 Zhaoshan Zhong1,2,3
Zhaoshan Zhong1,2,3 Minxiao Wang1,2,3
Minxiao Wang1,2,3 Hao Chen1,2,3Hao Wang1,2,3
Hao Chen1,2,3Hao Wang1,2,3 Huan Zhang1,2,3Li Zhou1,2,3
Huan Zhang1,2,3Li Zhou1,2,3 Yan Sun1,2,3Lei Cao1,2,3
Yan Sun1,2,3Lei Cao1,2,3 Chao Lian1,2,3Mengna Li1,2,3,4
Chao Lian1,2,3Mengna Li1,2,3,4 Chaolun Li1,2,3,4*
Chaolun Li1,2,3,4*- 1Center of Deep Sea Research, Chinese Academy of Sciences Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 4University of Chinese Academy of Sciences, Beijing, China
Gigantidas platifrons is one of the most dominant deep-sea mussels in cold seeps ecosystems in the South China sea. Studies have shown that deep-sea mussels are a gonochoristic species, however, little is known about the molecular mechanisms of sex determination in G. platifrons. In this study, RNA-seq and WGBS methylation analysis were performed on adult G. platifrons gonads to identify potential sex-related genes and generate a comprehensive analysis of sex determination in deep-sea mussels. A total of 5923 genes were identified as differentially expressed between the ovaries and testes, of which 2711 were female-biased and 3212 were male-biased. Among them, 161 genes may participate in the sex determination, and we found that DMRT2 may play an important role in male sex determination, and FOXL2, Wnt7, and β-catenin may have impact on female sex determination. Moreover, common expression patterns were found in majority of the sex-related genes such as FOXL2, β-catenin, and genes in SOX family, suggesting the sex determination mechanisms of mussels in different habitats were conserved. The 5mC levels of transcription start sites (TSS2K) were significantly higher in sex-related genes than other DEGs in both gonads. Positive correlation was observed between sex-related genes expression and methylation in male, however, the effects of the DNA methylation on gene expression were complex in female. In short, we argue that sex determination mechanisms of deep-sea mussel G. platifrons is mainly controlled by genetic, and the methylation may have a regulation role on male sex determination or differentiation.
Introduction
Deep-sea mussels belonging to the subfamily Bathymodiolinae (Mytilidae) are the dominant species in deep-sea chemosynthetic ecosystems such as cold seeps and hydrothermal vents (Jones et al., 2005). The population dynamics of deep-sea mussels and how they can thrive in these extreme environments have received much attention (Carney et al., 2006; Laming et al., 2018). The sex ratio of a species is one of the most important factors that could affect population dynamics (Dyson and Hurst, 2004). For mollusks for which sex determination can be influenced by the environment, the sex ratio has been observed to change with temperature and the seasons in shallow-water species (Stenyakina et al., 2009; Collin, 2013; Villamor and Yamamoto, 2015; Xu et al., 2016). In contrast to shallow-water ecosystems, the deep-sea environment has a stable and low temperature and no seasonal changes through the year (Boutet et al., 2008). Therefore, previously, deep-sea mussels were not expected to exhibit sex ratio changes, but from relatively recent investigations into deep-sea chemosynthetic ecosystems, many deep-sea mussels have been observed to have changed sex ratios with increasing age and sampling time in different seasons (Tyler et al., 2007; Laming et al., 2014; Zhong et al., 2020). Nonetheless, studies have shown that the sex determination mechanisms could change rapidly between phylogenetically closely-related species (Gempe and Beye, 2011). Based on phylogenetic studies and fossil evidence, it appears that deep-sea mussels entered that environment more than 85 million years ago (Lorion et al., 2013). Consequently, the mechanisms of sex determination or differentiation in deep-sea mussels may have differences from those for shallow water species. However, the same sex ratio changes observed in deep-sea mussels suggest that some sex determination or differentiation mechanisms are conserved between deep-sea mussels and their shallow water relatives. The similarities and differences between deep and shallow species need to be further researched.
Sex determination is the process that establishes the sex of an individual and regulates the differentiation of sex characteristics (Smith et al., 2009; Capel, 2017; Du et al., 2017; Lin et al., 2021). The sex determination mechanism is typically considered to include two sex-determining strategies, namely genetic sex determination (GSD) and environmental sex determination (ESD) (Conover and Heins, 1987). The sex of an individual is complex. The sex of most vertebrates was determined by sex chromosomes. However, the situation is much more complex in lower vertebrates like fishes and reptiles whose sex could be determined by multiple loci from different autosomal chromosomes or even by environments only (ESD) (Nagahama et al., 2021). As to mollusca, only gastropods have been reported with sex chromosomes and others including the bivalves which have not found sex chromosomes, and their sex can be determined by genetics or environment, or both (Breton et al., 2018). Bathymodioline mussels have been studied for over 40 years, and although various reproductive strategies have been observed such as hermaphroditism in the genus Idas (Tyler et al., 2009), gonochorism in the genera Bathymodiolus and Gigantidas (Pennec and Beninger, 1997; Comtet et al., 1999; Eckelbarger and Young, 1999; Kádár et al., 2006; Tyler et al., 2007; Rossi and Tunnicliffe, 2017; Zhong et al., 2020), the molecular mechanisms that determine hermaphroditism or gonochorism are not well understood.
With the development of sequencing technology, RNA sequencing (RNA-Seq) of the gonadal samples has also been used to examine sex determination or differentiation in bivalves (Teaniniuraitemoana et al., 2014; Li et al., 2016; Chen et al., 2017; Ghiselli et al., 2018; Li et al., 2018). Moreover, a set of conserved transcription factors which determine the direction of sex differentiation in bivalves has previously been identified, such as FOXL2 (forkhead box L2) in female-determining pathways and DMRT (double-sex- and mab3-related transcription factor) and SOX gene family (Sry-related HMG-box genes) in male-determining pathways (Naimi et al., 2009; Ghiselli et al., 2012; Teaniniuraitemoana et al., 2014; Li et al., 2016; Li et al., 2018). For shallow water mytilid species, through gonadal transcriptome of males Perumytilus purpuratus, some sperm-related transcripts have been found (Briones et al., 2018); by the comparison of female and male gonadal transcriptomes in Limnoperna fortunei, 187 sex-related genes including DMRT, SOX, and FOX were identified, which revealed target genes for sex determination or differentiation in L. fortunei (Afonso et al., 2019). Furthermore, DNA methylation has been shown to play an important role in gene expression, especially 5-methylcytosine (5mC) in the genome which have a regulative role in the transcription of sex-related genes and ultimately affect sex determination (Wittkopp et al., 2004; Feng et al., 2010; Tachibana, 2015; Li et al., 2021; Lin et al., 2021). For example, different 5mC levels have been determined for the two sexes of many animals including reptiles (turtles and alligators), fishes (half-smooth tongue sole and hybrid tilapia) and mollusca (oysters), by bisulfite sequencing (BS-Seq) (Olson and Roberts, 2014; Shao et al., 2014; Wan et al., 2016; Radhakrishnan et al., 2018; Lin et al., 2021). Through integrated analysis of transcriptome and methylome, (Lin et al., 2021) found some important sex maintenance and gonadal development pathways in adult alligators (e.g., DMRT1-SOX9-AMH pathway for males and oocyte meiotic maturation pathway for females). They also observed that DNA methylation density (calculated by the number of methylated C in a region divided by the C covered by more than 5X reads in that region) and level (calculated by the number of methylated C in a region divided by the sum of methylated C and unmethylated C) were higher in testes than in ovaries, and the hypermethylation in the gene bodies enhanced the expression of male-biased genes in the testes. All these findings indicate that gonadal transcriptome and methylome integrated analysis would be an effective tool for addressing sex determination in animals (Lin et al., 2021). Therefore, to better elucidate the sex determination or differentiation of deep-sea mussels, integrated transcriptome and methylome studies are urgently needed.
Gigantidas platifrons is an endemic and gonochoric species of the hydrothermal vents and cold seeps in the Northwest Pacific, and mainly gets nutrition from the methanotrophic endosymbionts in its gills (Barry et al., 2002; Sun et al., 2017b; Zhong et al., 2020). The genomic information of G. platifrons has been sequenced which make it a model species for investigating adaptations to extreme environments in deep sea (Sun et al., 2017a). In our study, we applied RNA-seq and whole-genome bisulfite (WGBS) methylation analysis on the gonad tissue of the adult deep-sea mussel G. platifrons to investigate the molecular mechanisms of sex determination. Further, through the comparison with shallow water relatives we investigate whether sex determination is conserved in deep-sea mussels.
Materials and Methods
Animal Material and Tissue Sampling
Deep-sea mussel G. platifrons specimens were obtained from Site F (22°06′N, 119°17′E), a cold seep which located at a depth of 1100m in the South China Sea (Figure 1A). The environmental temperatures of Site F are about 4°C and other physiochemical parameters were documented by (Cao et al., 2021). The mussel materials were captured using the TITAN 4 manipulator of the ROV Faxian, operated from the R/V Kexue during September 2017, and the mussels were transported in an isothermal bio-box (Figure 1B) in the underframe of the ROV, which kept the water temperature around 8–9°C. The gonads of adult G. platifrons were dissected immediately after ROV retrieval on deck and then split into two pieces. Each gonad sample was fixed in 4% paraformaldehyde (PFA) overnight, washed twice with 0.01 M phosphate-buffered saline (1 × PBS) and then stored in 70% ethanol for sex identification. The remainder were flash-frozen in liquid nitrogen and stored at -80°C for RNA and DNA extraction. RNA and DNA were extracted from the testes and ovaries using Trizol (Thermo, Waltham, USA) and E.Z.N.A.® Mollusc DNA kit (Omega, Norcross, USA), according to the manufacturer’s instructions, respectively.
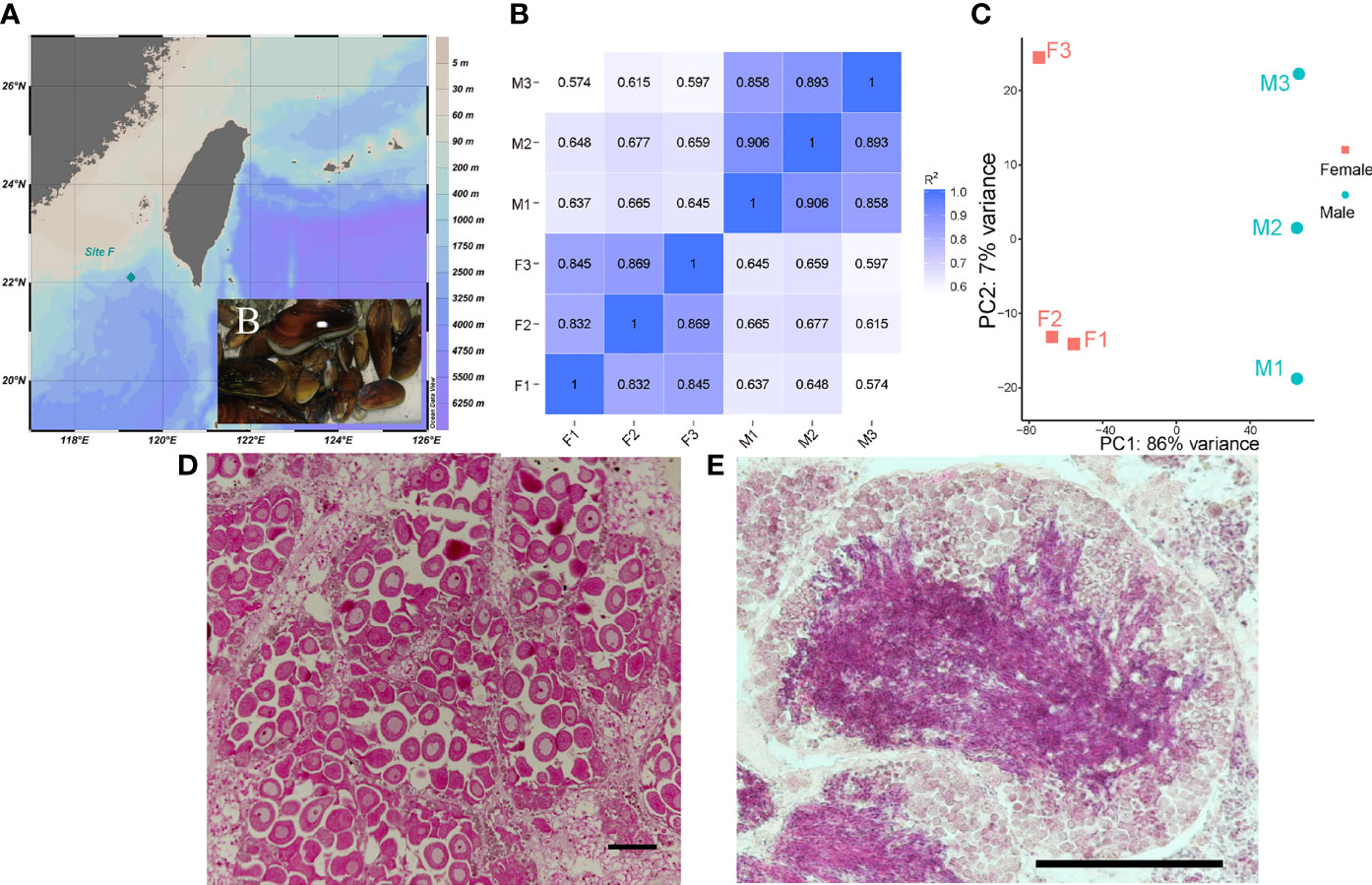
Figure 1 Sample site (A) and captured G. platifrons [insert picture (B)]; Person correlation between samples (C); PCA analysis of samples (D); Representative female ovary, scale bar=100 μm (E) and male testis, scale bar=100 μm (F) of deep-sea mussel G. platifrons.
