 Yuanri Hu
Yuanri Hu Yangzhen Li
Yangzhen Li Peng Cheng
Peng Cheng Songlin Chen
Songlin Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 01 April 2022
Sec. Marine Fisheries, Aquaculture and Living Resources
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.868987
Blind-side hypermelanosis is an emerging concern across the flatfish aquaculture industry including Chinese tongue sole (Cynoglossus semilaevis). Circular RNAs (circRNAs) as endogenous non-coding RNAs have been acknowledged to play important roles in various biological processes. However, the underlying regulatory mechanisms of circRNAs involved in flatfish blind-side hypermelanosis remain unclear. In this study, to profile the circRNA expression pattern and circRNA-microRNA-messenger RNA (mRNA) network, high-throughput sequencing was performed by using blind-side normal and hypermelanotic skins of tongue sole. A total of 73 differentially expressed circRNAs were identified, and the competing endogenous RNA (ceRNA) network was constructed. Furthermore, circRNA host genes and mRNAs involved in ceRNA network were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes enrichment analyses. Several GO terms and pathways of biological significance were identified and well addressed the generation of blind-side hypermelanosis to some extent. These interesting results extend the understanding of the functional profile of circRNAs and yield valuable insights into the molecular regulatory mechanisms of hypermelanosis in flatfish.
Chinese tongue sole (Cynoglossus semilaevis) is an indigenous flatfish species with high economic value in China. In the larvae stage, flatfishes undergo a morphological metamorphosis toward an asymmetric body plan (both eyes on the same side) to adapt the benthic dwelling lifestyle (Chen et al., 2014; Fox et al., 2018; Lü et al., 2021). Then, the body color of ocular and blind side exhibited as black-brown and pure white respectively in the wild. However, under commercial production environments, the coat color of the blind side is prone to pigment staining, which is referred to as blind-side hypermelanosis. The high proportion of hypermelanic individuals in production populations combined with their high area ratio of hypermelanosis has emerged as a major concern for fish farmers and consumers, as this yields an inferior product and substantially lower profit (Li et al., 2021a; Li et al., 2021b). Numerous key genes associated with blind-side hypermelanosis phenotype have been screened and identified, and single-nucleotide polymorphism mutations were detected in transcriptional level in flatfishes (Peng et al., 2020; Li et al., 2021a; Zhang et al., 2021). However, there is no clear evidence explaining the underlying regulatory mechanism of blind-side hypermelanosis especially regarding the regulation of non-coding RNA (ncRNA).
Evidence gathered over the past decade suggests that circular RNAs (circRNAs) as endogenous ncRNAs play crucial roles in various biological processes in eukaryotic cells (Memczak et al., 2013; Ebbesen et al., 2017). However, the functional mechanisms of circRNA are both complex and controversial. For a better understanding of the functions of circRNAs, firstly, the functions of their host genes should be focused. For example, circRNA can bind to its host gene locus through R-loop (RNA : DNA hybrid) formation, resulting in transcriptional pausing, which leads to phenotypic changes (Conn et al., 2017; Xu et al., 2020). Secondly, circRNAs could function as microRNA (miRNA) molecule sponges to counteract the inhibition of miRNA-mediated messenger RNA (mRNA) through competing endogenous RNA (ceRNA) networks (Salmena et al., 2011). In humans, recent studies detected thousands of circRNAs in melanocytes that were found to play key roles in cell proliferation and invasion (Wang et al., 2018b). Furthermore, circRNA can relieve the inhibition of miRNA on the tyr gene to promote melanogenesis through a ceRNA regulation network (Jiang et al., 2020). However, the specific roles and mechanisms of circRNAs in melanogenesis remain poorly understood, and further studies are needed (Zhou et al., 2021). Currently, pigmentation-related miRNAs have been identified in several fish species, including red tilapia (Oreochromis sp. red tilapia) (Wang et al., 2018a), common carp (Cyprinus carpio) (Yan et al., 2013), and Koi carp (Cyprinus carpio carpio) (Luo et al., 2018; Dong et al., 2020). Literature focusing on circRNA and ceRNA regulatory network involved in pigmentation is not available in fishes.
Therefore, in the current study, high-throughput sequencing and bioinformatics analyses were performed to decode the potential roles of circRNA in tongue sole blind-side hypermelanosis. Host genes of circRNAs were identified and functionally annotated. For the first time, the circRNA-miRNA-mRNA regulatory network associated with the blind-side hypermelanosis trait was predicted. This research expands the spectrum of non-coding regulatory mechanisms underpinning malpigmentation in flatfish.
Experimental fish (~50% pigmented area, body weight ~15 g) were randomly collected from our flatfish breeding center in Tangshan, China. Blind-side hypermelanotic (BH) and normal (BN) skin tissues were dissected from nine individuals. Each sample group used three pooled replicates (from three different individuals) that were named BH-1, BH-2, BH-3, BN-1, BN-2, and BN-3. Skin tissues were immediately frozen and stored in liquid nitrogen for later RNA isolation.
Total RNA from BH and BN samples was isolated using TRIzol reagent kit (Invitrogen, CA, USA) according to the manufacturer’s protocol. RNA degradation and contamination were assessed using RNase-free agarose gel electrophoresis. RNA quality was monitored using Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Then, mRNA was enriched by Oligo(dT) beads, and ribosomal RNA (rRNA) was removed by Ribo-ZeroTM Magnetic Kit (Epicentre, Madison, WI, USA). Subsequently, enriched mRNA was fragmented into short fragments by using fragmentation buffer and reverse transcribed into cDNA with random primers. DNA polymerase I, RNase H, dNTP, and buffer were used to synthesize second-strand cDNA. These cDNA fragments were purified with QiaQuick PCR extraction kit (Qiagen, Venlo, Netherlands), end repaired, poly(A) appended, and ligated to Illumina sequencing adapters. Finally, ligation products were collected by agarose gel electrophoresis, PCR amplified, and sequenced using Illumina HiSeq2500 by Gene Denovo Biotechnology Co. (Guangzhou, China). Raw data are available from the NCBI database under accession number PRJNA760350.
Raw data were filtered according to a series of procedures. Reads containing adapter, ploy-A, and low-quality reads were removed from raw data using fastp (v0.18.0) (Chen et al., 2018). Then, clean reads were aligned to the reference genome by HISAT2 (v2.1.0) (Kim et al., 2015). To obtain anchor reads, sequences from each end of unmapped reads (default 20 bp) were intercepted. These anchor reads were then aligned to the genome again, and the results were submitted to find_circ software (Memczak et al., 2013) to identify circRNAs. Host genes of circRNAs were identified by matching the genomic location of circRNAs to the location of genes detected by TopHat (Trapnell et al., 2009).
The expression level of circRNAs was normalized using reads per million mapped reads (RPM). The package edgeR (Robinson et al., 2010) was used to analyze circRNA differential expressions between two groups. CircRNAs displaying changes of >2-fold with p < 0.05 were considered differential expression circRNAs (DEcircRNAs). A total of four DEcircRNAs were randomly selected for quantitative real-time reverse transcription PCR (qRT-PCR) analysis and Sanger sequencing to verify the Illumina sequencing result. The PrimeScript 1st strand cDNA Synthesis Kit (Takara, Japan) was used for synthetizing cDNA, and elongation factor 1-alpha (EF1α) was set as a reference gene. Primer sets were designed by Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) based on their sequences. Specifically, the primers for circRNAs were designed to span the circRNA back-splicing junction. Information on all primers is listed in Table 1. qRT-PCR was performed with the 7500 Fast Real-Time PCR System (Applied Biosystems, USA) using SYBR® green I Premix Ex-Taq™ (TaKaRa, Japan). The relative expression of the circRNAs was calculated based on the comparative cycle threshold (Ct) method (2−ΔΔCt) (Livak and Schmittgen, 2001). Data are expressed as mean ± standard error. All statistical analyses were performed using R software (v4.1.0).

