 Yu Pan
Yu Pan Zhicheng Sun
Zhicheng Sun Tianxiang Gao
Tianxiang Gao Linlin Zhao
Linlin Zhao Na Song
Na Song
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 18 May 2022
Sec. Marine Molecular Biology and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.894821
Acanthogobius ommaturus is a large, fast-growing annual fish widely distributed in coastal and estuarine areas. The adults will die after breeding, and its life cycle is only 1 year. The first chromosome-level genome assembly of A. ommaturus was obtained by PacBio and Hi-C sequencing in this study. The final genome assembly after Hi-C correction was 921.49 Mb, with contig N50 and scaffold N50 values of 15.70 Mb and 40.99 Mb, respectively. The assembled sequences were anchored to 22 chromosomes by using Hi-C data. A total of 18,752 protein-coding genes were predicted, 97.90% of which were successfully annotated. Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment results for genome and gene annotations were 93.6% and 84.6%, respectively. A. ommaturus is phylogenetically closely related to Periophthalmodon magnuspinnatus and Boleophthalmus pectinirostris, diverging approximately 31.9 MYA with the two goby species. The A. ommaturus genome displayed 597 expanded and 3,094 contracted gene families compared with the common ancestor. A total of 1,155 positive selected genes (PSGs) (p < 0.05) were identified. Based on comparative genomic analyses, we obtained several expanded genes such as acsbg2, lrp1, lrp6, and znf638 involved in lipid metabolism. A total of twenty candidate genes were identified under positive selection, which associated with lifespan including ercc6, igf1, polg, and tert. Interspecific collinearity analysis showed a high genomic synteny between A. ommaturus and P. magnuspinnatus. The effective population size of A. ommaturus decreased drastically during 200–100 Ka because of Guxiang ice age and then increased gradually following warm periods. This study provides pivotal genetic resources for in-depth biological and evolutionary studies, and underlies the molecular basis for lipid metabolism.
The Gobiidae is the largest family of marine fishes, which consists of more than 200 genera and nearly 2,000 species, and is characterized by wide distribution, high diversity, and strong adaptability (Tassell, 2011). Acanthogobius ommaturus is a large, demersal, and fast-growing annual fish in the family of Gobiidae, which is widely distributed in coastal waters or brackish waters of the Northwest Pacific Ocean surrounding China, Korea, Japan, and Indonesia (Wu and Zhong, 2008). Individuals of this species grow rapidly, and have a nearly linear growth curve in the first 6 months during the year of its life cycle, and then the growth rate slows down. The standard length and body weight will continue to increase for spawning by the next April, after which the parents die (Wang et al., 2011). Studies have shown that annual fish species usually have rapid growth and sexual maturation to maximize reproduction (Lanés et al., 2014; Lanés et al., 2016). Annual fish, whether male or female, need to invest a large amount of energy for reproduction (Berois et al., 2015; Godoy et al., 2019). The body weight of A. ommaturus can reach more than 450 g in 1 year (Chen, 1978), which is completely rare in annual fish (Jakub et al., 2021). This suggests that the species has a strong growth potential and energy storage capacity. In addition, as a demersal fish inhabiting coastal region and estuaries, A. ommaturus often faces environmental changes, such as light, temperature, salinity, and other factors. To cope with complex and changing living environments, A. ommaturus needs to store energy and allocate energy reasonably. However, the energy metabolism or storage of A. ommaturus, especially in molecular level, has not been well studied.
Lipids with their constituent fatty acids (FAs) and proteins are the major organic constituents in fish, while carbohydrates are much less abundant in fish. Actually, the protein content is much less than the lipid content, which reflects that lipids are usually the major source of metabolism energy in fish for growth, reproduction, and movement (Tocher, 2003). Notably, FAs are an important composition of lipids, participating in a wide variety of metabolic pathways (Watkins et al., 2007). NADPH (nicotinamide adenine dinucleotide phosphate) produced by the FA oxidation can provide metabolism energy in the form of ATP through the oxidation phosphorylation process (FrOyland, 2015). The capelin, herring, and salmonids prove that FAs are the preferred source of metabolic energy (Henderson et al., 1984a; Henderson et al., 1984b; Henderson and Almatar, 1989; Tocher, 2003). Lipid homeostasis is the balance between lipid uptake, storage, biosynthesis, transportation, metabolism, and catabolism (Tocher, 2003), which plays an important role in maintaining normal life activities of fish. The lipids are only stored in the liver in the majority of gobiids (Akiyoshi and Inoue, 2004; Cuevas et al., 2016; Louiz et al., 2018), and gobies can maintain a lifelong high level of fat storage without pathological changes caused by abnormal lipid metabolism, suggesting that there may be a special mechanism to maintain lipid homeostasis in these fishes. Hence, elucidating the lipid metabolism of A. ommaturus is of great value for understanding the evolution of energy metabolism in teleost and the pathogenesis of abnormal lipid metabolism.
A high-quality, complete, and contiguous genome is essential to analyze the biological characteristics of ecology and evolution (Ravi and Venkatesh, 2018; Bian et al., 2019). Examples are sex-determination mechanisms (Cai et al., 2021), loss of pelvic fin (Lin et al., 2016), loss of adaptive immunity (Star et al., 2011), and genome compaction (Aparicio et al., 2002). Although many fish genome data were previously released at NCBI, most of them were assembled based on short reads with limited contiguity and quality. Compared to second-generation sequencing technologies, third-generation sequencing technologies, such as PacBio (Pacific Biosciences, Menlo Park, CA, USA) and Nanopore (ONT, Oxford, UK), can produce long reads and avoid many gaps (Ge et al., 2019). These long-read assembly approaches can improve contiguity and span repetitive regions, which provides a path forward for genome assembly at a high level (Gordon et al., 2016). Additionally, the high-throughput chromosome conformation capture (Hi-C)-assisted genome assembly technique has been used to assemble chromosome-level genome for many fishes, such as Epinephelus lanceolatus (Zhou et al., 2019), Oplegnathus fasciatus (Xiao et al., 2019), Lota lota (Han et al., 2021), Micropterus salmoides (Sun C. et al., 2021), and Mugilogobius chulae (Cai et al., 2021).
The high-quality complete genomes of fish have proliferated in recent years. Gobies are an important group in the ecosystem as the bait of many fish, and are one of the most diverse families of vertebrates on earth (Tassell, 2011). Unfortunately, little genomic information on gobies is available, and only a few goby genomes reach chromosomal level. In this study, the chromosome-level genome assembly of A. ommaturus was constructed by combining Illumina short reads, Pacbio long reads, and Hi-C sequencing data, and its phylogenic relationships with other fishes were elucidated by comparative genome analysis. Based on genome data, we aimed to identify the lipid metabolism and candidate aging genes. This work may provide important resources to study the mapping of traits with economic importance, evolutionary position, and it provides an accurate reference sequence for related species.
All animal experiments were conducted in accordance with the guidelines and approval of the respective Animal Research and Ethics Committees of Ocean University of China. In addition, frost anesthesia was used to minimize the suffering of A. ommaturus specimens.
One male A. ommaturus fish (Figure 1A) was sampled in October 2020 from offshore of Qingdao, China with a body weight of 142.10 g and a body length of 26.70 cm. Fresh muscle was collected and quickly frozen in liquid nitrogen before storage at −80°. Total genomic DNA was extracted from fresh muscle of A. ommaturus with the standard phenol/chloroform method (Sambrock and Russel, 2001). The extracted DNA was measured using a Nanodrop 2000 (Thermo Scientific, USA) and a Qubit 2.0 (Invitrogen, USA) bioanalyzer system.