Sex Identification of G. platifrons
The gonad samples were dehydrated with serial ethanol (80, 90, 100, and then 100%) and then transferred to xylene and embedded in paraffin wax. The 7 μm sections of gonad samples were cut on a Leica microtome and tiled on glass slides. Gonad sections were deparaffinized with xylene, hydrated with serial ethanol (100, 100, 90, 80, and then 70%), and stained with hematoxylin-eosin (HE) to determine the sex and development stages of the gonads (Figures 1E, F, other gonad sections of males and females are shown in Supplementary Figure 1).
RNA-Seq of G. platifrons Gonads
After sex identification, 12 female gonads and 12 male gonads were chosen for RNA extraction. RNA degradation and contamination were monitored on 1% agarose gels. RNA quality and integrity were checked using an Agilent 2100 Bioanalyzer, and the concentration of total RNA was quantified using a Nanodrop 1000 spectrophotometer. Qualified RNA was subsequently used for RNA-seq analysis. The RNA of each four female gonads was mixed in equal amounts to obtain three RNA pools of 12 female gonads and similar operation was performed for 12 male gonads. Herein, three female and three male gonadal RNA pools were prepared for RNA-seq. Six RNA samples were sent to Novogene Corporation (Beijing, China) for cDNA library construction. Then, library preparations were sequenced on an Illumina Hiseq 2500 platform and 125 bp paired-end reads were generated.
Gonad WGBS of G. platifrons
The WGBS sample scheme was same as RNA seq, and three female and three male gonadal DNA pools were obtained. For each DNA pool, 5.2 μg of genomic DNA spiked with 26 ng lambda DNA were fragmented by sonication to 200—300 bp with Covaris S220, followed by end repair and adenylation. These DNA fragments were treated twice with bisulfite using EZ DNA Methylation-GoldTM Kit (Zymo Research), before the resulting single-strand DNA fragments were PCR amplified using KAPA HiFi HotStart Uracil + ReadyMix (2X). Library concentration was quantified using Qubit® 2.0 Flurometer (Life Technologies, CA, USA) and quantitative PCR, and the insert size was assayed on an Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2500 platform and 125 bp paired-end reads were generated. Image analysis and base calling were performed with Illumina CASAVA pipeline, and generating 125bp paired-end reads.
Quality Control
Raw sequencing data were processed using fastp (version 0.20.0) with default parameters (Chen et al., 2018) to remove the adaptor sequences and low-quality reads of the RNA-seq and WGBS raw reads. Then the clean reads of RNA-seq and WGBS were prepared for downstream analysis.
RNA-Seq Data Analysis
The clean reads were mapped with an updated reference genome for G. platifrons (unpublished genome version, updated by Dr. Minxiao Wang) using HISAT2 (version 2.2.1). The mapped genes for G. platifrons were annotated in Nr, Swissprot, InterProscan, KEGG and the COG database. The transcripts of all the identified genes were quantified with featureCounts software (Version 2.0.1). Person correlation (Figure 1C) and principal component analysis (PCA) (Figure 1D) were then performed using custom-written R scripts to assess the relationships between gonad samples (Figure 1B). To identify sex-biased genes, the DESeq2 R package (version 1.10.1) was used. Only genes with | log2 (fold change) | > 1 and a corrected p value (q value) < 0.05 were regarded as differentially expressed genes (DEGs). Gene ontology (GO) and KEGG enrichment analysis of DEGs were conducted using Blast2GO and KOBAS software, GO terms and KEGG pathways with q values less than 0.05 were considered as significantly enriched. The results of enrichment analysis were visualized using ggplot2 R package (version 3.2.1) and OmicShare tools (www.omicshare.com/tools). Genes with GO annotation of 0007530 (sex determination) and 0007548 (sex differentiation) terms were selected as sex-related gene sets.
To find the similarities and differences in the gonadal transcriptomes and sex-related genes of deep-sea and shallow water mussels, the published transcriptome data of shallow water mussel gonads were selected for comparative transcriptome analysis. Transcriptome data for female and male gonads of only one shallow water mussel L. fortunei are published. The transcriptome and genome data for L. fortunei were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/bioproject/587212) and GIGADB (https://ftp.cngb.org/pub/gigadb/pub/10.5524/100001_101000/100386/), respectively. All data were reanalyzed in line with the method mentioned previously. In short, first, the DEGs among the two sexes of L. fortunei were determined by DESeq2 and the GO and KEGG enrichment analysis were conducted using Blast2GO and KOBAS software. We then used OrthoFinder (version 2.5.2) to find the homologous protein sequences of G. platifrons and L. fortunei. Next, we used the G. platifrons sex-related genes and screened out single copy orthologous genes to obtain the orthologue sex-related genes in both G. platifrons and L. fortunei. Finally, we compared the expression pattern of shared sex-related genes in female and male gonads between G. platifrons and L. fortunei.
WGBS Data Analysis
Bismark software (version 0.16.1) was used to perform alignments of bisulfite-treated reads to a reference genome with the default parameters. The G. platifrons reference genome was first transformed to a bisulfite-converted version (C-to-T and G-to-A converted) and then indexed using bowtie2. Sequence reads were also transformed into fully bisulfite-converted versions (C-to-T and G-to-A converted) before they are aligned to similarly converted versions of the genome in a directional manner. Sequence reads that produce a unique best alignment from the two alignment processes (original top and bottom strand) were then compared to the G. platifrons genomic sequence and the methylation state of all cytosine positions in the read was inferred. Read pairs that shared the same coordinates in the genome were regarded as duplicated, and removed before calling methylation state, thus avoiding potential methylation level calculation bias. The bisulfite non-coversion rate was calculated as the percentage of cytosines sequenced at cytosine reference positions in the lambda genome. To identify the methylation site, we modeled the sum of 5mC of methylated counts as a binomial (Bin) random variable with methylation rate (r), as 5mC ∼ Bin (5mC + umC*r). To calculate the methylation level of the sequence, we divided the sequence into multiple bins, with each bin size at 10 kb. The sum of methylated and unmethylated read counts in each window were calculated. The 5mC level (ML) for each window or C site shows the fraction of methylated Cs, and is defined as: ML(5mC) =reads (5mC)/(reads (5mC) + reads (umC)). The relative proportion of 5mCs in the three regions including 2kb upstream of the genes’ transcription start sites (TSS2K), the gene body, and 2kb downstream of transcription termination sites (TTS2K) were calculated as the proportion of mCG, mCHG, and mCHH of the total 5mC sites, respectively. Differentially methylated regions (DMRs) of the two sexes were identified using the DSS software (version 2.12.0). The regions with a p value less than 1e-05, length more than 50, average methylation level difference between groups greater than or equal to 20%, and at least three CpG sites were retained as a final DMR. We defined the genes related to DMRs as genes whose gene body region (from TSS to TTS) or TSS2K have an overlap (> 1bp) with the DMRs. Moreover, genes that were DEGs and had differential methylated promoter (DMP) regions were selected for the further analysis.
Integrative Analysis of the Expression Levels of Sex-Related Genes and Methylation
The average methylation level of all DEGs and sex-related genes in the three sequence regions (TSS2K, Gene body and TTS2K) were calculated and then compared between females and males. Conventionally, the major region of focus is promoter, so analyzed the 5mC level in the promoter of these genes between females and males.
Statistical Analyses
Statistical analyses of DEGs and sex-related genes methylation level between females and males were undertaken in GraphPad (version 7.04). The p values were calculated using non-parametric tests. P < 0.05 was considered statistically significant.
Results
Summary of Gonadal Transcriptome of G. platifrons
To provide a global view of transcriptional differentiation and determine the molecular mechanisms underlying sex maintenance between male and female G. platifrons, six cDNA libraries were constructed and utilized for Illumina paired-end sequencing to generate representative transcripts of a wide range of biological processes. As shown in Table 1, after eliminating the primer and adapter sequences as well as the low-quality reads by fastp, the number of clean reads ranged between 38391192–50489378 with a relatively high base quality (Q30 > 92.63% for all samples). The unique mapping rate of all samples ranged from 63.35 to 69.20% when mapped against the updated G. platifrons reference genome (unpublished data). We obtained a total of 32,918 expressed genes in female and male gonads according to featureCounts (Supplementary Table 1, genes with expression).
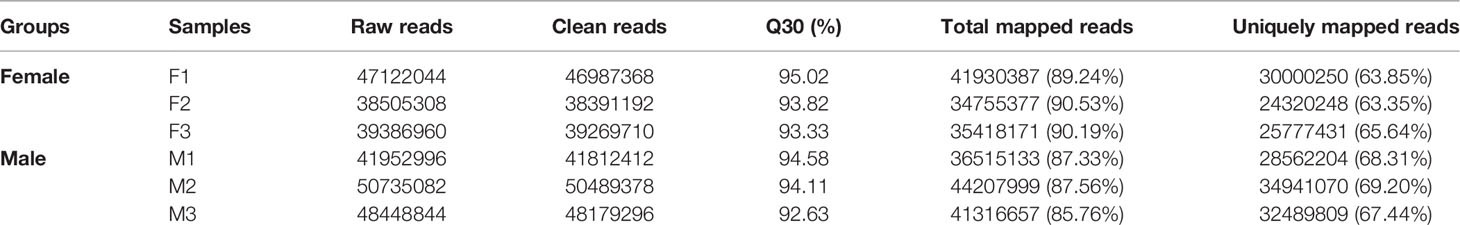
Table 1 Summary of clean reads of the transcriptome samples.
Enrichment Analysis of DEGs Between Ovaries and Testes
DEG analysis revealed 5923 sex-biased genes which were differentially expressed between the ovaries and testes (Figure 1B), 2711 of which were up-regulated in the ovaries (female-biased genes) and 3212 were up-regulated in the testes (male-biased genes) (Supplementary Table 1, all DEGs). To investigate the functions of DEGs, GO and KEGG analysis were implemented. For GO enrichment analysis, the results showed that 434 GO terms were enriched to 257 biological processes (BP), 96 to cellular components (CC) and 81 to molecular functions (MF) (q value < 0.05, Supplementary Table 2; GO_BP, GO_MF, and GO_CC). Among them, 3318 DEGs were enriched to BP, and most of the BP were enriched within single-organism processes and multicellular organismal processes. Altogether, 2995 DEGs were enriched to CC. In terms of CC, cell related terms contributed the greatest proportion. A total of 3738 DEGs were enriched within MF. For MF, protein binding and catalytic activity represented the most universal terms. 3318 DEGs assigned to BP and the top 20 enriched BP are showed in Figure 2A. The processes related to cell growth and development were extensively enriched, especially those related to cytoskeleton and cell motility. Additionally, 600, 116, and 11 DEGs were assigned to “reproduction”, “sex differentiation”, and “sex determination”, respectively, which account for 18.08, 3.5, and 0.33% of all BP terms, respectively. Furthermore, 2711 female-biased genes and 3212 male-biased genes were selected for separate GO enrichment analysis. We chose the top 10 enriched GO terms in BP, CC and MF for analyzing females and males (Supplementary Table 2, Top10 GO terms). Among them, nutrient metabolism related processes were the most enriched GO terms for female-biased genes while cytoskeletal correlation processes accounted for the dominant GO terms enriched for male-biased genes.
