
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 24 February 2022
Sec. Marine Biology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.795525
 Jialin Wang1,2
Jialin Wang1,2 Qian Yang1,2
Qian Yang1,2 Yuanri Hu1,2
Yuanri Hu1,2 Wenteng Xu2,3,4
Wenteng Xu2,3,4 Yingming Yang2,3,4
Yingming Yang2,3,4 Songlin Chen2,3,4*
Songlin Chen2,3,4* Na Wang2,3,4*
Na Wang2,3,4*
Chinese tongue sole (Cynoglossus semilaevis) is a flatfish species unique to Northeast Asia, exhibiting the typical female-biased sexual size dimorphism (SSD). To explore the possible regulatory roles of non-coding RNAs (ncRNAs) on this phenomenon, whole transcriptomic analysis was performed by using female, male, and pseudomale C. semilaevis to identify differentially expressed (DE) long ncRNAs (DE lncRNAs), microRNA (DE miRNAs), and differentially expressed genes (DEGs) from the brain, gonad, liver, and muscle tissues. Most of them were concentrated in the gonad and muscle, and the gene expression patterns of pseudomale individuals were similar to male individuals. The association of DE lncRNAs and target messenger RNAs (mRNAs) was predicted based on antisense, cis-, and trans-regulatory mechanisms, with enriched protein digestion and absorption, cyclic adenosine monophosphate (cAMP) signaling pathway, sulfur metabolism, cell cycle, and splicesome (p < 0.05). Furthermore, weighted gene co-expression network analysis (WGCNA) was employed to cluster the expression patterns of DE lncRNA, and two modules (greenyellow and blue) had the highest positive and negative correlations with growth traits, respectively. Importantly, the female-biased expression in the greenyellow module and the male- and pseudomale-biased expression in the blue module were observed in the gonad. The target gene analysis for DE miRNA revealed 3,034 mRNA-miRNA pairs with the opposite expression patterns. Finally, the lncRNA-miRNA-mRNA network, including 385 DE lncRNAs, 138 DE miRNAs, and 456 DEGs, was constructed. Among which, 78 DE lncRNAs, 12 DE miRNAs, and 13 DEGs involved in cell growth and death pathway were related to the SSD of C. semilaevis. This study described the lncRNA-miRNA-mRNA regulatory network in the SSD of C. semilaevis for the first time. The functional prediction analysis suggested that these DE lncRNAs and DE miRNAs might be involved in flatfish SSD by regulating several potential growth-related pathways (e.g., cell cycle, cAMP signaling, and Rap1 signaling). Further studies related to these ncRNAs will enlarge our understanding of the regulatory effects of ncRNAs on fish SSD.
Sexual size dimorphism (SSD), characterized by significant differences in the body size of male and female individuals, is a widespread phenomenon that existed in mammals, reptiles, fishes, insects, and spiders (Parker, 1992; Chen et al., 2012; O’Mara et al., 2012; Mei and Gui, 2015; Belhaoues et al., 2020; Kuntner and Coddington, 2020; Tracy et al., 2020). In aquaculture, the phenomenon of SSD exists in many teleost fish species. Nile tilapia (Oreochromis niloticus) and Mozambique tilapia (Oreochromis mossambicus) show male-biased SSD, and male individuals grow 40% faster than female individuals (Wan et al., 2021). Olive flounder (Paralichthys olivaceus) (Ryu et al., 2020), whiting (Merlangius merlangus) (Milić and Kraljević, 2011), Atlantic cod (Gadus morhua) (Gerritsen et al., 2003), and haddock (Melanogrammus aeglefinus) (Laurence, 1976) show female-biased SSD that female grow faster than male.
Chinese tongue sole (Cynoglossus semilaevis) is a flatfish species with a high economic value located in Northeast Asia (Chen et al., 2009). The sex determination mechanism of C. semilaevis is ZW/ZZ type (Zhou et al., 2005; Chen et al., 2014). In C. semilaevis, the phenomenon of female-biased SSD is also significant. Each female individual can achieve a body size of up to four times that of a male individual (Chen et al., 2009; Wang et al., 2016). In aquaculture, the research on the SSD of C. semilaevis is of great significance. Previous transcriptome data also showed multiple pathways related to the growth and sex differentiation of C. semilaevis, such as the steroid biosynthesis, growth factor binding, and cell growth pathways (Wang N. et al., 2018). A series of growth and sex-difference genes were detected. The growth hormone (gh) gene was identified as an important factor in the process of male and female growth (Qian et al., 2012), genes, such as forkhead box L2 (foxl2) (Dong et al., 2011) and folliculogenesis-specific basic helix-loop-helix (BHLH) transcription factor (figla) (Li et al., 2016) are related to sex differentiation of C. semilaevis.
From the perspective of epigenetics, the diversity of biological phenotypes is governed by many molecular mechanisms, such as genetic modification, transcription, and translation regulation. More than half of the DNA of higher organisms are transcribed into RNA, and most of them are non-coding RNAs (ncRNAs) (Holmes et al., 1972), including three types [microRNA (miRNA), long non-coding RNA (lncRNA), and circular RNA (circRNA)]. They can regulate biological processes (BPs) by participating in gene expression regulation (Gan et al., 2016). Previous studies have shown the biological function of lncRNAs, revealing the important role in transcriptional regulation, cell growth, and differentiation of humans and mammals (Yang et al., 2014; Liu et al., 2015, 2017). In fish species, lncRNA has been involved in many biological regulation processes, such as immune regulation (Wang M. et al., 2018), skin color differentiation (Luo et al., 2019), early development, and gender differentiation (Cai et al., 2019; Yuan et al., 2019). MiRNAs can regulate gene transcription by restraining the 3′ untranslated regions (3′UTRs) of messenger RNAs (mRNAs) to participate in BPs, such as cell development, transcription, translation, and occurrence of diseases (Hobert, 2008). At present, miRNA research in teleost fishes mainly focuses on growth and body development (Gan et al., 2016; Zhang et al., 2016; Zhao et al., 2017; Lan et al., 2019; Zhou et al., 2019). In addition, the competing endogenous RNA (ceRNA) theory has revealed a complex interacting mechanism between RNAs (Salmena et al., 2011). In ceRNA cross talk, lncRNAs and circRNAs could act as sponges to adsorb miRNAs and regulate target gene expression (Tay et al., 2014). Recently, it has shown extensive attention in tumor research, pathological research, growth and development, and other fields (Huang, 2018; Gao et al., 2019; Ghasemi et al., 2020; Liu et al., 2020; Huang et al., 2021). In C. semilaevis, lncRNAs play an important role in lipid metabolism (Xu et al., 2019), immune response (Wei et al., 2021), and ovarian development (Dong et al., 2021). There are few lncRNAs and ceRNA studies on the growth and SSD of C. semilaevis.
In this study, abundance of differentially expressed (DE) lncRNAs, DE miRNAs, and DEGs from female, male, and pseudomale C. semilaevis were identified by whole transcriptome sequencing and biological information analysis methods and predicted the lncRNA co-expression and lncRNA-miRNA-mRNA ceRNA regulatory networks. In addition, we analyzed the significant enrichment terms and pathways in networks and emphasized the potential implications in growth differences of C. semilaevis.
Healthy 1.5-year-old C. semilaevis individuals used in this study were obtained from Haiyang Huanghai Aquaculture Co., Ltd., China. The experimental fish were cultured in a circulating aquaculture system (water temperature of 23 ± 1°C) and fed two times a day with a compound feed pellet. Fish individuals were anesthetized with MS-222 (Sinopharm, Shanghai, China) to relieve pain. Four important somatotropic and reproductive tissues, namely, brain, gonad, liver, and muscle, were sampled from male (n = 9), female (n = 9), and pseudomale (n = 9) C. semilaevis and immediately stored in liquid nitrogen for total RNA extraction and sequencing. The genetic sex identification of the male and the pseudomale C. semilaevis was performed using the primers described in a previous study (Table 1; Liu et al., 2014). Every three tissues from different sexual individuals were pooled into one sample, generating 36 samples, including FB1-3, MB1-3, PMB1-3, FG1-3, MG1-3, PMG1-3, FL1-3, ML1-3, PML1-3, FM1-3, MM1-3, and PMM1-3 (FB, female brain; MB, male brain; PMB, pseudomale brain; FG, female gonad; MG, male gonad; PMG, pseudomale gonad; FL, female liver; ML, male liver; PML, pseudomale liver; FM, female muscle; MM, male muscle; and PMM, pseudomale muscle).

Table 1. All information about primers in this study.
All experimental methods and steps were carried out in accordance with the guidelines of the standard manual. The tissues collection and processing of all fish used in this study were approved by the Animal Care and Use Committee of the Chinese Academy of Fishery Sciences.
TRIzol Reagent Kit (Invitrogen, Carlsbad, CA, United States) was used to extract the total RNA according to the protocol of the manufacturer. Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States) and ribonuclease (RNase)-free agarose gel electrophoresis system were used to assess the quality and purity of RNA. Then, rRNAs were screened and eliminated to pure ncRNAs and mRNAs. The remaining reads were used for the assembly and analysis of the transcriptome. The reconstruction of transcripts was performed with StringTie software (version 1.3.6). To identify new transcripts, all reconstructed transcripts were aligned to the reference genome (NCBI GCA_000523025.1) and divided into twelve categories by Cuffcompare software. Transcripts with one of the class codes (u, i, j, x, c, e, and o), length more than 200 bp, and exon number more than 2 were defined as novel transcripts. Screened ncRNAs were fragmented into short fragments with fragment buffer, and random primers reverse-transcribed these fragments into complementary DNA (cDNA). RNase H, deoxynucleoside triphosphate (dNTP), DNA polymerase I, and polymerase buffer were used to synthesize the second-strand cDNA. Then, QIAQuick PCR Extraction Kit (QIAGEN, Venlo, the Netherlands) was used to purify the cDNA fragments and ligated to Illumina sequencing adapters. Meanwhile, the second-strand cDNA was digested with uracil-n-glycosylase (UNG). Then, the products were sequenced by Illumina HiSeq™ 4000 system (GeneDenovo Bio, Guangzhou, China). Specific raw data quality control methods were used based on a previous study (Chen et al., 2018).
