 Zhanpeng Sun1,2†
Zhanpeng Sun1,2† Li Lv
Li Lv Bin Wang
Bin Wang Tong Hao
Tong Hao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 28 July 2022
Sec. Aquatic Physiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.900160
Discontinuous muscle growth during molting is an important feature of Eriocheir sinensis. Molting is a physiological process completed by the cooperation of multiple organs. Signal transmission is critical for the accurate regulation of each step in molting. However, the knowledge of the signal transduction mechanism in the molting process of E. sinensis is presently very limited. In this work, the chromatin accessibility and gene expression of the muscle in E. sinensis in pre-molt (D) and post-molt (A) stages were sequenced by assay of transposase accessible chromatin sequencing (ATAC-seq) and RNA-seq, respectively. The differentially expressed genes (DEGs) in the muscle before and after molting were analyzed by combining ATAC-seq and RNA-seq, especially the G-protein coupled receptor (GPCR) genes in the process of signal transduction. The results showed that there were 616 common DEGs in ATAC-seq and RNA-seq in A vs. D stages, of which 538 were upregulated and 78 were downregulated. In the 19 DEGs included in the signaling transduction process, 13 were located in the GPCR signaling pathway and all were upregulated in A stages, which indicated that GPCRs play a leading role in muscle signal transmission during post-molt stage in molting. In these genes, the structure of the proteins encoded by 10 membrane-located genes with transmembrane activity was further analyzed. Six candidate GPCR genes were finally identified and further verified by real-time quantitative PCR (qRT-PCR). The GPCRs include metabotropic glutamate receptor 7, Mth-like 4, and Mth2 proteins. These results show the existence of GPCRs in the muscle of E. sinensis and, for the first time, found their dominant role in the signal transduction process during molting. It provides important clues for the study of muscle discontinuous growth and molting mechanism of E. sinensis.
Eriocheir sinensis, also named as Chinese mitten crab and river crab, is an important aquatic economic animal (Wang et al., 2020a). It goes through more than 10 times of ecdysis throughout its life, and each time of ecdysis is associated by a sharp increase in body size, weight, and development. The molting cycle of E. sinensis is divided into four stages, namely, inter-molt (C stage), pre-molt (D stage, including D0, D1, and D3–4), molt (E stage), and post-molt (A–B stage). In D0 stage, that is, the early period of pre-molt stage, the epithelial tissue retracts and separates from the exoskeleton, resulting in a transparent gap. This period marks the start of molting. D1 is the period of new seta formation. At this time, the epithelial tissue is further separated from the exoskeleton, the epithelium begins to fold inward, and new seta is formed. D3–4 is the late period of pre-molt stage, in which the crab is about to molt. The epithelium retracts, invaginates, and folds to the greatest extent to form new seta, which is mature in this period (Kang et al., 2012). In the process of molting, muscles show discontinuous growth, which is one of the important characteristics during the growth of E. sinensis. Previous studies have shown that the molting process is mainly regulated by the antagonism between molting hormone and molting inhibitory hormone (Zhang et al., 2011). The tissue structure of muscles and the content of calcium and phosphorus in claw muscles of E. sinensis change with the molting cycle (Wang et al., 2003; Tian et al., 2017). The claw muscles atrophy and the protein synthesis rate increase before molting (Tian and Jiao, 2016). In the molecular level, the myostatin gene Ers-MSTN related to molting was found in muscles (Wei et al., 2015). In addition, a transforming growth factor-β type I receptor (EsTGFBRI) might play roles in molting-related muscle growth in E. sinensis. EsTGFBRI in claw and abdominal muscles in the later pre-molt stage were significantly higher than those in the inter- and post-molt stages (Tian et al., 2020). However, the molecular mechanism of muscle discontinuous growth during molting is still unclear.
Assay of transposase accessible chromatin sequencing (ATAC-seq) has gained particular attention since it was first described in 2013 (Buenrostro et al., 2013). ATAC-seq is a sequencing technology that uses transposase to study chromatin accessibility. It uses DNA transposase Tn5 to cut open DNA regions and high-throughput sequencing to study the open state of chromatin (Buenrostro et al., 2015). Compared with the traditional MNase-seq and DNase-seq, it is widely used because of its advantages of strong repeatability, simple experimental steps, and less experimental sample size. An exponential increase in curated ATAC-seq datasets and publications indicates its value in a wide spectrum of biological questions (Yan et al., 2020). In addition, RNA-seq can also reflect the gene expression in cells at specific time points to a certain extent (Ozsolak and Milos, 2011). The comparison of gene expression at different time points can provide important reference information for the study of gene regulation mechanism in cells. ATAC-seq and RNA-seq can complement each other well and have good applications in analyzing gene regulation and expression (Ackermann et al., 2016; Koehl et al., 2019; Yang et al., 2020).
The pre- and post-molt stages are two special periods of molting with great differences in the molting circle of E. sinensis. The pre-molt stage represents the initiation of molting, and the post-molt stage represents the termination of molting. In this work, ATAC-seq and RNA-seq were used to analyze the differential expressed genes (DEGs) and their related chromatin open regions in E. sinensis between D and A stages, with emphasis on G protein-coupled receptors (GPCRs), to investigate the differences of signaling regulation between the initiation and termination process of molting. GPCRs are physiologically important membrane proteins that sense signaling molecules such as hormones and neurotransmitters, which widely spread in many organisms and participate in the regulation of various cellular activities (Venkatakrishnan et al., 2013; Wingler and Lefkowitz, 2020). About 800 GPCRs have been found in the human genome (Fredriksson et al., 2003). In many animals, the sequence of GPCR is quite conservative (Weis and Kobilka, 2014). However, knowledge of its roles in crustaceans is presently very limited. Here, we identified candidate GPCRs in the muscles of E. sinensis that may play roles in the molting process. The results provide important information for further exploring the molecular regulation mechanism of muscle discontinuous growth in E. sinensis and its role in the process of molting.
The healthy juvenile Chinese mitten crabs were taken from Tianjin Xieyuan Aquaculture Co., Ltd. The average body length, body width, and weight of crabs were 24.28 ± 1.54 mm, 26.97 ± 1.65 mm and 8.41 ± 1.47 g, respectively. The crabs were cultured in a plastic incubator (70 × 40 × 50 cm) at 20 ± 1°C with natural light for more than 7 days. The crabs were fed compound feed twice a day. The daily feeding amount was about 5% of the body weight. The siphon method was used to clean up the residual bait and inject new water. The daily water change is more than 50%. Three crabs were selected to collect the muscle samples from pre- and post-molt stages, respectively. The muscle tissues in the walking legs of crabs in pre- and post-molt stages were collected. The post-molt stage was determined by observing the crab’s exoskeleton. In the post-molt stage, the crab’s body is still soft, and the exoskeleton has not been completely calcified, which are obvious features that can be determined by direct observation. The pre-molt stage was determined with microscope observation. The third maxilliped was cut carefully with anatomical scissors and placed downward on a glass slide with clean water and then observed with an Olympus (OCKX41, Japan) optical microscope (magnification of 200–400×). The most obvious feature of the pre-molt stage is the separation of epithelium and exoskeleton, forming a transparent blank area, which marks the start of animal molting (Kang et al., 2012). Therefore, when the blank area between the epithelium and the exoskeleton is observed under the microscope, it is considered that the crab is in the pre-molt stage (Supplementary Figure S1). The crabs in pre- and post-molt stages were put on the ice plate for low-temperature anesthesia. The muscle tissue in the walking legs of the crabs was quickly collected and put in liquid nitrogen immediately and then preserved in a −80°C refrigerator. The muscle in one crab was separated into two samples for ATAC-seq and RNA-seq, respectively. Cell activity of the muscle sample was detected with Trypan blue assay and counted.