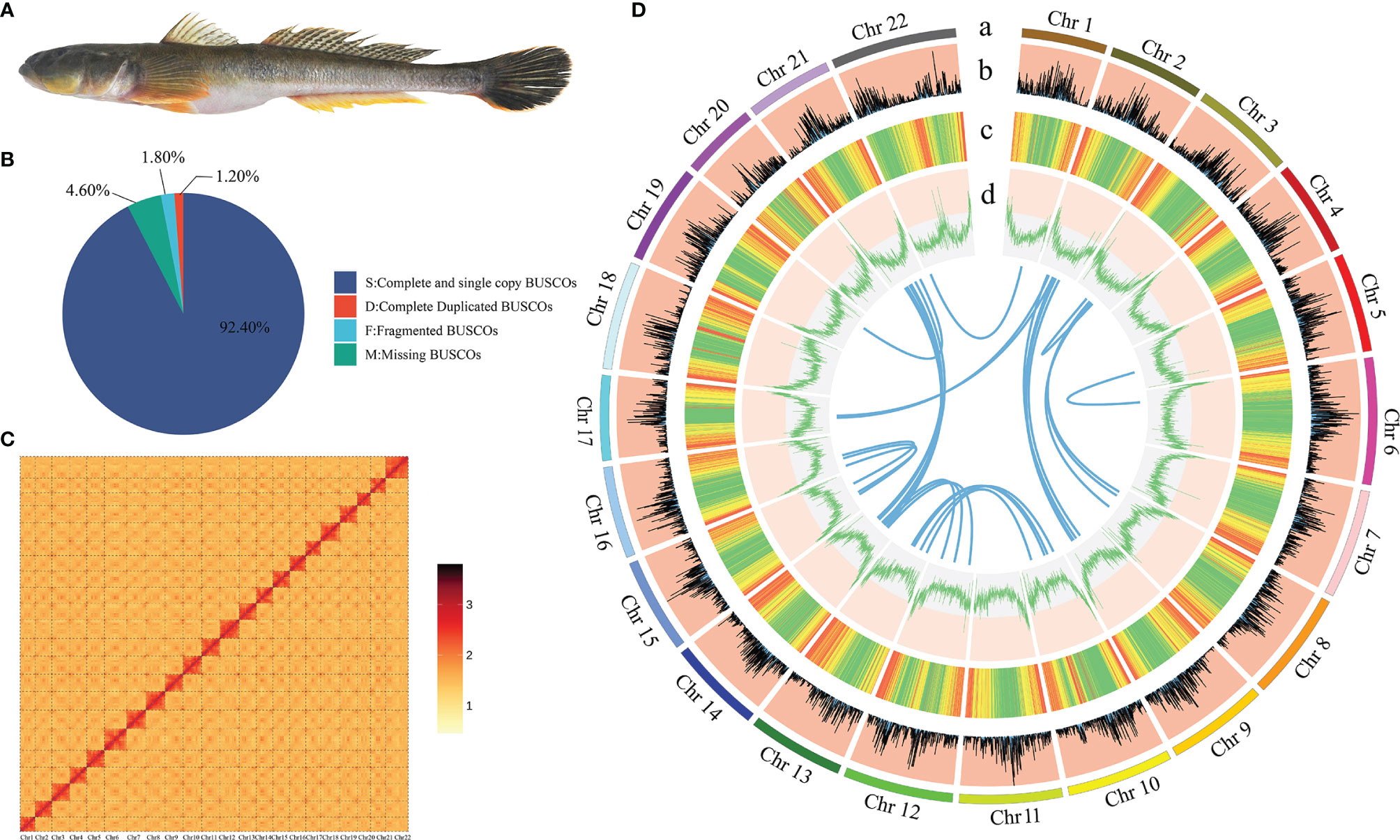
Figure 1 The genome assembly and circos atlas of A. ommaturus. (A) The figure of A. ommaturus. (B) BUSCO analysis result of the A. ommaturus genome. (C) Statistics of the Hi-C assembly of the A. ommaturus genome. (D) Genome characteristics of A. ommaturus. From the outer circle to the inner circle: (a) chromosome length; (b) distribution of gene density; (c) distribution of repetitive elements; (d) distribution of genomic GC content. The innermost syntenic blocks are connected with blue lines.
The Illumina sequencing libraries were prepared to estimate the genome size and correct the genome assembly. The paired-end library with an insert size of 350 was prepared using the Illumina Truseq Nano DNA Library Prep Kit (Illumina, United States) and then sequenced by the Illumina NovaSeq-6000 platform using 2 × 150 bp in paired-end mode. To obtain the clean reads, the reads with more than 10% N bases or low-quality bases ≤ 5, adapter sequences, and duplicated sequences were discarded. The clean reads were used for subsequent analysis.
An SMRTbell library with a fragment size of 20 kb was constructed for long-read sequencing by an SMRTbell Express Template Prep Kit (PacBio). PacBio Sequel II system was used to sequence the library to generate the data from the SMRT cell.
To obtain a chromosome-level genome assembly, the muscle tissue of A. ommaturus was used for Hi-C library construction. The method of Rao et al. (2014) was followed for library preparation. High-quality Hi-C fragment libraries were sequenced for the Illumina NovaSeq-6000 platform.
K-mer analysis was used to estimate the genome size, heterozygosity, and repeat content of A. ommaturus. The Jellyfish approach (Marçais and Kingsford, 2011) was used to obtain the k-mer depth distribution and the peak depth from the distribution from Jellyfish. The genome size estimation formula was applied: G = k-mer_number/k-mer_depth, where G is genome size, k-mer_number is total numbers of k-mers, and kmer_depth is peak depth. SOAPdenovo (Li et al., 2009) was applied for de novo pre-assembly of the A. ommaturus genome. The A. ommaturus genome was assembled by Next Denove package v2.3.1 (https://github.com/Nextomics/NextDenovo) with PacBio long reads. After the primary assembly, we applied the Illumina paired-end reads to polish the assembled genome by operating NextPolish v1.5 (Hu et al., 2020).
Hi-C clean reads were mapped to the draft genome with BWA v0.7.8 (Li and Durbin, 2009). ALLHIC v0.9.8 (Zhang et al., 2019) was applied to obtain the chromosomal-level genome assembly by using the corrected contigs. Genome completeness was estimated using Benchmarking Universal Single-Copy Orthologs (BUSCO v4.1.2, Manni et al., 2021) and Core Eukaryotic Genes Mapping Approach (CEGMA v2.5, Parra et al., 2007). Small fragment library reads were selected and compared to assembling genomes using BWA v0.7.8 (Li and Durbin, 2009), and the comparison rate of reads, the extent of genome coverage, and the distribution of depth were counted to evaluate assembly integrity and sequencing uniformity. Samtools v0.1.19 (Li et al., 2009) was used to process BWA results by chromosome coordinate sequencing and removing repetitive reads and then calculating the genome heterozygous and homologous SNP ratio.
Repeated sequence annotation was obtained based on homology alignment and de novo prediction approaches. Tandem repeat was extracted using TRF v4.07b (Benson, 1999) by ab initio prediction. The homolog prediction used the Repbase (Bao et al., 2015) database employing RepeatMasker v4.0.5 (Chen, 2004) and RepeatProteinMask v4.0.5 (Tarailo-Graovac and Chen, 2009) to extract repeat regions. RepeatModeler v1.0.8 (Tarailo-Graovac and Chen, 2009), RepeatScout v1.0.5 (http://www.repeatmasker.org/), and LTR_Finder v1.0.7 (Xu and Wang, 2007) were used for de novo identification of transposable elements (TEs). All TEs and repeats were combined into a repeat library, which was supplied for DNA-level repeat identification in the A. ommaturus genome.
Gene structure prediction used a combination of de novo prediction, homology-based prediction, and transcriptome-based strategy (transcriptome data were downloaded from NCBI databases with accession numbers PRJNA725985, PRJNA628563, and PRJNA725983; the detailed information is shown in Supplementary Table 1). Firstly, Augustus v3.2.3 (Stanke et al., 2006), GlimmerHMM v3.0.4 (Majoros et al., 2004), SNAP 2013-11-29 (http://korflab.ucdavis.edu/software.html), Geneid v1.4 (Blanco et al., 2007), and Genscan v1.0 (Burge and Karlin, 1997) were used for de novo predictions of genes. Secondly, TBLASTN (E-value ≤ 1e−5) was used to align protein sequences from Boleophthalmus pectinirostris, Periophthalmus magnuspinnatus, Larimichthys crocea, Collichthys lucidus, Gasterosteus aculeatus, and Danio rerio (Supplementary Table 2) to the assembled genome for homology-based prediction. Thirdly, transcriptomic data were aligned to the assembled genome sequences using TopHat v2.0.11 (Kim et al., 2013) to identify exon regions and splice positions. The alignment results were then used as input for Cufflinks v2.2.1 (Ghosh and Chan, 2016) for genome-based transcript assembly. The non-redundant reference gene set was generated by merging genes predicted by three methods with EVM v1.1.1 (Haas et al., 2008) using PASA (Program to Assemble Spliced Alignment) terminal exon support and including masked transposable elements as input into gene prediction.
The Gene Ontology (GO) IDs were assigned according to the corresponding InterPro entry. The Nr, Swissprot, Pfamily, and KEGG were used for the functional annotation and pathway information of protein-coding genes by using BLASTP v2.2.28 (E-value ≤ 1e-5) (McGinnis and Madden, 2004).
For non-coding RNA annotation, tRNAs were predicted using the program tRNAscan-SE v1.3.1 (Lowe and Chan, 2016) and rRNAs were predicted using Blast relative to the species’ rRNA sequence. Other ncRNAs, including miRNAs and snRNAs, were identified by searching against the Rfam database using the INFERNAL v1.1rc4 (Nawrocki and Eddy, 2013).
The protein sequences of 14 species of teleost fish (Supplementary Table 2) were downloaded from NCBI or Ensemble. The longest coding region transcript was selected from each gene locus. The genes that encode proteins with fewer than 50 amino acids were excluded. OrthoMCL v1.4 (Li et al., 2003) was used to construct the orthologous groups. The single-copy orthologous genes shared by all 15 species were aligned using MUSCLE v3.8.31 (Edgar, 2004), and alignment results were combined to form a super alignment matrix. We used RAXML v8.2.12 (Stamatakis, 2014) to construct a phylogenetic tree based on CDS (method: ML TREE; model: GTRGAMMA). The divergence time was estimated using memctree v4.9 of the PAML software package (Xu and Yang, 2013) and r8s v1.81 (Sanderson, 2003); the calibration time was selected from the TimeTree database (Kumar et al., 2017) and published articles. CAFÉ v3.1 was used in gene family expansion and contraction analyses (De Bie et al., 2006); p < 0.05 was used to screen out the significantly changed gene families. GO and KEGG were used to perform the enrichment of expanded and contracted gene families.