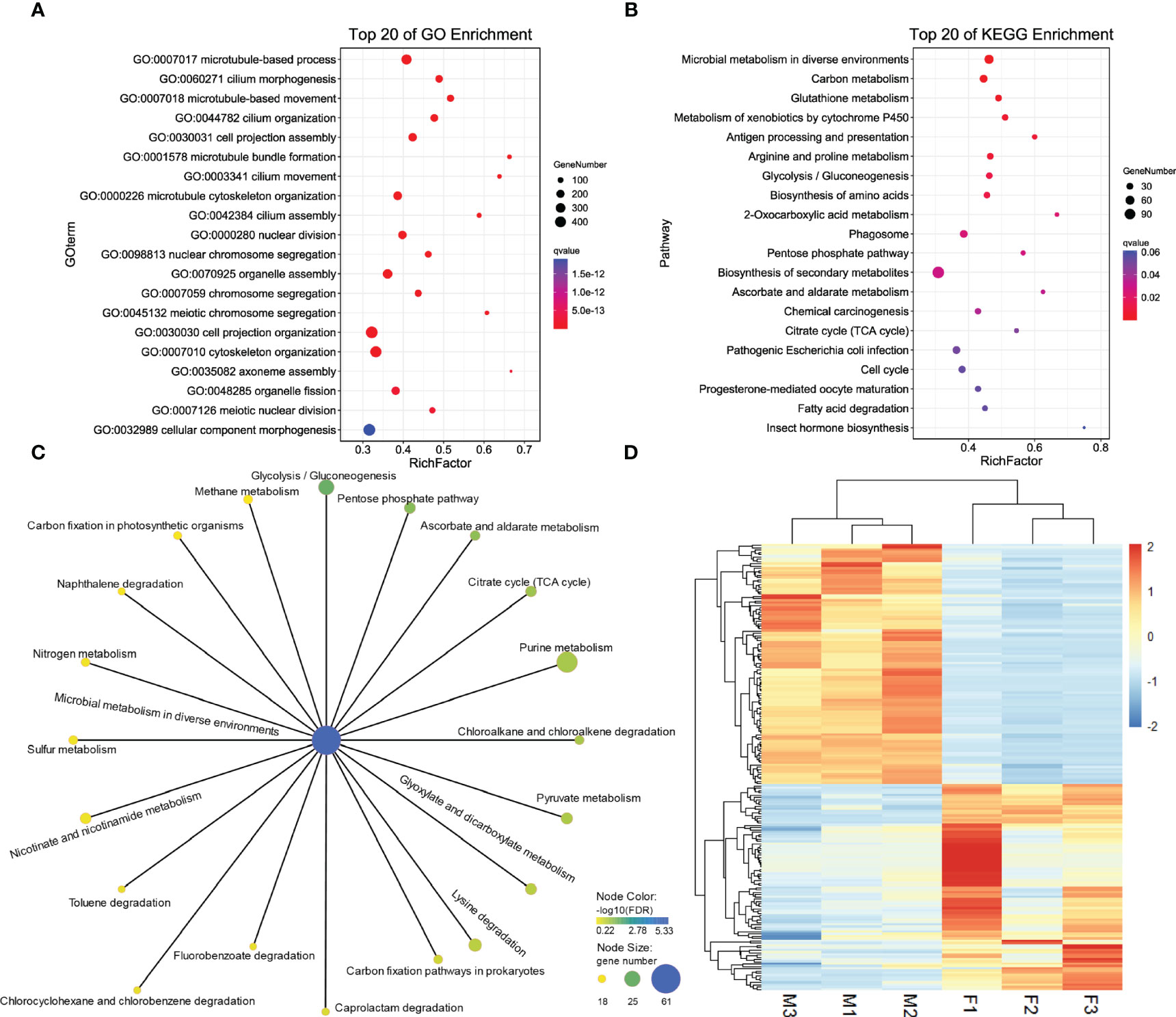
Figure 2 Top 20 enriched GO terms in BP of DEGs (A). Top 20 enriched KEGG pathways of DEGs (B). Pathways related microbial metabolism in diverse environments that have been enriched for DEGs (C). Expression patterns of 161 sex-related genes between two sexes of G. platifrons (D). For (A, B), the color represents the -log10 (q value), the bubble size represents the number of genes enriched into this term or pathway, and Rich Factor represents the ratio of the number of genes located in this term or pathway in the differentially expressed genes to the total number of genes located in this term or pathway in all the annotated genes. For (D), the heatmap were draw by “pheatmap” package (version 1.0.12) of R software. Each column represents a sample, each row represents a gene, the gene expression level was normalization by each row, red represents up-regulated while blue represents down-regulated.
For KEGG enrichment analysis, a total of 1120 candidate genes in DEGs were noted from KEGG. Of these, 470 DEGs were significantly mapped onto 15 pathways (q value < 0.05, Supplementary Table 2, KEGG). The KEGG enrichment results were comparable with GO results, nine were related to metabolism, including carbon, glutathione, cytochrome P450, amino acids, and glycometabolism, which indicated that metabolic process related pathways play a vital role in sex determination and differentiation (Figure 2B). We found that pathways of microbial metabolism in diverse environments were the most enriched pathways, the other 20 related microbial metabolism pathways are shown in Figure 2C. We also found that 17 DEGs mapped onto the WNT signaling pathway (Supplementary Table 2, WNT), including Wnt6 (CSbapl.23381) and Wnt7 (CSbapl.46743) which function as signaling molecules in sexually dimorphic development. Wnt6 and Wnt7 were up-regulated in female G. platifrons gonads and the canonical WNT signaling mediator, β-catenin, was also detected in transcriptome and up-regulated in female gonads, which indicated the conservatism of sex determination in the deep-sea mussel G. platifrons.
Search and Identification of Sex-Related Genes in G. platifrons
Of the total candidate genes, 161 were identified as sex-related genes generated using the “sex determination” and “sex differentiation” terms (Supplementary Table 2, sex-related genes). These genes could be categorized into transcriptional regulatory genes including the transcription factor, steroid hormone biosynthesis related genes, ovary and testis specific genes, gametogenesis related genes, some kinase genes related to cell differentiation, and development and other genes involved in sex determination or differentiation were mentioned in the literature. The expression patterns of 161 sex-related genes are shown in Figure 2D and Supplementary Table 2. Among these candidate genes, six vitelline envelope zona pellucida genes (VEZP type 6/9/14/17/24), one vitelline membrane outer layer protein 1 (VMO1), and one vitellogenin (VTG) were classified as ovary or oocyte-specific genes which were expressed more in females than males. Twenty-one genes had testis-specific expressions and 17 were sperm-specific genes such as testis-specific serine kinase (TSSK), testis-expressed protein (TEX), spermatogenesis-associated protein (SPATA), and sperm surface protein (SP); all of these genes were more highly expressed in males. Additionally, four genes annotated as the SOX gene family were found, including SOX2 in the SOXB1 group, SOX4 in the SOX C group, SOX5 in the SOX D group, and SOX9 in the SOX E group. The main transcription factors such as FOXL2 and DMRT2 involved in female and male sex determination, respectively were also identified, and the FOXL2 were expressed more in females, while DMRT2 were high expression in males. Moreover, sex steroidogenesis-related genes such as estradiol 17β-dehydrogenase (E2DH), 17β-hydroxysteroid dehydrogenase (17β-HSD type 6/10/12/14) and CYP17a were all significantly high expressed in females. The roles of these genes need further study in G. platifrons, but most of them have been identified as playing important roles in sex-determination/differentiation or gametogenesis in other animals and molluscan species.
Comparative Transcriptome Analysis Between Deep-Sea and Shallow Water Mussels
To find the similarities and differences between deep-sea mussels and shallow water relatives, we first used OrthoFinder (version 2.5.2) to screen the homologous genes of deep-sea mussel G. platifrons and shallow water mussel L. fortunei. A total of 2543 orthogroups were documented (Supplementary Table 3, othogroups), uncovering 35 shared sex-related genes by intersection with 161 sex-related genes in G. platifrons. Then, we compared the shared expression patterns of 35 sex-related genes between a deep-sea mussel and shallow water mussel. Thirty-five shared sex-related genes contained most of the genes that play an important role in sex determination and differentiation, such as SOX gene family, FOXL2, β-catenin and some testis-specific genes. For female high expressed genes, such as SOX2, FOXL2 and β-catenin, SOX2 high expressed in female than male about 10 times in both two species, FOXL2 were about 100 times more expressed in females than males of G. platifrons, and about 900 times more expressed in females than males of L. fortunei. The same expression patterns also found in β-catenin which were all more than 2 times expressed in females than males in both species. As similar expression pattern was observed in male-upregulated genes in both shallow water and deep-sea mussels, such as TEX36 and TEX47, which about 2-10 times more expressed in males than females (Figure 3 and Supplementary Table 3, shared sex-related genes).
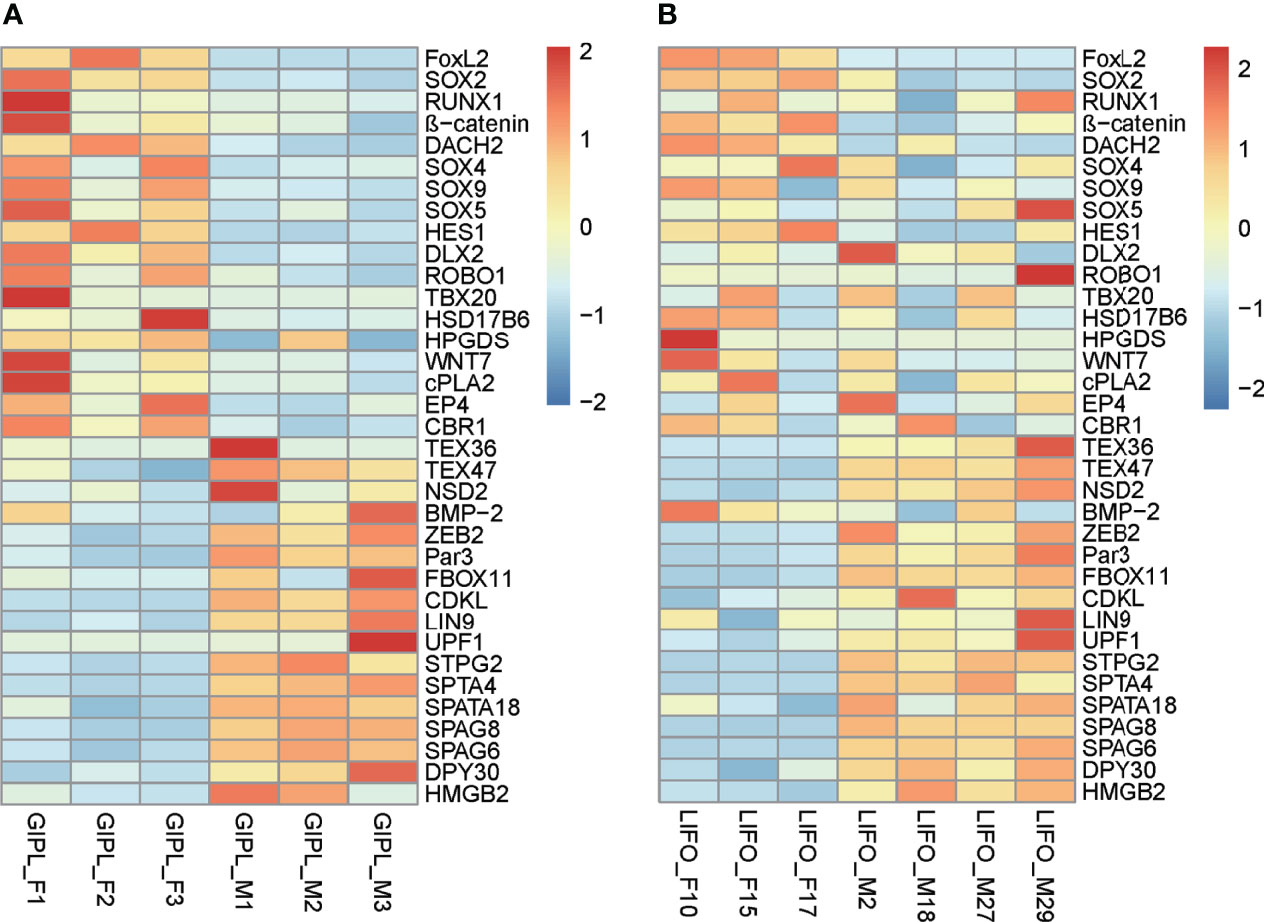
Figure 3 Expressed pattern of shared sex-related genes in female and male between G. platifrons (GIPL_F1, GIPL_F2, GIPL_F3 versus GIPL_M1, GIPL_M2, GIPL_M3) (A) and L. fortunei (LIFO_F10, LIFO_F15, LIFO_F17 versus LIFO_M2, LIFO_M18, LIFO_M27, LIFO_M29) (B) The heatmap were draw by “pheatmap” package (version 1.0.12) of R software. Each column represents a sample, each row represents a gene, the gene expression level was normalization by each row, red represents up-regulated while blue represents down-regulated.