Both CNCI (version 2) and CPC (version 0.9-r2) were used to assess the potential of transcripts to encode proteins by system default parameters (Kong et al., 2007; Sun et al., 2013). The intersections of both non-coding protein potential results were chosen as lncRNAs. The fragments per kilobase of transcript per million (FPKM) value was used to evaluate the expression levels of lncRNAs and mRNAs. LncRNAs and mRNAs differential expression analysis were assessed by DESeq2 software. The mRNAs and lncRNAs with the following parameters, including false discovery rate (FDR) < 0.05, absolute fold change (FC) ≥ 2, and p < 0.05, were considered DE mRNAs and DE lncRNAs.
The miRNA cDNA Synthesis Kit (Accurate biology, Changsha, China) was used to construct cDNA libraries with the standard manual. A poly (A) tail was added to the 3′ end of miRNA with Poly(A) Polymerase. Then, universal reverse transcription primers-Oligo (dT) primers with specific tags were used for reverse transcription reaction and generated cDNA.
The hairpin structure characteristics of miRNA precursors could predict novel miRNAs (Xiu et al., 2019). Software mirdeep2 and miREvo were used to identify novel miRNAs (Friedlander et al., 2012; Wen et al., 2012). The family information and sequences of miRNAs could be found from TargetScan.1 miRNAs with the parameters of absolute FC ≥ 2 and value of p < 0.05 were used as the evaluation standards of DE miRNA.
To further explore the functions of miRNAs and lncRNAs, Gene Ontology (GO) enrichment analysis with the GO database2 was used to analyze the enrichment of target genes and mainly referred to three aspects, namely, cellular component (CC), molecular function (MF), and BP. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with the KEGG database3 were referenced at the same time. In both GO terms and KEGG pathways, p < 0.05 was used as the evaluation standards.
To reveal the regulatory relationship and dynamic changes between lncRNAs and SSD of C. semilaevis, the weighted gene co-expression network analysis (WGCNA) was constructed using the WGCNA package for R (version 1.70) (Langfelder and Horvath, 2008). To improve the accuracy of the result, the low-quality data were filtered and then imported lncRNA expression values into WGCNA. The power was 6; mergeCutHeight was set as 0.2; minModuleSize was set as 50 in automatic network construction block-wise modules; the rest values were system defaults. We constructed a lncRNA cluster tree according to the relevance between the expression levels of lncRNAs and divided the modules according to the clustering relationship between them. Those lncRNAs with similar expression patterns were divided into the same modules. After the preliminary module division, the module result (Dynamic Tree Cut) of the preliminary division was obtained. Modules with similar expression patterns were combined according to the similarity of the feature values and got the final divided module Merged dynamic. To find out biologically significant modules, the correlation coefficient was calculated between module eigengenes and C. semilaevis growth traits. Pearson’s correlation between lncRNAs and trait data under the module was calculated to obtain the most relevant module (the most positive correlation and the most negative correlation) related to traits and gene significance (GS) value (GS-weight and GS-length). According to GS and weight values, the top-ranked lncRNAs were selected to draw the co-expression network by Gephi 0.9.2.
The lncRNA-miRNA-mRNA regulation network was constructed from potential functional relationships between DE lncRNAs, DE miRNAs, and DE mRNAs. Both DE lncRNAs and DE mRNAs were DE miRNAs target tags. The Spearman’s rank correlation coefficient (SCC) value assessed the expression relevance in miRNA-lncRNA and miRNA-mRNA pairs. The SCC value < −0.7 was used as the evaluation standard, and pairs that met this criterion were selected and retained. Then, the Pearson’s correlation coefficients (PCCs) between lncRNAs and mRNAs were calculated, and pairs with PCC > 0.9 were retained at the end (Chen et al., 2017). Both lncRNA and mRNA in this pair were targeted and co-expressed negatively with the same miRNA. Finally, the lncRNA-miRNA-mRNA regulation network was constructed and visualized by website tools4 and Cytoscape software (version 3.8.2).
To ensure the credibility of transcriptome sequencing data, the expression patterns of 4 lncRNAs, 4 miRNAs, and 4 mRNAs were validated by quantitative PCR (qPCR) using gonad, brain, muscle, and liver of female, male, and pseudomale C. semilaevis. PrimeScript™RT Reagent Kit (Takara, Shiga, Japan), miRNA 1st-Strand cDNA Synthesis Kit (Accurate Biology, Changsha, China), and SYBR® Green Premix HS qPCR Kit II (Accurate Biology, Changsha, China) were performed on 7500Fast RT PCR System (Applied Biosystems, United States). Gene-specific primers (Table 1) were designed using Primer 5 software. β-actin, 18s-rRNA, and 5s-rRNA were used as the internal references for determining mRNA, lncRNA, and miRNA of C. semilaevis, respectively. All the samples were made in triplicate, and the gene expression was calculated by the previously reported method (Livak and Schmittgen, 2001), and then log2 (FC)s of female/male (F/M), pseudomale/male (PM/M), and female/pseudomale (F/PM) C. semilaevis were calculated and compared with the transcriptome data to verify its reliability.
The expression results of lncRNAs, miRNAs, and mRNAs of C. semilaevis were detected by a high−throughput sequencing technique. The low-quality data were filtered by removing reads with adapters, and N sequence greater than 10%. The distribution map of data preprocessing was displayed. After filtering the data, the quality distribution and component of bases were analyzed, and the data quality is displayed in Supplementary Table 1.
A total of 21,063 genes including 20,373 known genes and 690 novel genes were identified. A total of 8,071 lncRNAs, including 1,673 known lncRNAs and 6,398 novel lncRNAs, and the specific lncRNA categories are shown in Supplementary Figure 1. A total of 2,822 miRNAs including 1,236 known miRNAs and 1,556 novel miRNAs were identified through further analysis of the transcriptome, based on the C. semilaevis genome. Also, miRNAs and lncRNAs were annotated separately using statistical methods (Supplementary Table 2).
From the basic histogram difference analysis, the DE mRNAs and DE lncRNAs were mainly concentrated in the gonad and muscle of C. semilaevis. In group MG vs. FG, the up-expressed and down-expressed DE lncRNAs were 1,290 and 2,693, respectively, and in group PMG vs. FG, the up-expressed and down-expressed DE lncRNAs were 1,310 and 2,879, respectively. In group MG vs. PMG, the up-expressed and down-expressed DE lncRNAs were 172 and 83, respectively. Pseudomale individuals were genetically closer to male individuals (Figure 1A). From the Venn diagrams, DE lncRNAs (Figure 1B) and DE miRNAs (Figure 1C) were concentrated in the gonad between different genders.
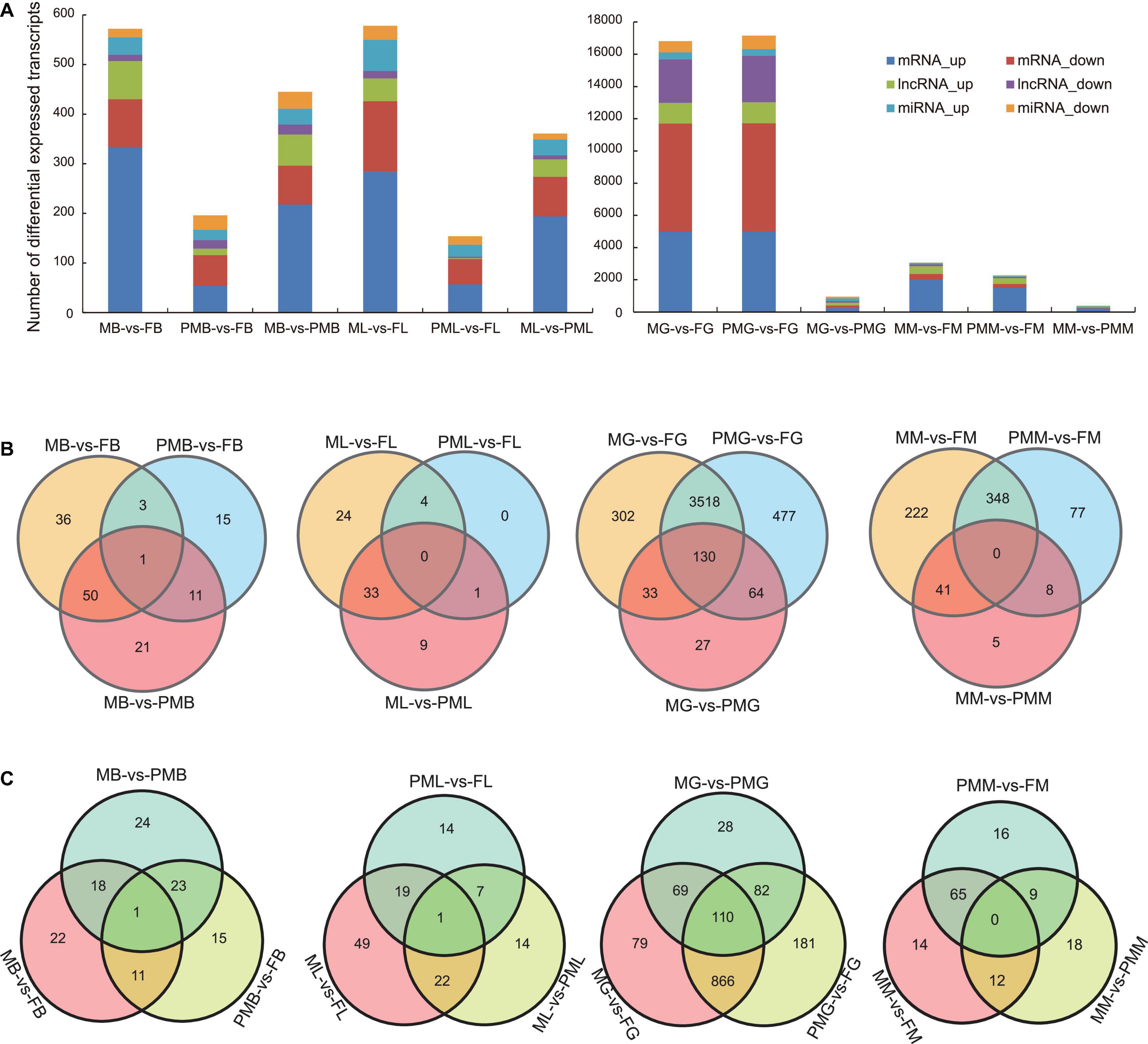
Figure 1. Identification and analysis of differentially expressed (DE) mRNAs, DE lncRNAs, and DE miRNAs. (A) Basic difference analysis histogram of DE mRNAs, DE lncRNAs, and DE miRNAs from the brain, gonad, liver, and muscle of M vs. F, M vs. PM, and PM vs. F groups. (B) Venn diagrams of DE lncRNAs. (C) Venn diagrams of DE miRNAs.