Positively selected genes (PSGs) in the A. ommaturus genome were detected using single-copy orthologous genes, in which A. ommaturus was used as a foreground branch, and D. rerio, Xiphias gladius, Seriola dorsalis, and Seriola dumerili were used as background branches. Muscle v3.8.31 (Edgar, 2004) was applied to perform multiple sequence alignment in positive selection analysis, and Gblocks (Castresana, 2000) was used to polish alignments. The branch-site model of the codeml program in PAML v4.9 (Xu and Yang, 2013) was applied to detect PSGs. The two-hypothesis likelihood ratio test was used to determine whether there was positive selection, rather than simply searching for genes with Ka/Ks > 1. p-values were adjusted for multiple testing using the false discovery rate (FDR) method. Genes with FDR < 0.05 were PSGs. Then, GO and KEGG enrichment were performed using Fisher’s exact test with FDR < 0.05. In addition, the candidate aging-related genes were identified by human and model organisms from GenAge and LongevityMap databases (de Magalhães et al., 2005). The tert gene associated with aging was selected for further analysis. The sequences of A. ommaturus (Ao), D. rerio (Dr), X. gladius (Xg), S. dorsalis (Sl), and S. dumerili (Sd) were obtained from this study. The sequences of Oryzias melastigma (Om, DQ286654), Oryzias latipes (Ol, DQ870623), and Epinephelus coioides (Ec, DQ317442) were downloaded from the NCBI database. The multiple sequences were aligned by ClustalX, and the conserved motif and domain regions were determined by MEME (https://meme-suite.org/meme/) and SMART (http://smart.embl-heidelberg.de/), respectively. Prediction of the functional impact of tert gene variants was analyzed by PROVEAN (http://provean.jcvi.org/seq_submit.php). The query sequence was obtained from the above comparison of multiple sequences.
To investigate the genomic collinearity of A. ommaturus with relative species, JCVI (Tang et al., 2015) was used to carry out the collinearity of A. ommaturus and P. magnuspinnatus.
A total of 50.50 Gb data (Coverage 54×) (Chen et al., 2020) about A. ommaturus genome survey sequencing were downloaded from NCBI (PRJNA658176). The Q20 and Q30 values of these data were over 96% and 90%, respectively. The sequencing quality is good and can be used for PSMC (pairwise sequentially Markovian coalescent) analysis. Therefore, these data were used to generate diploid consensus by bcftools v0.1.19 (Danecek et al., 2021). To obtain the input file for PSMC modeling, the “fq2pamcfa” and “splitfa” from the PSMC package (Li and Durbin, 2011) were used for analysis. Then, the parameter code was as follows: “seq 100 | xargs -i echo psmc -N25 -t15 -r5 -b -p “4+25*2+4+6” -o round-{}.psmc split.fa | sh “. The maturing age of A. ommaturus was set as 1 year, and the substitution rate was set as 2.5 × 10-8 per year for PSMC analysis.
In this study, a total of 63 Gb of filtered short-read sequencing data were obtained from the Illumina library, representing the 64.15-fold coverage of A. ommaturus (Supplementary Tables 3, 4). The size of the A. ommaturus genome was estimated to be about 982.02 Mb with a heterozygosity of 0.32% and a repeat content of 51.02%. The depth of k-mers peak was 47, and the total number of k-mers was 46,869,592,867 (Supplementary Figure 1 and Supplementary Table 5).
A total of 210 Gb of high-quality data were generated from the PacBio Sequel II platform, covering 213.85-fold of genome assembly (Supplementary Table 3). The size of the assembled genome was 921.47Mb with a contig N50 of 17.37 Mb (Table 1). This result was consistent with k-mer analysis. To evaluate the quality of initial genome assembly, the Illumina short reads and PacBio long reads were aligned to the A. ommaturus assembly. A total of 99.36% of Illumina reads and 95.89% of PacBio long reads were successfully mapped to the assembled genome (Supplementary Tables 6, 7).

Table 1 Summary of the assembled genome.
The BUSCO analysis is based on actinopterygii_odb10. A total of 93.6% (3,407/3,640) of the complete BUSCO were found in the genome assembly (Figure 1B). There were 243 CEGMA-identified core genes with 94.35% completeness (Supplementary Table 8) and a 0.1964% heterozygous SNP rate and a 0.0018% homologous SNP rate (Supplementary Table 9).
A total of ~98 Gb with 99.80× coverage (Supplementary Table 10) clean reads were obtained from the Hi-C library. The Q20 and Q30 of Hi-C data were 96.95% and 92.12%, respectively. The sequencing quality is good and can be used for subsequent analysis. Hi-C scaffolding approach was used to anchor and orient the draft assembly contigs into a chromosomal-scale assembly. A total of 90 assembled scaffolds were used for chromosomal-scale assembly, of which 22 assembled scaffolds were placed to chromosomes. The total length of placed scaffolds is 906.51 Mb; 98.38% of the assembled sequences were successfully clustered into 22 chromosome groups with chromosome lengths ranging from 31.13 Mb to 52.27 Mb (Supplementary Table 11, Figure 1C). The final genome assembly was 921.49 Mb in chromosomal scale, with contig N50 and scaffold N50 values of 15.70 Mb and 40.99 Mb, respectively (Table 2).

Table 2 Statistics of the Hi-C assembly of the A. ommaturus genome.
In this study, the size of repeat sequences was 418.38 Mb, accounting for 45.40% of the assembly genome (Table 3). The transposable elements mainly consisted of the long terminal repeats (LTR) (371.56 Mb; 40.32%), long interspersed elements (LINE) (64.28 Mb; 6.98%), and DNA transposable elements (DNA TE) in 4.70 Mb (0.51%) (Supplementary Table 12).
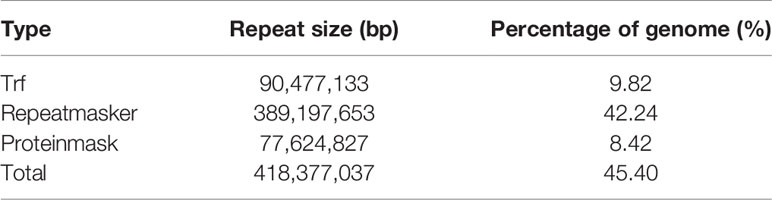
Table 3 Statistics of repetitive sequences in A. ommaturus genome.
A total of 18,752 protein-coding genes were predicted in this study. The average transcript length and average CDS length was 19,348.84 bp and 1,747.18, respectively. The average exon length and average intro length was 167.09 bp and 1,861.37 bp, respectively (Supplementary Table 13). Six other teleost species were used to compare with A. ommaturus to obtain the statistics of the predicted gene model, showing similar distribution patterns in CDS length, exon length, exon number, gene length, and intro length (Supplementary Figure 2). The summary of the genome characteristics of A. ommaturus is shown in Figure 1D. A total of 18,350 genes (97.90%) were successfully annotated by alignment to the nucleotide, protein, and annotation databases (Table 4). The statistics of the noncoding RNA of A. ommaturus is shown in Supplementary Table 14. Based on actinopterygii_odb10, BUSCO analysis showed that 84.6% (3,078/3,640) of the complete BUSCO were found in genome annotation (Supplementary Table 15).
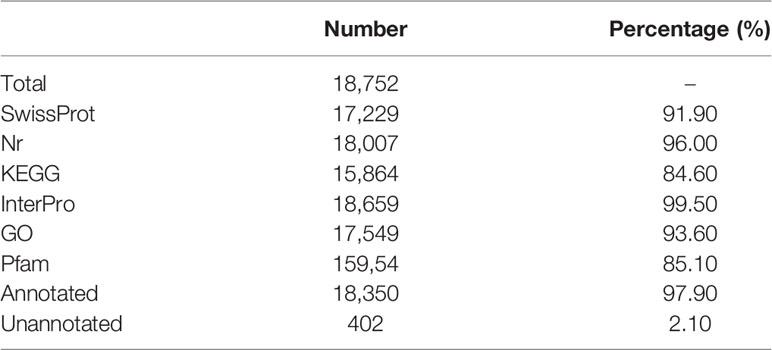
Table 4 Summary of functional annotations for predicted genes.
In this study, gene family was identified among 15 selected species. A total of 14,371 gene families were identified in A. ommaturus, including 167 unique gene families (Supplementary Table 16). On the basis of single-copy genes, the ML phylogenetic tree was constructed to investigate the phylogenetic evolutionary relationships of A. ommaturus with other species. A. ommaturus is phylogenetically closely related to P. magnuspinnatus and B. pectinirostris, diverging ~31.9 MYA with two goby species (Figure 2).
