 Luping Bi
Luping Bi Xiaoxin Zhang
Xiaoxin Zhang Songbao Zou
Songbao Zou Daode Ji
Daode Ji Qianqian Zhang
Qianqian Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 06 June 2022
Sec. Microbial Symbioses
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.879388
This article is part of the Research Topic Microbial Associations formed and hosted by Protists, Algae, and Fungi View all 6 articles
The interactions between symbiotic bacterial consortia and their protist hosts in benthic environments have attracted increasing interest in recent years. In the present study, we investigated the diversity of potentially associated bacteria for an astomatous ciliate, Kentrophoros flavus, collected in the intertidal zone of Yantai, China. For the first time, the diversity of the associated bacteria in the species K. flavus was examined using 16S rRNA-based techniques (clone libraries and PacBio sequencing) and the fluorescence in situ hybridization (FISH) technique. The 16S rRNA-based sequencing revealed a higher diversity of associated bacteria in K. flavus than previously expected. In addition to a genus-typical thiotrophic symbiont, the “Candidatus Kentron” stain YE, we provide evidence showing the consistent existence of one Muribaculaceae-like bacterium that was secondarily abundant among the bacterial operational taxonomic units (OTUs). Fluorescence in situ hybridization (FISH) with three specific probes and double-label FISH experiments with “Candidatus Kentron” probes showed that the Muribaculaceae-like bacterium was abundant and merged with the “Candidatus Kentron” stain YE on the cell surface of the host. A phylogenetic analysis of the bacterial 16S rRNA gene showed that the bacterium was a distinct branch in Muribaculaceae, members of which are primarily reported from gut microbiome. The name “Muribaculaceae-like bacterium associated with Kentrophoros flavus” (MLAKF) is proposed for the new bacterium. The higher 16S rRNA diversity in K. flavus and the discovery of MLAKF on the cell surface both suggest a potential bacterial consortium that interacts with the host K. flavus.
Ciliates are widespread unicellular organisms in marine benthic environments (Choi et al., 2012; Cai et al., 2013) that include some extreme habitats (Wilbert and Song, 2005; Lei and Xu, 2008). One important mechanism that makes ciliates thrive in extreme environments (e.g., micro-aerobic/anoxic, sulfide-rich, high-nitrate) or grow on inaccessible diets in the sediment is harboring and interacting with symbiotic bacteria (Nowack and Melkonian, 2010). Recently, studies of the diversity of symbiotic bacteria associated with benthic ciliates have revealed novel bacterial species that possess specialized physical or biochemical traits (Embley et al., 1992; Rosati, 2002; Beinart et al., 2018; Lind et al., 2018; Graf et al., 2021). One well-known representative is the association of benthic ciliates with thiotrophic bacteria in which ectosymbionts account for approximately half the biomass of the symbiotic consortium in some species (Fenchel and Finlay, 1989). Ciliates associated with thiotrophic bacteria are known typically from two genera—the periphyton Zoothamnium and the benthic Kentrophoros—both of which are densely covered by ectosymbionts on the cell surface (Raikov, 1971; Fenchel and Finlay, 1989; Edgcomb et al., 2011; Bright et al., 2014; Seah et al., 2017). Evidence has shown that thiotrophic symbionts can absorb and oxidize sulfide in sulfur-rich environments (Fenchel and Finlay, 1989; Volland et al., 2018; Seah et al., 2019). The symbionts are phagocytized by the host as a food resource (Foissner, 1995; Dubilier et al., 2008; Bright et al., 2014) or directly release soluble organic molecules that are uptaken into the host cytoplasm (Volland et al., 2018). Mutually, the ciliate host may carry the ectosymbionts to a depth in sediments where the chemical gradients are not too steep and where the bacterial metabolisms are optimized (Fenchel and Finlay, 1989).
Some ciliates host more than one symbiotic (or associated) species that may form structural and functional consortia (Edgcomb et al., 2011; Park and Yu, 2018; Lanzoni et al., 2019; Muñoz-Gómez et al., 2021). Functional consortia have been recovered in a geleiid karyorelictean ciliate that harbored two sulfate reducers, a methanogen, and possibly a Bacteroidetes and a Type I methanotroph (Edgcomb et al., 2011). A purple-green ciliate, Pseudoblepharisma tenue, was found simultaneously harboring green algae and anoxygenic photosynthesizers (e.g., purple bacteria) (Muñoz-Gómez et al., 2021). Boscaro et al. (2019) found that up to six different symbionts could co-occur in the cytoplasm of a single Euplotes species. In ciliates associated with thiotrophic bacteria, much less attention has been paid to the diversity and composition of the ectosymbionts, although single thiotrophic bacterial species have been recognized in Zoothamnium (“Candidatus Thiobios zoothamnicoli”, Gammaproteobacteria) and Kentrophoros (“Candidatus Kentron spp.”, Gammaproteobacteria) (Bright et al., 2014; Seah et al., 2017; Seah et al., 2019).
Members of the genus Kentrophoros (Ciliophora and Karyorelictea) are microaerobic ciliates that are typically distributed in the chemocline between the oxidized and anaerobic sulfide–reducing layers of marine sediments (Fenchel, 1969). They are typically characterized by a lack of the adoral zone for prey, but they are covered with dense rod-shaped chemolithotrophic bacteria on the cell surface (Foissner, 1995; Gao et al., 2010; Xu et al., 2011). It has been shown that this ciliate relies directly on engulfing symbionts (or adhering algae/particles) along the entire cell body as a food resource (Fenchel and Finlay, 1989; Foissner, 1995). Morphological observation and chemical measurement estimated that one Kentrophoros cell carries approximately 4500 bacteria that account for approximately half the biomass of the symbiont-ciliate consortium (Fenchel and Finlay, 1989). Earlier ultrastructural observations (Fenchel and Finlay, 1989; Foissner, 1995) and recent metagenomic methods (Seah et al., 2017) have both revealed sulfur-oxidizing bacteria among the symbionts. Using the meta-genome technique and 16S rDNA amplification, Seah et al. (2017; 2019) recognized one genus from 13 Kentrophoros spp. and proposed the name “Candidatus Kentron” (hereafter, “Ca. Kentron”). Despite being known as the primary food source for Kentrophoros, a genomic study of “Ca. Kentron” revealed that the thiotrophic bacteria lack genes that encode canonical enzymes for autotrophic CO2 fixation (Seah et al., 2019). This indicates that there must be extra energy and organic carbon sources for the “Kentron-Kentrophoros” system from the environment or microbial activities (Seah et al., 2019). Thus far, there have been no systematic studies investigating the bacterial diversity associated with the genus Kentrophoros (symbiotic and/or adhering). It is unclear if “Ca. Kentron” is the only genus among the ectosymbionts or whether there are other bacteria that interact with the “Kentron-Kentrophoros” system.
In recent decades, molecular approaches such as 16S rDNA cloning and sequencing and fluorescence in situ hybridization (FISH) have been increasingly used to detect and identify bacterial symbionts or associations with unicellular eukaryotes (Edgcomb et al., 2011; Gong et al., 2014; Seah et al., 2017; Plotnikov et al., 2019). Most recently, the newly developed PacBio platform was able to generate more than 5000 CCS 16S rDNA sequences from one sample (with >0.99 accuracy for 16S rDNA fragments). This allows for a much deeper sequencing depth than is the case with rDNA clone libraries. These approaches make it possible to discover the potentially associated prokaryotes on Kentrophoros outside of “Ca. Kentron”.
In this study, we collected and described a Kentrophoros species, K. flavus, from the intertidal region near the eastern gate of Yantai University (hereafter, K. flavus YE). The bacterial diversity associated with K. flavus YE was examined using 16S rRNA-based approaches, including the clone library and PacBio sequencing. We detected a genus-typical thiotrophic symbiont and named it the “Candidatus Kentron” stain YE (hereafter, “Ca. Kentron” YE). In addition, an undescribed bacterium belonging to the molecular family Muribaculaceae was also consistently detected in the PacBio data. The name “Muribaculaceae-like bacterium associated with Kentrophoros flavus” (MLAKF) is proposed for the bacterium. Three specific probes were designed, and in situ FISH was performed to determine the location of MLAKF on the ciliate cells.
Kentrophoros flavus YE was collected on different dates from May 2020 to March 2022 from the intertidal zone near the eastern gate of Yantai University, China (37°28′50″N, 121°27′41″E). Samples were scraped at the chemocline in fine, well-sorted sands with a reduced, sulfide layer at an approximate 15 cm beneath the sediment surface. Wet sands were transported to the laboratory and examined under a stereo microscope (Guiguang GL-6345BI, China). Living cells were observed using a bright field and differential interference contrast microscopy at 100-1000× magnifications (Zeiss Axio Scope A1, Germany). Counts and measurements of the living cells were conducted at a magnification of 1000×.
Approximately 10 cells were isolated from the raw cultures using a micropipette and washed three times with in situ water filtered by a millipore filter of 0.2 µm in pore diameter (Pall Laboratory, US) to remove the irrelevant bacteria in the environmental water. The ciliate cells were then starved for approximately 5 h in the sterilized water, allowing the hosts to digest the potential bacterial food in the cytoplasm. Each individual was transferred into 1.5 ml Eppendorf tubes with a small drop of water for DNA extraction. DNA was extracted using the REDExtract-N-Amp Tissue PCR Kit (Sigma, St. Louis, MO) according to Gong et al. (2016).
The host’s 18S rRNA gene was amplified via polymerase chain reaction (PCR) with the primers 82F (5′-GAAACTGCGAATGGCTC-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) according to Wang et al. (2019). The PCR reaction solution (25 µl) contained 2 µl of 10 µM primers, 1 µl of template, 12.5 µl of MegaFi™ Fidelity 2X PCR MasterMix with MegaFi™ Fidelity DNA Polymerase (ABM, Shanghai, China), and 9.5 µl of nuclease-free H2O. The cycling parameters were as follows: pre-run of 3 min at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 56°C, and 90 s at 72°C, with a final extension of 10 min at 72°C. The associating bacteria’s 16S rRNA gene was amplified via PCR with the primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1525R (5′-AAGGAGGTGATCCAGCC-3′). The PCR reaction solution (25 µl) contained 2 µl of 10 µM primers, 1 µl of template, 0.5 µl of dNTP Mix (10 mM of each), 1.25 units of Vazyme Lamp® DNA Polymerase (Vazyme, Nanjing, China), 2.5 µl 10X of Vazyme Lamp® buffer with MgCl2, and 18.75 µl of nuclease-free H2O. The cycling parameters were as follows: pre-run of 5 min at 94°C, followed by 30 cycles of 90 s at 94°C, 90 s at 53°C, and 3 min at 72°C, with a final extension of 10 min at 72°C. The PCR products were purified using a TIANgel Midi Purification Kit (Tiangen Bio. Co., Shanghai, China) and then inserted into pCE2 TA/Blunt-Zero vectors (5 min TA/Blunt-Zero Cloning Kit, Vazyme, Nanjing, China). The recombinant vectors were then transformed into competent cells Trans1-T1 (TransGen Biotech Co., Ltd., Beijing, China). The recombinants were screened using the blue/white selection. The white colonies were randomly selected and screened using PCR with the primers M13-20 and M13-26. Positive clones with the target inserts were sent for Sanger sequencing (Sangon Biotech Co., Ltd., Shanghai, China). A total of 132 clones were sent, and 46 sequences were successfully obtained, with an average length of 1500 bp.