To validate the credibility of RNA high-throughput sequencing data, 4 mRNAs [neurotrophic receptor tyrosine kinase 2 (ntrk2), growth hormone receptor (ghr), zona pellucida glycoprotein 4 (zp4), and minichromosome maintenance complex component 4 (mcm4)] (Figure 2A), 4 lncRNAs (MSTRG.7061.1, MSTRG.26730.1, MSTRG.25744.1, and MSTRG.26424.1) (Figure 2B), and 4 miRNAs (novel-m0995-3p, miR-205-X, miR-455-X, and novel-m0916-5p) (Figure 2C) were selected in the brain, liver, muscle, and gonad of C. semilaevis and verified by the qPCR method. Compared with the sequencing results, the selected RNAs displayed similar upregulation and downregulation patterns.
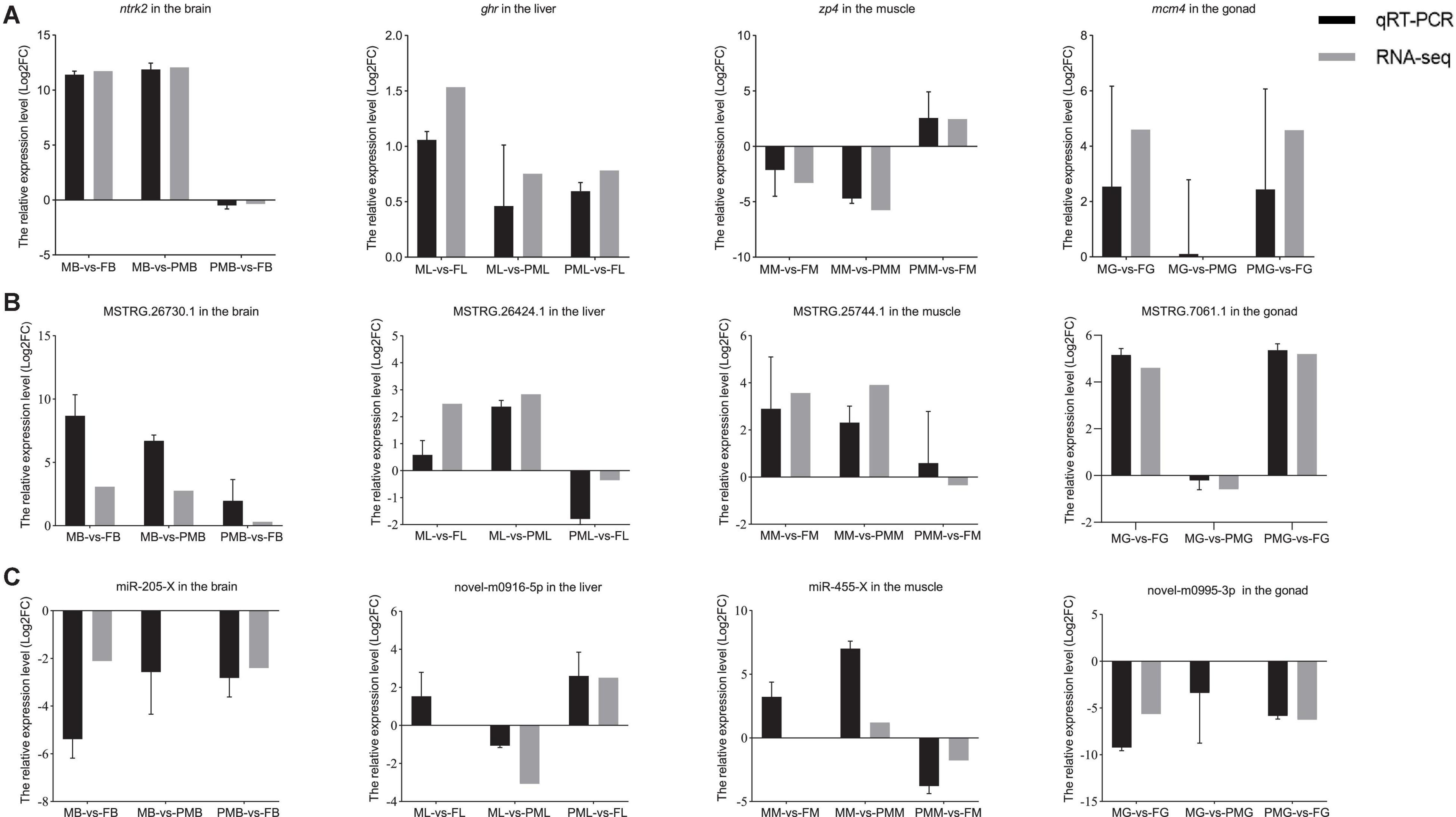
Figure 2. The quantitative PCR (qPCR) verification of mRNAs, lncRNAs, and miRNAs. (A) The relative expression levels of DE mRNAs. (B) The relative expression levels of DE lncRNAs. (C) The relative expression levels of DE miRNAs. Black and gray legends represent qPCR results and RNA-seq results, respectively. In qPCR, values are indicated as means ± SE derived from triplicate experiments.
To understand the possible function of lncRNA by targeting mRNA, three kinds of associations between lncRNA and mRNA, including antisense, cis, and trans, were predicted and analyzed.
First, 1,308 pairs of antisense lncRNA-mRNA were predicted with 1,150 DE lncRNAs and 932 mRNAs. The subsequent GO enrichment analysis revealed that protein metabolic process, homeostatic process, protein kinase activity, and so on were significantly enriched (p < 0.05). The KEGG enrichment analysis identified six pathways, namely, protein digestion and absorption, tight junction, bladder cancer, thyroid cancer, GABAergic synapse, and miRNAs in cancer (p < 0.05) (Figure 3A).
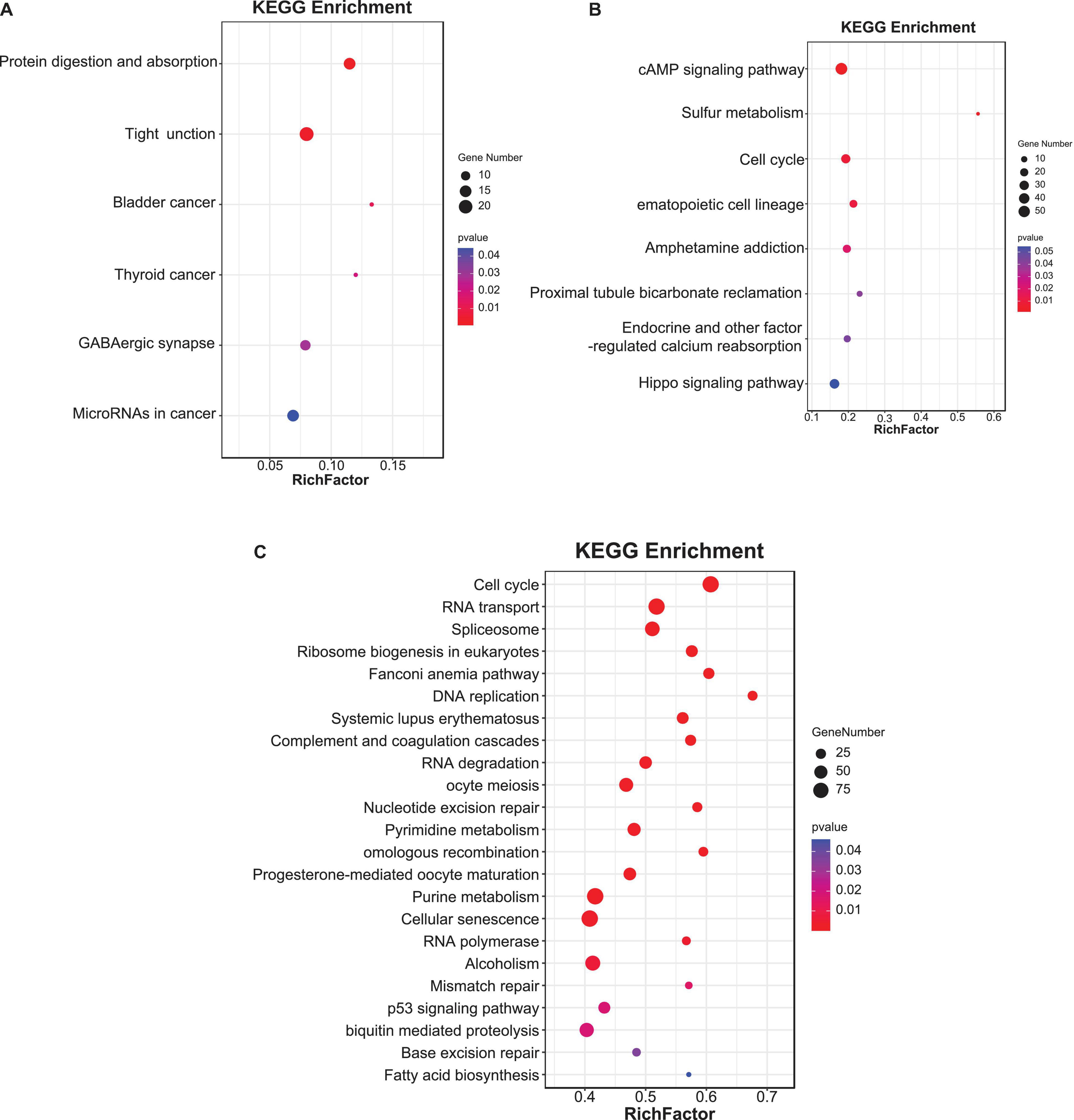
Figure 3. The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for genes involved in three kinds of lncRNA-mRNA relationships: (A) antisense-regulatory, (B) cis-regulatory, and (C) trans-regulatory. The color of the points refers to the p-value of the respective signaling pathway. The size of the point refers to the number of genes within each pathway. Same below.
