. Samples were homogenized at room temperature using a Precellys® 24 homogenizer (Bertin Technologies, France) set to 2x15 seconds (s) at 5,000 rpm. RNA was extracted from each specimen using the InviTrap Spin Tissue RNA Mini Kit (STRATEC Molecular GmbH, Berlin, Germany) according to the manufacturer`s protocol. Due to the large size of the winter solitaries, the homogenate was split into two extractions. Total RNA was eluted with 30 µL nuclease-free water and eluted twice. After extraction elutes of winter solitaries were pooled and reprecipitated: 0.1 volumes of 3M sodium acetate pH 5.2 and 2.5 volumes of ice-cold 100% ethanol were added to the eluted RNA and mixed thoroughly. RNA was allowed to precipitate overnight at -20°C. To collect the RNA pellets tubes were centrifuged for 30 min at 13000 g and 4°C. The pellet was washed twice with 500 µL ice cold 75 % ethanol and centrifuged for 10 min at 13000 g and 4°C after each wash step. A short centrifuge step was added at the end to remove any remaining ethanol. The RNA pellet was allowed to air dry for 5 min and resuspended in 30 µL of nuclease free water. Total RNA was quantified by NanoDropTM 2000 (Thermo Fisher Scientific, Waltham, MA, USA). RNA integrity of each sample was assessed by capillary electrophoresis with the RNA 6000 Nano LabChip using the Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Preferably, only high-quality RNA samples (RIN = 8-10) were used for library construction (Table S2). In case of lower RIN values, sample quality was evaluated on the electropherogram. The RNA-Seq of each individual sample was carried out from IGA Technology Services (Udine, Italy). cDNA libraries were constructed with 500 ng of total RNA by using the TruSeq Stranded mRNA Sample Prep kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Total RNA samples and final cDNA libraries were quantified with the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and quality tested by Agilent 2100 Bioanalyzer Nano assay. For cluster generation on the flow cell, libraries were processed with cBot (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Sequencing was carried out on paired-end mode by using HiSeq 2500 (Illumina) with a targeted sequencing depth of about 80 million reads per sample (125 bp) for specimens obtained during sampling campaigns (1) and (4), AMLR2016 and ANT XXVIII/3, respectively, and by using NovaSeq 6000 (Illumina) of about 40 million reads per sample (150 bp) for specimens caught during PS112, sampling campaigns (2) and (3). Raw data were processed with the software CASAVA v1.8.2 (Illumina) for both format conversion and de-multiplexing. Raw sequencing data are accessible within NCBI`s Sequence Read Archive (SRA) database under Bioproject Accession Number PRJNA822688.
The software Trimmomatic v. 0.36 (Bolger et al., 2014) was used for the removal of adapter sequences and for quality trimming of raw Illumina sequences. The quality of trimmed reads was checked using FASTQC v. 0.11.9 (Andrews, 2010). In addition to the sequence data obtained here [sampling campaign (1) to (4)], sequence data of Batta-Lona et al. (2017) were used for de novo transcriptome assembly (Bioproject Accession Number PRJNA279245). In order to achieve a high-quality transcriptome in terms of completeness and limited sequence redundancy, five different de novo assemblers were used (Nakasugi et al., 2014): Trinity v. 2.9.1 (Grabherr et al., 2011), BinPacker v. 1.1 (Liu et al., 2016), IDBA-tran v. 1.1.3 (Peng et al., 2013), rnaSPAdes v. 3.13.1 (Bushmanova et al., 2019), and TransAbySS v. 2.0.1 (Robertson et al., 2010). Each assembly was filtered independently before merging the outputs. A transcript quantification analysis was performed using Salmon software v. 1.3.0 (Patro et al., 2017). All transcripts with more than ten reads mapped per samples were retained. To select the best reconstruction of each assembler, the EvidentialGene tr2aacds pipeline v. 4 (Gilbert, 2013) was applied. Fragments were clustered and the presence and length of coding sequences were used to select the best reconstruction for a specific transcript among all available assemblies. Quality assessment of the assemblies was obtained using Trinity v. 2.9.1 (Grabherr et al., 2011) computing basic statistics including the number of total transcripts, percent guanine-cytosine (GC) content, the average fragment length, the total number of bases and the N50 index. In addition, the transcriptome completeness was assessed using eukaryotic BUSCO database v. 4.1.4 (Simão et al., 2015), calculating the number of complete (single-copy and duplicated), fragmented and missing genes. The abundance of transcripts in each of the samples was estimated using Salmon v. 1.3.0 (Patro et al., 2017). Following the suggestion of Soneson et al. (2016), transcript counts were summarized at the gene-level to improve the accuracy, power and interpretability of downstream analyses (Soneson et al., 2016). Functional annotation was performed using the Trinotate pipeline v. 3.2.1 (https://trinotate.github.io/), which includes BLAST+/SwissProt, HMMER/PFAM and signalP/tmHMM. Results were integrated into an SQLite database (provided by Trinotate) which includes generic data on SwissProt records and PFAM domains. For downstream analysis, transcript annotations were summarized and deduplicated to gene level.
Only samples from sampling campaigns (1) and (2) were considered for differential gene expression (DGE) analysis and gene ontology (GO) enrichment analysis (Figure 2 and Table 1). All analysis were performed in R Studio v. 2021.09.0 + 351 (RStudio Team, 2020) using R version 4.1.1 (R Core Team, 2021). Internal normalization of expression values and differential gene expression were analyzed using the Bioconductor R package DESeq2 v. 1.32.0 (Love et al., 2014). In order to estimate variation within and between groups, principal component analysis (PCA) was performed based on variance-stabilized normalized read counts generated via the vst function in DESeq2. Gene expression analysis was performed between the different seasons (winter vs. autumn) within aggregates as well as between forms (solitaries vs. aggregates) during winter using a test based on negative binomial distribution implemented in DESeq2 v. 1.32.0 (Love et al., 2014). Only genes with Benjamini- Hochberg adjusted p- value <0.001 and a twofold difference or a log2fold change (log2FC) ≥ 1 in gene expression levels, by setting the log fold change threshold (LFCT) to 1 were considered to be significantly differentially expressed.
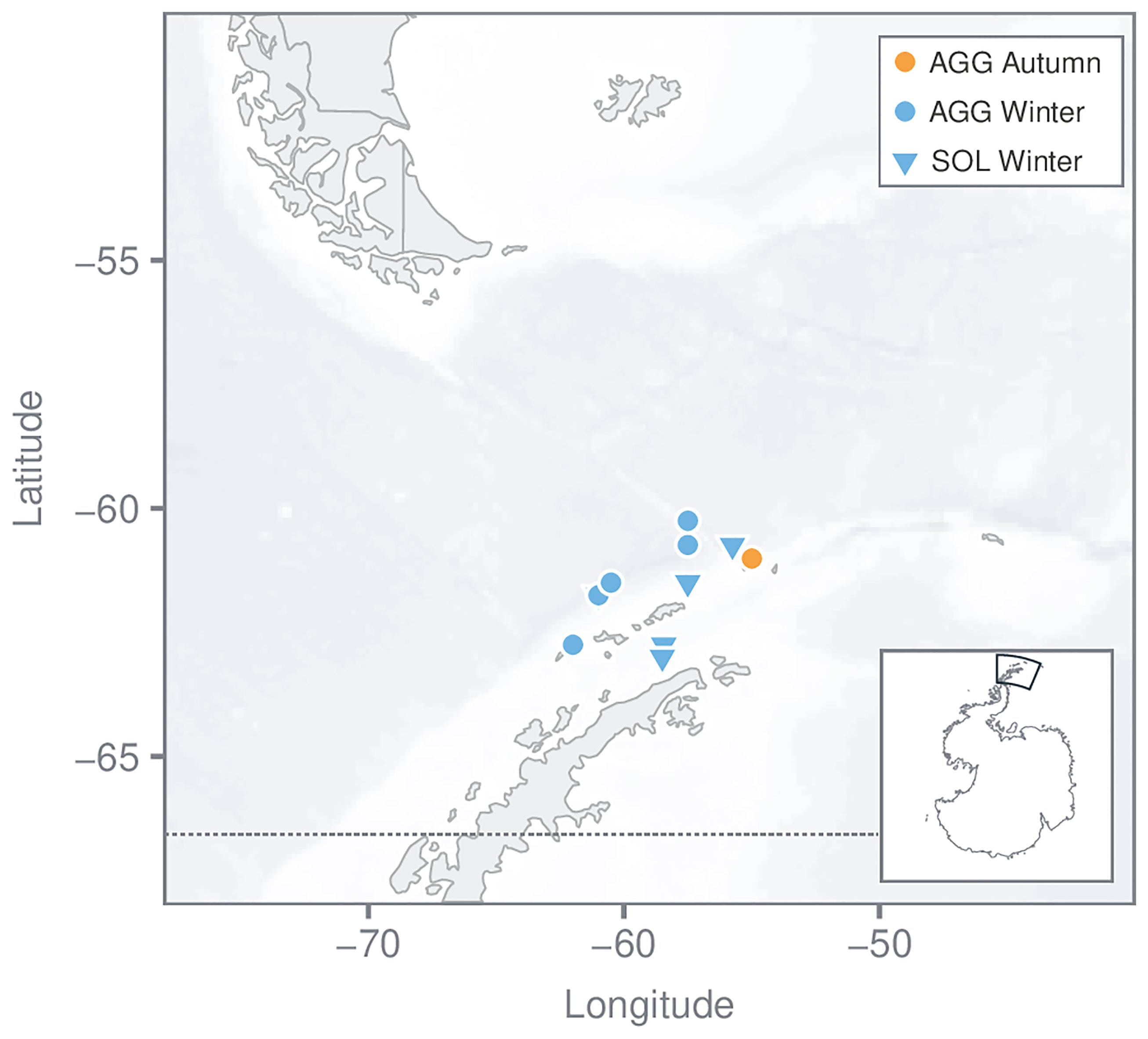
Figure 2 Map of sampling stations in/near the Bransfield Strait. Salp samples [Salpa thompsoni; aggregates (AGG) and solitaries (SOL)] were collected in winter (August 2016) and in autumn (April 2018) using an Isaacs-Kidd midwater trawl. Different symbol colors indicate the different sampling seasons, whereas different symbol shapes indicate different forms.