Summary Gonadal Methylome of G. platifrons
To examine the roles of DNA methylation in sex maintenance and gonad development in adult G. platifrons, the methylation patterns and levels of gonads for female and male G platifrons were detected with WGBS analysis (Supplementary File 1). We found that the 5mC levels of all genomic C sites for males (3.22%) were slightly higher than for females (3.09%) and the 5mC levels for the CG site (>96%) were higher than for the CHG (<1%) and CHH (~2%) sites. For the GC site, the average 5mC levels in the gene body (30-40%) were higher than that in other sequence regions such as promoter (~20%) and repeat regions (~10%) in the genome. We also found that the 5mC level showed a weak positive correlation with gene expression level (Spearman correlation r range from 0.313 to 0.543) at the three sequence regions (TSS2K, Gene body, TTS2K) and in both sexes (Supplementary Table 4, F_TTS2K_5mC_vs_gene_express, M_TTS2K_5mC_vs_gene_express, F_genebody_5mC_vs_gene_express, M_genebody_5mC_vs_gene_express, F_TSSK2k_5mC_vs_gene_express, and M_TSSK2k_5mC_vs_gene_express).
Integrative Analysis of Transcriptome and Methylome
The differential 5mC level of DEGs has the same trend with all genes being detected in the three sequence regions, and the methylation levels were all significantly higher in males than females (Figure 4A and Supplementary Table 4, 5mC_DEGs). To identify the role of methylation more clearly in sex determination or sex differentiation, we screened the 1903 shared genes of DMRs and DEGs for further analysis (Figure 4B, Supplementary Table 4, DMRs_vs_DEGs). GO functional enrichment analysis showed that 1180 shared genes were enriched in the BP category, and 256 BP GO terms were significantly enriched (q value < 0.05). The top 20 enriched BP terms are shown in Figure 4C. Additionally, BP terms related to “sex differentiation” and “male sex differentiation” were all significantly enriched (Supplementary Table 4, BP_shared_genes_ DMRs_vs_DEGs). For KEGG analysis, 419 shared genes between DMRs and DEGs were associated with 335 pathways. The top 20 enriched pathways are shown in Figure 4D. A total of 128 shared genes were significantly enriched in metabolism and hormone biosynthesis related pathways (q value < 0.05). Some of the shared genes were enriched in the “WNT signaling pathway”, “oocyte meiosis”, and “FoxO signaling pathway”, which play important roles in sex determination or differentiation and germ cell development.
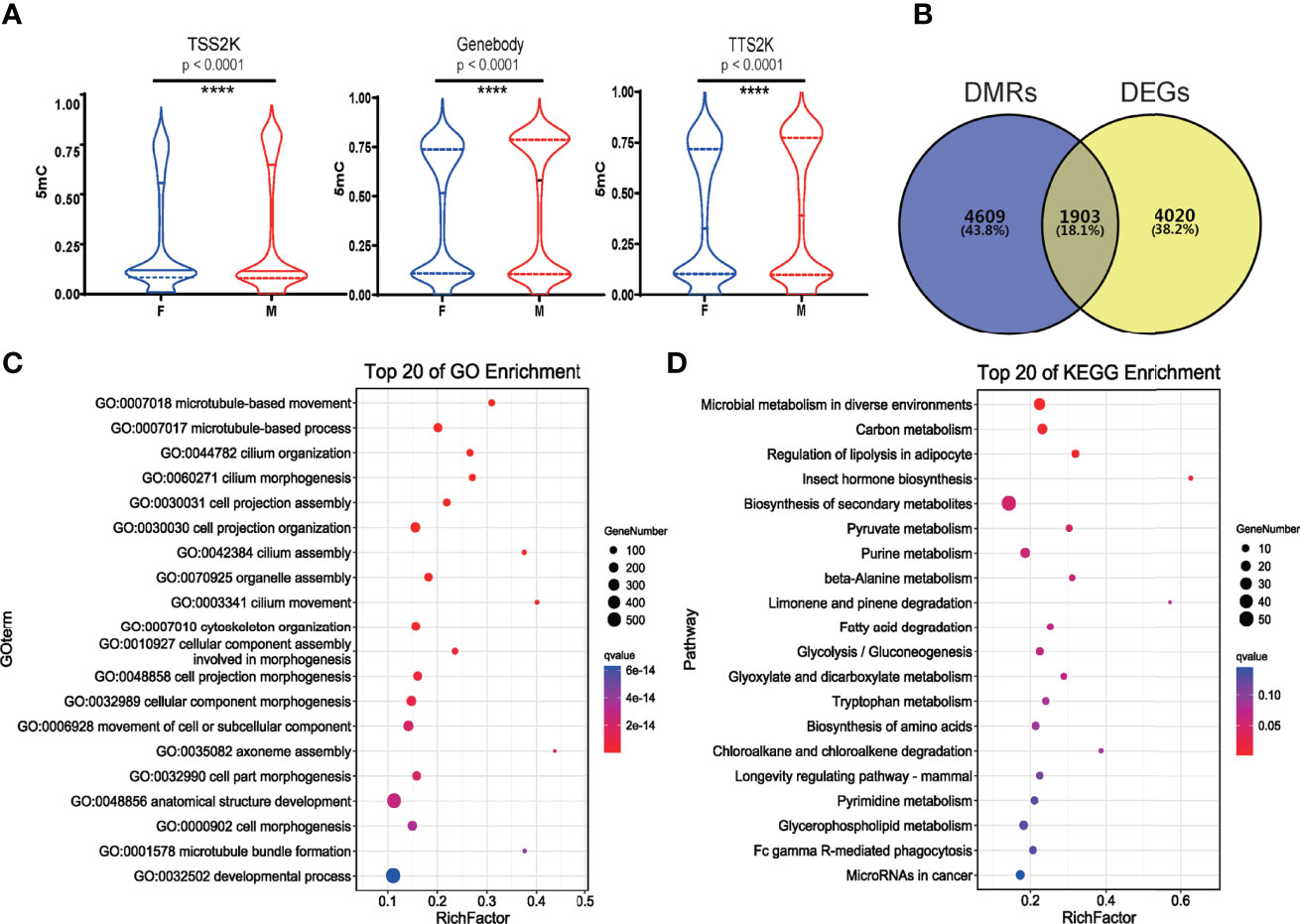
Figure 4 5mC levels of all genes in 3 sequence regions (TSS2K, Gene body and TTS2K) between female and male G. platifrons (Wilcoxon test, and p < 0.0001 (****) for all 3 sequence regions) (A); 1903 shared genes between DMRs and DEGs of two sexes (B); Top 20 enriched BP GO terms of shared genes between DMRs and DEGs (C); Top 20 enriched KEGG pathways of shared genes between DMRs and DEGs (D).
Comparing the 5mC Levels of Sex-Related Genes With Other DEGs, and the Different 5mC Level of Sex-Related Genes Between the Two Sexes
The 5mC levels of each sex-related gene and DEG were documented in Supplementary Table 4. In total, 59 of 161 (36.65%) sex-related genes in G. platifrons displayed significantly differential methylation patterns between females and males (Supplementary Table 4, DMR_vs_sex-related genes), which contrasts to ~ 20.97% (6904/32,918) over whole genome (Fisher’s exact test, p < 0.0001). To find out if the 5mC levels of sex-related genes were different from other DEGs, the 5mC levels of the TSS2K, the gene body and the TTS2K regions of 161 sex-related genes were compared with the other DEGs (5762 genes) using Kolmogorov-Smirnov tests. We defined the OF and OM as the other DEGs in females and other DEGs in males. The result shows that for the TSS2K region, the 5mC levels of sex-related genes for females (F) were significantly higher than for the other DEGs in females (OF) (Figure 5A, F vs OF, p=0.0169) while no significantly differences were found in males (Figure 5A, M vs OM, p=0.0977). For the gene body region, the 5mC levels of sex-related genes were not significantly different from OF (Figure 5B, F vs OF, p=0.0582), while 5mC levels of sex-related genes were significantly higher than OM (Figure 5B, M vs OM, p=0.0273). For the TTS2K region, 5mC levels of sex-related genes were not significantly different from OF (Figure 5C, F vs OF, p=0.2180) and OM (Figure 5C, M vs OMp=0.1567), respectively. Moreover, the 5mC levels of 161 sex-related genes for males were all significantly higher than for females in the gene body and TTS2K regions (Figure 5D, Gene body and TTS2K, p < 0.0001). However, for the TSS2K region, there were no significant differences between females and males (Figure 5D, TSS2K, p = 0.2113).
