 Teddy Namirimu
Teddy Namirimu Yun Jae Kim
Yun Jae Kim Mi-Jeong Park
Mi-Jeong Park Dhongil Lim
Dhongil Lim Jung-Hyun Lee
Jung-Hyun Lee Kae Kyoung Kwon
Kae Kyoung Kwon- 1Marine Biotechnology Research Center, Korea Institute of Ocean Science and Technology, Busan, South Korea
- 2Major of Ocean Science, University of Science and Technology, Daejeon, South Korea
- 3Library of Marine Samples, Korea Institute of Ocean Science and Technology, Busan, South Korea
Little is known about the community structure and metabolic potential of microbial communities in hydrothermal fields in the Central Indian Ridge (CIR). In this study, a metagenomic sequencing approach was conducted to explore the microbial diversity in three sediment samples collected during the 2019 expedition from two recently discovered hydrothermal vent fields; Invent E and Onnuri Vent Field (OVF). Analysis of unassembled metagenomic reads using the Metagenomic analysis server (MG-RAST) revealed that microbial communities of the two sampling sites were very similar, showing the dominance of Bacteria over Archaea. Proteobacteria, Firmicutes, Bacteroidetes, as well as Euryarchaeota were dominant in all samples. Functional annotation based on KEGG categories shows that the microbial populations in these vent fields possess metabolic capabilities for aerobic respiration, carbon fixation through the Calvin–Bassham–Benson (CBB) cycle, the reverse tricarboxylic acid (rTCA) cycle, and reductive acetyl-CoA pathway as well as sulfur and nitrogen metabolisms. Comparative metagenome analysis with different datasets obtained from different ocean ridges showed that microbial communities at low activity or hydrothermally influenced area differ from highly active hydrothermal communities. This study provides insights into the genetic diversity and functional capability of the microbial communities of slow to intermediate spreading hydrothermal systems.
Introduction
Deep-sea hydrothermal vents contain extreme thermal and chemical gradients, yet they harbor the most unique and diverse habitats for various microorganisms (Zeng et al., 2021). As seawater percolates into the ocean crust, volcanic water-rock reactions lead to hot, reduced metal-rich hydrothermal fluids venting from the seafloor. Biomass production in vent ecosystems is mainly fueled by oxidation of reduced compounds in hydrothermal fluids to fix carbon primarily by chemolithoautotrophic microorganisms (Fisher et al., 2007; Nakagawa and Takai, 2008) and consequently, they provide a source of nutrition for the higher organisms. In addition, many microbial populations play a crucial role in the global biogeochemical cycling of methane, nitrogen, sulfur, and carbon in these unique ecosystems (Zeng et al., 2021).
The discovery of deep hydrothermal vent fields in the 1970s (Francheteau et al., 1979) has attracted great attention to scientists with over 700 vent fields discovered and investigated along mid-ocean ridges, volcanic arcs, and tectonic settings (Dick, 2019; Beaulieu and Szafrański, 2020). However, until recently, majority of the hydrothermal system discoveries and investigation have exclusively focused on the Pacific and Atlantic Oceans (Nakamura and Takai, 2015), thus, the Indian Ocean hydrothermal systems remain understudied. To date a limited number of hydrothermal fields have been identified in the Indian Ocean including the MESO Mineral Zone (Halbach et al., 1998), Kairei (Gamo et al., 2001), Edmond (Van Dover et al., 2001), Solitaire, Dodo (Nakamura et al., 2012), and Onnuri fields (Kim et al., 2020) in the Central Indian Ridge (CIR), Daxi and Wocan fields in the Carlsberg Ridge (Wang et al., 2017, 2020), Mount Jourdanne (Münch et al., 2001), Yuhuang-1 (Liao et al., 2018), Longqi (Tao et al., 2012), Duanqiao (Yang et al., 2017), and Tiancheng (Zhou et al., 2018) in the South West Indian Ridge (SWIR), and Pelagia vent field (Han et al., 2018) in the South East Indian Ridge (SEIR).