For the analysis of the GO term distribution of all annotated genes in the transcriptome and differentially expressed genes, all GO terms were grouped into GO categories of the second level of biological processes (BP) using R package GO.db v. 3.8.2 (Carlson, 2021), taking into account the hierarchical organization, e.g., the fact that a term can have more than one parent term (Tables S3–S5). GO term enrichment analysis was performed for significantly up- and downregulated genes together using the weight01 algorithm implemented in topGO v. 2.44.0 (Alexa and Rahnenfuhrer, 2021) after limiting nodes to BP. We pruned the GO hierarchy by having at least ten annotated genes in the reference list (nodeSize=10). Only GO terms with p-values < 0.05 (Fishers exact test) were considered significant.
Underlying bathymetric data were accessed using R package marmap v. 1.0.6 (Pante and Simon-Bouhet, 2013), and data processing and visualization was done with help of tidyverse v. 1.3.1 (Wickham et al., 2019).
The final assembly contained 204,806 transcripts and a total number of 107,680 genes with an N50 value of 2830 bp (Table 2). Most reads (82.5%) were mapped back to the transcriptome. The GC content of the final transcriptome assembly was 40.72%. By using eukaryotic database as a reference BUSCO results suggested an almost complete de novo assembly, as 99.2% of essential genes are represented [(single-copy: 20.4%; duplicated: 78.8%), Fragmented: 0.0%, Missing: 0.8%]. In total, 38,313 (35.6%) genes were associated with known and predicted proteins and 37,601 (34.9%) were annotated with GO terms. Within biological processes, most genes were related to cellular (78.7%) and metabolic processes (63.2%; Figure 3). Genes annotated within metabolic processes (23,768) were assigned to 146 terms at the fourth hierarchical level (Table S5). Comparatively more genes were categorized as belonging to proteins (50.64% of 23,768 genes) and nucleobase-containing compounds (44.88%) than to carbohydrates or lipids (Figure 3). A complete list of assigned categories can be found in the Supplementary Material (Tables S3-S5).

Table 2 Basic de novo transcriptome statistics for S. thompsoni.
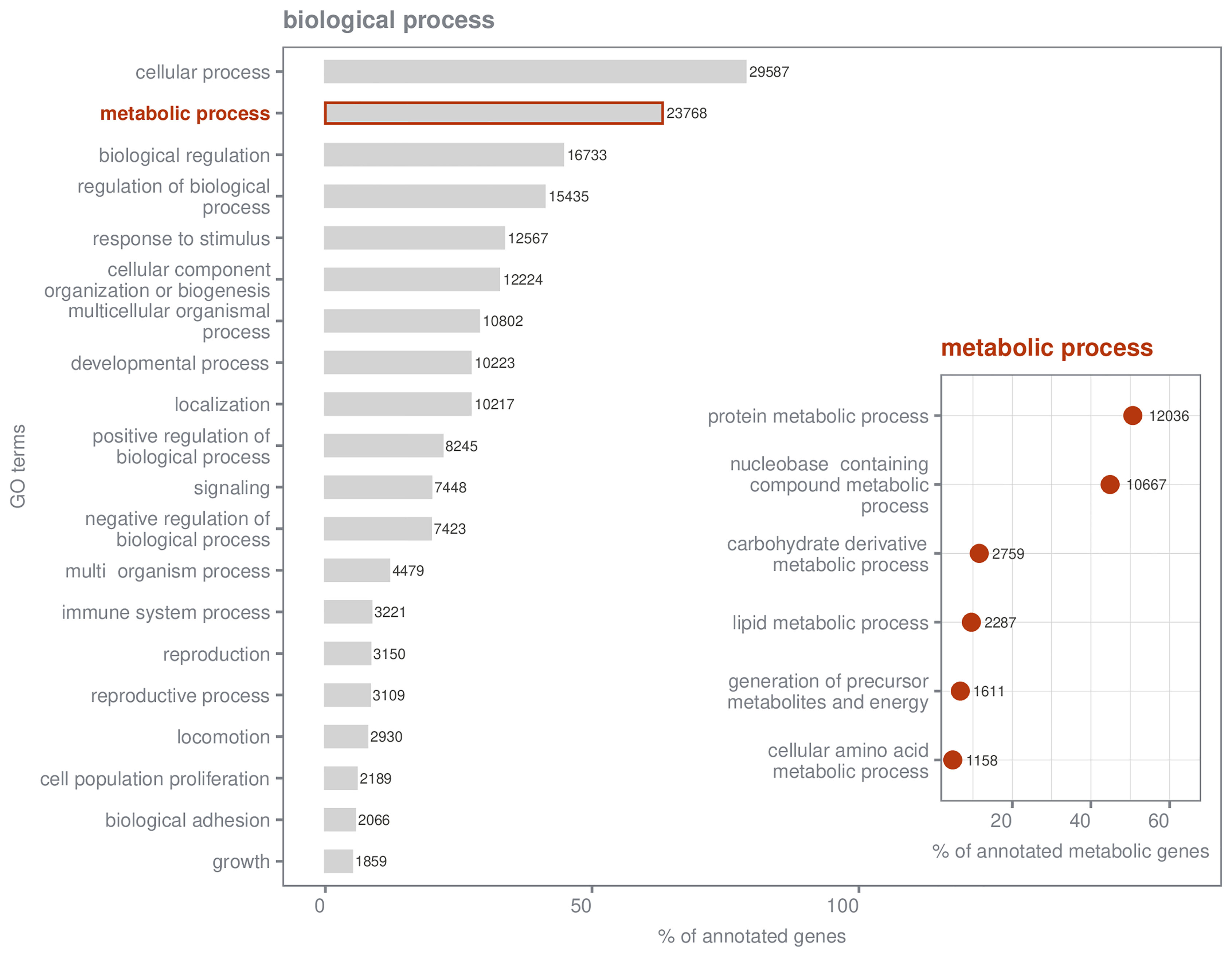
Figure 3 Gene ontology (GO) term distribution of annotated genes present in the transcriptome. Numbers next to the bar/dot indicate the absolute number of genes assigned to each category, while the x- axis indicates the respective percentage. Classification of biological processes of all annotated genes in the reference transcriptome (=37,601), summarized at second level, is shown. Only categories that were assigned to at least 4% of all annotated genes are displayed. A classification of genes within metabolic processes (23,768 in total) is also provided, summarized at the fourth level of biological hierarchy.
Gene expression signatures were obtained from samples collected in/near the Bransfield Strait during autumn and winter at a sampling depth of ~170 m (Figure 2, source (1) and (2), Table 1). In winter, all sampling areas in the region of the Bransfield Strait (Elephant Island, south and north) were characterized by a homogeneous environment with a mean sea surface temperature (SST) of - 1.73 ± 0.13 °C and low Chl a concentration (0.16 ± 0.07 mg m-3). Sea ice was present at all sampling stations. Total length was similar among individuals within each form (solitaries 60.00 ± 1.58 mm, n = 5; aggregates 14.80 ± 0.84 mm, n = 5), and except for a single aggregate, all individual salps had a developing embryo or chain. The autumn samples included only aggregates (22.00 ± 1.23 mm, n=5, stage=1) which were captured at a station with SST of 0.91 °C and Chl a concentration of 0.30 mg m-3 near Elephant Island (Table 1 and Table S1).
