 Tianhang Zhang1,2,3,4
Tianhang Zhang1,2,3,4 Rui Liu
Rui Liu Chaomin Sun
Chaomin Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 14 April 2022
Sec. Deep-Sea Environments and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.848136
This article is part of the Research TopicDeep-sea Chemosynthetic Ecosystems: Living in Extreme EnvironmentsView all 13 articles
As a member of Planctomycetes–Verrucomicrobia–Chlamydia (PVC) superphylum, the phylum Lentisphaerae is broadly distributed in various environments. Lentisphaerae members have been recognized as being typically difficult to isolate in pure culture—only five strains belonging to this phylum were cultured up to now—and therefore their major physiology and ecology are largely unknown. Here, using a polysaccharide degradation-driven strategy, we successfully cultured a representative of Lentisphaerae bacteria (Candidatus Coldseepensis marina zth2) from the deep-sea cold seep. Based on physiological and phylogenetic analyses, strain zth2 is identified as a type strain of a novel family. Notably, strain zth2 divides by a budding manner and a unique feature seen in ultrathin-section electron microscopic observation is the occurrence of narrow protrusions, namely, both cell wall and cytoplasm on the surface of strain zth2. Through growth assays and transcriptomic analyses, strain zth2 was verified to efficiently utilize polysaccharides, phosphate, and iron for growth. Strikingly, the utilization of polysaccharide, phosphate, and iron is correlative and jointly contributes to carbon, phosphorus, and iron metabolisms and thereby facilitating the production of energy. By mimicking the ocean environment and using operational taxonomic units (OTUs) analysis, we demonstrated that strain zth2 significantly enrich the microbial diversity by metabolizing polysaccharides or other substances. Overall, we propose that Lentisphaerae members are potential contributors to the cycling of nutrient, carbon, phosphorus, and other elements in the deep biosphere.
The PVC superphylum gathers bacteria from seven phyla: namely, the three founder phyla (Planctomycetes, Verrucomicrobia, Chlamydiae) that it is named after, Lentisphaerae and Kirimatiellaeota and also some uncultured candidate phyla, such as the Candidatus Omnitrophica (previously known as OP3 and WWE2) (Pinos et al., 2016; Rivas-Marin and Devos, 2018). Members within the PVC superphylum present different lifestyles, cell plans, and environments (Pinos et al., 2016), and show diverse interesting characteristics (Pilhofer et al., 2008), such as, some Planctomycetes are implicated in carbon and nitrogen cycles (Strous et al., 2006), or synthesize special sterols (Pearson et al., 2003), numerous Chlamydiae are pathogens of mammals (Corsarol and Greub, 2006), some members of Chlamydiae and Verrucomicrobiae are symbionts (Horn, 2008). With the superphylum status now broadly accepted, it is clear that PVC bacteria are fascinating new model organisms for bacterial and evolutionary cell biology (Rivas-Marin et al., 2016), biogeochemical cycles (Strous et al., 2006), biotechnology (Devos and Ward, 2014; Wiegand et al., 2020), and human health (Devos and Ward, 2014).
Among the PVC superphylum, the Lentisphaerae phylum was proposed in 2004 after the isolation of two marine strains (Cho et al., 2004). Members belonging to the phylum Lentisphaerae have been recognized as being typically difficult to isolate in pure culture, therefore only five pure cultures are available at the time of writing (Choi et al., 2015) and few progresses except taxonomic characteristics associated with the pure culture of this phylum have been disclosed. Nevertheless, the phylum Lentisphaerae bacteria are ubiquitously distributed, such as seawater (Cho et al., 2004; Choi et al., 2013) and sediments (Polymenakou et al., 2009), anaerobic sludge (Qiu et al., 2013), landfill leachate (Limam et al., 2010), and animal and human gut (Peng et al., 2015; Liu et al., 2018). In addition, Lentisphaerae bacteria are predicted to be closely associated with biogeochemical cycles (Jiang et al., 2015), bioremediation (Das and Kazy, 2014), polysaccharide degradation (Yan et al., 2012), and human health issues, namely, type 2 diabetes (T2D) (Maskarinec et al., 2021), inflammation in the terminal ileum (Fan et al., 2020), and autoimmune hepatitis (Lou et al., 2020). However, the majority of above achievements associated with Lentisphaerae bacteria were predicted based on the results of metagenomic. Despite the surge of microbial metagenome data, experimental testing is necessary to confirm inferences about the cell biology, ecological roles, and evolution of Lentisphaerae bacteria. Therefore, cultivation of more Lentisphaerae members is a priority to understand the unique biology of this kind of difficult-to-culture bacteria.
To date, the phylum Lentisphaerae is classified into two classes, namely, Lentisphaeria (Choi et al., 2015) and Oligosphaeria (Qiu et al., 2013). There are two orders (Lentisphaerales and Victivallales) and two families (Lentisphaeraceae and Victivallaceae) (Zoetendal et al., 2003; Choi et al., 2013) within the Lentisphaeria class, and there are only one order (Oligosphaerales) and one family (Oligosphaeraceae) within the Oligosphaeria class (Qiu et al., 2013). Correspondingly, three genera (Victivallis, Lentisphaera, and Oligosphaera) have been proposed. The five cultured species include Victivallis vadensis ATCC BAA-548T (Zoetendal et al., 2003), Lentisphaera araneosa HTCC2155T (Cho et al., 2004), Lentisphaera marina IMCC11369T (Choi et al., 2013), Oligosphaera ethanolica 8KG-4T (Qiu et al., 2013), and Lentisphaera profundi SAORIC-696T (Choi et al., 2015). Notably, among these five pure cultures, V. vadensis ATCC BAA-548T was a strictly anaerobic cellobiose-degrading strain (Zoetendal et al., 2003); L. marina IMCC11369T was found to have a capability of degrading starch (Choi et al., 2013); and O. ethanolica 8KG-4T could grow fermentatively on glucose, ribose, xylose, galactose, and sucrose (Qiu et al., 2013). Therefore, Lentisphaerae bacteria possess potentials of degrading polysaccharides as those reported in other PVC superphylum members (van Vliet et al., 2019).
As the primary biomass production, polysaccharides are a ubiquitous energy source for microbes in both terrestrial and marine ecosystems including the deep ocean (Zheng et al., 2021a). Many members of the Bacteroidetes phylum are specialized in polysaccharide degradation and contribute to the carbon and nutrient cycling of the deep biosphere (Zheng et al., 2021a). Notably, many PVC superphylum bacteria also evolved prominent polysaccharides degradation capabilities: two novel Kiritimatiellales strains isolated from Black Sea sediment could anaerobically degrade sulfated polysaccharides; and Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan (Sichert et al., 2020). Up to now, it is obscure whether Lentisphaerae members possess the polysaccharide degradation capability. When we enriched microorganisms with deep-sea cold seep samples using a basal medium supplemented with various polysaccharides (Zheng et al., 2021a), we found a small amount of Lentisphaerae bacterial 16S rRNA gene sequences in addition to Bacteroidetes bacteria based on the operational taxonomic units (OTUs) analysis of the bacterial community within the enrichment. However, the potentials of these deep-sea Lentisphaerae members toward polysaccharides degradation were unknown due to the lack of pure cultures.
Here, we successfully cultured a novel Lentisphaerae strain zth2 from deep-sea cold seep samples by using a polysaccharide-degradation approach. Using growth assay and transcriptomic methods, we further investigated the degradative details of two polysaccharides (starch and laminarin) by strain zth2. We also found the metabolism of polysaccharide and the utilization of phosphorus and iron were closely correlative. Lastly, its contribution to the environmental micro-biodiversity and oceanic carbon/phosphorus cycling were also investigated and discussed.
The deep-sea samples were collected by the RV KEXUE from a typical cold seep at the Formosa Ridge (Site F) in the South China Sea (E119°17’07.322”, N22°06’58.598”) as described by Cao et al. (2021). The relevant chemical parameters of the cold seep are shown in Supplementary Table 1. The temperature, salinity, and underwater depth of sampling sites were recorded in real time by an SBE 25plus Sealogger CTD (SBE, USA). Concentrations of CO2 and CH4 in surface sediments were measured in situ with CONTROSHydroCO2 (CONTROS, Norway) and HydroCH4 (CONTROS, Norway) sensors, respectively. All sensors were mounted on the Discovery ROV (Cao et al., 2021). Dissolved oxygen was measured above the vent site and the measurement method of dissolved oxygen can refer to the article of Cao et al. (2021) and more details can be found in Materials and Methods. Specific site information is shown in Supplementary Table 2. To enrich the polysaccharide degrading bacteria, the sediment needs to be suspended with seawater sterilized three times to ensure that the sediment is not contaminated. A temperature gradient is then set before the culture is carried out, with the aim of slowly reviving the microbes in the sediment, which is very important for the whole isolation and culture process. Later, the sediment samples were cultured at 28°C for 6 months in an anaerobic inorganic medium supplemented with 1.0 g/L of mixed seaweed polysaccharides, namely, porphyra polysaccharide (PP), agar, carrageenan, laminarin, fucoidan, starch, alginate, ulvan, and enteromorpha polysaccharide (EP). The compositions of inorganic medium are 1.0 g NH4Cl, 1.0 g NaHCO3, 1.0 g CH3COONa, 0.5 g KH2PO4, 0.2 g MgSO4, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin in 1 L filtered seawater, pH 7.0. This medium was prepared anaerobically as previously described (Fardeau et al., 1997). After the 6-month enrichment, 50 µl dilution portions were spread on Hungate tubes covered by a modified organic medium (1.0 g NH4Cl, 1.0 g NaHCO3, 1.0 g CH3COONa, 0.5 g KH2PO4, 0.2 g MgSO4, 1.0 g yeast extract, 1.0 g peptone, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin in 1 L filtered seawater, pH 7.0) supplemented with 15.0 g/L agar. The Hungate tubes were anaerobically incubated at 28°C for 7 days. Individual colonies with distinct morphology were selected using sterilized bamboo sticks and then cultured in the anaerobic liquid 2216E medium (5.0 g/L yeast extract, 1.0 g/L peptone, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin, 1 L filtered seawater, pH 7.0). Strain zth2 was isolated and purified with 2216E medium by repeated use of the Hungate roll-tube methods for several rounds until it was considered to be pure. The purity of strain zth2 was conventionally confirmed repeated partial sequencing of the 16S rRNA gene. Strain zth2 is preserved at −80°C in 2216E medium supplemented with 20% (v/v) glycerol.