Six muscle samples were used for ATAC-seq, numbered as Mu_A_1, Mu_A_2, and Mu_A_3 (samples for post-molt stages) and Mu_D_1, Mu_D_2, and Mu_D_3 (samples for pre-molt stages). ATAC-seq was performed as previously reported (Corces et al., 2017). Briefly, nuclei were extracted from muscle samples, and the nuclei pellet was resuspended in the Tn5 transposase reaction mix. The transposition reaction was incubated at 37°C for 30 min. Equimolar adapter 1 and 2 were added after transposition. PCR was then performed to amplify the library. After the PCR reaction, libraries were purified with the AMPure beads, and library quality was assessed with Qubit. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina NovaSeq platform, and 150 bp paired-end reads were generated. Nextera adaptor sequences were first trimmed from the reads using a skewer (0.2.2). Q20 and Q30 of the raw data and clean data were calculated. These reads were aligned to the reference genome downloaded from the NCBI BioProject database (accession number PRJNA555707) using BWA, with standard parameters. These reads were then filtered for high-quality (MAPQ ≥ 13), non-mitochondrial chromosome and properly paired reads (longer than 18 nt). All peak calling was performed with MACS2 using “MACS2 call-peak – nomodel – keepdup all – call-summits.” For simulations of peaks called per input read, aligned and de-duplicated BAM files were used without any additional filtering.
Differential analysis of the two molting stages was performed using the DESeq2 R package (1.20.0). The resulting p-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Peaks with p-value ≤ 0.05 and |log2.FoldEnrich| ≥1 were assigned as differential peaks.
ChIPseeker software was used to stat the distribution of peak in each functional area. The correspondence between peak and each functional area is in the priority order of promoter, untranslated region (UTR), UTR, exon, intron, downstream TTS, and distal intergenic regions. That is, if the position of a peak may be the exon of one gene and the intron of another gene, according to the above priority, it will be considered as the exon rather than the intron. Goseq and KOBAS software were used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation for the genes associated with differential peaks.
Total RNA from E. sinensis muscle samples was sequenced using the Illumina high-throughput sequencing technology by Novogene Inc. The total RNA was extracted using the TRIzol method (Invitrogen) for transcriptome analysis. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations. Library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s protocols. After cluster generation, the library preparations were sequenced on an Illumina NovaSeq platform, and 150 bp paired-end reads were generated.
The original image data were transferred into sequencing data by base calling, which were defined as raw reads. Clean data (clean reads) were obtained by removing reads containing an adapter, reads containing poly-N, and low-quality reads from the raw data. At the same time, Q20, Q30, and GC contents of the clean data were calculated. All the downstream analysis was based on the clean data with high quality. Then, E. sinensis genome was downloaded from the NCBI BioProject database (accession number PRJNA555707) and used as the reference genome. Index of the reference genome was built using Hisat2 v2.0.5, and paired-end clean reads were aligned to the reference genome using Hisat2 v2.0.5. FeatureCounts v1.5.0-p3 was used to count the number of reads mapped to each gene. Then, FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. FPKM is the expected number of fragments per kilobase of transcript sequence per millions base pairs sequenced.
Differential expression analysis of two molting stages was performed using the DESeq2 R package (1.20.0). The resulting p-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with padj ≤ 0.05 and |log2.Fold_change| ≥ 1 were assigned as differentially expressed. Goseq and KOBAS software were used for GO and KEGG annotation for the DEGs. The ATAC-seq and RNA-seq data have been deposited to Gene Expression Omnibus (GEO) database with the accession number GSE206233.
The expression profile of the differential peak-related genes in ATAC-seq results was compared with the DEGs in RNA-seq results. The upregulated DEGs in RNA-seq were compared with the associated genes with the upregulated peaks in ATAC-seq. The downregulated DEGs in RNA-seq were compared with the associated genes with the downregulated peaks in ATAC-seq. The genes with the same expression profile in the two methods were taken for GO enrichment analysis.
TMHMM (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) (Krogh et al., 2001) was used for the prediction of the TMHs for the candidate GPCR genes. PreidictProtein (https://predictprotein.org/) (Bernhofer et al., 2021) and SWISS-MODEL (https://swissmodel.expasy.org/) (Waterhouse et al., 2018) were used to predict the secondary and three-dimensional structure of the proteins encoded by the candidate genes.
The expressions of candidate GPCRs were verified with real-time quantitative PCR (qRT-PCR). Three muscle samples were used in the qPR-PCR validation for each gene as the biological duplication, with each sample containing the mixed muscle tissues from three crabs to avoid individual differences. The primer sequences used in qRT-PCR are listed in Supplementary File S1. The RNA was extracted with TRIzol reagent (Invitrogen, San Diego, CA, USA). The reverse transcription of RNA was conducted with PrimeScriptTM RT Master Mix (Perfect Real Time) test kit (Takara, Japan). Fluorescence quantitative detection TB Green® Premix Ex TaqTM II(Tli RNaseH Plus)*2 test kit (Takara, Japan) was used for fluorescence quantitative detection. ABI Applied Biosystems was used in this experiment. The housekeeping gene is β-actin. The 20-μl reaction system contains 1 μl cDNA, 0.4 μl each primer (10 μM), 10 μl TB Green® Premix Ex TaqTM II (2×), 0.4 μl ROX Reference Dye (50×), and ddH2O up to 20 μl. The reaction program was set as 95°C for 30 s, followed by 40 cycles of 5 s at 95°C, 30 s at 60°C, and 60°C for 1 min. The data were quantitatively analyzed with 2–ΔΔCt method.
ATAC-seq was used to detect the landscape of genomic chromatin accessibility in the muscle during molting. After the raw data have been obtained from ATAC-seq, the data were trimmed to get the clean data for the following analysis. The result of quality control for raw and clean data is shown in Table 1. The clean data were then mapped to the genome, and the results are shown in Table 2. The mappability is above 80%, which is a quite high ratio. The unique mapped reads is 63% on average for all the samples. The results of mapped read distributions across the gene bodies and transcriptional start site (TSS) showed the good quality of the ATAC-seq (Figures 1A, B).
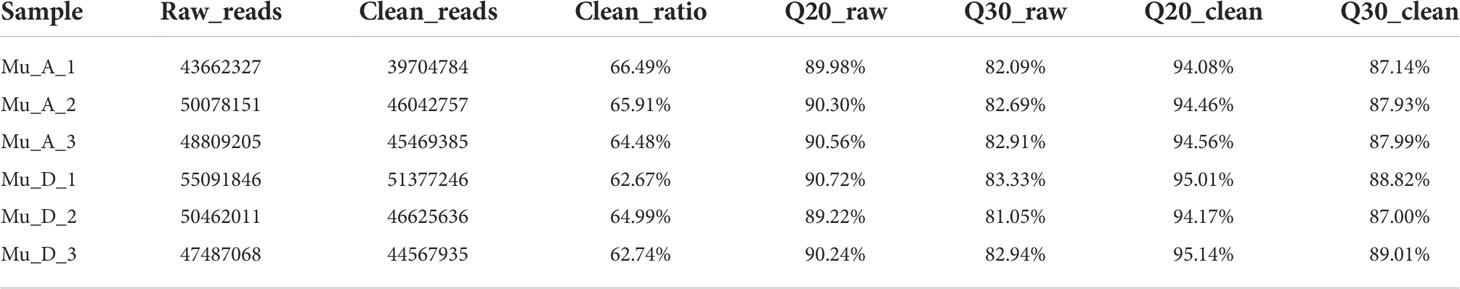
Table 1 Results of quality control for raw data and clean data.

Table 2 Results of mapping to reference genome and peak calling.