After filtering for a minimum of ten total counts per gene, 61,313 genes were retained for subsequent analyses. Principal component analysis revealed that large parts of the variance could be explained by the differences in gene expression between the two reproductive forms (PC1: 38%, solitaries vs. aggregates; Figure 4). Differences in gene expression between seasons were also present but much less pronounced (PC2: 24%, winter vs. autumn). Due to the pronounced effect between aggregates and solitaries as well as the absence of autumn solitary samples in our study, comparison between seasons was only conducted within aggregates, whereas only winter samples (aggregates and solitaries) were considered for the comparison between the two reproductive forms.
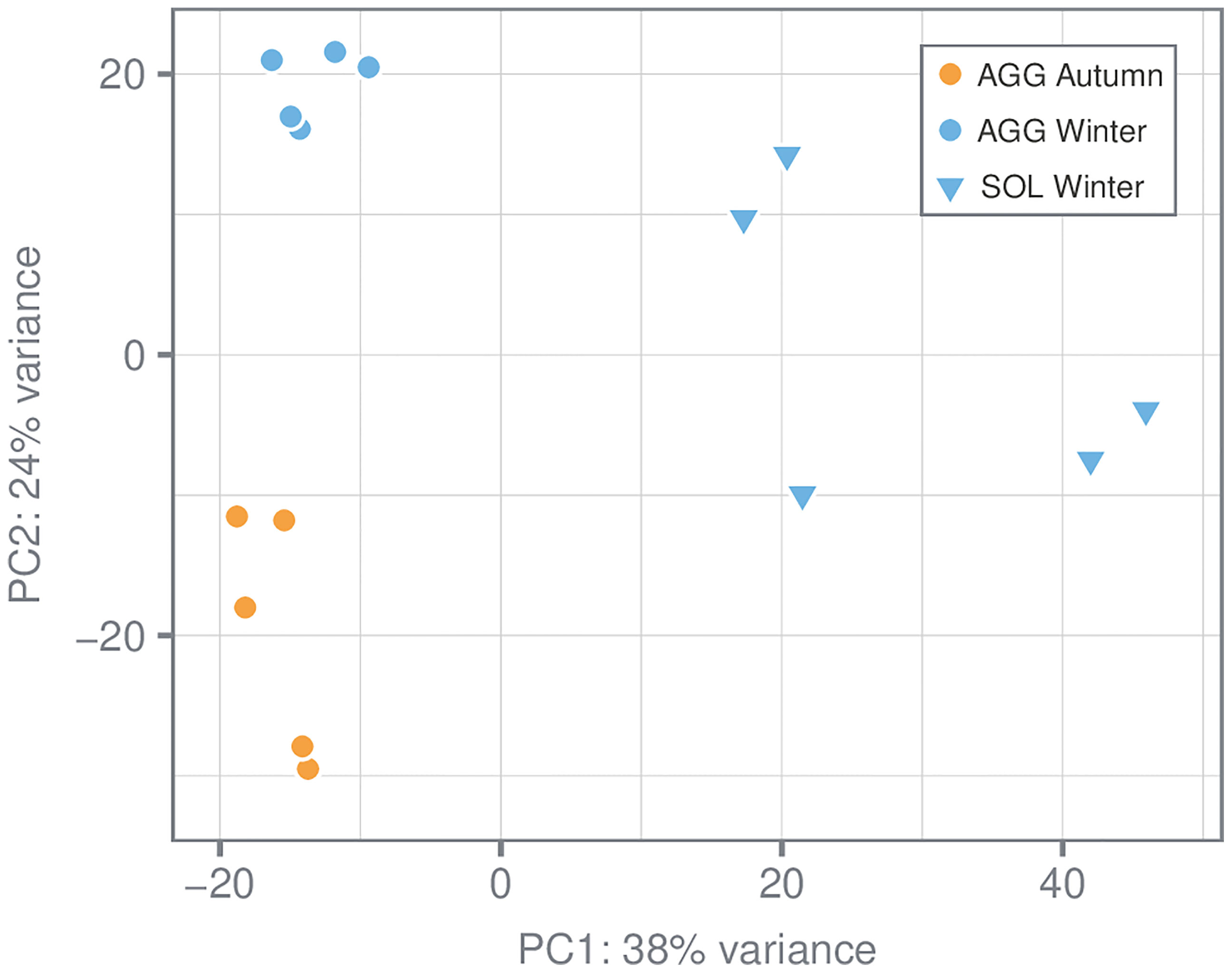
Figure 4 Principal Component Analysis (PCA) of the samples analyzed from sampling campaigns (1) and (2). The PCA shows the variance- stabilized gene expression dataset containing 15 samples of S. thompsoni with each dot representing an individual sample. Different symbol colors indicate the different sampling seasons (autumn and winter, respectively), whereas different symbol shapes indicate the different forms [aggregates (AGG) and solitaries (SOL)]. The explained variance [%] for PC1 and PC2 are also given.
Differential gene expression (DGE) analysis revealed 209 (LFCT=1, BH adjusted p-value < 0.001) differentially expressed genes in aggregates between autumn (n=5) and winter (n=5), of which 34.93% (73 genes) were annotated (Table S6). The majority of these genes (61.24%) were mainly upregulated in winter compared to autumn. By analyzing GO term distribution, GO terms describing genes of interest were assigned to multiple second levels of hierarchy of biological processes (BP), and revealed that most differentially expressed genes with annotation were assigned to cellular (83.56%) and metabolic processes (60.27%) (Figure 5A). GO term enrichment analysis revealed 56 significant GO terms within BP category (p < 0.05, see Table S8 for the complete list of enriched terms). Most genes within this category were related to translation (14 of 15 genes were upregulated in winter), with the majority encoding for 40S and 60S ribosomal proteins. No GO term related to reproductive processes was found to be enriched in the differentially expressed genes. Nevertheless, analysis of the distribution of GO terms identified 6 genes associated with reproduction including four genes that were upregulated in winter compared to autumn and that were related to germ cell (Peroxiredoxin 1), male (Nuclear autoantigenic sperm protein) and female gonad development (Cofilin/actin-depolymerizing factor homolog) (Figure 5A and Table S6).
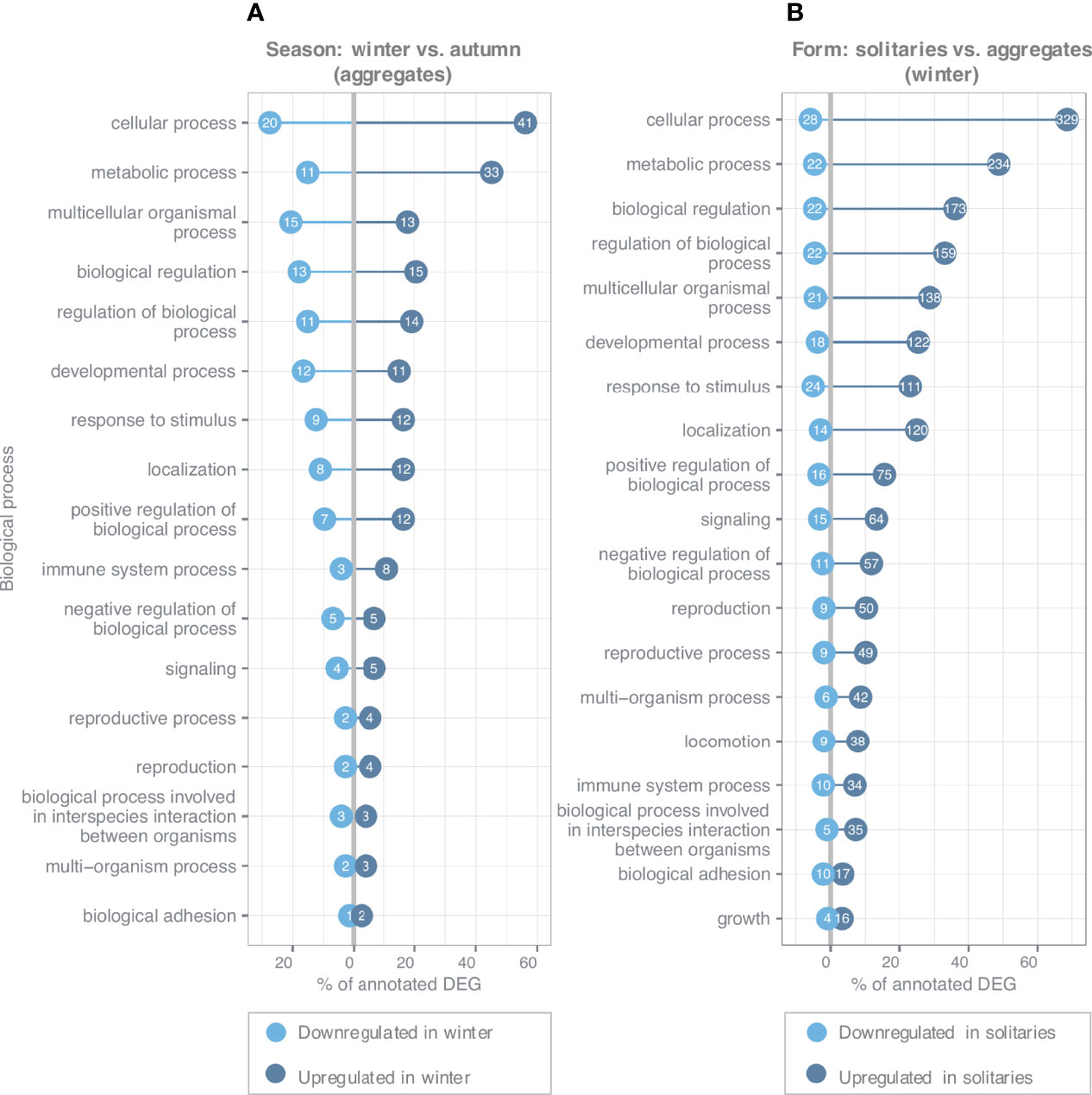
Figure 5 Distribution of Gene Ontology terms summarized at second level of biological processes for (A) differentially expressed genes (DEG) between seasons (winter vs. autumn) in aggregates and (B) differentially expressed genes between forms (solitaries vs. aggregates) in winter. Up- and downregulated refer to the number of genes which were found up- or down- regulated in (A) winter compared to autumn or in (B) solitaries compared to aggregates within the respective term. Numbers in circles indicate the absolute number of genes assigned to each category, while the x-axis indicates the respective percentage of all annotated differentially expressed genes [(A) 73/34.93% annotation and (B) 480/62.18% annotation]. Only categories that were assigned to at least 4% of annotated genes are shown.