To observe the morphological characteristics of strain zth2, its cell pellets were washed with 10 mM phosphate buffer solution (PBS, pH 7.4) and then centrifuged at 5,000 g for 5 min, and taken by immersing copper grids coated with a carbon film for 20 min, washed for 10 min in PBS and dried for 20 min at room temperature (Buchan et al., 2014). Ultrathin-section electron microscopic observation was performed as described previously (Sekiguchi et al., 2003; Graham and Orenstein, 2007). Briefly, the samples were firstly preserved in 2.5% (v/v) glutaraldehyde overnight at 4°C and then washed three times using PBS in the next day. Later, samples were dehydrated in ethanol solutions of 30, 50, 70, 90, and 100% for 10 min each time, and then the dehydrated samples were embedded in a plastic resin. Ultrathin sections (50–70 nm) of cells were prepared with an ultramicrotome (Leica EM UC7, Germany), stained with uranyl acetate and lead citrate. All samples were examined using TEM (HT7700, Hitachi, Japan) with a JEOL JEM 12000 EX (equipped with a field emission gun) at 100 kV.
The temperature, pH, and NaCl concentration ranges for the growth of strain zth2 were determined in the anaerobic liquid 2216E medium incubated for 5 days. Growth assays were performed at different temperatures (4, 28, 30, 37, and 60°C). The pH range for growth was tested from pH 2.0 to 10.0 with increments of 1 pH unit. Salt tolerance was tested with 0–10% (w/v) NaCl (1.0% intervals) in 2216E medium prepared with sterile water instead of sea water. Substrates utilization of strain zth2 was tested in the medium consisting of (L−1): 1.0 g NH4Cl, 1.0 g NaHCO3, 1.0 g CH3COONa, 0.5 g KH2PO4, 0.2 g MgSO4, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin in 1 L filtered seawater, pH 7.0. Single substrate (namely, glucose, mannitol, laminarin, fructose, dextrin, fucoidan, maltose, mannose, glycine, pullulan, lactose, polyethylene glycol, butyrate, sucrose, acetate, formate, starch, trehalose, galactose, cellulose, xylose, lactate, ethanol, D-mannose, glycerin, phosphate, and sorbitol) was added from sterile filtered stock solutions to the final concentration at 1.0 g/L, respectively. Cell culture containing basic medium without adding any other substrates was used as a control. For each substrate, three biological replicates were performed.
According to the instructions of the manufacturer, genomic DNA of strain zth2 was extracted using a QIAGEN Genomic-tip (QIAGEN Biotech Co., Ltd., Germany). For genome sequencing, 15 ml strain zth2 culture was inoculated in a 2 L anaerobic bottle containing 1.5 L anaerobic 2216E medium. After cell growth reached the logarithmic phase, the culture solution was centrifuged at 8,000 g for 10 min to obtain cell pellets.
Its cell pellets were washed with 10 mM phosphate buffer solution (PBS, pH 7.4) and then centrifuged at 8,000 g for 5 min. Then the supernatant was poured out and the cell pellets were preserved at −80°C for sequencing. Quality inspection of DNA purity, concentration, and integrity was performed by NanoDrop (Thermo Scientific, USA), and 0.35% agarose gel electrophoresis, respectively. Then, a genomic DNA library for Nanopore sequencing was constructed by using a ligation sequencing kit (Oxford Nanopore Technologies Ltd., UK). The library was sequenced using a Promethion sequencer 48. Canu v1.5 (Koren et al., 2017) was used to assemble the filtered subreads and Pilon v1.22 (Walker et al., 2014) was used to correct the assembly sequence with higher accuracy. Sequencing work was conducted by Biomarker Technologies Co., Ltd. (Beijing, China).
Genome related values are calculated by several methods: Average Nucleotide Identity (ANI) based on the MUMMER ultra-rapid aligning tool (ANIm), ANI based on the BLASTN algorithm (ANIb), the tetra-nucleotide signatures (Tetra), and in silico DNA–DNA similarity. ANIm, ANIb, and Tetra frequencies were calculated using JSpecies WS (http://jspecies.ribohost.com/jspeciesws/). The recommended species criterion cut-offs for the ANIb and ANIm were respectively used as 95% and 0.99 for the Tetra signature. The calculation of in silico DNA–DNA similarity values was performed by the Genome-to-Genome Distance Calculator (GGDC) (http://ggdc.dsmz.de/) (Meier-Kolthoff et al., 2013). The isDDH results were calculated based on the recommended formula 2.
The 16S rRNA gene based tree was constructed with the full-length 16S rRNA sequences by the maximum likelihood method. In short, the full-length 16S rRNA (1,361 bp) gene sequence of strain zth2 was obtained from its whole genome (accession number MW729759), and other 16S rRNA sequences of related taxa used for phylogenetic analysis were obtained from NCBI (www.ncbi.nlm.nih.gov/). The multiple sequence alignment of sequences was performed by MAFFT ver.7 (https://mafft.cbrc.jp/alignment/server/). The method of Gappyout was used to trim sequences. Phylogenetic trees were constructed using W-IQ-TREE web server (http://iqtree.cibiv.univie.ac.at/) (Trifinopoulos et al., 2016) with GTR + F + I + G4 model. The edition of the phylogenetic tree was performed by the online tool Interactive Tree of Life (iTOL v4) (Letunic and Bork, 2019).
The genome tree was constructed from a concatenated alignment of 37 protein-coding genes (Wu et al., 2013) that extracted from each genome by Phylosift (v1.0.1) (Darling et al., 2014) with automated setting. The genomes used to construct the genome tree included both draft and finished genomes from NCBI. Aligned sequences were trimmed using TrimAl (version 1.2) with gappyout function. The phylogenetic trees were constructed using W-IQ-TREE web server (http://iqtree.cibiv.univie.ac.at) (Trifinopoulos et al., 2016) with GTR + F + I + G4 model, and the online tool Interactive Tree of Life (iTOL v6) was used for the tree edition (Letunic and Bork, 2019).
Growth assays of strain zth2 were performed at atmospheric pressure. To detect the effect of polysaccharides on the growth of strain zth2, the modified 1/5 2216E medium (containing 1 g/L yeast extract, 0.2 g/L peptone, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin, 1 L seawater, pH 7.0) supplemented either with or without 1 g/L of different polysaccharides, namely, PP (porphyra polysaccharide), carrageenan, laminarin, starch or ulvan. For all the growth assays, 100 µl strain zth2 culture was inoculated in 15 ml Hungate tubes containing 10 ml medium. All of the Hungate tubes were anaerobically incubated at 28°C. Bacterial growth status was monitored by measuring the OD600 value via a microplate reader (Infinite M1000 Pro; Tecan, Mannedorf, Switzerland) every day until cell growth reached the stationary phase.
To explore the effects of phosphate and iron on bacterial growth, strain zth2 was grown in basic medium (containing 10 g/L glucose, 0.3 g/L NaCl, 0.5 g/L (NH4)2SO4, 0.3 g/L MgSO4, 0.2 g/L KCl, 0.03 g/L MnSO4, 0.2 g/L peptone, 0.7 g cysteine hydrochloride, 500 µl 0.1% (w/v) resazurin, 1 L seawater, pH 6.0–6.5) supplemented with Na3PO4 (2.6 mM), or FeSO4 (0.03 g/L), or both FeSO4 (0.03 g/L) and Ca3(PO4)2(1.0 g/L), or both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM). Similarly, the growth assays were performed as described above.
Transcriptomic analysis was performed by Novogene (Tianjin, China). For the transcriptomic analysis of the effects of polysaccharides on the growth of strain zth2, strain zth2 was cultured in the above modified 1/5 2216E medium supplemented either with or without 1 g/L of polysaccharides (laminarin or starch) in 2 L anaerobic bottles for 4 days. For the transcriptomic analysis of the effects of phosphate and iron on the growth of strain zth2, cells suspension of strain zth2 was cultured in the above basic medium supplemented with Na3PO4 (2.6 mM), or FeSO4 (0.03 g/L), or both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM) in 2 L anaerobic bottles for 5 days. After incubation, the cells were collected by centrifugation at 8,000 g, 4°C, 10 min. For transcriptomic analyses, total RNAs of strain zth2 were extracted using TRIzol reagent (Invitrogen, USA) and DNA contamination was removed using the MEGA clear™Kit (Life technologies, USA). Detailed protocols of the following procedures, namely, library preparation, clustering and sequencing and data analyses were described in the Supplementary Information.
To better understand the ecological role of strain zth2, we mimicked the ocean environment in the anaerobic bottles. Briefly, dialysis bags (containing 50 ml sterilized seawater, named SW; 50 ml sterilized seawater with strain zth2 cells, named SWL; 50 ml sterilized seawater with 1 g/L of laminarin, named SWP; 50 ml sterilized seawater with 1 g/L of laminarin and strain zth2 cells, named SWLP) were respectively put into four anaerobic bottles filled with filtered sea water using medium speed double circle qualitative filter paper from the First Sea Bathing Beach, Qingdao, China. After a month of cultivation, bacterial cells grown outside of the dialysis bags were respectively collected and used for OTUs sequencing that was performed by Novogene (Tianjin, China). Total DNAs from the above four samples were extracted by the CTAB/SDS method (Murray and Thompson, 1980) and were diluted to 1 ng/µl with sterile water and used for PCR template. 16S rRNA genes of distinct regions (16S V3/V4) were amplified using specific primers (341F: 5′-CCTAYGGGRBGCASCAG and 806R: 5′-GGACTACNNGGGTATCTAAT) with the barcode. The PCR products were purified with a Gel Extraction Kit (Qiagen, Germany) and prepared to construct libraries. Sequencing libraries were generated using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the instructions of the manufacturer. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific, USA) and Agilent Bioanalyzer 2100 system (Agilent, USA). The library was sequenced on an Illumina NovaSeq platform. Paired-end reads (250 bp) were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/) (Magoc and Salzberg, 2011). Quality filtering on the raw tags was performed according to the QIIME (V1.9.1, http://qiime.org/scripts/split_libraries_fastq.html) quality controlled process (Bokulich et al., 2013).
The tags were compared with the reference database (Silva database, release 111-2012_07, https://www.arbsilva.de/) using UCHIME algorithm (UCHIME Algorithm, http://www.drive5.com/usearch/manual/uchime_algo.html) (Edgar et al., 2011) to detect chimera sequences, and then the chimera sequences were removed (Haas et al., 2011). Sequence analyses were performed by Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/) (Edgar, 2013). Sequences with ≥97% similarity were assigned to the same OTUs. The representative sequence for each OTU was screened for further annotation. For each representative sequence, the Silva Database (release 111-2012_07, http://www.arb-silva.de/) (Quast et al., 2013) was used based on Mothur algorithm to annotate taxonomic information
In our previous work, we successfully isolated a novel Bacteroidetes species through a polysaccharide degradation-driven strategy from the deep-sea cold seep (Zheng et al., 2021a). Beyond our expectation, we found there was a small amount of Lentisphaerae bacteria within the enrichment based on the OTUs analysis. Given that members of Lentisphaerae are typical difficult-to-culture microbes (Choi et al., 2015), we spent much effort for a couple of rounds of purification and 16S rRNA sequencing confirmation and eventually obtained the pure culture of a Lentisphaerae bacterium (strain zth2) (Figure 1A). Under the TEM observation, strain zth2 possessed an irregular coccoid cell (~400–500 nm in size, no flagellum) with the extracellular slime layer (Figure 1B), which was very similar to the morphologies of other Lentisphaerae members reported previously (Zoetendal et al., 2003; Cho et al., 2004).
