
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 29 March 2022
Sec. Marine Biogeochemistry
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.858860
This article is part of the Research TopicChemical and Biogeochemical Processes at Methane and Other Cold SeepsView all 15 articles
 Meng Jin1,2,3
Meng Jin1,2,3 Dong Feng2,4*
Dong Feng2,4* Kangjun Huang5
Kangjun Huang5 Shanggui Gong4
Shanggui Gong4 Min Luo4
Min Luo4 Jörn Peckmann6
Jörn Peckmann6 Xudong Wang4
Xudong Wang4 Yu Hu4
Yu Hu4 Duofu Chen3,4
Duofu Chen3,4The magnesium (Mg) isotopic composition of marine authigenic carbonates is considered as promising archive of ancient seawater geochemistry and paleoenvironments. Previous experimental and theoretical work has shown that Mg isotope fractionation during carbonate mineral formation is a function of mineralogy and precipitation rate. However, information on Mg isotope fractionation is limited for well-defined precipitation rates in natural settings. Here, we investigate pore waters from sediments of an area of active methane seepage in the South China Sea. Low δ13C values (< −48.3‰ VPDB) of dissolved inorganic carbon (DIC) near the sulfate-methane transition zone (SMTZ) indicate that sulfate-driven anaerobic oxidation of methane (SD-AOM) is the predominant biogeochemical process. Pore water composition of dissolved Mg, calcium (Ca), and strontium (Sr) agrees with aragonite as the dominant carbonate mineral at the site ROV1, and high Mg-calcite at sites ROV2 and ROV4. Calculated carbonate precipitation rates are 0.92 μmol cm−2 yr−1 for site ROV2 and 1.24 μmol cm−2 yr−1 for site ROV4; these estimates are similar to previous calculations for seeps from other areas. The pore water δ26Mg values (−0.88‰ to −0.71‰) obtained for the three study sites are similar to those of seawater, in accord with a minor effect of Rayleigh fractionation due to abundant supply of Mg from seawater and insignificant consumption of Mg during carbonate precipitation. The modeled Mg isotope fractionation (ϵ = −2.0‰ to −1.0‰ for core ROV2; ϵ = −1.3‰ to −0.3‰ for core ROV4) can be explained by kinetic isotope fractionation during carbonate precipitation. The calculated carbonate precipitation rates and the degree of fractionation of Mg isotopes support the notion that fractionation is small at high precipitation rates. However, the carbonate precipitation rates calculated for the studied seep environments are much smaller than those in laboratory experiments, documenting a discrepancy of isotopic fractionation between carbonate authigenesis in laboratory experiments and natural environments. These results, including the modeled precipitation rates, provide new constraints for Mg isotope fractionation in natural settings.
Carbonate precipitation, representing a sink of Mg in the ocean, is an important part of the oceanic Mg cycle (Higgins and Schrag, 2015). The Mg isotopic composition of marine carbonates is used as a proxy to constrain the composition of seawater on geological timescales and to trace the global Mg cycle (e.g. Tipper et al., 2006; Fantle and Higgins, 2014; Kasemann et al., 2014; Pogge von Strandmann et al., 2014; Higgins and Schrag, 2015; Gothmann et al., 2017). A large number of theoretical calculations and laboratory experiments have been conducted to determine the fractionation of Mg isotopes during precipitation of different types of carbonate minerals (e.g. Saenger and Wang, 2014; Li et al., 2015; Pinilla et al., 2015; Wang et al., 2017). Fractionation exhibits a temperature dependence of only approximately 0.01‰ °C−1 (Li et al., 2012; Pearce et al., 2012; Wang et al., 2013), while it largely depends on carbonate mineralogy and precipitation rate (Immenhauser et al., 2010; Mavromatis et al., 2013; Wang et al., 2013). The mineralogical control is expressed in a general trend toward the enrichment of heavy Mg isotopes from calcite over magnesite over dolomite to aragonite (Wang et al., 2013). Moreover, small fractionation of Mg isotopes was observed in laboratory experiments with high carbonate precipitation rates (Immenhauser et al., 2010; Wombacher et al., 2011; Mavromatis et al., 2013), opposite to trends reported for Ca isotopes in carbonate minerals (Tang et al., 2008; DePaolo, 2011). This was attributed to the high free energy of hydration of Mg2+ ions (Mavromatis et al., 2013). However, such rate-dependent Mg isotope fractionation was not observed in some other calcite precipitation experiments, although precipitation rates varied over a wide range (Li et al., 2012; Chen et al., 2020). It follows that the influence of carbonate precipitation rate on fractionation of Mg isotopes may not be straightforward. Remarkably, research on the effect of precipitation rate on Mg isotope fractionation was mostly based on laboratory experiments to date. Yet, the conditions of carbonate precipitation in the environment are more complex than conditions during laboratory experiments (Li et al., 2015; Jin et al., 2021). Research investigating Mg isotope fractionation during carbonate precipitation in the environment is still limited (e.g. Lu et al., 2017; Pogge von Strandmann et al., 2019). Such discrepancy adds to the uncertainties in our understanding of the behavior of Mg isotopes during carbonate precipitation.
Organoclastic sulfate reduction (OSR) and sulfate-driven anaerobic oxidation of methane (SD-AOM) are two widespread biogeochemical processes in marine sediments commonly favoring carbonate precipitation (Jørgensen and Kasten, 2006; Schrag et al., 2013). It has been proposed that the rates of carbonate precipitation during SD-AOM are generally faster than those during OSR, owing to the higher rate of SD-AOM and the resulting higher alkalinity compared to OSR (Karaca et al., 2010). High rates of SD-AOM result in high precipitation rates of methane-derived seep carbonates, making these authigenic carbonates a significant component of overall marine sedimentary carbonates (Bradbury and Turchyn, 2019). Previously, Mg isotope fractionation during precipitation has been deduced from the analysis of the mineral products, i.e., seep carbonates (Lu et al., 2017; Jin et al., 2021). These studies suggested that besides precipitation rate, hydrogen sulfide produced by SD-AOM may also affect the fractionation of Mg isotopes (Lu et al., 2017; Jin et al., 2021). However, precipitation rate, which is believed to have a major influence on Mg isotope fractionation based on previous laboratory experiments, cannot be accurately constrained using rock samples. The lack of documented Mg isotope fractionation at well-constrained natural precipitation rates limits our understanding of Mg isotope variability in ancient carbonate rocks (cf. Blättler et al., 2021).
This study presents Mg isotope compositions of pore fluid from the Haima methane seeps of the South China Sea, a well-studied area where seep carbonate is forming today. The precipitation rate of the authigenic seep carbonate was constrained by pore fluid measurements and by the use of a diffusion-advection-reaction (DAR) model. Obtained precipitation rates are significantly lower than those observed in laboratory experiments (Immenhauser et al., 2010; Li et al., 2012; Mavromatis et al., 2013; Chen et al., 2020). The Sr/Ca and Mg/Ca ratios of pore water were used to constrain carbonate mineralogy (Nöthen and Kasten, 2011). Since the mineralogy of the nodules does not necessarily reflect today’s pore water composition – the nodules may have formed in the past under a different pore water regime (cf. Blättler et al., 2021) – we decided to assess the current diagenetic environment from the cation composition of pore waters. For one core, aragonite was found to be the dominate calcium carbonate mineral, for the other two cores it was high-Mg calcite. These results were then used to evaluate the degree of fractionation, enabling us to provide new constraints on the extent of Mg isotope fractionation during carbonate mineral formation in natural environments.
Three gravity piston cores (ROV1, ROV2, and ROV4; water depth approximately 1300 to 1400 m) were collected at the Haima seeps from the northwest South China Sea during the Haiyang-6 cruise in 2020 (Figure 1). The presence of gas hydrates is inferred from widely distributed bottom simulating reflectors and gas chimneys (Hui et al., 2016). Gas hydrates, authigenic carbonates, and living chemosynthesis-based communities were recovered from the study area (Liang et al., 2017).