A total of 772 genes (480 (62.18%) annotated) were found to be differentially expressed (LFCT=1, BH adjusted p-value < 0.001) between aggregates (n=5) and solitaries (n=5) in winter (Table S7). The majority (90.93%) of differentially expressed genes identified were significantly upregulated in solitaries compared to aggregates. GO term distribution revealed that most genes were mainly related to cellular (74.37% of all annotated differentially expressed genes) and metabolic processes (53.33% of all annotated differentially expressed genes) within BP (Figure 5B).
GO enrichment analysis identified 143 enriched GO terms (p < 0.05) (Table S9). A total of 32 terms related to metabolic processes were found to be significantly enriched and, besides cellular processes, mostly contribute to the differences in gene expression found between the two forms in winter (Figure 6A). The majority of genes (107) were related to protein metabolic processes with translation being the major process involved: 47 out of 67 genes encoding for 60S and 40S ribosomal subunits were significantly upregulated in solitaries compared to aggregates. Similarly, protein catabolic processes (modification-dependent protein catabolic process, regulation of endopeptidase activity) were represented within the top enriched terms. Mainly genes encoding for different types of proteasome subunits (alpha/beta) and ubiquitin- protein ligase were upregulated in solitaries. Additionally, terms involved in processes such as energy metabolism (ATP metabolic process, ATP synthesis coupled proton transport, tricarboxylic acid cycle (TCA) and mitochondrial electron transport) were found to be enriched: Many genes encoding for enzymes involved directly or indirectly in the TCA cycle were found to be upregulated in solitaries (Probable citrate synthase 1, mitochondrial; Isocitrate dehydrogenase [NADP]; Succinate–CoA ligase; Succinate dehydrogenase [ubiquinone] cytochrome b small subunit; Malate dehydrogenase, cytoplasmic). Besides protein and energy metabolism terms, the term “negative regulation of metabolic process” was also present among enriched terms, including 35 of 42 upregulated genes identified in solitaries.
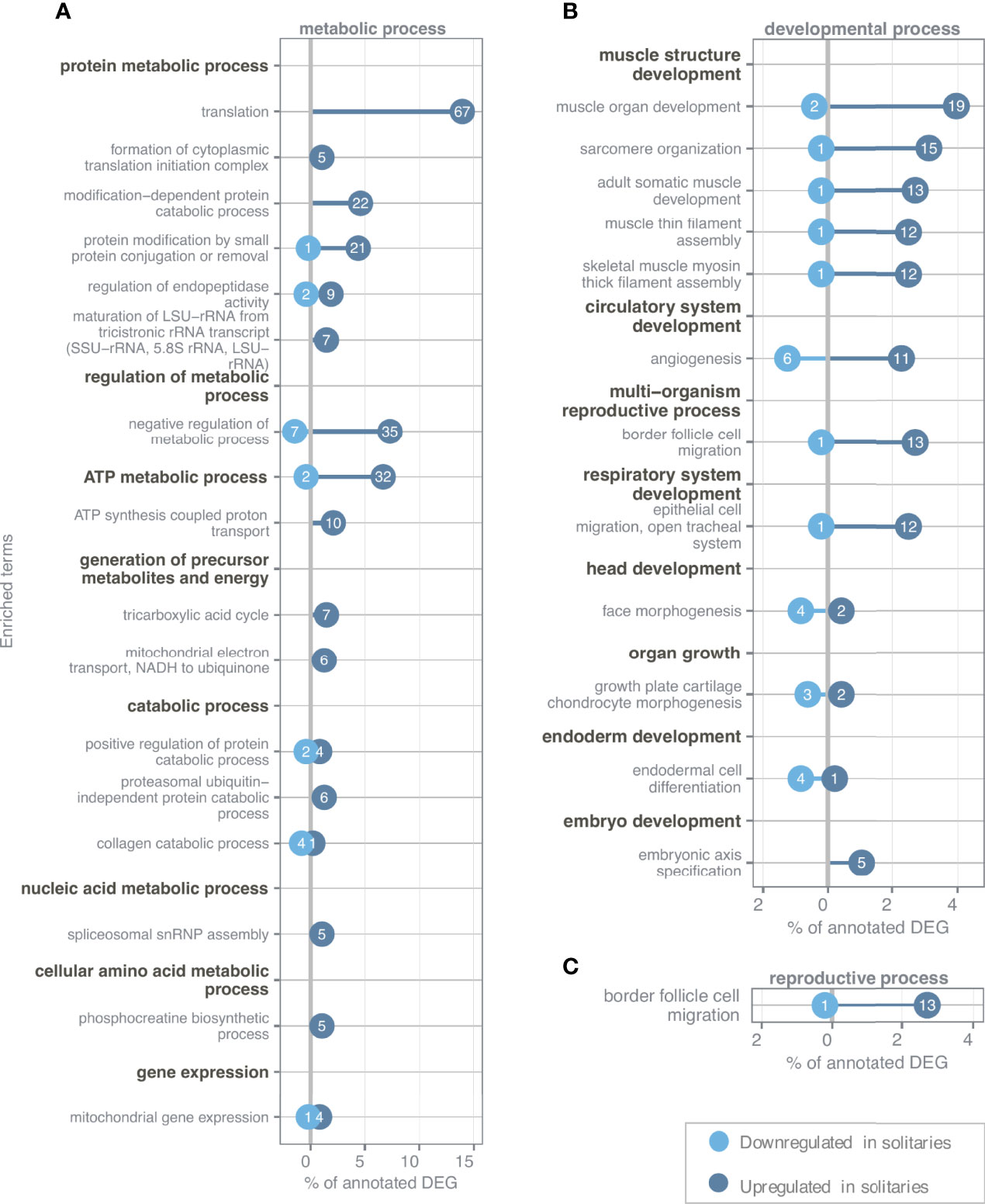
Figure 6 Significant Gene Ontology terms within (A) metabolic processes, (B) developmental processes and (C) reproductive processes (within biological processes) for differentially expressed genes (DEG) between forms (solitaries vs aggregates) in winter are shown. Up- and downregulated refer to up-or downregulation in solitaries in comparison to aggregates. Only significant terms covering at least 4 DEG are shown. Numbers in circles indicate the absolute number of genes assigned to each category, while the x- axis indicates the respective percentage of all annotated DEG (480/62.18% annotation). For the sake of clarity, the terms have been manually assigned to higher-level processes (marked in bold).
Within reproduction and developmental processes, 29 developmental (Figure 6B) and eight reproductive terms (Figure 6C) were enriched. Most of the genes (23) assigned to developmental processes were related to muscle structure development and upregulated in solitaries. In particular, the enriched processes involved mostly genes encoding for myosin (heavy chain, muscle). The same genes were also associated with other highly enriched processes such as the reproductive term “border follicle cell migration”. In addition to these remodeling processes, genes related to angiogenesis were also found significantly upregulated in solitaries (11 of 17 genes).