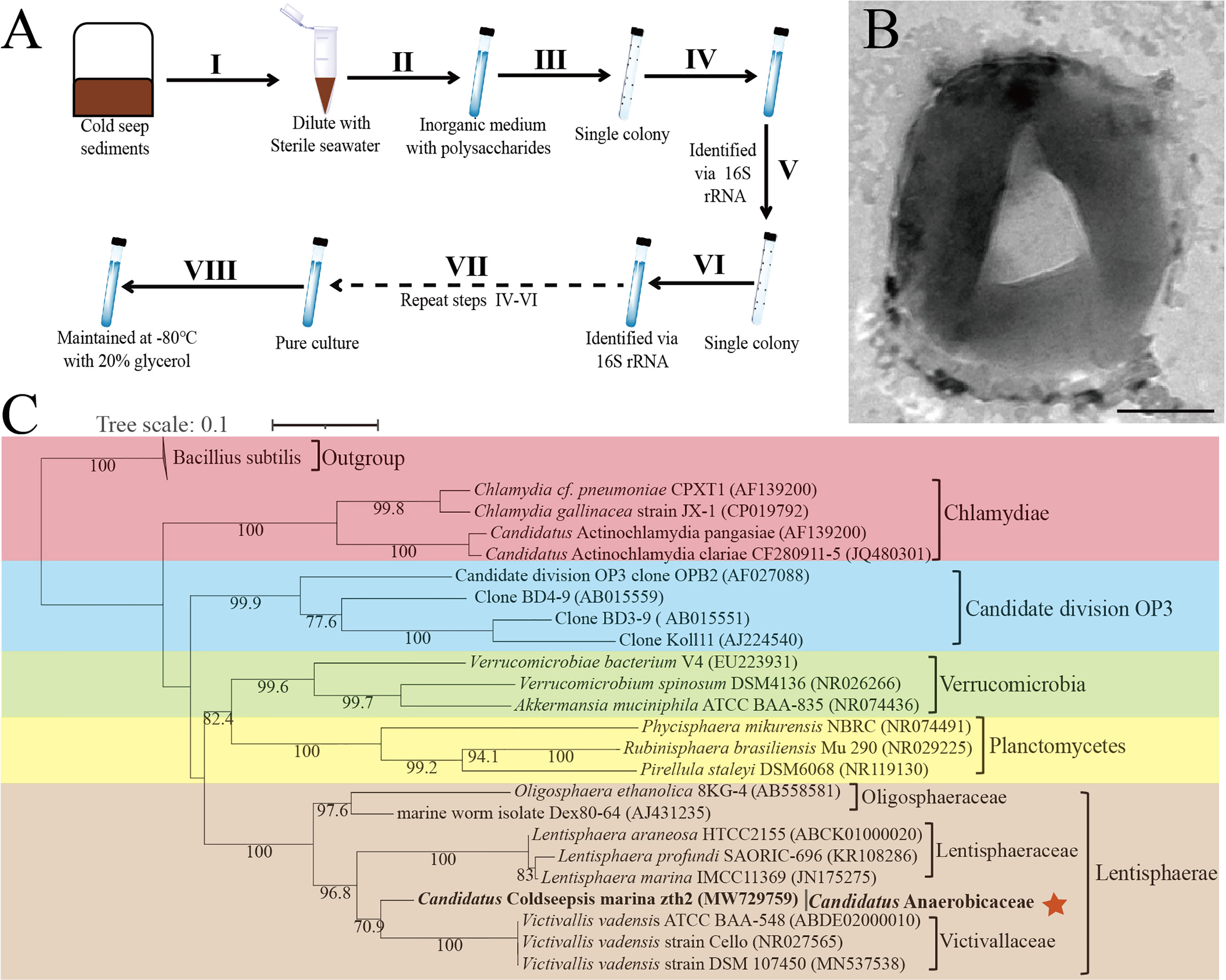
Figure 1 Isolation, morphology and phylogeny of Candidatus Coldseepensis marina zth2. (A) Diagram of enrichment and isolation of strain zth2 through a polysaccharide degradation-driven strategy. (B) Observation of the cell morphology of strain zth2 through the transmission electron microscopy. Bar is 250 nm. (C) Maximum likelihood 16S rRNA phylogenetic tree showed the positions of the phylum Lentisphaerae (including strain zth2 and other cultured type strains) together with other members within the PVC superphylum. The numbers on the node indicate statistical support for bootstrap values, respectively. The position of Candidatus Coldseepensis marina zth2 and its attributive family Candidatus Anaerobicaceae is indicated with red star. Some Bacillus members were used as outgroup. The accession number of each 16S rRNA is shown in the parentheses after corresponding strain name. Scale bar, 0.1 substitutions per nucleotide position.
To gain more insights into strain zth2, the whole genome was sequenced and analyzed. The genome size of strain zth2 is 3,198,720 bp with a DNA G+C content of 43.77% and it is complete (Supplementary Figure S1A and Supplementary Table 3). Annotation of the genome of strain zth2 revealed 2,683 predicted genes that included 66 RNA genes (6 rRNA genes, 52 tRNA genes and 8 ncRNAs). The genome contains 146 CAZymes that may be related to its capability of polysaccharides degradation (Supplementary Figure S1B). The genome relatedness values were calculated by the average nucleotide identity (ANI), in silico DNA–DNA similarity (isDDH) and the tetranucleotide signatures (Tetra), against two available genomes of type strains of Lentisphaerae bacteria: V. vadensis ATCC BAA-548T (Zoetendal et al., 2003) and L. araneosa HTCC2155T (Cho et al., 2004) (Supplementary Table 3). The average nucleotide identities (ANIb) of strain zth2 with BAA-548T and HTCC2155T were 65.32 and 65.45%, respectively. The average nucleotide identities (ANIm) of strain zth2 with BAA-548T and HTCC2155T were 89.31 and 83.99%, respectively. The tetra values of strain zth2 with BAA-548T and HTCC2155T were 0.69176 and 0.48857, respectively. Based on digital DNA–DNA hybridization employing the Genome-to-Genome Distance Calculator GGDC, the in silico DDH estimates for strain zth2 with BAA-548T and HTCC2155T were 26.1 and 25.5%, respectively. These results together demonstrated the genome of strain zth2 to be clearly below established ‘cut-off’ values (ANIb: 95%, ANIm: 95%, AAI: 95%, isDDH: 70%, Tetra: 0.99) (Goris et al., 2007; Meier-Kolthoff et al., 2013) for defining bacterial species (Supplementary Table 3), suggesting it represents a novel taxon of Lentisphaerae.
To clarify the taxonomic status of strain zth2, we further performed the phylogenetic analysis with 16S rRNA genes of some cultured and uncultured representatives of PVC superphylum. The maximum likelihood tree of 16S rRNA placed strain zth2 as a distinct cluster separating from other three identified families, namely, Victivallaceae (from order Victivallales), Lentisphaeraceae (from order Lentisphaerales) and Oligosphaeraceae (from order Oligosphaerales) (Figure 1C). Consistently, a genome-based tree (Supplementary Figure S2) further confirmed the position of strain zth2, located between the branches of families of Victivallaceae and Lentisphaeraceae. Moreover, based on the 16S rRNA gene sequence of strain zth2, a sequence similarity calculation using the NCBI server indicated that the closest relatives of strain zth2 were V. vadensis ATCC BAA-548T (88.77%, family Victivallaceae), L. araneosa HTCC2155T (86.09%, Lentisphaeraceae), L. marina IMCC11369T (85.73%, Lentisphaeraceae), L. profundi SAORIC-696T (85.72%, Lentisphaeraceae), and O. ethanolica 8KG-4T (83.87%, Oligosphaeraceae). Recently, the proposed median and minimum sequence identity values of 16S rRNA sequence for a new family have been revealed as 92.25 and 87.65% (Yarza et al., 2014), respectively. Therefore, combining the results of 16s rRNA phylogenetic tree, a genome-based tree and ANI analysis, we propose that strain zth2 is classified as the type strain of a novel family. Given its isolated site, we proposed the name Candidatus Coldseepensis marina gen. nov., sp. nov. for strain zth2. In addition, we also proposed the associated genus and family as Candidatus Coldseepensis gen. nov. and Candidatus Anaerobicaceae fam. nov., respectively.
As previously reported, members of the PVC superphylum have very complex and specific cytoarchitectural features (Rivas-Marin and Devos, 2018). Ultrathin TEM sections showed that strain zth2 possessed distinctive membrane and compartmentalization systems (Figures 2A, B). In particular, its outer membrane possessed a lot of twists and turns (Figure 2A), which effectively increased the surface area of the cell membrane. Notably, strain zth2 divided by budding (Figure 2A) but not the usual binary fission manner that adopted by most bacteria. In the division process, daughter cells always originated from one site of the mother cell (Figure 2A, panel I), and then the bud became bigger and bigger along with the cellular content distributing into the daughter cell (Figure 2A, panels II to VI). Once the content and size of daughter cell were equal to those of mother cell, the daughter cell would separate from the mother cell (Figure 2A, panels VII to VIII), and a cell division cycle was finished.