Table 1 Information of qRT-PCR primers.
To better understand the underlying molecular functions of circRNAs, the host genes of circRNAs were annotated by Gene Ontology (GO) (http://geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000). The significance for GO terms and KEGG pathways was set at a threshold p-value <0.05.
In combination with our previously reported transcriptome result of differentially expressed miRNA (DEmiRNA) (Li et al., 2022), the DEmiRNAs targeted by DEcircRNAs were predicted by three software packages: RNAhybrid (Rehmsmeier et al., 2004), miRanda (http://www.microrna.org/microrna/home.do), and TargetScan (http://www.targetscan.org/). To better assess the potential functions of circRNAs, a circRNA-mediated ceRNA network was constructed with previously reported miRNA–mRNA pair data (Li et al., 2022). The standard steps of network construction were as follows: 1) candidate genes included only DEcircRNAs, DEmiRNAs, and DEmRNAs (i.e., differentially expressed genes); 2) the expression of miRNAs and target genes (circRNAs or mRNAs) followed opposite trends (Spearman rank correlation coefficient <-0.7); 3) expression correlation (Pearson correlation coefficient) between circRNA and mRNA >0.9; 4) co-expressed circRNA–mRNA pairs shared the same miRNA. Cytoscape (v3.9.1) software (http://www.cytoscape.org/) was used to visualize the ceRNA network.
To further identify the potential roles of circRNAs and circRNA-miRNA-mRNA triplets involved in blind-side hypermelanosis in tongue sole, GO enrichment (http://geneontology.org/) and KEGG (Kanehisa and Goto, 2000) pathway analyses were performed to annotate the function and signaling pathways of DEmRNAs involved in ceRNA networks. The significance for GO terms and KEGG pathways was set at a threshold p-value <0.05.
Six rRNA-depleted RNA libraries from normal and melanistic skin in tongue sole were sequenced to analyze the profiles of circRNAs using RNA sequencing (RNA-Seq). A total of 1,556 unique circRNAs between blind-side normal skin and blind-side hypermelanotic skin were identified. According to the positions of the circRNAs in the genome, in both groups, the most abundant circRNA type was exonic circRNA (76%–77%), whereas intergenic and intronic circRNAs only accounted for 23%–24% (Figures 1A, B). In this study, 91% (1,414/1,556) of circRNAs were less than 2,000 bp in length, and most lengths ranged from 0 to 500 bp (54%) (Figure 1C). circRNAs with respect to their host genes showed that the great majority of host genes produce only one circRNA isoform (1,556 circRNA candidates from 1,147 host genes) (Figure 1D).
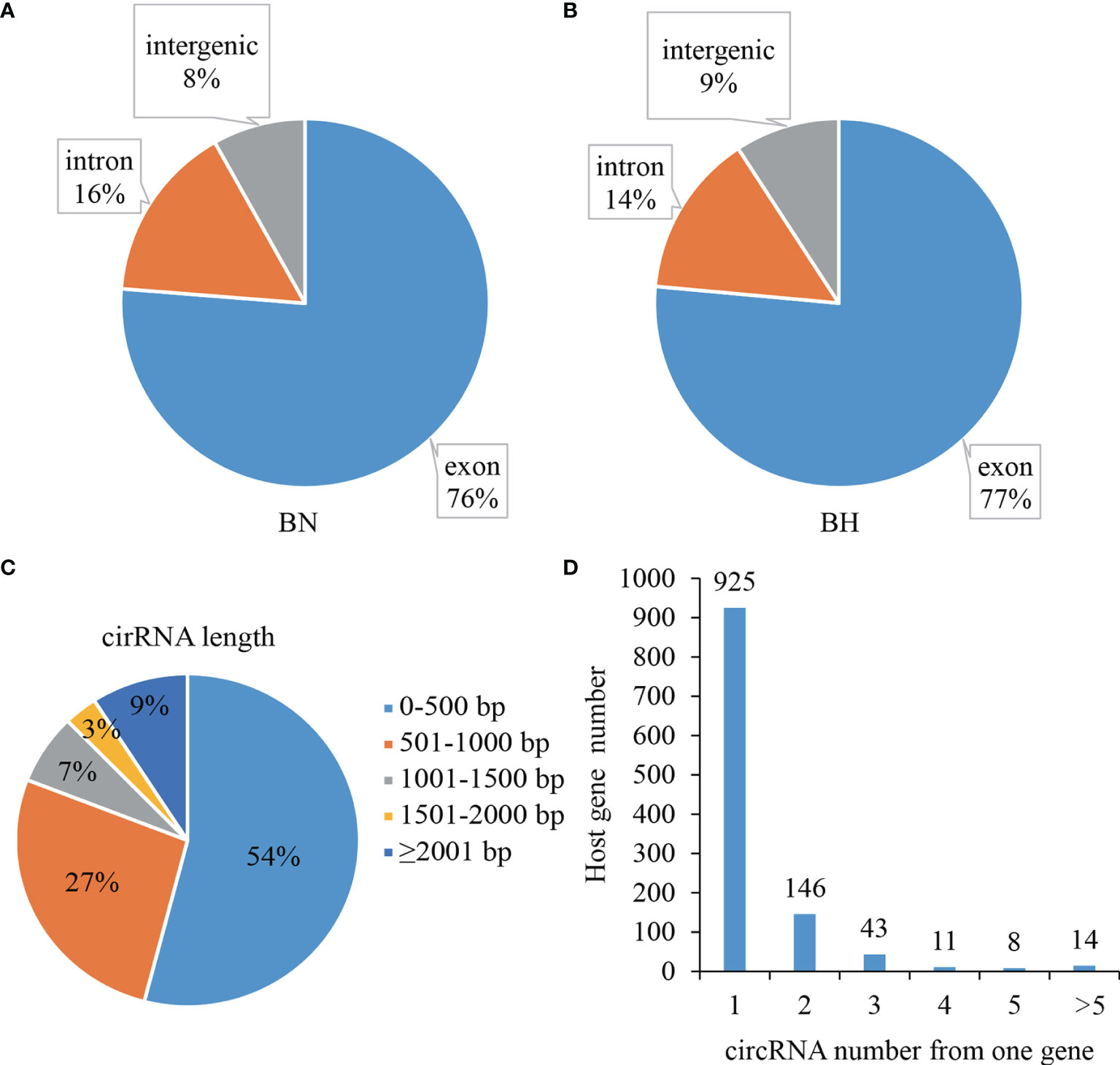
Figure 1 Summary statistics of circular RNA (circRNA) in blind-side skin tissues of tongue sole. (A) Derivation distribution of circRNAs in blind-side normal (BN) and blind-side hypermelanotic (BH) groups. (B) Length distribution of circRNAs. (C) CircRNA numbers from host gene. (D) CircRNA numbers from host genes.
