 Chao Deng1†
Chao Deng1† Caihuan Ke
Caihuan Ke Weiwei You
Weiwei You Ying Wang
Ying Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Mar. Sci. , 16 February 2022
Sec. Marine Fisheries, Aquaculture and Living Resources
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.841561
This article is part of the Research Topic Mollusk Breeding and Genetic Improvement View all 25 articles
Aquaculture is a rapidly growing industry that brings huge economic benefits. Genome-wide association study (GWAS) is critical for aquaculture species’ productivity, sustainability, and product quality. The current integrated GWAS pipeline either includes only specific limited steps or requires a complex prerequisite environment and configurations. In this study, we developed AquaGWAS, a highly user-friendly graphical user interface (GUI) GWAS pipeline, by integrating four well-known GWAS models. AquaGWAS is a complete GWAS pipeline from preprocessing, multiple choice of GWAS models, postprocessing to visualizations. AquaGWAS offers GUI easy running on Linux and automatically generates running command lines for high-performance computing (HPC) or non-GUI servers. AquaGWAS is free from installation, configurations, and complicated augment inputs. It offers whole packages of required reference files for 27 common aquatic species. Furthermore, aiming at the issue that the availability of genomic reference sequences limits single-nucleotide polymorphism (SNP) detection, we attempted to detect SNPs in Pacific abalone using classical alignment-based reference-required strategy and k-mer-based reference-free strategy combined with downstream AquaGWAS. On 222 resequencing data of Pacific abalone, two strategies detected 221,061 and 230,213 variants, respectively, with 180,161 common variants. The two strategies emphasized different variant situations: capturing variants missed by incomplete or inaccurate reference genomic sequence (k-mer-based) and capturing the indel variants having the baseline of genomic sequence (alignment-based). Combining the two strategies offers a complementary framework to obtain the accurate and complete GWAS analysis for non-model organism species. AquaGWAS is available at https://github.com/Ying-Lab/AquaGWAS.
Genome-wide association study (GWAS) effectively identifies genotypic variants associated with particular phenotypic traits. Lots of related tools have been developed to improve the efficiency of GWAS analysis. However, many tools were designed using a specific model or for a certain processing step, which led to fussy file format conversions between various interfaces and frequent switches among different running environments (C/C++, Python, R, etc.). In contrast, the integrated pipeline, such as iPat (Chen and Zhang, 2018), HAPPI GWAS (Slaten et al., 2020), either prerequire a JAVA environment or includes only a single GWAS model, and the complicated augments and options in command lines limit the feasibility for non-computational researchers.
Aquaculture is a rapidly growing industry that brings huge economic benefits (FAO, 2017). GWAS, which is used for exploring molecular markers and mechanisms related to traits, is critical to improving aquaculture species’ productivity, sustainability, and product quality. Meanwhile, species-specific single-nucleotide polymorphism (SNP) arrays are still not widely used for aquaculture species. In most cases, species were sequenced and aligned to reference genomic sequence and then genotyped using a vcf format file (Jiang et al., 2019; Wu et al., 2019). Three obstructions limit the feasibility of the GWAS study: (1) Most marine biologists have a limited background in modeling and bioinformatics. At the same time, it is hard for them to switch among different running environments, convert fussy file formats, and handle command lines with complex augments and different models. (2) Due to the diversity of aquaculture species, there is no common data format and there are a few available public databases. In that way, annotation and reference information requires extra manual operations. (3) A large amount of non-model aquaculture animals, novel species, or species are hard to detect SNPs because of lack of reference genomes or incomplete genome. Therefore, it is necessary to develop environment-friendly software to resolve these problems.
This study developed AquaGWAS, a highly integrated GWAS analysis pipeline with a full reference package, including 27 aquatic species. AquaGWAS offers an easy-to-use graphic user interface (GUI) for stand-alone Linux server running mode and an automatic command-generator on Windows for HPC job-submission running mode. Free from complex installation and configurations, AquaGWAS is compatible with various file formats and highly friendly to users with a non-computational background.