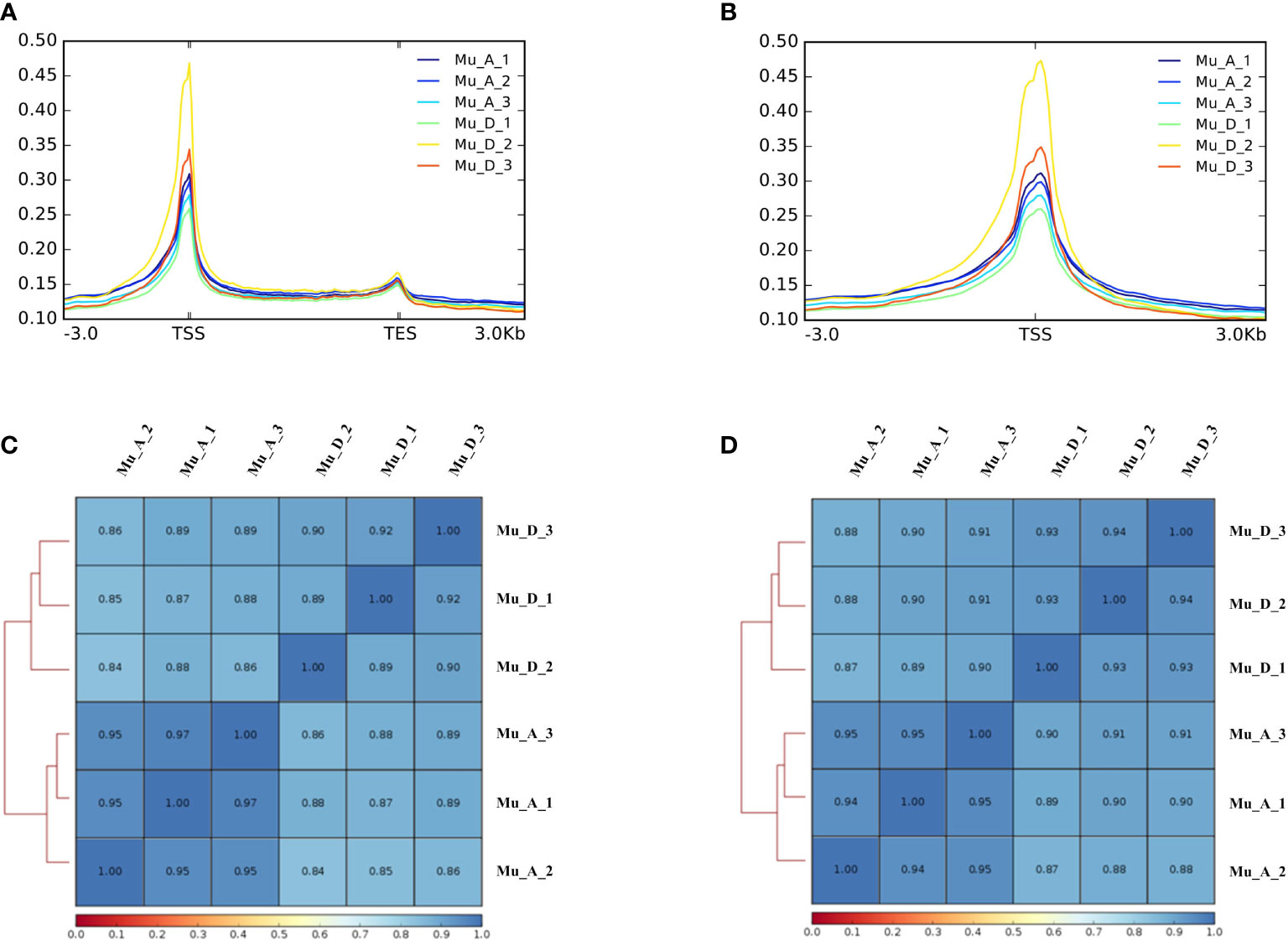
Figure 1 Results of the assay for ATAC. A represents the post-molt stage, and D stands for the pre-molt stage. (A) Mapped read distributions across the gene bodies; (B) mapped read distributions across the TSS; (C) heatmap for Pearson correlation analysis; (D) heatmap for Spearman correlation analysis.
To further confirm the quality of ATAC-seq, Pearson and Spearman correlation analysis was performed based on the signals of merged peaks from all samples. The closer the correlation coefficient is to 1, the higher the similarity of expression patterns between samples. As shown in Figures 1C, D, the similarity of the three samples in each group (D and A group) was quite high. After the mapped reads were obtained, the duplicate reads were deleted, and the peaks were called. The results of peak calling are shown in Table 2.
The “FoldEnrich” values in the two groups (D and A groups) were compared, and 7,163 differential peaks were found, in which 3,404 were upregulated and 3,759 were downregulated. The functional regions of each peak on the genome were annotated. The genome-wide functional regions were divided into promoter, UTR, exon, intron, downstream gene start site (TTS), and distal intergenic regions. The TSS are usually enriched in the promoter region. The functional distribution of differential peaks is shown in Figure 2. The distal intergenic and intron regions took the largest part in the differential peaks. The percentages of peaks enriched near the TSS region were 14% and 15% in all the up- and downregulated peaks.

Figure 2 The genome-wide distribution of differential peaks.
The results of GO enrichment analysis for the differential peak-related genes showed that in the biological process, these genes were mainly concentrated in the regulation of metabolic process and gene expression; in the cellular component, the genes were mainly located in the membrane and extracellular; and in the molecular function, the genes were enriched in the catalytic activity, phosphotransferase activity, and protein kinase activity. In the KEGG enrichment analysis, a large number of genes were distributed in the endocytosis, protein processing in endoplasmic reticulum, purine metabolism, and many signaling pathways (Figure 3). The enriched functions of these differential peak-related genes indicate that plenty of specific signaling regulations occur in the muscle during molting, and the regulatory process mainly takes place in the membrane and extracellular. In addition, the large amount of genes with catalytic activity reflects that the metabolic process also undergoes great changes.
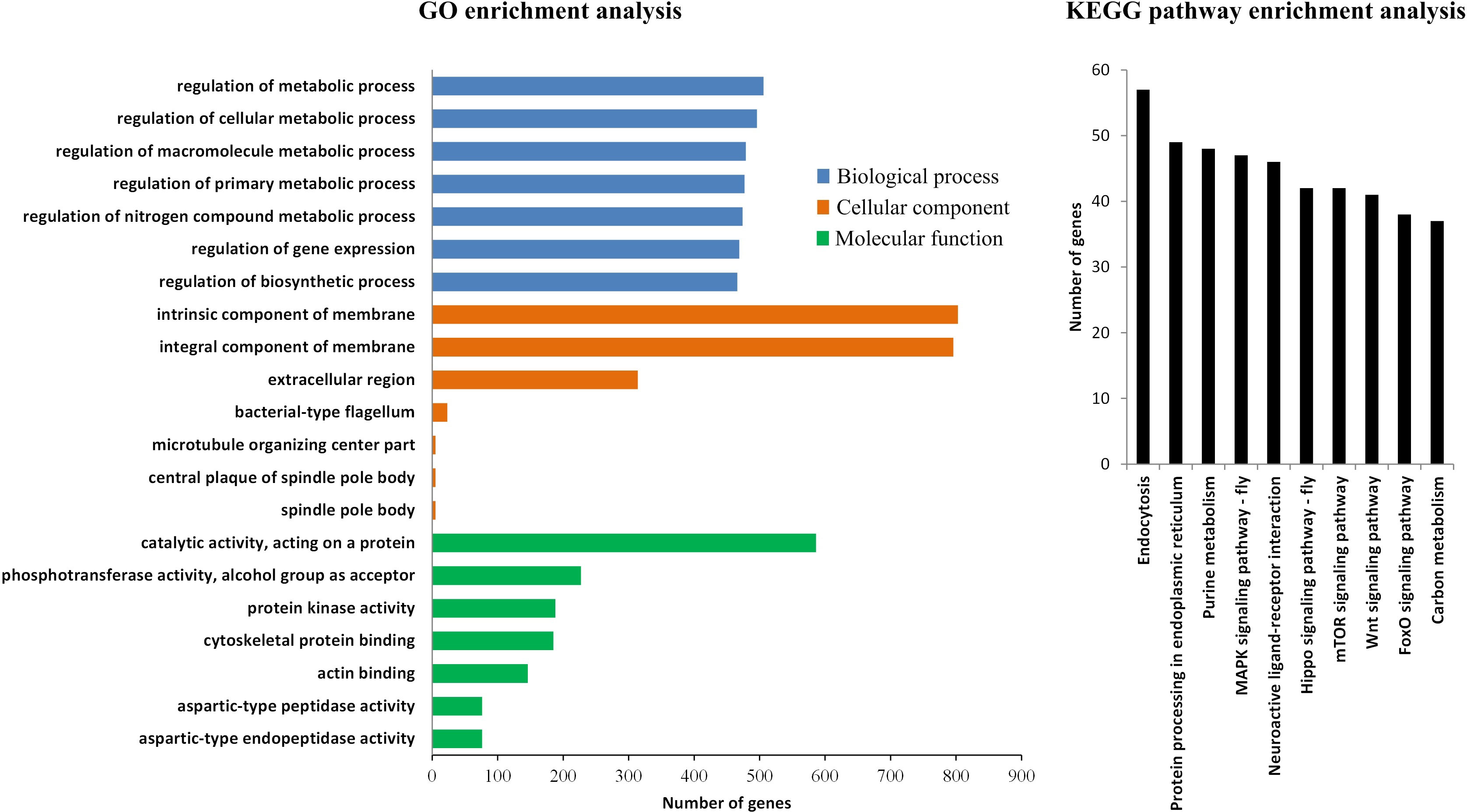
Figure 3 The GO and KEGG pathway enrichment analysis of the differential peak related genes.