Abundance and vertical distribution of S. thompsoni are thought to depend on the season and reproductive generation (Foxton, 1966; Loeb and Santora, 2012). In recent decades, new interest in the ecological importance of S. thompsoni within the SO ecosystem has driven research on seasonal and form-specific patterns (Chiba et al., 1999; Ross et al., 2008; Pakhomov et al., 2011; von Harbou et al., 2011; Dubischar et al., 2012). However, few data are available on the physiological response of salps to seasonal environmental changes and no transcriptomic study to date has considered both reproductive generations, which is critical in understanding the factors driving seasonal patterns in population dynamics. We constructed the most complete de novo transcriptome of S. thompsoni currently available and examined seasonal as well as form-specific gene expression profiles.
In this study, we created a near complete de novo transcriptome (99.2%) with slightly higher annotation success (35.6% of all genes were associated with known and predicted proteins) than generated by Batta-Lona et al. (2017) (18%). The difference in annotation may be explained by the low fragmentated transcript percentage (0.0%) in our transcriptome compared to the highly fragmented transcript rates stated by Batta-Lona et al. (2017). The functional annotation of our full de novo transcriptome revealed that the most common terms were “cellular process” and “metabolic processes” in the biological process (BP) category. This was also found in the reference genome of S. thompsoni (Jue et al., 2016) as well as in other transcriptomic studies of major grazers of the Southern Ocean, such as the Antarctic krill (Meyer et al., 2015; Biscontin et al., 2019). The low number of genes belonging to lipid and carbohydrate metabolism may reflect the biochemical composition of S. thompsoni, which is characterized by low lipid (2.9% of dry weight) and carbohydrate (2.1% of dry weight) content. In contrast, protein metabolic process represented the major category in the distribution of gene ontology terms which may be related to the high protein content of salps, accounting for at least 10% of dry weight (Dubischar et al., 2012).
The PCA analysis suggests (1) a seasonally driven effect on gene expression in aggregates, and (2) form- specific gene expression signatures during winter. The processes discovered in both comparisons will be placed within the complex life cycle of salps in the following.
It is generally assumed that the peak abundance of aggregates in the upper water column mainly occurs between spring and autumn due to massive chain release under favorable conditions this time of year (Foxton, 1966; Loeb and Santora, 2012; Henschke and Pakhomov, 2019). S. thompsoni abundances decrease as the year progresses towards winter and it is still unclear if and how these salps overwinter. While it has been hypothesized that immigration and passive advection are key factors for seeding the salp blooms in spring, there are also indications that solitaries are able to successfully overwinter at greater depths (Pakhomov et al., 2011; Groeneveld et al., 2020). At least some aggregates should theoretically also be able to survive the winter season in order to fertilize newly released female aggregates in spring. Observations during a 2013 Polarstern winter cruise (ANT-XXIX/7) indicate that S. thompsoni may successfully reproduce during winter even under ice (Pakhomov and Hunt, 2014). However, if and how aggregates overwinter is still under debate.
In this study a majority of differentially expressed genes between autumn and winter aggregates were found to be related to translation. More specifically, genes related to the synthesis of ribosomal proteins (14 of 15 genes) were upregulated in winter compared to autumn, indicating a higher demand for ribosomal capacities during winter. Batta-Lona et al. (2017) also detected significant upregulation of genes involved in translational processes in samples obtained in spring compared to summer, and concluded that upregulation was characteristic of rapid growth associated with early-stage aggregates analyzed during spring (Batta-Lona et al., 2017). Unfortunately, findings on in situ growth rates of aggregates are mainly limited to the summer season (Loeb and Santora, 2012; Henschke et al., 2018), but there is evidence to support the notion that aggregates may be able to continue growth and development during winter (Pakhomov and Hunt, 2014). In our study, aggregates from autumn and winter were of similar size and developmental stage (female) (Table 1). We can therefore exclude rapid growth as a driving factor for increased translational capacities in winter found, suggesting that the upregulation of genes encoding for ribosomal proteins might represent a more complex response here. An increased number of ribosomal proteins may indicate a response to lower temperatures experienced during winter, compensating for reduced translation efficiency as observed in other cold water planktonic species (Toseland et al., 2013; Jue et al., 2016). The observed drop from 0.91°C in autumn to -1.73 ± 0.13°C in winter, as in our study, may therefore explain the observed pattern (Table 1).
Another explanation for the upregulation of ribosomal genes in winter aggregates may be preparation for the upcoming spring. Gene transcription and translation as a measure of core metabolic activity may begin early in preparation for the more active part of the life cycle in spring when they have to grow (Kumar, 2017).
Moreover, Jue et al. (2016) detected a small number of ribosomal genes undergoing rapid evolution in the genome of S. thompsoni, which were previously found in other urochordates. These fast-evolving genes may reflect the complex seasonal life cycle of salps in the polar environment. However, whether the abundances of ribosomal genes in Jue et al. (2016) and in our analysis represents an adaptive advantage to the extreme seasonality of Antarctic regions or reflects the general nature of pelagic tunicates remains controversial (Jue et al., 2016).
No GO terms related to reproductive processes were enriched among the differentially expressed genes between both seasons, although autumn (March-April) has been considered the active embryo release period (Foxton, 1966). The reproduction of S. thompsoni is negatively affected by low temperatures and generation times increase with low food concentrations and unfavorable temperatures (Heron, 1972; Pakhomov et al., 2011; Ono and Moteki, 2017; Henschke and Pakhomov, 2019). The number of failed embryos already increases in autumn when temperatures and food decrease (Chiba et al., 1999; Henschke and Pakhomov, 2019). In our study, SST in autumn (0.91°C) was slightly below the suggested thermal threshold for successful reproduction (1°C) and Chl a concentration (0.30 mg m-3) was comparable to winter conditions (0.16 ± 0.07 mg m-3) (Table 1) (Pakhomov et al., 2011; Ono and Moteki, 2013; Henschke et al., 2018). Therefore, similar to the findings made by Chiba et al. (1999), our data suggest that sexual reproduction was deactivated or in the process of deactivation, driven by autumn water temperature and phytoplankton decline. Low energetic investment in reproduction may sustain vital processes during unfavorable conditions and could indicate anticipation for the more favorable spring time.
Although enrichment of processes involved in reproduction was not detected we were able to identify 6 differentially expressed genes related to male and female gonad development by GO term distribution analysis (Figure 5A). This may be indicative for different reproductive stages (within aggregates) in both seasons. The aggregates obtained in autumn were females with the beginnings of embryo development, classified as stage 1 (Foxton, 1966). We were unable to obtain information on the developmental stage of the winter aggregate samples. However, we assumed that the winter aggregates reached at least the same stage as those from autumn since every individual contained an embryo except one. The complex protogynous nature of aggregates may further complicate the genetic response here: generally, it was assumed that aggregate testes do not mature until the embryo is released to prevent self-fertilization (Godeaux et al., 1998). However, observations of S. thompsoni during the Polarstern PS112 cruise in 2018 and of its close Mediterranean relative S. fusiformis at the Oceanological Observatory in Villefranche-sur-mer (France) in spring 2021 showed that aggregates can simultaneously exhibit female (presence of the embryo) and male (development of the testes) physiology (Figure S1). Considering the complex nature of hermaphrodism, differentially expressed genes involved in reproduction may imply that the aggregates obtained in winter already began male development even though they still contained an embryo. This hypothesis may be supported by limited observations made by Pakhomov & Hunt (2014) who concluded that salps could continue growing and developing during winter. Further evidence is needed to support this potential strategy.
The Southern Ocean is characterized by strong seasonal fluctuations of temperature, light and food availability, and so far, there have been no findings on overwintering strategies in salps. Other zooplankton grazers that occur in polar regions such as different copepod species or Antarctic krill (Euphausia superba) developed unique strategies to survive the winter season. For example, E. superba survive low winter temperature and food availability by accumulating lipid stores, using alternative food sources, and regressing their sexual organs (Hagen and Auel, 2001; Kawaguchi et al., 2007). Salps are unlikely to build up sufficient lipid reserves for winter survival because of their exceptionally low lipid content (<3% of dry weight) (Dubischar et al., 2006; Dubischar et al., 2012). Feeding dynamics for S. thompsoni were investigated by von Harbou et al. (2011) in the Lazarev Sea and the analysis of fatty acid trophic biomarkers showed low seasonality. However, the analysis of winter samples was restricted to solitaries only (von Harbou et al., 2011). While there is no evidence of sexual regression during winter in salps as observed for krill, a low investment in reproduction may also represent an energy conservation strategy, which, according to our findings, may begin in autumn. The low number of differentially expressed genes between autumn and winter may also indicate that aggregates are not able to adapt their physiology to the adverse conditions in winter, which would explain the lower aggregates to solitaries ratio observed in winter (Pakhomov et al., 2011). Although our results should be interpreted with caution due to the difference in sampling years, 2018 (autumn) and 2016 (winter), based on our data it seems that translation and, more specifically, higher ribosomal capacities in winter aggregates compared to autumn is the main process involved in facing the challenge of surviving the cold, food scarce winter season.