Furthermore, to implement GWAS analysis for aquaculture species without a reference genome or only with an incomplete genome, we attempted to detect SNPs for Pacific abalone Haliotis discus hannai using classical alignment-based reference-required strategy and k-mer-based reference-free strategy. The two strategies were implemented using AquaGWAS combined with upstream alignment-based [Bowtie2 (Langmead and Salzberg, 2012) SAMtools and BCFtools (Danecek et al., 2021)] and k-mer-based [DISCOSNP (Uricaru et al., 2015)] pipelines. Based on 222 genomic resequencing data of Pacific abalones, the two strategies detected 221,061 and 230,213 variants, respectively, with 180,161 common variants. The detected variants belonged to 2,048 and 2,068 genes, respectively, with 2,046 common gene resolutions. The two strategies were highly consistent in both gene and nucleotide resolutions. With MWR and SLW as traits, the two strategies captured four and five common-associated SNPs using AquaGWAS. The two strategies have clear advantages and limitations due to their basic principle and have explicit complementation during the GWAS analysis.
The Pacific abalone used in this study contained a total of 222 individuals randomly selected from 10 families (Peng et al., 2021). Two growth-related traits (the ratio of foot muscle weight divided by wet weight, MWR, and the ratio of shell length divided by shell width, SLW) were measured and calculated after 2.5 years of culture. The genotypic data were generated using whole-genome sequencing (WGS) technology. Sequence reads for each sample were quality controlled using an in-house Perl script with a total of 2,408.162 Gb clean data remaining. The average sequencing depth for each sample was 7.75×.
Currently, AquaGWAS integrates widely used GWAS tools within a GUI on Linux to perform a complete GWAS analysis pipeline for aquatic animals using various SNP files, vcf, ped/map, tped/tfam, and bed/bim/fam as inputs. AquaGWAS contains five functional modules: (1) GWAS; (2) linkage disequilibrium (LD) decay analysis; (3) visualization with principal component analysis (PCA), Manhattan plot, and quantile-quantile (Q-Q) plot; (4) gene structural annotation; and (5) gene functional annotation. After integrating the reference sequences, structural annotation, and function annotation information from 27 aquatic species, AquaGWAS can implement the complete GWAS analysis for these aquatic species without extra reference databases.