Despite their inaccessible locations, recent advances in sampling equipment and NGS approaches have revealed that the geochemical and biological communities of the Indian Ocean hydrothermal vent systems are quite diverse (Cao et al., 2014; Ding et al., 2017; Yang et al., 2020). Using an integrated approach, the predominance of formate- and CO-utilizing bacteria among others was observed in the serpentinite-hosted Old City hydrothermal field in SWIR (Lecoeuvre et al., 2021). A previous 16S rRNA analysis of microbial communities at a black smoker in Dodo field showed that it is dominated by Bacteria with dominance of Gammaproteobacteria, Alphaproteobacteria, and Bacteroidetes (Kawagucci et al., 2016). In the high-temperature Edmond sulfide chimney, representative taxa included Epsilonproteobacteria and Aquificales as well as archaeal genera Aciduliprofundus and Thermococcus (Hoek et al., 2003) whereas H2-enriched fluids from Kairei field were dominated by (hyper)thermophilic hydrogenotrophic methanogens (Takai et al., 2004). These chemolithotrophic microbial communities involved in hydrogenotrophic, thiotrophic, and methanotrophic metabolisms have been reported to mediate biogeochemical processes in deep-sea hydrothermal systems (Nakamura and Takai, 2014).
Previous studies in the CIR listed above have solely relied on 16S rRNA gene libraries, however, 16S rRNA gene surveys offer limited information on understanding biological patterns of microbial communities within an environment. In this study, we describe the microbial structure and functional diversity in hydrothermally influenced sediments from the newly discovered vent fields in the CIR. This is the first study using metagenomic approach, to report in detail the metabolic potential of microbial communities from hydrothermal vent fields in the CIR. Comparative metagenomic analysis of hydrothermally influenced and hydrothermal samples from different locations and/or with different geochemistry would give us a better understanding of the metabolic potential of microbial communities inhabiting slow to intermediate spreading hydrothermal systems in the Indian Ocean.
Materials and Methods
Study Site and Sample Collection
The Onnuri vent field (OVF) (Kim et al., 2020) is an ultramafic-hosted active vent located more than ∼800 km north of previously known active vent fields (i.e., Dodo and Solitaire fields) (Son et al., 2014). It is located about 11 km off axis, 1,990–2,170 m deep, characterized with small chimneys, and low-temperature clear fluid effusing from rock fissures supporting a wide range of vent fauna. The OVF vent fluids are enriched in CH4 (52.5 nM max) and low metal concentrations (Kim et al., 2020). In contrast, segment 1 (Invent B) appears to be a magmatic basaltic-hosted system with several inactive vent chimneys structures. Diffuse venting was observed characterized with high temperature, high concentration of Fe (366 nM max), Mn (46 nM max), and dissolved methane (13.0 nM max) (Kim et al., 2020). Invent E sediments geochemical compositions included Fe2O3 (45.6%), MnO (7.8%), CaO (1.4%), TN (313 μg/g), TOC (1865 μg/g), and TS (1374 μg/g) in total concentration (Table 1 and Supplementary Table 1). These unique venting styles and different geological properties of the newly identified vent fields provide an interesting opportunity to understand the microbial community structure and metabolic potentials of hydrothermal vents in slow to intermediate spreading ridges.