Total RNA from muscles in pre- and post-molt stages was extracted to obtain the E. sinensis transcriptome data. The RNA samples had no genomic contamination, protein and impurity contamination, and color abnormality. The final quality testing result was level A, which was the highest level for eukaryotic transcriptome RNA. Total RNA was used as the input material for the RNA sample preparations and then sequenced. The results of RNA-seq are shown in Table 3. The average percentages at Q20 and Q30 of all the samples are 95.97% and 90.68%, respectively. The average GC content is 55.08%. More than 80% of the clean reads of each sample were mapped to E. sinensis genome. The distribution of the mapped reads on the genome is shown in Figure 4. The proportion of reads aligned to the exon region was the highest in all the samples. The reads aligned to the intron region may come from the precursor mRNA or the intron retained by variable splicing events. The reads aligned to the intergenic region may be influenced by ncRNA or a few DNA fragments, or the imperfect genome annotation.
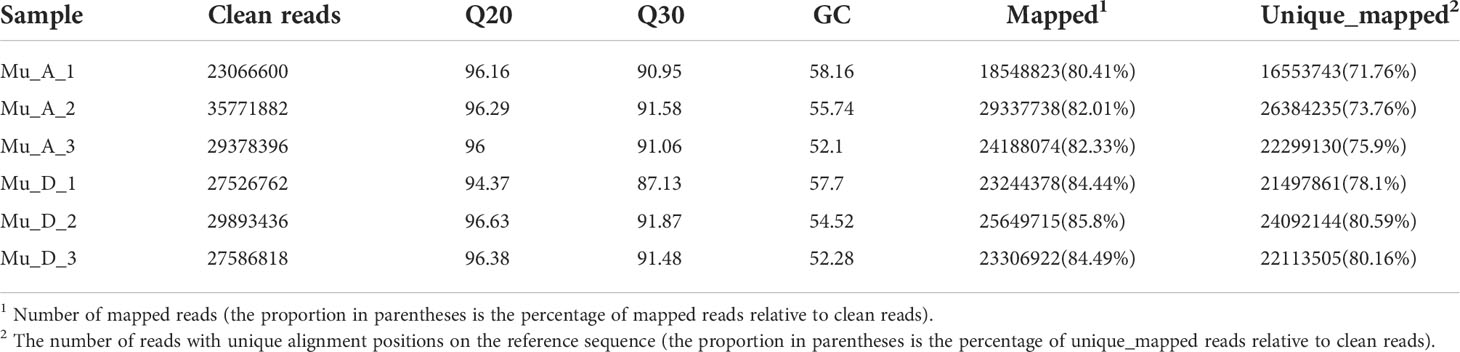
Table 3 Results of RNA-seq.
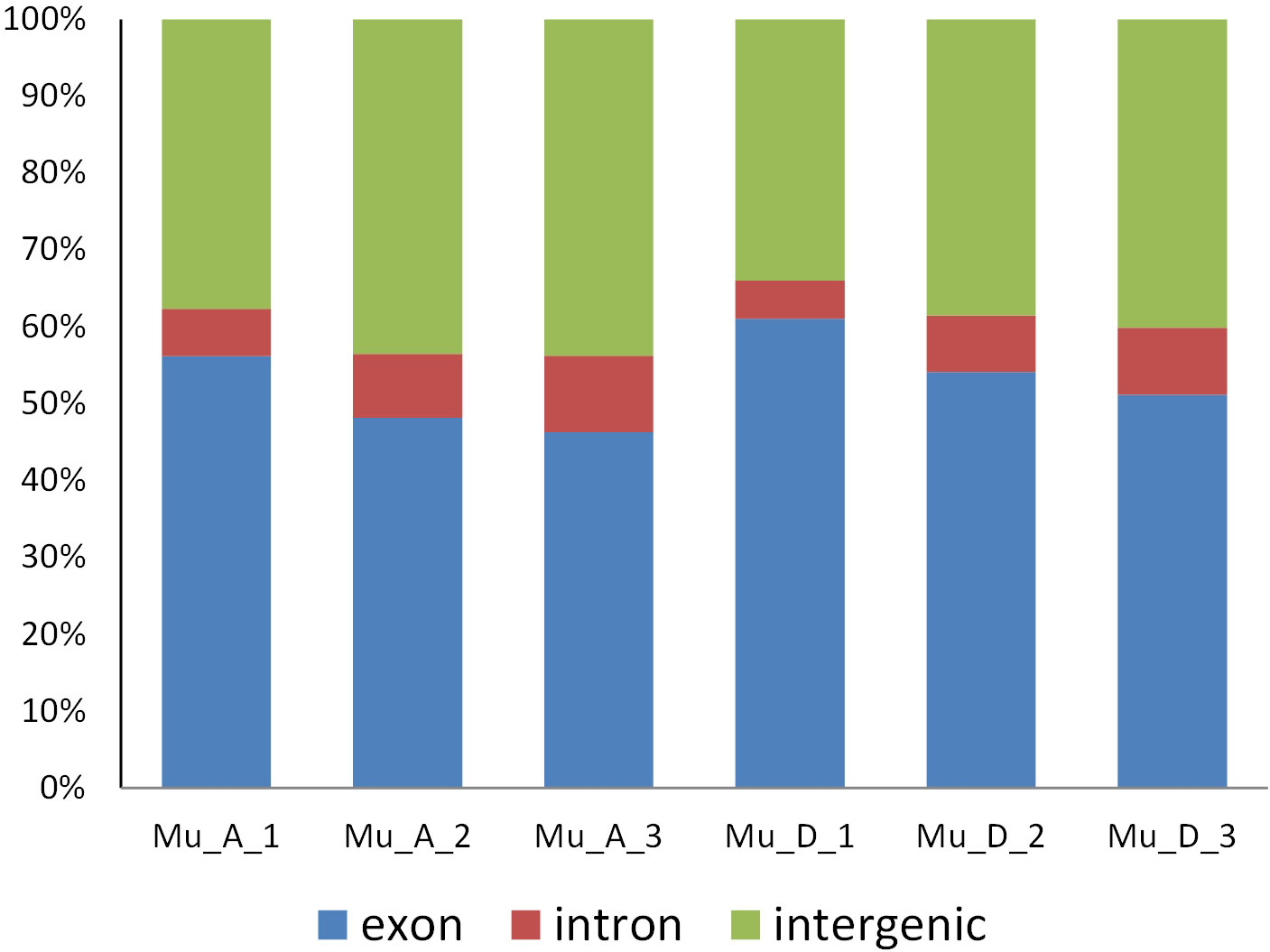
Figure 4 Distribution of mapped reads on E. sinensis genome.