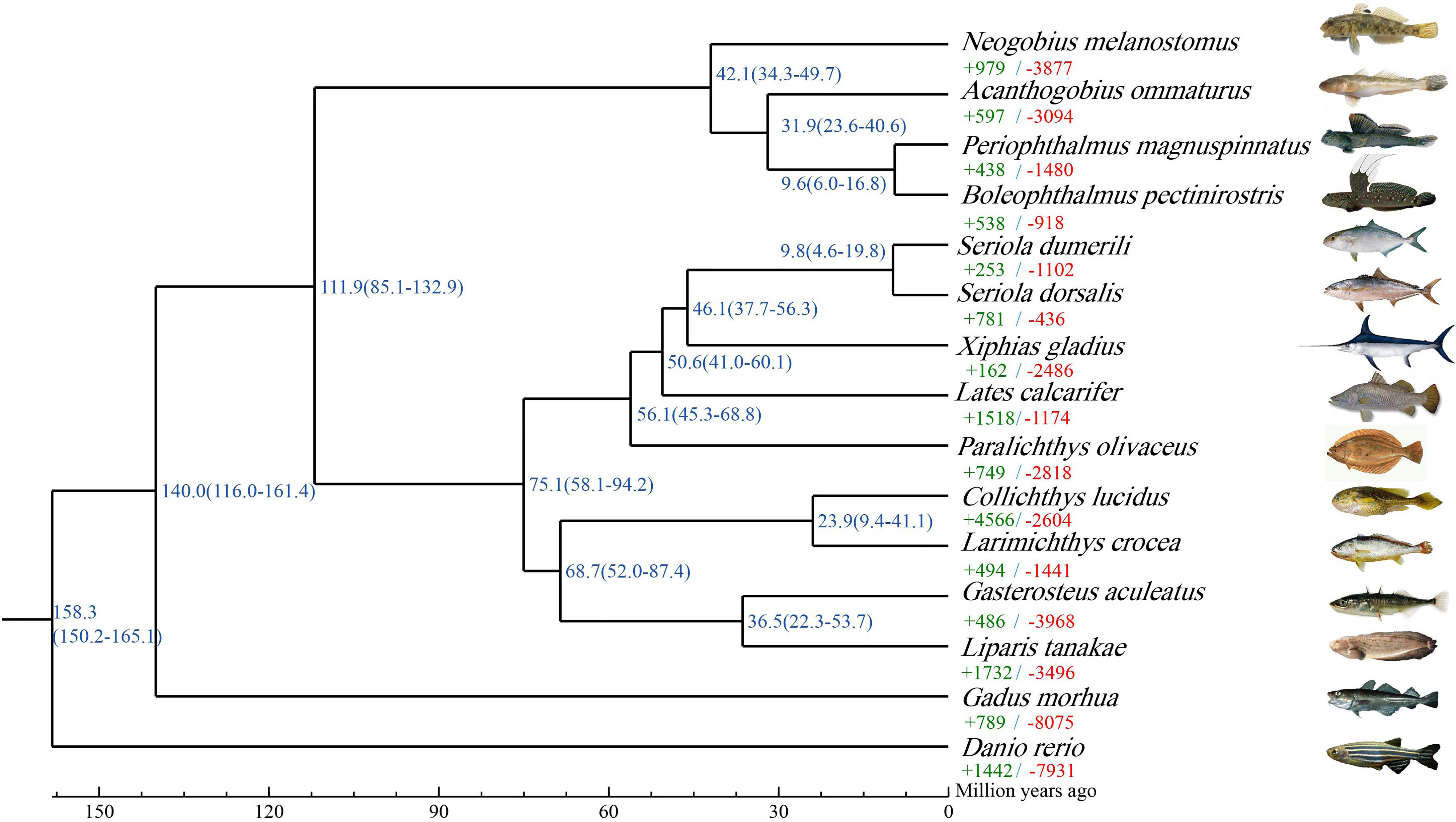
Figure 2 Phylogenetic analysis and divergence time tree of A. ommaturus with 14 other fish species. Each branch site shows the estimated species divergence time (million years ago). Each branch shows the number of expanded (+, green) and contracted (−, red) gene families. The source of species images were shown in Supplementary Table 2.
The A. ommaturus genome displayed 597 expanded and 3,094 contracted gene families compared with a common ancestor (Figure 2). Among these families, 64 expanded and 68 contracted gene families were significantly (p < 0.05) changed (Supplementary Figure 3). Notably, A. ommaturus exhibited the expansion of genes related to FA biosynthesis (ko00061), FA degradation (ko00071), FA metabolism (ko01212), PPAR signaling pathway (ko03320), and adipocytokine signaling pathway (ko04920) (Supplementary Figure 4). There were eight copies of the Acyl-CoA synthetase bubblegum family member 2 (acsbg2) gene (Supplementary Table 17). Eight acsbg2 genes were localized on two chromosomes (Chr9 and Chr11) (Figure 3A). The low-density lipoprotein receptor (ldlr) gene family and Zinc finger protein 638 (znf638) gene were expanded. The KEGG enrichment analysis of contracted gene families is shown in Supplementary Figure 5.
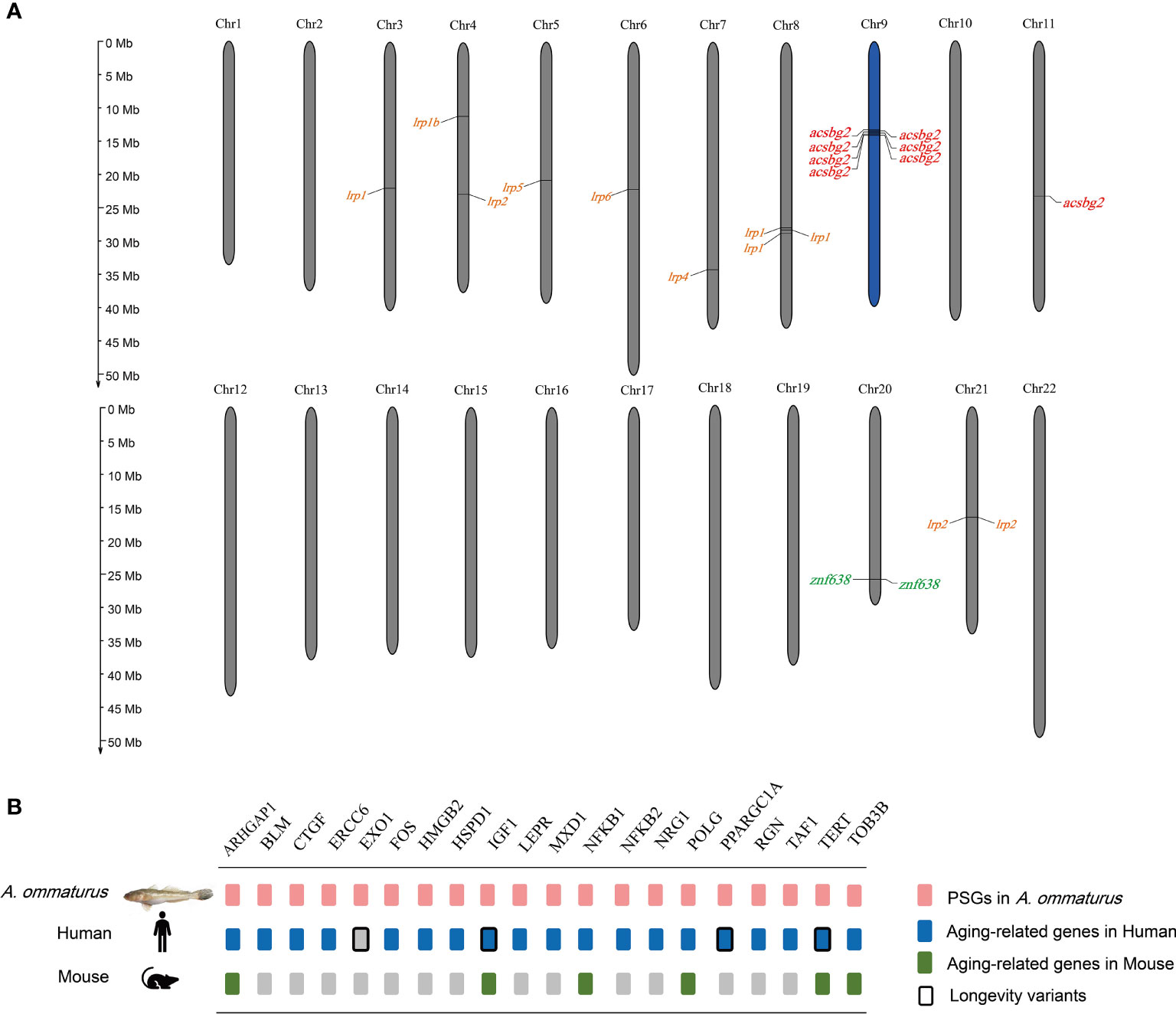
Figure 3 (A) The location of expanded genes related to lipid metabolism in A. ommaturus chromosomes. (B) Candidate aging-related genes under positive selection in A. ommaturus.
Using A. ommaturus as the foreground branch and D. rerio, X. gladius, S. dorsalis, and S. dumerili as the background branches, we incorporated the branch-site model of PAML package to detect positively selected genes (PSGs). A total of 1,155 PSGs (p < 0.05) were identified in A. ommaturus, which were related to autophagy–animal (ko04140), p53 signaling pathway (ko04115), cellular senescence (ko04218), cell cycle (ko04110), and apoptosis (ko04210) (Supplementary Figure 6). Nine PSGs (acsl6, acsf3, hrasls, stard3, fads2, fabp6, pparγ, acat1, and apoe) were related to lipid metabolism. Based on the GenAge and LongevityMap databases, 20 PSGs were identified as being associated with aging (Figure 3B). The multiple sequence alignments of tert are shown in Supplementary Figure 7. The conserved motif and domain results are shown in Supplementary Figures 8, 9. The prediction of the functional impact of tert gene variants is shown in Supplementary Table 19.