To screen the key circRNAs regulating melanotic mechanisms, the package edgeR (Robinson et al., 2010) was used to identify the DEcircRNAs. Based on the differential expression analysis, 73 DEcircRNAs (71 downregulated and two upregulated) were identified (p < 0.05) (Figure 2 and Supplementary Table S1). qRT-PCR was performed to confirm the reliability of RNA-Seq data. Sanger sequencing was employed to verify the existence of the back-splicing junction sites (Figure 3A). The results showed that the expression trends of the four randomly selected DEcircRNAs were similar to those obtained by sequencing data (Figure 3B).
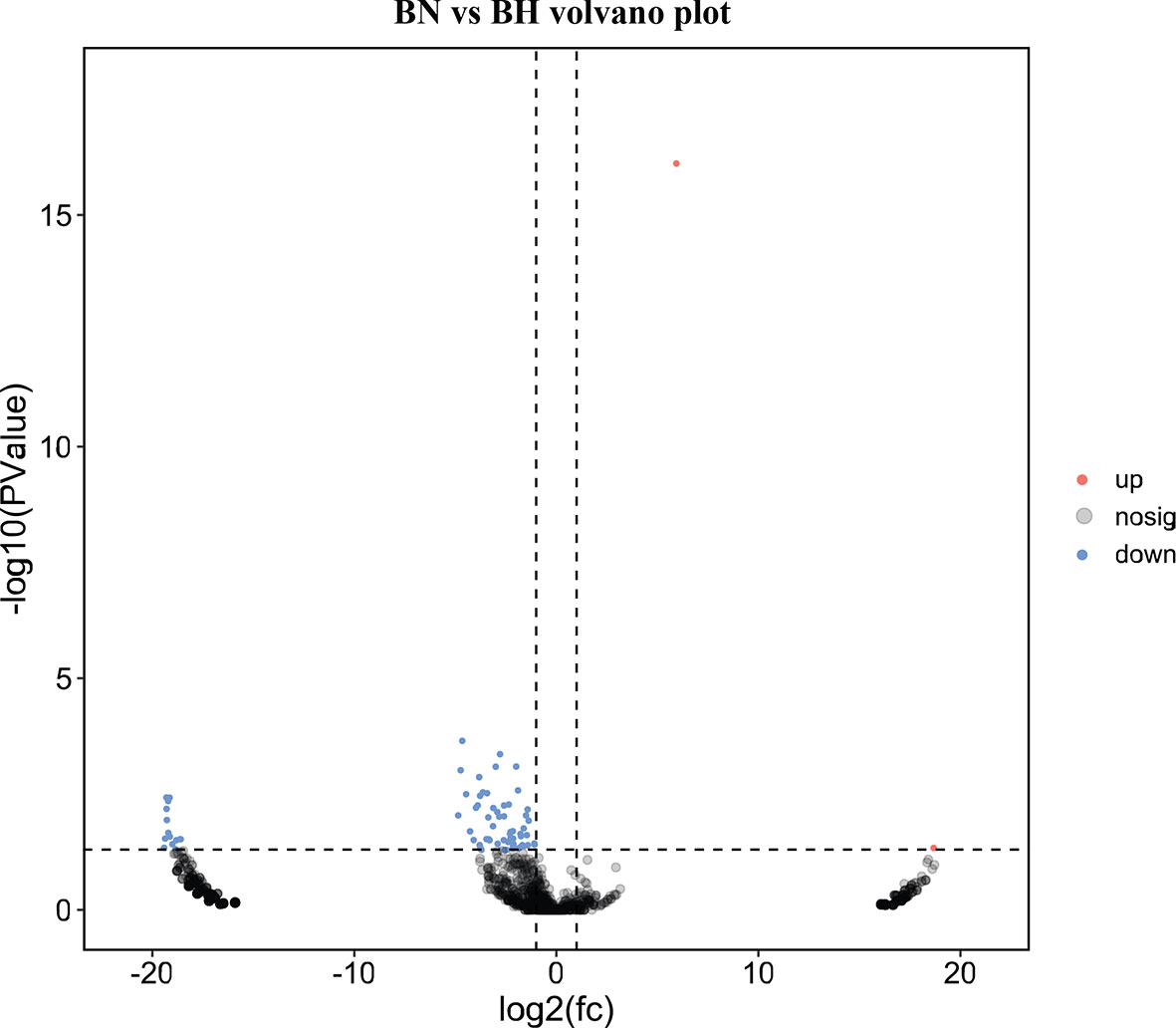
Figure 2 Volcano plot of differentially expressed circular RNAs (DEcircRNAs) between blind-side normal (BN) and blind-side hypermelanotic (BH) groups in tongue sole.
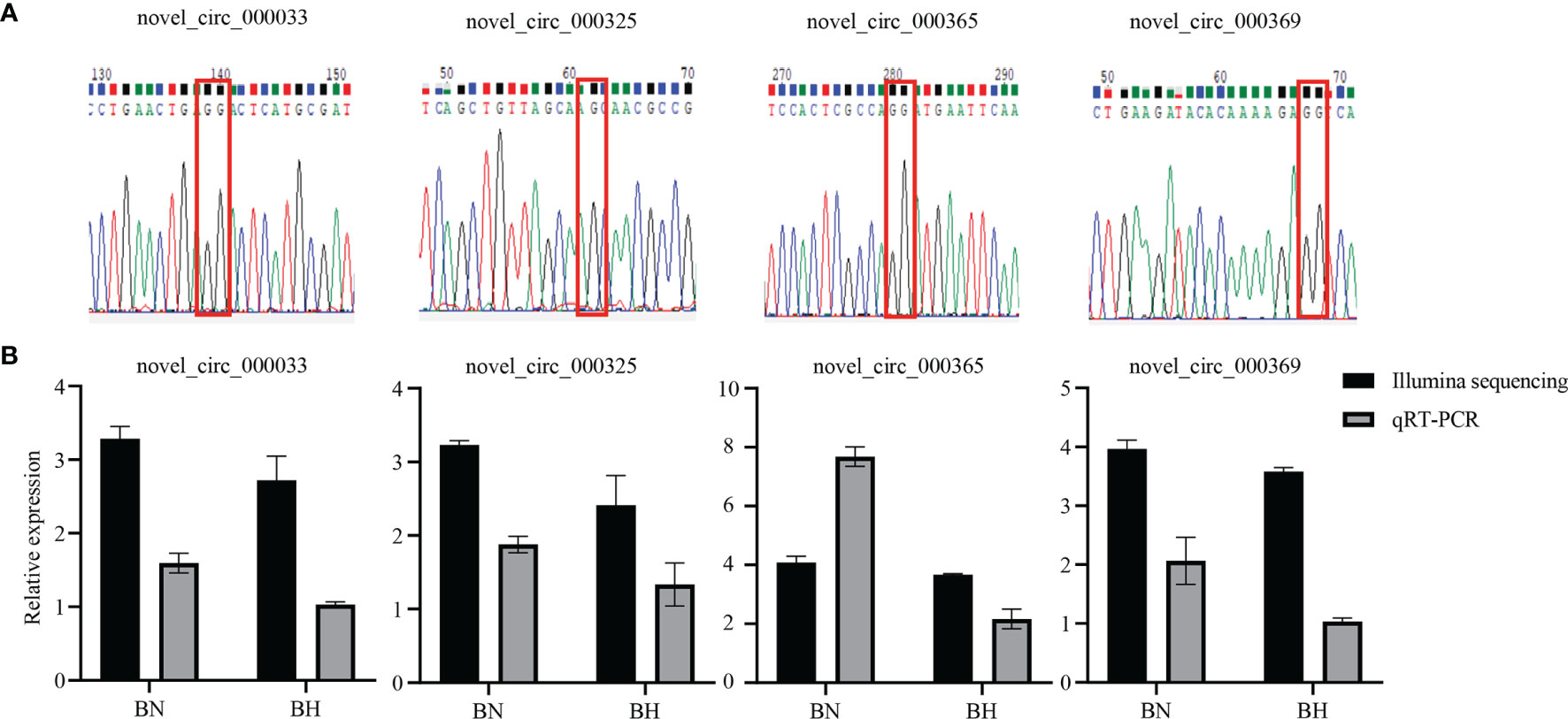
Figure 3 Validation of circular RNAs (circRNAs) by Sanger sequencing and qRT-PCR. (A) Sanger sequencing for the head-to-tail back-splicing of circRNAs. The nucleotides highlighted by red boxes indicate the junction sites. (B) The qRT-PCR analysis results and data obtained from RNA sequencing (RNA-Seq) in blind-side normal (BN) and blind-side hypermelanotic (BH) groups.