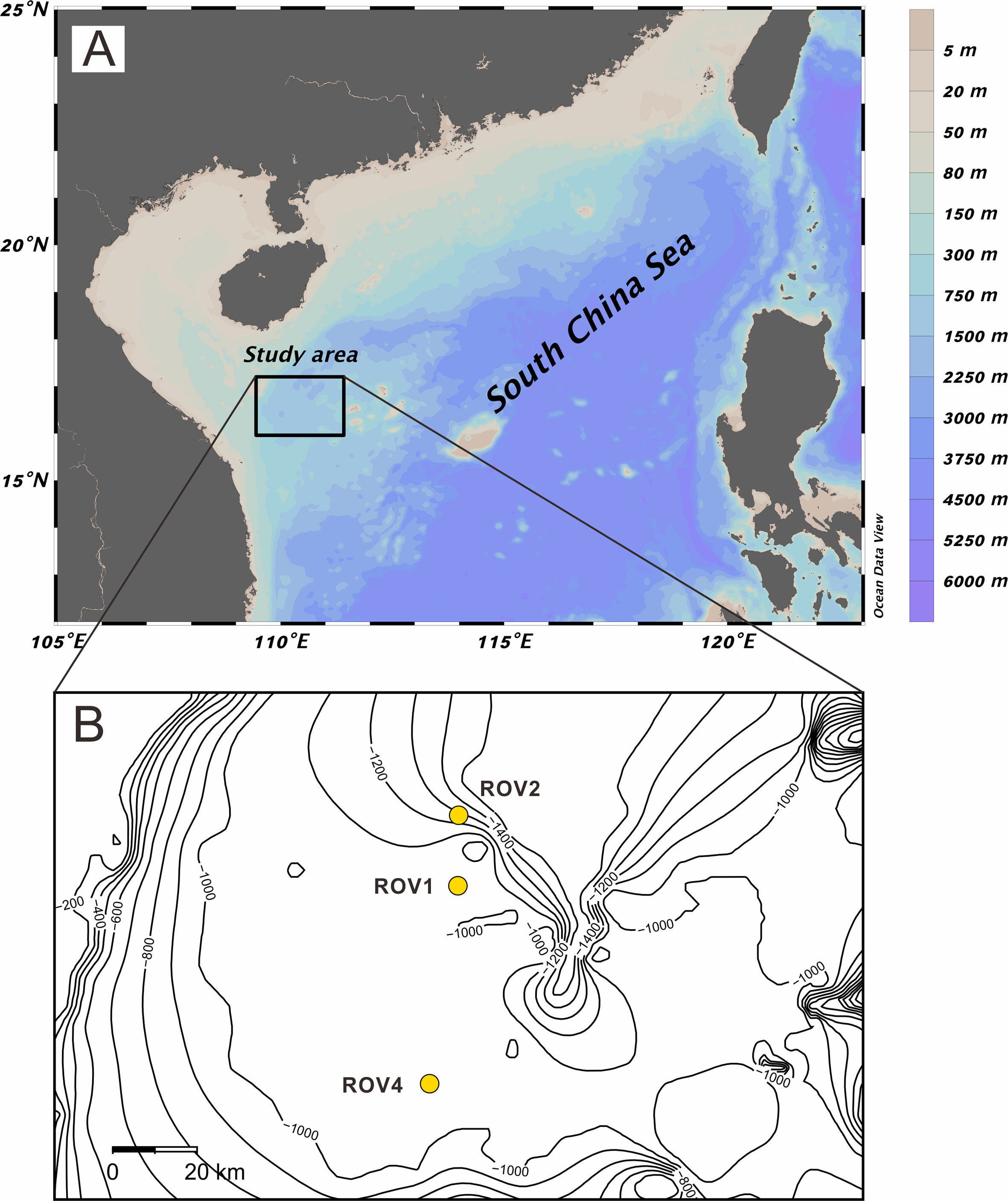
Figure 1 Map showing the location of study area (Schlitzer, Reiner, Ocean Data View, https://odv.awi.de, 2021) (A) and sampling sites (B).
Immediately after recovery, the retrieved cores were brought to the onboard laboratory for pore water extraction. Pore water was collected at 5 to 20 cm intervals using Rhizon samplers with pore size of 0.28 μm. Subsamples for pore water DIC concentration were preserved with saturated HgCl2 solution. For ions, dissolved elements, and Mg isotope analyses, subsamples were acidified with ultrapure concentrated HNO3. All pore water subsamples were stored at 4°C. Carbonate nodules ranging in size from about 5 mm to 35 mm were distributed throughout the core ROV1, but were limited to the upper 230 cm in core ROV2. No nodules were observed in core ROV4.
Sulfate concentrations were measured on a Dionex ICS-5000 ion chromatograph with an analytical precision of < 2%. Calcium (Ca2+), magnesium (Mg2+), and strontium (Sr2+) concentrations were determined by ICP-OES. The analytical precision was better than 3%. Concentration and isotope composition of DIC was determined with a Gas-bench continuous flow Delta-V Plus mass spectrometer after acidifying the sample with pure H3PO4. About 0.5 ml pore water was treated with 6 drops of pure H3PO4 in a glass vial at 25°C. After reacting for 18 h, the produced CO2 was separated through a gas chromatographic column and was transferred to the mass spectrometer for δ13C measurement. The isotopic ratio is expressed relative to the Vienna-Pee Dee Belemnite (V-PDB) standard and the analytical precision was better than ±0.1‰. NaHCO3 lab-standard samples with concentration ranges from 1 to 30 mM were prepared for the determination of DIC concentration. Calculation of DIC concentration was based on the excellent linear correlation (R2>0.999, N=7) between the intensity of CO2 gas produced and the DIC concentration of the NaHCO3 lab-standard samples. The analytical precision of the DIC concentration was <2%. The analyses were conducted at Shanghai Ocean University.
The analytical procedure for Mg isotope analysis was based on Bao et al. (2019). Two columns were used to purify Mg from other matrix metals. Column #1 (loaded with 2 ml of Bio-Rad 200–400 mesh AG50W-X12 resin) was designated to separate Mg from Ca. Column #2 (loaded with 0.5 ml of Bio-Rad 200–400 mesh AG50W-X12 resin) was used to separate Mg from all other matrices (Na, Al, Fe, Ti). To acquire a pure Mg fraction, each sample was passed through column #1 twice, followed by two passages through column #2. The recovery of Mg after the whole procedure of column chemistry was better than 99%, and the total blank for the complete analysis was <10 ng Mg, which is insignificant compared to the mass of the sample. One in-house standard Alfa Mg, one USGS standard (BCR-2), and seawater from the South China Sea were processed with samples for the whole procedure of column chemistry to assess the accuracy of column chemistry. Magnesium isotope ratios were measured with a Thermo Scientific Neptune Plus high-resolution MC-ICPMS at Northwest University, China. Measurements were conducted with the standard-sample bracketing method to correct for the instrumental mass bias and drift. Analyses were performed in low mass resolution mode, simultaneously measuring 26Mg, 25Mg, and 24Mg. The measured Mg isotope ratios are reported in the delta notation as per mil (‰) deviation relative to the DSM3 standard (Young and Galy, 2004): δxMg = [(xMg/24Mg) sample/(xMg/24Mg) DSM3] × 103, where x refers to 25 or 26. All samples were analyzed three times within an analytical session. The internal precision determined on the basis of ≥3 repeated runs of the same sample solution during a single analytical session was better than ±0.10‰ (2SD). Analyses of the Alfa Mg, BCR-2, and seawater standards yielded δ26Mg values of –3.92 ± 0.09‰ (2SD), –0.22 ± 0.07‰ (2SD), and –0.86 ± 0.04‰ (2SD), respectively. When the equivalent 2SD uncertainties are considered, the Mg isotopic compositions reported herein are consistent with previously published values (Foster et al., 2010; Huang et al., 2015; Teng, 2017).
Depth profiles of dissolved sulfate, DIC concentrations, and δ13CDIC values are shown in Figure 2. Sulfate concentrations remain unchanged from 0 to 28 cm depth and linearly decrease to 140 cm for core ROV1, while sulfate concentrations decline from the topmost sediments to 200 cm below the seafloor for core ROV2. At site ROV4, sulfate reveals bottom-water concentration down to 240 cm below the seafloor and then linearly decreases to a depth of 460 cm. The approximate depths of the SMTZ at sites ROV1, ROV2, and ROV4 are 140 cm, 200 cm, and 460 cm below the seafloor, respectively. Concentrations of DIC steadily increase from 4 mM at 20 cm depth to 18 mM at the SMTZ at site ROV1, while rising from 3.3 mM in the topmost sediments to 18 mM at the SMTZ at site ROV2. For core ROV4, concentrations of DIC increase from 3.8 mM at 210 cm to 21 mM at the SMTZ. The δ13CDIC values decrease with depth toward the SMTZ. The lowest δ13CDIC values for the ROV1, ROV2, and ROV4 study sites are –48.3‰, –54.1‰, and –50.6‰, respectively.