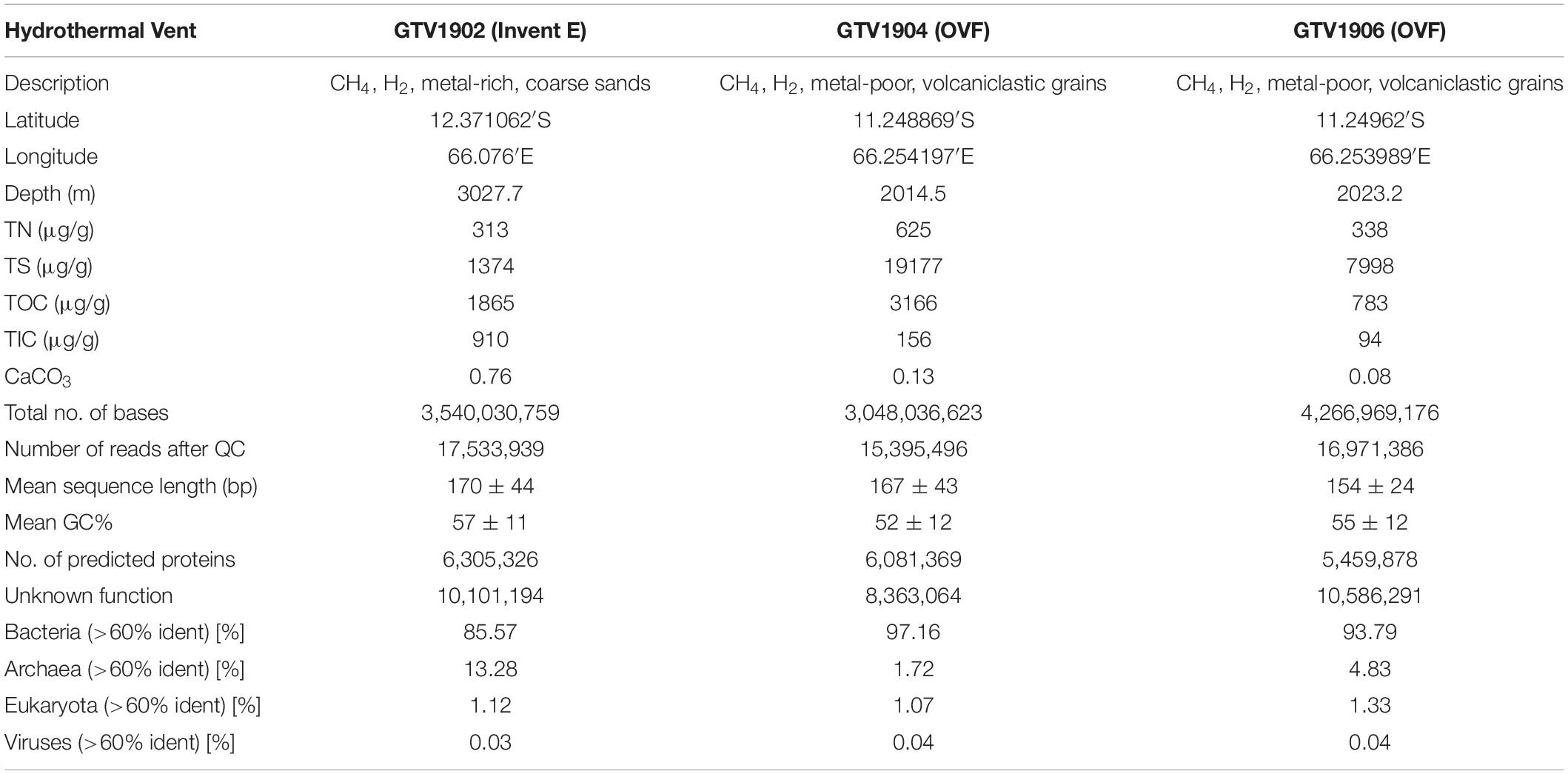
Table 1. Sampling information, physico-chemical characteristics of samples, and summary statistics of the three metagenomes studied obtained from MG-RAST.
Deep-sea sediments were obtained from three different vent fields along the CIR during the deep-sea expedition aboard R/V ISABU of Korea Institute of Ocean Science and Technology (KIOST) in June 2019. Sediment samples were collected using either a TV-grab (GTV), multiple corer (MC) and/or box corer (BC). Eight sediment samples BC1902, BC1903, GTV1904, GTV1907, GTV1906, MC1906, and MC1912 were collected around Onnuri Vent Field (OVF), a newly identified and the sixth hydrothermal vent system discovered in the Central Indian Ridge (Kim et al., 2020). Two other sediment samples GTV1902 and GTV1908 were collected from Invent E and Invent B fields respectively, currently inferred hydrothermal vent fields in the CIR (Figure 1). In case of OVF samples were retrieved from the venting areas (∼100–150 m diameter), however, samples from Invents B and E were obtained from areas that did not show signature of plume. Once on board, samples were subsampled, preserved at −80°C until further use.