The framework of AquaGWAS is shown in Figure 1. The input to AquaGWAS was phenotype data and SNP information data. The SNP information data, generally containing genotype and map, were compatible with the following commonly used file formats: (1) vcf, (2) ped/map, (3) tped/tfam, and (4) bed/bim/fam. Quality control (QC) was performed for SNP input files. The optional files included covariate and kinship files, which would be the additional information for GWAS analysis models and postprocessing for gene structural or functional annotation, respectively. The output included p-values of SNPs, Manhattan plot, Q-Q plot, PCA plot, LD decay plot, and the following structural annotations and functional annotations.
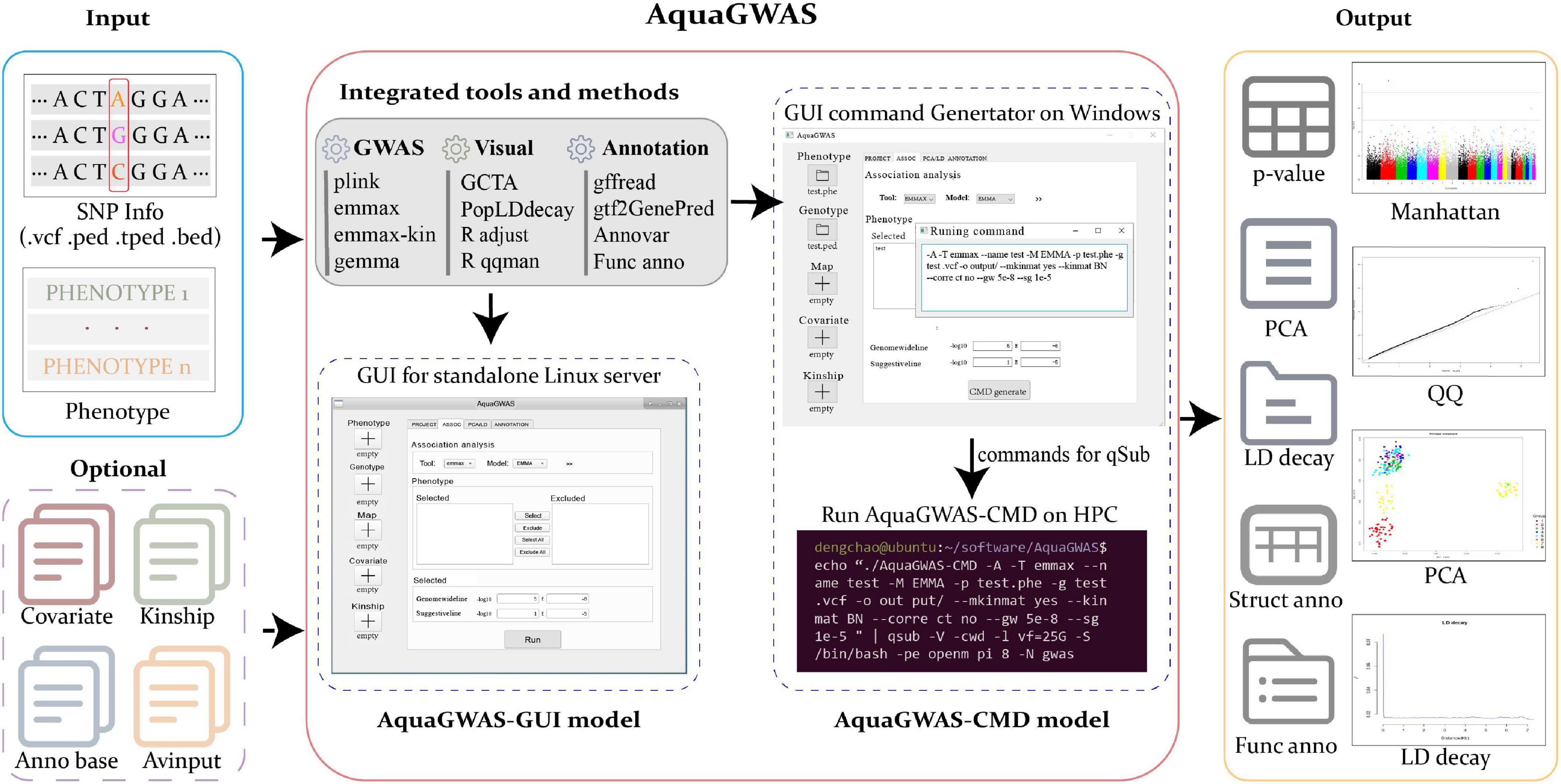
Figure 1. The framework of AquaGWAS. AquaGWAS is compatible with several SNP information file formats, vcf, ped/map, tped/tfam, and bed/bim/fam. Several widely used GWAS tools, preprocessing and postprocessing, visualizations, and structural and functional annotations are integrated seamlessly, and output textual and visual analyzing results.
The GWAS of AquaGWAS offered a linear model and a regression model from PLINK (Purcell et al., 2007), a linear mixed model (LMM, also called MLM) from GEMMA (Zhou and Stephens, 2012), and an efficient mixed-model association (EMMA) from EMMAX (Kang et al., 2010). Meanwhile, QC was achieved through PLINK. The PopLDdecay (Zhang et al., 2018) and GCTA (Yang et al., 2011) were used for LD decay analysis and PCA analysis of AquaGWAS, respectively. Regarding the structural annotation, it was achieved using Annovar (Wang et al., 2010). In addition, the gffread and gtfToGenePred (Pertea and Pertea, 2020) helped to automatically generate the input file of Annovar. Then, some custom functions were used for functional annotation to extract the response gene annotation information directly from the gene functional annotation database file by gene ID.
The tools integrated by AquaGWAS, such as PLINK and GEMMA, include multiple small functions. Additionally, some of them might not be required for GWAS analysis. In AquaGWAS, we only integrated the required function packages and offered the corresponding parameter options in GUI. The fixed parameters are predetermined in the code. For more flexible parameters and options, we welcome more voices, and we will always update and improve AquaGWAS according to the user’s feedback.