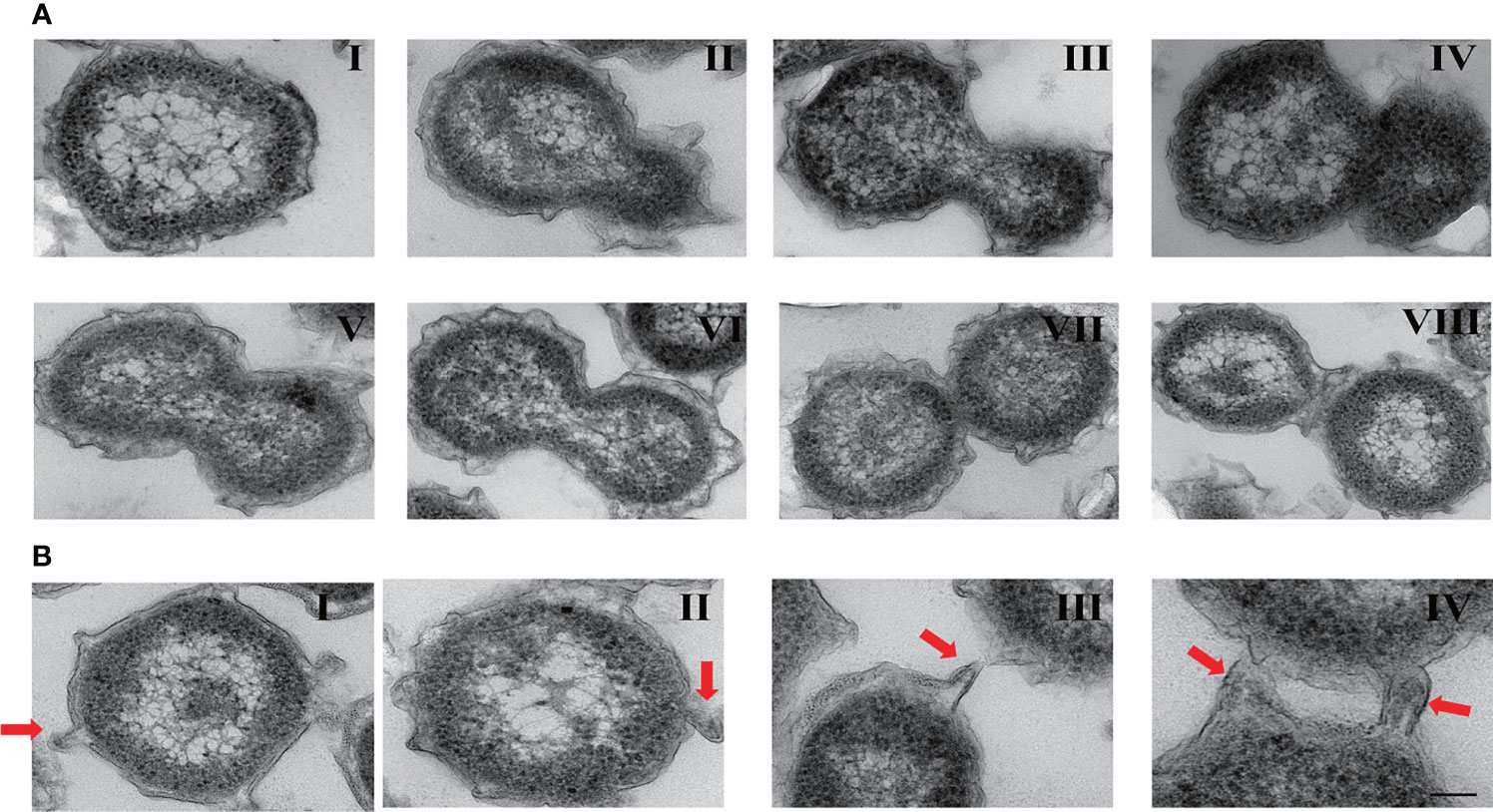
Figure 2 Ultrathin-section electron microscopic observation of cell biological characteristics of strain zth2. (A) Ultrathin TEM sections showing the process of polar budding division (panels I–VIII) conducted by strain zth2. (B) Ultrathin TEM sections showing the narrow protrusions around the cell (panels I–IV, the red arrow indicates).
Another dramatic feature of strain zth2 was that there were several narrow protrusions around the cells (Figure 2B, panels I–IV, the red arrow points). These protruded 80–114 nm from the cell and were 40–80 nm in diameter measured at the base of the structure preserved slender shape. They seemed to be coincided with the definition of prosthecae as cellular appendages or extensions of the cell containing cytoplasm (Staley, 1968).
Based on our enrichment and isolation strategy of strain zth2 (Figure 1A), this strain was proposed to possess a prominent capability of degrading polysaccharides. To test the potential polysaccharide substrate spectra of strain zth2, we checked the growth status of strain zth2 in the medium supplemented with different kinds of polysaccharides at a final concentration of 1 g/L. The results showed the supplementation of porphyra polysaccharide (PP), carrageenan and ulvan could only modestly promote the growth of strain zth2 (Figure 3A). In contrast, the supplement of starch or laminarin could significantly facilitate the growth of strain zth2, resulting in an almost 4-fold increased growth compared to the control group (Figure 3A).
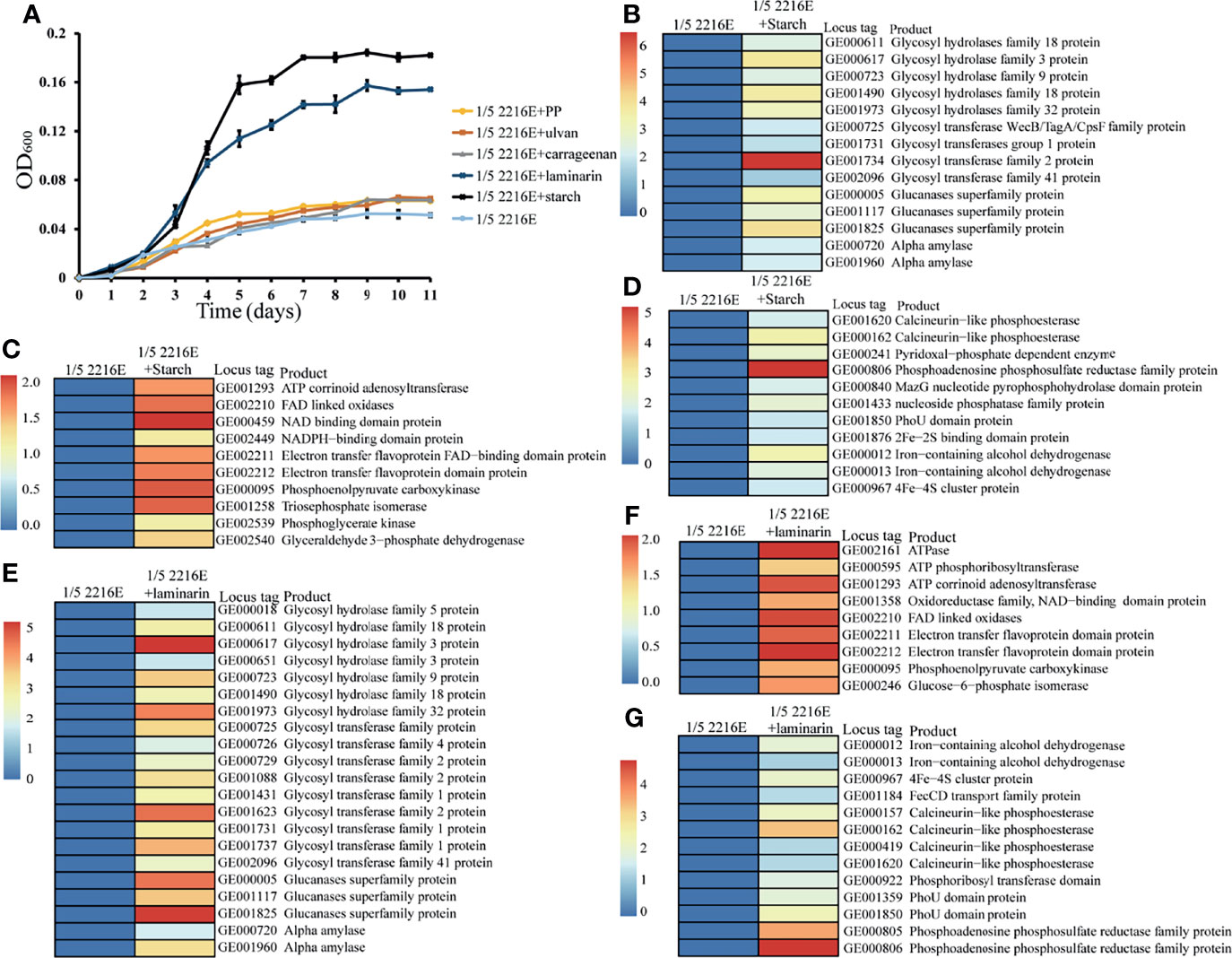
Figure 3 Strain zth2 could effectively degrade and utilize starch and laminarin for growth. (A) Growth assays of strain zth2 in the 1/5 2216E medium supplemented either without or with 1 g/L of laminarin, starch, PP (porphyra polysaccharide), carrageenan, and ulvan, respectively. (B–D) Transcriptomics based heat map showing all upregulated genes encoding proteins associated with polysaccharide degradation (B), energy generation (C), phosphorus and iron metabolisms (D) supplemented with 1 g/L of starch. (E–G) Transcriptomics based heat map showing all upregulated genes encoding proteins associated with polysaccharide degradation (E), energy generation (F), phosphorus and iron metabolisms (G) supplemented with of 1 g/L of laminarin.
To further validate the mechanism of polysaccharides degradation mediated by strain zth2, we performed transcriptomic analysis of strain zth2 grown in the medium supplemented with starch or laminarin. Indeed, the expression of many genes encoding glycan degradation was evidently upregulated when 1 g/L of starch was added in the medium. These included genes encoding amylase, glycosyl hydrolases (GH families 3, 9, 18, and 32), glycosyl transferases (GT1, GT2, and GT41), and glucanases (Figure 3B). Meanwhile, many proteins associated with ATP production (namely, ATP corrinoid adenosyltransferase, NAD/NADPH binding domain proteins, electron transfer flavoprotein, etc.) were also significantly upregulated in the presence of starch (Figure 3C), which might contribute to the growth promotion of strain zth2. Out of our expectation, the supplementation of starch could also dramatically upregulate the expression of many proteins responsible for phosphorus and iron metabolisms (Figure 3D). These included phosphoadenosine phosphosulfate reductase, PhoU, Fe–S binding protein and so on. Similarly, the supplement of 1 g/L of laminarin in the medium also markedly upregulated the expression of proteins responsible for glycan degradation (Figure 3E), ATP production (Figure 3F) and phosphorus and iron metabolisms (Figure 3G). Significantly, GH9 family proteins were annotated as laminarinase in CAZy database, which may play the key role in the degradation of laminaria.