Under low temperatures and Chl a conditions, solitaries are thought to be the “overwintering stage”, slowly growing at depth until conditions are favorable for chain release in spring (Foxton, 1966; Loeb and Santora, 2012). Loeb & Santora (2012) and Pakhomov & Hunt (2014) suggested that asexual reproduction may begin already in mid to early winter, while sexual reproduction appears to suffer from low temperatures and Chl a concentrations (Chiba et al., 1999; Henschke and Pakhomov, 2019). The overwintering strategy of S. thompsoni is still under debate due to limited observations during winter (Henschke et al., 2018; Groeneveld et al., 2020). Theoretically some of male aggregates should survive winter to fertilize young female chains released heading into spring (Pakhomov and Hunt, 2014). Here, we compared the effect of winter conditions on both forms at the transcriptomic level.
Gene expression patterns during winter strongly differed between aggregates and solitaries, suggesting form- specific gene expression signatures and/or potential form-specific overwintering strategies (Figures 5B, 6). As already observed in the seasonal comparison (see Seasonal Transition From Autumn to Winter in Aggregates), translation appears to not only play an important role during preparation for the winter season in aggregates but also constitutes a major difference between gene expression signatures of the two forms during winter (Figure 6A). In addition, protein catabolic genes encoding for different types of proteasome subunits were upregulated in solitaries compared to aggregates, indicating higher protein modification and breakdown rates in solitaries (Tanaka and Chiba, 1998). Higher translation and protein modification in solitaries compared to aggregates may be linked to the different timing of reproduction within their alternating seasonal life cycle: Solitaries release chains in spring and occasionally in winter when the conditions are suitable. Therefore, a greater investment in ribosomal protein production and modification in winter may indicate preparation for upcoming or ongoing chain release in early spring and winter (Foxton, 1966; Loeb and Santora, 2012; Pakhomov and Hunt, 2014). In addition, the body size of the solitaries used here suggests that they have already reached stage 3 or 4 (Foxton, 1966) implying that individual buds of the first aggregate chain are visible, which would make an early chain release in spring or even winter likely.
Nearly all enzymes involved directly or indirectly in the TCA and ATP metabolic processes were identified, implying different demands for energy production between the two forms. Translation is an energy-consuming process that can take up to nearly 75% of a cell’s energy budget (Lane and Martin, 2010). The upregulation of ribosomal proteins and thus protein synthesis capacity observed in solitaries fits with their potentially higher energy demand. Few studies compared the metabolic rates (oxygen consumption and ammonia excretion) of both forms, aggregates and solitaries. No differences in metabolic activity between forms were found in S. thompsoni during austral summer (Iguchi and Ikeda, 2004). In related species the metabolic rate was only slightly lower in aggregates compared to solitaries (Biggs, 1977; Cetta et al., 1986). However, comparative data from winter samples are lacking. The marked difference in metabolic gene expression between forms observed here may indicate differences in metabolic activity in winter and/or form-specific characteristics. The maintenance of costly metabolic activity in solitaries compared to aggregates is also in contrast to other organisms such as krill and copepods in the Southern Ocean, which can cope with low food concentrations in winter and tend to downregulate their metabolism to reduce energy demands (Hagen, 1999; Meyer et al., 2010; Höring et al., 2020).
The expression of several genes encoding for myosin (heavy chain muscle, MHC) was also significantly higher in solitaries (Figure 6B). Myosins play an important role in the development of muscle structure, and MHC isoforms are known to be differentially expressed across species, tissues and developmental stages in tunicates (Razy-Krajka and Stolfi, 2019). Generally, the shape and number of muscle bands differ in size and arrangement between both stages, due to their different locomotory behavior. Aggregates are usually linked in a chain whereas the solitary stage occurs as a single individual, resulting in differences in maneuverability and speed. Solitaries have many more muscle bands and therefore exhibit higher activity rates as well as swimming speeds which could serve as an explanation for the generally higher expression of muscle related genes in solitaries found here (Godeaux et al., 1998). Interestingly, myosins play also an important role in border cell migration, a process important in reproduction (Figure 6C). The high expression levels of genes encoding for myosin proteins in solitaries may also indicate a more active reproductive state of solitaries during winter. Genes involved in angiogenesis or blood vessel formation were upregulated in solitaries as well. Since salps do not have “true” blood vessels this GO term is not appropriate, and the upregulation of these genes may rather underscore a general anatomical restructuring in solitaries during winter.
In contrast, it is likely that aggregates slow down their metabolism compared to solitaries and proceed to hibernation during winter to reduce energy demands. If they are able to continue growth and development in winter [according to Pakhomov & Hunt (2014)], the question arises whether aggregates must reach a certain developmental stage to successfully overwinter.
We detected a different response of genes involved in metabolic activity, energy demand and development between the reproductive forms during winter. Whether the observed differences in gene expression patterns are unique to winter, representing different overwintering strategies between the forms, or whether the differences are general form effects independent of the season remains unclear. During the complex life cycle of salps, aggregates and solitaries differ in reproduction and morphology. Furthermore, the protogynous nature of aggregates may further complicate the interpretation of responses. A general form effect may therefore be assumed. On the other hand, differences in distribution and abundance of both forms throughout the year as part of their complex life cycle would account for different strategies to cover their metabolic needs between the seasons. Nevertheless, our study highlights the importance and urgency to include both forms (and preferably several developmental stages within) in future research, not only to provide a more complete picture of salp biology over the course of the year, but to discern general form and stage effects from seasonal effects.
Transcriptomic studies provide insight into physiological processes involved in an organism`s response to different factors, such as environmental change, and are especially advantageous when studying fragile organisms that are difficult to maintain in aquaria. However, we also have to be aware of inherent limitations as those studies reflect the transcriptional regulatory level only, and not the higher levels of biological organization. Limited functional annotation of non-model organisms tends to identify well-studied processes discovered in well-studied species. Performing GO enrichment analyses on non-model organisms likely underestimates or hides actual responses, compromising mechanistic understanding of complex reactions in progress.
By creating a high-quality de novo transcriptome of S. thompsoni we were able to perform GO term distribution and enrichment analysis. We examined seasonal and form-specific differences in gene expression patterns, focusing on genes involved in metabolism, development and reproduction. Seasonal differences between aggregates were mainly characterized by pronounced upregulation of genes involved in translation processes, indicating higher ribosomal capacities in winter compared to autumn. In turn, solitaries appear to have a higher need for translation capacities compared to aggregates during winter possibly fueled by elevated energy production. It remains unclear if the observed patterns are due to different strategies in response to winter conditions or if they represent a more general form effect, detectable throughout the year. Nevertheless, our study suggests a form-specific response to winter, which should be considered in future research on salps `physiology and provides a basis for the identification of form-specific marker genes and key processes in the life cycle of S. thompsoni. By providing this resource we contribute to a better understanding of S. thompsoni`s response to environmental changes and it´s underlying molecular mechanisms, which is of particular interest under predicted climate change scenarios.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA822688.
SM, KM, WW, and BM designed the study. WW conducted all laboratory procedures and produced preliminary results with LS. Bioinformatic analyses were performed by SM, IU, GS, and CP. SM wrote the manuscript and KM, EP, and BM contributed significantly to revisions. All authors reviewed and approved the final version of the manuscript.
This work was supported by project phases I+II of “The performance of krill vs. salps to withstand in a warming Southern Ocean” (PEKRIS (FKZ 03F0746A), PEKRIS II (FKZ 03F0828A)) of the German Ministry for Education and Research (BMBF) and the Programma Nazionale di Ricerche in Antartide – PNRA (grant 2016_00225 and PNRA19_00065). Financial support for open-access publication has been given by the Open Access Publication Funds of Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank chief scientist Dr. Christian Reiss as well as the captain, crew, and technical staff of RVIB Nathaniel B. Palmer for providing salp samples and environmental data from Antarctic winter 2016. In addition, we thank the captain and crew of R.V. Polarstern for providing salps from the Antarctic summer (ANT XXVIII/3, 2012) and autumn (PS112, 2018) seasons. We are also indebted to Lutz Auerswald for his great help in maintaining freshly caught salps in the laboratory, under different temperature regimes during PS112. Many thanks also to Sara Driscoll for editing our manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.914095/full#supplementary-material
Alexa A., Rahnenfuhrer J. (2021). topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.44.0.
Andersen V. (1998). “Salp and Pyrosomid Blooms and Their Importance in Biogeochemical Cycles,” in The Biology of Pelagic Tunicates. Ed. Bone Q. (Oxford: Oxford University Press), 125–138.