Second, 4,200 cis-lncRNA-mRNA pairs were predicted to explore the function of lncRNA in local gene regulation. Notably, 2,677 DE lncRNAs and 2,752 mRNAs were contained. Based on the classification of GO term, these 2,752 mRNAs were significantly enriched to the spindle, plasma membrane part, membrane region, response to a steroid hormone, and cytoplasmic transport. These mRNAs were significantly enriched to cyclic adenosine monophosphate (cAMP) signaling pathway, sulfur metabolism, cell cycle, hematopoietic cell lineage, endocrine, and other factor-regulated calcium reabsorption (p < 0.05) (Figure 3B). The Hippo signaling pathway was also enriched with p = 0.054.
Finally, a total of 838,748 trans-lncRNA-mRNA pairs, including 3,412 lncRNAs and 7,299 mRNAs, were predicted based on the expression level analysis. In total, 7,299 mRNAs were enriched in intracellular organelle part, macromolecular complex, organelle part, small molecular binding, transferase activity, DNA metabolic process, chromosome organization, and nucleic acid metabolic process GO terms. Subsequently, 23 pathways, including cell cycle, RNA transport, spliceosome, ribosome biogenesis in eukaryotes, DNA replication, cellular senescence, and so on, were significantly enriched (p < 0.05) (Figure 3C).
After removing 150 lncRNAs with no expression in all tissues, a total of 7,921 lncRNAs were selected for the subsequent network. According to the correlation coefficients of the remaining lncRNAs, a merged dynamic tree was constructed with parameters, such as power value = 6, similarity = 0.8, and minModuleSize = 50 (Figure 4). The numbers of lncRNAs in different modules are shown in Table 2. To find the module most relevant to the growth traits and search for the hub lncRNAs, all modules were analyzed in pairs, and the lncRNAs in all modules were clustered. Meanwhile, the data of module characteristic value and growth traits (weight, length, and width) were used for correlation analysis, and the results showed that the greenyellow and blue modules had the highest positive and negative correlations with growth traits, respectively (Figure 4B). We displayed the expression pattern of each lncRNA contained in each module with heatmaps and used histograms to show the changes of the module feature value in different samples. In the blue module, the male- and pseudomale-biased expression levels of lncRNA were observed in the gonad (Supplementary Figure 2A). In the greenyellow module, the female-biased expression of lncRNA was observed in the gonad (Supplementary Figure 2B).
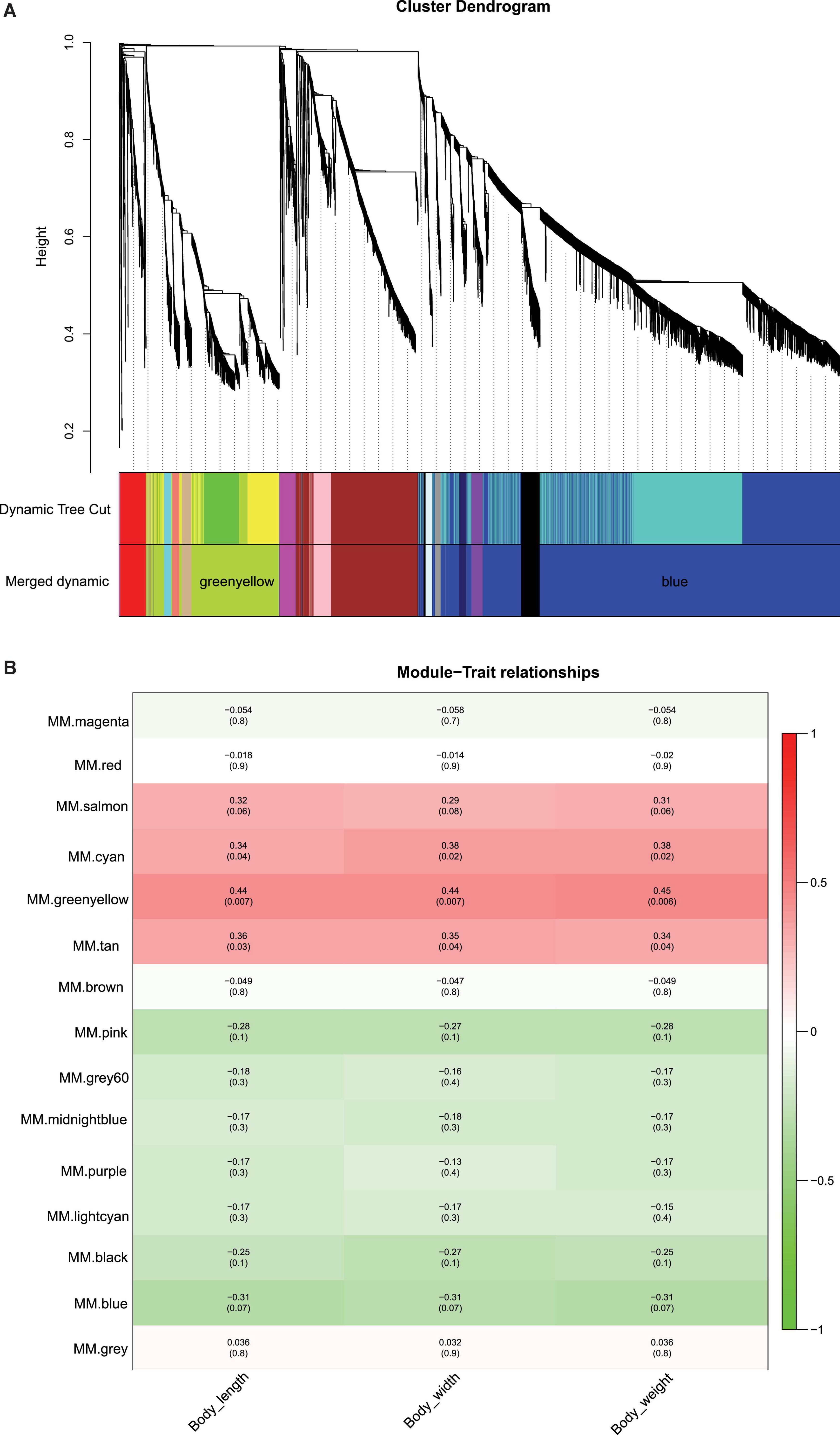
Figure 4. The weighted gene co-expression network analysis (WGCNA) for lncRNAs. (A) Cluster dendrogram for lncRNAs. The longitudinal distance represents the distance between two nodes. Dynamic Tree Cut indicates the divided modules based on the clustering result of lncRNAs. Merged dynamic indicates the divided modules by combining modules with similar expression patterns. (B) The correlation of module and growth traits. Red represents positive correlation, and green represents negative correlation, the darker the color, the stronger the correlation. The number in parentheses below represents the significant p-value; the smaller the value, the stronger the significance.

Table 2. Long non-coding RNA (lncRNA) numbers in different modules.
Finally, in the blue module, the top 202 pairs, including 59 lncRNAs with GS-weight and GS-length < −0.40 and weight value > 0.32, were selected to construct a co-expression network. Of these, 5 lncRNAs exhibited a close relationship with others, including MSTRG.9203.3, MSTRG.20720.1, MSTRG.17756.1, MSTRG.11690.1, and XR.521356 (Figure 5A). In the greenyellow module, the top 403 pairs, including 48 lncRNAs with GS-weight and GS-length > 0.48 and weight value > 0.64, were selected to construct the co-expression network. There were 5 lncRNAs (MSTRG.1489.4, MSTRG.15905.6, MSTRG.17586.2, MSTRG.7942.1, MSTRG.2826.1) that exhibited a close relationship with other lncRNAs (Figure 5B). At the same time, 59 lncRNAs and 48 lncRNAs selected in the blue and greenyellow modules were used to make heatmaps of expression. The 59 lncRNAs were mainly expressed in the male and pseudomale gonads (Figure 5C), and the 48 lncRNAs were mainly expressed in the female gonad (Figure 5D).
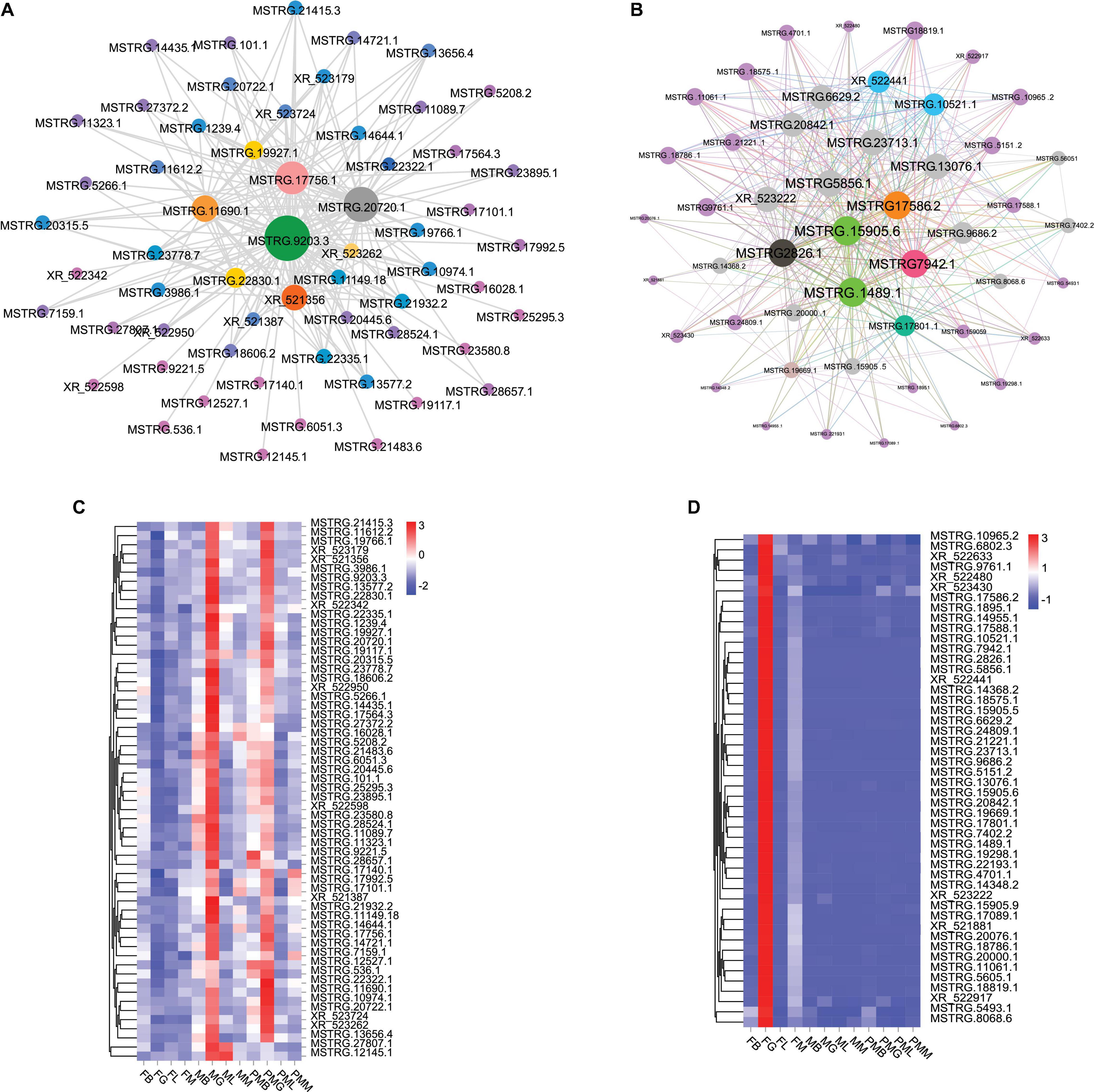
Figure 5. The lncRNAs co-expression networks of two modules. (A) The network in the blue module. (B) The network in the green-yellow module. The size of the ellipse represents the degree of relevance, a larger ellipse means a higher degree of relevance. (C) The heatmap of 59 lncRNAs in the blue module. (D) The heatmap of 48 lncRNAs in the green-yellow module. Red and blue denote high and low expression levels, respectively.