circRNAs participated in transcriptional control by cis-regulation of their host genes (Li et al., 2015). Functional enrichment analyses were conducted for host genes against GO and KEGG databases. The corresponding host genes of DEcircRNAs were significantly enriched in 60 GO terms (p < 0.05), some of which functioned in ion export and Golgi-related processes, such as “calcium ion export”, “Golgi membrane”, and “Golgi apparatus part” (Figure 4A and Supplementary Table S2). KEGG enrichment analysis showed that these host genes were significantly enriched in seven pathways (p < 0.05), including “Ribosome biogenesis in eukaryotes”, “Wnt signaling pathway”, and “tight junction” (Figure 5A and Supplementary Table S3).
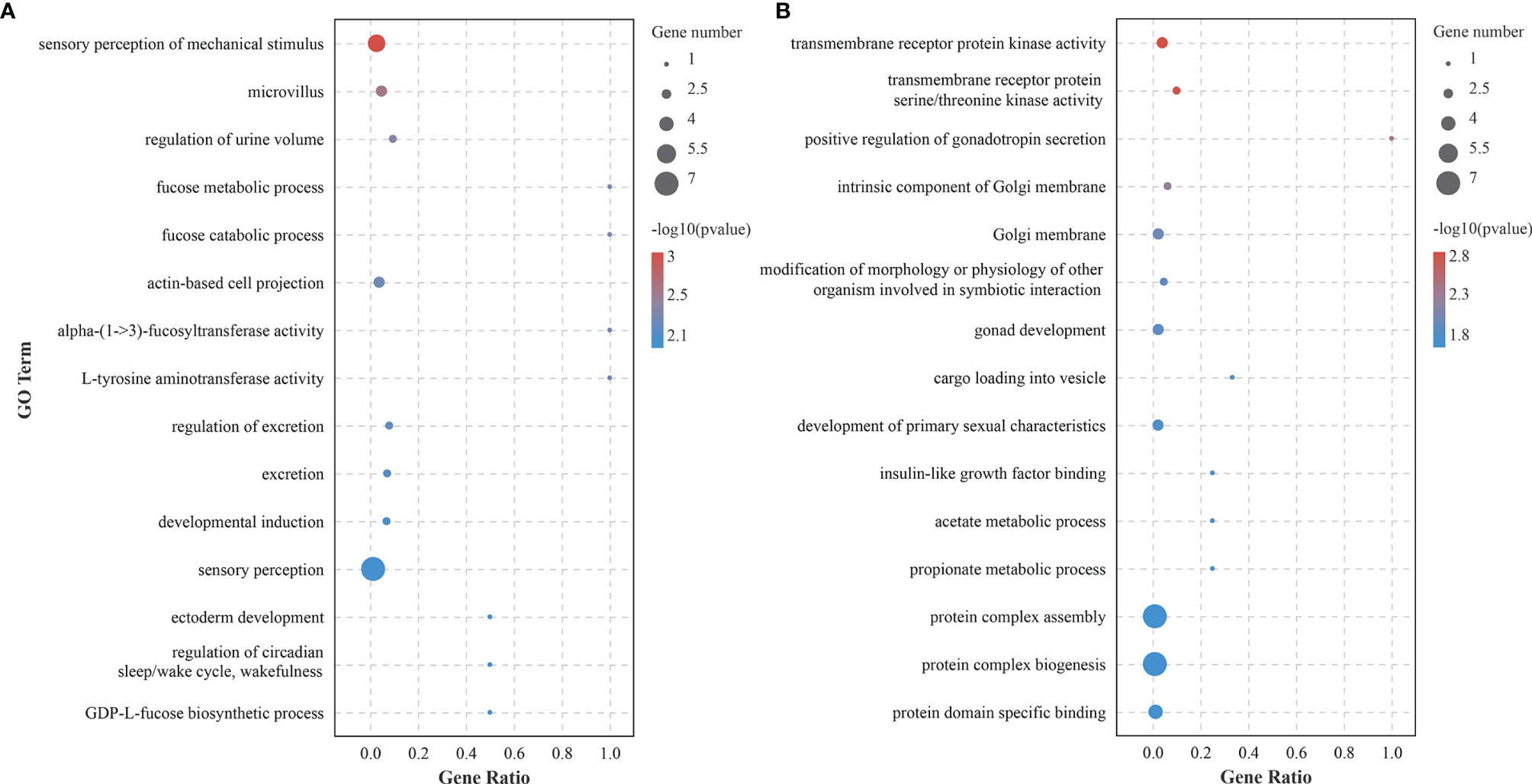
Figure 4 Gene Ontology (GO) enrichment analysis of host genes of differentially expressed circular RNAs (circRNAs) (A) and mRNAs (B) involved in the competing endogenous RNA (ceRNA) network.