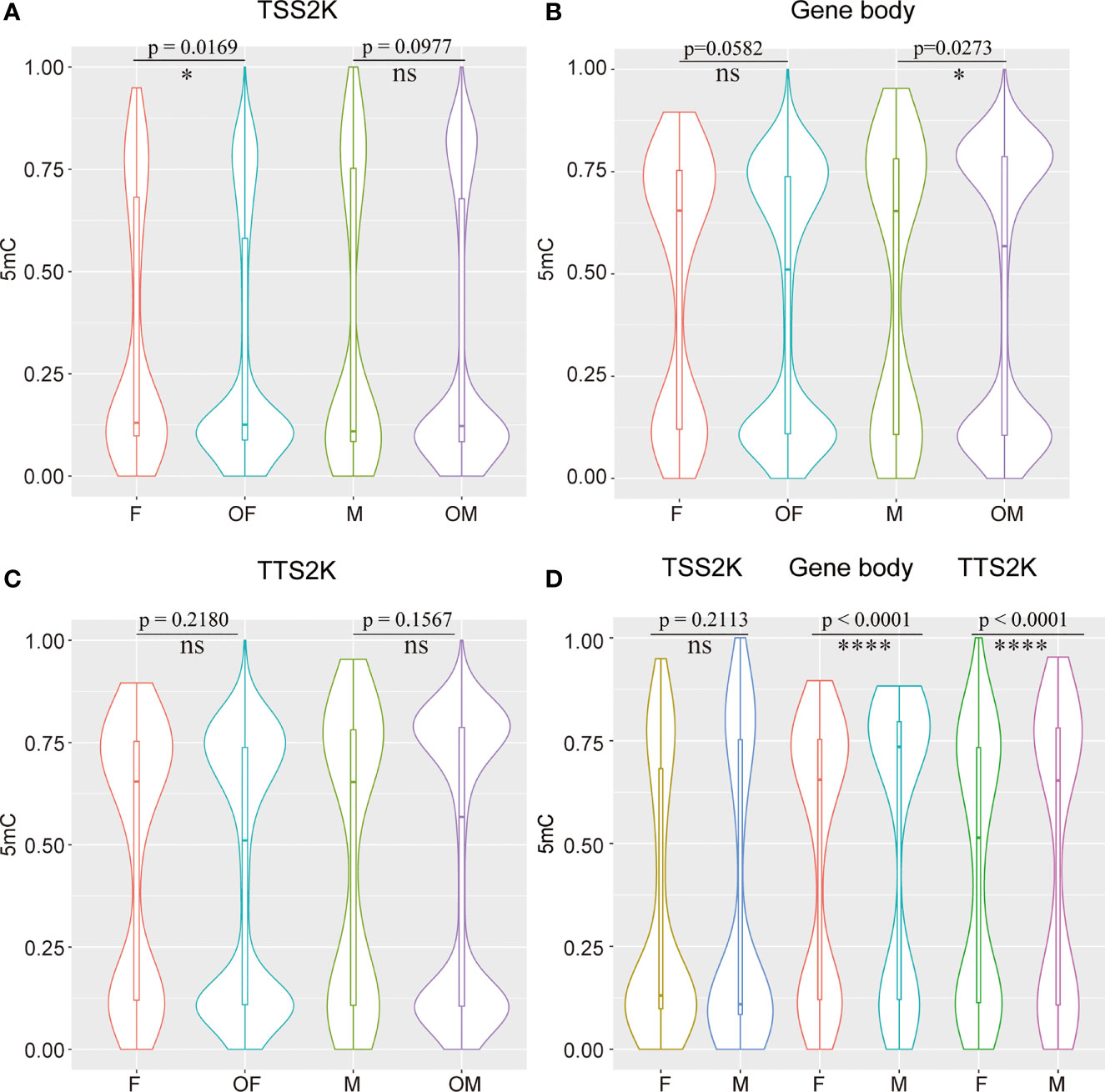
Figure 5 Compare 5mC levels between 161 sex-related genes and the other DEGs in two sexes among TSS 2K, Gene body, and TTS 2K regions (A–C) and compare 5mC levels of 161 sex-related genes between females and males in TSS 2K, Gene body, and TTS 2K regions (D); Kolmogorov-Smirnov test for (A, C), and Wilcoxon test for (D), ns, *, and **** represent p > 0.05, p < 0.05 and p < 0.0001 respectively, OF and OM represent the other DEGs in female and the other DEGs in male.
We then divided 161 sex-related genes into 73 female-biased sex-related genes (FBSGs) and 88 male-biased sex-related genes (MBSGs) according to expression levels. We found that 5mC levels were all significantly higher in MBSGs than FBSGs for the two sexes and the three regions (Figure 6, p < 0.0001). To find if the 5mC levels of FBSGs and MBSGs were significantly different in females and males, we conducted Wilcoxon matched-pairs signed rank tests for the three regions (Figure 6). The results showed that, except for the levels of FBSG 5mC levels in the gene body regions, there were no significant differences between the two sexes (Figure 6, p = 0.0554). Other 5mC levels of FBSGs and MBSGs were all higher in males than females in all three regions (p < 0.0001).
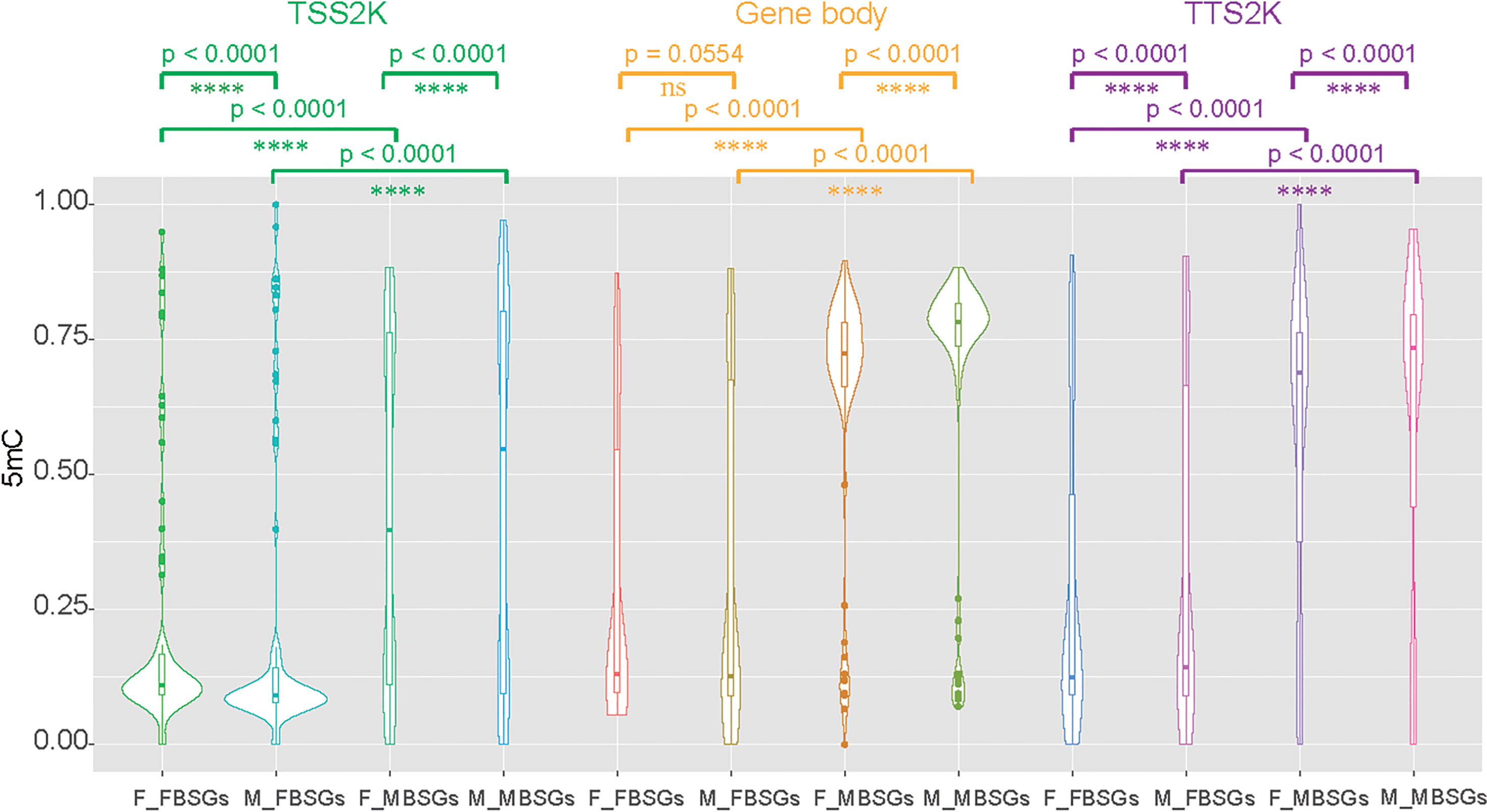
Figure 6 Compare 5mC levels between female-biased sex-related genes (FBSGs) and male-biased sex-related genes (MBSGs) in the TSS 2K, Gene body, and TTS 2K regions by Kolmogorov-Smirnov test and compare 5mC levels of FBSGs and MBSGs in two sexes by Wilcoxon test. ns and **** represent p > 0.05 and p < 0.0001 respectively.
We subsequently investigated whether the sex-related genes overlapped with the DMPs (Supplementary Table 4, DMP_vs_sex-related genes). Only 12 sex-related genes were found by transcriptome and methylation associated analysis. These genes include male-biased genes such as testis-specific serine kinase (CSbapl.702), a WNT pathway gene β-catenin (CSbapl.3788) which is believed to be essential for female sex determination, and the transcription factor SOX family gene SOX5. Except for gene Prx, all 5mC levels of females were lower than those of males, which is in line with the trend of 5mC levels of all sex-related genes. For five female-biased genes, the hypomethylation of promoter regions corresponded with high methylation levels in promoter regions, which would suppress gene expression. However, for six male-biased genes except Prx, the hypermethylation of promoter regions resulted in high gene expression.
Discussion
Sex determination mechanisms are widely divergent in animals and they can evolve rapidly (Bachtrog et al., 2014; Beukeboom and Perrin, 2014). Genetic and environmental sex determination systems are now considered as two ends of a continuum rather than separate mechanisms in vertebrate temperature-dependent sex determination species (Sarre et al., 2004; Quinn et al., 2011). As for bivalve species, sex may be determined by genes in combination with environment (Breton et al., 2018). Deep-sea mussels entered the deep-sea chemosynthetic environment at least 85 million years ago (Lorion et al., 2013), therefore the mechanisms of sex determination or differentiation may not be the same as for shallow water species. Sex determination or sex differentiation are intricate processes not always clearly distinguishable because their signaling cascades can be integrated in invertebrates (Matson and Zarkower, 2012; Huang et al., 2017). As with previous studies, in the present study the genes involved in sex determination or sex differentiation are described as ‘sex-related genes’ (Li et al., 2016; Chen et al., 2017; Gonzalez-Castellano et al., 2019; Yao et al., 2021). In the present work we used transcriptomic analysis to unravel sex-related genes featured in sex determination or differentiation, and we also tried to find out if epigenetics would affect the expression levels of sex-related genes. This is the first integrated analysis of the gonads of deep-sea mussels, providing data about sex-related genes and to study the correlation between gene methylation levels and expression levels.
Putative Mechanisms of Sex Determination or Differentiation in G. platifrons
Many genes related to sex determination are transcription factors, especially in terms of the DMRT, FOX and SOX, gene families (Smith et al., 2009; Matson et al., 2011; Huang et al., 2017; Li et al., 2018). By analyzing the female and male gonad transcriptome of G. platifrons, we identified at least 161 sex-related genes which were all belonged to DEGs and annotated to sex determination or sex differentiation GO term. Sixteen transcription factors were detected, including the common genes DMRT2, FOXL2, and SOX9, which were shown to be deeply conserved in sex determination or differentiation in animals (Liu et al., 2012; Li et al., 2018; Afonso et al., 2019).
The DMRT family of genes contains a highly conserved DNA binding domain called the DM domain. There are seven DMRT genes that have been shown to play critical roles in sexual regulation, including sex differentiation, sexual dimorphism, and spermatogenesis in a broad variety of metazoans, including nematodes, insects, and vertebrates (Zarkower, 2013). Another famous sex determination transcription factor is FOXL2. FOXL2 is one of the FOX gene family members and preferentially expressed in the ovary of vertebrates, controlling ovarian differentiation and maintenance by repression of testis-specific genes from the early embryonic gonad throughout adult life (Uhlenhaut et al., 2009). Therefore, FOXL2 and DMRT are the key genes for female and male sex determination and maintenance, respectively. In addition, they are mutually antagonistic in determining the sex of mammals (Matson et al., 2011). In G. platifrons, the DMRT2 gene was examined and found to be up-regulated in male gonads (q < 0.05), and FOXL2 was at higher levels in females than males (q < 0.05). As in mammals, the balance between FOXL2 and DMRT2 is suggested to be important for sex determination in G. platifrons. This phenomenon was also reported in other mollusca species such as the blacklip pearl oyster Pinctada margaritifera, Pacific oyster Crassostrea gigas, Yesso scallop Patinopecten yessoensis and razor clam Sinonovacula constricta, although the genes in the DMRT family and FOX family are not always the same among different species, for example DMRT2 and FOXL2 in P. margaritifera, Dsx and FOXL2 in C. gigas, DMRT1L and FOXL2 in P. yessoensis, FOXA1, FOXD2, and DMRTA2 in S. constricta (Teaniniuraitemoana et al., 2014; Zhang et al., 2014; Li et al., 2018; Yao et al., 2021).