Different from the plant and livestock, many aquatic animals do not have genomic reference sequences or only have an incomplete genomic sequence, which limits the accurate and complete GWAS analysis for these species. Therefore, using Pacific abalone as an example, we implemented classical alignment-based reference-required GWAS (called alignment-based) and k-mer-based reference-free GWAS (called k-mer-based) and combined AquaGWAS with two different upstream strategies, as shown in Figure 2. The alignment-based strategy refers to the classical GWAS processing pipeline. It aligns the genomic sequencing reads to reference genomic sequences using sequence alignment tools, such as Bowtie (Langmead et al., 2009) or BWA (Li and Durbin, 2009). The alignment results were then sorted and filtered using SAMtools and BCFtools to call the candidate genetic variants. The k-mer-based strategy was implemented using DISCOSNP, which calls the candidate variants free from the genomic reference sequence. DISCOSNP detects SNPs with a k-mer comparison based on a de Bruijn graph. It is a directed graph that contains all the k-mers present in the read dataset as vertices, and all the possible (k − 1) overlaps between k-mers as edges. Then, for each such couple of k-mers starting with the same k − 1 length prefix, if both paths cannot be right extended with the same nucleotide, the bubble was discarded, and only isolated SNPs can be detected. Then, the detected k-mers with SNPs were assembled into contigs, and the whole procedure was free from the genomic reference sequence. The detail of DISCOSNP can be found in the study by Uricaru et al. (2015). The variants calling files from the above two strategies were input into AquaGWAS for GWAS, with MWR and SLW as traits.

Figure 2. The two different upstream strategies, namely, alignment-based reference-required and k-mer-based reference-free, combined AquaGWAS for genome-wide association study (GWAS). The alignment-based strategy aligns the genomic sequencing reads to reference genomic sequences and then calls the candidate genetic variants. Additionally, the k-mer-based strategy calls the candidate variants free from reference genomic sequence with a k-mer comparison based on a de Bruijn graph. The variants calling files from the above two strategies are input into AquaGWAS for GWAS.
There are several functions for AquaGWAS. First, AquaGWAS offers the thresholds to exclude individuals with too much missing genotype data, SNPs with too small minor allele frequency (MAF), and SNPs with too large missing genotype rate. Users are free to select none/some/all these parameters, and the parameters (window size, step length, and r2 threshold) for data filtering using linkage disequilibrium are also allowed to be selected and set.
Second, PLINK, GEMMA, and EMMAX are integrated by AquaGWAS for GWAS. Users can input single phenotype or multiple phenotype files for association analysis, and the kinship and covariates can be used as optional inputs to control false positive analysis results. The analysis results are visualized using the Manhattan plot and Q-Q plot. Users can also get the results of population genetic diversity of samples and linkage disequilibrium decay analysis, which are subsequently implemented using GCTA and PopLDdecay, and then visualized using PCA and LD plots.
Third, the process of annotation was divided into two steps, structure annotation and function annotation. Among these, the first step abstracted information of SNPs above the threshold [–log (10–5) by default] from the vcf file and then converted it into the file needed for gene structural and functional annotation. Structural annotations offer exon regions such as variant functions, types, amino acid changes, and all the genes and positions of the mutations. Additionally, the functional annotation is realized on the gene functional annotation database of corresponding species.
AquaGWAS is an easy and user-friendly software for analyzing aquatic animals with non-commonly used reference genome databases and rare information in public databases, which makes it difficult for analyses by regular software and pipeline. Several features of the AquaGWAS are as follows.
First, AquaGWAS is free from any installation and environmental configuration, which offers the executable file to be run directly.
Second, AquaGWAS offers a user-friendly GUI. To begin with, encapsulating the entire tedious process, AquaGWAS offers an intuitive parameter setting interface. Furthermore, in AquaGWAS, users can switch among various tools smoothly and seamlessly during the analysis process, free from complicated file format conversions between interfaces, complicated command lines with tedious running arguments, and file folder switching.
Third, AquaGWAS automatically generate a command line in HPC. For HPC users, jobs are required to submit with command lines, where the direct running on the graphic interface is not available. Therefore, AquaGWAS also offers GUI automatic command-line generator on Windows so that users can submit the job with the generated command line on Windows conveniently.
Finally, AquaGWAS is compatible with various file formats. Because different GWAS tools support various file formats, AquaGWAS automatically converts the input file according to the selected tool. For example, although EMMAX only supports tped/tfam files, users can choose vcf, ped/map, or bed/bim/fam in AquaGWAS for analysis using EMMAX through automatic file format conversion. AquaGWAS also supports the automatic loading of multiple files with the same prefix filename under the same folder. For example, if the input file is of ped/map or tped/tfam format, users can choose to input only ped or tped file. AquaGWAS will automatically load the associated map or tfam file under the same path with the same prefix of the filename.