For the other two ciliate individuals, the PacBio Sequel II platform was selected to complete the collection of the 16S rRNA full-length amplicons. The 16S rRNA gene was amplified using the same primers and PCR cycling parameters as those applied in the clone library construction. The PCR amplicons derived from two individuals were purified separately using the DNA Clean & Concentrator™-5 Kit (Zymo Research, Beijing, China), and each library was constructed according to the size of the amplified fragments (approximately 1500 bp). Sequencing was performed on a PacBio sequel II platform (Frasergen Co., Ltd., Wuhan, China), using a Hifi mode for small-insert (1.6kb) libraries, which allowed the insert being corrected by circular consensus sequences (CCS) for 30-40 times (with CCS≥3) and ensured an accuracy higher than 99.9%. More than 5000 raw CCS were generated from each sample.
Data derived from the clone libraries were examined manually to remove vector sections and low-qualified sequences using Bioedit (Hall et al., 2011). Data generated from PacBio sequencing were pretreated and qualified using the Pacific Biosciences’ SMRT Link v8.0 workflow (https://www.pacb.com/support/software-downloads). Subreads were generated using the lima program integrated in SMRT Link inferring the barcode information of each sample. The subreads were converted to raw CCS using the CCS program with a strict threshold (minPasses≥3, minPredictedAccuracy≥0.99). Cutadapt V3.2 (Martin, 2011) was used to identify and trim the adapters from the raw CCS with a maximum error rate of 0.1. Chimeras and raw CCS shorter than 1300 bp were also filtered by Cutadapt V3.2.
The qualified clone sequences and clean CCS were pretreated following the methods of Fu et al. (2020). Briefly, chimeras were further removed using the vsearch program integrated in QIIME v.1.8.0 (Caporaso et al., 2010), and the singletons were discarded using Mothur v.1.34.4 (Schloss et al., 2009). The resulting data were clustered according to operational taxonomic units (OTUs) at a sequence similarity of 97% using QIIME v.1.8.0 (Caporaso et al., 2010). The OTUs were blasted, aligned, and classified against the SILVA ribosomal RNA gene database (SILVA 128 database, accessed October 2021) (Quast et al., 2012). Analysis of the species composition and abundance was conducted to examine the major taxa of the associated bacteria using the Pacman package (Peng et al., 2021) appended in R (v3.6.2).
For the phylogenetic analyses of the 18S rRNA gene (ciliate host), 34 sequences from the ciliate class Karyorelictea and 11 sequences of the class Heterotrichea were downloaded from the GenBank database and were appended to the new 18S rRNA sequences. Members of class Heterotrichea were selected as outgroups. Two separate phylogenetic analyses of 16S rRNA were performed for the bacterial OTUs “Ca. Kentron” YE and MLAKF. For “Ca. Kentron” YE, the reference sequences were revised from the dataset of Seah et al. (2017). A total of 42 16S rRNA sequences from the class Gammaproteobacteria were appended to the new sequences, and the classes Coxiellaceae and Ectothiorhodospiraceae were used as outgroups (Seah et al., 2017). For the OTU MLAKF, 44 sequences from the classes Muribaculaceae, Bacteroidaceae, Prevotellaceae, and Porphyromonadaceae of the phylum Bacteroidetes were appended. The classes Bacteroidaceae, Prevotellaceae, and Porphyromonadaceae were selected as outgroups (Lagkouvardos et al., 2019).
The assembled 16S and 18S rRNA datasets were first aligned using SINA v.1.2.11 integrated in the SILVA Web server (https://www.arb-silva.de/aligner/) with default parameters (Pruesse et al., 2012). The resulting alignments were refined manually to excise poorly aligned sites in SeaView v4.0 (Galtier et al., 1996). For the dataset of K. flavus YE, “Ca. Kentron” YE, and MLAKF, the final matrix used for the tree analysis comprised 1524, 1580, and 1505 nucleotide positions, respectively. The maximum likelihood (ML) and MrBayes v.3.2.2 (BI) algorithms were used to perform the phylogenetic tree analyses. In all three datasets, the GTR + I + Г model was selected as the best model for both algorithms by jModelTest 2.1.7 (Posada, 2008) using the Akaike Information Criterion (AIC). The ML trees of the rRNA genes were constructed with RAxML V. 8 (Stamatakis, 2014) with 100 random taxon additions to generate the starting trees. Bootstrap support was inferred from 1000 pseudo replicate datasets. The Bayesian inference (BI) approach was implemented by MrBayes v.3.2.2 (Ronquist and Huelsenbeck, 2003). Markov chain Monte Carlo (MCMC) simulations were run for 5,000,000 generations (default temperature parameter and sampling frequency 0.01) with two parallel runs and each run with four simultaneous chains. The mean standard deviation of the split frequencies based on the last 75% of generations was 0.004 (< 0.01) at the end of the analysis. The first 12,500 trees (corresponding to 25% of generations) were removed as burn-in. MEGA v.6 (Tamura et al., 2013) was used to visualize tree topologies and adjust the branches.
Three 16S rRNA-targeted oligonucleotide probes targeting MLAKF were newly designed according to the method of Gong et al. (2014), and they were named C01-612, C01-824, and C01-1125. The specificities of the probes were confirmed using the Test Probe 3.0 tool against the SILVA NR 99 database (https://www.arb-silva.de/search/testprobe/). The non-sense probe NON338, complementary to C01-612/824/1125, was used as a negative control for the hybridization protocol. The specific probe chr4Ca for “Ca. Kentron” (Seah et al., 2017) was also employed in our FISH analysis to virtualize “Ca. Kentron” YE. The primary sequence and specificity of the four probes are shown in Table 1.

Table 1 Oligonucleotide probes used in FISH for the detection of the MLAKF bacteria and “Candidatus Kentron”.
The whole-cell hybridization procedure followed the protocol of Omar et al. (2017). Starved ciliates were transferred on microscopic slides (Frontier FRC-05) and dried at room temperature after fixation using Bouin’s solution (50%, final concentration). Prior to the FISH assay, the slides were washed three times in distilled water and then progressively dehydrated via ethanol gradients (30%, 50%, 80%, and 100%). The concentration of formamide for each probe was estimated in the mathFISH webserver (http://mathfish.cee.wisc.edu/formamide.html) (Yılmaz and Noguera, 2007). The highest concentration (30%) among the three was applied to maximally remove unspecific hybridization (Gong et al., 2014). There was 36 μl of the hybridization buffer (20 mM Tris–HCl at pH 8.0, 0.9 M NaCl, 0.01% SDS = sodium dodecyl sulfate, and 30% formamide) added to the slides together with 4 μl of the targeting fluorescent probes (5 ng/μl final concentration). “Cy3” fluorescence was applied to all MLAKF probes (C01-612/824/1125). For the double label experiment, the MLAKF and ”Ca. Kentron” YE probes were labeled with “Cy3” and “AF488”, respectively. Considering the disparity in the bacterial abundance, the two probes were mixed in various ratios (3.5 μl Cy3-C01-612/824:0.5 μl AF488-chrCa and 2 μl Cy3-C01-824: 2 μl AF488-chrCa) when applied in the experiment. The slides were incubated at 46°C for 3 h. After hybridization, the slides were dipped in a washing buffer (20 mM of Tris-HCl at pH 8.0, 450 mM of NaCl, 0.01% SDS) for 15 min at 48°C and then washed in chilled distilled water three times for 30 s. The specimens on the slides were covered with anti-fade mounting medium (Beyotime, China) and DAPI (50 ng/ml) for 5 min in darkness prior to observations under an epifluorescence microscope (Olympus BX61, Japan) using a SPOT RT3 digital camera (SPOT Imaging Solutions, Sterling Heights, US) and mercury lamp accessories (Olympus U-RFL-T, Japan). Photomicrographs were colored using Image-pro plus 5.1.
The size of the host ciliate was 200-460 × 16-36 μm in vivo, with a length:width ratio of 8-12:1 when fully extended (n=20; SD=71.5 and 7.8 for the length and width, respectively). The cells were highly flattened and had long ribbon shapes (Figure 1A). The body was curved (Figure 1A). The cells crawled along the bottom of the petri dish and attached to the substrate when stimulated. The ciliate had no cytostome. Nearly the entire dorsal surface of the cell was covered by rod-shaped bacteria that attached to the host erectly with one end (Figures 1B–E). Refractile globules (approximately 1 μm in diameter) were found within nearly all the rod-shaped bacteria (Figures 1C, D). The ventral side of the ciliates showed no symbiotic bacteria (Figure 1E).