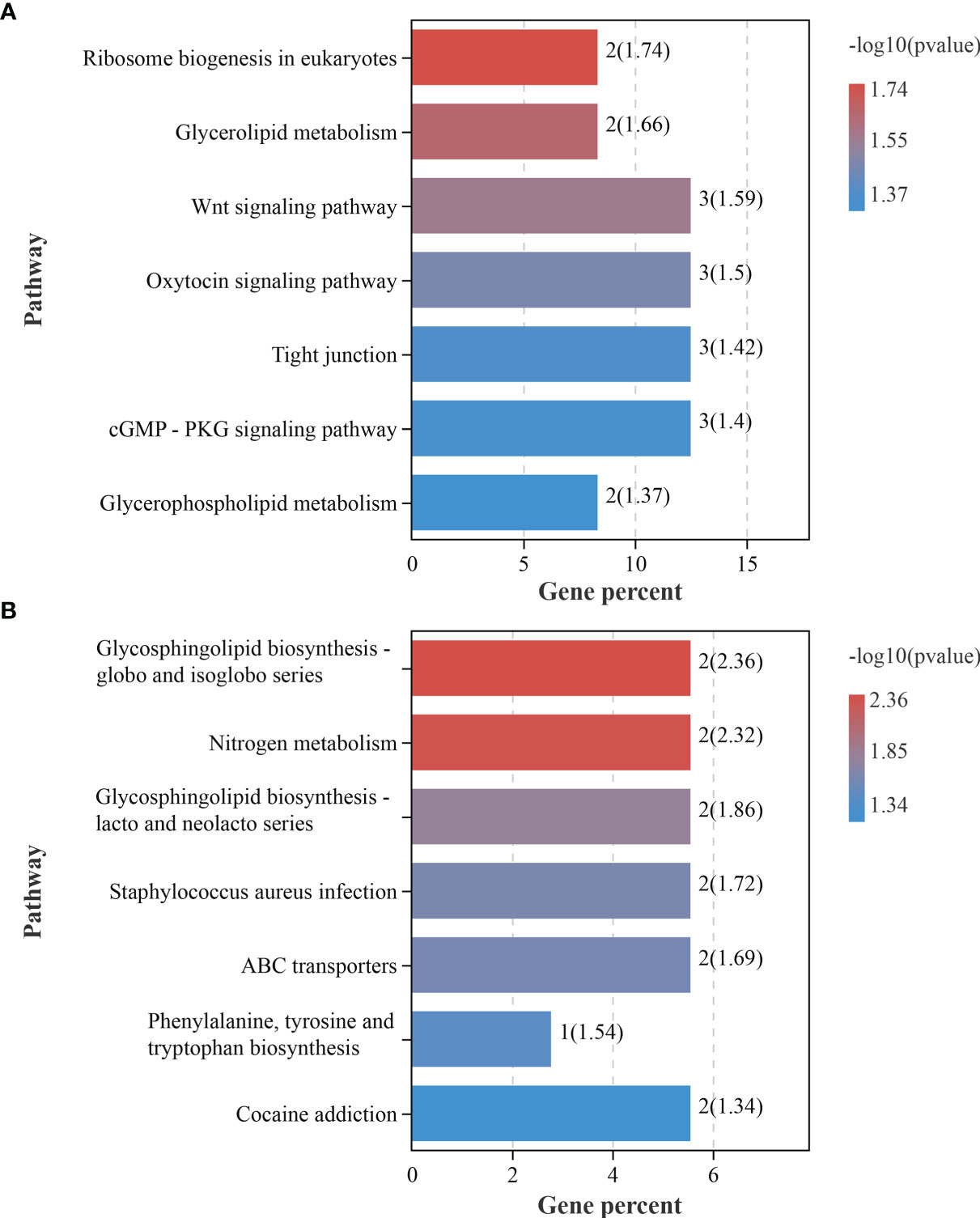
Figure 5 Kyoto Encyclopedia of Genes and Genomes pathway analysis of the differentially expressed circular RNA (circRNA) host genes (A) and mRNAs (B) involved in the competing endogenous RNA (ceRNA) network.
To further identify the hypermelanosis regulatory network of mRNAs and ncRNAs, ceRNA networks based on the ceRNA hypothesis were constructed using DEcircRNAs, DEmiRNAs, and DEmRNAs. The relationship between DEcircRNA and DEmiRNA was predicted by RNAhybrid, miRanda, and TargetScan. A total of 241 DEcircRNA–DEmiRNA pairs with rho (Spearman rank correlation coefficient) <-0.7 were obtained, including 52 DEcircRNAs and 99 DEmiRNAs (Supplementary Table S4). According to previously reported sequence data, there are 2,274 DEmRNA–DEmiRNA pairs. After the above series of screening, 186 DEcircRNA-DEmiRNA-DEmRNA triplets (ceRNA network) were established (Figure 6 and Supplementary Table S5). The network contained 40 circRNAs, 76 miRNAs, and 101 mRNAs. These results show that there is a complicated regulatory mechanism of blind-side hypermelanosis in tongue sole, further suggesting that circRNAs may act as miRNA sponges, thus playing key roles in regulating mRNA expression for blind-side pigment generation and maintenance. Three circRNAs (i.e., novel_circ_000176, novel_circ_000301, and novel_circ_001038) with high degrees in the network are considered to be hub circRNAs, which are worthy of intensive study in the future.
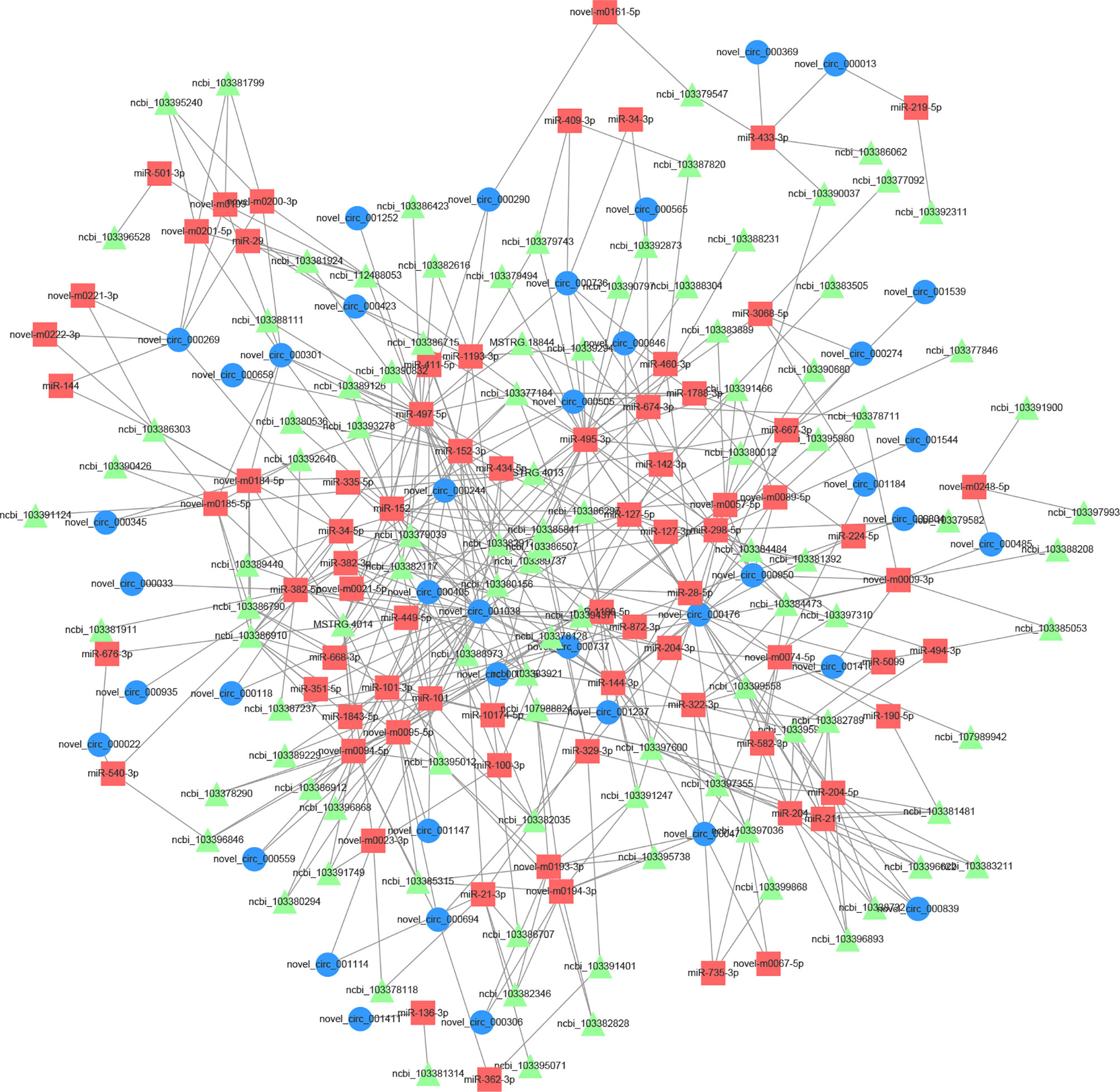
Figure 6 The predicted biomathematical circular RNA (circRNA)-microRNA (miRNA)-messenger RNA (mRNA) networks associated with blind-side hypermelanosis in tongue sole.