Interspecific collinearity analysis showed that there were high collinearity in A. ommaturus and P. magnuspinnatus (Figure 4A). Compared with P. magnuspinnatus, A. ommaturus has 22 chromosomes. The Chr6 of A. ommaturus corresponds to two chromosomes Chr12 and Chr23 of P. magnuspinnatus, and Chr22 corresponds to Chr2 and Chr24 as shown in Figure 4A.
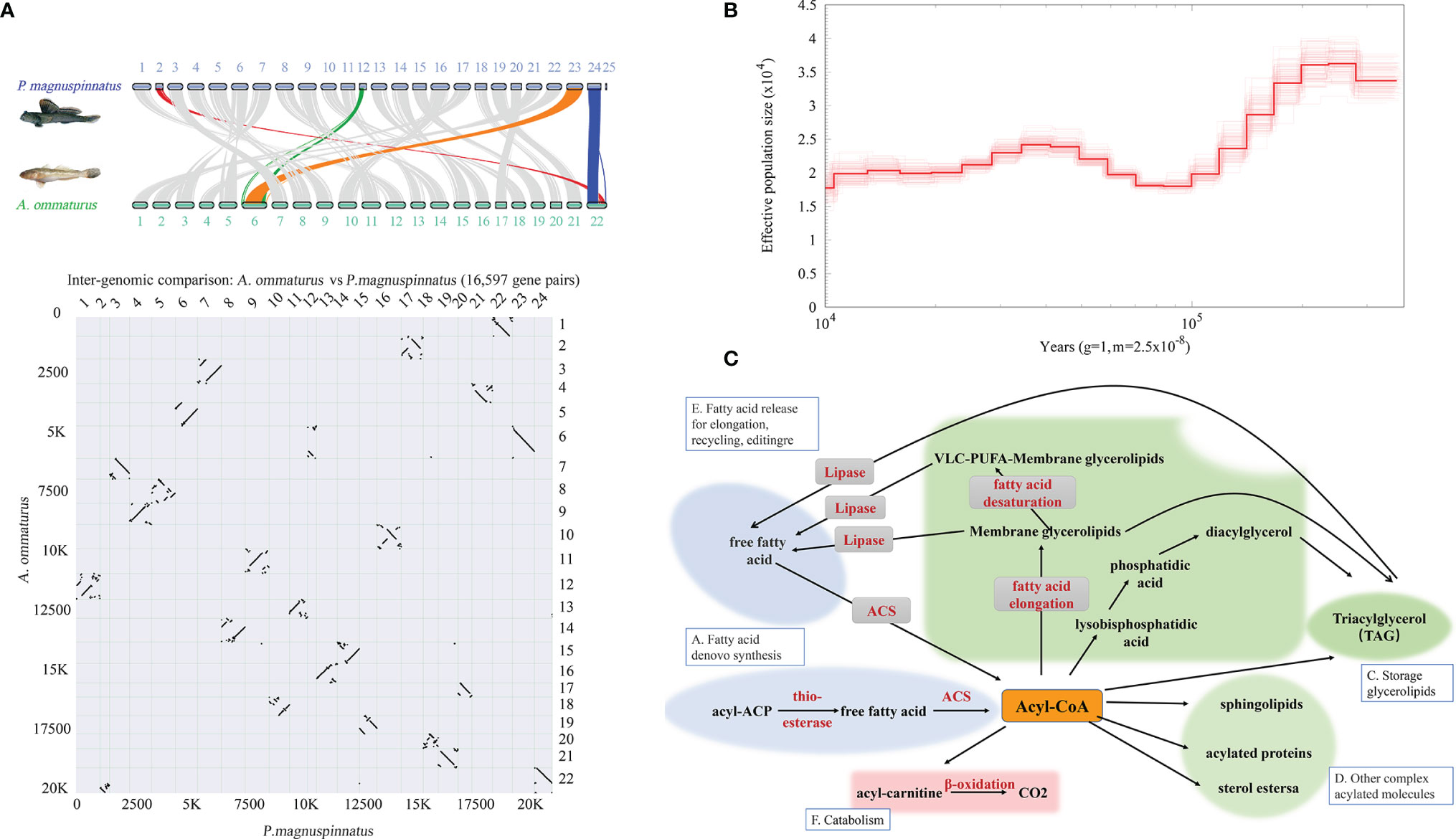
Figure 4 Genome synteny between A. ommaturus and P. magnuspinnatus and population history of A. ommaturus. (A) Chromosomal syntenic relationships between A. ommaturus and P. magnuspinnatus. (B) The effective population size of A. ommaturus. (C) Central role of acyl-CoAs in cell metabolism. Adapted from Coleman et al. (2002).
The effective population size (Ne) of A. ommaturus varied in the range of ~1.75×104–3.6×104 from 400 to 10 Ka. The effective population size of A. ommaturus experienced a bottleneck event from 200 to 100 Ka (Figure 4B).
In the present study, the whole genome of A. ommaturus was generated by PacBio sequencing combined with the Hi-C approach, yielding high-quality genome annotations and a chromosome-level genome for this economically important species. A total of 33 million Hi-C raw reads were finally clustered into 22 chromosomes, which is consistent with a previous karyotype study of A. ommaturus (Wang and Zhao, 1993). In addition, we sorted out the chromosome number of some fishes in Gobiidae based on literature, the NCBI database, and the Fish Karyome database. The chromosome number of Gobiidae fish varies from 22 to 38 (Supplementary Table 20). The chromosome number of A. ommaturus is the same as its relative species, Acanthogobius flavimanus. The genome size of 921.49 Mb of A. ommaturus was in the middle of the genome size of its relative species (M. chulae: 1.002 Gb, Neogobius melanostomus: 1.00 Gb, B. pectinirostris: 955.75 Mb, Rhinogobius similis: 890.10 Mb). We identified 18,752 protein-coding genes in A. ommaturus, which was similar to those in Scartelaos histophorus (18,156) (You et al., 2014) and Lepisosteus oculatus (18,328) (Braasch et al., 2016), but lower than those in M. chulae (20,531) (Cai et al., 2021), B. pectinirostris (20,798) (You et al., 2014), and D. rerio (26,260) (Howe et al., 2013). The annotation data of relative species in databases were less, which may affect the homology prediction results. Moreover, the gene copy number of this species may decrease during evolution. A previous study has shown that the functions of genes are redundant after gene and genome duplications during evolution. Therefore, a single-copy gene is sufficient to perform its function, while the sub-copies of gene may accumulate harmful mutations and loss (Meyer and Schartl, 1999). Notably, the functionality and number of genes have to be further confirmed, and the current finding could only be the first preliminary step towards identifying key genes.
We discovered that the effective population size of A. ommaturus experienced a bottleneck event from 200 to 100 Ka, which may be related to the Guxiang Glacial Stage (ShangZhe et al., 2007). Glacial stage can affect atmospheric circulation, monsoon strength, animal and plant changes, soil development, and sea level rise and fall, resulting in a decline in population size. The population size of A. ommaturus gradually increased from 70 to 30 Ka, which may have something to do with several warm periods during this time and the influence of the Kuroshio (Hu et al., 2021).
Annual fish often demand great energy for rapid growth and sexual maturity in the juvenile stage. A. ommaturus feeds on a variety of shrimps in the juvenile stage (Sun et al., 1996), which may be associated with energy requirements. Previous studies have shown that A. ommaturus enhanced energy metabolism to increase their ability to survive in salinity changes (Sun et al., 2020), and the energy-related pathways were significantly enriched in response to temperature changes (Sun Z. et al., 2021). To facilitate rapid energy mobilization, A. ommaturus may have evolved a regulatory mechanism. In the A. ommaturus genome, acsbg2, znf638, and the ldlr gene family were expanded compared to 14 other fishes, and there were eight copies of the acsbg2 gene. Nine genes (acsl6, acsf3, hrasls, stard3, fads2, fabp6, pparγ, acat1, and apoe) with essential roles in lipid metabolism were identified as PSGs.
Lipids play important roles in numerous biological processes, such as inflammation response, biofilms, reproduction, and energy source and storage (Lopes-Marques et al., 2018). FAs are important components of many lipids, including structural lipids, storage molecules, and signaling molecules. By now, FA catabolism is the main energy source of many fish (Tocher, 2003). They can be degraded for energy production to serve many essential functions in living organisms (Watkins et al., 2007). Notably, any anabolic and catabolic process of FA participation has a critical initial step, the “activation” of FAs. The activation reaction is catalyzed by ACS to produce a thioester with CoA (Watkins, 1997). The ACS is essential to lipid metabolism (Figure 4C). The acsbg2 gene has been demonstrated to convert FAs to active Acyl-CoA using C18:1 and C18:2 as substrates (Pei et al., 2006). acsls typically activate C16:0 and C18:1 (Watkins, 1997). Huang et al. (2014) have researched the FAs in the muscle of A. ommaturus, and the results showed that the FAs were mainly C22:6 (~24.77%), C16:0 (~20.62%), C18:1 (~17.31%), C20:5 (~14.78%), C18:0 (~7.74%), and C18:2 (~3.84%) (Huang et al., 2014). The total content of these six FAs is up to 90%, which shows that the acsbg2 and acsl6 genes play an important role in A. ommaturus FA metabolism and synthesis.