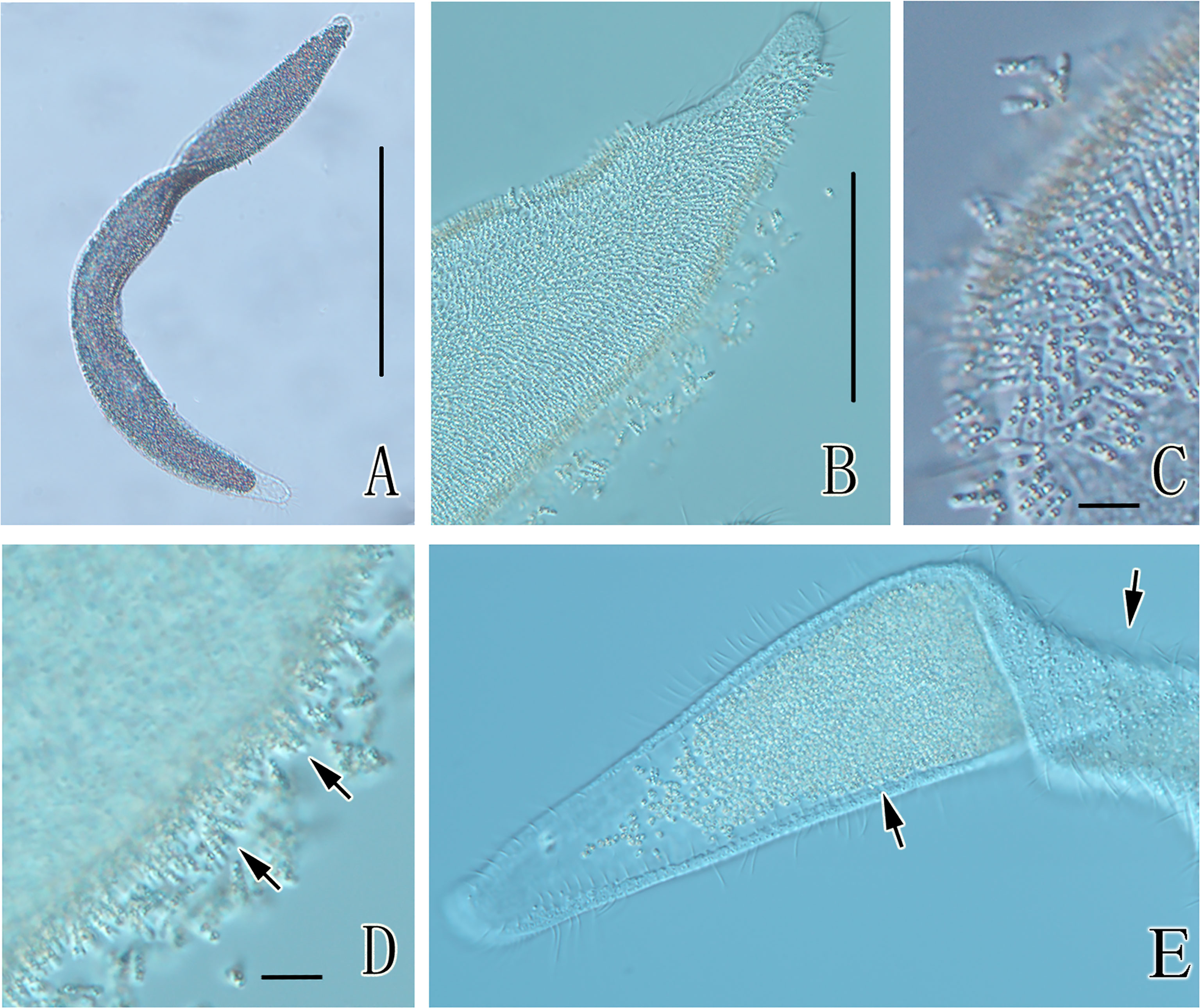
Figure 1 Photomicrographs of Kentrophoros flavus YE from life. (A) Typical individual showing fold-over shape while gliding on the substrate; (B) showing the dense rod-shaped bacteria on the cell surface; (C) showing refractile globules within the rod-shaped bacteria; (D) details of the body margin showing rod-shaped bacteria (arrows); and (E) folded individual shows symbiotic bacteria on only one side (arrows). Scale bars = 100 μm (A); 30 μm (B, E); 5 μm (C, D).
The 18S rRNA gene sequence of Kentrophoros flavus YE was deposited in the GenBank database with the accession number OM421748. The acquired 18S rRNA gene fragment was 1566 bp long and had a GC content of 48.98%. The BLAST search on NCBI showed that this 18S rRNA gene fragment had the highest identity (99.68%) with that of the K. flavus isolate QD (the accession number FJ467505) (Gao et al., 2010). The ML and BI trees based on the 18S rRNA gene showed identical topologies (Figure 2) in which the new sequence of K. flavus YE clustered with two species, the K. flavus QD and one isolate of K. gracilis, with high support from the ML (97%) and BI (1.00).
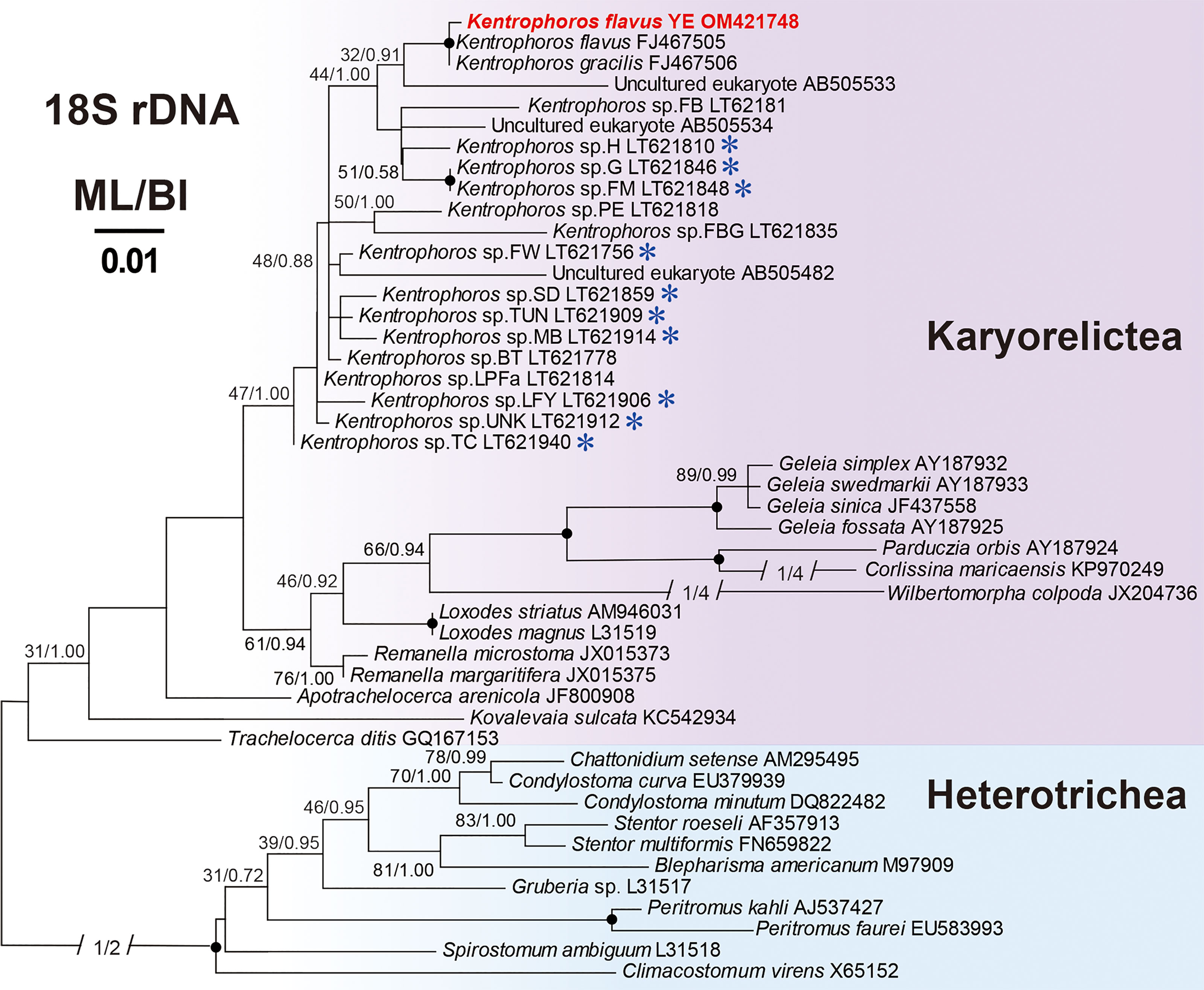
Figure 2 Maximum likelihood (ML) tree based on the 18S rRNA genes showing the positions of Kentrophoros flavus YE (in red) with a GTR + I + Г model. The numbers on the nodes represent the bootstrap values of the ML and the posterior probability of the Bayesian inference (BI). The black dot on the node represents the bootstrap value >95%. The species marked with blue asterisks have been reported with thiotrophic ectosymbionts. GenBank accession numbers are given following the species names. All branches are drawn to scale.
A total of 46 clean 16S rRNA gene sequences were obtained from the clone library technique and were assigned to 17 OTUs (Figure 3). Proteobacteria accounted for a sizable proportion of the assemblages (80.43%, 37 sequences). The most abundant OTU was affiliated with the genus “Candidatus Kentron” (41.30%, 19 sequences). The other OTUs comprised only a few sequences that were closely related to the genera Thalassolituus (10.87%, 5 sequences), Neisseria (8.7%, 4 sequences), Escherichia (4.35%, 2 sequences), Sphingobium (4.35%, 2 sequences), Streptococcus (4.35%, 2 sequences), and “Candidatus Nitrososphaera” (4.35%, 2 sequences).
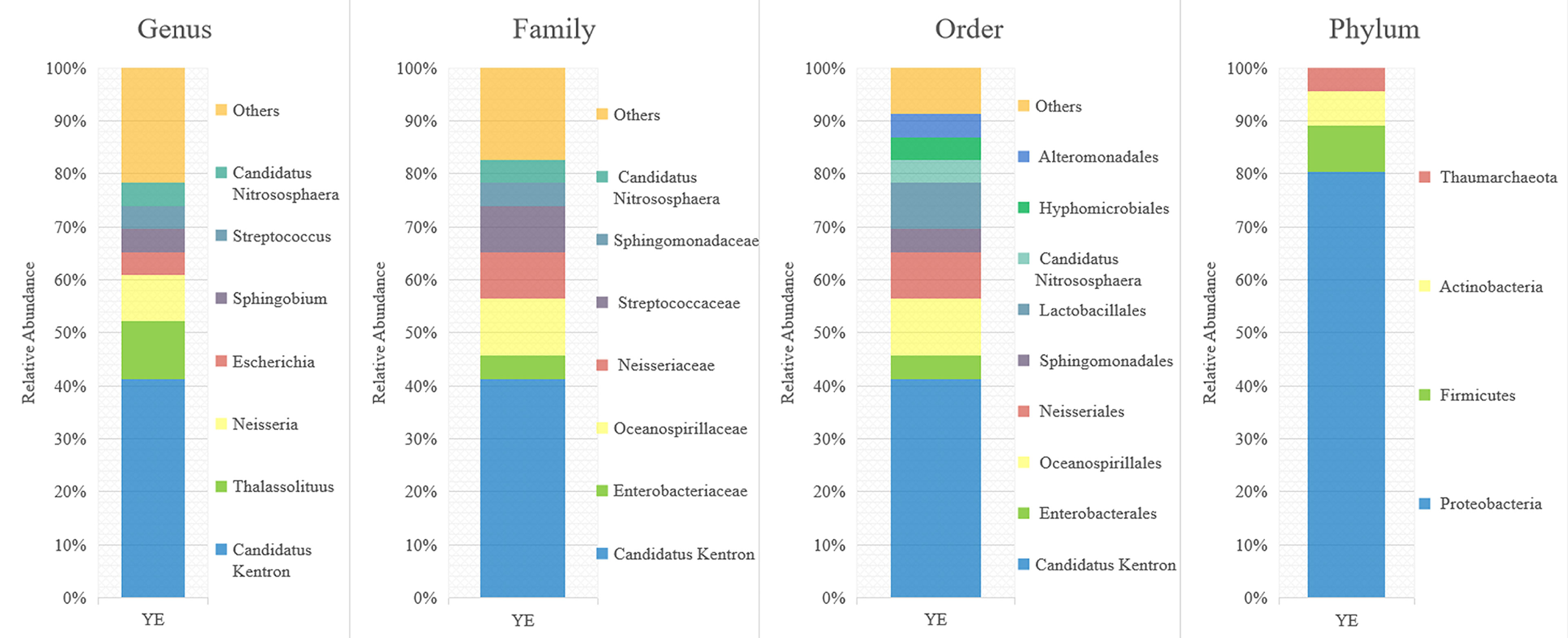
Figure 3 Relative abundances of the bacterial major taxa in Kentrophoros flavus YE are suggested by the clone library data. Members of the genus, “Candidatus Kentron”, are shown as the most frequently occurring bacteria.