Atkinson A., Hill S. L., Pakhomov E. A., Siegel V., Anadon R., Chiba S., et al. (2017). KRILLBASE: A Circumpolar Database of Antarctic Krill and Salp Numerical Densities 1926-2016. Earth System Sci. Data 9 (1), 193–210. doi: 10.5194/essd-9-193-2017
Batta-Lona P. G., Maas A. E., O’Neill R. J., Wiebe P. H., Bucklin A. (2017). Transcriptomic Profiles of Spring and Summer Populations of the Southern Ocean Salp, Salpa Thompsoni, in the Western Antarctic Peninsula Region. Polar Biol. 40 (6), 1261–1276. doi: 10.1007/s00300-016-2051-6
Biggs D. C. (1977). Respiration and Ammonium Excretion by Open Ocean Gelatinous Zooplankton. Limnol. Oceanography 22 (1), 108–117. doi: 10.4319/lo.1977.22.1.0108
Biscontin A., Martini P., Costa R., Kramer A., Meyer B., Kawaguchi S., et al. (2019). Analysis of the Circadian Transcriptome of the Antarctic Krill Euphausia Superba. Sci. Rep. 9 (1), 1–11. doi: 10.1038/s41598-019-50282-1
Böckmann S., Koch F., Meyer B., Pausch F., Iversen M., Driscoll R., et al. (2021). Salp Fecal Pellets Release More Bioavailable Iron to Southern Ocean Phytoplankton Than Krill Fecal Pellets. Curr. Biol. 31 (13), 2737–2746.e3. doi: 10.1016/j.cub.2021.02.033
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bushmanova E., Antipov D., Lapidus A., Prjibelski A. D. (2019). RnaSPAdes: A De Novo Transcriptome Assembler and its Application to RNA-Seq Data. GigaScience 8 (9), 1–13. doi: 10.1093/gigascience/giz100
Carlson M. (2021). GO.db: A Set of Annotation Maps Describing the Entire Gene Ontology. R Package Version 3.13.0.
Casareto B. E., Nemoto T. (1986). National Institute of Polar Research NII-Electronic Library Service (National Institute of Polar Research).
Cetta C. M., Madin L. P., Kremer P. (1986). Respiration and Excretion by Oceanic Salps. Marine Biol. 91 (4), 529–537. doi: 10.1007/BF00392605
Chiba S., Ishimaru T., Hosie G. W., Wright S. W. (1999). Population Structure Change of Salpa Thompsoni From Austral Mid-Summer to Autumn. Polar Biol. 22 (5), 341–349. doi: 10.1007/s003000050427
Dubischar C. D., Pakhomov E. A., von Harbou L., Hunt B. P. V., Bathmann U. V. (2012). Salps in the Lazarev Sea, Southern Ocean: II. Biochemical Composition and Potential Prey Value. Marine Biol. 159 (1), 15–24. doi: 10.1007/s00227-011-1785-5
Dubischar C. D., Pakhomov E. A., Bathmann U. V. (2006). The Tunicate Salpa Thompsoni Ecology in the Southern Ocean. II. Proximate and Elemental Composition. Marine Biol. 149, 625–632. doi: 10.1007/s00227-005-0226-8
Foxton P. (1966). “The Distribution and Life History of Salpa Thompsoni Foxton With Observations on a Related Species S.gerlachei, Foxton,” in Discovery Reports, vol. 34. , 1–116. Discovery Expedition Collections
Gilbert D. (2013) EvidentialGene: Tr2aacds, mRNA Transcript Assembly Software. Available at: http://arthropods.eugenes.org/EvidentialGene/about/EvidentialGene_trassembly_pipe.html.
Godeaux J. E. A., Bone Q., Braconnot J.-C. (1998). “Anatomy of Thaliacea,” in The Biology of Pelagic Tunicates. Ed. Bone Q. (Oxford: Oxford University Press: Oxford), 1–24.
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-Length Transcriptome Assembly From RNA-Seq Data Without a Reference Genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Groeneveld J., Berger U., Henschke N., Pakhomov E. A., Reiss C. S., Meyer B. (2020). Blooms of a Key Grazer in the Southern Ocean – An Individual-Based Model of Salpa Thompsoni. Prog. Oceanogr. 185(2019), 102339. doi: 10.1016/j.pocean.2020.102339
Hagen W. (1999). Reproductive Strategies and Energetic Adaptations of Polar Zooplankton. Invertebrate Reprod. Dev. 36 (1–3), 25–34. doi: 10.1080/07924259.1999.9652674
Hagen W., Auel H. (2001). Seasonal Adaptations and the Role of Lipids in Oceanic Zooplankton. Zoology 104 (3–4), 313–326. doi: 10.1078/0944-2006-00037
Henschke N., Pakhomov E. A., Groeneveld J., Meyer B.. (2018). Modelling the Life Cycle of Salpa Thompsoni. Ecol. Modell. 387, 17–26. doi: 10.1016/j.ecolmodel.2018.08.017
Henschke N., Pakhomov E. A. (2019). Latitudinal Variations in Salpa Thompsoni Reproductive Fitness. Limnol. Oceanogr. 64 (2), 575–584. doi: 10.1002/lno.11061
Heron A. C. (1972). Population Ecology of a Colonizing Species: The Pelagic Tunicate Thalia Democratica. Oecologia 10 (4), 269–293. doi: 10.1007/bf00345733
Höring F., Biscontin A., Harms L., Sales G., Reiss C. S., De Pittà C, et al. (2020). Seasonal Gene Expression Profiling of Antarctic Krill in Three Different Latitudinal Regions. Mar. Genomics 56. doi: 10.1016/j.margen.2020.100806
Iguchi N., Ikeda T. (2004). Metabolism and Elemental Composition of Aggregate and Solitary Forms of Salpa Thompsoni ( Tunicata : Thaliacea ) in Waters Off the Antarctic Peninsula During Austral Summer 1999. Journal of Plankton Research 26(9), 1025–37. doi: 10.1093/plankt/fbh093
Jue N. K., Batta-Lona P. G., Trusiak S., Obergfell C., Bucklin A., O’neill M. J., et al. (2016). Rapid Evolutionary Rates and Unique Genomic Signatures Discovered in the First Reference Genome for the Southern Ocean Salp, Salpa Thompsoni (Urochordata, Thaliacea). Genome Biol. Evol. 8 (10), 3171–3186. doi: 10.1093/gbe/evw215
Kawaguchi S., Yoshida T., Finley L., Cramp P., Nicol S. (2007). The Krill Maturity Cycle: A Conceptual Model of the Seasonal Cycle in Antarctic Krill. Polar Biol. 30 (6), 689–698. doi: 10.1007/s00300-006-0226-2
Kumar V. (2017). Biological Timekeeping: Clocks, Rhythms and Behaviour, Biological Timekeeping: Clocks, Rhythms and Behaviour. New Delhi: Springer India. doi: 10.1007/978-81-322-3688-7
Lane N., Martin W. (2010). The Energetics of Genome Complexity. Nature 467 (7318), 929–934. doi: 10.1038/nature09486
Lenz P. H., Roncalli V., Cieslak M. C., Tarrant A. M., Castelfranco A. M., Hartline D. K. (2021). Diapause vs. Reproductive Programs: Transcriptional Phenotypes in a Keystone Copepod. Commun. Biol. 4 (1), 1–13. doi: 10.1038/s42003-021-01946-0
Liu J., Li G., Chang Z., Yu T., Liu B., McMullen R., et al. (2016). BinPacker: Packing-Based De Novo Transcriptome Assembly From RNA-Seq Data. PloS Comput. Biol. 12 (2), 1–15. doi: 10.1371/journal.pcbi.1004772
Loeb V. J., Siegel V., Holm-Hansen O., Hewitt R., Fraser W., Trivelpiece W., et al. (1997). Effects of Sea-Ice Extent and Krill or Salp Dominance on the Antarctic Food Web. Nature 387 (6636), 897–900. doi: 10.1038/43174
Loeb V. J., Santora J. A. (2012). Population Dynamics of Salpa Thompsoni Near the Antarctic Peninsula: Growth Rates and Interannual Variations in Reproductive Activity, (1993-2009). Prog. Oceanogr. 96 (1), 93–107. doi: 10.1016/j.pocean.2011.11.001
Love M. I., Huber W., Anders S. (2014). Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol. 15 (12), 1–21. doi: 10.1186/s13059-014-0550-8
Lüskow F., Pakhomov E. A., Stukel M. R., Décima M. (2020). Biology of Salpa Thompsoni at the Chatham Rise, New Zealand: Demography, Growth, and Diel Vertical Migration. Marine Biol. 167 (12), 175. doi: 10.1007/s00227-020-03775-x
Meyer B., Auerswald L., Siegel V., Spahić S., Pape C., Fach B. A., et al. (2010). Seasonal Variation in Body Composition, Metabolic Activity, Feeding, and Growth of Adult Krill Euphausia Superba in the Lazarev Sea. Mar. Ecol. Prog. Ser. 398, 1–18. doi: 10.3354/meps08371