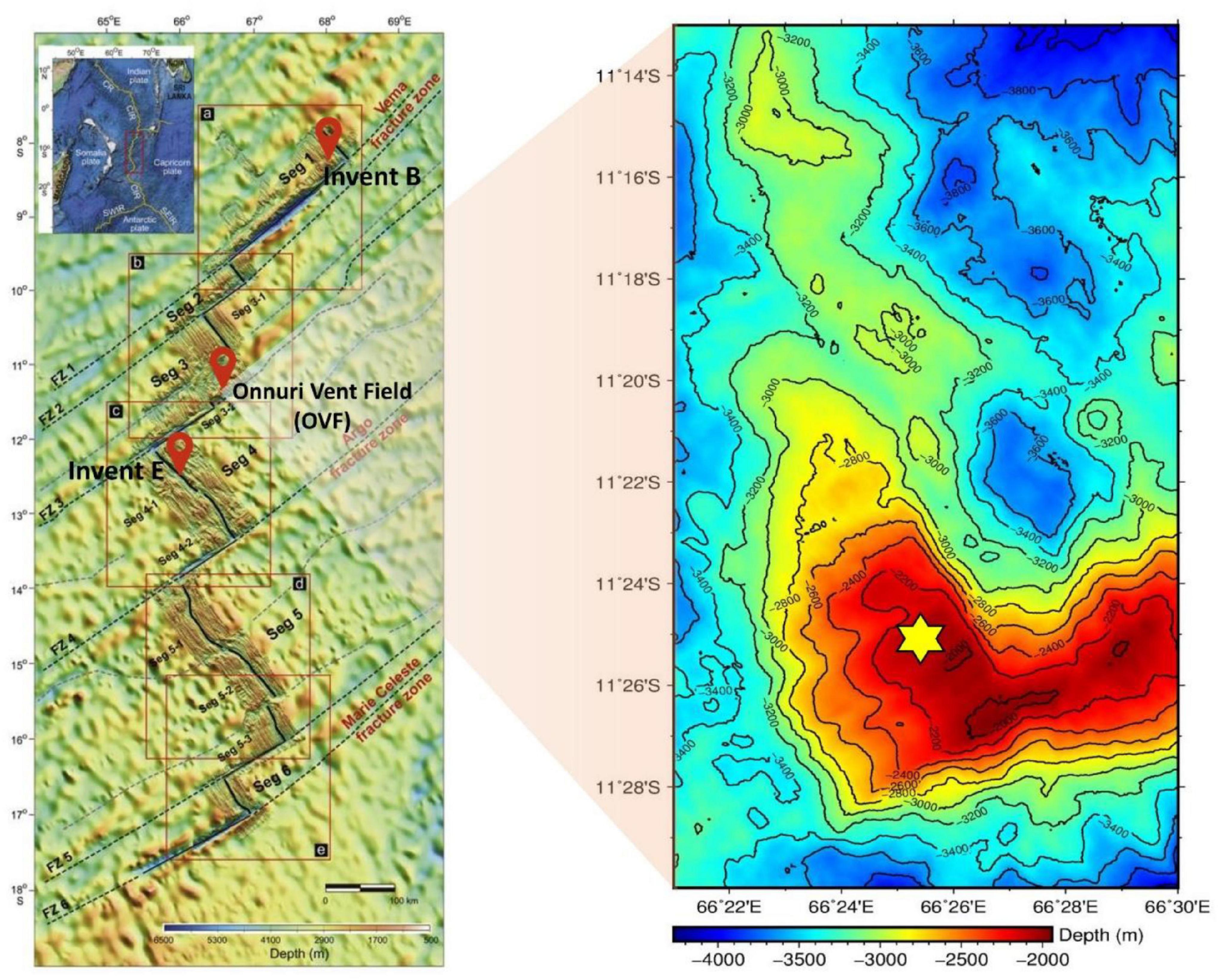
Figure 1. Location of the studied hydrothermal vent fields. Bathymetric map of Invent B, Invent E and Onnuri Vent Field (OVF) located on segment 1, segment 3 and Segment 4 along the Central Indian Ridge [map modified from Pak et al. (2017)]. Sampling site at OVF is noted with yellow star.