Meanwhile, because the AquaGWAS is a pipeline that integrates the current widely used GWAS analyzing tools, the upper limit of the amount of running data is determined by the selected tool during the AquaGWAS running and the hardware of the running server.
The alignment-based and k-mer-based strategies were applied for GWAS analysis of Pacific abalone using 222 genomic resequencing data. Using Chromosome 1 as an example, the alignment-based and k-mer-based methods detected 221,061 and 230,213 variants, respectively. To compare the results from the two strategies, the contigs from the k-mer-based strategy were aligned to the reference genome of the Pacific abalone, and the corresponding position in Chromosome 1 was obtained. Compared with the genetic variants from the alignment-based method, 180,161 common variants were detected by the two strategies, as shown in the Venn diagram in Figure 3A. The 50,052 variants detected by k-mer-based but missed from alignment-based were partly caused by incomplete or inaccurate genomic reference sequences. In contrast, the k-mer-based method also missed 40,900 variants found by the alignment-based method, which is probably caused by the structural insert and delete due to the de Bruijn assembly errors caused by low reads coverage, repeat sequences, and short overlaps. Considering the genes corresponding to SNP, the intersection number of genes detected by kmer-based (2048) and alignment-based (2068) is 2046, as shown in the Venn diagram in Figure 3B.
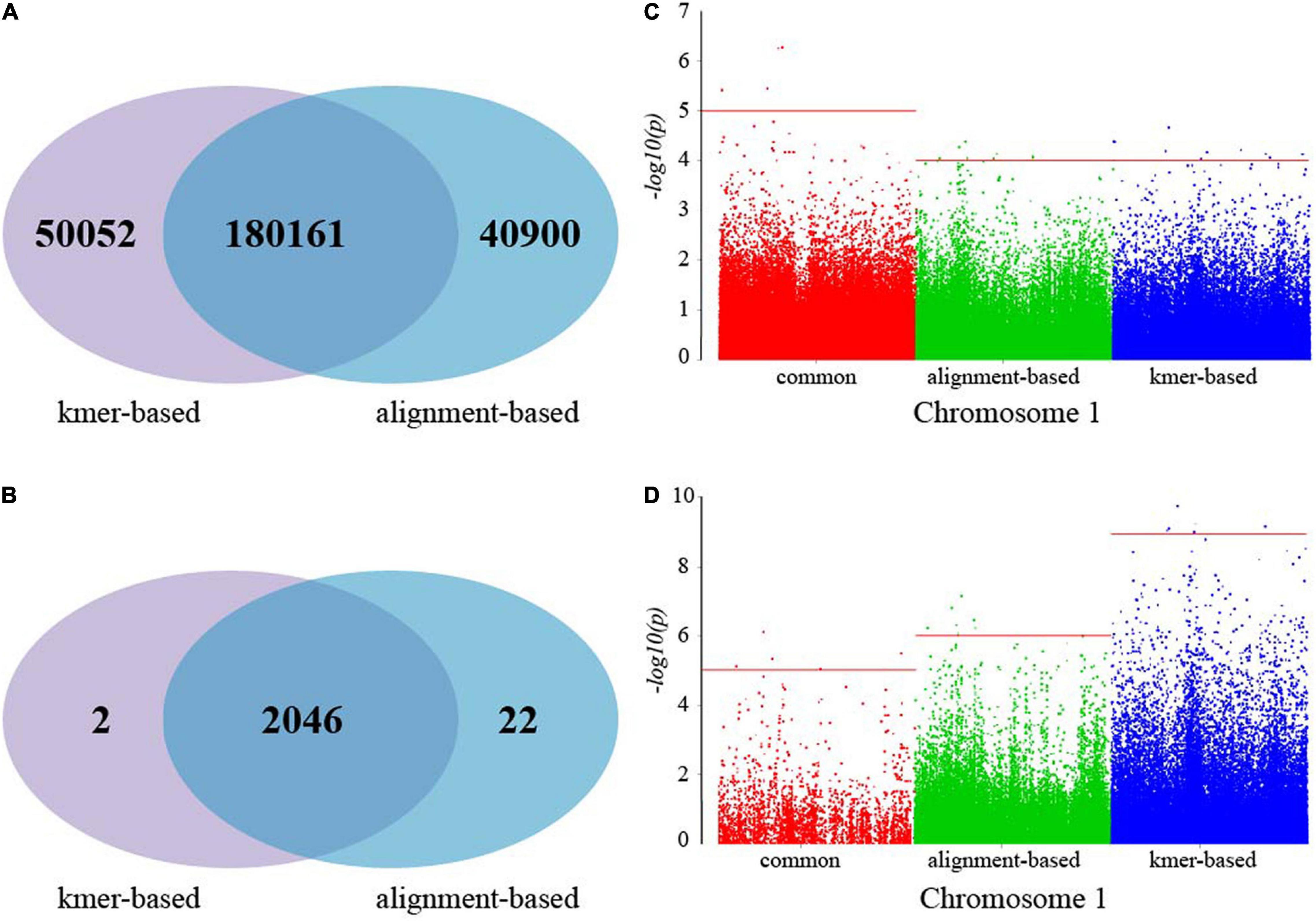
Figure 3. The comparison of the results from the alignment-based and the k-mer-based strategies. (A) The Venn diagram of candidate variants from k-mer-based and alignment-based strategies; (B) the Venn diagram of genes including variants from k-mer-based and alignment-based strategies; (C) the Manhattan plots for MWR traits from common, alignment-based, and k-mer-based SNPs; (D) the Manhattan plots for SLW traits from common, alignment-based, and k-mer-based SNPs.