Meyer B., Martini P., Biscontin A., De Pittà C., Romualdi C., Teschke M., et al. (2015). Pyrosequencing and De Novo Assembly of Antarctic Krill (Euphausia Superba) Transcriptome to Study the Adaptability of Krill to Climate-Induced Environmental Changes. Mol. Ecol. Resour. 15 (6), 1460–1471. doi: 10.1111/1755-0998.12408
Nakasugi K., Crowhurst R., Bally J., Waterhouse P. (2014). Combining Transcriptome Assemblies From Multiple De Novoassemblers in the Allo-Tetraploid Plant Nicotiana Benthamiana. PloS One 9(3), e91776. doi: 10.1371/journal.pone.0091776
Ono A., Moteki M. (2013). ‘Spatial Distributions and Population Dynamics of Two Salp Species, Ihlea Racovitzai and Salpa Thompsoni, in the Waters North of Lützow-Holm Bay (East Antarctica) During Austral Summers of 2005 and 2006. Polar Biol. 36 (6), 807–817. doi: 10.1007/s00300-013-1305-9
Ono A., Moteki M. (2017). Spatial Distribution of Salpa Thompsoni in the High Antarctic Area Off Adélie Land, East Antarctica During the Austral Summer 2008. Polar Sci. 12, 69–78. doi: 10.1016/j.polar.2016.11.005
Pakhomov E. A., Dubischar C. D., Strass V., Brichta M., Bathmann U. V. (2006). The Tunicate Salpa Thompsoni Ecology in the Southern Ocean. I. Distribution, Biomass, Demography and Feeding Ecophysiology. Mar. Biol. 149 (3), 609–623. doi: 10.1007/s00227-005-0225-9
Pakhomov E. A., Dubischar C. D., Hunt B. P. V., Strass V., Cisewski B., Siegel V., et al. (2011). Biology and Life Cycles of Pelagic Tunicates in the Lazarev Sea, Southern Ocean. Deep Sea Res. Part II 58 (13–16), 1677–1689. doi: 10.1016/j.dsr2.2010.11.014
Pakhomov E. A., Hunt B. P. V. (2014). “Macrozooplankton and micronekton dynamics during the Antarctic winter,” in The Expedition of the Research Vessel Polarstern to the Antarctic in 2013 (ANT-XXIX/7) In Berichte zur Polar- und Meeresforschung = Reports on Polar and Marine Research. Eds. Meyer B., Auerswald L., 31–36. (Berichte zur Polar- und Meeresforschung)doi: 10.2312/BzPM_0674_2014
Pakhomov E. A., Hunt B. P. V. (2017). Trans-Atlantic Variability in Ecology of the Pelagic Tunicate Salpa Thompsoni Near the Antarctic Polar Front. Deep Sea Res. Part II 138 (March), 126–140. doi: 10.1016/j.dsr2.2017.03.001
Pante E., Simon-Bouhet B. (2013). Marmap: A Package for Importing, Plotting and Analyzing Bathymetric and Topographic Data in R. PloS One 8 (9), e73051. doi: 10.1371/journal.pone.0073051
Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C.. (2017). Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 14 (4), 417–419. doi: 10.1038/nmeth.4197
Pauli N.-C., Flintrop C. M., Konrad C., Pakhomov E. A., Swoboda S., Koch F., et al. (2021a). Krill and Salp Faecal Pellets Contribute Equally to the Carbon Flux at the Antarctic Peninsula. Nat. Commun. 12 (1), 1–12. doi: 10.1038/s41467-021-27436-9
Pauli N.-C., Metfies K., Pakhomov E. A., Neuhaus S., Graeve M., Wenta P., et al. (2021b). Selective Feeding in Southern Ocean Key Grazers—Diet Composition of Krill and Salps. Commun. Biol. 4 (1), 1–12. doi: 10.1038/s42003-021-02581-5
Peng Y., Leung H. C. M., Yiu S. M., Lv M. J., Zhu X. G., Chin F. Y. L. (2013). IDBA-Tran: A More Robust De Novo De Bruijn Graph Assembler for Transcriptomes With Uneven Expression Levels. Bioinformatics 29 (13), 326–334. doi: 10.1093/bioinformatics/btt219
Razy-Krajka F., Stolfi A. (2019). Regulation and Evolution of Muscle Development in Tunicates. EvoDevo 10 (1), 1–34. doi: 10.1186/s13227-019-0125-6
R Core Team (2021). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (Vienna, Austria). Available at: https://www.r-project.org/.
Robertson G., Schein J., Chiu R., Corbett R., Field M., Jackman S. D., et al. (2010). De Novo Assembly and Analysis of RNA-Seq Data. Nat. Methods 7 (11), 909–912. doi: 10.1038/nmeth.1517
Ross R. M., Quetin L. B., Martinson D. G., Iannuzzi R. A., Stammerjohn S. E., Smith R. C.. (2008). Palmer LTER: Patterns of Distribution of Five Dominant Zooplankton Species in the Epipelagic Zone West of the Antarctic Peninsula 1993-2004. Deep Sea Res. Part II: Topical Stud. Oceanogr. 55 (18–19), 2086–2105. doi: 10.1016/j.dsr2.2008.04.037
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness With Single-Copy Orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Soneson C., Love M. I., Robinson M. D. (2016). Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 4, 1–23. doi: 10.12688/F1000RESEARCH.7563.2
Strass V. H., Leach H., Prandke H., Donnelly M., Bracher A. U., Wolf-Gladrow D. A. (2017). The Physical Environmental Conditions for Biogeochemical Differences Along the Antarctic Circumpolar Current in the Atlantic Sector During Late Austral Summer 2012. Deep Sea Res. Part II: Topical Stud. Oceanography 138, 6–25. doi: 10.1016/j.dsr2.2016.05.018
Tanaka K., Chiba T. (1998). The Proteasome : A Protein-Destroying Machine. Genes to Cells pp, 499–510. doi: 10.1046/j.1365-2443.1998.00207.x
Toseland A., Daines S. J., Clark J. R., Kirkham A., Strauss J., Uhlig C., et al. (2013). The Impact of Temperature on Marine Phytoplankton Resource Allocation and Metabolism. Nat. Climate Change 3 (11), 979–984. doi: 10.1038/nclimate1989
von Harbou L., Dubischar C. D., Pakhomov E. A., Hunt B. P. V., Hagen W. (2011). Salps in the Lazarev Sea, Southern Ocean: I. Feeding Dynamics. Marine Biol. 158 (9), 2009–2026. doi: 10.1007/s00227-011-1709-4
Voronina N. M. (1998). Comparative Abundance and Distribution of Major Filter-Feeders in the Antarctic Pelagic Zone. J. Marine Syst. 17 (1–4), 375–390. doi: 10.1016/S0924-7963(98)00050-5
Walsh J., Reiss C. (2020). Lipid Content and Stable Isotopes of Zooplankton During Five Winters Around the Northern Antarctic Peninsula. Sci. Data 7 (1), 1–6. doi: 10.1038/s41597-020-00722-9
Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., et al. (2019). Welcome to the Tidyverse. J. Open Source Softw. 4 (43), 1686. doi: 10.21105/joss.01686
Windisch H. S., Kathöver R., Pörtner H. O., Frickenhaus S., Lucassen M.. (2011). Thermal Acclimation in Antarctic Fish: Transcriptomic Profiling of Metabolic Pathways. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 301 (5), R1453–66. doi: 10.1152/ajpregu.00158.2011
Keywords: Southern Ocean, Salpa thompsoni, overwintering, aggregates, solitaries, season
Citation: Müller SJ, Michael K, Urso I, Sales G, De Pittà C, Suberg L, Wessels W, Pakhomov EA and Meyer B (2022) Seasonal and Form-Specific Gene Expression Signatures Uncover Different Generational Strategies of the Pelagic Tunicate Salpa thompsoni During the Southern Ocean Winter. Front. Mar. Sci. 9:914095. doi: 10.3389/fmars.2022.914095
Received: 06 April 2022; Accepted: 09 May 2022;
Published: 16 June 2022.
Edited by:
Sandie M. Degnan, The University of Queensland, AustraliaReviewed by:
Lucia Pita, Institute of Marine Sciences (CSIC), SpainCopyright © 2022 Müller, Michael, Urso, Sales, De Pittà, Suberg, Wessels, Pakhomov and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svenja J. Müller, c3ZlbmphLmp1bGljYS5tdWVsbGVyQHVuaS1vbGRlbmJ1cmcuZGU=; Bettina Meyer, YmV0dGluYS5tZXllckBhd2kuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.