A total of 6154 and 4729 clean CCS reads were generated from the PacBio sequencing data for the two ciliate individuals, the total of which was clustered to 121 OTUs (Figure 4). Based on the total reads, the phylum Proteobacteria accounted for a considerable proportion of the assemblages (72.12%), followed by Bacteroidetes (10.63%) and Firmicutes (9.78%). The top three abundant bacterial orders were “Ca. Kentron” (53.89%), Bacteroidales (10.63%), and Pseudomonas (6.63%). The measurable OTUs (>2% relative abundance) were affiliated with 7 genera: “Ca. Kentron” (53.89%), Muribaculaceae-like bacteria (the formerly S24–7 group of Bacteroidales) (10.34%), Psychrobacter (5.53%), Cutibacterium (4.73%), Lactobacillus (3.74%), Staphylococcus (3.07%), and Thalassolituus (2.01%). Of the different ciliate individuals, the diversity of the most least abundant OTUs (<10%) varied obviously, for example, Psychrobacter (5.53% vs. 0.01%), Cutibacterium (4.32% vs. 0.41%), Lactobacillus (2.26% vs. 1.48%), Staphylococcus (2.69% vs. 0.38%), and Thalassolituus (1.86% vs. 0.15%). Nevertheless, the OTUs affiliated with “Ca. Kentron” and Muribaculaceae-like bacteria consistently occurred with the highest proportions in both individuals (Figure 4). Sequence of the former was nearly identical to the OTU affiliated with “Ca. Kentron” revealed in the Clone Library (differed by two bases). Accordingly, we named the two consistently occurring OTUs “Ca. Kentron” YE and “Muribaculaceae-like bacteria associated with K. flavus” (MLAKF). For the measurable OTUs in the PacBio data, the sequence length, GC content of the representative 16S rRNA, and the closely related sequences in the NCBI database gene fragments are provided in Table 2.
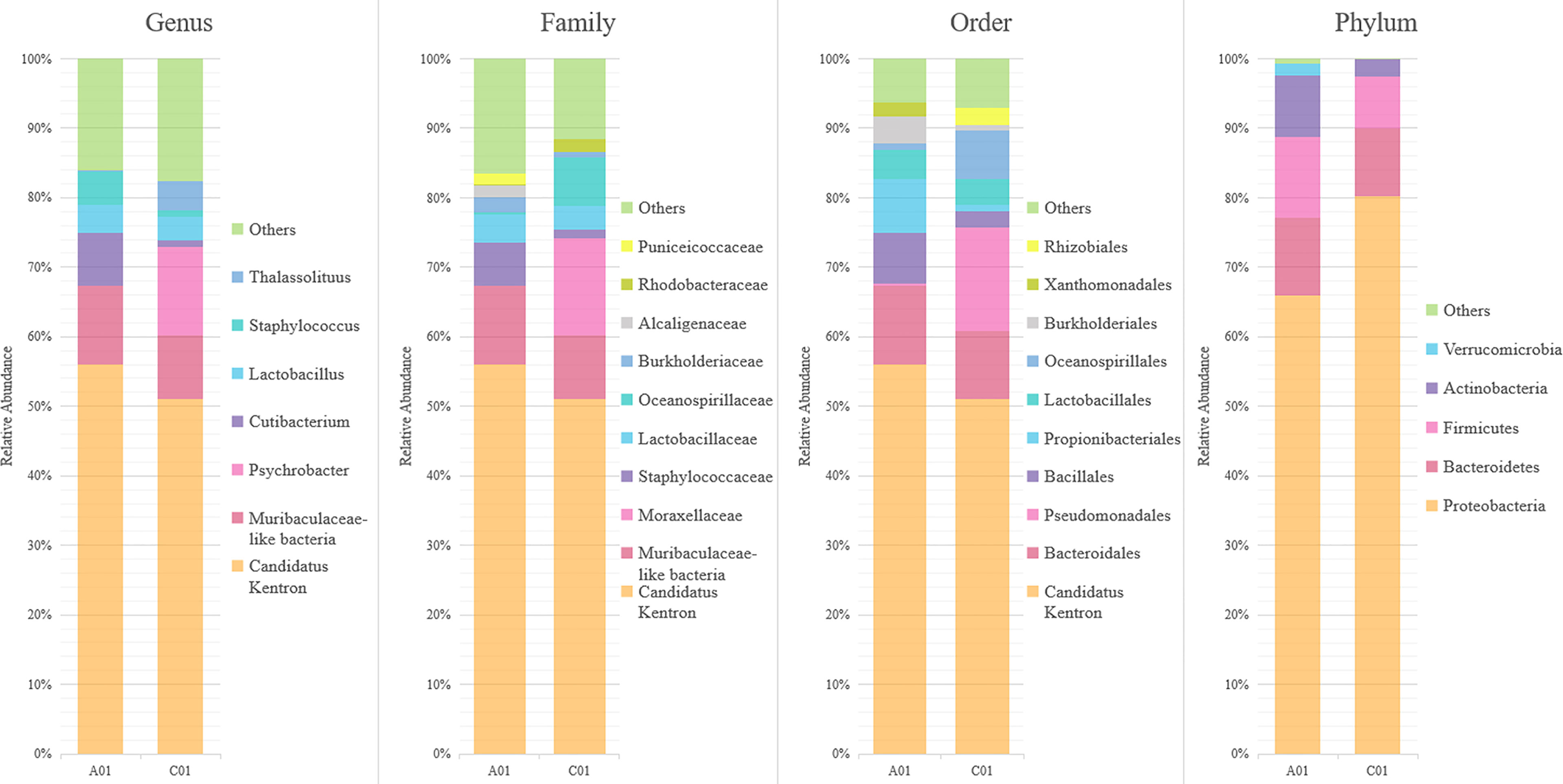
Figure 4 Relative abundances of the bacterial major taxa in Kentrophoros flavus YE, inferred by the PacBio data. Note that the genera, “Candidatus Kentron” and Muribaculaceae-like bacteria, are shown as the most frequently occurring taxa. A01 and C01 represent different individuals of Kentrophoros flavus YE.
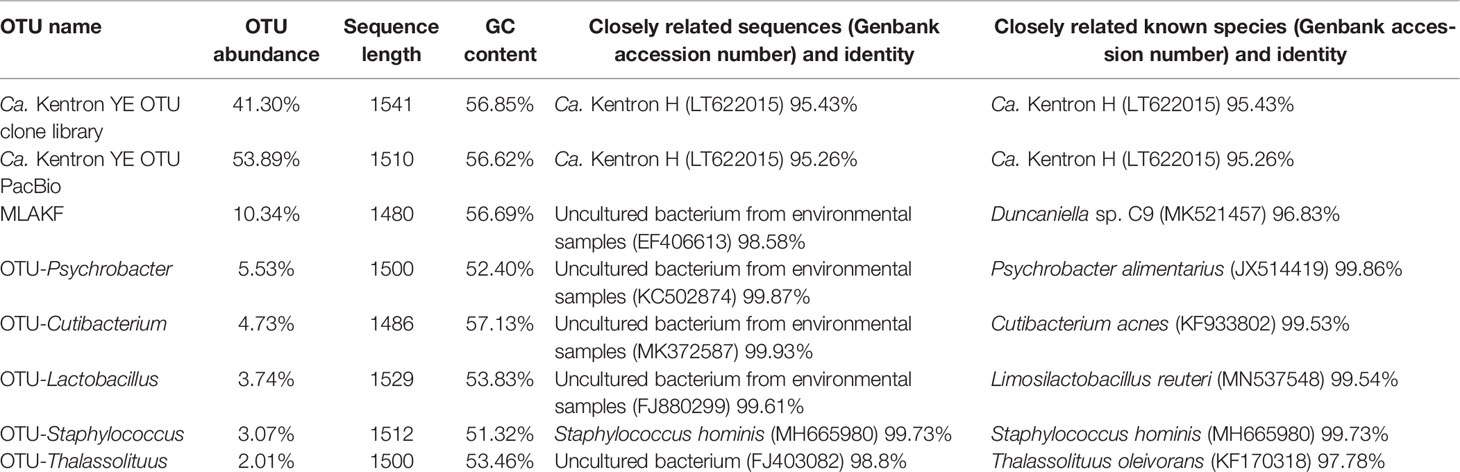
Table 2 Details of 16S rRNA gene fragments of “Ca. Kentron” YE and the measurable bacterial OTUs revealed by PacBio sequencing.
The 16S rRNA gene sequences of “Ca. Kentron” YE (derived from the Clone Library and the PacBio platform) and MLAKF were deposited in the GenBank database with accession numbers OM501125, OM501126, and OM468468. The read sequences derived from the PacBio sequencing were also deposited in GenBank, with the bioproject number PRJNA801354.
Phylogenetic analyses based on the 16S rRNA genes were performed to assess the accurate affiliations of the OTU “Ca. Kentron” YE (Figure 5) and the OTU MLAKF (Figure 6). In the phylogenetic trees, “Ca. Kentron” YE fell into the clade of the genus “Ca. Kentron”, belonging to the class Gammaproteobacteria (Figure 5). The BLAST search on the NCBI showed that this 16S rRNA gene fragment had the highest identity (95.43%) with that of “Ca. Kentron” H (accession number LT622015) (Seah et al., 2017). The OTU MLAKF fell into the family Muribaculaceae in phylum Bacteroidetes (100% ML/1.00 BI) (Figure 6). Within Muribaculaceae, MLAKF appeared to be a distinct taxon, as a sister branch to a clade comprising Duncaniella species and several uncultured bacteria (98% ML/1.00 BI) (Figure 6).