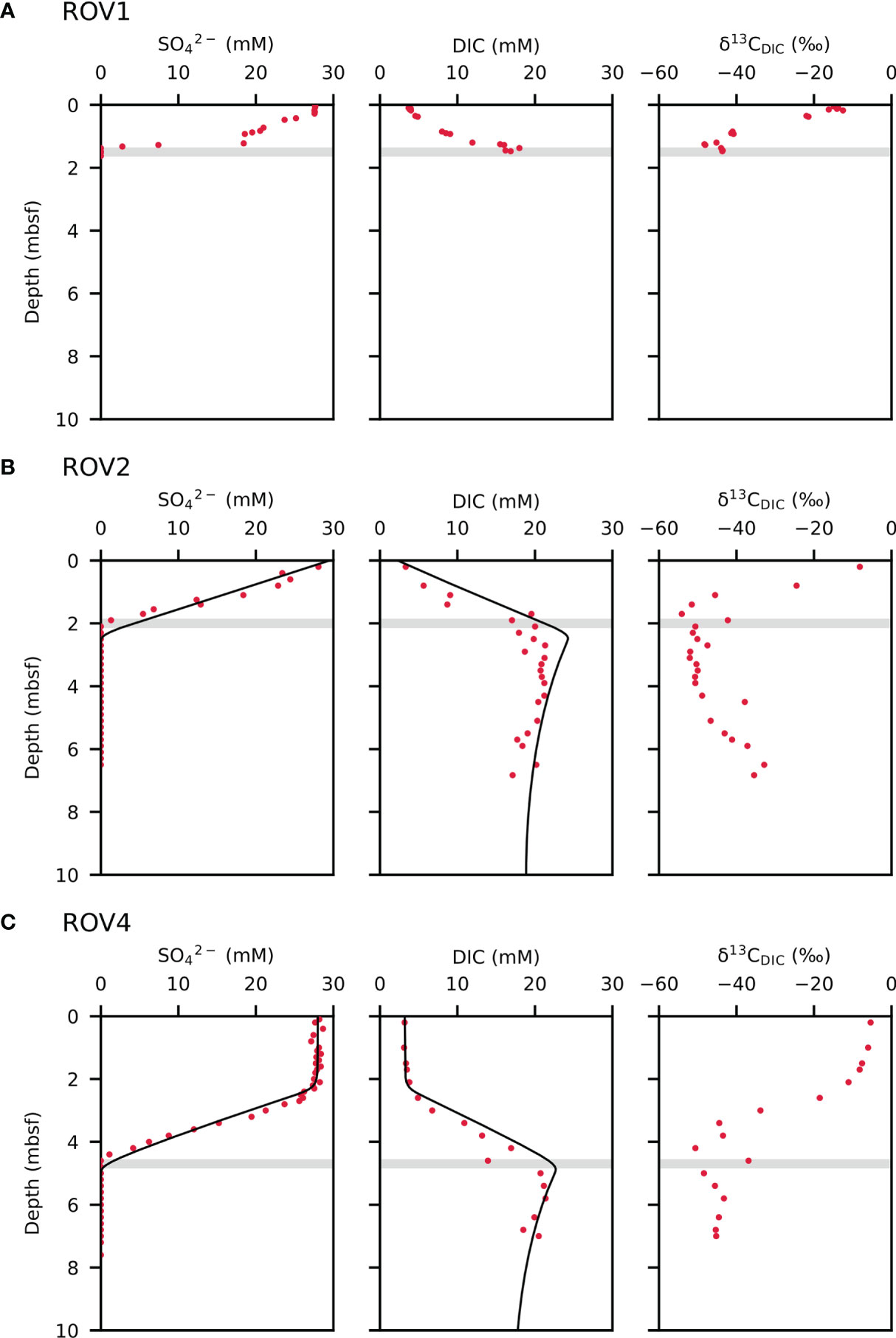
Figure 2 Pore water profiles of sulfate and dissolve inorganic carbon (DIC) concentrations and δ13CDIC values for cores ROV1 (A), ROV2 (B), and ROV4 (C). Dots are measured data and solid lines are model results. The “mbsf” refers to meters below seafloor. The shaded bar marks the sulfate-methane transition zone (SMTZ).
Depth profiles of Ca2+, Mg2+, and Sr2+ concentrations are presented in Figure 3. Calcium concentration drops rapidly from the surface layer to the SMTZ at sites ROV1 and ROV2, while Ca concentration remains constant down to 240 cm below the seafloor before declining toward the SMTZ at the ROV4 site. Magnesium concentrations decline from 52.2 mM to 49.9 mM and from 53.9 mM to 48.2 mM with depth in the measured profiles at the ROV1 and ROV2 sites, respectively. For core ROV4, Mg2+ exhibits bottom water concentration down to a depth of 240 cm below the seafloor and before concentration decreases by 7 mM toward the base of the profile. For Sr2+ concentrations, only pore water at the ROV1 site shows a significant decline of approximately 70 μM within the upper 150 cm. Strontium concentrations at sites ROV2 and ROV4 decrease only little with depth (< 20 μM).
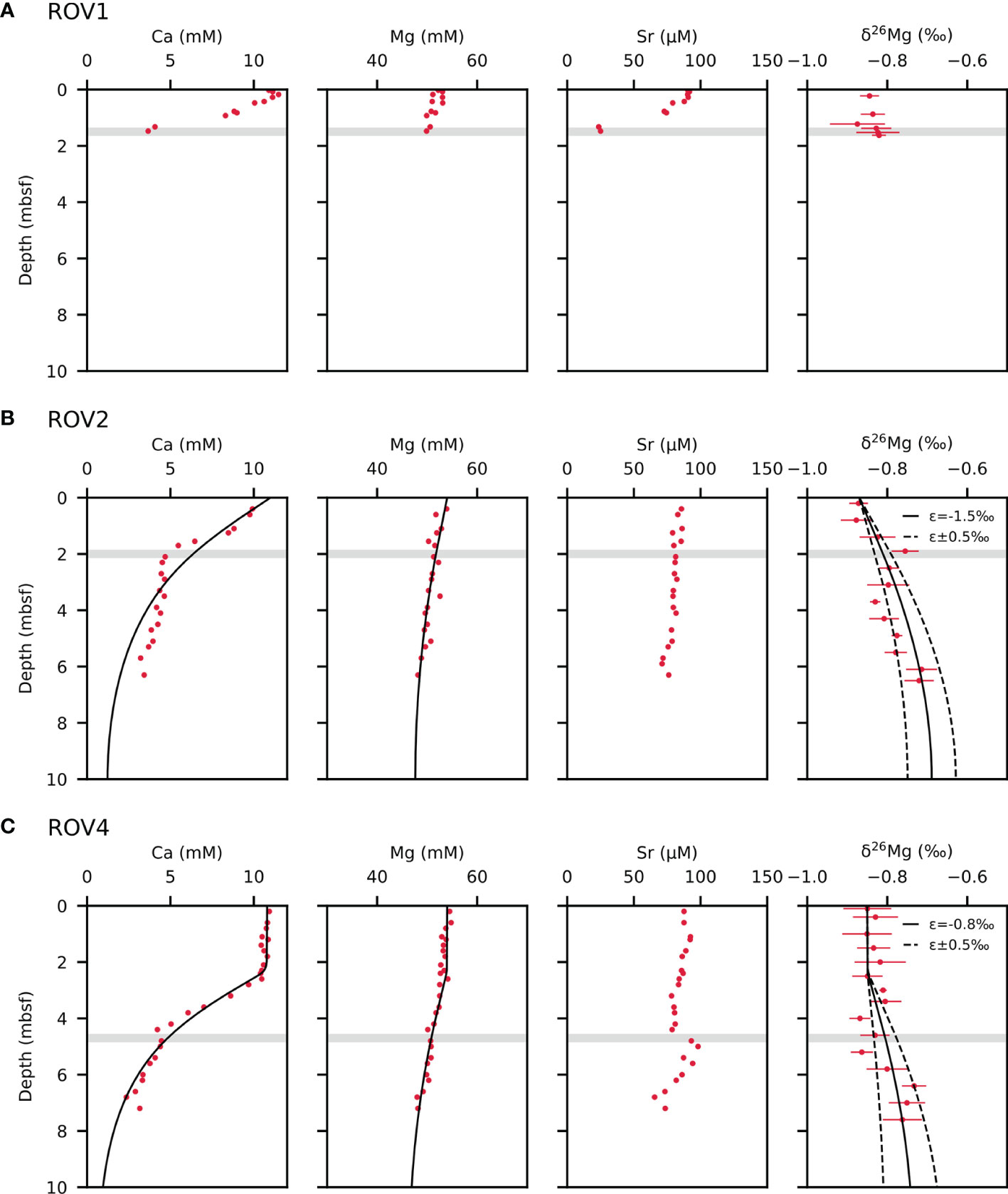
Figure 3 Pore water profiles of calcium (Ca2+), magnesium (Mg2+), strontium (Sr2+) concentrations, and δ26Mg values for cores ROV1 (A), ROV2 (B), and ROV4 (C). Dots are measured data and solid lines are model results. Uncertainties in modeled fractionation factors (ϵ) are calculated by varying fractionation factors by ±0.5‰ indicated by the dotted lines. The “mbsf” refers to meters below seafloor. The shaded bar marks the sulfate-methane transition zone (SMTZ).
Depth profiles of δ26Mg values are represented in Figure 3. The δ26Mg and δ25Mg values are converted to δ26Mg′ and δ25Mg′ values (Young and Galy, 2004), which are then presented in a δ25Mg′ vs. δ26Mg′ plot (Figure 4). All samples and standards analyzed in this study fall on a single mass-dependent fractionation line with a slope of 0.5169. The Mg isotopic composition of pore water from the three sites is close to seawater composition. The Mg isotopic composition of pore water from core ROV1 remains almost constant, ranging from −0.87‰ to −0.82‰. The δ26Mg values of pore water extracted from the ROV2 and ROV4 cores increase with depth from −0.88‰ to −0.71‰, and −0.87‰ to −0.73‰, respectively.