Based on the above transcriptomic results of polysaccharide utilization, we speculate that the utilization of polysaccharide could promote phosphorus and iron metabolisms (Figure 3D). Therefore, we next sought to explore the effects of the supplementation of phosphorus and iron on the growth of strain zth2. First, we analyzed the growth status of strain zth2 in a basic medium supplemented with Na3PO4 (2.6 mM), or FeSO4 (0.03 g/L), or both FeSO4 (0.03 g/L) and Ca3(PO4)2 (1 g/L), or both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM). The growth curves showed that strain zth2 could only grow to an OD600 value about 0.06 in the basic medium amended with FeSO4 after two weeks incubation (Figure 4A). In contrast, in the presence of FeSO4, the growth of strain zth2 could be significantly promoted to OD600 values about 0.20 and 0.68 (about 11-fold increase) when insoluble Ca3(PO4)2 or soluble Na3PO4 was added into the basic medium (Figure 4A). These results indicated the soluble phosphate was necessary for the growth of strain zth2 and this bacterium could transform insoluble Ca3(PO4)2 to soluble phosphate for utilization to some extent. On the other hand, in the presence of Na3PO4, the absence of FeSO4 in the medium could decrease about a quarter of the growth rate (from OD600 value 0.68 to 0.53) (Figure 4A), suggesting that iron is also a key factor for the growth of strain zth2.
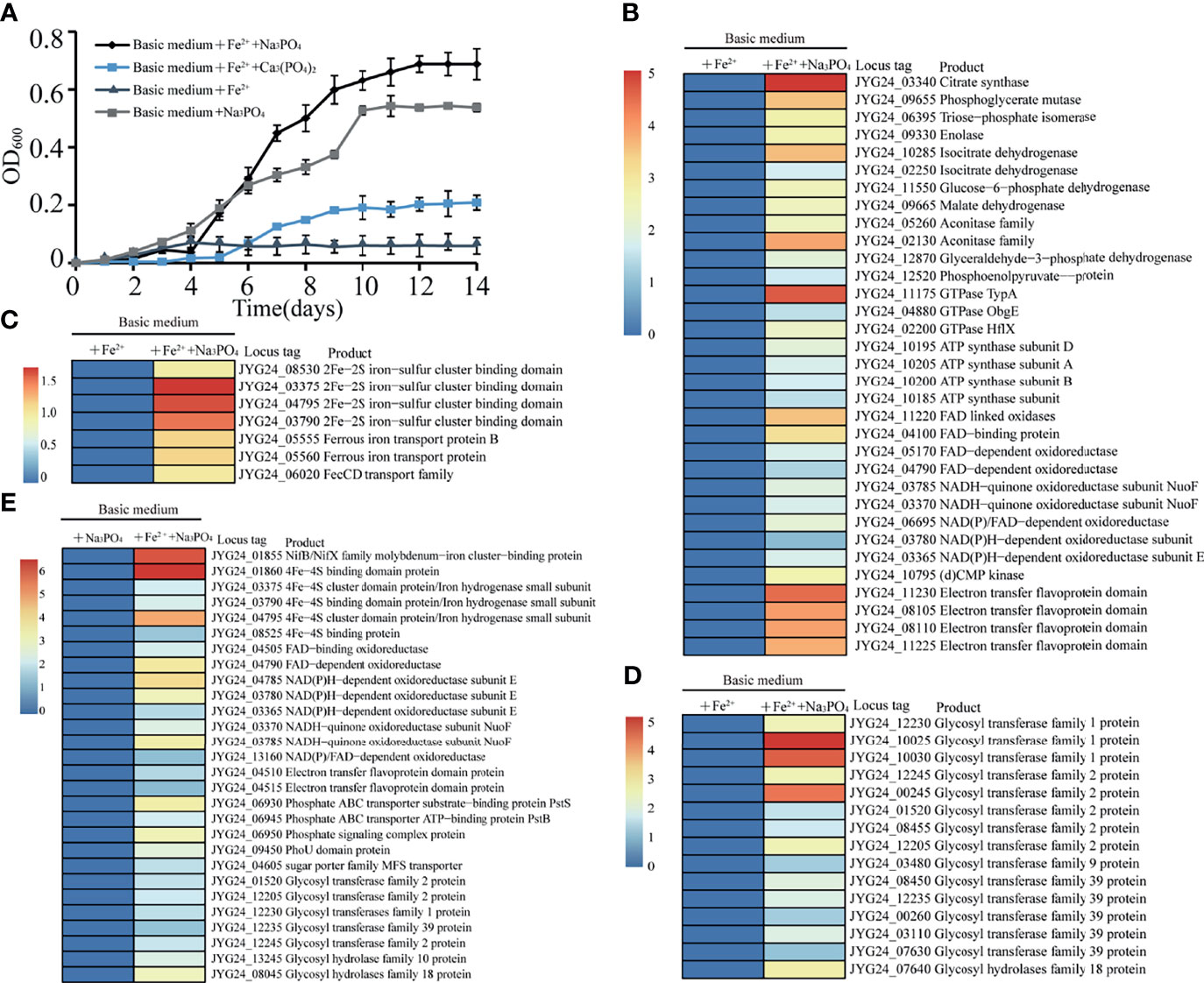
Figure 4 Strain zth2 could effectively utilize phosphate and iron for growth. (A) Growth assays of strain zth2 in basic medium supplemented with FeSO4 (0.03 g/L), or Na3PO4 (2.6 mM), or both FeSO4 (0.03 g/L) and Ca3(PO4)2 (1 g/L), or both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM). (B, C, D) Transcriptomics based heat map showing all upregulated genes encoding proteins associated with energy generation (B), iron metabolism (C), polysaccharide degradation (D) when compared the growth of strain zth2 in basic medium added both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM) with that in basic medium added only FeSO4 (0.03 g/L). (E) Transcriptomics based heat map showing all upregulated genes encoding proteins associated with iron metabolism, energy generation, phosphorus metabolism and polysaccharide degradation when compared the growth of strain zth2 in basic medium added both FeSO4 (0.03 g/L) and Na3PO4 (2.6 mM) with that in basic medium added only Na3PO4 (2.6 mM).
We next analyzed the effects of Na3PO4 or FeSO4 on the growth of strain zth2 through a transcriptomic approach. The results showed that, compared to the medium only amended with FeSO4, the supplement of both Na3PO4 and FeSO4 could upregulate the expression of proteins responsible for energy production, such as citrate synthase, malate dehydrogenase, GTPase, ATP synthase, FAD/NADH dependent enzymes and electron transfer flavoprotein (Figure 4B). This result was well consistent with the growth cure shown in Figure 4A. Strikingly, many proteins associated with iron metabolisms (Figure 4C) and polysaccharide degradation (Figure 4D) were also evidently upregulated when Na3PO4 was supplemented in the medium beside FeSO4, suggesting the presence of soluble phosphate could facilitate iron metabolisms and polysaccharide degradation. On the other hand, the absence of FeSO4 in the medium indeed weakened the metabolisms of phosphorus, iron, and polysaccharide and the eventual energy generation even though Na3PO4 was present in the medium (Figure 4E). This result is a good explanation that the absence of FeSO4 in the medium could decrease a half of growth rate as shown in Figure 4A. In the combined growth assay and transcriptomics results, we conclude that the utilization of polysaccharide, phosphate, and iron by strain zth2 is correlative and mutually promoted, and these three nutrient sources are essential factors supporting the best growth of strain zth2.
Given its prominent capability of polysaccharide degradation and acquiring phosphorus and iron, we next sought to ask whether strain zth2 could provide extra nutrient to other bacteria and thereby promote microbial diversity as shown in the previous report of the ecological function of Bacteroides (Zheng et al., 2021a). We therefore mimicked the natural ecological environment for bacterial growth and survival in the laboratory (Figure 5A). Correspondingly, four anaerobic dialysis bags (namely, SW, SWL, SWP, and SWLP) were respectively incubated in different anaerobic bottles filled with filtered but non-sterilized sea water using medium speed double circle qualitative filter paper for one month (Figure 5A). After 30-day incubation, the cells growing in the non-sterilized sea water were collected and used for OTUs analysis to determine the variety and abundance of bacteria in each sample. The OTUs analysis results revealed that the bacterial diversity of samples SW and SWP was very low: at the phylum level, only 3–4 phyla including Proteobacteria, Bacteroides and Firmicutes (or Gracilibacteria) were identified in the sea water (Figure 5B). In contrast, the bacterial diversity of samples SWL and SWPL was significantly increased (Figure 5B). Especially, the bacterial variety was increased to 8–9 phyla in sample SWL, and many bacteria belonging to unidentified bacteria and different known phyla (namely, Tenericutes, Fusobacteria, Actinobacteria, and Verrucomicrobia) were enriched, indicating that strain zth2 is indeed able to increase the environmental bacteria diversity. Similarly, at the family level, the abundance and variety of novel members belonging to other phyla rather than Proteobacteria were obviously enhanced in both SWL and SWLP samples, further confirming the proposal that strain zth2 contributes to the increase and maintainance of microbial diversity regardless of the presence of polysaccharide.
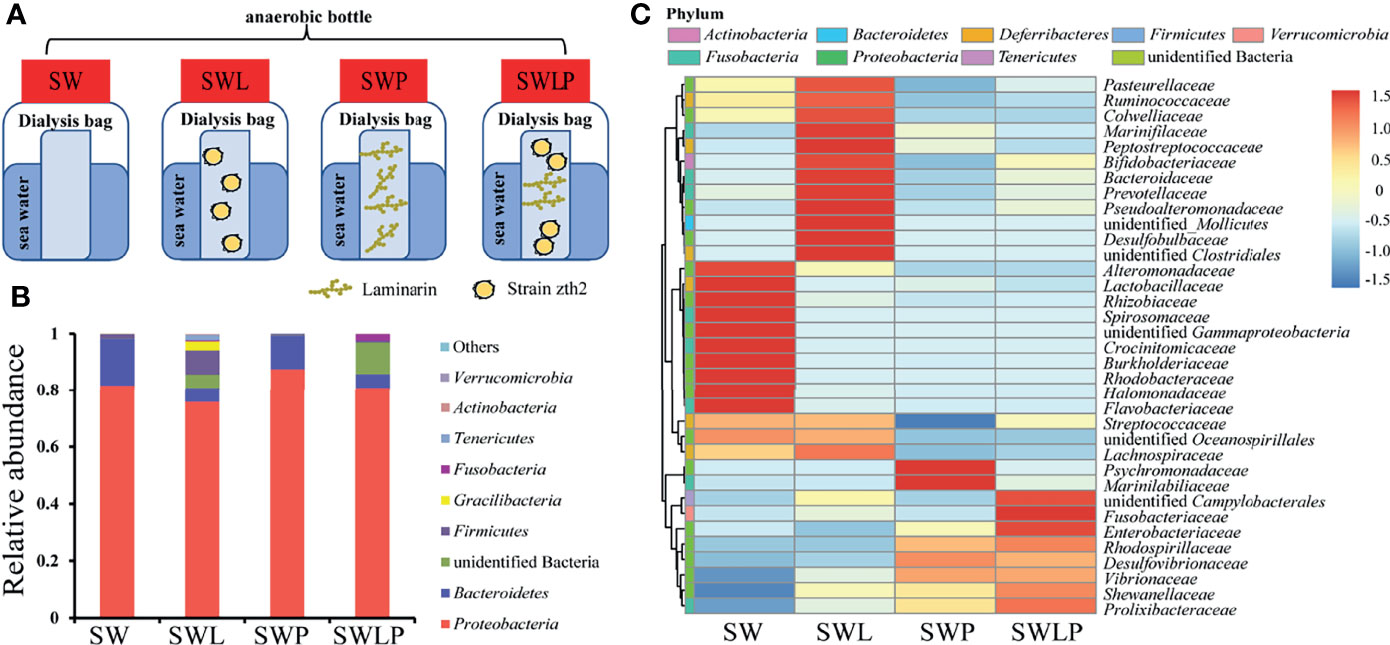
Figure 5 Strain zth2 evidently increases marine microbial diversity in a system that mimics natural environment. (A) Diagrammatic scheme of the experimental design for analyzing the ecological role of strain zth2 in a system that mimics natural environment. Briefly, four anaerobic dialysis bags (containing sterile sea water, named SW; sterile sea water supplemented with strain zth2 cells, named SWL; sterile sea water supplemented with 1 g/L of laminarin, named SWP; sterile sea water supplemented with both strain zth2 cells and 1 g/L of laminarin, named SWLP) were respectively incubated in different anaerobic bottles filled with filtered but non-sterilized sea water. (B, C) The relative abundances of operational taxonomic units (OTUs) representing different bacteria in the sea water are shown at the phylum (B) and family (C) levels.