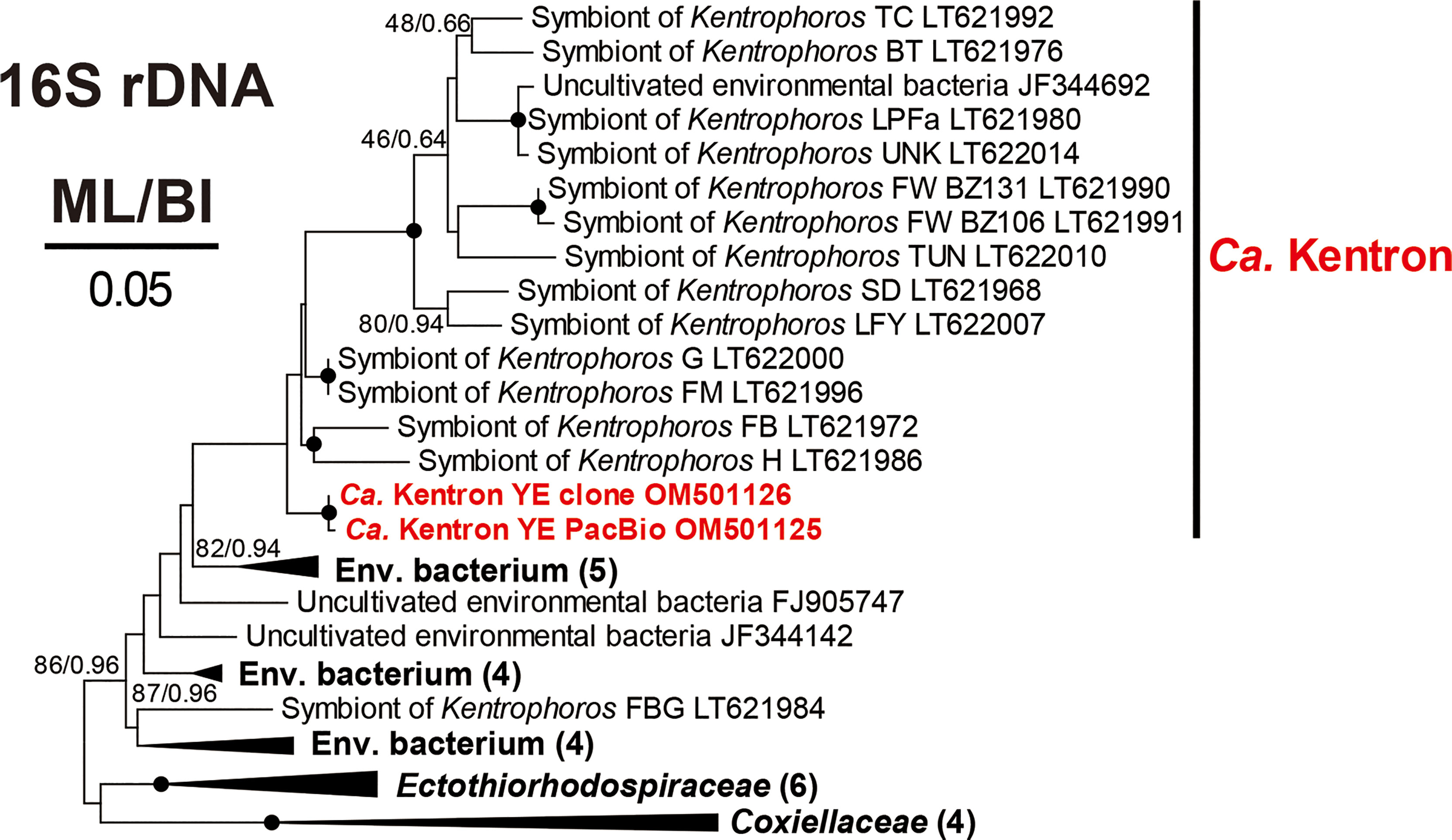
Figure 5 Maximum likelihood (ML) tree based on the 16S rRNA genes showing the positions of “Candidatus Kentron” YE (in red) with a GTR + I + Г model. The numbers on the nodes represent the bootstrap values of ML and the posterior probability of the Bayesian inference (BI). The black dot on the node represents the bootstrap value > 95%. Genbank accession numbers are given following the species names. All branches are drawn to scale.
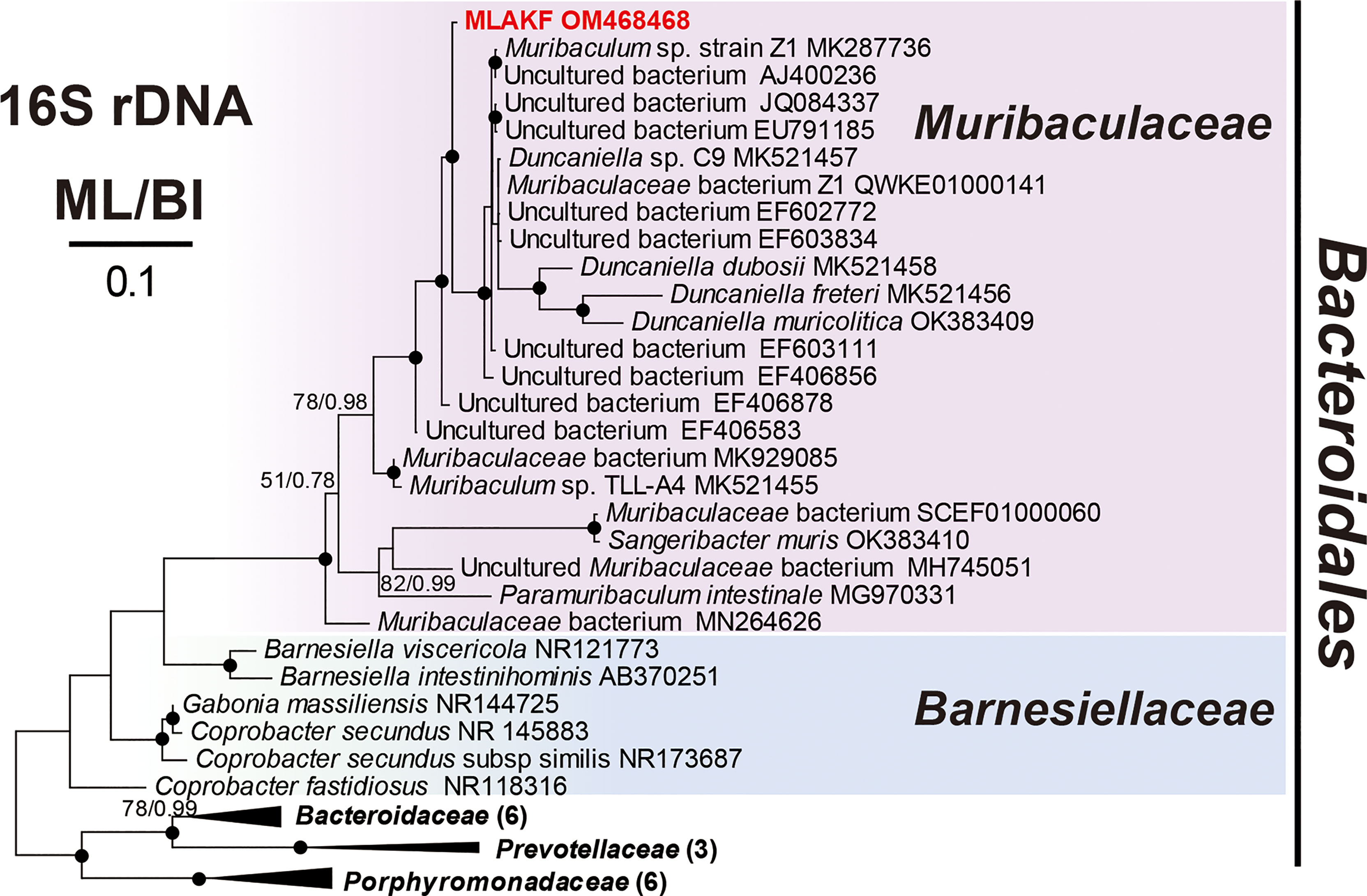
Figure 6 Maximum likelihood (ML) tree based on the 16S rRNA genes showing the positions of MLAKF (in red) with a GTR + I + Г model. The numbers on the nodes represent the bootstrap values of the ML and the posterior probability of the Bayesian inference (BI). The black dot on the node represents the bootstrap value > 95%. Genbank accession numbers are given following the species names. All branches are drawn to scale.
The newly designed probes C01-824 and C01-612 for OTU MLAKF have been shown in silico to be highly Muribaculaceae specific in the SILVA NR 99 database (accessed March 2022). C01-824 completely matched the family Muribaculaceae (126/126). C01-612 primarily matched the family Muribaculaceae (151/154), except for one sequence of the family Rikenellaceae (Bacteroidetes) and two sequences of Firmicutes (Bacteria). The third probe, C01-1125, was more MLAKF-specific but matched a smaller range of Muribaculaceae (62/72). It also matched 10 sequences of the family Prolixibacteraceae (Bacteroidetes) and one sequence of Thiomicrospiraceae (Gammaproteobacteria). All matched non-Muribaculaceae families above were not detected in our clone library (Figure 3) and PacBio data (Figure 4), implying their absence in the samples. Compared to the 16S rDNA of “Ca. Kentron” YE, the three probes C01-612, C01-824, and C01-1125 were mismatched by 6, 7, and 7 sites, respectively, excluding the possibility of nonspecific binding to “Ca. Kentron” YE.
The three probes were applied separately to a total of 10 cells of K. flavus (4 cells with C01-612, 3 with C01-824, and 3 with C01-1125). Strong positive signals were observed on all specimens, with rod-shaped structures distinguishable on several slides (Figures 7C, G, K; Figures 8C, H, M). The overlays of the DAPI and probe’s signals showed that the MLAKF bacteria were primarily located on the surface of the ciliate cells (Figures 7D, H, L). To distinguish MLAKF and “Ca. Kentron” YE, double-label experiments were conducted on the same ciliate with specific probes labeled with different fluorophores (Figure 8). For two sets of probe combinations, namely C01-612 + chr4Ca and C01-824 + chr4Ca, the red fluorescence (Cy3) of MLAKF (Figures 8C, H, M), and the green ones (AF488) of “Ca. Kentron” YE (Figures 8D, I, N) were simultaneously observed, both appearing on the host’s surface. Negative control was conducted by using the non-sense probe NON338 (Supplementary Figure S1) in the FISH protocol, in which neither Cy3/AF488 fluorescence nor autofluorescence could be observed. Overlays of the signals on the same cell showed that the MLAKF and “Ca. Kentron” YE were mixed but were distinguishable from each other, with the former appearing more peripheral on the ciliate’s cell surface (Figures 8E, J, O).