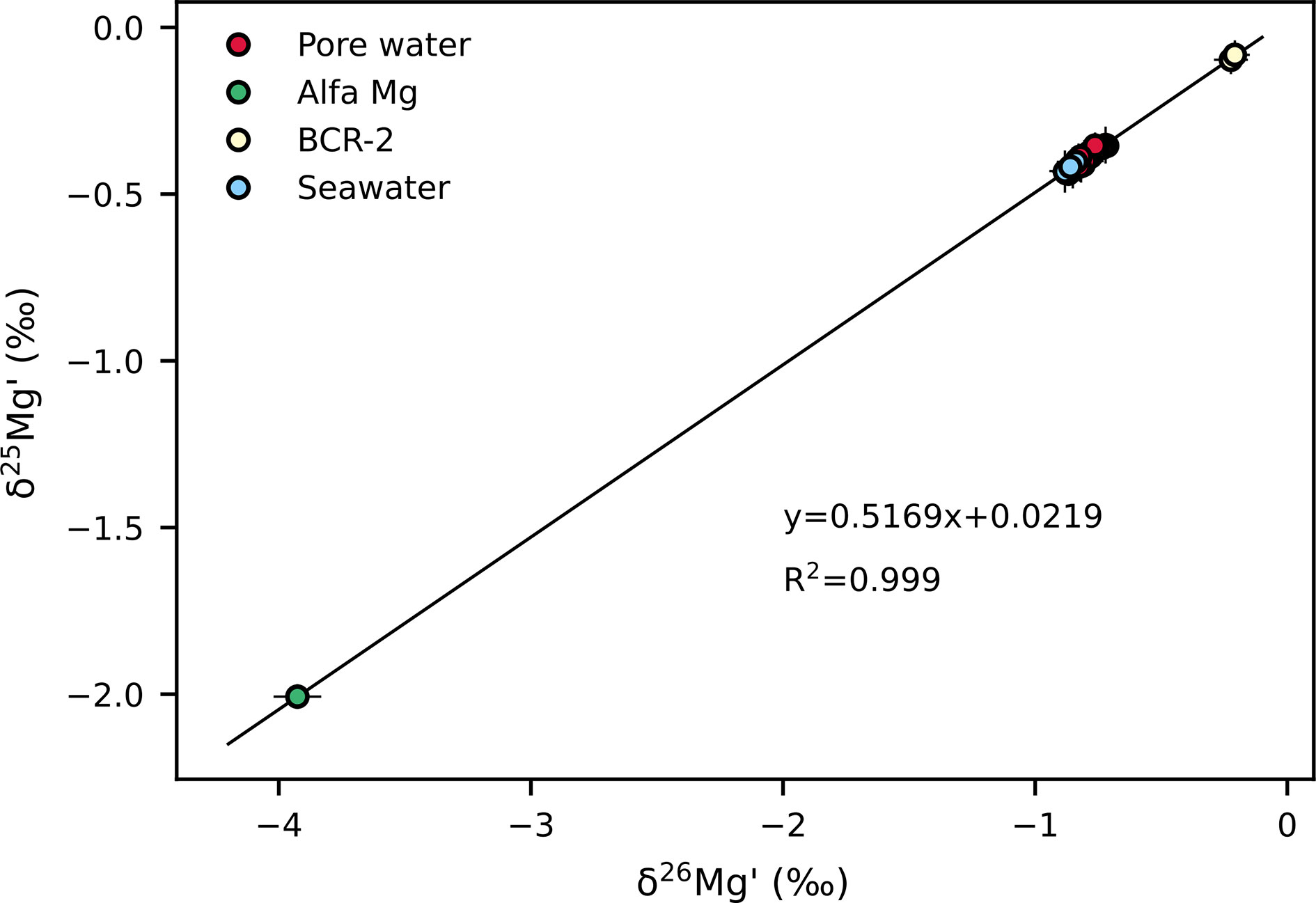
Figure 4 Plot of δ25Mg’ vs. δ26Mg’ illustrating Mg isotopic composition of pore water and standards (Alfa Mg, BCR-2, seawater). Solid line represents the regression line of all data.
The δ13C values of DIC generated from SD-AOM and OSR are close to δ13C values of parent methane and organic matter, respectively (cf. Whiticar, 1999). The δ13C values of methane in the study area range from −72.3‰ to −71.5‰ (Wei et al., 2020). The δ13C value of South China Sea organic matter is approximately −20‰ (Chen et al., 2012). Therefore, the low δ13CDIC values of pore water from the study sites (Figure 2) indicates that SD-AOM is the dominant biogeochemical process. This is confirmed by the observed rapid depletion of sulfate and the shallow depth of the SMTZ. Intense SD-AOM resulted in a DIC concentration, which is known to facilitate carbonate precipitation at methane seeps (Berner, 1980; Aloisi et al., 2000; Naehr et al., 2007). The decrease of Ca2+, Mg2+, and Sr2+ concentrations with depth agrees with carbonate authigenesis in the local sedimentary environment (Figure 3), with the decline of Sr concentration best explained by aragonite formation (Snyder et al., 2007).
Changes of Ca2+, Mg2+, and Sr2+ concentrations in pore water can be used to identify the mineralogy of the carbonate minerals deemed to precipitate under current conditions (e.g. Nöthen and Kasten, 2011). Formation of high-Mg calcite, which has lower Sr/Ca ratios than seawater, results in an increase of the Sr/Ca ratios in pore water. Conversely, aragonite has higher Sr/Ca ratios but lower Mg/Ca ratios than seawater; its removal from pore water will lead to the decrease of Sr/Ca ratios and the increase of Mg/Ca ratios in pore water. Accordingly, the current pore water conditions favor aragonite formation at the ROV1 site, but high-Mg calcite formation at the ROV2 and ROV4 sites (Figure 5).

Figure 5 Plot of Sr/Ca versus Mg/Ca (weight ratios) in pore water of cores ROV1, ROV2, and ROV4 as well as in seawater (de Villiers, 1999). The two bold lines indicate changes in the Sr/Ca to Mg/Ca relationship in pore water with respect to the composition of seawater that occur during precipitation of either aragonite or high Mg-calcite. The Sr/Ca and Mg/Ca ratios of the precipitating aragonite and high Mg-calcite were taken from Bayon et al. (2007) and Nöthen and Kasten (2011).
The one-dimensional, steady state, diffusion-advection-reaction (1D-DAR) model was applied to quantify the geochemical profiles of sediment pore water. This model has been widely used in simulations of early diagenetic processes (e.g. Wallmann et al., 2006; Fantle and DePaolo, 2007; Higgins and Schrag, 2010; Hu et al., 2018).
The simplified 1D-DAR model is expressed as:
where C (mol m−3) is the concentration of a certain element, Φ is porosity, z (m) is the depth below the seafloor, ω (m yr−1) corresponds to advective velocities, D (m2 yr−1) is the vertical diffusivity coefficient of bulk sediment, and ∑R is the first-order rate constant for chemical reactions that remove elements from pore water.
Depth-dependent molecular diffusion coefficients of dissolved species were calculated after Oelkers and Helgeson (1991) and corrected for the effect of tortuosity:
where D0 is the molecular diffusion coefficient in free seawater at the in-situ temperature, salinity, and pressure.
Since porosity and sedimentation rate data are not known, we assume a constant porosity and adopt the sedimentation rate of Wang et al. (2000) within the modeled domain. As a result, in the current absence of externally imposed fluid advection at the seafloor, the advective velocities of pore water are equivalent to the sedimentation rate, s (m yr−1), at the seafloor.
Gas bubble irrigation is considered in the model to fit the obtained pore water data for the ROV4 site above 240 cm. Similar to bioirrigation, gas bubble irrigation promotes the exchange of pore water and bottom water, but its influence may extend to greater depth than bioirrigation (e.g. Haeckel et al., 2007; Hu et al., 2019). According to previous studies (Haeckel et al., 2007; Chuang et al., 2013; Hu et al., 2019), gas bubble irrigation is described by parameters α1 (yr−1) and α2 (cm) that define the irrigation intensity and its attenuation below the irrigation depth Lirr (cm), respectively. The variable α1 is a model fitting parameter, and irrigation depth can be determined with the aid of pore water data. For simplification, α2 is assumed to be a constant value.
Given that SD-AOM is the dominant biogeochemical process at the study sites, carbonate precipitation driven by SD-AOM is assumed to explain the consumption of Mg2+ and Ca2+:
Thus, it is inapplicable to calculate the reaction rate of Mg independently. Precipitation rates (RP) of carbonate were calculated by saturation state and kinetic constant (kCa) in previous studies (e.g. Luff and Wallmann, 2003; Hu et al., 2019):
where Ksp refers to the solubility product of pure calcite for simplification, ignoring Mg contained in carbonate. The reaction rate of Mg was tuned to fit the concentration profiles in Higgins and Schrag (2010). In this study, we calculated the reaction rate of Ca and Mg respectively in order to model the fractionation of Mg isotopes during carbonate precipitation at the ROV2 and ROV4 study sites:
where Ksp1 and Ksp2 refers to the solubility product of CaCO3 and MgCO3, respectively, and kCa and kMg are tuned to fit the measured data. A typical pore water pH value of 7.6 was used to calculate from the modeled DIC concentration (Zeebe and Wolf-Gladrow, 2001).
To model the Mg isotopic profiles (i.e. δ26Mg) of pore water, we treat the isotopes 24Mg and 26Mg separately. Eq. (1) can be re-expressed as follows:
where iMg is the concentration of 24Mg or 26Mg. We use the same diffusion coefficient (DMg) for both 24Mg and 26Mg owing to limited fractionation of Mg isotopes during diffusion ( = 1.00003 ± 0.00006; Richter et al., 2006). Here, carbonate precipitation is the chemical reaction involving Mg ions.
The fractionation of Mg isotopes is represented as:
where
The length of the simulated model domain was set to 1000 cm and 1200 cm for cores ROV2 and ROV4, respectively. Upper boundary conditions for all species were imposed as fixed concentrations (Dirichlet boundary), using measured values for the uppermost sediment layer where available. A zero-concentration gradient (Neumann-type boundary) was imposed at the lower boundary for all species. Model parameters are listed in Table 1.

Table 1 Parameters used in the model to assess Mg isotope fractionation.
The calculated reaction rates of Ca and Mg reach a peak near the SMTZ, owing to the highest DIC concentrations and the highest carbonate saturation in pore water. The depth-integrated rates and converted carbonate precipitation rates are listed in Table 2. The depth-integrated rates refer to the downward fluxes of Ca or Mg per unit surface area. Both the fluxes of Ca and Mg for the study sites are much higher than those calculated for ODP Site 1082, where SD-AOM-driven carbonate precipitation occurs in shallow sediments (Moore et al., 2004). RMg calculated in this study is also higher than that of ODP Site 1082 given by Higgins and Schrag (2010). The SMTZ at ODP Site 1082 is situated between 18 to 24 mbsf (meters below seafloor; Moore et al., 2004), which is much deeper than the position of the SMTZ at our study sites. Higher fluxes of Ca and Mg at the South China Sea sites agree with higher carbonate precipitation rates than those at ODP Site 1082.