The most important simple lipid is cholesterol (Tocher, 2003). ldlr is a kind of lipoprotein-carrying cholesterol, which plays a role for essential energy production, cell membrane, and cholesterol homeostasis (Brown and Goldstein, 1986; Go and Mani, 2012). ldlr gene variants with impaired function result in early-onset atherosclerosis known as familial hypercholesterolemia (FH) (Go and Mani, 2012). The ldlr gene knockout zebrafish showed moderate hypercholesterolemia when fed a normal diet (Liu et al., 2017). The ldlr gene family including lrp1, lrp2, lrp4, and lrp6 were expanded in A. ommaturus. lrp1 is involved in FA uptake, and lrp6 is involved in the synthesis of TG and FAs, which both suggested that this species has a strong ability to regulate lipid homeostasis. In addition, znf638 is a key regulator of adipogenic differentiation (Meruvu et al., 2011). Pparγ and apoe are also essential to lipid metabolism (Ren et al., 2002; Huang and Mahley, 2014). The expansion of lipid metabolism genes in A. ommaturus may help it to adapt to the complex environment, for instance, by allocating energy properly to respond to changes in external environmental factors.
Aging is broadly defined as the time-dependent irreversible physiological decline that affects most organisms, which has been demonstrated into nine hallmarks. These hallmarks are as follows: loss of proteostasis, telomere attrition, stem cell exhaustion, epigenetic alterations, mitochondrial dysfunction, cellular senescence, genomic instability, deregulated nutrient sensing, and altered intercellular communication (López-Otín et al., 2013). A. ommaturus that has a life span of 1 year may be a good model for aging studies. Analysis of positive selection in the genome of A. ommaturus identified 20 genes associated with aging, such as ercc6, hspd1, igf1, polg, taf1, and tert. These genes can be mainly divided into two categories based on the characteristics of aging.
(I) Genomic instability. The accumulation of various types of genetic damage throughout lifetime is the common feature of aging (Moskalev et al., 2013). Cells may undergo a series of phenotypic changes, from cell cycle arrest, cellular senescence, to malignant transformation when DNA damage reaches a certain level (Erol, 2011). Progeria aging syndromes are often the result of increasing DNA damage accumulation (Burtner and Kennedy, 2010). ercc6, known as CSB, is involved in DNA unwinding and DNA repair (Licht et al., 2003). A previous study showed that ercc6 and xpa mutant mice die before weaning and display some premature aging phenotypes, such as attenuated growth, retinal degeneration, kyphosis, progressive neurological dysfunction, and cachexia (Van et al., 2007). Cockayne syndrome (CS) is a rare progeroid syndrome caused by mutations of ercc6 (CSB) genes (Karikkineth et al., 2017), which has characteristics of normal aging including vision and hearing loss, early-onset neurodegeneration, and impaired mitophagy in early life, ultimately leading to premature death (Lee et al., 2019). Thus, the ercc6 (FDR = 0.032) under positive selection may be a candidate aging gene in A. ommaturus.
(II) Telomere attrition. Telomerase consists of the protein component tert and the RNA component terc, which prolong telomeres after replication to maintain the telomere length (Harel et al., 2015). Telomeres are considered as a good biomarker in age research (Boonekamp et al., 2013), because they shorten during vertebrate aging, including Nothobranchius furzeri (Hartmann et al., 2009; Artandi and DePinho, 2010). The syndromes of telomere shortening, such as dyskeratosis, were caused by mutations in tert or other genes in the telomere-protecting complex (Armanios, 2009). The clinical presentations of dyskeratosis congenita are characterized by premature aging, such as pulmonary fibrosis, marrow failure (Armanios, 2009), reduced fertility (Bessler et al., 2010), and several cancers (Alter et al., 2009). A previous study produced tert-deficient fish by CRISPR/Cas9 technology, and the results showed that tert-deficient fish exhibit the loss of telomerase function, reduced fertility, and defects in highly proliferative tissues, and have genetic ability (Harel et al., 2015). Thus, mutations in tert could lead to a series of premature aging in fish. In this study, the variants of F to S in the telomerase RNA binding domain might have consequences in lifespan regulation. We speculated that tert under positive selection may be associated with the short lifespan of A. ommaturus. In addition, there are other candidate genes in A. ommaturus PSGs within the hallmarks of aging pathways (López-Otín et al., 2013), including mitochondrial dysfunction (polg and ppargc1a), deregulated nutrient sensing (igf1), loss of proteostasis (hsdpd1), and cellular senescence (taf1). In summary, A. ommaturus is an annual fish with a high-quality genome in the chromosomal level, which will be valuable for the identification of conserved genes important for lifespan, and the candidate genes related to aging can provide insight into the evolutionary power of lifespan strategies.
In this study, we presented a high-quality chromosome-level genome assembly of A. ommaturus. The final size of the A. ommaturus genome assembly was 921.49 Mb, with a contig N50 size of 15.70 Mb. The assembled sequences were clustered into 22 chromosomes by using Hi-C data. A total of 18,752 protein-coding genes were predicted. Phylogenetic analysis showed that A. ommaturus is closely related to P. magnuspinnatus and B. pectinirostris; the divergence time was appropriately 31.9 MYA. Several genes related to lipid metabolism such as acsbg2, znf638, and the ldlr gene family were expanded in comparative genomic analyses. Several candidate genes involved in aging were identified in positive selection of A. ommaturus. The high-quality genome assembly and annotation information supplied important genomic data to further investigate the evolution of A. ommaturus with other species, and it will be important for conservation applications and determining the location of important traits in this fish.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA795319; https://ngdc.cncb.ac.cn/, GWHBHRM00000000.
The animal study was reviewed and approved by the Ocean University of China Academic Committee.
NS conceived and managed the project. YP and ZS collected the sequencing samples. YP and LZ performed the experiment and analysis. YP wrote the manuscript, and TG, NS, and LZ revised the manuscript. All authors reviewed and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (2018YFD0900905) and the National Natural Science Foundation of China (No.U20A2087).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.894821/full#supplementary-material
Akiyoshi H., Inoue A. (2004). Comparative Histological Study of Teleost Livers in Relation to Phylogeny. Zool. Sci. 21, 841–850. doi: 10.2108/zsj.21.841
Alter B. P., Giri N., Savage S. A., Rosenberg P. S. (2009). Cancer in Dyskeratosis Congenita. Blood 113, 6549–6557. doi: 10.1182/blood-2008-12-192880
Aparicio S., Chapman J., Stupka E., Putnam N., Chia J. M., Dehai I. P., et al (2002). Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu Rubripes. Science 297, 1301–1310. doi: 10.1126/science.1072104
Armanios M. (2009). Syndromes of Telomere Shortening. Annu. Rev. Genomics Hum. Genet. 10, 45–61. doi: 10.1146/annurev-genom-082908-150046
Artandi S. E., DePinho R. A. (2010). Telomeres and Telomerase in Cancer. Carcinogenesis 31, 9–18. doi: 10.1093/carcin/bgp268
Bao W., Kojima K. K., Kohany O. (2015). Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 6, 11. doi: 10.1186/s13100-015-0041-9
Benson G. (1999). Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Berois N., García G., de Sa R. O. (2015). Annual Fishes: Life History Strategy, Diversity, and Evolution (CRC Press: Lund University Libraries). doi: 10.1201/b19016-16
Bessler M., Wilson D. B., Mason P. J. (2010). Dyskeratosis Congenita. FEBS Lett. 584, 3831–3838. doi: 10.1016/j.febslet.2010.05.019
Bian C., Huang Y., Li J., You X., Yi Y., Ge W., et al (2019). Divergence, Evolution and Adaptation in Ray-Finned Fish Genomes. Sci. China Life Sci. 62, 1003–1018. doi: 10.1007/s11427-018-9499-5
Blanco E., Parra G., Guigó R. (2007). Using Geneid to Identify Genes. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.3. doi: 10.1002/0471250953.bi0403s18