A total of 3,034 mRNA-miRNA pairs with the opposite expression patterns were found, including 657 miRNAs and 1,225 target mRNAs (Supplementary Table 3). The 1,225 target DEGs were enriched and analyzed, and specific GO terms (p < 0.05) were involved in cell components (e.g., organelle membrane), MF (e.g., catalytic activity), and BP (e.g., single-organism catabolic process) (Supplementary Figures 3A–C). Of these, pairs with SCC < −0.90 were selected to build a regulatory network (Figure 6A). Similar methods were used to construct the lncRNA-miRNA network. Also, 1,443 lncRNA-miRNA pairs were predicted, including 301 miRNAs and 555 lncRNAs. Pairs with SCC < −0.86 were selected to build a regulatory network (Figure 6B). In addition, the KEGG analysis of 1,225 target DEGs revealed that the peroxisome proliferator-activated receptor (PPAR) signaling pathway and the biosynthesis of secondary metabolism pathways were enriched with statistical significance (p < 0.05), and the top 20 pathways are shown in Figure 7A.

Figure 6. Illustration of miRNA-mRNA and lncRNA-miRNA regulatory network. (A) miRNA-mRNA regulatory network. (B) lncRNA-miRNA regulatory network. The lncRNAs, mRNAs, and miRNAs are indicated by pink vees, purple triangles, and green ellipses, respectively.
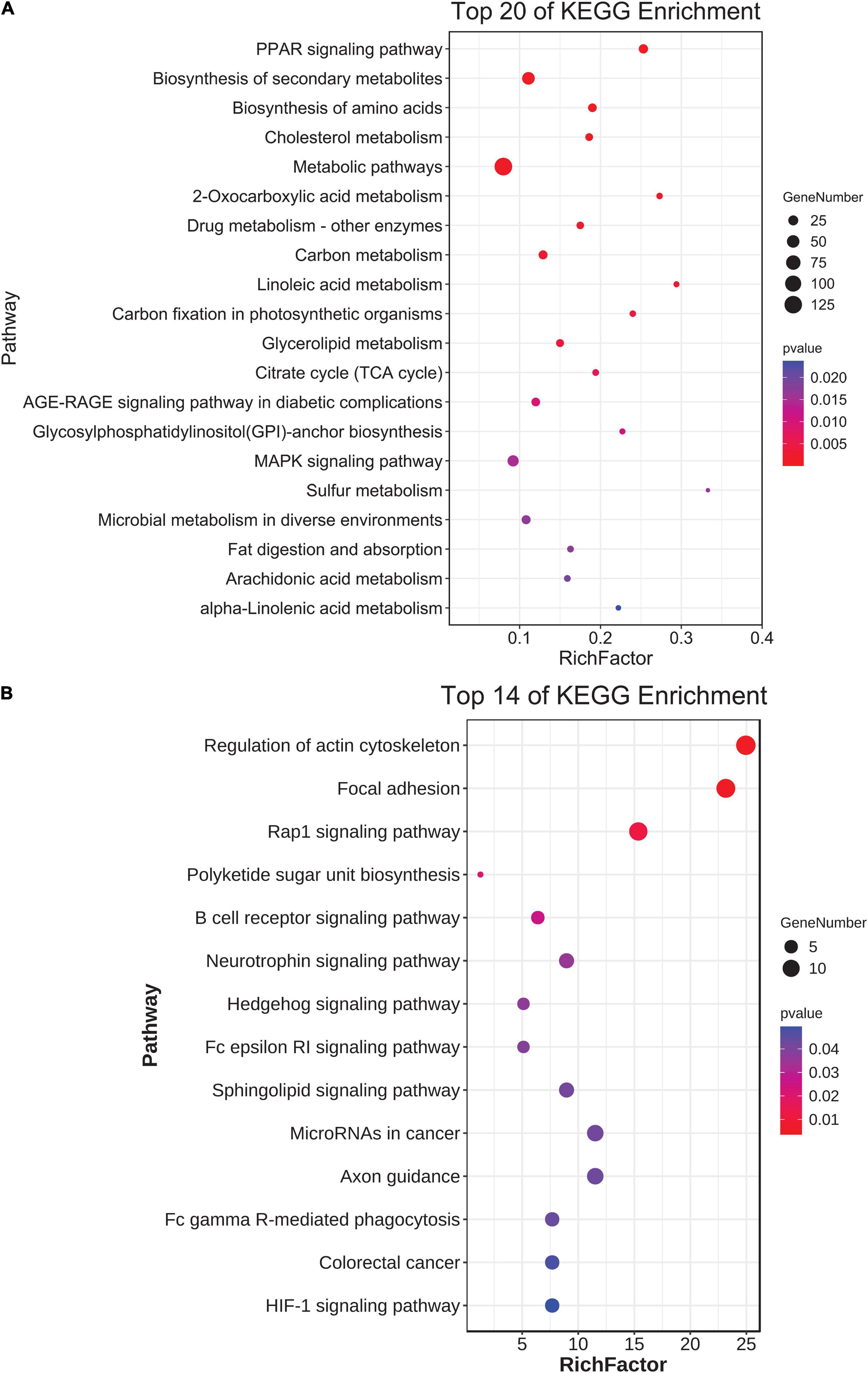
Figure 7. The KEGG analyses of the differentially expressed genes (DEGs) in the miRNA-mRNA pairs and lncRNA-miRNA-mRNA network. (A) Top 20 of KEGG enrichments of the target DEGs of miRNAs. (B) Statistics of the enrichment pathways of DEGs in the lncRNA-miRNA-mRNA network.
To show the potential lncRNA-miRNA-mRNA regulatory network, the DE lncRNAs and DE miRNAs were analyzed for co-expression. A total of 1,443 miRNA-lncRNA pairs, including 301 miRNAs and 555 lncRNAs, were generated (PCC ≤ −0.7). A total of 21,766 ceRNA pairs were generated potential pairs (PCC > 0.9), and then, the final pairs were determined by the hypergeometric cumulative distribution function test (p < 0.05). There were 3,497 ceRNA pairs, including 385 lncRNAs, 138 miRNAs, and 456 mRNAs (Supplementary Table 3).
Then, the GO analysis revealed that there were some DEGs involved in the biological roles in the SSD of C. semilaevis. Specific GO items (p < 0.05) were found in cell components (e.g., endomembrane system), MF (e.g., transmembrane receptor protein tyrosine kinase activity), and BPs (e.g., phosphorylation) (Supplementary Figures 3D–F). The KEGG analysis revealed that the top 14 pathways were significant (p < 0.05), including focal adhesion, Ras-proximate-1 (Rap1) signaling pathway, and so on (Figure 7B).
A total of 13 DEGs in cell growth and death pathway, including transferrin receptor 1b (tfr1b), diablo homolog, mitochondrial-like 1 (dhmtl1), homeodomain-interacting protein kinase 2 (hipk2), cohesin subunit SA-2-like (cssa2), phosphoinositide-3-kinase, regulatory subunit 2 (pik3r2), DNA fragmentation factor, alpha (dffa), mitogen-activated protein 3 kinase 5 (map3k5), solute carrier family 25 member 4 (slc25a4), BUB3 mitotic checkpoint protein (bub3), adenylate cyclase 9 (adcy9), cyclin-dependent kinase 6 (cdk6), cyclin-dependent kinase 2 (cdk2), and BCL2 apoptosis regulator (bcl2), were determined to be involved. According to these genes, their corresponding lncRNAs and miRNAs were found simultaneously. We constructed the Sankey diagram of the ceRNA network, including 85 lncRNA-miRNA-mRNA pairs with 78 lncRNAs, 12 miRNAs, and 13 mRNAs (Figure 8). According to the result of the lncRNA-miRNA-mRNA network, the heatmaps of the expression patterns of these lncRNAs, miRNAs, and mRNAs in different genders and tissues were drawn. The heatmaps indicated that mRNAs and lncRNAs in the same pairs showed the same pattern of expression, but miRNAs in the same pairs showed an expression pattern that was completely opposite to them (Figure 9).