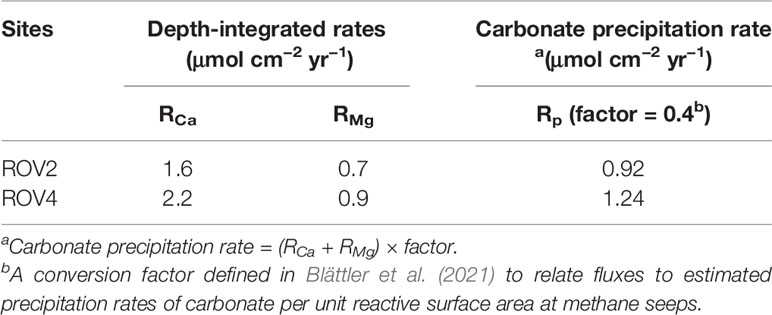
Table 2 Depth-integrated rates of downward flux of Ca and Mg and converted carbonate precipitation rates.
Note that RCa and RMg are in the same order of magnitude, which is consistent with the consumption of Ca and Mg apparent in the depth profiles (Figure 3). A similar situation has been reported by Blättler et al. (2021), who observed a 26% decrease of Mg within 1.7 m of sediment depth. In case of aragonite precipitation, the consumption of Mg in pore water can be neglected owing to the small amount of Mg in aragonite compared to high-Mg calcite (Bayon et al., 2007). Carbonate precipitation rates were typically calculated from the depth profiles of Ca concentrations in previous studies (e.g. Luff and Wallmann, 2003; Karaca et al., 2010). However, in cases of high-Mg calcite precipitation with significant decrease of Mg in depth profiles, carbonate precipitation rates may be underestimated when based on the flux of Ca alone. In this study, RCa and RMg were therefore added up to calculate carbonate precipitation rates. In order to compare the carbonate precipitation rates in this study to that obtained from laboratory experiments, the sum of fluxes of Ca and Mg were converted into the precipitation rate of carbonate per unit reactive surface area. This requires several assumptions and rough estimates relevant to a roughness factor and a surface reactivity factor (Beckingham et al., 2016). A factor of 0.4 was yielded in Blättler et al. (2021) to convert Ca fluxes to carbonate precipitation rates on available mineral surface areas at methane seeps, which was adopted in this study (see Table 2). The resultant carbonate precipitation rates for sites ROV2 and ROV4 are 0.92 μmol cm−2 yr−1 and 1.24 μmol cm−2 yr−1, respectively (Table 2). Uncertainties on estimating were considered to be an order of magnitude in either direction according to Blättler et al. (2021). The intensity of seepage in this study is similar to that in Blättler et al. (2021), and the calculated carbonate precipitation rates are in the same order of magnitude too.
The modeled Mg isotopic compositions of pore water are represented in Figure 3. The model was run with a constant seawater Mg isotopic composition and a varying Mg isotope fractionation factor (α = 0.998 to 0.999, ϵ = −2.0 to −1.0‰ for core ROV2; α = 0.9987 to 0.9997, ϵ = −1.3 to −0.3‰ for core ROV4). Our modeled results match the measured data well within the analytical error.
The Mg concentrations of pore water are typically related to Mg concentration of seawater, carbonate authigenesis, and formation/dissolution of clay minerals (Higgins and Schrag, 2010). Considering that the samples were collected from continental slopes, terrigenous clasts rather than authigenic clay minerals were the main source of silicate minerals in sediment at seeps. The formation of clay minerals was characterized by negative ΔMg/ΔCa (~ −0.95) in pore water profiles and mainly occurs in deeper layers of sediments (Higgins and Schrag, 2010). Likewise, silicate weathering, which may exist at seeps, generally occurs at greater depth (e.g. Torres et al., 2020). Authigenic carbonate precipitation caused by intense SD-AOM tends to occur in the shallow sediments where the decline in Mg is accompanied by a decline in Ca. Since Mg isotope fractionation during formation of clay minerals and carbonate precipitation head in opposite directions (Galy et al., 2002; Higgins and Schrag, 2010), the increase of pore water δ26Mg values with depth helps to eliminate the influence of clay minerals. Therefore, we suggest that the concentration of Mg and change in isotopic composition at a depth of several meters mainly account for the effect of carbonate precipitation rather than the formation of clay minerals. Although carbonate precipitation consumes Ca2+ and Mg2+ in pore water and preferentially incorporates light Mg isotopes, the δ26Mg value of pore water at the study sites of the South China Sea is still close to that of seawater (Figure 3). The invariance of δ26Mg values observed for core ROV1 can be attributed to the minimal decrease in pore water Mg2+ concentration (~2 mM). Because aragonite is the dominant carbonate mineral precipitating in sediment at the ROV1 site and aragonite accommodates very little Mg in its crystal lattice (Snyder et al., 2007), the concentration of Mg2+ in pore water does not significantly decline with depth. The shallow depth of the SMTZ at this site also facilitates sufficient replenishment of Mg2+ ions by downward diffusion of seawater. Although the precipitation of high-Mg calcite consumes much more Mg2+ in pore water, the δ26Mg values of pore water only decreased slightly with depth at the ROV2 and ROV4 sites compared to seawater (Figure 3). Bubble irrigation at the ROV4 seep site apparently promoted the supply of Mg from seawater to an extent similar to the amount of Mg taken up by high-Mg calcite, although the SMTZ at this site is deeper than at the other two sites. We ascribe the small observed variation of pore water δ26Mg values at the ROV seeps to a low rate of carbonate precipitation and sufficient downward replenishment of Mg from seawater. The concentration of Mg2+ in seawater is about five times that of Ca2+, and the precipitation of carbonate minerals at seeps consumes more Ca2+ than Mg2+ (Himmler et al., 2013; Zwicker et al., 2018). Therefore, carbonate precipitation typically does not cause large variation of δ26Mg compared to δ44Ca values of pore water at seeps (Blättler et al., 2021). At seeps with carbonate authigenesis, the Rayleigh effect in pore water is consequently much weaker for Mg isotopes than for Ca isotopes, and authigenic carbonate derived from SD-AOM can be assumed to form from pore water with a Mg isotopic composition similar to that of seawater. Therefore, the Mg isotopic composition of seep carbonates is probably more affected by fractionation during precipitation than by a variation of pore water Mg isotopic composition caused by a Rayleigh effect. Understanding the behavior of Mg isotopes during carbonate precipitation at seeps may provide the means to use seep carbonate as a proxy of seawater Mg isotope composition and geochemistry in Earth’s history.
Laboratory experiments have been conducted to investigate the fractionation of Mg isotopes between calcite and solution (Δ26Mgcal-sol) during carbonate precipitation (e.g. Immenhauser et al., 2010; Li et al., 2012; Mavromatis et al., 2013; Chen et al., 2020). Immenhauser et al. (2010) and Mavromatis et al. (2013) reported a correlation between Δ26Mgcal-sol and precipitation rate of higher Δ26Mgcal-sol values at higher precipitation rates. This trend is opposite to the rate-dependent Ca isotope fractionation during calcite precipitation (Tang et al., 2008). It was attributed to the incorporation of partially dehydrated Mg2+ ions into the calcite crystal lattice at faster precipitation rates, given that the hydration energy of Mg2+ is close to 4 orders of magnitude higher than that of Ca2+ (Mavromatis et al., 2013). In contrast, no such correlation was observed in precipitation experiments conducted by Li et al. (2012) and Chen et al. (2020), although the precipitation rates in their experiments varied widely. An equilibrium Δ26Mgcal-sol value of −2.47 ± 0.09‰ at 25°C was reported by Chen et al. (2020). Our study now documents higher carbonate precipitation rates than those at ODP Site 1082 and higher Δ26Mgcal-sol values, corresponding to a smaller degree of Mg isotope fractionation at the studied seeps. This is consistent with the conclusion drawn from laboratory experiments and travertine calcite precipitation that the extent of Mg isotope fractionation decreases with increasing carbonate growth rate (Immenhauser et al., 2010; Mavromatis et al., 2013; Pogge von Strandmann et al., 2019). However, the carbonate precipitation rates calculated herein, based on pore water geochemistry at seeps, are generally lower than those in laboratory experiments (Figure 6). According to Mavromatis et al. (2013), such low carbonate formation rates should lead to equilibrium fractionation of Mg isotopes. The equilibrium Δ26Mgcal-sol would be ~ −2.47‰ at 25°C (Chen et al., 2020). However, our modeled results show a smaller fractionation compared to that in calcite equilibrium experiments, indicating kinetically controlled fractionation. This discrepancy may be caused by several factors. Firstly, conversion of the fluxes of Ca and Mg in natural environments to precipitation rates of carbonate per unit reactive surface area requires several assumptions (Blättler et al., 2021). It is currently unrealistic to accurately constrain all the physical and chemical conditions in the natural environment where carbonate formation occurs. Mineralogy, choices of the roughness factor, and the error in the estimation of grain size distribution at seeps may result in large uncertainties in the estimation of carbonate precipitation rates in natural environments (Beckingham et al., 2016). The second factor is the uncertainty in the calculation of the extent of Mg isotope fractionation. Carbonate precipitation was the only process considered in our model outlined above to fit the concentration and isotopic composition of pore water profiles. The real situation is probably more complex due to the precipitation-dissolution equilibrium of carbonate minerals (cf. Karaca et al., 2010) and other processes that influence both concentration and isotopic composition of Mg (e.g. dissolution and desorption of exchangeable Mg from clay minerals; Mavromatis et al., 2014). In our model, the finite consumption of Mg2+, leading to a limited variation of the Mg isotopic composition in pore waters, resulted in a relatively large error of the estimated isotope fractionation. The third factor is the difference between the experimental conditions in the laboratory and the conditions in the natural environment. Calcite seeds are usually used in experiments, and experimental conditions are generally controlled by a chemostat, free drift, and constant addition of ions (e.g. Li et al., 2012; Mavromatis et al., 2013; Chen et al., 2020). Other possible factors not considered in experiments such as dissolved sulfide in pore water and microbial activity (e.g. methanogens and sulfate-reducing bacteria) may also influence the behavior of Mg isotopes in nature (Lu et al., 2017; Jin et al., 2021). Hence, it is difficult to reproduce carbonate precipitation in the environment during laboratory experiments. More research is needed to better constrain the physical and chemical conditions in natural environments and the array of factors that may influence the behavior of Mg isotopes during carbonate precipitation.