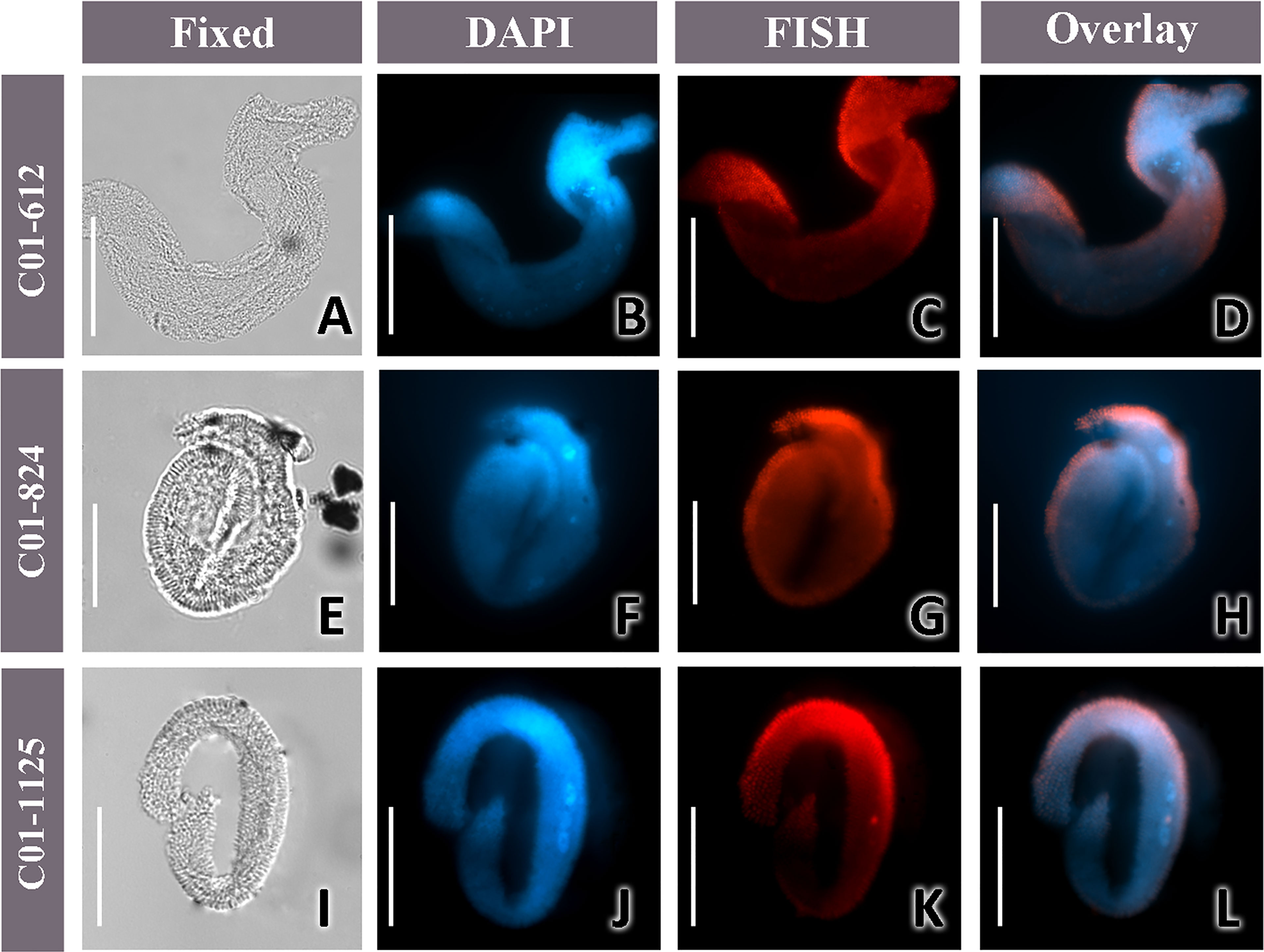
Figure 7 Micrographs of K. flavus with MLAKF. (A, E, I) Microphotographs of the fixed cells in the bright field; (B, F, J) DAPI staining; (C, G, K), and fluorescence in situ hybridization (FISH) using Cy3-C01-612 (C), Cy3-C01-824 (G), and Cy3-C01-1125 probes (K); (D, H, L) overlay of the DAPI and FISH. Scale bars = 50 μm (A–L). The result for C01-824 was derived from a double-label experiment with a probe solution of 3.5 μl of Cy3-C01-824 plus 0.5 μl of AF488-chrCa, while others were generated from a single-probe (4-μl probe solution) hybridization.
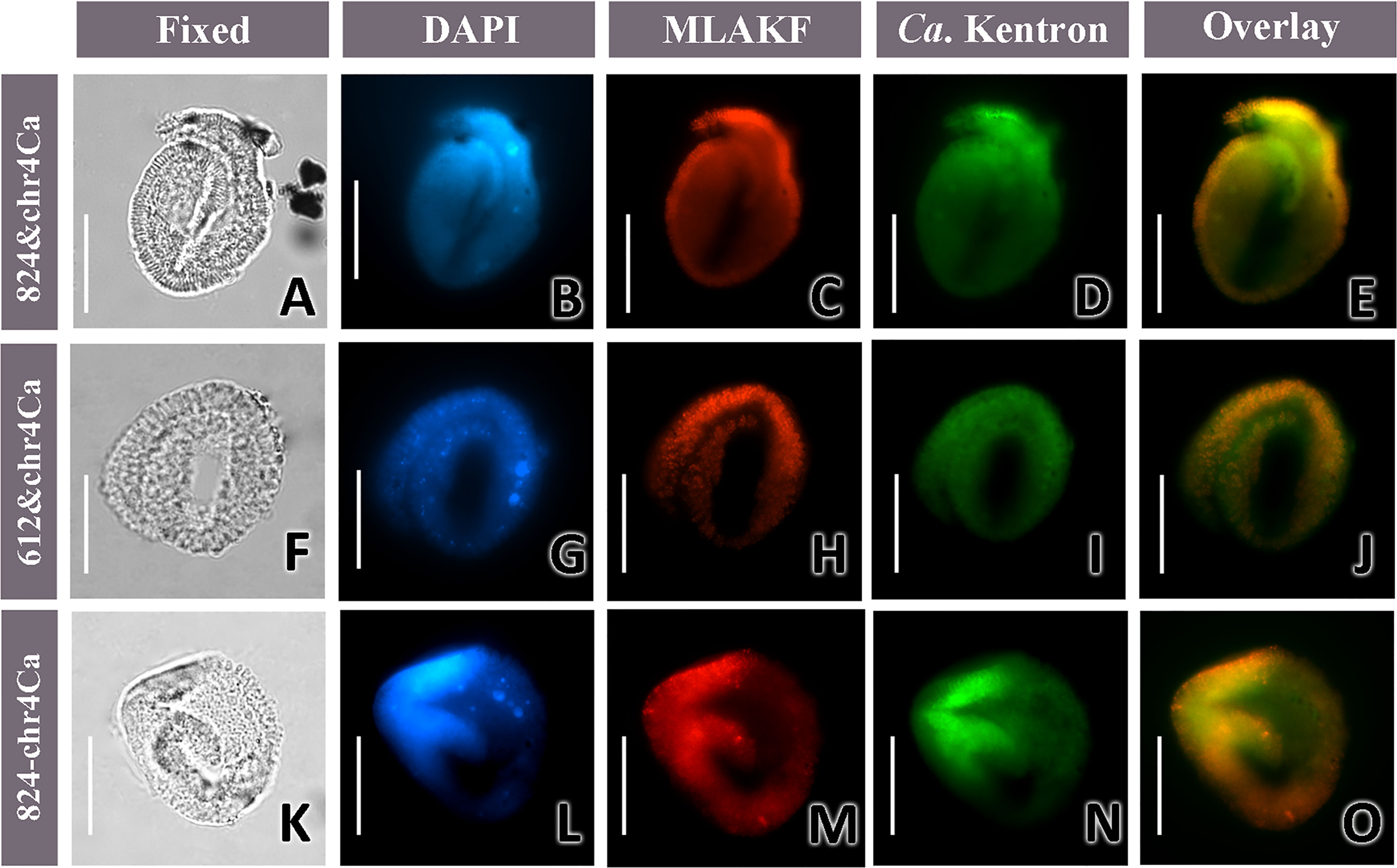
Figure 8 Double-label experiments on the same cell with both the Cy3-C01-612/824 and AF488-chr4Ca probes; (A, F, K) microphotographs of the fixed cells in the bright field; (B, G, L) DAPI staining; (C, H, M) species-specific probes Cy3-C01-824 (C, M) and Cy3-C01-612 (H); (D, I, N) species-specific probe AF488-chr4Ca; and (E, J, O) overlays of Cy3 and AF488 images. Scale bars = 50 μm (A–O). For the cells in images A and F, the probe solutions of 3.5 μl of Cy3-C01-824/612 plus 0.5 μl of AF488-chrCa were applied, while for the one in K, 2 μl of Cy3-C01-824 plus 2 μl of AF488-chrCa were applied.
The newly obtained 18S rRNA gene sequence for Kentrophoros flavus YE was closely clustered with Kentrophoros gracilis and Kentrophoros flavus (97% ML and 1.00 BI in both, Figure 2). In a comparison of the primary sequences, K. flavus YE differed from K. flavus and K. gracilis by 4 and 5 sites, respectively. In vivo, the Yantai populations matched the K. flavus population isolate in Qingdao (Xu et al., 2011) in terms of body shape and general appearance. In addition, the living cell characteristics of K. flavus YE were different from the original description of K. gracilis (Xu et al., 2011) in cell size (200–460 × 16–36 μm vs. 250–600 × 30–60 μm) and shape (K. gracilis was more elongated). The cells of K. flavus YE we observed were ciliated on the ventral side and contained ectosymbionts on the dorsal side (Figure 1E) corresponding to the “open” type among the four different body involution types recognized by Seah et al. (2017) for the genus Kentrophoros.
The OTU “Ca. Kentron” YE always displayed the highest proportion (41.3% in the clone library data and 53.89% in the PacBio data) among the K. flavus-associating bacteria (Figures 3, 4). This aligns with the previous study that used the meta-genomic technique to detect the genus “Ca. Kentron” in 13 Kentrophoros morphospecies (Seah et al., 2017). Compared with the most similar relative, “Ca. Kentron” H (Table 2), “Ca. Kentron” YE was different by 70 bases in the primary 16S rRNA sequence, indicating that “Ca. Kentron” YE may represent a new ectosymbiotic species of the genus “Ca. Kentron”.