The variant calling files from the two strategies were input into AquaGWAS. The k-mer-based method used p − value ≤ 10−4 and p − value ≤ 10−9 to screen the significant SNPs of MWR and SLW and obtained 10 and 6 significant SNPs, respectively. For the alignment-based method, 11 and 8 significant SNPs were obtained by using a p − value ≤ 10−4 and p − value ≤ 10−5 of MWR and SLW. Using p − value ≤ 10−5, there are 4 and 5 common SNPs associated with MWR and SLW traits detected by both alignment-based and k-mer-based methods, which are shown in Figures 3C,D. The detailed analysis results are presented in Supplementary Table 1.
To compare the variants detected by two strategies elaborately, the contigs, including variants detected by alignment-based strategy and the k-mers detected by variants from the k-mer-based strategy, are aligned to the genomic sequences of Pacific abalone. Because a reference sequence is required, we only gave the situations that both the k-mers and reads can be aligned to the reference genomic sequences shown in Figure 4. The genomic region from chromosome 1:21634620-21634760 is selected and viewed on Integrative Genomics Viewer (IGV) (Robinson et al., 2011). The variant in the red border is detected by both strategies. Additionally, the variants in yellow borders are detected only by k-mer-based strategy. The variant and insertion in the green border are detected only by alignment-based strategy. The two strategies emphasized different variant situations. After tracing back to the original variant detection process, we found k-mer-based method can capture variants missed by incomplete or inaccurate reference genomic sequences. In contrast, an alignment-based strategy can capture the insertion or deletion having the baseline of genomic sequence. Therefore, the two strategies have explicit complementation for GWAS analysis.

Figure 4. The detailed comparison of genetic variants detected by the alignment-based and k-mer-based strategies. Both strategies detect the variant in the red border. Furthermore, the variants in yellow borders are detected only by k-mer-based strategy. The variant and insertion in the green border are detected only by alignment-based strategy.
The current integrated GWAS pipeline either includes only specific limited steps or requires a complex prerequisite environment and configurations. AquaGWAS offers GUI easy running for stand-alone Linux servers and an automatic command line generator for HPC or non-GUI servers. AquaGWAS integrated complete GWAS pipeline from preprocessing, multiple choice of GWAS models, postprocessing to visualizations. Furthermore, AquaGWAS is highly friendly to users of non-computational background, which is free from installation, configurations, and complicated augments input. AquaGWAS is compatible with various frequently used SNP file formats and can be applied to several species, especially aquatic animals. AquaGWAS offers whole packages of required reference files for 27 common aquatic species to implement the express aquaculture GWAS analysis without any extra reference inputs.
The experiment shows that the k-mer-based strategy can capture the variants, which are missed because of the incomplete or inaccurate reference genomic sequences. However, the k-mer-based reference-free strategy also missed SNPs, which are found by the classical reference-alignment strategy. The two strategies have clear advantages and limitations due to their basic principle and have explicit complementation during the GWAS analysis.