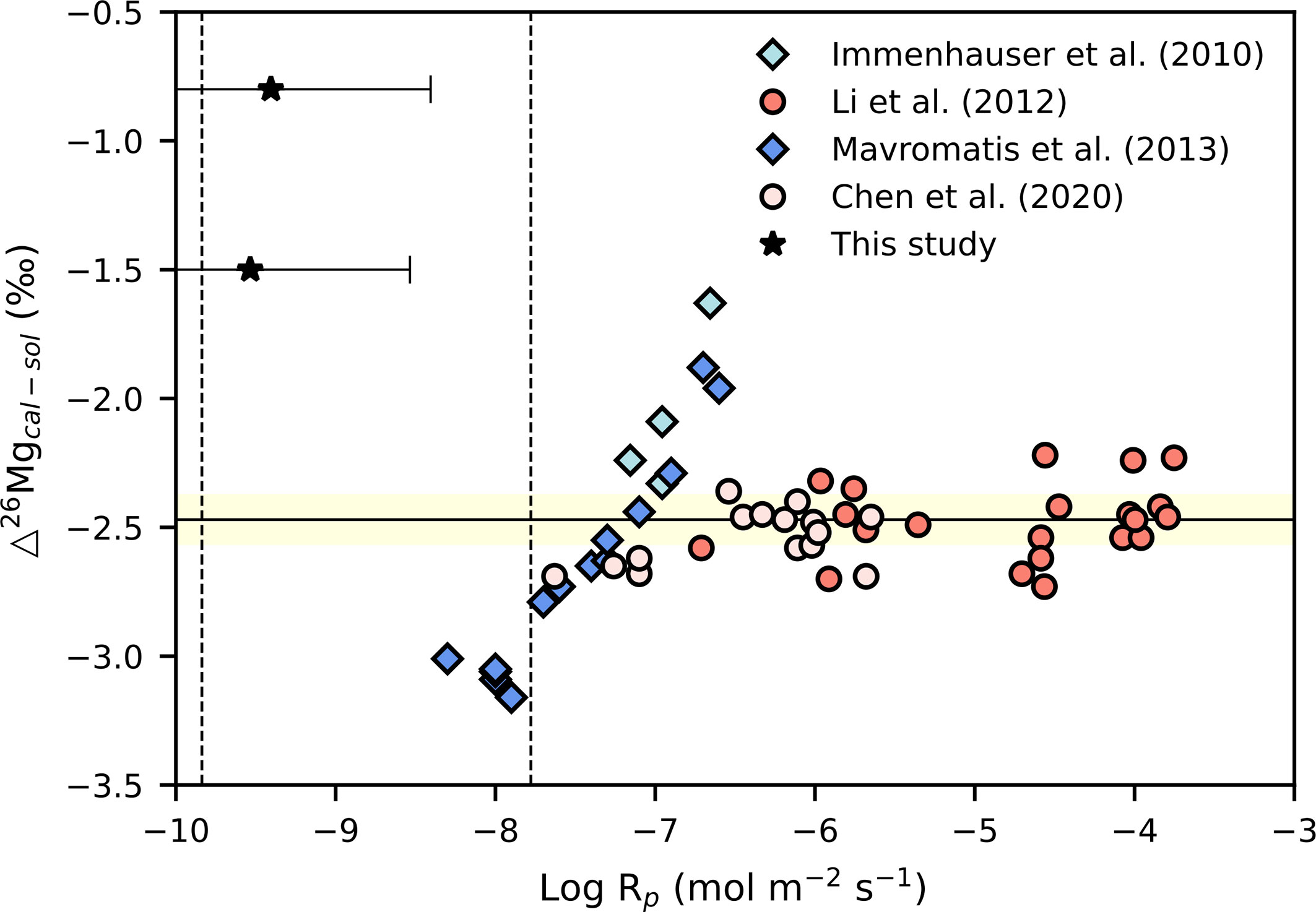
Figure 6 Cross-plot of Δ26Mgcal-sol versus calcite precipitation rate (Log Rp). The dashed line represents the range of precipitation rates of seep carbonates (Karaca et al., 2010). A factor of 0.4 was also applied to data from literature to convert Ca fluxes to carbonate precipitation rates (for details see Rates of Carbonate Precipitation and Mg Isotope Composition). The estimated equilibrium Δ26Mgcal-sol value of −2.47 ± 0.09‰ from literature at 25°C is represented by the solid line and yellow error envelope (Chen et al., 2020). Note that ϵ values in this study are plotted in the figure to describe the fractionation of Mg isotopes and to allow for comparison with Δ26Mgcal-sol values obtained from literature.
Pore waters from sediments of the Haima methane seeps of the South China Sea were investigated for (1) their cation inventory affecting the mineralogy the seep carbonate precipitating, (2) carbonate precipitation rates, and (3) the relationship between these parameters and Mg isotope fractionation. The different degrees of consumption of Ca, Mg, and Sr with depth suggest that carbonate minerals precipitating are aragonite at the ROV1 site and high-Mg calcite at the ROV2 and ROV4 sites. Precipitation rates between 0.92 and 1.24 μmol cm−2 yr−1 are similar to rates at seeps from other areas, but significantly lower than rates in laboratory experiments looking at Mg isotope fractionation during carbonate formation. Pore water at all study sites reveals Mg isotope composition close to that of seawater, indicating a weak Rayleigh fractionation effect due to sufficient replenishment of cations from seawater and moderate consumption during carbonate authigenesis in pore water. The modeled low carbonate precipitation rates and small Mg isotope fractionation are in discrepancy to laboratory experiments. The reason for this discrepancy is unknown, but it likely results from uncertainties in the conversion of precipitation rates, the calculation of isotope fractionation, and the complex conditions in natural environments. This study provides first insight into the link between rates of carbonate precipitation in a natural environment at seeps and Mg isotope fractionation. The results help to constrain the multiple controls on Mg isotope fractionation in natural environments.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
MJ: conceptualization, methodology, data analysis, and writing−original manuscript. DF: conceptualization, supervision, funding acquisition, and writing−review and editing. KH: writing−review and editing. SG: sample collection and data analysis. ML: numerical simulation. JP: writing−review and editing. XW: data analysis. YH: writing−review and editing. DC: resources, supervision. All authors contributed to manuscript preparation. All authors contributed to the article and approved the submitted version.
This study was partially supported by the National Natural Science Foundation of China (Grants: 42176056, 41773091 and 41973008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the crew of Haiyang-6 research vessel for sample collection. Comments of Pogge von Strandmann and an anonymous reviewer helped to improve this article.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.858860/full#supplementary-material
Aloisi G., Pierre C., Rouchy J. M., Foucher J. P., Woodside J., the MEDINAUT Scientific Party. (2000). Methane-Related Authigenic Carbonates of Eastern Mediterranean Sea Mud Volcanoes and Their Possible Relation to Gas Hydrate Destabilisation. Earth Planet. Sci. Lett. 184 (1), 321–338. doi: 10.1016/S0012-821x(00)00322-8
Bao Z., Huang K., Huang T., Shen B., Zong C., Chen K., et al. (2019). Precise Magnesium Isotope Analyses of High-K and Low-Mg Rocks by MC-ICP-MS. J. Anal. At. Spectrom. 34 (5), 940–953. doi: 10.1039/c9ja00002j
Bayon G., Pierre C., Etoubleau J., Voisset M., Cauquil E., Marsset T., et al. (2007). Sr/Ca and Mg/Ca Ratios in Niger Delta Sediments: Implications for Authigenic Carbonate Genesis in Cold Seep Environments. Mar. Geol. 241 (1-4), 93–109. doi: 10.1016/j.margeo.2007.03.007
Beckingham L. E., Mitnick E. H., Steefel C. I., Zhang S., Voltolini M., Swift A. M., et al. (2016). Evaluation of Mineral Reactive Surface Area Estimates for Prediction of Reactivity of a Multi-Mineral Sediment. Geochim. Cosmochim. Acta 188, 310–329. doi: 10.1016/j.gca.2016.05.040
Berner R. A. (1980). Early Diagenesis – A Theoretical Approach (Princeton: Princeton University Press).