The male-determining gene in mammals is the SRY gene. SRY is a member of the SOX gene family of DNA-binding proteins. SRY complexed with the SF1 protein would upregulate other transcription factors including the important SOX9 which is the up-stream regulator of male sex determination (Sinclair et al., 1990; Kashimada and Koopman, 2010). SOX9 is one gene in the SOXE group that has an expression in the Sertoli cells necessary for testis determination, and is reinforced by FGF9 and DMRT1 which are thus essential to this network (Matson and Zarkower, 2012). In mammals, once SRY expression begins, other genes start to express only in one sex, so the SOX9 would not be active in female gonads (Gross, 2006). However, in our study, although no SRY was detected, the downstream gene SOX9 was found in the gonad transcriptome, and SOX9 was not only active in male testes but also in female ovaries. Moreover, SOX9 was not only highly expressed in males but also female. High expression in females may explain the phenomenon of hermaphroditism previously discovered in G. platifrons (Zhong et al., 2020). For other gonadal transcriptomes of shallow water bivalves, SOX9 expression levels were also determined, such as in P. yessoensis, C. gigas and L. fortunei, but for these bivalves the SOX9 expression levels were not significantly different between males and females (Zhang et al., 2014; Li et al., 2016; Afonso et al., 2019). Overall, we suggest that SOX9 could be a key factor in sex determination or differentiation in G. platifrons, but it may not function in the way that it does in mammals or indeed other shallow bivalves.
We also identified the β-catenin gene in G. platifrons. In mammals, β-catenin, the center in the canonical WNT signaling pathway, is a key transcriptional regulator and is essential for ovarian development and maintenance (Liu et al., 2009). RSPO1-WNT4-β-catenin in the canonical WNT signaling pathway is independent and complementary to the FOXL2-leading pathway (Kocer et al., 2008). β-catenin presents in the gonads of both sexes of G. platifrons but was expressed significantly higher in ovaries, while RSPO1 was not found, and Wnt4 was not a DEG in the transcriptome of G. platifrons. Therefore, the existence and specific function of β-catenin for female sex determination needs further investigation.
In non-mammalian vertebrates, sterol hormones play an important role in sex determination and sex differentiation (Navarro-Martin et al., 2011). Aromatase CYP19a (Cytochrome P450 family 19) can irreversibly converts androgens into estrogens, which influences the ratio of androgen-to-estrogen, and the different ratio then determines undifferentiated gonad sexually differentiates into a testis or ovary. Sixteen Cytochrome P450 family genes were found in the deep-sea mussel gonadal transcriptome and eight were found in females and males’ DEGs, however, CYP19a were not found in these data, but another important Cytochrome P450 family 17, one of the sex-related genes CYP17a was found both in two sexes of G. platifrons which were high expression in female. CYP17a has been shown to play important roles in female sex determination and male fertility by regulating sex steroid biosynthesis in fish (Yang et al., 2021). For G. platifrons, 9.88% were methylated in the testes and 8.21% were methylated in the ovaries for the TSS2K region of CYP17a, however, CYP17a was not defined as a DMR. So, the effect of methylation on CYP17a is not as pronounced as it is on CYP19a which hypermethylation occurs in testis and gene high expression in ovaries of fish (Wen et al., 2014), and it also possible that the mechanism of sterol hormones in mollusks is obviously different from that in vertebrates.
Specifically, we compared the sex-related genes between the deep-sea mussel G. platifrons and the shallow water mussel L. fortunei. The results showed that the common expression pattern of the male-biased sex-related genes and the expression level of those genes were all coincident among male samples, while for female-biased sex-related genes, the expression pattern and gene expression levels were inconsistent in L. fortunei. We suggest that the common expression pattern for female G. platifrons may be caused by the stable deep sea environment or the same development stages of gonads. Therefore, there are two factors that may lead to differences in the expression patterns of female-biased sex-related genes between G. platifrons and L. fortunei. First, the development cycle is different at the time of sampling for female gonads of L. fortunei. Second, the shallow water environment is not stable, and the maternal immunity may be influential, given that the SOX family genes are important genes that cause differential expression patterns in female individuals of L. fortunei, and the SOX family genes are reported to be related to immunity (Furukawa et al., 2021). The ovary is an important source of maternal immunity, therefore the differential expression between individuals may be caused by their different responses to the changing environment (Wang et al., 2015). In addition, the other important sex determination genes like FOXL2, β-catenin, and Wnt7 all have same the expression patterns between G. platifrons and L. fortunei (Figure 3), suggesting some conserved sex determination mechanisms between these two mussels.Based on previous studies and our transcriptome data, all the aforementioned genes which are shared among G. platifrons and other animals are thought to be involved in sex determination, and thus we speculate that the sex determination or differentiation mechanism may be conserved in some degree among deep-sea mussels and shallow water species or other vertebrates, but there are also significant differences in the male and female expression patterns of specific sex-related genes. As for G. platifrons, based on sequence homology and functions inferred from transcriptome data, we suggest that although traditionally identified sex-related genes are not strictly expressed in male and female gonads, the expression of these genes or homologues, especially SOX, FOXL2, and DMRT genes, which are commonly referred to in other mollusks, are significantly different between males and females. We suggest that SOX may have high expression in the early developmental stage before G. platifrons gonadal maturation and in the up-stream regulator leading to male sex determination. However, in the gonadal maturation stage, the SOX gene would not highly express in testes. Thus, SOX would directly or indirectly activate DMRT2 for testis development in early stages and DMRT2 may keep testis functional through a life. FOXL2 may be highly expressed in ovaries and complemented by β-catenin leading to female sex-determination pathways in G. platifrons.
The Relationship Between DNA Methylation and Sex-Related Genes Expression
In the present study, we have shown that G. platifrons has some sex determination or differentiation mechanisms common with the shallow water mussel L. fortunei and other bivalves. Additionally, G. platifrons is shown to be a gonochoric species (Zhong et al., 2020), while the sex ratio is not coincident among groups of different shell lengths. Additionally, while hermaphroditism was observed in our samples, this was only the case in two large individuals, so we suspect that this species may not exhibit large-scale sex-reversal and sex maintenance. We recorded higher levels of global DNA methylation level in the testis (3.22%) than ovary (3.09%), suggesting that male germ cells may be more heavily methylated than female germ cells, which concurs with findings reported for P. yessoensis (Li et al., 2019). Additionally, we also found that 5mC is predominantly in the gene body (30-40%) in both sexes, which is in agreement with previous studies of C. gigas (Gavery and Roberts, 2013; Olson and Roberts, 2014; Wang et al., 2014). Through enrichment analysis of the intersection of DMRs and DEGs in two sexes of G. platifrons, we found that methylation mainly affects metabolic processes and morphological differentiation of organisms (Figure 4C, D). Therefore, we hypothesize that methylation plays an important role in the differential metabolism of males and females in deep-sea mussels, in particular high 5mC levels may have a vital effect on male sex determination.
To further explore the effect of methylation on sex determination or differentiation, we chose to study mainly the sex-related genes. The methylation levels presented negative correlations with expression levels of sex-related genes in the ovaries of G. platifrons, which corroborates the concept that increased methylation corresponds to decreased gene expression (Riviere et al., 2013). In males, our results indicated the positive correlation between 5mC and sex-related gene expression, where genes with high expression levels had high 5mC levels (Supplementary Figure 2). This is similar to the observations reported for oyster male gamete (Gavery and Roberts, 2013; Olson and Roberts, 2014). We also found that the tendency for 5mC levels of all sex-related genes and MBSGs or FBSGs were all higher in males than females in the three sequence regions, although some comparisons were not statistically significantly different (Figures 5D and 6). Moreover, similar patterns were found at the level of the single sex-related gene (Supplementary Table 4, 5mC_sex-related genes) indicating that females present more hypomethylation than males, but in males hypermethylation correlated with high levels of gene expression in all sequence regions (Supplementary Figure 2). We hypothesize that DNA methylation may act as an enhancer rather than a repressor in testis development. As show in Figure 7, we integrated the differential methylation of several key sex-related genes to speculate the sex-determining mechanism of deep-sea mussel G. platifrons.
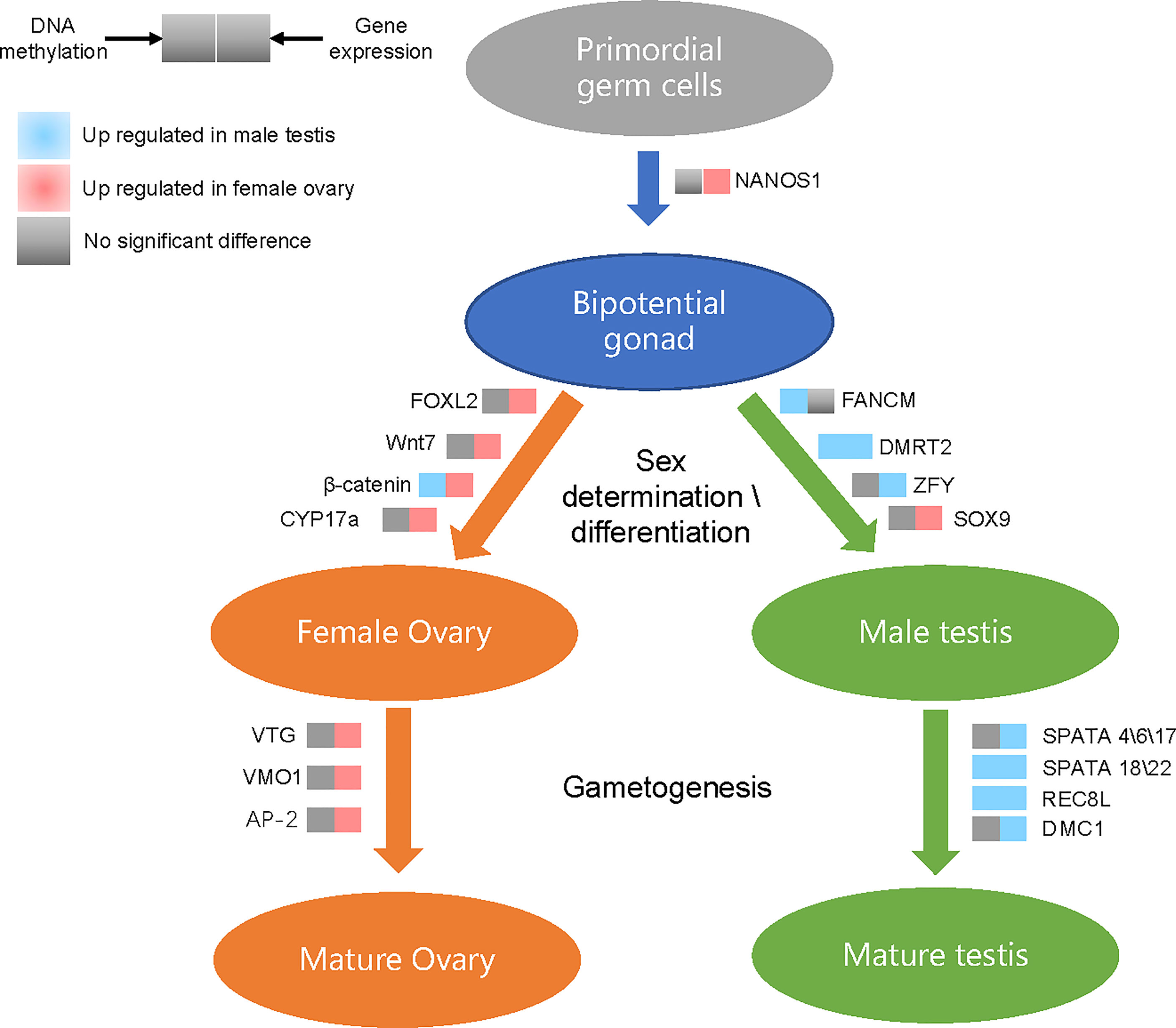
Figure 7 Differentially methylated and differentially expressed genes in the putative sex determination pathway of G. platifrons. For each gene presented in the pathway, the methylation (left square) or expression (right square) changes when comparing testes with ovaries are shown by different colors.
For the result stated above, we suggest that the different methylation levels of sex-related genes in the two sexes and the inconsistent correlation between methylation level and sex-related genes expression level may play an important role for sex determination or differentiation of G. platifrons in the deep-sea.