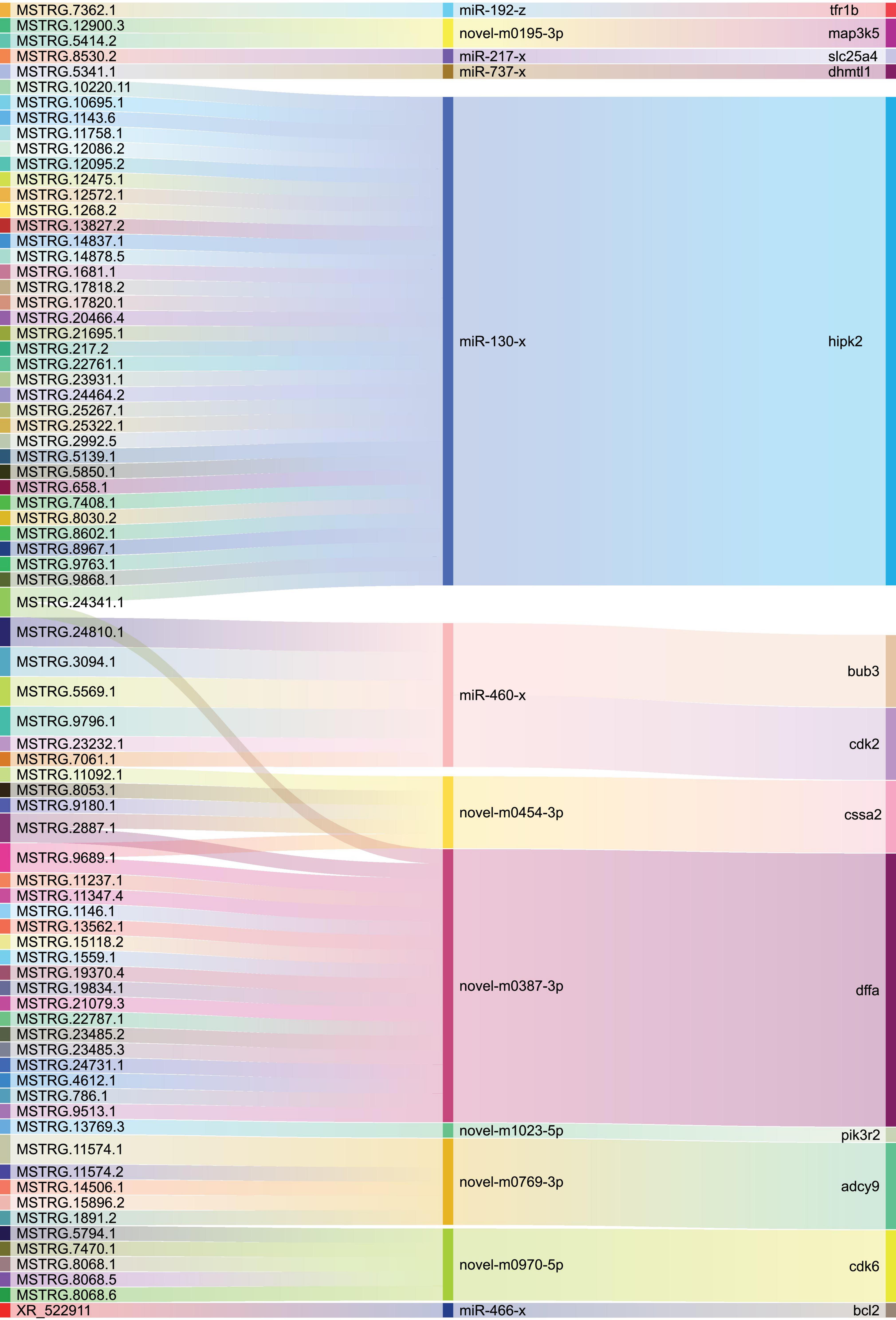
Figure 8. Functional lncRNA-miRNA-mRNA regulatory modules in cell growth and death pathway. The lncRNA, miRNA, and mRNA are on the left, middle, and right, respectively.
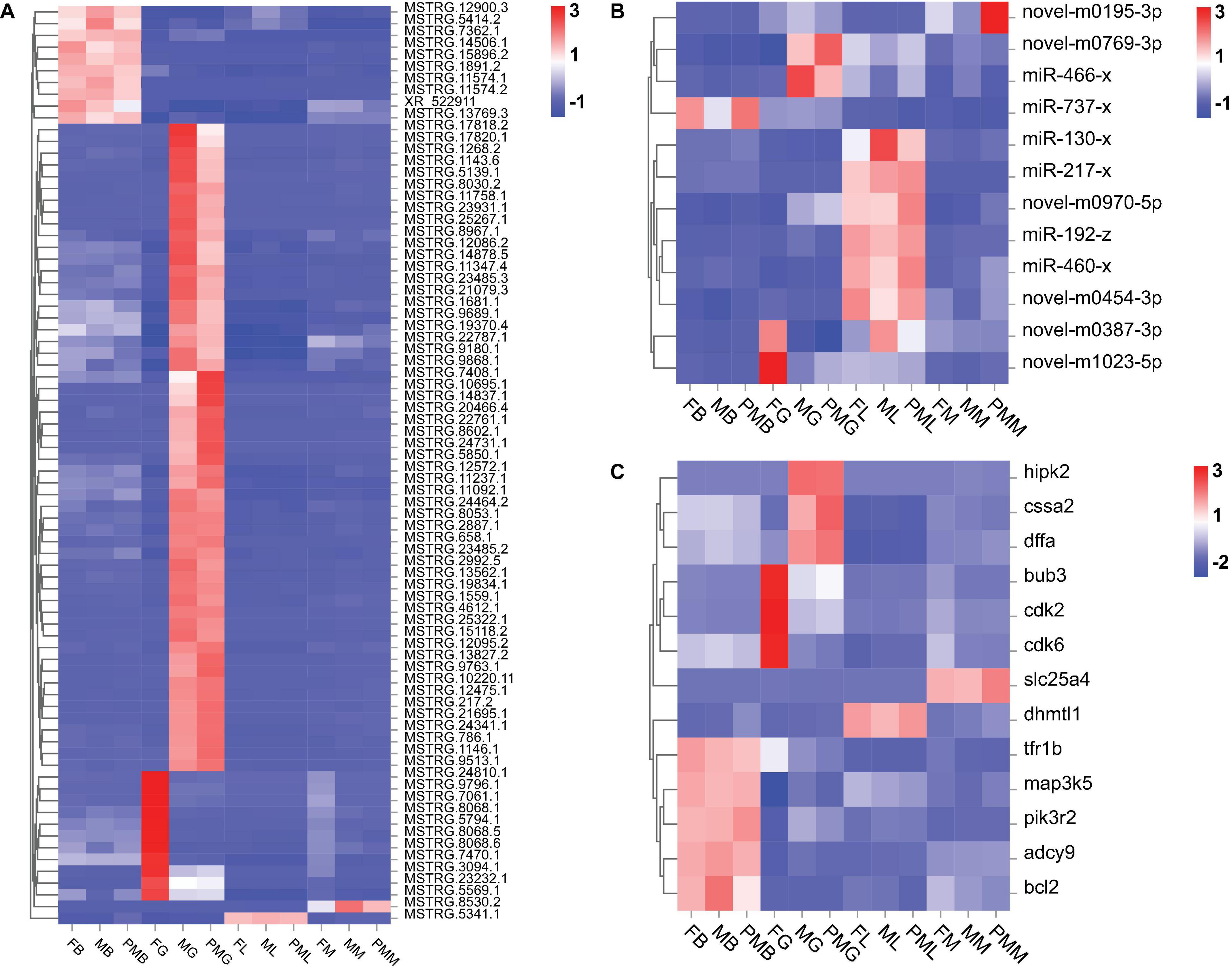
Figure 9. The expression patterns of lncRNAs, miRNAs, and mRNAs in the lncRNA-miRNA-mRNA network. (A) The heatmap of expression patterns of lncRNAs. (B) The heatmap of expression patterns of miRNAs. (C) The heatmap of expression patterns of mRNAs.
In aquaculture, growth traits have been the focus of attention. For species with SSD, such as C. semilaevis, the research on the mechanism of growth difference is important. After 9 months of sexual maturity in male individuals, there was a significant difference in weight between the sexes (Ji et al., 2011). At present, high−throughput sequencing has been widely used in marine fish. Previous C. semilaevis transcriptome sequencing revealed some growth-related pathways and genes (Wang N. et al., 2018; Wang et al., 2021). However, the study for growth-related ncRNAs identification in C. semilaevis is still limited.
The miRNAs can inhibit mRNAs transcription, and each miRNA may repress up to hundreds of transcripts (Salmena et al., 2011). In addition to regulating multiple mRNAs, each miRNA was related to multiple lncRNAs. In humans and model organisms, the mechanisms and functions of miRNA have been identified (Bartel, 2004). However, the role of miRNA in fish growth regulation is unclear. In our study, many miRNAs were related to the growth of C. semilaevis, such as miR-130-x and miR-460-x. Previous studies showed that the miR-130 family was closely related to cell migration, cell development, and the occurrence of cancer through the regulation of target genes and signaling pathways (Egawa et al., 2016; Wang et al., 2020). The miR-460 is related to growth in Nile tilapia (O. niloticus), and the expression is negatively correlated with skeletal muscle growth (Huang et al., 2012). In the lncRNA-miRNA-mRNA network, miR-460-x could regulate two genes, namely, cdk2 and bub3, which play important roles in cell proliferation and cell cycle (Choi and Anders, 2014; Li et al., 2018; Poon, 2021). It suggested that miR-460-x might be involved in cell proliferation by targeting and inhibiting its target genes.
Many studies have shown that lncRNAs can sponge miRNAs to protect target mRNAs from repression (Tay et al., 2014). In this study, miR-460-x showed low expression in the female gonad; however, its target genes (cdk2 and bub3) and lncRNAs (MSTRG.24810.1, MSTRG.3094.1, MSTRG.5569.1, MSTRG.9796.1, MSTRG.23232.1, and MSTRG.7061.1) exhibited high expression in the female gonad, suggesting that these lncRNAs might be implicated in the competition between miRNA and genes to regulate SSD. Similarly, as a female-biased expression of miRNA in the gonad, novel-m0387-3p targeted gene dffa and lncRNAs all showed high expression in the male and pseudomale gonads. Notably, dffa, a classic negative growth regulation gene, has been reported to trigger DNA fragmentation during apoptosis (Wu et al., 2014). Moreover, the results of high-throughput sequencing showed that intergenic lncRNAs occupied 39.5% in all DE lncRNAs, antisense lncRNAs occupied 35.5%, and the smallest proportion was intronic lncRNAs, which is only 2.45% of total DE lncRNAs. It indicated that most DE lncRNAs were located in the exons and intergenic region. In the study of lncRNA-mRNA associations, only a few lncRNAs were defined as antisense and cis-regulators, and most annotated lncRNAs were defined as trans-regulators. It showed that lncRNAs directly activated the transcription of adjacent mRNAs, which was relatively rare in lncRNA-mRNA regulation (Wang and Chang, 2011). Regulating chromatin states and gene expression at regions distant from their transcription site, influencing nuclear structure and organization, and interacting with other RNA molecules are the main regulation methods (Kopp and Mendell, 2018). Previous lncRNA regulation research in Koi carp (Cyprinus carpio L.) has also proven this mechanism (Luo et al., 2019). Herein, a few of hub lncRNAs were identified (MSTRG.9203.3, MSTRG.20720.1, MSTRG.11690.1, MSTRG.17756.1, XR521356, MSTRG.15905.6, MSTRG.1489.1, MSTRG.17586.2, MSTRG.7942.1, and MSTRG.2826.1). They had higher weight in DE lncRNAs of male and female gonads of C. semilaevis, indicating that there was a close relationship between these hub lncRNAs and growth, gonadal differentiation, or SSD.