Our exploration not only offers an option to study the genetic variants for non-model organism species, novel species, or species without genomic reference sequence but also provides a complementary framework for accurate and complete GWAS analysis.
The data analyzed in this study is subject to the following licenses/restrictions: The dataset is on another ongoing manuscript, which will be public available after that manuscript is published. Requests to access these datasets should be directed to WY, MTA5ODUyMzgzQHFxLmNvbQ==.
YW, WY, and CK planned the project. CD and ZM finished the programming. WP designed the processing steps of GWAS. WP, CD, and ZM tested the software. YW and CD designed the experiments. YW, CD, and WP wrote the main manuscript. All the authors read and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (62173282, U1605213, and 31872564), the National Key Research and Development Program of China (2018YFD0901401), Fujian Provincial S&T Project (2019N0001 and 2017FJSCZY02), Open Fund of Engineering Research Center for Medical Data Mining and Application of Fujian Province (MDM2018002), and Natural Science Foundation of Fujian (2018J01097).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank W. He, S. Fang, and T. Wu from the Information and Network Center of Xiamen University for their help with GPU computing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.841561/full#supplementary-material
Chen, C. J., and Zhang, Z. (2018). iPat: intelligent prediction and association tool for genomic research. Bioinformatics 34, 1925–1927. doi: 10.1093/bioinformatics/bty015
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10:8. doi: 10.1093/gigascience/giab008
FAO (2017). Genome-Based Biotechnologies in Aquaculture. Rome: Food and Agriculture Organization of the United Nations, 2–3.
Jiang, D. L., Gu, X. H., Li, B. J., Zhu, Z. X., Qin, H., Meng, Z. N., et al. (2019). Identifying a long QTL cluster across chrLG18 associated with salt tolerance in tilapia using GWAS and QTL-seq. Mar. Biotechnol. 21, 250–261. doi: 10.1007/s10126-019-09877-y
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S.-Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Peng, W., Yu, F., Wu, Y., Zhang, Y., Lu, C., Wang, Y., et al. (2021). Identification of growth-related SNPs and genes in the genome of the Pacific abalone (Haliotis discus hannai) using GWAS. Aquaculture 541:736820. doi: 10.1016/j.aquaculture.2021.736820
Pertea, G., and Pertea, M. (2020). GFF utilities: GffRead and GffCompare. F1000Research 9:23297. doi: 10.12688/f1000research.23297.1
Purcell, S., Neale, B. Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Slaten, M. L., Chan, Y. O., Shrestha, V., Lipka, A. E., and Angelovici, R. (2020). HAPPI GWAS: holistic analysis with Pre- and Post-integration GWAS. Bioinformatics 36, 4655–4657. doi: 10.1093/bioinformatics/btaa589
Uricaru, R., Rizk, G., Lacroix, V., Quillery, E., Plantard, O., Chikhi, R., et al. (2015). Reference-free detection of isolated SNPs. Nucleic Acids Res. 43:e11. doi: 10.1093/nar/gku1187
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wu, L., Yang, Y., Li, B., Huang, W., Wang, X., Liu, X., et al. (2019). First genome-wide association analysis for growth traits in the largest coral reef-dwelling bony fishes, the giant grouper (Epinephelus lanceolatus). Mar. Biotechnol. 21, 707–717. doi: 10.1007/s10126-019-09916-8
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Zhang, C., Dong, S., Xu, J., He, W., and Yang, T. (2018). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi: 10.1093/bioinformatics/bty875
Keywords: GWAS, aquatic animal, integrated pipeline, abalone, k-mer-based GWAS
Citation: Deng C, Peng W, Ma Z, Ke C, You W and Wang Y (2022) AquaGWAS: A Genome-Wide Association Study Pipeline for Aquatic Animals and Its Application to Reference-Required and Reference-Free Genome-Wide Association Study for Abalone. Front. Mar. Sci. 9:841561. doi: 10.3389/fmars.2022.841561
Received: 22 December 2021; Accepted: 17 January 2022;
Published: 16 February 2022.
Edited by:
Yinghui Dong, Zhejiang Wanli University, ChinaReviewed by:
Xiaoting Huang, Ocean University of China, ChinaCopyright © 2022 Deng, Peng, Ma, Ke, You and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiwei You, d3d5b3VAeG11LmVkdS5jbg==; Ying Wang, d2FuZ3lpbmdAeG11LmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.