In the combined data on the utilization of polysaccharide, phosphate and iron, and also the contribution to the microbial diversity conducted by strain zth2, we proposed a model representing the central metabolism and ecological function of strain zth2 (Figure 6). In this model, different polysaccharides were degraded and then transported into the cells by special transporters; then the oligosaccharide could be further transformed into glucose (or other simple sugar) and enters into the Embden–Meyerhof–Parnas (EMP) glycolysis pathway, which thereby joining to the tricarboxylic acid cycle (TCA pathway) and producing energy; the degradation products can also promote the growth of other microorganisms; on the other hand, different kinds of phosphate could be effectively obtained and utilized as a nutrient source for phosphorus cycling and ATP production; meanwhile, iron could be acquired and transported into the cells to activate essential iron-associated enzymes (e.g., Fe–Fe hydrogenase and Fe–S protein) for various metabolic pathways and thereby generating energy. As mentioned above, the utilization of polysaccharide, phosphate, and iron by strain zth2 is correlative and jointly contributes to carbon, phosphorus, and iron metabolisms and cycling, which thereby supports bacterial growth and promotes the environmental microbial diversity.
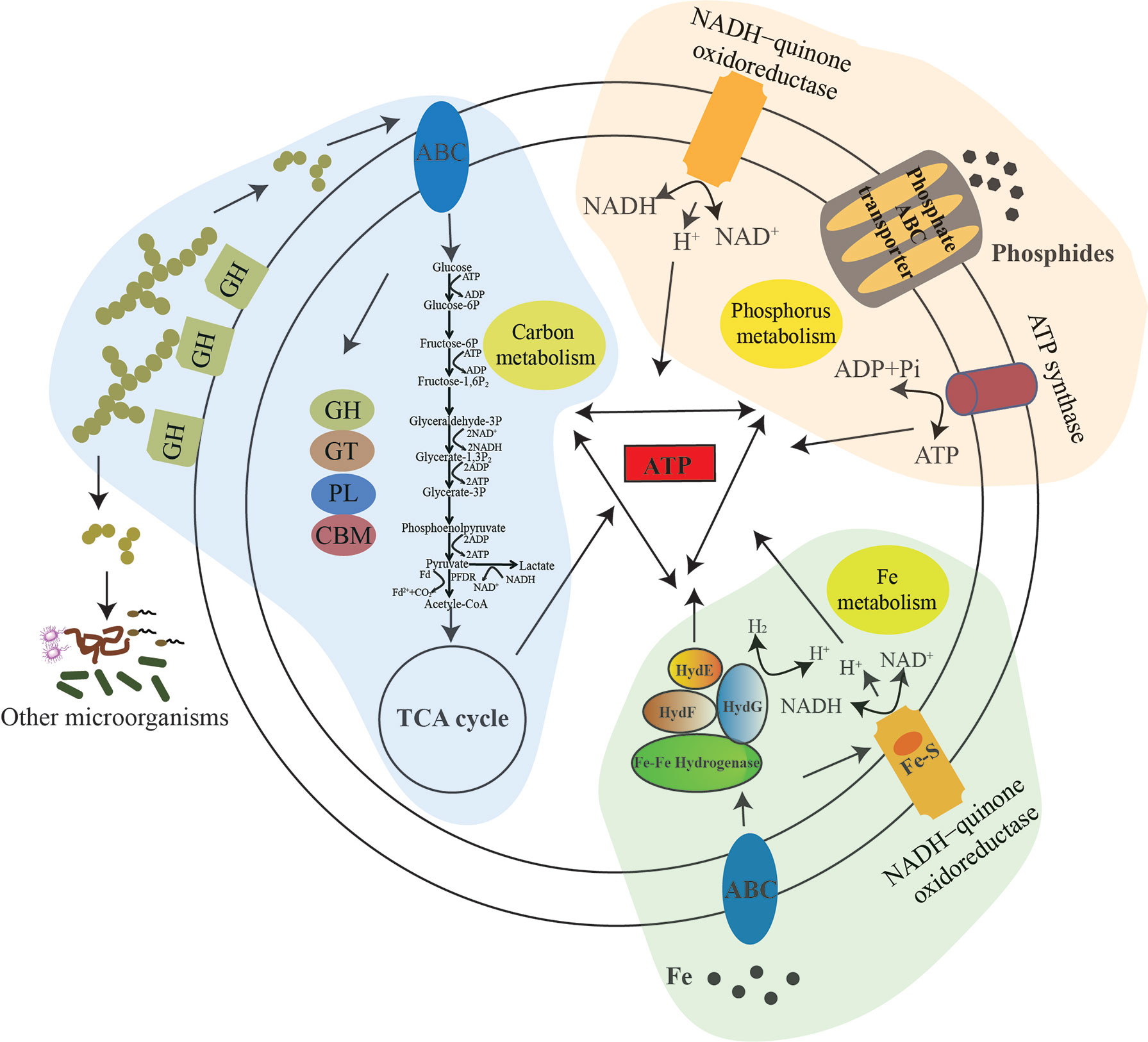
Figure 6 A proposed model showing the utilization of polysaccharide, phosphate and iron for energy production and growth of strain zth2, and also its contribution to the promotion of microbial diversity. In this model, three different pathways for ATP generation are shown: tricarboxylic acid cycle (TCA pathway) driven by the polysaccharides degradation and the Embden–Meyerhof–Parnas (EMP) glycolysis pathway; utilization of phosphate as a nutrient source for phosphorus cycling and ATP production; acquisition of iron to activate essential iron-associated enzymes (e.g., Fe–Fe hydrogenase and Fe–S protein) for various metabolic pathways and thereby generating energy. Meanwhile, the cycling of carbon, phosphorus and iron is activated to facilitate the central metabolism of strain zth2. Obviously, the utilization of polysaccharide, phosphate and iron by strain zth2 is correlative and jointly contributes to promote bacterial growth and environmental microbial diversity.
Candidatus Anaerobicaceae (An.ae’ro.bica.ce.ae. N.L. fem. adj. Candidatus Coldseepensis type genus of the family; suff. -aceae, ending to denote a family; N.L. fem. pl. adj. Candidatus Anaerobicaceae the family of the genus Candidatus Coldseepensis.) The type genus is Candidatus Coldseepensis.
For the genus name Candidatus Coldseepensis, cold.seep’ensis. N.L. fem. n. Given its isolated site, we proposed this name. The type and only species of the genus is Candidatus Coldseepensis marina.
Candidatus Coldseepensis marina (ma.ri’na. L. fem. adj. marina of the sea, marine). Cells are coccoid, ~400–700 nm in size, and have no flagellum; strictly anaerobic; the temperature range for growth is 28–37°C with an optimum at 28°C (Supplementary Figure S3A); growing at pH values of 6.0–8.0 (optimum, pH 7.0) (Supplementary Figure S3B); growth occurs at NaCl concentrations between 2.0 and 4.0% with an optimum growth at 3% NaCl (Supplementary Figure S3C); growth is stimulated by using glucose, maltose, fructose, sucrose, mannose, starch, cellulose, laminarin, dextrin, lactose, galactose, xylose, mannitol or sorbitol as a sole carbon source (Supplementary Table 4). The type strain, zth2T, was isolated from the sediment of deep-sea cold seep, P.R. China.
Microbial communities in marine sediments are known to play a key role in global elemental and nutrient cycling (Zhang et al., 2020; Baker et al., 2021). However, the vast majority of microbes present in sediments have not been successfully cultured in a laboratory setting (Baker et al., 2021). In fact, the large proportion of uncultured strains is estimated to account for more than 75% of sediment genera (Lloyd et al., 2018), impacting that our understanding of marine sediment microbiology is limited. However, a detailed comparison of the metabolic potential of uncultured strains suggests that these new microbes are capable of directing previously undescribed fundamental pathways in elemental and nutrient cycling (Baker et al., 2021; Zheng et al., 2021a; Zheng et al., 2021b). Given the important role that uncultured communities play in the oceans, there is an urgent need to better resolve the diversity and ecological roles of these uncultured taxa (Baker et al., 2021). It is accepted that members belonging to the phylum Lentisphaerae are typically difficult to obtain in pure culture in the laboratory condition (Choi et al., 2015). In the present study, we developed an innovative approach by using a polysaccharide degradation-directed approach to enrich and purify Lentisphaerae bacteria (Figure 1A). With this, the first representative (Candidatus Coldseepensis marina zth2) belonging to a novel family (Candidatus Anaerobicaceae) was cultured in laboratory from a deep-sea cold seep sedimentary sample (Figure 1C). Actually, we also obtained a novel species belonging to Bacteroidetes phylum through the same enrichment method from deep-sea cold seep sediments, and the prominent polysaccharide degradation capability of this Bacteroidetes strain was verified (Zheng et al., 2021a). Therefore, it will be a promising approach to isolate more uncultured microbes from deep-sea samples that enriched in an inorganic medium supplemented with different polysaccharides given that polysaccharides are deemed to be ubiquitously distributed in deep-sea sediments (Gao et al., 2017).