As seen from the PacBio results (Figure 4), the OTU MLAKF accounted for the second-largest number (10.34%, approximately 1125 reads) among the associated bacteria in two K. flavus individuals. On 10 host cells that were sampled on different dates, the FISH results with three specific probes all produced positive signals of MLAKF on the surface of K. flavus (Figures 7C, G, K; Figures 8C, H, M), suggesting that their presence on the ciliate was relatively stable.
The 16S rRNA gene sequence of MLAKF was 21 bases different from an uncultured bacterium with 98.58% identity (Table 2), and 47 bases were different from the most similar species Duncaniella sp. C9 with 96.83% identity (accession number: MK521457). Combined with its distinct position in the phylogenetic trees (Figure 6), this suggests that MLAKF should represent a new bacterial species in the family Muribaculaceae. The family Muribaculaceae comprises mainly molecular taxa, which are known as prevalent and abundant bacterial components in the mammal gut microbiome (Lagkouvardos et al., 2019). Members of Muribaculaceae have been widely detected in the intestines of mice, humans, ruminants, and homeothermic animals (Van Valkenburgh, 1991; Salzman et al., 2002; Lagkouvardos et al., 2016; Lagkouvardos et al., 2019; Miyake et al., 2020), with only a few species abundant in the natural environment, including manure, wastewater treatment plants, and coral reefs (Lagkouvardos et al., 2019). Our study adds to this list but with another scenario. Muribaculacid bacteria are abundant on the surface of the ciliated protist K. flavus and merge with thiotrophic ectosymbionts. This is the first time a muribaculacid bacterium has been found to be consistently associated with unicellular eukaryotic organisms.
It is worth noting that MLAKF was consistently present in large numbers on the cell surface of the host (Figure 7), emerging with or on an outlayer of the ectosymbiotic “Ca. Kentron” YE (Figure 8). The two symbionts and the ciliate host may have formed a three-partner consortium. First, MLAKF may offer extra organic carbon for the host-symbiont system. Genomic studies of “Ca. Kentron” have revealed that “Ca. Kentron” lacks genes encoding canonical enzymes for autotrophic CO2 fixation, despite being a food source for Kentrophoros (Seah et al., 2019). It has been suggested that “Ca. Kentron” relies more on the uptake of exogenous organic substrates, heterotrophic carboxylation, and the recycling of host waste for growth (Seah et al., 2019). Extra energy and organic carbon sources should be required for the “Kentron-Kentrophoros” symbiote, and these should come from the environment or microbial activities (Seah et al., 2019). Coincidently, bacteria of Muribaculaceae have been predicted to play important roles in degrading dietary carbohydrates in the animal gut (Lagkouvardos et al., 2019). Genomic studies on a wide range of muribaculacid members have highlighted their prevalent versatility in degrading complex carbohydrates, benzoate resistance, and nitrogen utilization (>90% prevalence) (Ormerod et al., 2016; Lagkouvardos et al., 2019). Therefore, it is possible that MLAKF on the ciliate cells may play similar roles, degrading a wide range of substrate in the sediment (this might also include host waste) for growth, whereby the bio-product may provide accessible organic carbon for “Ca. Kentron”. Simultaneously, MLAKF themselves may offer an extra food source for the ciliate to support the overall growth of the host-symbiont system.
Second, it is known that Kentrophoros likely shuttles between oxic and anoxic zones in marine sediment with the ectosymbionts (Fenchel and Finlay, 1989; Seah et al., 2019). Under anoxic conditions, thiotrophic bacteria would not completely oxidize sulfide to sulfate, leading to less efficient energy generation (Seah et al., 2019). MLAKF may contribute as a compensated food source under anoxic conditions. Members of Muribaculaceae have been revealed to be well equipped with anti-oxygen strategies to support potential microaerobic growth (Ormerod et al., 2016). Nevertheless, the nature of the relationship between the MLAKF bacterium, “Ca. Kentron” YE, and K. flavus remains to be resolved, and single-cell genome sequencing can be a powerful tool for revealing the interactions between ciliated protists and their bacterial symbionts (Martinez-Garcia et al., 2012).
Aside from the thiotrophic bacteria “Ca. Kentron” YE, we detected a high diversity of associated bacteria using 16S rDNA-based approaches (Figures 3 and 4; Table 2). This result accords with a previous morphological study of the species K. latum that recorded ectosymbiotic spirochaetes and intracellular prokaryotic symbionts (Raikov, 1971). The existence of particles adhering to thiotrophic bacteria has also been observed (Fenchel, 1969; Fenchel and Finlay, 1989) (e.g., diatoms and other ingested particles within Kentrophoros), which may be another explanation for the high bacterial diversity being detected. Except for the overwhelming OTU “Ca. Kentron” YE, the revealed diversity of the associating bacteria in K. flavus was variable between the clone library and PacBio sequencing (Figures 3, 4). This might be attributable to the significant difference in sequencing depths between the two techniques (46 clones vs. 10,883 CCS).
In addition to the consistently associated MLAKF bacterium, the comprehensive sequencing by the PacBio platform also detected OTUs related to members in the genera Psychrobacter, Cutibacterium, Lactobacillus, Staphylococcus, and Thalassolituus, all with measurable abundance (2–5.53%) (Figure 4; Table 2). Most of these 16S rDNA fragments are most similar to the known species in GenBank (Table 2), indicating that these might represent common taxa. Among these, members of Cutibacterium and Psychrobacter are obligate symbionts in human skin and the anogenital gland of captive pandas (Scholz and Kilian, 2016; Zhou et al., 2021), suggesting their symbiotic potential. However, the sequences might also derive from particles adhering to “Ca. Kentron”, as assumed by previous morphological studies (Fenchel, 1969). Because of the variable result among the different individuals and a lack of FISH analysis, we must refrain from predicting the stable existence or roles of these OTUs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: OM421748, OM501125, OM501126, OM468468, and PRJNA801354].
LB performed the microscopy observation, PCR experiment, data processing, and drafted the manuscript. LB, SZ, and XZ conducted the FISH and phylogenetic analysis. QZ and DJ designed the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (project numbers: 31672251, 31772413, and 42176163), the Youth Innovation Promotion Association, CAS (No. 2019216), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA23050303), and the Key Research Program of Frontier Science, CAS (No. QYZDB-SSW-DQC013-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.879388/full#supplementary-material
Beinart R. A., Rotterová J., Čepička I., Gast R. J., Edgcomb V. P. (2018). The Genome of an Endosymbiotic Methanogen is Very Similar to Those of its Free-Living Relatives. Environ. Microbiol. 20 (7), 2538–2551. doi: 10.1111/1462-2920.14279
Boscaro V., Husnik F., Vannini C., Keeling P. J. (2019). Symbionts of the Ciliate Euplotes: Diversity, Patterns and Potential as Models for Bacteria–Eukaryote Endosymbioses. Proc. R. Soc. B: Biol. Sci. 286 (1907), 20190693. doi: 10.1098/rspb.2019.0693
Bright M., Espada-Hinojosa S., Lagkouvardos I., Volland J. M. (2014). The Giant Ciliate Zoothamnium Niveum and its Thiotrophic Epibiont Candidatus Thiobios Zoothamnicoli: A Model System to Study Interspecies Cooperation. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00145
Cai L.-Z., Hwang J.-S., Dahms H.-U., Fu S.-J., Chen X.-W., Wu C. (2013). Does High Organic Matter Content Affect Polychaete Assemblages in a Shenzhen Bay Mudflat, China? J. Marine Sci. Technol. 21 (7), 37. doi: 10.6119/jmst-013-1223-5
Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 7 (5), 335–336. doi: 10.1038/nmeth.f.303
Choi K.-H., Kim Y.-O., Lee J.-B., Wang S.-Y., Lee M.-W., Lee P.-G., et al. (2012). Thermal Impacts of a Coal Power Plant on the Plankton in an Open Coastal Water Environment. J. Marine Sci. Technol. 20 (2), 9. doi: 10.51400/2709-6998.1837
Dubilier N., Bergin C., Lott C. (2008). Symbiotic Diversity in Marine Animals: The Art of Harnessing Chemosynthesis. Nat. Rev. Microbiol. 6 (10), 725–740. doi: 10.1038/nrmicro1992
Edgcomb V., Leadbetter E., Bourland W., Beaudoin D., Bernhard J. (2011). Structured Multiple Endosymbiosis of Bacteria and Archaea in a Ciliate From Marine Sulfidic Sediments: A Survival Mechanism in Low Oxygen, Sulfidic Sediments? Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00055
Embley T. M., Finlay B. J., Brown S. (1992). RNA Sequence Analysis Shows That the Symbionts in the Ciliate Metopus Contortus are Polymorphs of a Single Methanogen Species. FEMS Microbiol. Lett. 97 (1-2), 57–61. doi: 10.1111/j.1574-6968.1992.tb05439.x
Fenchel T. (1969). The Ecology of Marine Microbenthos IV. Structure and Function of the Benthic Ecosystem, its Chemical and Physical Factors and the Microfauna Commuities With Special Reference to the Ciliated Protozoa. Ophelia 6 (1), 1–182. doi: 10.1080/00785326.1969.10409647
Fenchel T., Finlay B. J. (1989). Kentrophoros: A Mouthless Ciliate With a Symbiotic Kitchen Garden. Ophelia 30 (2), 75–93.
Foissner W. (1995). Kentrophoros (Ciliophora, Karyorelictea) has Oral Vestiges: A Reinvestigation of K. Fistulosus (FaurÉ-Fremiet 1950) Using Protargol Impregnation. Archiv Für Protistenkunde 146 (2), 165–179. doi: 10.1016/S0003-9365(11)80107-7
Fu Y., Zheng P., Zhang X., Zhang Q., Ji D. (2020). Protist Interactions and Seasonal Dynamics in the Coast of Yantai, Northern Yellow Sea of China as Revealed by Metabarcoding. J. Ocean Univ. China 19 (4), 961–974. doi: 10.1007/s11802-020-4461-x
Galtier N., Gouy M., Gautier C. (1996). SEAVIEW and PHYLO_WIN: Two Graphic Tools for Sequence Alignment and Molecular Phylogeny. Bioinformatics 12 (6), 543–548. doi: 10.1093/bioinformatics/12.6.543
Gao S., Strüder-Kypke M. C., Al-Rasheid K. A. S., Lin X., Song W. (2010). Molecular Phylogeny of Three Ambiguous Ciliate Genera: Kentrophoros, Trachelolophos and Trachelotractus (Alveolata, Ciliophora). Zool Scripta 39 (3), 305–313. doi: 10.1111/j.1463-6409.2010.00416.x
Gong J., Qing Y., Guo X., Warren A. (2014). Candidatus Sonnebornia Yantaiensis a Member of Candidate Division OD1, as Intracellular Bacteria of the Ciliated Protist Paramecium Bursaria (Ciliophora, Oligohymenophorea). Systematic Appl. Microbiol. 37 (1), 35–41. doi: 10.1016/j.syapm.2013.08.007
Gong J., Qing Y., Zou S., Fu R., Su L., Zhang X., et al. (2016). Protist-Bacteria Associations: Gammaproteobacteria and Alphaproteobacteria Are Prevalent as Digestion-Resistant Bacteria in Ciliated Protozoa. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00498
Graf J. S., Schorn S., Kitzinger K., Ahmerkamp S., Woehle C., Huettel B., et al. (2021). Anaerobic Endosymbiont Generates Energy for Ciliate Host by Denitrification. Nature 591 (7850), 445–450. doi: 10.1038/s41586-021-03297-6
Hall T., Biosciences I., Carlsbad C. (2011). BioEdit: An Important Software for Molecular Biology. GERF Bull. Biosci. 2 (1), 60–61.