To determine the biological roles of DEcircRNAs, GO and KEGG analyses were performed based on the DEmRNAs in the ceRNA crosstalk network. The results showed that these genes were significantly enriched into 87 GO terms (p < 0.05), among which “sensory perception of mechanical stimulus” was the most significant term (Figure 4B and Supplementary Table S6), which is interesting. Moreover, we astutely noted that two GO terms “L-tyrosine aminotransferase activity” and “L-phenylalanine aminotransferase activity” may be of biological significance. KEGG pathway analysis identified seven significantly enriched pathways, of which “glycosphingolipid biosynthesis-globo and isoglobo series” was the most involved (Figure 5B and Supplementary Table S7).
Despite the contribution of genetic and plastic traits to environmental adaptation, phenotypic plasticity allows species to survive in a wider range of conditions and respond quicker to environmental changes (Agrawal, 2001). One significant example of phenotypic plasticity in animals is body color change. In flatfish, blind-side pigment distribution abnormalities are often observed in farming populations. To understand the processes responsible for this adaptation and evolution, research linking to genetic factor and phenotype of body color is necessary (Chen et al., 2019). Recently, an increasing number of studies have focused on circRNAs because of their involvement in pigmentation regulatory function (Zhu et al., 2020). However, research on circRNAs in non-model organisms such as flatfish species is still lacking, especially studies on melanogenesis mechanism.
In the present paper, an overview of circRNAs in the blind-side skins of tongue sole is provided. A total of 73 DEcircRNAs were found between BN and BH. GO enrichment and KEGG pathway analyses for host genes of these DEcircRNAs showed that the variation in hypermelanosis was related to cellular components, such as “Golgi membrane”, “Golgi apparatus part”, and “intrinsic component of Golgi membrane”. Tyrosinase family proteins (e.g., TYR and TYRP1) play important roles in the melanin biosynthetic pathway. These two enzymes can be trafficked through the Golgi apparatus and then join type II melanosomes, thus promoting the deposition of melanin that finally develops into a mature melanosome (Dooley et al., 2012). On this basis, it can be speculated that circRNAs, such as novel_circ_000033, novel_circ_000485, and novel_circ_000748, might regulate their host genes to affect TYR and TYRP1 transport on the Golgi. Additionally, the host gene (Slc8a2) of novel_circ_000935 was significantly enriched in “calcium ion export” and “cGMP-PKG signaling pathway”. Slc8a2 is a member of the solute carrier 8 family and encodes a Na+/Ca2+ exchanger to mediate cellular Ca2+ efflux/influx (Quednau et al., 2004). The cGMP-PKG signaling pathway can inhibit Ca2+ currents (Sandoval et al., 2017). Increasing intracellular Ca2+ can mediate pigment aggregation (Aspengren et al., 2003). Therefore, the downregulated novel_circ_000935 and Slc8a2 may partially explain the blind-side hypermelanosis in tongue sole.
Understanding concerning details on the body color change depends on knowledge of where pigment translocations originate on isolated melanophores from fish (Sköld et al., 2016). The aggregation of pigment in fish is regulated by hormonal stimuli or direct innervation (Aspengren et al., 2003). When melanogenesis is activated, molecular motors carry melanosomes along the cytoskeleton, dispersing them throughout the cell or aggregating them in the cell center (Aspengren et al., 2009). During melanogenesis, pigment translocation depends on actin filaments and microtubules. In this study, it was found that circRNAs (novel_circ_000176, novel_circ_000290, novel_circ_000737, novel_circ_000839, novel_circ_000846, novel_circ_001038, novel_circ_001147, novel_circ_001237, and novel_circ_001416) mediated in the ceRNA network were associated with stimulus/perception-related biological processes. Examples are “sensory perception of mechanical stimulus”, “sensory perception”, and “neurological system process”. The relevant genes (e.g., slc17a8, Espn, MYO15A, and PJVK) mined in this study have been suggested to play important roles in mediating sensory transduction in various mechanosensory and chemosensory processes (Sekerkova et al., 2008; Zhang et al., 2015; Ryu et al., 2017; Zhu et al., 2018; Zhang et al., 2019). Empirically, blind-side skin of tongue sole would show varying degrees of abrasions (Figure 7A) because of long-term captivity in aquaculture tanks (that are relatively smooth). A plausible explanation for this is that to better adapt to the benthic life, sensory perception-related genes are downregulated, and tongue sole fish may have “tired” of the smooth environment.

Figure 7 (A) Blind-side hypermelanosis of tongue sole (15-month-old male) with wounds (red box). (B) Blind-side cycloid scale (from 15-month-old female tongue sole). (C) Blind-side ctenoid scale (from 15-month-old female tongue sole).
Coincidentally, blind-side hypermelanosis is accompanied by a morphological change of scales in the corresponding parts, where the scales cannot develop into cycloid scales (Figure 7B) but remain ctenoid scales (Figure 7C). Normally, after metamorphosis in the larva stage, the blind side is initially covered by ctenoid scales (without pigmentation); then, ctenoid scales gradually change into cycloid scales in the juvenile stage. While, after metamorphosis, the ocular side is covered by ctenoid scales throughout the later life stages. This demonstrates that the morphological structure of scales may be closely associated with hypermelanosis. Fish scales originate from the dermis of the skin (Sire and Akimenko, 2004). In nature, flatfish bury themselves in sediment such as sand for camouflage, predation, and protection. To avoid removal of sand from the ocular-side skin by water currents, ctenoid scales lead to an increase of friction forces (Spinner et al., 2016). From this perspective, the interruption of morphological change of scales on the blind side may imply that tongue sole fish could tackle the relatively smooth artificial environment by maintaining ctenoid scales on the blind side. In this study, it was found that certain host genes of circRNAs were significantly enriched in “Wnt signaling pathway” and “tight junction” pathway. Previous studies reported that these two pathways are related to the development of fish scales (Aman et al., 2018; Wang et al., 2021). However, the causal relationship between ctenoid scale maintenance and hypermelanosis is still unclear.
In fishes, morphological color change requires weeks or months, which implies that substantial increases or decreases in the numbers of chromatophores are needed (Sköld et al., 2016). A previous report regarding tongue sole has indicated that the melanocyte is a requisite for blind-side pigmentation (Shi et al., 2015). Melanin is enriched in melanocytes and mainly synthesized from tyrosine. Tyrosine aminotransferase (TAT) can catalyze phenylalanine into tyrosine (Mehere et al., 2010). Increased or reduced expression of TAT affects the synthesis of tyrosine and melanin. In the present study, it was predicted that novel_circ_000405 may act as a sponge for miR-1193-3p and miR-495-3p to regulate TAT expression. The expression level of TAT in the BH group was significantly lower than that in the BN group. This suggests that novel_circ_000405 may also play an important role in blind-side hypermelanosis in tongue sole.