Amplicon Sequencing and Analysis
DNA in duplicate was extracted from each of the sediments using the FastDNA™ SPIN Kit for soil (MP Biomedicals, LLC, Solon, OH, United States) following the manufacturer’s instructions. Microbial communities were characterized by sequencing the 16S rRNA gene using prokaryotic universal primers 314F (5′-CCTACGGGRBGCASCAG-3′)/805R (5′-GACTACNVGGGTATCTAAT-3′) which target the V3-V4 regions (Takahashi et al., 2014). The concentration of the amplicons was quantified using NanoDrop™ 2000 spectrophotometer and visualized by gel electrophoresis. The purified PCR products were pooled at equimolar concentrations and paired-end sequenced using Illumina Miseq platform at Chunlab, Inc., South Korea. 16S rRNA gene reads were processed using Mothur version 1.41.0 (Schloss et al., 2009), and operational taxonomic units (OTUs) at 97% similarity were classified using the SILVA v128 database (Quast et al., 2013). Sequence data were normalized by randomly sub-sampling all datasets to the lowest sample size. Statistical analyses were carried out in R version 4.1.0 (R Core Team, 2021). Diversity analyses (alpha and beta diversity) were calculated using the packages phyloseq (McMurdie and Holmes, 2013) and microeco (Liu et al., 2021). Visualization was performed using the ggplot2 package (Wickham et al., 2016).
Metagenome DNA Extraction and Sequencing
Based on the amplicon preliminary results, three representative samples (GTV1902, GTV1904, and GTV1906) were selected for shotgun sequencing. GTV1902 presented the most contrasting microbial community from the other studied sediments while GTV1904 and GTV1906 from which high-quality DNA could be obtained were the most diverse representative community among OVF samples. DNA purification from the three sediment samples was carried out using sterilized spatulas, separate pipettes, and reagents to avoid cross contamination. Sediment samples (0.5 g each) were subjected to DNA extraction using the FastDNA™ SPIN Kit for soil (MP Biomedicals, LLC, Solon, OH, United States). Briefly, each sample was transferred into separate lysing matrix E tubes and then used for community DNA extraction. Two to four DNA extractions were pooled into a single sample and further concentrated by ethanol precipitation to obtain sufficient DNA for each sediment sample. DNA quality was verified by agarose gel electrophoresis and quantified using NanoDrop™ 2000 spectrophotometer. Paired-end read sequencing of 2 × 250 bp was performed using Illumina MiSeq platform 2500 at Chunlab, Seoul (South Korea).
Metagenome Analysis, Assembly, and Annotation
Unassembled reads from the three datasets (GTV1902, GTV1904, and GTV1906) were submitted to the MG-RAST webserver version 4.0.31 (Meyer et al., 2019) for processing following their standard protocol. Briefly, the mate-pairs were joined and then pre-processed for quality control. Adapter sequences were identified and trimmed using skewer (Jiang et al., 2014). Using duplicate read inferred sequencing error estimation (DRISEE) (Keegan et al., 2012) artificial duplicate reads (ADRs) (Gomez-Alvarez et al., 2009) were analyzed and de-replicated. Host DNA contamination was removed using Bowtie2 (Langmead and Salzberg, 2012). Coding regions within the sequences were predicted using FragGeneScan (Rho et al., 2010) and clustered at 90% homology using CD-HIT (Fu et al., 2012). Sequences similarity searches were then performed using BLAST-like alignment tool (BLAT) (Kent, 2002) against the M5NR non-redundant protein database (Wilke et al., 2012) in MG-RAST version 4.0.3 (Meyer et al., 2019). Taxonomic and functional classification was performed using the RefSeq (O’Leary et al., 2016) and Kyoto Encyclopedia of Genes and Genome (KEGG) Orthology (KO) (Kanehisa et al., 2007) or SEED (Overbeek et al., 2005) databases with default parameters, respectively. The resulting profiles were downloaded for further analyses in R version 4.1.0 (R Core Team, 2021). The taxonomic composition and relative abundance of the key metabolic genes in the metagenomes were visualized using the package ggplot2 (Wickham et al., 2016).