Boonekamp J. J., Simons M. J., Hemerik L., Verhulst S. (2013). Telomere Length Behaves as Biomarker of Somatic Redundancy Rather Than Biological Age. Aging Cell 12, 330–332. doi: 10.1111/acel.12050
Braasch I., Gehrke A. R., Smith J. J., Kawasaki K., Manousaki T., Pasquier J., et al (2016). The Spotted Gar Genome Illuminates Vertebrate Evolution and Facilitates Human-Teleost Comparisons. Nat. Genet. 48, 427–437. doi: 10.1038/ng.3526
Brown M. S., Goldstein J. L. (1986). A Receptor-Mediated Pathway for Cholesterol Homeostasis. Science 232, 34–47. doi: 10.1126/science.3513311
Burge C., Karlin S. (1997). Prediction of Complete Gene Structures in Human Genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Burtner C. R., Kennedy B. K. (2010). Progeria Syndromes and Ageing: What Is the Connection? Nat. Rev. Mol. Cell. Biol. 11, 567–578. doi: 10.1038/nrm2944
Cai L., Liu G., Wei Y., Zhu Y., Li J., Miao Z., et al (2021). Whole-Genome Sequencing Reveals Sex Determination and Liver High-Fat Storage Mechanisms of Yellowstripe Goby (Mugilogobius Chulae). Commun. Biol. 4, 15. doi: 10.1038/s42003-020-01541-9
Castresana J. (2000). Selection of Conserved Blocks From Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen N. (2004). Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.10. doi: 10.1002/0471250953.bi0410s05
Chen B., Sun Z., Lou F., Gao T. X., Song N. (2020). Genomic Characteristics and Profile of Microsatellite Primers for Acanthogobius Ommaturus by Genome Survey Sequencing. Biosci. Rep. 40, BSR20201295. doi: 10.1042/BSR20201295
Coleman R. A., Lewin T. M., Van Horn C. G., Gonzalez-Baró M. R. (2002). Do Long-Chain acyl-CoA Synthetases Regulate Fatty Acid Entry Into Synthetic Versus Degradativegradative Pathways? J. Nutr. 132, 2123–2126. doi: 10.1093/jn/132.8.2123
Cuevas N., Zorita I., Franco J., Costa P. M., Larreta J. (2016). Multi-Organ Histopathology in Gobies for Estuarine Environmental Risk Assessment: A Case Study in the Ibaizabal Estuary (Se Bay of Biscay). Estuar. Coast. Shelf Sci. 179, 145–154. doi: 10.1016/j.ecss.2015.11.023
Danecek P., Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., et al (2021). Twelve years of SAMtools and BCFtools. Gigascience. 10, giab008. doi: 10.1093/gigascience/giab008
De Bie T., Cristianini N., Demuth J. P., Hahn M. W. (2006). CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
de Magalhães J. P., Costa J., Toussaint O. (2005). HAGR: The Human Ageing Genomic Resources. Nucleic Acids Res. 33, D537–D543. doi: 10.1093/nar/gki017
Edgar R. C. (2004). MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/molbev/msw054
Erol A. (2011). Deciphering the Intricate Regulatory Mechanisms for the Cellular Choice Between Cell Repair, Apoptosis or Senescence in Response to Damaging Signals. Cell. Signal. 23, 1076–1081. doi: 10.1016/j.cellsig.2010.11.023
FrOyland L. (2015). Mitochondrial and Peroxisomal β-Oxidation Capacities in Various Tissues From Atlantic Salmon Salmo Salar. Aquac. Nutr. 6, 85–89. doi: 10.1046/j.1365-2095.2000.00130.x
Ge H., Lin K., Shen M., Wu S., Wang Y., Zhang Z., et al (2019). De Novo Assembly of a Chromosome-Level Reference Genome of Red-Spotted Grouper (Epinephelus Akaara) Using Nanopore Sequencing and Hi-C. Mol. Ecol. Resour. 19, 1461–1469. doi: 10.1111/1755-0998.13064
Ghosh S., Chan C. K. (2016). Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol. Biol. 1374, 339–361. doi: 10.1007/978-1-4939-3167-5_18
Godoy R. S., Lanés L., Weber V., Stenert C., Nóblega H. G., Oliveira G. T., et al (2019). Age-Associated Liver Alterations in Wild Populations of Austrolebias Minuano, a Short-Lived Neotropical Annual Killifish. Biogerontology 20, 687–698. doi: 10.1007/s10522-019-09822-5
Go G. W., Mani A. (2012). Low-Density Lipoprotein Receptor (LDLR) Family Orchestrates Cholesterol Homeostasis. Yale J. Biol. Med. 85, 19–28. doi: 10.1038/scibx.2011.77
Gordon D., Huddleston J., Chaisson M. J., Hill C. M., Kronenberg Z. N., Munson K. M., et al (2016). Long-Read Sequence Assembly of the Gorilla Genome. Science 352, aae0344. doi: 10.1126/science.aae0344
Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., et al (2008). Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Han Z., Liu M., Liu Q., Zhai H., Xiao S., Gao T. (2021). Chromosome-Level Genome Assembly of Burbot (Lota Lota) Provides Insights Into the Evolutionary Adaptations in Freshwater. Mol. Ecol. Resour. 21, 2022–2033. doi: 10.22541/au.160218269.98008776/v1
Harel I., Benayoun B. A., Machado B., Singh P. P., Hu C. K., Pech M. F., et al (2015). A Platform for Rapid Exploration of Aging and Diseases in a Naturally Short-Lived Vertebrate. Cell 160, 1013–1026. doi: 10.1016/j.cell.2015.01.038
Hartmann N., Reichwald K., Lechel A., Graf M., Kirschner J., Dorn A., et al (2009). Telomeres Shorten While Tert Expression Increases During Ageing of the Short-Lived Fish Nothobranchius Furzeri. Mech. Ageing Dev. 130, 290–296. doi: 10.1016/j.mad.2009.01.003
Henderson R. J., Almatar S. M. (1989). Seasonal Changes in the Lipid Composition of Herring (Clupea Harengus) in Relation to Gonad Maturation. J. Mar. Biol. Assoc. U. K. 69, 323–334. doi: 10.1017/S0025315400029441
Henderson R. J., Sargent J. R., Hopkins C. (1984a). Changes in the Content and Fatty Acid Composition of Lipid in an Isolated Population of the Capelin Mallotus Villosus During Sexual Maturation and Spawning. Mar. Biol. 78, 255–263. doi: 10.1007/BF00393011
Henderson R. J., Sargent J. R., Pirie B. J. S. (1984b). Fatty Acid Catabolism in the Capelin Mallottus Villosus (Muller) During Sexual Maturation. Mar. Biol. Lett. 5, 115–126.
Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., et al (2013). The Zebrafish Reference Genome Sequence and its Relationship to the Human Genome. Nature 496, 498–503. doi: 10.1038/nature12111
Huang Y., Mahley R. W. (2014). Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 72, 3–12. doi: 10.1016/j.nbd.2014.08.025
Huang W., Zhang Z. H., Shi Y. H., Zhang G. Y., Zhang H. M., Lu G. H., et al (2014). Analysis and Evaluation of Nutritional Components in Muscle of Cultured Synechogobius Ommaturus. Chin. J. Anim. Nutr. 26, 2866–2873.
Hu J., Fan J., Sun Z., Liu S. (2020). NextPolish: A Fast and Efficient Genome Polishing Tool for Long-Read Assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Hu Y., Lu L., Zhou T., Sarker K. K., Huang J., Xia J., et al (2021). A High-Resolution Genome of an Euryhaline and Eurythermal Rhinogoby (Rhinogobius Similis Gill 1895). G3 (Bethesda) 12, jkab395. doi: 10.1093/g3journal/jkab395
Jakub A., Milan V., Polaik M., Radim B., Martin R. (2021). Short-Lived Fishes: Annual and Multivoltine Strategies. Fish Fish. 22, 546–561. doi: 10.1111/faf.12535
Karikkineth A. C., Scheibye-Knudsen M., Fivenson E., Croteau D. L., Bohr V. A. (2017). Cockayne Syndrome: Clinical Features, Model Systems and Pathways. Ageing Res. Rev. 33, 3–17. doi: 10.1016/j.arr.2016.08.002
Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L. (2013). TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 14 (4), R36. doi: 10.1186/gb-2013-14-4-r36
Kumar S., Stecher G., Suleski M., Hedges S. B. (2017). Timetree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Lanés L., Godoy R. S., Maltchik L., Polačik M., Blažek R., Vrtílek M., et al (2016). Seasonal Dynamics in Community Structure, Abundance, Body Size and Sex Ratio in Two Species of Neotropical Annual Fishes. J. Fish Biol. 89, 2345–2364. doi: 10.1111/jfb.13122
Lanés L. E.K., Keppeler F. W., Maltchik L. (2014). Abundance Variations and Life History Traits of Two Sympatric Species of Neotropical Annual Fish (Cyprinodontiformes: Rivulidae) in Temporary Ponds of Southern Brazil. J. Nat. Hist. 48, 1971–1988. doi: 10.1080/00222933.2013.862577
Lee J. H., Demarest T. G., Babbar M., Kim E. W., Okur M. N., De S., et al (2019). Cockayne Syndrome Group B Deficiency Reduces H3K9me3 Chromatin Remodeler SETDB1 and Exacerbates Cellular Aging. Nucleic Acids Res. 47, 8548–8562. doi: 10.1093/nar/gkz568
Licht C. ,. L., Stevnsner T., Bohr V. A. (2003). Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 73, 1217–1239. doi: 10.1086/380399
Li H., Durbin R. (2009). Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Durbin R. (2011). Inference of Human Population History From Individual Whole-Genome Sequences. Nature 475, 493–496. doi: 10.1038/nature10231