Lagkouvardos I., Lesker T. R., Hitch T. C. A., Gálvez E. J. C., Smit N., Neuhaus K., et al. (2019). Sequence and Cultivation Study of Muribaculaceae Reveals Novel Species, Host Preference, and Functional Potential of This Yet Undescribed Family. Microbiome 7 (1), 28. doi: 10.1186/s40168-019-0637-2
Lagkouvardos I., Pukall R., Abt B., Foesel B. U., Meier-Kolthoff J. P., Kumar N., et al. (2016). The Mouse Intestinal Bacterial Collection (miBC) Provides Host-Specific Insight Into Cultured Diversity and Functional Potential of the Gut Microbiota. Nat. Microbiol. 1 (10), 16131. doi: 10.1038/nmicrobiol.2016.131
Lanzoni O., Plotnikov A., Khlopko Y., Munz G., Petroni G., Potekhin A. (2019). The Core Microbiome of Sessile Ciliate Stentor Coeruleus is Not Shaped by the Environment. Sci. Rep. 9 (1), 11356. doi: 10.1038/s41598-019-47701-8
Lei Y., Xu K. (2008). Research Progress in the Ecology of Benthic Ciliated Protozoa. Acta Hydrobiol Sin. 32, 155–160. doi: 10.3724/sp.j.0000.2008.70155
Lind A. E., Lewis W. H., Spang A., Guy L., Embley T. M., Ettema T. J. G. (2018). Genomes of Two Archaeal Endosymbionts Show Convergent Adaptations to an Intracellular Lifestyle. ISME J. 12 (11), 2655–2667. doi: 10.1038/s41396-018-0207-9
Martin M. (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet J. 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Martinez-Garcia M., Brazel D., Poulton N. J., Swan B. K., Gomez M. L., Masland D., et al. (2012). Unveiling in Situ Interactions Between Marine Protists and Bacteria Through Single Cell Sequencing. ISME J. 6 (3), 703–707. doi: 10.1038/ismej.2011.126
Miyake S., Ding Y., Soh M., Low A., Seedorf H. (2020). Cultivation and Description of Duncaniella Dubosii Sp. Nov., Duncaniella Freteri Sp. Nov. And Emended Description of the Species Duncaniella Muris. Int. J. Syst. Evol. Microbiol. 70 (5), 3105–3110. doi: 10.1099/ijsem.0.004137
Muñoz-Gómez S. A., Kreutz M., Hess S. (2021). A Microbial Eukaryote With a Unique Combination of Purple Bacteria and Green Algae as Endosymbionts. Sci. Adv. 7 (24), eabg4102. doi: 10.1126/sciadv.abg4102
Nowack E. C., Melkonian M. (2010). Endosymbiotic Associations Within Protists. Philosophical Transactions of the Royal Society of London. Ser. B: Biol. Sci. 365 (1541), 699–712. doi: 10.1098/rstb.2009.0188
Omar A., Zhang Q., Zou S., Gong J. (2017). Morphology and Phylogeny of the Soil Ciliate Metopus Yantaiensis N. Sp. (Ciliophora, Metopida), With Identification of the Intracellular Bacteria. J. Eukaryotic Microbiol. 64 (6), 792–805. doi: 10.1111/jeu.12411
Ormerod K. L., Wood D. L. A., Lachner N., Gellatly S. L., Daly J. N., Parsons J. D., et al. (2016). Genomic Characterization of the Uncultured Bacteroidales Family S24-7 Inhabiting the Guts of Homeothermic Animals. Microbiome 4 (1), 36. doi: 10.1186/s40168-016-0181-2
Park T., Yu Z. (2018). Do Ruminal Ciliates Select Their Preys and Prokaryotic Symbionts? Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01710
Peng Z. H., Jiao S., Wei G. H. (2021). Research Methods of Microbial Biogeography. Bio-protocol, e2003930. doi: 10.21769/BioProtoc.2003930
Plotnikov A. O., Balkin A. S., Gogoleva N. E., Lanzoni O., Khlopko Y. A., Cherkasov S. V., et al. (2019). High-Throughput Sequencing of the 16S rRNA Gene as a Survey to Analyze the Microbiomes of Free-Living Ciliates Paramecium. Microbial Ecol. 78 (2), 286–298. doi: 10.1007/s00248-019-01321-x
Posada D. (2008). Jmodeltest: Phylogenetic Model Averaging. Mol. Biol. Evol. 25 (7), 1253–1256. doi: 10.1093/molbev/msn083
Pruesse E., Peplies J., Glöckner F. O. (2012). SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinformatics 28 (14), 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41 (1), 590–596. doi: 10.1093/nar/gks1219
Raikov I. B. (1971). Bactéries Épizoiques Et Mode De Nutrition Du Cilié Psammophile Kentrophoros Fistulosum Fauré-Fremiet (Étude Au Microscope Électronique). Protistologica 7 (3), 365–378.
Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian Phylogenetic Inference Under Mixed Models. Bioinformatics 19 (12), 1572–1574. doi: 10.1093/bioinformatics/btg180
Rosati G. (2002). “Ectosymbiosis in Ciliated Protozoa,” in Symbiosis: Mechanisms and Model Systems. Ed. Seckbach J. (Dordrecht: Springer Netherlands), 475–488.
Salzman N. H., de Jong H., Paterson Y., Harmsen H. J. M., Welling G. W., Bos N. A. (2002). Analysis of 16S Libraries of Mouse Gastrointestinal Microflora Reveals a Large New Group of Mouse Intestinal Bacteria. Microbiology 148 (11), 3651–3660. doi: 10.1099/00221287-148-11-3651
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 75 (23), 7537–7541. doi: 10.1128/AEM.01541-09
Scholz C. F. P., Kilian M. (2016). The Natural History of Cutaneous Propionibacteria, and Reclassification of Selected Species Within the Genus Propionibacterium to the Proposed Novel Genera Acidipropionibacterium Gen. Nov., Cutibacterium Gen. Nov. And Pseudopropionibacterium Gen. Nov. Int. J. Systematic Evol Microbiol. 66 (11), 4422–4432. doi: 10.1099/ijsem.0.001367
Seah B. K. B., Antony C. P., Huettel B., Zarzycki J., Schada von Borzyskowski L., Erb T. J., et al. (2019). Sulfur-Oxidizing Symbionts Without Canonical Genes for Autotrophic CO2 Fixation. mBio 10 (3), e01112–e01119. doi: 10.1128/mBio.01112-19
Seah B. K. B., Schwaha T., Volland J.-M., Huettel B., Dubilier N., Gruber-Vodicka H. R. (2017). Specificity in Diversity: Single Origin of a Widespread Ciliate-Bacteria Symbiosis. Proc. R. Soc. B: Biol. Sci. 284 (1858), 20170764. doi: 10.1098/rspb.2017.0764
Stamatakis A. (2014). RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30 (12), 2725–2729. doi: 10.1093/molbev/mst197
Van Valkenburgh B. (1991). Iterative Evolution of Hypercarnivory in Canids (Mammalia: Carnivora): Evolutionary Interactions Among Sympatric Predators. Paleobiology 17 (4), 340–362. doi: 10.1017/S0094837300010691
Volland J.-M., Schintlmeister A., Zambalos H., Reipert S., Mozetič P., Espada-Hinojosa S., et al (2018). NanoSIMS and Tissue Autoradiography Reveal Symbiont Carbon Fixation and Organic Carbon Transfer To Giant Ciliate Host. ISME J. 12(3), 714–727. doi: 10.1038/s41396-018-0069-1
Wang Y., Ji D., Yin J. (2019). Morphology and Phylogeny of Two Phialina Species (Ciliophora, Haptoria) From Northern China. Eur. J. Protistol 67, 46–58. doi: 10.1016/j.ejop.2018.10.002
Wilbert N., Song W. (2005). New Contributions to the Marine Benthic Ciliates From the Antarctic Area, Including Description of Seven New Species (Protozoa, Ciliophora). J. Natural History 39 (13), 935–973. doi: 10.1080/00222930400001509
Xu Y., Huang J., Warren A., Al-Rasheid K. A. S., Al-Farraj S. A., Song W. (2011). Morphological and Molecular Information of a New Species of Geleia (Ciliophora, Karyorelictea), With Redescriptions of Two Kentrophoros Species From China. Eur. J. Protistol 47 (3), 172–185. doi: 10.1016/j.ejop.2011.03.003
Yılmaz L.Ş., Noguera D. R. (2007). Development of Thermodynamic Models for Simulating Probe Dissociation Profiles in Fluorescence in Situ Hybridization. Biotechnol. Bioengineering 96 (2), 349–363. doi: 10.1002/bit.21114
Keywords: Kentrophoros, symbiosis, bacterial diversity, ciliate, Muribaculaceae
Citation: Bi L, Zhang X, Zou S, Ji D and Zhang Q (2022) Detection of Prokaryotes on the Astomatous Ciliated Protist Kentrophoros flavus (Ciliophora, Karyorelictea) Revealed A Consistently Associated Muribaculaceae-Like Bacterium. Front. Mar. Sci. 9:879388. doi: 10.3389/fmars.2022.879388
Received: 19 February 2022; Accepted: 03 May 2022;
Published: 06 June 2022.
Edited by:
Alexey Potekhin, Saint Petersburg State University, RussiaReviewed by:
Valentina Serra, University of Pisa, ItalyCopyright © 2022 Bi, Zhang, Zou, Ji and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daode Ji, ZGFvZGVqaUAxMjYuY29t; Qianqian Zhang, cXF6aGFuZ0B5aWMuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.