In the differential analysis, a total of 1,684 genes were found to be differentially expressed in the A vs. D group, in which 1,388 were upregulated and 296 were downregulated. Through the GO enrichment analysis of these DEGs, it was found that in the biological process, the DEGs were mainly concentrated in several metabolic processes, biological regulation, and gene expression; in the cellular components, the DEGs were mainly located in the membrane; and in the molecular function, the DEGs were mostly enriched in binding, catalytic activity, protein binding, and structural molecule activity. In the KEGG pathway enrichment analysis, lots of genes were enriched in ribosome, amino sugar and nucleotide sugar metabolism, carbon metabolism, biosynthesis of amino acids, purine metabolic, and mitogen-activated protein kinase (MAPK) signaling pathway (Figure 5). The enriched functions of the DEGs reflect that the metabolic process changes a lot during molting. In addition, some specific signaling regulations such as MAPK signaling pathway were quite different in pre- and post-molt stages.

Figure 5 The GO and KEGG pathway enrichment analysis of the DEGs.
The associate genes related to differential peaks from ATAC-seq were compared with the DEGs from RNA-seq. Finally, 784 common genes were found, in which 616 had the same expression profile (Table 4). The other 168 genes were common but inconsistent in expression profile, in which 22 genes were upregulated in ATAC-seq but downregulated in RNA-seq, and 78 genes were downregulated in ATAC-seq but upregulated in RNA-seq. Compared to D stage, most of the DEGs were upregulated in A stage. The GO enrichment analysis of the consistent DEGs in biological process, molecular function, and cellular components is shown in Figure 6. The metabolic process contains the most DEGs, most of which are concentrated in organic substance, nitrogen compound, and macromolecule metabolic processes. The regulation of cellular process contains 44 differential genes, including regulation of metabolic process, gene expression, and transcription. In terms of the cellular component, the most DEGs were distributed in the cell membrane. For the molecular function, the number of DEGs with protein binding, ion binding, and catalytic activities is the largest. All these indicate that the expression levels of many genes involved in metabolism, information transmission and transcriptional regulation have changed during molting.

Table 4 Comparison of DEGs from ATAC-seq and RNA-seq.

Figure 6 GO enrichment analysis of consistent DEGs.
The molting process of E. sinensis is a physiological process completed by the coordination of multiple organs. Different organs exchange information through signal transmission at each molting stage, so as to accurately control the activities and launch time of multiple organs participating in the molting process. In order to explore the molecular regulation mechanism on the change of muscles during molting, it is important to investigate its signaling transduction process. Therefore, among the DEGs between D and A stages, we focused on the genes related to the signaling transduction. According to GO enrichment analysis, in the consistent DEGs, 19 genes were annotated to the “signal transduction (GO:0007165),” of which 13 are located in the G-protein-coupled receptor signaling pathway. Further analysis on the molecular function of these genes showed that they all had transmembrane signaling receptor activity, of which 10 genes were located on the membrane. These genes were all upregulated, which suggests that some specific GPCRs may play an important role in the process of receiving and transmitting signals in the post-molt stage in muscle.
GPCRs mediate the majority of cellular responses to external stimuli, including light, odors, hormones, and growth factors. GPCRs are integral membrane proteins that contain seven transmembrane α-helices (TMHs) (Weis and Kobilka, 2014). In order to further analyze whether the above 10 membrane-located genes encode GPCRs, TMHMM, PredictProtein, and SWISS-MODEL were used to predict their transmembrane structure, secondary structure, and three-dimensional structure, respectively. The prediction results and FPKM of these genes are shown in Table 5 (Supplementary Figures S2–4). According to the analysis of transmembrane structure predicted by TMHMM, five of these genes encode proteins with seven or more TMHs. Through the analysis of secondary structure and three-dimensional structure, U165 and U196 have seven TMHs, which is consistent with the prediction results of TMHMM. U116 is predicted to have seven TMHs according to TMHMM and PredictProtein, while it is predicted by SWISS-MODEL to have two adjacent seven-TMH structures, which is consistent with the characteristics of family C GPCRs. Family C GPCRs are unusual in that, besides the GPCR-defining seven-THM domain, they possess relatively large amino-terminal extracellular domains that form obligate dimers (Koehl et al., 2019). The protein encoded by U120 is predicted to have eight TMHs by TMHMM, but in the prediction of the PredictProtein and SWISS-MODEL, only seven of the α-helices are THMs; the other one does not have the transmembrane characteristics. The protein encoded by U258 is predicted to have eight THMs according to TMHMM. While similar as U120, in the predictions of PredictProtein and SWISS-MODEL, only seven α-helices are TMHs. In addition, although U33 is predicted to have six THMs by TMHMM and SWISS-MODEL, it should have seven THMs according to PredictProtein. It is worth noting that the similarity between the reference template sequence and U33 is <25% in SWISS-MODEL prediction. Therefore, the three-dimensional structure prediction results of U33 is of little reference value. There are a total of six genes predicted to have seven THMs by at least one sort of software. Therefore, we considered these genes as the candidate GPCRs for further experimental validation. The other four proteins (encoded by U519, U241, U9, and U387) are excluded as GPCRs, as they are impossible to have seven THMs in the prediction results of all the three sort of software.

Table 5 Prediction of TMHs and FPKM for the candidate genes.
The above six candidate GPCRs were verified by qRT-PCR. As Figure 7 shows, all the seven genes were expressed in both A and D stages. The relative expression levels of all these genes in the A stage were higher than those in the D stage, showing an upregulated trend in A vs. D group, which was consistent with the results from ATAC-seq and RNA-seq. The ratio of standard deviation to mean value is in the range of 12%–26%, which is in a reasonable scale. qRT-PCR results verified the results of high-throughput sequencing. Therefore, these proteins can further verify their role as GPCRs in muscle discontinuous growth during molting by gene knockout in subsequent experiments.
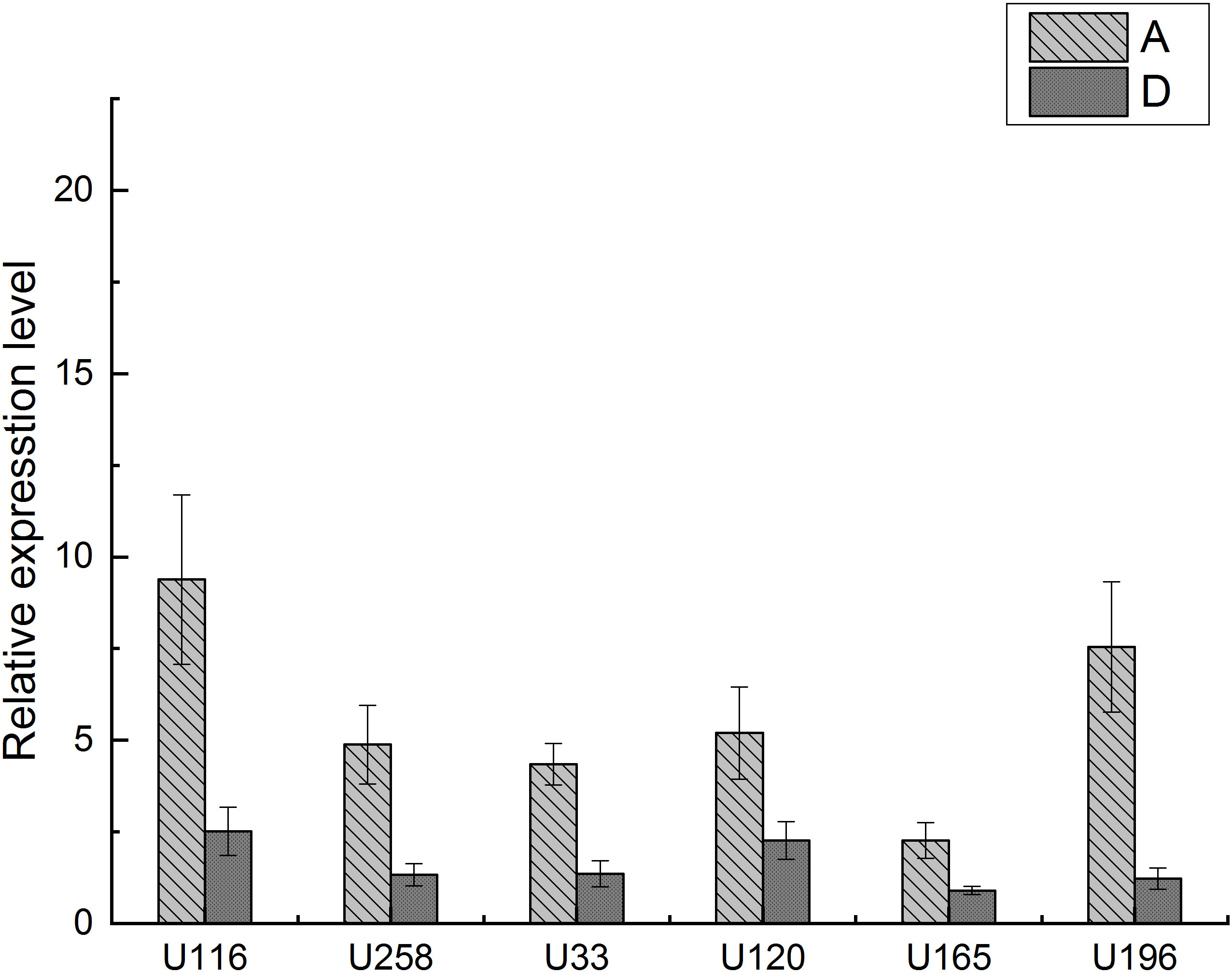
Figure 7 Relative expression levels of the DEGs. X-axes are the gene codes of the DEGs, and Y-axes are the relative expression levels of the DEGs. A represents post-molt group, and D represents pre-molt group.