We sorted out the three relationships between lncRNAs and target mRNAs; of these, the KEGG enrichment analysis presented that there were many overlapping pathways in the associations of cis and trans, such as the cell cycle pathway. It indicated that whether in cis-regulation or trans-regulation, the effects of the cell cycle were all remarkable. The cell cycle contains a series of complex processes, such as genome replication, growth, and production of two daughter cells (Schafer, 1998). The integration of transcriptome and methylome based on the same individuals as the present study, have highlighted that the activation of the cell cycle was implicated in C. semilaevis female-biased SSD (Wang et al., 2021). In the network of lncRNA-miRNA-mRNA, two cell cycle-related genes (cdk2 and cdk6) were also identified, which further implied that these ncRNAs might be involved in the growth regulation by interacting with the cell cycle. In addition, there were many signaling pathways identified in this study. Among them, cAMP signaling pathway, Rap1 signaling pathway, and hippo signaling pathway caught our attention. The cAMP, as a second messenger, is an extremely important signaling molecule in the cell. A previous study showed that cAMP was involved in growth regulation in mouse fibroblasts and exhibited rhythmic changes during the cell cycle (Seifert and Rudland, 1974). Rap1 is a small guanosine triphosphate (GTP)-binding protein involved in intracellular signaling, cell structure, and the development of rat dendrites (Chen et al., 2005). In addition, there was a close connection between cAMP signaling pathway and Rap1 signaling pathway. Rap1 couples extracellular stimulation with intracellular signaling through cAMP, diacylglycerol (DAG), and calcium ion and controls the important cellular functions, including shaping neurons (Kosuru and Chrzanowska, 2020). The hippo signaling pathway is a typical pathway that inhibits cell growth. Relevant results of the research in the past decade have shown that the hippo signaling pathway can regulate the size of organs by regulating cell proliferation, apoptosis, and stem cell self-renewal capabilities (Pan, 2010; Zhao et al., 2011). Herein, the hippo signaling pathway was identified in lncRNA-mRNA cis-regulation. However, the detailed mechanism of ncRNA implicated in these growth-related pathways deserves further exploration.
This study provided lncRNA-miRNA-mRNA network for exploring C. semilaevis female-biased SSD for the first time. However, key miRNAs and lncRNAs predicted by the bioinformatics analysis were not verified in the growth and gender differences of C. semilaevis, which should be studied in our subsequent study.
The datasets presented in this study can be found in the NCBI SRA repository with accession number PRJNA743138 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA743138/).
The animal study was reviewed and approved by the Animal Care and Use Committee at the Chinese Academy of Fishery Sciences. Written informed consent was obtained from the owners for the participation of their animals in this study.
NW and SC conceived the experiments. NW, WX, YY, and JW performed fish tissue sampling. NW, JW, and QY conducted qPCR validation for ncRNAs and genes. JW and NW analyzed the data and wrote the manuscript. YH participated in the transcriptomic analysis. All authors read and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (31730099 and 31873037), the National Key R&D Program of China (2018YFD0900205), the Central Public-Interest Scientific Institution Basal Research Fund, the CAFS (2020TD20), and the Taishan Scholar Project of Shandong Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to Guangzhou Genedenovo Biotechnology Co., Ltd., for assisting in sequencing and bioinformatics analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.795525/full#supplementary-material
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/s0092-8674(04)00045-5
Belhaoues, F., Breit, S., Forstenpointner, G., and Gardeisen, A. (2020). Sexual dimorphism in limb long bones of the german shepherd dog. Anat. Histol. Embryol. 49, 464–477. doi: 10.1111/ahe.12550
Cai, J., Li, L., Song, L., Xie, L., Luo, F., Sun, S., et al. (2019). Effects of long term antiprogestine mifepristone (RU486) exposure on sexually dimorphic lncRNA expression and gonadal masculinization in nile tilapia (Oreochromis niloticus). Aquat. Toxicol. 215:105289. doi: 10.1016/j.aquatox.2019.105289
Chen, I. P., Stuart-Fox, D., Hugall, A. F., and Symonds, M. R. (2012). Sexual selection and the evolution of complex color patterns in dragon lizards. Evolution 66, 3605–3614. doi: 10.1111/j.1558-5646.2012.01698.x
Chen, S., Zhang, G., Shao, C., Huang, Q., Liu, G., Zhang, P., et al. (2014). Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 46, 253–260. doi: 10.1038/ng.2890
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chen, S. L., Tian, Y. S., Yang, J. F., Shao, C. W., Ji, X. S., Zhai, J. M., et al. (2009). Artificial gynogenesis and sex determination in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. 11, 243–251. doi: 10.1007/s10126-008-9139-0
Chen, X., Ma, C., Chen, C., Lu, Q., Shi, W., Liu, Z., et al. (2017). Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees. PeerJ 5:e3881. doi: 10.7717/peerj.3881
Chen, Y., Wang, P. Y., and Ghosh, A. (2005). Regulation of cortical dendrite development by Rap1 signaling. Mol. Cell. Neurosci. 28, 215–228. doi: 10.1016/j.mcn.2004.08.012
Choi, Y. J., and Anders, L. (2014). Signaling through cyclin D-dependent kinases. Oncogene 33, 1890–1903. doi: 10.1038/onc.2013.137
Dong, X., Chen, S., and Xiangshan, J. I. (2011). Molecular cloning, characterization and expression analysis of sox9a and foxl2 genes in half-smooth tongue sole(Cynoglossussemilaevis). Acta Oceanol. Sin. 12, 57–59.
Dong, Y., Lyu, L., Zhang, D., Li, J., Wen, H., and Shi, B. (2021). Integrated lncRNA and mRNA transcriptome analyses in the ovary of cynoglossus semilaevis reveal genes and pathways potentially involved in reproduction. Front. Genet. 12:671729. doi: 10.3389/fgene.2021.671729
Egawa, H., Jingushi, K., Hirono, T., Ueda, Y., Kitae, K., Nakata, W., et al. (2016). The miR-130 family promotes cell migration and invasion in bladder cancer through FAK and Akt phosphorylation by regulating PTEN. Sci. Rep. 6:20574. doi: 10.1038/srep20574
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Gan, S., Huang, Z., Liu, N., Su, R., Xie, G., Zhong, B., et al. (2016). MicroRNA-140-5p impairs zebrafish embryonic bone development via targeting BMP-2. FEBS Lett. 590, 1438–1446. doi: 10.1002/1873-3468.12190
Gao, Z., Fu, P., Yu, Z., Zhen, F., and Gu, Y. (2019). Comprehensive analysis of lncRNA-miRNA- mRNA network ascertains prognostic factors in patients with colon cancer. Technol. Cancer Res. Treat. 18:1533033819853237. doi: 10.1177/1533033819853237
Gerritsen, H. D., Armstrong, M. J., Allen, M., Mccurdy, W. J., and Peel, J. (2003). Variability in maturity and growth in a heavily exploited stock: cod (Gadus morhua L.) in the Irish Sea. J. Sea Res. 49, 69–82.
Ghasemi, T., Khalaj-Kondori, M., Hosseinpour Feizi, M. A., and Asadi, P. (2020). LncRNA-miRNA-mRNA interaction network for colorectal cancer; an in silico analysis. Comput. Biol. Chem. 89:107370. doi: 10.1016/j.compbiolchem.2020.107370
Hobert, O. (2008). Gene regulation by transcription factors and microRNAs. Science 319, 1785–1786. doi: 10.1126/science.1151651
Holmes, D. S., Mayfield, J. E., Sander, G., and Bonner, J. (1972). Chromosomal RNA: its properties. Science 177, 72–74. doi: 10.1126/science.177.4043.72
Huang, C. W., Li, Y. H., Hu, S. Y., Chi, J. R., Lin, G. H., Lin, C. C., et al. (2012). Differential expression patterns of growth-related microRNAs in the skeletal muscle of nile tilapia (Oreochromis niloticus). J. Anim. Sci. 90, 4266–4279. doi: 10.2527/jas.2012-5142
Huang, M., Long, Y., Jin, Y., Ya, W., Meng, D., Qin, T., et al. (2021). Comprehensive analysis of the lncRNA-miRNA-mRNA regulatory network for bladder cancer. Transl. Androl. Urol. 10, 1286–1301. doi: 10.21037/tau-21-81
Huang, Y. (2018). The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J. Cell. Mol. Med. 22, 5768–5775. doi: 10.1111/jcmm.13866
Ji, X. S., Liu, H. W., Chen, S. L., Jiang, Y. L., and Tian, Y. S. (2011). Growth differences and dimorphic expression of growth hormone (GH) in female and male Cynoglossus semilaevis after male sexual maturation. Mar. Genomics 4, 9–16. doi: 10.1016/j.margen.2010.11.002
Kong, L., Zhang, Y., Ye, Z. Q., Liu, X. Q., Zhao, S. Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Kopp, F., and Mendell, J. T. (2018). Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407. doi: 10.1016/j.cell.2018.01.011
Kosuru, R., and Chrzanowska, M. (2020). Integration of Rap1 and Calcium signaling. Int. J. Mol. Sci. 21, 11–13. doi: 10.3390/ijms21051616
Kuntner, M., and Coddington, J. A. (2020). Sexual size dimorphism: evolution and perils of extreme phenotypes in spiders. Annu. Rev. Entomol. 65, 57–80. doi: 10.1146/annurev-ento-011019-025032
Lan, T., Chen, Y. L., Gul, Y., Zhao, B. W., and Gao, Z. X. (2019). Comparative expression analysis of let-7 microRNAs during ovary development in Megalobrama amblycephala. Fish Physiol. Biochem. 45, 1101–1115. doi: 10.1007/s10695-019-00624-7
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Laurence, G. C. (1976). Effects of temperature and salinity on comparative embryo development and mortality of Atlantic cod (Gadus morhua L.) and haddock (Melanogrammus aeglefinus (L.)). ICES J. Mar. Sci. 36, 220–228.