The phylum Lentisphaerae is one of the members of PVC superphylum. With the superphylum status now amply accepted, it is clear that PVC bacteria are fascinating new model organisms for bacterial and evolutionary cell biology (Rivas-Marin and Devos, 2018). For example, they have features that separate them from other bacteria, such as extensive bacterial endomembrane systems and atypical modes of cell division (Rivas-Marin et al., 2016). Consistently, strain zth2 divided by budding (Figure 2A) but not the usual binary fission manner that is adopted by most bacteria. Actually, budding is also employed by some species of Planctomycetes phylum (another important PVC member) (Wiegand et al., 2018; Wiegand et al., 2020). To our knowledge, this is the first report that Lentisphaerae bacterium performs budding fission, suggesting the budding division might be an alternative manner adopted by many PVC bacteria besides binary fission. However, the universal bacterial cell division protein FtsZ was found to exist in strain zth2, indicating that strain zth2 might conduct a different budding division pathway compared to Planctomycetes bacteria which lack FtsZ-like proteins (Wiegand et al., 2020). Another distinct cell biological trait of strain zth2 is that there were several narrow protrusions around the cells like appendages or prosthecae as previously reported (Butler et al., 2002). This similar structure has been found in some members of Planctomycetes and Verrucomicrobia. Butler et al. found strain ATCC 35122 belonging to membership of the Planctomycetales possessed a unique feature seen in both negatively stained cells and in thin-sectioned cells that was described as the occurrence of ‘hump’ protrusions (Butler et al., 2002). Kulichevskaya et al. reported the presence of crateriform pits and numerous fibrillar appendages on the cell surface and an unusual spur-like projection on one pole of the cell by phase-contrast micrographs of strain MPL7 belonging to the Planctomycetales (Kulichevskaya et al., 2007). Also, Dobro et al. observed novel extracellular appendages along the prosthecae on the cell surface of Prosthecobacter debontii belonging to Verrucomicrobia through using electron cryotomography (ECT) (Dobro et al., 2017). However, the specific function of this prosthecae structure is not clear. Notably, Wagner et al. showed that prosthecae, cylindrical extensions of the Caulobacter crescentus cell envelope, could take up and hydrolyze organic phosphate molecules (Wagner et al., 2006), which indicated that strain zth2 may be capable of phosphate uptake through the protrusions of its cells. Taken together, it will be interesting to understand its unusual cell division and disclose the detailed function of this narrow structure on the cell of strain zth2 in the future.
It is well accepted that nutrient and element cycling driven by microbes is essential for the whole deep-sea ecological system (Baker et al., 2021). Various polysaccharides (e.g., glycogen, starch, laminarin) are stored as energy reserve by animals, plants and microorganisms (van Vliet et al., 2019), and the degradation and utilization of algal polysaccharides greatly contribute to the deep-sea element and nutrient cycling (Zheng et al., 2021a). Through growth assays and transcriptomic analysis, we found that strain zth2 was able to efficiently utilize not only polysaccharides (Figure 3) but also phosphate and iron (Figure 4) for growth, indicating its potentials to drive diverse element and nutrient cycles. Phosphorus is an essential element to all life, being a structural and functional component of all organisms inhabiting across the ocean surface to the sediments (Paytan and McLaughlin, 2007). Previous studies indicated a coupling between iron cycling and phosphate cycling in suboxic environments (Paytan and McLaughlin, 2007). Indeed, in strain zth2, the utilization and metabolisms of phosphate and iron are correlative (Figure 4), moreover, this coupling process was also demonstrated to be closely associated with polysaccharides metabolism (Figure 4), highlighting contributions of strain zth2 to the cycling of nutrient, carbon, phosphorus, and iron (Figures 3, 4, and 6). With this, we speculate that the metabolisms of organic (like polysaccharide) and inorganic (e.g., phosphate, iron) substances are correlated and more complicated than our present cognitive.
Given that strain zth2 possesses versatile metabolic pathways and is a potential contributor to the nutrient and element cycling (Figure 6), it is not a surprise to observe that it significantly increased the microbial diversity in a simulative natural ecological system (Figure 5). Therefore, we propose that Lentisphaerae members might play underestimated roles toward the cycling of nutrient, carbon, phosphorus, iron, and other elements in the deep biosphere. In summary, this study extends our understanding of the cultivation and unique physiological characterizations and also ecological roles of Lentisphaerae bacteria.
The whole 16S rRNA sequence and complete genome sequence of strain zth2 have been respectively deposited in the GenBank database under accession numbers MW729759 and CP071032. The raw sequencing reads for transcriptomic analysis and OTUs analysis of microbial diversity have been respectively deposited to NCBI Short Read Archive under accession numbers PRJNA756144, PRJNA754499 and PRJNA756866 Strain zth2 has been preserved in the Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China (accession number: MCCC1K06937).
TZ and CS conceived and designed the study. TZ conducted most of the experiments. RZ helped to isolate and identify the Lentisphaerae strain, RL (Rui Liu) collected the samples from the deep-sea cold seep. RL (Ronggui Li) provided suggestions for the experiments design. TZ and CS lead the writing of the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work is funded by the Key Deployment Projects of Center of Ocean Mega-Science of the Chinese Academy of Sciences (Grant No. COMS2020Q04), China Ocean Mineral Resources R&D Association Grant (Grant No. DY135-B2-14), Shandong Provincial Natural Science Foundation (Grant No.ZR2021ZD28), Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA22050301), Major Research Plan of the National Natural Science Foundation (Grant No. 92051107) and Taishan Young Scholar Program of Shandong Province (Grant No. tsqn20161051) for CS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.848136/full#supplementary-material
Baker B. J., Appler K. E., Gong X. Z. (2021). New Microbial Biodiversity in Marine Sediments. Annu. Rev. Mar Sci. 13, 161–175. doi: 10.1146/annurev-marine-032020-014552
Bokulich N. A., Subramanian S., Faith J. J., Gevers D., Gordon J. I., Knight R., et al. (2013). Quality-Filtering Vastly Improves Diversity Estimates From Illumina Amplicon Sequencing. Nat. Methods 10, 57–59. doi: 10.1038/NMETH.2276
Buchan A., LeCleir G. R., Gulvik C. A., Gonzalez J. M. (2014). Master Recyclers: Features and Functions of Bacteria Associated With Phytoplankton Blooms. Nat. Rev. Microbiol. 12, 686–698. doi: 10.1038/nrmicro3326
Butler M. K., Wang J., Webb R. I., Fuerst J. A. (2002). Molecular and Ultrastructural Confirmation of Classification of ATCC 35122 as a Strain of Pirellula Staleyi. Int. J. Syst. Evol. Microbiol. 52, 1663–1667. doi: 10.1099/ijs.0.02167-0
Cao L., Lian C., Zhang X., Zhang H., Wang H., Zhou L., et al. (2021). In Situ Detection of the Fine Scale Heterogeneity of Active Cold Seep Environment of the Formosa Ridge, the South China Sea. J. Mar. Syst. 218, 611–21. doi: 10.1016/j.jmarsys.2021.103530
Choi A., Song J., Joung Y., Kogure K., Cho J. C. (2015). Lentisphaera Profundi Sp Nov., Isolated From Deep-Sea Water. Int. J. Syst. Evol. Microbiol. 65, 4186–4190. doi: 10.1099/ijsem.0.000556
Choi A., Yang S. J., Rhee K. H., Cho J. C. (2013). Lentisphaera Marina Sp Nov., and Emended Description of the Genus Lentisphaera. Int. J. Syst. Evol. Microbiol. 63, 1540–1544. doi: 10.1099/ijs.0.046433-0
Cho J. C., Vergin K. L., Morris R. M., Giovannoni S. J. (2004). Lentisphaera Araneosa Gen. Nov., Sp Nov, a Transparent Exopolymer Producing Marine Bacterium, and the Description of a Novel Bacterial Phylum, Lentisphaerae. Environ. Microbiol. 6, 611–621. doi: 10.1111/j.1462-2920.2004.00614.x
Corsarol D., Greub G. (2006). Pathogenic Potential of Novel Chlamydiae and Diagnostic Approaches to Infections Due to These Obligate Intracellular Bacteria. Clin. Microbiol. Rev. 19, 283–297. doi: 10.1128/CMR.19.2.283-297.2006
Darling A. E., Jospin G., Lowe E., Matsen F. I. V., Bik H. M., Eisen J. A. (2014). PhyloSift: Phylogenetic Analysis of Genomes and Metagenomes. Peerj 2, e243. doi: 10.7717/peerj.243
Das R., Kazy S. K. (2014). Microbial Diversity, Community Composition and Metabolic Potential in Hydrocarbon Contaminated Oily Sludge: Prospects for in Situ Bioremediation. Environ. Sci. Pollut. R. 21, 7369–7389. doi: 10.1007/s11356-014-2640-2
Devos D. P., Ward N. L. (2014). Mind the PVCs. Environ. Microbiol. 16, 1217–1221. doi: 10.1111/1462-2920.12349
Dobro M. J., Oikonomou C. M., Piper A., Cohen J., Guo K., Jensen T., et al. (2017). Uncharacterized Bacterial Structures Revealed by Electron Cryotomography. J. Bacteriol. 199, e00100–e00117. doi: 10.1128/JB.00100-17
Edgar R. C. (2013). UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads. Nat. Methods 10, 996–998. doi: 10.1038/NMETH.2604
Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fan H. N., Zhu P., Lu Y. M., Guo J. H., Zhang J., Qu G. Q., et al. (2020). Mild Changes in the Mucosal Microbiome During Terminal Ileum Inflammation. Microb. Pathog. 142, 104104. doi: 10.1016/j.micpath.2020.104104
Fardeau M. L., Ollivier B., Patel B. K., Magot M., Thomas P., Rimbault A., et al. (1997). Thermotoga Hypogea Sp. Nov., a Xylanolytic, Thermophilic Bacterium From an Oil-Producing Well. Int. J. Syst. Bacteriol. 47, 1013–1019. doi: 10.1099/00207713-47-4-1013