Molting is a critical developmental process for crustacean, which is a complex process regulated by the combination of multiorgans. The molting frequency directly influences the growth rate of E. sinensis. Currently, many organs have been found to participate in the molting process, such as hepatopancreas (Huang et al., 2015), eyestalk (Li et al., 2016), muscle (Wang et al., 2003; Tian et al., 2017), Y organ (Stewart et al., 2013), etc. Some genes were also reported to be involved in the molting process (Zhang et al., 2011; Li et al., 2015). However, the regulation mechanism of the molting process is still unclear. Muscle is one of the organs with obvious changes in the molting process. The study of the mechanism on the initiation and termination of molting in the muscle is important to understand and control the molting cycle of E. sinensis. Presently, the research on muscle changes mostly focuses on the observation and analysis at the tissue level. There is little research at the molecular level, especially for the research on the signal transmission of muscle in the molting process.
In this work, the chromatin accessibility and gene expression of muscle during pre- and post-molt stages were investigated with ATAC-seq and RNA-seq, respectively. ATAC-seq is used for the detection of chromatin accessibility, which reflects the transcriptional activity of chromatin. It is an important method to study the regulation of gene expression and has been widely used in human, plant, animals, and yeast (Hendrickson et al., 2018; Liu et al., 2019; Shashikant and Ettensohn, 2019; Yang et al., 2020). The precise identification of specific open DNA regions in the genome is not only capable of exploring the regulatory elements in the genome but also an effective way to analyze the genes expression by identifying the peak-related genes. The integration of ATAC-seq and RNA-seq is capable of providing more accurate expression profile of genes and proposing more instructive suggestions based on the high-throughput data analysis. It also supplies a better solution for the in-depth analysis of the mechanism on cellular activities in the process of biological growth and development (Lowe et al., 2019). There have been some related studies on the integration of ATAC-seq and RNA-seq to analyze the gene expression of various organisms. For example, Yang et al. established the differential expression profile of light-induced primordia formation in Sparassis latifolia by integrating the assay for ATAC-seq and RNA-seq (Yang et al., 2020). Ackermann et al. determined the genetic expression of human alpha- and beta-cells based on chromatin accessibility and transcript levels, which allowed for the detection of novel alpha- and beta-cell signature genes not previously known to be expressed in islets (Ackermann et al., 2016). In this work, ATAC-seq and RNA-seq techniques were used to analyze the muscle samples in the pre- and post-molt stages of molting circle and determine the specific genes functioning in the muscle before and after molting.
The results of enrichment analysis based on ATAC-seq and RNA-seq reflect that the metabolism and regulation in muscle both undergo great changes during molting. Some enriched GO annotations and KEGG pathways obtained by these two methods are consistent. Lots of the DEGs were enriched in biological regulation process. Most DEGs were located in the membrane, and many of them have catalytic and binding activities. For the pathway distribution, the DEGs were commonly enriched in endocytosis, protein processing in endoplasmic reticulum, carbon metabolism, purine metabolism, and MAPK signaling pathway. The enrichment of DEGs in the biological regulation process and signaling pathway indicates that the regulation mechanisms in pre- and post-molt stages are quite different. The initiation and termination of molting are regulated by different regulatory elements.
The integration analysis of ATAC-seq and RNA-seq showed 538 upregulated genes and 78 downregulated genes, which indicates that more specific genes were active in post-molt stage. The result of GO analysis showed that these DEGs were distributed in a variety of biological process, especially in regulation of metabolic process, gene expression, and transcription. Most DEGs were located in the membrane with protein binding, ion binding, or catalytic activity. In the results of ATAC-seq and RNA-seq, the common DEGs with the same expression profile account for 78.57% (616/784) of all the common DEGs identified in the two methods. This indicates that ATAC-seq and RNA-seq may have a common problem of high-throughput sequencing, that is, false positive results. It further demonstrates that the integration analysis of the two high-throughput sequencing techniques can effectively reduce false positives and obtain more accurate results.
In order to explore the signal transduction mechanism of muscles in the molting process, the DEGs in the signal transduction process were further analyzed. We found that in the 19 DEGs annotated to the signal transduction process, 13 are located in the G-protein-coupled receptor signaling pathway and all upregulated. This result shows that GPCRs play a leading role in muscle signal transmission during post-molt stage. The other six genes are zonadhesin, ankyrin-3, probable nuclear hormone receptor, Toll-like receptor Tollo, hormone receptor, and mitochondrial uncoupling protein 4. Ankyrin-3 and Toll-like receptor Tollo are upregulated in D stage, which indicates that they are specifically active in pre-molt stage, while the other four genes are upregulated in A stage like GPCRs, indicating that they are specifically functioning in post-molt stage.
Currently, there are few studies on the existence of GPCR in E. sinensis. Only Xu et al. cloned a gene belonging to the GPCR family named EsGPCR89 in E. sinensis and found that it played roles in cerebral antimicrobial function via hemocytes (Qin et al., 2019). In terms of the signal transduction, Tian et al. suggested that a transforming growth factor-β type I receptor, named EsTGFBRI, probably play roles in the molting-related muscle growth in E. sinensis (Wei et al., 2015). Wang et al. cloned an activin receptor IIB (ActRIIB) gene in E. sinensis and showed that ActRIIB signaling pathway might be involved in the regulation of growth mass and protein and lipid metabolism in muscle during molting (Wang et al., 2020b). Some studies have found that ecdysone signal transduction in Drosophila is mediated by GPCR (Srivastava et al., 2005), but there is no such research in crustaceans. In this work, it is found for the first time that GPCRs are involved in the molting process of E. sinensis and play an important role in muscle signal transduction.
As a result, six candidate GPCR genes (U116, U165, U196, U120, U258, and U33) were identified by structural analysis and qRT-PCR verification after the integration analysis of ATAC-seq and RNA-seq. U116 encodes metabotropic glutamate receptor 7, which is a GPCR activated by glutamate. Its ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of downstream effectors in human (Wu et al., 1998; Song et al., 2021). U258 is a probable GPCR Mth-like 4 gene, which were found negatively regulating oxidative stress in Tribolium castaneum (Li et al., 2014). Besides, methuselah (Mth) is associated with lifespan, stress resistance, and reproduction in Drosophila melanogaster (Araujo et al., 2013). U165 and U196 are both GPCR Mth2 genes, which are involved in maintaining genome stability in human cells against oxidative stress (Hashiguchi et al., 2018). U33 and U120 were predicted to be GPCRs by GO annotation, while their specific functions have not been determined. The effects of these candidate GPCR genes can be further determined by gene knockout to analyze their specific functions during molting.