This is the first paper to describe the comprehensive expression of circRNAs of the blind-side hypermelanosis in tongue sole and predict the functional profile of circRNAs based on host gene analysis and ceRNA regulatory network construction. Numerous key circRNAs of functional significance were identified. The results suggest that circRNAs may play vital roles in blind-side pigmentation, and valuable insights into the molecular mechanism of hypermelanosis in flatfish are provided. It is concluded that blind-side hypermelanosis is an interesting game between fish and aquaculture environment. The findings also reveal adaptive trait evolution to the aquaculture environment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: PRJNA760350].
The present study was approved by the Institutional Animal Care and Use Ethics Committee of the Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China. When sampling, fishes were anesthetized with MS-222 to minimize suffering.
YH Methodology, software, and writing—original draft preparation. YL Conceptualization, methodology, data curation, visualization, validation, and writing—reviewing and editing. PC Visualization and investigation. SC Supervision and writing—reviewing and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the China Agriculture Research System of MOF and MARA (CARS-47-G03), National Natural Science Foundation of China (31702333), the Innovative Team Project of Chinese Academy of Fishery Sciences (2020TD20), and Taishan Scholar Climbing Program of Shandong Province China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.868987/full#supplementary-material
Agrawal A. A. (2001). Ecology - Phenotypic Plasticity in the Interactions and Evolution of Species. Science 294, 321–326. doi: 10.1126/science.1060701
Aman A. J., Fulbright A. N., Parichy D. M. (2018). Wnt/beta-Catenin Regulates an Ancient Signaling Network During Zebrafish Scale Development. Elife 7, e37001. doi: 10.7554/eLife.37001
Aspengren S., Sköld H. N., Quiroga G., Martensson L., Wallin M. (2003). Noradrenaline- and Melatonin-Mediated Regulation of Pigment Aggregation in Fish Melanophores. Pigment Cell Res. 16, 59–64. doi: 10.1034/j.1600-0749.2003.00003.x
Aspengren S., Sköld H. N., Wallin M. (2009). Different Strategies for Color Change. Cell Mol. Life Sci. 66, 187–191. doi: 10.1007/s00018-008-8541-0
Chen S., Xu W., Liu Y. (2019). Fish Genomic Research: Decade Review and Prospect. J. Fish. China 43, 1–14. doi: 10.11964/jfc.20181211599
Chen S., Zhang G., Shao C., Huang Q., Liu G., Zhang P., et al. (2014). Whole-Genome Sequence of a Flatfish Provides Insights Into ZW Sex Chromosome Evolution and Adaptation to a Benthic Lifestyle. Nat. Genet. 46, 253–260. doi: 10.1038/ng.2890
Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Conn V. M., Hugouvieux V., Nayak A., Conos S. A., Capovilla G., Cildir G., et al. (2017). A circRNA From SEPALLATA3 Regulates Splicing of its Cognate mRNA Through R-Loop Formation. Nat. Plants 3, 5. doi: 10.1038/nplants.2017.53
Dong Z., Luo M., Wang L., Yin H., Zhu W., Fu J. (2020). MicroRNA-206 Regulation of Skin Pigmentation in Koi Carp (Cyprinus Carpio L.). Front. Genet. 11. doi: 10.3389/fgene.2020.00047
Dooley C. M., Schwarz H., Mueller K. P., Mongera A., Konantz M., Neuhauss S. C. F., et al. (2012). Slc45a2 and V-ATPase are Regulators of Melanosomal pH Homeostasis in Zebrafish, Providing a Mechanism for Human Pigment Evolution and Disease. Pigment Cell Res. 26, 205–217. doi: 10.1111/pcmr.12053
Ebbesen K. K., Hansen T. B., Kjems J. (2017). Insights Into Circular RNA Biology. RNA Biol. 14, 1035–1045. doi: 10.1080/15476286.2016.1271524
Fox C. H., Gibb A. C., Summers A. P., Bemis W. E. (2018). Benthic Walking, Bounding, and Maneuvering in Flatfishes (Pleuronectiformes: Pleuronectidae): New Vertebrate Gaits. Zoology 130, 19–29. doi: 10.1016/j.zool.2018.07.002
Jiang L., Huang J., Hu Y., Lei L., Ouyang Y., Long Y., et al. (2020). Identification of the ceRNA Networks in α-MSH-Induced Melanogenesis of Melanocytes. Aging 13, 2700–2726. doi: 10.18632/aging.202320
Kanehisa M., Goto S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim D., Landmead B., Salzberg S. L. (2015). HISAT: A Fast Spliced Aligner With Low Memory Requirements. Nat. Method 12, 357–360. doi: 10.1038/NMETH.3317
Li Y., Cheng P., Li M., Hu Y., Cui Z., Zhang C., et al. (2021a). Transcriptome Analysis and Candidate Gene Identification Reveals Insights Into the Molecular Mechanisms of Hypermelanosis in Chinese Tongue Sole (Cynoglossus Semilaevis). Aquacult Fish. doi: 10.1016/j.aaf.2021.02.003
Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., et al. (2015). Exon-Intron Circular RNAs Regulate Transcription in the Nucleus. Nat. Struct. Mol. Biol. 22, 256–264. doi: 10.1038/nsmb.2959
Li Y., Hu Y., Cheng P., Chen S. (2022). Identification of Potential Blind-Side Hypermelanosis Related lncRNA-miRNA-mRNA Regulatory Network in a Flatfish Species, Chinese Tongue Sole (Cynoglossus Semilaevis). Front. Genet. 12. doi: 10.3389/fgene.2021.817117
Li Y., Hu Y., Zheng W., Cui Z., Cheng J., Cheng P., et al. (2021b). Insights Into the Heritable Variation of Hypermelanosis in Chinese Tongue Sole (Cynoglossus Semilaevis): Potential for Future Selective Breeding. Aquaculture 539, 736617. doi: 10.1016/j.aquaculture.2021.736617
Livak K. J., Schmittgen T. D. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2–ΔΔct Method. Methods 25, 402–408. doi: 10.1006/meth.2001
Lü Z., Gong L., Ren Y., Chen Y., Wang Z., Liu L., et al. (2021). Large-Scale Sequencing of Flatfish Genomes Provides Insights Into the Polyphyletic Origin of Their Specialized Body Plan. Nat. Genet. 53, 742–751. doi: 10.1038/s41588-021-00836-9
Luo M., Wang L., Zhu W., Fu J., Song F., Fang M., et al. (2018). Identification and Characterization of Skin Color microRNAs in Koi Carp (Cyprinus Carpio L.) by Illumina Sequencing. BMC Genom. 19, 779. doi: 10.1186/s12864-018-5189-5