Gao B. L., Jin M., Li L., Qu W., Zeng R. Y. (2017). Genome Sequencing Reveals the Complex Polysaccharide-Degarding Ability of Novel Deep-Sea Bacterium Flammeovirga Pacifica WPAGA1. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00600
Goris J., Konstantinidis K. T., Klappenbach J. A., Coenye T., Vandamme P., Tiedje J. M. (2007). DNA-DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int. J. Syst Evol Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Graham L., Orenstein J. M. (2007). Processing Tissue and Cells for Transmission Electron Microscopy in Diagnostic Pathology and Research. Nat. Protoc. 2, 2439–2450. doi: 10.1038/nprot.2007.304
Haas B. J., Gevers D., Earl A. M., Feldgarden M., Ward D. V., Giannoukos G., et al. (2011). Chimeric 16s rRNA Sequence Formation and Detection in Sanger and 454-Pyrosequenced PCR Amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Horn M. (2008). Chlamydiae as Symbionts in Eukaryotes. Annu. Rev. Microbiol. 62, 113–131. doi: 10.1146/annurev.micro.62.081307.162818
Jiang X. X., Dang H. Y., Jiao N. Z. (2015). Ubiquity and Diversity of Heterotrophic Bacterial nasA Genes in Diverse Marine Environments. PloS One 10, e0117473. doi: 10.1371/journal.pone.0137817
Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kulichevskaya I. S., Ivanova A. O., Belova S. E., Baulina O. I., Bodelier P. L. E., Rijpstra W. I. C., et al. (2007). Schlesneria Paludicola Gen. Nov., Sp. Nov., the First Acidophilic Member of the Order Planctomycetales, From Sphagnum-Dominated Boreal Wetlands. Int. J. Syst. Evol. Microbiol. 57, 2680–2687. doi: 10.1099/ijs.0.65157-0
Letunic I., Bork P. (2019). Interactive Tree Of Life (iTOL) V4: Recent Updates and New Developments. Nucleic Acids Res. 47, 256–259. doi: 10.1093/nar/gkz239
Limam R. D., Bouchez T., Chouari R., Li T. L., Barkallah I., Landoulsi A., et al. (2010). Detection of WWE2-Related Lentisphaerae by 16S rRNA Gene Sequencing and Fluorescence in Situ Hybridization in Landfill Leachate. Can. J. Microbiol. 56, 846–852. doi: 10.1139/W10-065
Liu Y. H., Song X. B., Zhou H. M., Zhou X., Xia Y. L., Dong X., et al. (2018). Gut Microbiome Associates With Lipid-Lowering Effect of Rosuvastatin in Vivo. Front. Microbiol. 9, 0530. doi: 10.3389/fmicb.2018.00530
Lloyd K. G., Steen A. D., Ladau J., Yin J. Q., Crosby L. (2018). Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 3, e00055–e00018. doi: 10.1128/mSystems.00055-18
Lou J., Jiang Y., Rao B., Li A., Ding S., Yan H., et al. (2020). Fecal Microbiomes Distinguish Patients With Autoimmune Hepatitis From Healthy Individuals. Front. Cell Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.00342
Magoc T., Salzberg S. L. (2011). FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinform 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maskarinec G., Raquinio P., Kristal B. S., Setiawan V. W., Wilkens L. R., Franke A. A., et al. (2021). The Gut Microbiome and Type 2 Diabetes Status in the Multiethnic Cohort. PloS One 16, e0250855. doi: 10.1371/journal.pone.0250855
Meier-Kolthoff J. P., Auch A. F., Klenk H. P., Goker M. (2013). Genome Sequence-Based Species Delimitation With Confidence Intervals and Improved Distance Functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Murray M. G., Thompson W. F. (1980). Rapid Isolation of High Molecular-Weight Plant DNA. Nucleic Acids Res. 8, 4321–4325. doi: 10.1093/nar/8.19.4321
Paytan A., McLaughlin K. (2007). The Oceanic Phosphorus Cycle. Chem. Rev. 107, 563–576. doi: 10.1021/cr0503613
Pearson A., Budin M., Brocks J. J. (2003). Phylogenetic and Biochemical Evidence for Sterol Synthesis in the Bacterium Gemmata Obsuriglobus. PNAS 100, 15352–15357. doi: 10.1073/pnas.2536559100
Peng S., Yin J. G., Liu X. L., Jia B. Y., Chang Z. G., Lu H. J., et al. (2015). First Insights Into the Microbial Diversity in the Omasum and Reticulum of Bovine Using Illumina Sequencing. J. Appl. Genet. 56, 393–401. doi: 10.1007/s13353-014-0258-1
Pilhofer M., Rappl K., Eckl C., Bauer A. P., Ludwig W., Schleifer K. H., et al. (2008). Characterization and Evolution of Cell Division and Cell Wall Synthesis Genes in the Bacterial Phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and Phylogenetic Comparison With rRNA Genes. J. Bacteriol. 190, 3192–3202. doi: 10.1128/Jb.01797-07
Pinos S., Pontarotti P., Raoult D., Baudoin J. P., Pagnier I. (2016). Compartmentalization in PVC Super-Phylum: Evolution and Impact. Biol. Direct 11, 38. doi: 10.1186/s13062-016-0144-3
Polymenakou P. N., Lampadariou N., Mandalakis M., Tselepides A. (2009). Phylogenetic Diversity of Sediment Bacteria From the Southern Cretan Margin, Eastern Mediterranean Sea. Syst. Appl. Microbiol. 32, 17–26. doi: 10.1016/j.syapm.2008.09.006
Qiu Y. L., Muramatsu M., Hanada S., Kamagata Y., Guo R. B., Sekiguchi Y. (2013). Oligosphaera Ethanolica Gen. Nov., Sp Nov., an Anaerobic, Carbohydrate-Fermenting Bacterium Isolated From Methanogenic Sludge, and Description of Oligosphaeria Classis Nov in the Phylum Lentisphaerae. Int. J. Syst. Evol. Microbiol. 63, 533–539. doi: 10.1099/ijs.0.039545-0
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rivas-Marin E., Canosa I., Devos D. P. (2016). Evolutionary Cell Biology of Division Mode in the Bacterial Planctomycetes-Verrucomicrobia-Chlamydiae Superphylum. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01964
Rivas-Marin E., Devos D. P. (2018). The Paradigms They Are a-Changin': Past, Present and Future of PVC Bacteria Research. Anton Leeuw Int. J. G 111, 785–799. doi: 10.1007/s10482-017-0962-z
Sekiguchi Y., Yamada T., Hanada S., Ohashi A., Harada H., Kamagata Y. (2003). Anaerolinea Thermophila Gen. Nov., Sp. Nov. And Caldilinea Aerophila Gen. Nov., Sp. Nov., Novel Filamentous Thermophiles That Represent a Previously Uncultured Lineage of the Domain Bacteria at the Subphylum Level. Int. J. Syst. Evol. Microbiol. 53, 1843–1851. doi: 10.1099/ijs.0.02699-0
Sichert A., Corzett C. H., Schechter M. S., Unfried F., Markert S., Becher D., et al. (2020). Verrucomicrobia Use Hundreds of Enzymes to Digest the Algal Polysaccharide Fucoidan. Nat. Microbiol. 5, 1026–1039. doi: 10.1038/s41564-020-0720-2
Staley J. T. (1968). Prosthecomicrobium and Ancalomicrobium - New Prosthecate Freshwater Bacteria. J. Bacteriol. 95, 1921–1942. doi: 10.1128/Jb.95.5.1921-1942.1968
Strous M., Pelletier E., Mangenot S., Rattei T., Lehner A., Taylor M. W., et al. (2006). Deciphering the Evolution and Metabolism of an Anammox Bacterium From a Community Genome. Nature 440, 790–794. doi: 10.1038/nature04647
Trifinopoulos J., Nguyen L. T., von Haeseler A., Minh B. Q. (2016). W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 44, 232–235. doi: 10.1093/nar/gkw256
van Vliet D. M., Palakawong Na Ayudthaya S., Diop S., Villanueva L., Stams A. J. M., Sanchez-Andrea I. (2019). Anaerobic Degradation of Sulfated Polysaccharides by Two Novel Kiritimatiellales Strains Isolated From Black Sea Sediment. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00253
Wagner J. K., Setayeshgar S., Sharon L. A., Reilly J. P., Brun Y. V. (2006). A Nutrient Uptake Role for Bacterial Cell Envelope Extensions. PNAS 103, 11772–11777. doi: 10.1073/pnas.0602047103
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wiegand S., Jogler M., Boedeker C., Pinto D., Vollmers J., Rivas-Marin E., et al. (2020). Cultivation and Functional Characterization of 79 Planctomycetes Uncovers Their Unique Biology. Nat. Microbiol. 5, 126–140. doi: 10.1038/s41564-019-0588-1
Wiegand S., Jogler M., Jogler C. (2018). On the Maverick Planctomycetes. FEMS Microbiol. Rev. 42, 739–760. doi: 10.1093/femsre/fuy029
Wu D. Y., Jospin G., Eisen J. A. (2013). Systematic Identification of Gene Families for Use as “Markers” for Phylogenetic and Phylogeny-Driven Ecological Studies of Bacteria and Archaea and Their Major Subgroups. PloS One 8, e77033. doi: 10.1371/journal.pone.0077033
Yan L., Gao Y. M., Wang Y. J., Liu Q., Sun Z. Y., Fu B. R., et al. (2012). Diversity of a Mesophilic Lignocellulolytic Microbial Consortium Which is Useful for Enhancement of Biogas Production. Bioresour Technol. 111, 49–54. doi: 10.1016/j.biortech.2012.01.173
Yarza P., Yilmaz P., Pruesse E., Glockner F. O., Ludwig W., Schleifer K. H., et al. (2014). Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Zhang J., Liu R., Xi S. C., Cai R. N., Zhang X., Sun C. M. (2020). A Novel Bacterial Thiosulfate Oxidation Pathway Provides a New Clue About the Formation of Zero-Valent Sulfur in Deep Sea. ISME J. 14, 2261–2274. doi: 10.1038/s41396-020-0684-5
Zheng R. K., Cai R. N., Liu R., Liu G., Sun C. M. (2021a). Maribellus Comscasis Sp. Nov., A Novel Deep-Sea Bacteroidetes Bacterium, Possessing a Prominent Capability of Degrading Cellulose. Environ. Microbiol. 23, 4561-4575. doi: 10.1111/1462-2920.15650
Zheng R. K., Liu R., Shan Y. Q., Cai R. N., Liu G., Sun C. M. (2021b). Characterization of the First Cultured Free-Living Representative of Candidatus Izemoplasma Uncovers its Unique Biology. ISME J. 15, 2676–2691. doi: 10.1038/s41396-021-00961-7
Keywords: Lentisphaerae, cold seep, polysaccharide, phosphorus, cultivation, deep-sea
Citation: Zhang T, Zheng R, Liu R, Li R and Sun C (2022) Cultivation and Functional Characterization of a Deep-Sea Lentisphaerae Representative Reveals Its Unique Physiology and Ecology. Front. Mar. Sci. 9:848136. doi: 10.3389/fmars.2022.848136
Received: 04 January 2022; Accepted: 14 March 2022;
Published: 14 April 2022.
Edited by:
Jian-Wen Qiu, Hong Kong Baptist University, Hong Kong SAR, ChinaReviewed by:
Hideyuki Tamaki, National Institute of Advanced Industrial Science and Technology (AIST), JapanCopyright © 2022 Zhang, Zheng, Liu, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaomin Sun, c3VuY2hhb21pbkBxZGlvLmFjLmNu; Ronggui Li, bHJnQHFkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.