Blättler C. L., Hong W.-L., Kirsimäe K., Higgins J. A., Lepland A. (2021). Small Calcium Isotope Fractionation at Slow Precipitation Rates in Methane Seep Authigenic Carbonates. Geochim. Cosmochim. Acta 298, 227–239. doi: 10.1016/j.gca.2021.01.001
Bradbury H. J., Turchyn A. V. (2019). Reevaluating the Carbon Sink Due to Sedimentary Carbonate Formation in Modern Marine Sediments. Earth Planet. Sci. Lett. 519, 40–49. doi: 10.1016/j.epsl.2019.04.044
Chen F., Chen J., Jin H., Li H. (2012). Correlation of δ13Corg in Surface Sediments With Sinking Particulate Matter in South China Sea and Implication for Reconstructing Paleo-Environment. Acta Sedimentol. Sin. 30 (2), 340–345. (in Chinese with English abstract).
Chen X.-Y., Teng F.-Z., Sanchez W. R., Romanek C. S., Sanchez-Navas A., Sánchez-Román M. (2020). Experimental Constraints on Magnesium Isotope Fractionation During Abiogenic Calcite Precipitation at Room Temperature. Geochim. Cosmochim. Acta 281, 102–117. doi: 10.1016/j.gca.2020.04.033
Chuang P.-C., Dale A. W., Wallmann K., Haeckel M., Yang T. F., Chen N.-C., et al. (2013). Relating Sulfate and Methane Dynamics to Geology: Accretionary Prism Offshore SW Taiwan. Geochem. Geophys. Geosyst. 14 (7), 2523–2545. doi: 10.1002/ggge.20168
de Villiers S. (1999). Seawater Strontium and Sr/Ca Variability in the Atlantic and Pacific Oceans. Earth Planet Sci. Lett. 171 (4), 623–634. doi: 10.1016/S0012-821x(99)00174-0
DePaolo D. J. (2011). Surface Kinetic Model for Isotopic and Trace Element Fractionation During Precipitation of Calcite From Aqueous Solutions. Geochim. Cosmochim. Acta 75 (4), 1039–1056. doi: 10.1016/j.gca.2010.11.020
Fantle M. S., DePaolo D. J. (2007). Ca Isotopes in Carbonate Sediment and Pore Fluid From ODP Site 807a: The Ca2+(aq)–calcite Equilibrium Fractionation Factor and Calcite Recrystallization Rates in Pleistocene Sediments. Geochim. Cosmochim. Acta 71 (10), 2524–2546. doi: 10.1016/j.gca.2007.03.006
Fantle M. S., Higgins J. (2014). The Effects of Diagenesis and Dolomitization on Ca and Mg Isotopes in Marine Platform Carbonates: Implications for the Geochemical Cycles of Ca and Mg. Geochim. Cosmochim. Acta 142, 458–481. doi: 10.1016/j.gca.2014.07.025
Foster G. L., Pogge von Strandmann P. A. E., Rae J. W. B. (2010). Boron and Magnesium Isotopic Composition of Seawater. Geochem. Geophys. Geosyst. 11 (8), Q08015. doi: 10.1029/2010GC003201
Galy A., Bar-Matthews M., Halicz L., O’Nions R. K. (2002). Mg Isotopic Composition of Carbonate: Insight From Speleothem Formation. Earth Planet. Sci. Lett. 201 (1), 105–115. doi: 10.1016/S0012-821x(02)00675-1
Gothmann A. M., Stolarski J., Adkins J. F., Higgins J. A. (2017). A Cenozoic Record of Seawater Mg Isotopes in Well-Preserved Fossil Corals. Geology 45 (11), 1039–1042. doi: 10.1130/G39418.1
Haeckel M., Boudreau B. P., Wallmann K. (2007). Bubble-Induced Porewater Mixing: A 3-D Model for Deep Porewater Irrigation. Geochim. Cosmochim. Acta 71 (21), 5135–5154. doi: 10.1016/j.gca.2007.08.011
Higgins J. A., Schrag D. P. (2010). Constraining Magnesium Cycling in Marine Sediments Using Magnesium Isotopes. Geochim. Cosmochim. Acta 74 (17), 5039–5053. doi: 10.1016/j.gca.2010.05.019
Higgins J. A., Schrag D. P. (2015). The Mg Isotopic Composition of Cenozoic Seawater – Evidence for a Link Between Mg-Clays, Seawater Mg/Ca, and Climate. Earth Planet. Sci. Lett. 416, 73–81. doi: 10.1016/j.epsl.2015.01.003
Himmler T., Haley B. A., Torres M. E., Klinkhammer G. P., Bohrmann G., Peckmann J. (2013). Rare Earth Element Geochemistry in Cold-Seep Pore Waters of Hydrate Ridge, Northeast Pacific Ocean. Geo-Marine Lett. 33 (5), 369–379. doi: 10.1007/s00367-013-0334-2
Huang K.-J., Shen B., Lang X.-G., Tang W.-B., Peng Y., Ke S., et al. (2015). Magnesium Isotopic Compositions of the Mesoproterozoic Dolostones: Implications for Mg Isotopic Systematics of Marine Carbonates. Geochim. Cosmochim. Acta 164, 333–351. doi: 10.1016/j.gca.2015.05.002
Huang C. Y., Wu S. F., Zhao M. X., Chen M. T., Wang C. H., Tu X., et al. (1997). Surface Ocean and Monsoon Climate Variability in the South China Sea Since the Last Glaciation. Mar. Micropaleontol. 32 (1-2), 71–94. doi: 10.1016/S0377-8398(97)00014-5
Hui G., Li S., Guo L., Zhang G., Gong Y., Somerville I. D., et al. (2016). Source and Accumulation of Gas Hydrate in the Northern Margin of the South China Sea. Mar. Pet. Geol. 69, 127–145. doi: 10.1016/j.marpetgeo.2015.10.009
Hu Y., Luo M., Chen L., Liang Q., Feng D., Tao J., et al. (2018). Methane Source Linked to Gas Hydrate System at Hydrate Drilling Areas of the South China Sea: Porewater Geochemistry and Numerical Model Constraints. J. Asian Earth Sci. 168, 87–95. doi: 10.1016/j.jseaes.2018.04.028
Hu Y., Luo M., Liang Q., Chen L., Feng D., Yang S., et al. (2019). Pore Fluid Compositions and Inferred Fluid Flow Patterns at the Haima Cold Seeps of the South China Sea. Mar. Pet. Geol. 103, 29–40. doi: 10.1016/j.marpetgeo.2019.01.007
Immenhauser A., Buhl D., Richter D., Niedermayr A., Riechelmann D., Dietzel M., et al. (2010). Magnesium-Isotope Fractionation During Low-Mg Calcite Precipitation in a Limestone Cave - Field Study and Experiments. Geochim. Et Cosmochim. Acta 74 (15), 4346–4364. doi: 10.1016/j.gca.2010.05.006
Jørgensen B. B., Kasten S. (2006). “Sulfur Cycling and Methane Oxidation,” in Marine Geochemistry. Eds. Horst D. S., Matthias Z. (Verlag: Springer), 271–309.