The raw sequence reads were also assembled de novo into contigs using metaSPAdes (v 3.10) (Nurk et al., 2017) implemented in ATLAS (Kieser et al., 2020), using the default parameters for the three metagenomes (GTV1902, GTV1904, and GTV1906). Genome binning was carried out individually for each sample using two binning methods MetaBat v2.12.1 (Kang et al., 2019) and MaxBin v2.2.6 (Wu et al., 2016). The resulting bins were further refined using the DAS Tool (Sieber et al., 2018) into metagenome-assembled genomes (MAGs) and the quality of each MAG was assessed using CheckM (Parks et al., 2015). Open reading frames (ORFs) within MAGs from each sample were predicted using Prodigal v2.6.3 (Hyatt et al., 2010), clustered using mmseqs (Steinegger and Soding, 2017) and the resulting gene catalogs were mapped to the eggNOG catalog v5 (Huerta-Cepas et al., 2019) using eggNOG mapper implemented in ATLAS (Kieser et al., 2020). Taxonomic annotation of the resulting MAGs was inferred using the genome taxonomy database tool kit (GTDB-tk) (Parks et al., 2018).
Phylogenetic Analysis of Dissimilatory Sulfite Reductase Genes
The dissimilatory sulfite reductase (dsr) is a key enzyme in both the reduction of sulfate in sulfate-reducing microorganisms or functions in reverse in sulfide-oxidizing microorganism. To distinguish the pathway directionality of dissimilatory sulfate reduction or oxidation, dsrAB phylogeny was conducted (Müller et al., 2015). The gene sequences encoding dsrAB (KO number K11180, K11181) were retrieved manually from all metagenomic contigs based on KEGG annotations. Reference protein sequences for phylogenetic analyses were obtained by BLAST searches against the NCBI protein database. Using several reference sequences, phylogenetic trees were reconstructed using the maximum-likelihood treeing method with Jones–Taylor–Thornton (JTT) amino acid substitution model (Jones et al., 1992). All processes were conducted by using MEGA ver. 7 (Kumar et al., 2016).
Comparative Metagenomic Analysis
The CIR metagenomic datasets were compared to seven hydrothermal vent metagenomes available within the MG-RAST pipeline. Two datasets from Mid Atlantic Ridge (MAR) were retrieved by searching the MG-RAST database using appropriate keywords (e.g., “hydrothermal vent,” “shotgun metagenome,” “sediment”). The other five Southwest Indian ridge metagenome datasets were downloaded from the NCBI’s SRA, uploaded into MG-RAST server, and processed according to the methods described above. A double hierarchical heatmap was constructed using the RefSeq, SEED, and KEGG profiles of each metagenome to compare the taxonomic and functional diversity in different vent systems. The number of sequence hits in each metagenome was normalized, and their Euclidean distances were calculated and visualized in R using the package pheatmap (Kolde, 2019).
Data Availability
The raw reads from this study were deposited to NCBI under the Bio project accession number PRJNA751690 The metagenomic sequencing data are also publicly available from the MG-RAST server version 4.0.3 (see text footnote 1) (Meyer et al., 2008) under the following IDs: mgm4898492.3 for GTV1902, mgm4898484.3 for GTV1904, and mgm4898483.3 for GTV1906.
Results
Physico-Chemical Composition of the Sediments
The geochemical compositions of the sediments were found to vary across sediment samples. TN, TS, and TOC were enriched in GTV1904 (OVF) than in the other two studied sediments, whereas TIC and CaCO3 were found to be higher in GTV1902 (Table 1). The detailed geochemical and elemental compositions of the three sediment samples studied are given in Table 1 and Supplementary Table 1, respectively. The concentration of Ba was the highest in GTV1904 and GTV1906, whereas that of Cu was the highest in GTV1902. In addition, sediment GTV1902 from Invent E presented higher concentrations of Fe2O3, MnO, CaO, K2O, Na2O, V, and Zn than those of sediments from OVF (GTV1904 and GTV1906); however, SiO2, Sn, and Pb concentrates were lower (Supplementary Table 1). These differences in physicochemical compositions may be reflected in the microbial community composition of the three sediment samples.