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al (2009). The Sequence Alignment/Map Format and Samtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin Q., Fan S., Zhang Y., Xu M., Zhang H., Yang Y., et al (2016). The Seahorse Genome and the Evolution of its Specialized Morphology. Nature 540, 395–399. doi: 10.1038/nature20595
Li L., Stoekert C. J. Jr, Roos D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liu C., Kim Y. S., Kim J., Pattison J., Kamaid A., Miller Y. I. (2017). Modeling Hypercholesterolemia and Vascular Lipid Accumulation in LDL Receptor Mutant Zebrafish. J. Lipid Res. 59, 391–399. doi: 10.1194/jlr.D081521
Lopes-Marques M., Machado A. M., Ruivo R., Fonseca E., Carvalho E., Castro L. (2018). Expansion, Retention and Loss in the Acyl-CoA Synthetase “Bubblegum” (Acsbg) Gene Family in Vertebrate History. Gene 664, 111–118. doi: 10.1016/j.gene.2018.04.058
López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013). The Hallmarks of Aging. Cell 153, 1194–1217. doi: 10.1016/j.nbd.2014.08.025
Louiz I., Palluel O., Ben-Attia M., Ait-Aissa S., Hassine O. K. B. (2018). Liver Histopathology and Biochemical Biomarkers in Gobius Niger and Zosterisessor Ophiocephalus From Polluted and Non-Polluted Tunisian Lagoons (Southern Mediterranean Sea). Mar. Pollut. Bull. 128, 248–258. doi: 10.1016/j.marpolbul.2018.01.028
Lowe T. M., Chan P. P. (2016). Trnascan-SE On-line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Majoros W. H., Pertea M., Salzberg S. L. (2004). TigrScan and GlimmerHMM: Two Open Source Ab Initio Eukaryotic Gene-Finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Manni M., Berkeley M. R., Seppey M., Simão F. A., Zdobnov E. M. (2021). Busco Update: Novel and Streamlined Workflows Along With Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654. doi: 10.1093/molbev/msab199
Marçais G., Kingsford C. (2011). A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27 (6), 764–770. doi: 10.1093/bioinformatics/btr011
McGinnis S., Madden T. L. (2004). BLAST: at the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Meruvu S., Hugendubler L., Mueller E. (2011). Regulation of Adipocyte Differentiation by the Zinc Finger Protein ZNF638. J. Biol. Chem. 286, 26516–26523. doi: 10.1074/jbc.M110.212506
Meyer A., Schartl M. (1999). Gene and Genome Duplications in Vertebrates: The One-to-Four (-to-Eight in Fish) Rule and the Evolution of Novel Gene Functions. Curr. Opin. Cell Biol. 11 (6), 699–704. doi: 10.1016/s0955-0674(99)00039-3
Moskalev A. A., Shaposhnikov M. V., Plyusnina E. N., Zhavoronkov A., Budovsky A., Yanai H., et al (2013). The Role of DNA Damage and Repair in Aging Through the Prism of Koch-like Criteria. Ageing Res. Rev. 12, 661–684. doi: 10.1016/j.arr.2012.02.001
Nawrocki E. P., Eddy S. R. (2013). Infernal 1.1: 100-Fold Faster RNA Homology Searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Parra G., Bradnam K., Korf I. (2007). CEGMA: A Pipeline to Accurately Annotate Core Genes in Eukaryotic Genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Pei Z., Jia Z., Watkins P. A. (2006). The Second Member of the Human and Murine Bubblegum Family Is a Testis- and Brainstem-Specific acyl-CoA Synthetase. J. Biol. Chem. 281, 6632–6641. doi: 10.1074/jbc.M511558200
Rao S. S., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., et al (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Ravi V., Venkatesh B. (2018). The Divergent Genomes of Teleosts. Annu. Rev. Anim. Biosci. 6, 47–68. doi: 10.1146/annurev-animal-030117-014821
Ren D., Collingwood T. N., Rebar E. J., Wolffe A. P., Camp H. S. (2002). Ppargamma Knockdown by Engineered Transcription Factors: Exogenous PPARgamma2 But Not PPARgamma1 Reactivates Adipogenesis. Genes 16, 27–32. doi: 10.1101/gad.953802
Sanderson M. J. (2003). r8s: Inferring Absolute Rates of Molecular Evolution and Divergence Times in the Absence of a Molecular Clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
ShangZhe Z., LiuBing X., Colgan P. M., Mickelson D. M. (2007). Cosmogenic ~(10) Be Dating of Guxiang and Baiyu Glaciations. Chin. Sci. Bull., 1387–1393. doi: 10.1007/s11434-007-0208-y
Stamatakis A. (2014). RaxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. (2006). AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Star B., Nederbragt A. J., Jentoft S., Grimholt U., Malmstrom M., Gregers T. F., et al (2011). The Genome Sequence of Atlantic Cod Reveals a Unique Immune System. Nature 477, 207–210. doi: 10.1038/nature10342
Sun G. Y., Chen J. Y., Wu Y. Z. (1996). Ovarian Maturation and Spawning of Synechogobius Ommaturus (Richardson). Fish. Sci. Technol. Inf. 23, 99–107.
Sun C., Li J., Dong J., Niu Y., Hu J., Lian J., et al (2021). Chromosome-Level Genome Assembly for the Largemouth Bass Micropterus Salmoides Provides Insights Into Adaptation to Fresh and Brackish Water. Mol. Ecol. Resour. 21, 301–315. doi: 10.1111/1755-0998.13256
Sun Z., Lou F., Zhang Y., Song N. (2020). Gill Transcriptome Sequencing and De Novo Annotation of Acanthogobius Ommaturus in Response to Salinity Stress. Genes 11, 631–648. doi: 10.3390/genes11060631
Sun Z., Lou F., Zhao X., Song N. (2021). Characterization and Analysis of Transcriptome Complexity Using SMRT-Seq Combined With RNA-Seq for a Better Understanding of Acanthogobius Ommaturus in Response to Temperature Stress. Int. J. Biol. Macromol. 193, 1551–1561. doi: 10.1016/j.ijbiomac.2021.10.218
Tang H., Krishnakumar V., Li J. (2015). Jcvi: JCVI Utility Libraries. Zenodo. doi: 10.5281/zenodo.31631
Tarailo-Graovac M., Chen N. (2009). Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.10. doi: 10.1002/0471250953.bi0410s25
Tocher D. R. (2003). Metabolism and Functions of Lipids and Fatty Acids in Teleost Fish. Rev. Fish. Sci. 11, 107–184. doi: 10.1080/713610925
Van I., Garinis G., Brandt R., Gorgels T., Wijnhoven S. W., Diderich K. E., et al (2007). Impaired Genome Maintenance Suppresses the Growth Hormone–Insulin-Like Growth Factor 1 Axis in Mice With Cockayne Syndrome. PLoS Biol. 5, e2. doi: 10.1371/journal.pbio.0050002
Wang J. Y., Ye Z. J., Liu Q., Cao L. (2011). Stock Discrimination of Spottedtail Goby (Synechogobius Ommaturus) in the Yellow Sea by Analysis of Otolith Shape. Chin. J. Oceanol. Limnol. 29, 192–198. doi: 10.1007/s00343-011-9087-9
Wang J. X., Zhao X. F. (1993). Chromosome Studies of Synechogobius Ommaturus (OSTEICHTHYES: GOBIIDAE). Mar. Sci. 1994, 47–50.
Watkins P. A. (1997). Fatty Acid Activation. Prog. Lipid Res. 36, 55–83. doi: 10.1016/S0163-7827(97)00004-0
Watkins P. A., Maiguel D., Jia Z., Pevsner J. (2007). Evidence for 26 Distinct Acyl-Coenzyme A Synthetase Genes in the Human Genome. J. Lipid Res. 48, 2736–2750. doi: 10.1194/jlr.M700378-JLR200
Wu H. L., Zhong J. S. (2008). Fauna Sinica. Ostichthyes. Perciformes. Gobioidei (Beijing: Science Press), 211–215.
Xiao Y., Xiao Z., Ma D., Liu J., Li J. (2019). Genome Sequence of the Barred Knifejaw Oplegnathus Fasciatus (Temminck & Schlegel 1844): The First Chromosome-Level Draft Genome in the Family Oplegnathidae. Gigascience 8, giz013. doi: 10.1093/gigascience/giz013
Xu Z., Wang H. (2007). LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Xu B., Yang Z. (2013). PAMLX: A Graphical User Interface for PAML. Mol. Biol. Evol. 30, 2723–2724. doi: 10.1093/molbev/mst179
You X., Bian C., Zan Q., Xu X., Liu X., Chen J., et al (2014). Mudskipper Genomes Provide Insights Into the Terrestrial Adaptation of Amphibious Fishes. Nat. Commun. 5, 5594. doi: 10.1038/ncomms6594
Zhang X., Zhang S., Zhao Q., Ming R., Tang H. (2019). Assembly of Allele-Aware, Chromosomal-Scale Autopolyploid Genomes Based on Hi-C Data. Nat. Plants 5, 833–845. doi: 10.1038/s41477-019-0487-8
Keywords: Acanthogobius ommaturus, genome sequencing, chromosomal assembly, comparative genomics, PSMC (pairwise sequentially Markovian coalescent) analysis
Citation: Pan Y, Sun Z, Gao T, Zhao L and Song N (2022) Chromosome-Level Genome Assembly of Acanthogobius ommaturus Provides Insights Into Evolution and Lipid Metabolism. Front. Mar. Sci. 9:894821. doi: 10.3389/fmars.2022.894821
Received: 12 March 2022; Accepted: 14 April 2022;
Published: 18 May 2022.
Edited by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Xinhui Zhang, Beijing Genomics Institute (BGI), ChinaCopyright © 2022 Pan, Sun, Gao, Zhao and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linlin Zhao, emhhb2xpbmxpbkBmaW8ub3JnLmNu; Na Song, c29uZ25hNjI0QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.