Mehere P., Han Q., Lemkul J. A., Vavricka C. J., Robinson H., Bevan D. R., et al. (2010). Tyrosine Aminotransferase: Biochemical and Structural Properties and Molecular Dynamics Simulations. Protein Cell 1, 1023–1032. doi: 10.1007/s13238-010-0128-5
Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., et al. (2013). Circular RNAs are a Large Class of Animal RNAs With Regulatory Potency. Nature 495, 333–338. doi: 10.1038/nature11928
Peng K., Zhang B., Xu J., Zhao N., Jia L., Chen J., et al. (2020). Identification of SNPs Related to Hypermelanosis of the Blind Side by Transcriptome Profiling in the Japanese Flounder (Paralichthys Olivaceus). Aquaculture 519, 734906. doi: 10.1016/j.aquaculture.2019.734906
Quednau B. D., Nicoll D. A., Philipson K. D. (2004). The Sodium/Calcium Exchanger Family - SLC8. Pflugers Arch. - Eur. J. Phy. 447, 543–548. doi: 10.1007/s00424-003-1065-4
Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. (2004). Fast and Effective Prediction of microRNA/Target Duplexes. RNA 10, 1507–1517. doi: 10.1261/rna.5248604
Robinson M. D., McCarthy D. J., Smyth G. K. (2010). Edger: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Ryu N., Lee S., Park H. J., Lee B., Kwon T. J., Bok J., et al. (2017). Identification of a Novel Splicing Mutation Within SLC17A8 in a Korean Family With Hearing Loss by Whole-Exome Sequencing. Gene 627, 233–238. doi: 10.1016/j.gene.2017.06.040
Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P. (2011). A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 146, 353–358. doi: 10.1016/j.cell.2011.07.014
Sandoval A., Duran P., Gandini M. A., Andrade A., Almanza A., Kaja S., et al. (2017). Regulation of L-Type Ca(v)1.3 Channel Activity and Insulin Secretion by the cGMP-PKG Signaling Pathway. Cell Calcium 66, 1–9. doi: 10.1016/j.ceca.2017.05.008
Sekerkova G., Zheng L., Mugnaini E., Bartles J. R. (2008). Espin Actin-Cytoskeletal Proteins are in Rat Type I Spiral Ganglion Neurons and Include Splice-Isoforms With a Functional Nuclear Localization Signal. J. Comp. Neurol. 509, 661–676. doi: 10.1002/cne.21755
Shi X., Xu Y., Wu N., Liu X., Yang H., Zang K., et al. (2015). Preliminary Studies on Blind-Side Hypermelanosis of Cynoglossus Semilaevis: Chromatophores Observation and Expression of Proopiomelanocortin. Prog. Fishery Sci. 36, 45–54. doi: 10.11758/yykxjz.20150206
Sire J. Y., Akimenko M. A. (2004). Scale Development in Fish: A Review, With Description of Sonic Hedgehog (Shh) Expression in the Zebrafish (Danio Rerio). Int. J. Dev. Biol. 48, 233–247. doi: 10.1387/ijdb.15272389
Sköld H. N., Aspengren S., Cheney K. L., Wallin M. (2016). Chapter Four - Fish Chromatophores—From Molecular Motors to Animal Behavior. Int. Rev. Cel. Mol. Bio. 321, 171–219. doi: 10.1016/bs.ircmb.2015.09.005
Spinner M., Kortmann M., Traini C., Gorb S. N. (2016). Key Role of Scale Morphology in flatfshes (Pleuronectiformes) in the Ability to Keep Sand. Sci. Rep. 6, 26308. doi: 10.1038/srep26308A.J
Trapnell C., Pachter L., Salzberg S. L. (2009). TopHat: Discovering Splice Junctions With RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Wang Q., Chen J., Wang A., Sun L., Qian L., Zhou X., et al. (2018b). Differentially Expressed circRNAs in Melanocytes and Melanoma Cells and Their Effect on Cell Proliferation and Invasion. Oncol. Rep. 39, 1813–1824. doi: 10.3892/or.2018.6263
Wang Z., Jia Y., Huang X., Zhu D., Liu H., Wang W. (2021). Transcriptome Profiling Towards Understanding of the Morphogenesis in the Scale Development of Blunt Snout Bream (Megalobrama Amblycephala). Genomics 113, 983–991. doi: 10.1016/j.ygeno.2020.12.043
Wang L., Zhu W., Dong Z., Song F., Dong J., Fu J. (2018a). Comparative microRNA-Seq Analysis Depicts Candidate miRNAs Involved in Skin Color Differentiation in Red Tilapia. Int. J. Mol. Sci. 19, 1209. doi: 10.3390/ijms19041209
Xu X., Zhang J., Tian Y., Gao Y., Dong X., Chen W., et al. (2020). CircRNA Inhibits DNA Damage Repair by Interacting With Host Gene. Mol. Cancer 19, 1. doi: 10.1186/s12943-020-01246-x
Yan B., Liu B., Zhu C., Li K., Yue L., Zhao J., et al. (2013). MicroRNA Regulation of Skin Pigmentation in Fish. J. Cell Sci. 126, 3401–3408. doi: 10.1242/jcs.125831
Zhang J., Guan J., Wang H., Yin L., Wang D., Zhao L., et al. (2019). Genotype-Phenotype Correlation Analysis of MYO15A Variants in Autosomal Recessive Non-Syndromic Hearing Loss. BMC Med. Genet. 20, 60. doi: 10.1186/s12881-019-0790-2
Zhang Q. J., Lan L., Li N., Qi Y., Zong L., Shi W., et al. (2015). Identification of a Novel Mutation of PJVK in the Chinese non-Syndromic Hearing Loss Population With Low Prevalence of the PJVK Mutations. Acta Otolaryngol. 135, 211–216. doi: 10.3109/00016489.2014.985799
Zhang B., Peng K., Che J., Zhao N., Jia L., Zhao D., et al. (2021). Single-Nucleotide Polymorphisms Responsible for Pseudo-Albinism and Hypermelanosis in Japanese Flounder (Paralichthys Olivaceus) and Reveal Two Genes Related to Malpigmentation. Fish Physiol. Biochem. 47, 339–350. doi: 10.1007/s10695-020-00916-3
Zhou S., Zeng H., Huang J., Lei L., Tong X., Li S., et al. (2021). Epigenetic Regulation of Melanogenesis. Aging Res. Rev. 69, 101349. doi: 10.1016/j.arr.2021.101349
Zhu Z., Ma Y., Li Y., Li P., Cheng Z., Li H., et al. (2020). The Comprehensive Detection of miRNA, lncRNA, and circRNA in Regulation of Mouse Melanocyte and Skin Development. Biol. Res. 53, 4. doi: 10.1186/s40659-020-0272-1
Keywords: circRNA, ceRNA network, body color, ncRNA, flatfish, malpigmentation
Citation: Hu Y, Li Y, Cheng P and Chen S (2022) Comprehensive Analysis of Circular RNAs to Decipher the Potential Roles in Blind-Side Hypermelanosis in Chinese Tongue Sole (Cynoglossus semilaevis). Front. Mar. Sci. 9:868987. doi: 10.3389/fmars.2022.868987
Received: 03 February 2022; Accepted: 07 March 2022;
Published: 01 April 2022.
Edited by:
Rodrigo Vidal, University of Santiago, ChileReviewed by:
Yunji Xiu, Qingdao Agricultural University, ChinaCopyright © 2022 Hu, Li, Cheng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangzhen Li, bGl5ekB5c2ZyaS5hYy5jbg==; Songlin Chen, Y2hlbnNsQHlzZnJpLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.