Although the effect of GCPRs on ecdysis is unclear in E. sinensis, it has been explored in other animals. Bai et al. identified 111 GPCRs through genome analysis of T. castaneum and screened 8 GPCR encoding genes through large-scale RNA interference experiments. The knockdown of these GPCRs resulted in serious development stagnation and molting failure, which shows that GPCRs are important for molting and development of T. castaneum. These eight GPCRs include D2R, cirl, Stan, mthl, FZ, SMO, bursicon, and ccapr (Bai et al., 2011), where mthl is encoded by U258 in this study. In addition, the 111 GPCRs identified by them also include several metabotropic glutamate receptors, which are encoded by U116 in this study. Zhao et al. found that GPCRs are related to the regulatory function of 20-hydroxyecdysone (20E) through the study on the Lepidoptera insect Helicoverpa armigera (Cai et al., 2014). GPCRs control the transduction of 20E in cell membrane, transmit 20E signal, and control 20E to enter cells (Wang et al., 2015). In another study, they found that 20E activates phospholipase Cγ1 via the GPCRs and cytosolic tyrosine kinases to regulate Ca2+ influx and protein kinase C phosphorylation of transcription factor ultraspiracle and subsequently influence gene transcription for molting and metamorphosis (Liu et al., 2014). These studies provide clues for the further study of the GCPRs in E. sinensis.
In addition, through RNA-seq analysis, the researchers also found the existence of GPCRs in the Y-organ of Gecarcinus lateralis (Tran et al., 2019) and the central nervous system tissue and Y-organ and epidermal tissue of Carcinus maenas (Oliphant et al., 2018), indicating that GPCR may be a widely existing regulatory element in crabs, but their regulatory mechanism in crabs is still unclear.
The study of intramuscular signal transduction mechanism is the key factor to analyze the mechanism of molting and muscle discontinuous growth. In this work, ATAC-seq and RNA-seq were used to jointly analyze the chromosome accessibility and gene expression in the muscle of E. sinensis in pre-and post-molt stages, and the DEGs in the two periods were determined. Through the analysis of signal transduction process, it was found that the regulation in initiation and termination of molting in the muscle is quite different, and GPCRs play a leading role in the signal transduction of the muscle during post-molt stages. Six candidate GPCRs were identified by structural analysis and qRT-PCR validation. The results of this work show the advantages of integration analysis with different high-throughput experiments. Furthermore, it provides important clues for analyzing the intramuscular signal transduction mechanism in the molting process and supplies significant information for the in-depth study of the molting mechanism in E. sinensis.
The data presented in the study are deposited in the GEO DataSets, accession number GSE206233.
The animal study was reviewed and approved by Tianjin Normal University.
JL generated the data. ZS organized the data. BW, LL, and YG analyzed the data. JL and ZS wrote the manuscript. TH designed the experiment and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Research and Development Program of China (2018YFD0901301), National Natural Science Foundation of China (31770904), Tianjin Development Program for Innovation and Entrepreneurship team (ITTFRS2017007), Program for Innovative Research Team in University of Tianjin (TD13-5076), and Doctoral Foundation of Tianjin Normal University (52XB1915).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.900160/full#supplementary-material
Supplementary Table 1 | The primer sequences used in qRT-PCR.
Supplementary Figure 1 | The microscope observation of the third maxilliped in pre-molt stage.
Supplementary Figure 2 | The TMHs of proteins encoded by candidate genes predicted by TMHMM.
Supplementary Figure 3 | The secondary structure of proteins encoded by candidate genes predicted by PreidictProtein.
Supplementary Figure 4 | The three-dimensional structure of proteins encoded by candidate genes predicted by SWISS-MODEL.
Ackermann A. M., Wang Z., Schug J., Naji A., Kaestner K. H. (2016). Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 5 (3), 233–244. doi: 10.1016/j.molmet.2016.01.002
Araujo A. R., Reis M., Rocha H., Aguiar B., Morales-Hojas R., Macedo-Ribeiro S., et al. (2013). The drosophila melanogaster methuselah gene: A novel gene with ancient functions. PloS One 8 (5), e63747. doi: 10.1371/journal.pone.0063747
Bai H., Zhu F., Shah K., Palli S. R. (2011). Large-Scale RNAi screen of G protein-coupled receptors involved in larval growth, molting and metamorphosis in the red flour beetle. BMC Genomics 12, 388. doi: 10.1186/1471-2164-12-388
Bernhofer M., Dallago C., Karl T., Satagopam V., Heinzinger M., Littmann M., et al. (2021). PredictProtein - predicting protein structure and function for 29 years. Nucleic Acids Res. 49 (W1), W535–W540. doi: 10.1093/nar/gkab354
Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10 (12), 1213–1218. doi: 10.1038/nmeth.2688
Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J. (2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109 (1), 21.29.21–21.29.29. doi: 10.1002/0471142727.mb2129s109
Cai M. J., Dong D. J., Wang Y., Liu P. C., Liu W., Wang J. X., et al. (2014). G-Protein-coupled receptor participates in 20-hydroxyecdysone signaling on the plasma membrane. Cell Commun. Signal 12, 9. doi: 10.1186/1478-811X-12-9
Corces M. R., Trevino A. E., Hamilton E. G., Greenside P. G., Sinnott-Armstrong N. A., Vesuna S., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14 (10), 959–962. doi: 10.1038/nmeth.4396
Fredriksson R., Lagerström M. C., Lundin L.-G., Schiöth H. B. (2003). The G-Protein-Coupled receptors in the human genome form five main families. phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63 (6), 1256–1272. doi: 10.1124/mol.63.6.1256
Hashiguchi K., Hayashi M., Sekiguchi M., Umezu K. (2018). The roles of human MTH1, MTH2 and MTH3 proteins in maintaining genome stability under oxidative stress. Mutat. Res. 808, 10–19. doi: 10.1016/j.mrfmmm.2018.01.002
Hendrickson D. G., Soifer I., Wranik B. J., Kim G., Robles M., Gibney P. A., et al. (2018). A new experimental platform facilitates assessment of the transcriptional and chromatin landscapes of aging yeast. eLife 7, e39911. doi: 10.7554/eLife.39911
Huang S., Wang J., Yue W., Chen J., Gaughan S., Lu W., et al. (2015). Transcriptomic variation of hepatopancreas reveals the energy metabolism and biological processes associated with molting in Chinese mitten crab, Eriocheir sinensis. Sci. Rep. 5 (1), 14015. doi: 10.1038/srep14015
Kang X., Tian Z., Wu J., Mu S.. (2012). Molt stages and changes in digestive enzyme activity in hepatopancreas during molt cycle of Chinese mitten crab, Eriocheir sinensis. J. Fisheries. China 19 (5), 806–812. doi: 10.3724/SP.J.1118.2012.00806
Koehl A., Hu H., Feng D., Sun B., Zhang Y., Robertson M. J., et al. (2019). Structural insights into the activation of metabotropic glutamate receptors. Nature 566 (7742), 79–84. doi: 10.1038/s41586-019-0881-4
Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. L. (2001). Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by f. Cohen. J. Mol. Biol. 305 (3), 567–580. doi: 10.1006/jmbi.2000.4315
Li R., Tian J.-Z., Zhuang C.-H., Zhang Y.-C., Geng X.-Y., Zhu L.-N., et al. (2016). CHHBP: A newly identified receptor of crustacean hyperglycemic hormone. J. Exp. Biol. 219 (8), 1259–1268. doi: 10.1242/jeb.133181
Liu W., Cai M. J., Zheng C. C., Wang J. X., Zhao X. F. (2014). Phospholipase Cgamma1 connects the cell membrane pathway to the nuclear receptor pathway in insect steroid hormone signaling. J. Biol. Chem. 289 (19), 13026–13041. doi: 10.1074/jbc.M113.547018
Liu C., Wang M., Wei X., Wu L., Xu J., Dai X., et al. (2019). An ATAC-seq atlas of chromatin accessibility in mouse tissues. Sci. Data 6 (1), 65–65. doi: 10.1038/s41597-019-0071-0
Li X., Xu Z., Zhou G., Lin H., Zhou J., Zeng Q., et al. (2015). Molecular characterization and expression analysis of five chitinases associated with molting in the Chinese mitten crab, Eriocheir sinensis. Comp. Biochem. Physiol. Part B.: Biochem. Mol. Biol. 187, 110–120. doi: 10.1016/j.cbpb.2015.05.007
Li C., Zhang Y., Yun X., Wang Y., Sang M., Liu X., et al. (2014). Methuselah-Like genes affect development, stress resistance, lifespan and reproduction in tribolium castaneum. Insect Mol. Biol. 23 (5), 587–597. doi: 10.1111/imb.12107
Lowe E. K., Cuomo C., Voronov D., Arnone M. I. (2019). “Chapter 3 - using ATAC-seq and RNA-seq to increase resolution in GRN connectivity,” in Methods in Cell Biology. Eds. Hamdoun A., Foltz K. R. (Academic Press), 115–126.