Conclusion
In this study, we offer the first report of the gonadal transcriptome and methylome underlying sex determination or differentiation of G. platifrons which is the representative deep-sea bivalve in chemosynthetic ecosystem. We identified 161 sex-related genes that could be involved in sex determination or differentiation, along with the molecular mechanisms of sex determination in G. platifrons. Moreover, through the 5mC levels of genes detected, we suggest that hypermethylation phenomenon of male-biased genes are important for male sex differentiation and keeping the sex ratio balanced in deep-sea chemosynthetic ecosystems.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA789090.
Author Contributions
ZZ and MW, conceptualization, methodology, investigation, formal analysis, and writing – original draft. HC, HW, HZ, LZ, LC, CL and ML, samples collection, inverstigation. CLL, conceptualization, supervision, project administration, funding acquisition, writing – review and editing. All authors gave their final approval for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant No. 42106100, 42076091 and 42030407), the Open Research Project of National Major Science and Technology infrastructure (RV KEXUE) (NMSTI-KEXUE2017K01), the Key Research Program of Frontier Sciences, CAS, (No. QYZDB-SSW-DQC036), the Senior User Project of R/V Kexue (No. KEXUE2019GZ02) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB42000000).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the crews onboard the R/V Kexue for their assistance in sample collection and all the laboratory staff for continuous technical advice and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.856291/full#supplementary-material
References
Afonso L. F., Americo J. A., Soares-Souza G. B., Torres A. L. Q., Wajsenzon I. J. R., de Freitas Rebelo M. (2019). Gonad Transcriptome of Golden Mussel Limnoperna Fortunei Reveals Potential Sex Differentiation Genes. bioRxiv doi: 10.1101/818757
Bachtrog D., Mank J. E., Peichel C. L., Kirkpatrick M., Otto S. P., Ashman T. L., et al. (2014). Sex Determination: Why So Many Ways of Doing It? PloS Biol. 12 (7), e1001899. doi: 10.1371/journal.pbio.1001899
Barry J. P., Buck K. R., Kochevar R. K., Nelson D. C., Fujiwara Y., Goffredi S. K., et al. (2002). Methane-Based Symbiosis in a Mussel, Bathymodiolus Platifrons, From Cold Seeps in Sagami Bay, Japan. Invertebrate Biol. 121 (1), 47–54. doi: 10.1111/j.1744-7410.2002.tb00128.x
Beukeboom L. W., Perrin N. (2014). The Evolution of Sex Determination (USA: Oxford University Press).
Boutet I., Tanguy A., Le Guen D., Piccino P., Hourdez S., Jollivet D., et al. (2008). Transcriptomic Approach to Study the Response to Rapid Changes of Temperature in the Hydrothermal Vent Mussel Bathymodiolus Azoricus. Comp. Biochem. Physiol. Part A.: Mol. Integr. Physiol. 151, S38–S39. doi: 10.1016/j.cbpa.2008.05.143
Breton S., Capt C., Guerra D., Stewart D. (2018). “Sex-Determining Mechanisms in Bivalves,” in Transitions Between Sexual Systems: Understanding the Mechanisms of, and Pathways Between, Dioecy, Hermaphroditism and Other Sexual Systems. Ed. Leonard J. L. (Cham, Switzerland: Springer International Publishing), 165–192.
Briones C., Nuñez J. J., Pérez M., Espinoza-Rojas D., Molina-Quiroz C., Guiñez R. (2018). De Novo Male Gonad Transcriptome Draft for the Marine Mussel Perumytilus Purpuratus With a Focus on its Reproductive-Related Proteins. J. Genomics 6, 127–132. doi: 10.7150/jgen.27864
Cao L., Lian C., Zhang X., Zhang H., Wang H., Zhou L., et al. (2021). In Situ Detection of the Fine Scale Heterogeneity of Active Cold Seep Environment of the Formosa Ridge, the South China Sea. J. Mar. Syst. 218, 103530. doi: 10.1016/j.jmarsys.2021.103530
Capel B. (2017). Vertebrate Sex Determination: Evolutionary Plasticity of a Fundamental Switch. Nat. Rev. Genet. 18 (11), 675–689. doi: 10.1038/nrg.2017.60
Carney S. L., Formica M. I., Divatia H., Nelson K., Fisher C. R., Schaeffer S. W. (2006). Population Structure of the Mussel “Bathymodiolus” Childressi From Gulf of Mexico Hydrocarbon Seeps. Deep Sea Res. Part I: Oceanographic Res. Papers 53 (6), 1061–1072. doi: 10.1016/j.dsr.2006.03.002
Chen H., Xiao G., Chai X., Lin X., Fang J., Teng S. (2017). Transcriptome Analysis of Sex-Related Genes in the Blood Clam Tegillarca Granosa. PloS One 12 (9), e0184584. doi: 10.1371/journal.pone.0184584
Chen S., Zhou Y., Chen Y., Gu J. J. B. (2018). Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 34, 17, i884–i890. doi: 10.1093/bioinformatics/bty560
Collin R. (2013). Phylogenetic Patterns and Phenotypic Plasticity of Molluscan Sexual Systems. J. Integr. Comp. Biol. 53 (4), 723–735. doi: 10.1093/icb/ict076
Comtet T., Le Pennec M., Desbruyères D. (1999). Evidence of a Sexual Pause in Bathymodiolus Azoricus (Bivalvia: Mytilidae) From Hydrothermal Vents of the Mid-Atlantic Ridge. J. Marine Biol. Assoc. U. K. 79 (6), 1149–1150. doi: 10.1017/S0025315499001514
Conover D. O., Heins S. W. J. N. (1987). Adaptive Variation in Environmental and Genetic Sex Determination in a Fish 326 (6112), 496–498. doi: 10.1038/326496a0
Du X., Wang B., Liu X., Liu X., He Y., Zhang Q., et al. (2017). Comparative Transcriptome Analysis of Ovary and Testis Reveals Potential Sex-Related Genes and Pathways in Spotted Knifejaw Oplegnathus Punctatus. Gene 637, 203–210. doi: 10.1016/j.gene.2017.09.055
Dyson E. A., Hurst G. D. D. (2004). Persistence of an Extreme Sex-Ratio Bias in a Natural Population. Proc. Natl. Acad. Sci. U.S.A. 101 (17), 6520–6523. doi: 10.1073/pnas.0304068101
Eckelbarger K. J., Young C. M. (1999). Ultrastructure of Gametogenesis in a Chemosynthetic Mytilid Bivalve (Bathymodiolus Childressi ) From a Bathyal, Methane Seep Environment (Northern Gulf of Mexico). Mar. Biol. 135 (4), 635–646. doi: 10.1007/s002270050664
Feng S., Cokus S. J., Zhang X., Chen P.-Y., Bostick M., Goll M. G., et al. (2010). Conservation and Divergence of Methylation Patterning in Plants and Animals. Proc. Natl. Acad. Sci. 107 (19), 8689–8694. doi: 10.1073/pnas.1002720107
Furukawa F., Doshimo Y., Sodeyama G., Adachi K., Mori K., Mori Y., et al. (2021). Hemocyte Migration and Expression of Four Sox Genes During Wound Healing in Pacific Abalone, Haliotis Discus Hannai. Fish & Shellfish Immunology 117, 24–35. doi: 10.1016/j.fsi.2021.07.011
Gavery M. R., Roberts S. B. (2013). Predominant Intragenic Methylation Is Associated With Gene Expression Characteristics in a Bivalve Mollusc. PeerJ 1, e215. doi: 10.7717/peerj.215
Gempe T., Beye M. (2011). Function and Evolution of Sex Determination Mechanisms, Genes and Pathways in Insects. BioEssays: News Rev. Mol. Cell. Dev. Biol. 33 (1), 52–60. doi: 10.1002/bies.201000043
Ghiselli F., Iannello M., Puccio G., Chang P. L., Plazzi F., Nuzhdin S. V., et al. (2018). Comparative Transcriptomics in Two Bivalve Species Offers Different Perspectives on the Evolution of Sex-Biased Genes. Genome Biol. Evol. 10 (6), evy082–evy082. doi: 10.1093/gbe/evy082
Ghiselli F., Milani L., Chang P. L., Hedgecock D., Davis J. P., Nuzhdin S. V., et al. (2012). De Novo Assembly of the Manila Clam Ruditapes Philippinarum Transcriptome Provides New Insights Into Expression Bias, Mitochondrial Doubly Uniparental Inheritance and Sex Determination. Mol. Biol. Evol. 29 (2), 771–786. doi: 10.1093/molbev/msr248
Gonzalez-Castellano I., Manfrin C., Pallavicini A., Martinez-Lage A. (2019). De Novo Gonad Transcriptome Analysis of the Common Littoral Shrimp Palaemon Serratus: Novel Insights Into Sex-Related Genes. BMC Genomics 20 (1), 757. doi: 10.1186/s12864-019-6157-4
Gross L. (2006). Male or Female? It Depends on the Dose. PloS Biol. 4 (6), e211–e211. doi: 10.1371/journal.pbio.0040211
Huang S., Ye L., Chen H. (2017). Sex Determination and Maintenance: The Role of DMRT1 and FOXL2. Asian J. Androl. 19 (6), 619–624. doi: 10.4103/1008-682X.194420
Jones W. J., Won Y. J., Maas P. A. Y., Smith P. J., Lutz R. A., Vrijenhoek R. C. (2005). Evolution of Habitat Use by Deep-Sea Mussels. Marine Biol. 148 (4), 841–851. doi: 10.1007/s00227-005-0115-1
Kádár E., Lobo-da-Cunha A., Santos R. S., Dando P. (2006). Spermatogenesis of Bathymodiolus Azoricus in Captivity Matching Reproductive Behaviour at Deep-Sea Hydrothermal Vents. J. Exp. Marine Biol. Ecol. 335 (1), 19–26. doi: 10.1016/j.jembe.2006.02.016
Kashimada K., Koopman P. (2010). Sry: The Master Switch in Mammalian Sex Determination. Development 137 (23), 3921–3930. doi: 10.1242/dev.048983
Kocer A., Pinheiro I., Pannetier M., Renault L., Parma P., Radi O., et al. (2008). R-Spondin1 and FOXL2act Into Two Distinct Cellular Types During Goat Ovarian Differentiation. BMC Dev. Biol. 8 (1), 36. doi: 10.1186/1471-213X-8-36
Laming S. R., Duperron S., Cunha M. R., Gaudron S. M. (2014). Settled, Symbiotic, Then Sexually Mature: Adaptive Developmental Anatomy in the Deep-Sea, Chemosymbiotic Mussel Idas Modiolaeformis. Mar. Biol. 161 (6), 1319–1333. doi: 10.1007/s00227-014-2421-y
Laming S. R., Gaudron S. M., Duperron S. (2018). Lifecycle Ecology of Deep-Sea Chemosymbiotic Mussels: A Review. Front. Mar. Sci. 5 (282). doi: 10.3389/fmars.2018.00282
Li S. F., Lv C. C., Lan L. N., Jiang K. L., Zhang Y. L., Li N., et al. (2021). DNA Methylation Is Involved in Sexual Differentiation and Sex Chromosome Evolution in the Dioecious Plant Garden Asparagus. Hortic. Res. 8 (1), 198. doi: 10.1038/s41438-021-00633-9
Lin J. Q., Yu J., Sun L., Fang S. G. (2021). Genome-Wide DNA Methylation and Transcriptome Analyses Reveal Epigenetic and Genetic Mechanisms Underlying Sex Maintenance of Adult Chinese Alligator. Front. Genet. 12. doi: 10.3389/fgene.2021.655900
Liu C. F., Bingham N., Parker K., Yao H. H. (2009). Sex-Specific Roles of Beta-Catenin in Mouse Gonadal Development. Hum. Mol. Genet. 18 (3), 405–417. doi: 10.1093/hmg/ddn362