Li, F., Kim, H., Ji, Z., Zhang, T., Chen, B., Ge, Y., et al. (2018). The BUB3-BUB1 complex promotes telomere DNA replication. Mol. Cell 70, 395–407.e4. doi: 10.1016/j.molcel.2018.03.032
Li, H., Xu, W., Zhang, N., Shao, C., Zhu, Y., Dong, Z., et al. (2016). Two figla homologues have disparate functions during sex differentiation in half-smooth tongue sole (Cynoglossus semilaevis). Sci. Rep. 6:28219.
Liu, S., Wang, Z., Chen, D., Zhang, B., Tian, R. R., Wu, J., et al. (2017). Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res. 27, 1608–1620. doi: 10.1101/gr.217463.116
Liu, Y., Chen, S., Gao, F., Meng, L., Hu, Q., Song, W., et al. (2014). SCAR-transformation of sex-specific SSR marker and Its application in half-smooth tongue sole(Cynoglossus semiliaevis). J. Agric. Biotechnol. 22, 787–792.
Liu, Y., Zhang, R., and Ying, K. (2015). Long noncoding RNAs: novel links in respiratory diseases (review). Mol. Med. Rep. 11, 11–15.
Liu, Z., Xu, S., Dao, J., Gan, Z., and Zeng, X. (2020). Differential expression of lncRNA/miRNA/mRNA and their related functional networks during the osteogenic/odontogenic differentiation of dental pulp stem cells. J. Cell. Physiol. 235, 3350–3361. doi: 10.1002/jcp.29223
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, M., Wang, L., Yin, H., Zhu, W., Fu, J., and Dong, Z. (2019). Integrated analysis of long non-coding RNA and mRNA expression in different colored skin of koi carp. BMC Genomics 20:515. doi: 10.1186/s12864-019-5894-8
Mei, J., and Gui, J. F. (2015). Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci. China Life Sci. 58, 124–136. doi: 10.1007/s11427-014-4797-9
Milić, D., and Kraljević, M. (2011). Biometry analysis of the whiting, Merlangius merlangus (Linneaus, 1758) from the northern Adriatic Sea. Acta Adriat. 1, 60–63.
O’Mara, M. T., Gordon, A. D., Catlett, K. K., Terranova, C. J., and Schwartz, G. T. (2012). Growth and the development of sexual size dimorphism in lorises and galagos. Am. J. Phys. Anthropol. 147, 11–20. doi: 10.1002/ajpa.21600
Pan, D. (2010). The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505. doi: 10.1016/j.devcel.2010.09.011
Parker, G. A. (1992). The evolution of sexual size dimorphism in fish. J. Fish Biol. 41, 1–20. doi: 10.1111/j.1095-8649.1992.tb03864.x
Poon, R. Y. C. (2021). Cell cycle control: a system of interlinking oscillators. Methods Mol. Biol. 2329, 1–18. doi: 10.1007/978-1-0716-1538-6_1
Qian, M., Liu, S. F., Zhuang, Z. M., Lin, L., Sun, Z. Z., Liu, C. L., et al. (2012). Genomic structure, polymorphism and expression analysis of the growth hormone (GH) gene in female and male half-smooth tongue sole (Cynoglossus semilaevis). Gene 493, 92–104.
Ryu, J. W., Jung, J., Park, K., Lee, S., Park, I., Kim, W. J., et al. (2020). Characterization of sexual size dimorphism and sex-biased genes expression profile in the olive flounder. Mol. Biol. Rep. 47, 8317–8324. doi: 10.1007/s11033-020-05843-3
Salmena, L., Poliseno, L., Tay, Y., Kats, L., and Pandolfi, P. P. (2011). A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146, 353–358. doi: 10.1016/j.cell.2011.07.014
Schafer, K. A. (1998). The cell cycle: a review. Vet. Pathol. 35, 461–478. doi: 10.1177/030098589803500601
Seifert, W., and Rudland, P. S. (1974). Cyclic nucleotides and growth control in cultured mouse cells: correlation of changes in intracellular 3’:5’ cGMP concentration with a specific phase of the cell cycle. Proc. Natl. Acad. Sci. U. S. A. 71, 4920–4924. doi: 10.1073/pnas.71.12.4920
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41:e166. doi: 10.1093/nar/gkt646
Tay, Y., Rinn, J., and Pandolfi, P. P. (2014). The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–352. doi: 10.1038/nature12986
Tracy, C. B., Nguyen, J., Abraham, R., and Shirangi, T. R. (2020). Evolution of sexual size dimorphism in the wing musculature of Drosophila. PeerJ 8:e8360. doi: 10.7717/peerj.8360
Wan, Z. Y., Lin, V. C. L., and Hua, Y. G. (2021). Pomc plays an important role in sexual size dimorphism in tilapia. Mar. Biotechnol. 23, 201–214. doi: 10.1007/s10126-020-10015-2
Wang, J., Zhao, L., Peng, X., Liu, K., Zhang, C., Chen, X., et al. (2020). Evaluation of miR-130 family members as circulating biomarkers for the diagnosis of bladder cancer. J. Clin. Lab. Anal. 34:e23517. doi: 10.1002/jcla.23517
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Wang, M., Jiang, S., Wu, W., Yu, F., Chang, W., Li, P., et al. (2018). Non-coding RNAs function as immune regulators in teleost fish. Front. Immunol. 9:2801. doi: 10.3389/fimmu.2018.02801
Wang, N., Wang, R., Wang, R., and Chen, S. (2018). Transcriptomics analysis revealing candidate networks and genes for the body size sexual dimorphism of chinese tongue sole (Cynoglossus semilaevis). Funct. Integr. Genomics 18, 327–339. doi: 10.1007/s10142-018-0595-y
Wang, N., Yang, Q., Wang, J., Shi, R., Li, M., Gao, J., et al. (2021). Integration of transcriptome and methylome highlights the roles of cell cycle and hippo signaling pathway in flatfish sexual size dimorphism. Front. Cell Dev. Biol. 9:743722. doi: 10.3389/fcell.2021.743722
Wang, P., Zheng, M., Liu, J., Liu, Y., Lu, J., and Sun, X. (2016). Sexually dimorphic gene expression associated with growth and reproduction of tongue sole (Cynoglossus semilaevis) revealed by brain transcriptome analysis. Int. J. Mol. Sci. 17:1402. doi: 10.3390/ijms17091402
Wei, S., Chen, Y., Huang, L., Ma, H., Qi, L., Wang, Q., et al. (2021). Analysis of lncRNA and mRNA expression profiles in peripheral blood leukocytes of the half-smooth tongue sole (Cynoglossus semilaevis) treated with chitosan oligosaccharide. Dev. Comp. Immunol. 120:104043. doi: 10.1016/j.dci.2021.104043
Wen, M., Shen, Y., Shi, S., and Tang, T. (2012). MiREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13:140. doi: 10.1186/1471-2105-13-140
Wu, X., Lee, E. M., Hammack, C., Robotham, J. M., Basu, M., Lang, J., et al. (2014). Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J. Virol. 88, 8433–8444. doi: 10.1128/JVI.00081-14
Xiu, Y., Jiang, G., Zhou, S., Diao, J., Liu, H., Su, B., et al. (2019). Identification of potential immune-related circRNA-miRNA-mRNA regulatory network in intestine of paralichthys olivaceus during edwardsiella tarda infection. Front. Genet. 10:731. doi: 10.3389/fgene.2019.00731
Xu, H., Cao, L., Sun, B., Wei, Y., and Liang, M. (2019). Transcriptomic analysis of potential “lncRNA-mRNA” interactions in liver of the marine teleost Cynoglossus semilaevis fed diets with different DHA/EPA ratios. Front. Physiol. 10:331. doi: 10.3389/fphys.2019.00331
Yang, G., Lu, X., and Yuan, L. (2014). LncRNA: a link between RNA and cancer. Biochim. Biophys. Acta 11, 538–542.
Yuan, W., Jiang, S., Sun, D., Wu, Z., Wei, C., Dai, C., et al. (2019). Transcriptome profiling analysis of sex-based differentially expressed mRNAs and lncRNAs in the brains of mature zebrafish (Danio rerio). BMC Genomics 20:830. doi: 10.1186/s12864-019-6197-9
Zhang, G., Yin, S., Mao, J., Liang, F., Zhao, C., Li, P., et al. (2016). Integrated analysis of mRNA-seq and miRNA-seq in the liver of Pelteobagrus vachelli in response to hypoxia. Sci. Rep. 6:22907. doi: 10.1038/srep22907
Zhao, B., Tumaneng, K., and Guan, K. L. (2011). The hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883. doi: 10.1038/ncb2303
Zhao, B. W., Zhou, L. F., Liu, Y. L., Wan, S. M., and Gao, Z. X. (2017). Evolution of fish let-7 microRNAs and their expression correlated to growth development in blunt snout bream. Int. J. Mol. Sci. 18:646.
Zhou, J., Zhao, H., Zhang, L., Liu, C., Feng, S., Ma, J., et al. (2019). Integrated analysis of RNA-seq and microRNA-seq depicts miRNA-mRNA networks involved in stripe patterns of Botia superciliaris skin. Funct. Integr. Genomics 19, 827–838. doi: 10.1007/s10142-019-00683-2
Keywords: Cynoglossus semilaevis, sexual size dimorphism, lncRNA, miRNA, mRNA, lncRNAs co-expression network, lncRNA-miRNA-mRNA network
Citation: Wang J, Yang Q, Hu Y, Xu W, Yang Y, Chen S and Wang N (2022) Identification of lncRNA-miRNA-mRNA Network Involved in Sexual Size Dimorphism of Chinese Tongue Sole (Cynoglossus semilaevis). Front. Mar. Sci. 9:795525. doi: 10.3389/fmars.2022.795525
Received: 15 October 2021; Accepted: 03 January 2022;
Published: 24 February 2022.
Edited by:
Zhitao Qi, Yancheng Institute of Technology, ChinaReviewed by:
Jie Mei, Huazhong Agricultural University, ChinaCopyright © 2022 Wang, Yang, Hu, Xu, Yang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Wang, d2FuZ25hQHlzZnJpLmFjLmNu; Songlin Chen, Y2hlbnNsQHlzZnJpLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.