Jin M., Feng D., Huang K., Peckmann J., Li N., Huang H., et al. (2021). Behavior of Mg Isotopes During Precipitation of Methane-Derived Carbonate: Evidence From Tubular Seep Carbonates From the South China Sea. Chem. Geol. 567, 120101. doi: 10.1016/j.chemgeo.2021.120101
Karaca D., Hensen C., Wallmann K. (2010). Controls on Authigenic Carbonate Precipitation at Cold Seeps Along the Convergent Margin Off Costa Rica. Geochem. Geophys. Geosyst. 11 (8), Q08S27. doi: 10.1029/2010GC003062
Kasemann S. A., Pogge von Strandmann P. A. E., Prave A. R., Fallick A. E., Elliott T., Hoffmann K.-H. (2014). Continental Weathering Following a Cryogenian Glaciation: Evidence From Calcium and Magnesium Isotopes. Earth Planet. Sci. Lett. 396, 66–77. doi: 10.1016/j.epsl.2014.03.048
Liang Q. Y., Hu Y., Feng D., Peckmann J., Chen L. Y., Yang S. X., et al. (2017). Authigenic Carbonates From Newly Discovered Active Cold Seeps on the Northwestern Slope of the South China Sea: Constraints on Fluid Sources, Formation Environments, and Seepage Dynamics. Deep Sea Res. Part I: Oceanogr. Res. Papers 124, 31–41. doi: 10.1016/j.dsr.2017.04.015
Li W., Beard B. L., Li C., Xu H., Johnson C. M. (2015). Experimental Calibration of Mg Isotope Fractionation Between Dolomite and Aqueous Solution and its Geological Implications. Geochim. Cosmochim. Acta 157, 164–181. doi: 10.1016/j.gca.2015.02.024
Li W. Q., Chakraborty S., Beard B. L., Romanek C. S., Johnson C. M. (2012). Magnesium Isotope Fractionation During Precipitation of Inorganic Calcite Under Laboratory Conditions. Earth Planet. Sci. Lett. 333, 304–316. doi: 10.1016/j.epsl.2012.04.010
Luff R., Wallmann K. (2003). Fluid Flow, Methane Fluxes, Carbonate Precipitation and Biogeochemical Turnover in Gas Hydrate-Bearing Sediments at Hydrate Ridge, Cascadia Margin: Numerical Modeling and Mass Balances. Geochim. Cosmochim. Acta 67 (18), 3403–3421. doi: 10.1016/s0016-7037(03)00127-3
Lu Y., Liu Y., Sun X., Lin Z., Xu L., Lu H., et al. (2017). Intensity of Methane Seepage Reflected by Relative Enrichment of Heavy Magnesium Isotopes in Authigenic Carbonates: A Case Study From the South China Sea. Deep Sea Res. Part I: Oceanogr. Res. Papers 129, 10–21. doi: 10.1016/j.dsr.2017.09.005
Mavromatis V., Gautier Q., Bosc O., Schott J. (2013). Kinetics of Mg Partition and Mg Stable Isotope Fractionation During its Incorporation in Calcite. Geochim. Et Cosmochim. Acta 114, 188–203. doi: 10.1016/j.gca.2013.03.024
Mavromatis V., Prokushkin A. S., Pokrovsky O. S., Viers J., Korets M. A. (2014). Magnesium Isotopes in Permafrost-Dominated Central Siberian Larch Forest Watersheds. Geochim. Cosmochim. Acta 147, 76–89. doi: 10.1016/j.gca.2014.10.009
Moore T. S., Murray R. W., Kurtz A. C., Schrag D. P. (2004). Anaerobic Methane Oxidation and the Formation of Dolomite. Earth Planet. Sci. Lett. 229 (1-2), 141–154. doi: 10.1016/j.epsl.2004.10.015
Naehr T. H., Eichhubl P., Orphan V. J., Hovland M., Paull C. K., Ussler W., et al. (2007). Authigenic Carbonate Formation at Hydrocarbon Seeps in Continental Margin Sediments: A Comparative Study. Deep Sea Res. Part II: Topical Stud. Oceanogr. 54 (11), 1268–1291. doi: 10.1016/j.dsr2.2007.04.010
Nöthen K., Kasten S. (2011). Reconstructing Changes in Seep Activity by Means of Pore Water and Solid Phase Sr/Ca and Mg/Ca Ratios in Pockmark Sediments of the Northern Congo Fan. Mar. Geol. 287 (1-4), 1–13. doi: 10.1016/j.margeo.2011.06.008
Oelkers E. H., Helgeson H. C. (1991). Calculation of Activity Coefficients and Degrees of Formation of Neutral Ion Pairs in Supercritical Electrolyte Solutions. Geochim. Cosmochim. Acta 55 (5), 1235–1251. doi: 10.1016/0016-7037(91)90303-M
Pearce C. R., Saldi G. D., Schott J., Oelkers E. H. (2012). Isotopic Fractionation During Congruent Dissolution, Precipitation and at Equilibrium: Evidence From Mg Isotopes. Geochim. Cosmochim. Acta 92, 170–183. doi: 10.1016/j.gca.2012.05.045
Pinilla C., Blanchard M., Balan E., Natarajan S. K., Vuilleumier R., Mauri F. (2015). Equilibrium Magnesium Isotope Fractionation Between Aqueous Mg2+ and Carbonate Minerals: Insights From Path Integral Molecular Dynamics. Geochim. Cosmochim. Acta 163, 126–139. doi: 10.1016/j.gca.2015.04.008
Pogge von Strandmann P. A. E., Forshaw J., Schmidt D. N. (2014). Modern and Cenozoic Records of Seawater Magnesium From Foraminiferal Mg Isotopes. Biogeosciences 11 (18), 5155–5168. doi: 10.5194/bg-11-5155-2014
Pogge von Strandmann P. A. E., Olsson J., Luu T.-H., Gislason S. R., Burton K. W. (2019). Using Mg Isotopes to Estimate Natural Calcite Compositions and Precipitation Rates During the 2010 Eyjafjallajökull Eruption. Front. Earth Sci. 7. doi: 10.3389/feart.2019.00006
Richter F. M., Mendybaev R. A., Christensen J. N., Hutcheon I. D., Williams R. W., Sturchio N. C., et al. (2006). Kinetic Isotopic Fractionation During Diffusion of Ionic Species in Water. Geochim. Cosmochim. Acta 70 (2), 277–289. doi: 10.1016/j.gca.2005.09.016
Saenger C., Wang Z. R. (2014). Magnesium Isotope Fractionation in Biogenic and Abiogenic Carbonates: Implications for Paleoenvironmental Proxies. Quat. Sci. Rev. 90, 1–21. doi: 10.1016/j.quascirev.2014.01.014
Schrag D. P., Higgins J. A., Macdonald F. A., Johnston D. T. (2013). Authigenic Carbonate and the History of the Global Carbon Cycle. Science 339 (6119), 540–543. doi: 10.1126/science.1229578
Snyder G. T., Hiruta A., Matsumoto R., Dickens G. R., Tomaru H., Takeuchi R., et al. (2007). Pore Water Profiles and Authigenic Mineralization in Shallow Marine Sediments Above the Methane-Charged System on Umitaka Spur, Japan Sea. Deep Sea Res. Part II: Topical Stud. Oceanogr. 54 (11-13), 1216–1239. doi: 10.1016/j.dsr2.2007.04.001
Tang J., Dietzel M., Böhm F., Köhler S. J., Eisenhauer A. (2008). Sr2+/Ca2+ and 44Ca/40Ca Fractionation During Inorganic Calcite Formation: II. Ca Isotopes. Geochim. Cosmochim. Acta 72 (15), 3733–3745. doi: 10.1016/j.gca.2008.05.033
Teng F.-Z. (2017). Magnesium Isotope Geochemistry. Rev. Mineral. Geochem. 82 (1), 219–287. doi: 10.2138/rmg.2017.82.7
Tipper E., Galy A., Gaillardet J., Bickle M., Elderfield H., Carder E. (2006). The Magnesium Isotope Budget of the Modern Ocean: Constraints From Riverine Magnesium Isotope Ratios. Earth Planet. Sci. Lett. 250 (1-2), 241–253. doi: 10.1016/j.epsl.2006.07.037
Torres M. E., Hong W.-L., Solomon E. A., Milliken K., Kim J.-H., Sample J. C., et al. (2020). Silicate Weathering in Anoxic Marine Sediment as a Requirement for Authigenic Carbonate Burial. Earth-Science Rev. 200, 102960. doi: 10.1016/j.earscirev.2019.102960
Wallmann K., Aloisi G., Haeckel M., Obzhirov A., Pavlova G., Tishchenko P. (2006). Kinetics of Organic Matter Degradation, Microbial Methane Generation, and Gas Hydrate Formation in Anoxic Marine Sediments. Geochim. Cosmochim. Acta 70 (15), 3905–3927. doi: 10.1016/j.gca.2006.06.003
Wang Z. R., Hu P., Gaetani G., Liu C., Saenger C., Cohen A., et al. (2013). Experimental Calibration of Mg Isotope Fractionation Between Aragonite and Seawater. Geochim. Et Cosmochim. Acta 102, 113–123. doi: 10.1016/j.gca.2012.10.022
Wang P., Prell W., Blum P. (2000). Proceeding of the Ocean Drilling Program: Initial Reports 184 (College Station, TX, USA: Texas A&M University).
Wang W., Qin T., Zhou C., Huang S., Wu Z., Huang F. (2017). Concentration Effect on Equilibrium Fractionation of Mg-Ca Isotopes in Carbonate Minerals: Insights From First-Principles Calculations. Geochim. Cosmochim. Acta 208, 185–197. doi: 10.1016/j.gca.2017.03.023
Wei J., Li J., Wu T., Zhang W., Li J., Wang J., et al. (2020). Geologically Controlled Intermittent Gas Eruption and its Impact on Bottom Water Temperature and Chemosynthetic Communities—A Case Study in the “HaiMa”, Cold Seeps South China Sea. Geol. J. 55 (9), 6066–6078. doi: 10.1002/gj.3780
Whiticar M. J. (1999). Carbon and Hydrogen Isotope Systematics of Bacterial Formation and Oxidation of Methane. Chem. Geol. 161 (1), 291–314. doi: 10.1016/S0009-2541(99)00092-3
Wombacher F., Eisenhauer A., Bohm F., Gussone N., Regenberg M., Dullo W. C., et al. (2011). Magnesium Stable Isotope Fractionation in Marine Biogenic Calcite and Aragonite. Geochim. Et Cosmochim. Acta 75 (19), 5797–5818. doi: 10.1016/j.gca.2011.07.017
Young E. D., Galy A. (2004). The Isotope Geochemistry and Cosmochemistry of Magnesium. Geochem. Non-Traditional Stable Isotopes 55, 197–230. doi: 10.2138/gsrmg.55.1.197
Zeebe R. E., Wolf-Gladrow D. A. (2001). CO2 in Seawater: Equilibrium, Kinetics and Isotopes (U. K: Elsevier).
Keywords: Mg isotopes, authigenic carbonate, pore water geochemistry, methane seep, South China Sea
Citation: Jin M, Feng D, Huang K, Gong S, Luo M, Peckmann J, Wang X, Hu Y and Chen D (2022) Magnesium Isotopes in Pore Water of Active Methane Seeps of the South China Sea. Front. Mar. Sci. 9:858860. doi: 10.3389/fmars.2022.858860
Received: 20 January 2022; Accepted: 08 March 2022;
Published: 29 March 2022.
Edited by:
Ed Hathorne, Helmholtz Association of German Research Centres (HZ), GermanyReviewed by:
Philip Pogge von Strandmann, University College London, United KingdomCopyright © 2022 Jin, Feng, Huang, Gong, Luo, Peckmann, Wang, Hu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Feng, ZGZlbmdAc2hvdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.