Liu X.-L., Zhang Z.-F., Shao M.-Y., Liu J.-G., Muhammad F. (2012). Sexually Dimorphic Expression of Foxl2 During Gametogenesis in Scallop Chlamys Farreri, Conserved With Vertebrates. Dev. Genes Evol. 222 (5), 279–286. doi: 10.1007/s00427-012-0410-z
Li Y., Zhang L., Li Y., Li W., Guo Z., Li R., et al. (2019). Dynamics of DNA Methylation and DNMT Expression During Gametogenesis and Early Development of Scallop Patinopecten Yessoensis. Mar. Biotechnol. (NY) 21 (2), 196–205. doi: 10.1007/s10126-018-09871-w
Li R., Zhang L., Li W., Zhang Y., Li Y., Zhang M., et al. (2018). FOXL2 and DMRT1L Are Yin and Yang Genes for Determining Timing of Sex Differentiation in the Bivalve Mollusk Patinopecten Yessoensis. Front. Physiol. 9, 1166. doi: 10.3389/fphys.2018.01166
Li Y., Zhang L., Sun Y., Ma X., Wang J., Li R., et al. (2016). Transcriptome Sequencing and Comparative Analysis of Ovary and Testis Identifies Potential Key Sex-Related Genes and Pathways in Scallop Patinopecten Yessoensis. Marine Biotechnol. 18 (4), 453–465. doi: 10.1007/s10126-016-9706-8
Lorion J., Kiel S., Faure B., Kawato M., Ho S. Y., Marshall B., et al. (2013). Adaptive Radiation of Chemosymbiotic Deep-Sea Mussels. Proc. Biol. Sci. 280 (1770), 20131243. doi: 10.1098/rspb.2013.1243
Matson C. K., Murphy M. W., Sarver A. L., Griswold M. D., Bardwell V. J., Zarkower D. (2011). DMRT1 Prevents Female Reprogramming in the Postnatal Mammalian Testis. Nature 476 (7358), 101–104. doi: 10.1038/nature10239
Matson C. K., Zarkower D. (2012). Sex and the Singular DM Domain: Insights Into Sexual Regulation, Evolution and Plasticity. Nat. Rev. Genet. 13 (3), 163–174. doi: 10.1038/nrg3161
Nagahama Y., Chakraborty T., Paul-Prasanth B., Ohta K., Nakamura M. (2021). Sex Determination, Gonadal Sex Differentiation, and Plasticity in Vertebrate Species. Physiol. Rev. 101 (3), 1237–1308. doi: 10.1152/physrev.00044.2019
Naimi A., Martinez A.-S., Specq M.-L., Diss B., Mathieu M., Sourdaine P. (2009). Molecular Cloning and Gene Expression of Cg-Foxl2 During the Development and the Adult Gametogenetic Cycle in the Oyster Crassostrea Gigas. Comp. Biochem. Physiol. Part B.: Biochem. Mol. Biol. 154 (1), 134–142. doi: 10.1016/j.cbpb.2009.05.011
Navarro-Martin L., Vinas J., Ribas L., Diaz N., Gutierrez A., Di Croce L., et al. (2011). DNA Methylation of the Gonadal Aromatase (cyp19a) Promoter Is Involved in Temperature-Dependent Sex Ratio Shifts in the European Sea Bass. PloS Genet. 7 (12), e1002447. doi: 10.1371/journal.pgen.1002447
Olson C. E., Roberts S. B. (2014). Genome-Wide Profiling of DNA Methylation and Gene Expression in Crassostrea Gigas Male Gametes. Front. Physiol. 5. doi: 10.3389/fphys.2014.00224
Pennec M. L., Beninger P. G. (1997). Ultrastructural Characteristics of Spermatogenesis in Three Species of Deep-Sea Hydrothermal Vent Mytilids. Can. J. Zool. 75 (75), 308–316. doi: 10.1139/z97-039
Quinn A. E., Sarre S. D., Ezaz T., Marshall Graves J. A., Georges A. (2011). Evolutionary Transitions Between Mechanisms of Sex Determination in Vertebrates. Biol. Lett. 7 (3), 443–448. doi: 10.1098/rsbl.2010.1126
Radhakrishnan S., Literman R., Neuwald J. L., Valenzuela N. (2018). Thermal Response of Epigenetic Genes Informs Turtle Sex Determination With and Without Sex Chromosomes. Sex Dev. 12 (6), 308–319. doi: 10.1159/000492188
Riviere G., Wu G.-C., Fellous A., Goux D., Sourdaine P., Favrel P. J. M. B. (2013). DNA Methylation is Crucial for the Early Development in the Oyster C. Gigas 15 (6), 739–753. doi: 10.1007/s10126-013-9523-2
Rossi G. S., Tunnicliffe V. (2017). Trade-Offs in a High CO2 Habitat on a Subsea Volcano: Condition and Reproductive Features of a Bathymodioline Mussel. Marine Ecol. Prog. Ser. 574, 49–64. doi: 10.3354/meps12196
Sarre S. D., Georges A., Quinn A. (2004). The Ends of a Continuum: Genetic and Temperature-Dependent Sex Determination in Reptiles. Bioessays 26 (6), 639–645. doi: 10.1002/bies.20050
Shao C., Li Q., Chen S., Zhang P., Lian J., Hu Q., et al. (2014). Epigenetic Modification and Inheritance in Sexual Reversal of Fish. Genome Res. 24 (4), 604–615. doi: 10.1101/gr.162172.113
Sinclair A. H., Berta P., Palmer M. S., Hawkins J. R., Griffiths B. L., Smith M. J., et al. (1990). A Gene From the Human Sex-Determining Region Encodes a Protein With Homology to a Conserved DNA-binding Motif. Nature 346 (6281), 240–244. doi: 10.1038/346240a0
Smith C. A., Roeszler K. N., Ohnesorg T., Cummins D. M., Farlie P. G., Doran T. J., et al. (2009). The Avian Z-linked Gene DMRT1 Is Required for Male Sex Determination in the Chicken. Nature 461 (7261), 267–271. doi: 10.1038/nature08298
Stenyakina A., Walters L., Hoffman E., Calestani C. (2009). Food Availability and Sex Reversal in Mytella Charruana, an Introduced Bivalve in the Southeastern United States. Mol. Reprod. Dev. 77, 222–230. doi: 10.1002/mrd.21132
Sun Y., Wang M., Li L., Zhou L., Wang X., Zheng P., et al. (2017b). Molecular Identification of Methane Monooxygenase and Quantitative Analysis of Methanotrophic Endosymbionts Under Laboratory Maintenance in Bathymodiolus Platifrons From the South China Sea. PeerJ 5, e3565. doi: 10.7717/peerj.3565
Sun J., Zhang Y., Xu T., Zhang Y., Mu H., Zhang Y., et al. (2017a). Adaptation to Deep-Sea Chemosynthetic Environments as Revealed by Mussel Genomes. Nat. Ecol. Evol. 1 (5). doi: 10.1038/s41559-017-0121
Tachibana M. (2015). Epigenetic Regulation of Mammalian Sex Determination. J. Med. Invest. 62 (1-2), 19–23. doi: 10.2152/jmi.62.19
Teaniniuraitemoana V., Huvet A., Levy P., Klopp C., Lhuillier E., Gaertner-Mazouni N., et al. (2014). Gonad Transcriptome Analysis of Pearl Oyster Pinctada Margaritifera: Identification of Potential Sex Differentiation and Sex Determining Genes. BMC Genomics 15 (1), 491. doi: 10.1186/1471-2164-15-491
Tyler P. A., Marsh L., Baco-Taylor A., Smith C. R. (2009). Protandric Hermaphroditism in the Whale-Fall Bivalve Mollusc Idas Washingtonia. Deep Sea Res. Part II: Topical Stud. Oceanography 56 (19-20), 1689–1699. doi: 10.1016/j.dsr2.2009.05.014
Tyler P., Young C. M., Dolan E., Arellano S. M., Brooke S. D., Baker M. (2007). Gametogenic Periodicity in the Chemosynthetic Cold-Seep Mussel “Bathymodiolus” Childressi. Marine Biol. 150 (5), 829–840. doi: 10.1007/s00227-006-0362-9
Uhlenhaut N. H., Jakob S., Anlag K., Eisenberger T., Sekido R., Kress J., et al. (2009). Somatic Sex Reprogramming of Adult Ovaries to Testes by FOXL2 Ablation. Cell 139 (6), 1130–1142. doi: 10.1016/j.cell.2009.11.021
Villamor S., Yamamoto T. (2015). Reproductive Seasonality of Monetaria Annulus (Linnaeus 1758) (Mollusca: Gastropoda: Cypraeidae) in a Temperate Area. Molluscan Res. 35 (2), 95–101. doi: 10.1080/13235818.2014.954660
Wang X., Li Q., Lian J., Li L., Jin L., Cai H., et al. (2014). Genome-Wide and Single-Base Resolution DNA Methylomes of the Pacific Oyster Crassostrea Gigas Provide Insight Into the Evolution of Invertebrate CpG Methylation. BMC Genomics 15 (1), 1–12. doi: 10.1186/1471-2164-15-1119
Wang L., Yue F., Song X., Song L. J. D., Immunology C. (2015). Maternal Immune Transfer in Mollusc 48, 2, 354–359. doi: 10.1016/j.dci.2014.05.010
Wan Z. Y., Xia J. H., Lin G., Wang L., Lin V. C., Yue G. H. (2016). Genome-Wide Methylation Analysis Identified Sexually Dimorphic Methylated Regions in Hybrid Tilapia. Sci. Rep. Developmental & Comparative Immunology 6, 35903. doi: 10.1038/srep35903
Wen A. Y., You F., Sun P., Li J., Xu D. D., Wu Z. H., et al. (2014). Cpg Methylation of Dmrt1 and cyp19a Promoters in Relation to Their Sexual Dimorphic Expression in the Japanese Flounder Paralichthys Olivaceus. J. Fish Biol. 84 (1), 193–205. doi: 10.1111/jfb.12277
Wittkopp P. J., Haerum B. K., Clark A. G. (2004). Evolutionary Changes in Cis and Trans Gene Regulation. Nature 430 (6995), 85–88. doi: 10.1038/nature02698
Xu M., Fang M., Yang Y., Dick J. T. A., Song H., Luo D., et al. (2016). Spatial Variation in Adult Sex Ratio Across Multiple Scales in the Invasive Golden Apple Snail, Pomacea Canaliculata. Ecol. Evol. 6 (8), 2308–2317. doi: 10.1002/ece3.2043
Yang L., Zhang X., Liu S., Zhao C., Miao Y., Jin L., et al. (2021). Cyp17a1 is Required for Female Sex Determination and Male Fertility by Regulating Sex Steroid Biosynthesis in Fish. Endocrinology 162 (12). doi: 10.1210/endocr/bqab205
Yao H., Lin Z., Dong Y., Kong X., He L., Xue L. (2021). ). Gonad Transcriptome Analysis of the Razor Clam (Sinonovacula Constricta) Revealed Potential Sex-Related Genes. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.725430
Zarkower D. (2013). “"Chapter Twelve - DMRT Genes in Vertebrate Gametogenesis,",” in Current Topics in Developmental Biology. Ed. Wassarman P. M. (San Diego, CA, USA: Academic Press), 327–356.
Zhang N., Xu F., Guo X. (2014). Genomic Analysis of the Pacific Oyster (Crassostrea Gigas) Reveals Possible Conservation of Vertebrate Sex Determination in a Mollusc. G3 (Bethesda Md) 4 (11), 2207. doi: 10.1534/g3.114.013904
Keywords: deep-sea mussel, Gigantidas platifrons, gonad transcriptome, methylation, sex determination and differentiation
Citation: Zhong Z, Wang M, Chen H, Wang H, Zhang H, Zhou L, Sun Y, Cao L, Lian C, Li M and Li C (2022) Gonad Transcriptome and Whole-Genome DNA Methylation Analyses Reveal Potential Sex Determination/Differentiation Mechanisms of the Deep-Sea Mussel Gigantidas platifrons. Front. Mar. Sci. 9:856291. doi: 10.3389/fmars.2022.856291
Received: 17 January 2022; Accepted: 30 March 2022;
Published: 28 April 2022.
Edited by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Dafni Anastasiadi, The New Zealand Institute for Plant and Food Research Ltd, New ZealandKazue Nagasawa, Tohoku University, Japan
Copyright © 2022 Zhong, Wang, Chen, Wang, Zhang, Zhou, Sun, Cao, Lian, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaolun Li, bGNsQHFkaW8uYWMuY24=