Oliphant A., Alexander J. L., Swain M. T., Webster S. G., Wilcockson D. C. (2018). Transcriptomic analysis of crustacean neuropeptide signaling during the moult cycle in the green shore crab, carcinus maenas. BMC Genomics 19 (1), 711. doi: 10.1186/s12864-018-5057-3
Ozsolak F., Milos P. M. (2011). RNA Sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 12 (2), 87–98. doi: 10.1038/nrg2934
Qin X., Jin X., Zhou K., Li H., Wang Q., Li W., et al. (2019). EsGPCR89 regulates cerebral antimicrobial peptides through hemocytes in Eriocheir sinensis. Fish. Shellfish. Immunol. 95, 151–162. doi: 10.1016/j.fsi.2019.10.015
Shashikant T., Ettensohn C. A. (2019). Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 151, 219–235. doi: 10.1016/bs.mcb.2018.11.002
Song J. M., Kang M., Park D. H., Park S., Lee S., Suh Y. H. (2021). Pathogenic GRM7 mutations associated with neurodevelopmental disorders impair axon outgrowth and presynaptic terminal development. J. Neurosci. 41 (11), 2344–2359. doi: 10.1523/JNEUROSCI.2108-20.2021
Srivastava D. P., Yu E. J., Kennedy K., Chatwin H., Reale V., Hamon M., et al. (2005). Rapid, nongenomic responses to ecdysteroids and catecholamines mediated by a novel drosophila G-protein-coupled receptor. J. Neurosci. 25 (26), 6145–6155. doi: 10.1523/JNEUROSCI.1005-05.2005
Stewart M. J., Stewart P., Sroyraya M., Soonklang N., Cummins S. F., Hanna P. J., et al. (2013). Cloning of the crustacean hyperglycemic hormone and evidence for molt-inhibiting hormone within the central nervous system of the blue crab portunus pelagicus. Comp. Biochem. Physiol. Part A.: Mol. Integr. Physiol. 164 (2), 276–290. doi: 10.1016/j.cbpa.2012.10.029
Tian Z., Cheng Y., Wu X., Liu Q. (2017). Changes of histology, ultrastructure and main protein in muscles of Chinese mitten crab, Eriocheir sinensis during the molt cycle. Acta Hydrobiol. Sin. 41 (05), 1036–1041. doi: 10.7541/2017.129
Tian Z., Jiao C. (2016). Muscle atrophy and growth induced by molting in crustacean: A review. Aquacult. Fishery. 35 (05), 603–606. doi: 10.16378/j.cnki.1003-1111.2016.05.026
Tian Z., Peng H., Deng W., Jiao C. (2020). Identification of a transforming growth factor-β type I receptor transcript in Eriocheir sinensis and its molting-related expression in muscle tissues. Mol. Biol. Rep. 47 (1), 77–86. doi: 10.1007/s11033-019-05108-8
Tran N. M., Mykles D. L., Elizur A., Ventura T. (2019). Characterization of G-protein coupled receptors from the blackback land crab gecarcinus lateralis y organ transcriptome over the molt cycle. BMC Genomics 20 (1), 74. doi: 10.1186/s12864-018-5363-9
Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013). Molecular signatures of G-protein-coupled receptors. Nature 494 (7436), 185–194. doi: 10.1038/nature11896
Wang S., Wei Y., Shen D. (2003). Calcium and phosphorus levels in the muscle, hepatopancreas and carapace of Eriocheir sinensis in different stages of moulting cycle. J. Fisheries. China 03), 219–224. doi: 000-615(2003)03-0219-06
Wang B., Yang J., Gao C., Hao T., Li J., Sun J. (2020a). Reconstruction of Eriocheir sinensis y-organ genome-scale metabolic network and differential analysis after eyestalk ablation. Front. Genet. 11 (1179). doi: 10.3389/fgene.2020.532492
Wang J., Zhang K., Hou X., Yue W., Yang H., Chen X., et al. (2020b). Molecular characteristic of activin receptor IIB and its functions in growth and nutrient regulation in Eriocheir sinensis. PeerJ 8, e9673. doi: 10.7717/peerj.9673
Wang D., Zhao W. L., Cai M. J., Wang J. X., Zhao X. F. (2015). G-Protein-coupled receptor controls steroid hormone signaling in cell membrane. Sci. Rep. 5, 8675. doi: 10.1038/srep08675
Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–W303. doi: 10.1093/nar/gky427
Weis W. I., Kobilka B. K. (2014). The molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem. 87 (1), 897–919. doi: 10.1146/annurev-biochem-060614-033910
Wei W., Xugan W., Lei X., Qinqin Y., Yognxu C. (2015). Expression analysis of myostatin during molting cycle in Eriocheir sinensis. J. Shanghai. Ocean. Univ. 24 (05), 662–667. doi: 1674-5566(2015)05-0662-06
Wingler L. M., Lefkowitz R. J. (2020). Conformational basis of G protein-coupled receptor signaling versatility. Trends Cell Biol. 30 (9), 736–747. doi: 10.1016/j.tcb.2020.06.002
Wu S., Wright R. A., Rockey P. K., Burgett S. G., Arnold J. S., Rosteck P. R. Jr., et al. (1998). Group III human metabotropic glutamate receptors 4, 7 and 8: molecular cloning, functional expression, and comparison of pharmacological properties in RGT cells. Brain Res. Mol. Brain Res. 53 (1-2), 88–97. doi: 10.1016/s0169-328x(97)00277-5
Yang C., Ma L., Xiao D., Ying Z., Jiang X., Lin Y. (2020). Integration of ATAC-seq and RNA-seq identifies key genes in light-induced primordia formation of sparassis latifolia. Int J Mol Sci 21, 1, 185. doi: 10.3390/ijms21010185
Yan F., Powell D. R., Curtis D. J., Wong N. C. (2020). From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 21 (1), 22. doi: 10.1186/s13059-020-1929-3
Zhang Y., Sun Y., Liu Y., Geng X., Wang X., Wang Y., et al. (2011). Molt-inhibiting hormone from Chinese mitten crab (Eriocheir sinensis): Cloning, tissue expression and effects of recombinant peptide on ecdysteroid secretion of YOs. Gen. Comp. Endocrinol. 173 (3), 467–474. doi: 10.1016/j.ygcen.2011.07.010
Keywords: ATAC-seq, RNA-seq, Eriocheir sinensis, G-protein coupled receptor, molting
Citation: Sun Z, Li J, Lv L, Gou Y, Wang B and Hao T (2022) Integration of ATAC-seq and RNA-seq identifies active G-protein coupled receptors functioning in molting process in muscle of Eriocheir sinensis. Front. Mar. Sci. 9:900160. doi: 10.3389/fmars.2022.900160
Received: 20 March 2022; Accepted: 05 July 2022;
Published: 28 July 2022.
Edited by:
Khor Waiho, University of Malaysia Terengganu, MalaysiaCopyright © 2022 Sun, Li, Lv, Gou, Wang and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tong Hao, c2t5aHRAdGpudS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.