16S rRNA Sequencing Diversity
Initially, we assessed the microbial community diversity associated with hydrothermally influenced sediments from different vent fields along the CIR by targeting the 16S rRNA gene V3-V4 region. Bacteria were the most diverse across all samples dominated by Gammaproteobacteria (26.7% in the Invent B; 40.8%–12.5% in the OVF samples) except GTV1902 (Invent E) where Thermoplasmata (18.2%) were the most dominant OTUs observed (Supplementary Figure 1). Gammaproteobacterial marine benthic group JTB255, the OM1 clade (Actinobacteria), SAR202 (Chloroflexi), and Epsilonproteobacteria members Sulfurovum and Sulfurimonas were some of the abundant OTUs identified (Supplementary Figure 2). The NMDS plot showed that sediment from Invent B clustered together with OVF sediments while Invent E sample clustered further apart from the rest of the samples (Supplementary Figure 3). Sample GTV1902 had the most contrasting microbial community composition and GTV1904 presented the highest abundance of Bacteroidetes while GTV1906 was the most diverse and representative sample among OVF samples. These preliminary findings prompted us to further investigate the microbial communities of GTV1902, GTV1904, and GTV1906 by shotgun metagenome sequencing.
Microbial Community Composition
Shotgun metagenomic sequencing resulted in a total of 65,672,806 raw sequencing reads from the three samples with an average length of 166 bases. Quality processing produced 17,533,939 sequence reads from GTV1902, 15,395,496 for GTV1904, and 16,971,386 from GTV1906. Of the sequence reads that passed QC, 6,305,326 (GTV1902), 6,081,369 (GTV1904), and 5,459,878 (GTV1906) sequences contained predicted proteins with known functions while 10,101,194 (GTV1902), 8,363,064 (GTV1904), and 10,586,291 (GTV1906) sequence contained predicted proteins with unknown functions. The metagenome sequence statistics for the three metagenomes and the publicly available metagenomes used in this study are shown in Table 1 and Supplementary Table 2.
Taxonomic classification of protein-coding genes was assigned using the NCBI RefSeq (O’Leary et al., 2016) protein database as implemented in MG-RAST. Bacterial sequences dominated in all metagenomes with 85.6% (GTV1902), 97.2% (GTV1904), and 93.8% (GTV1906) of the total assigned reads (Table 1). GTV1902 metagenome exhibited the highest archaeal representatives accounting for 13.3% of the total hits. Eukaryotic reads were also detected while less than 1% of the reads were assigned to viruses (Table 1). Sequences from all datasets were assigned to 28 different bacterial and 5 archaeal phyla.
All three samples shared Proteobacteria as the most dominant phylum. GTV1902 was dominated by Deltaproteobacteria (12.9%), within those the genus Geobacter, was the most abundant taxa (Supplementary Figure 4). Bacteria belonging to the phylum Firmicutes had the second highest relative abundance (17.6%), followed by Chloroflexi (8.7%), Actinobacteria (7.5%), and Bacteroidetes (4.8%) (Figure 2A). Members of the genera Clostridium and Bacillus were the representatives of the phylum Firmicutes while the most abundant Chloroflexi genera detected in GTV1902 were Roseiflexus and Dehalococcoides (Supplementary Figure 4). In contrast, the bacterial community in GTV1904 and GTV1906 were mainly composed of Gammaproteobacteria (29.6% and 22.3%, respectively; Figure 2A). Thioalkalivibrio, Pseudomonas, and Nitrosococcus were the dominant gammaproteobacterial genera found in GTV1904 and GTV1906 metagenomes (Supplementary Figure 4 and Supplementary Table 3). Other dominant bacterial phyla within GTV1904 and GTV1906 included Bacteroidetes, Firmicutes, Actinobacteria, and Planctomycetes (Figure 2A).
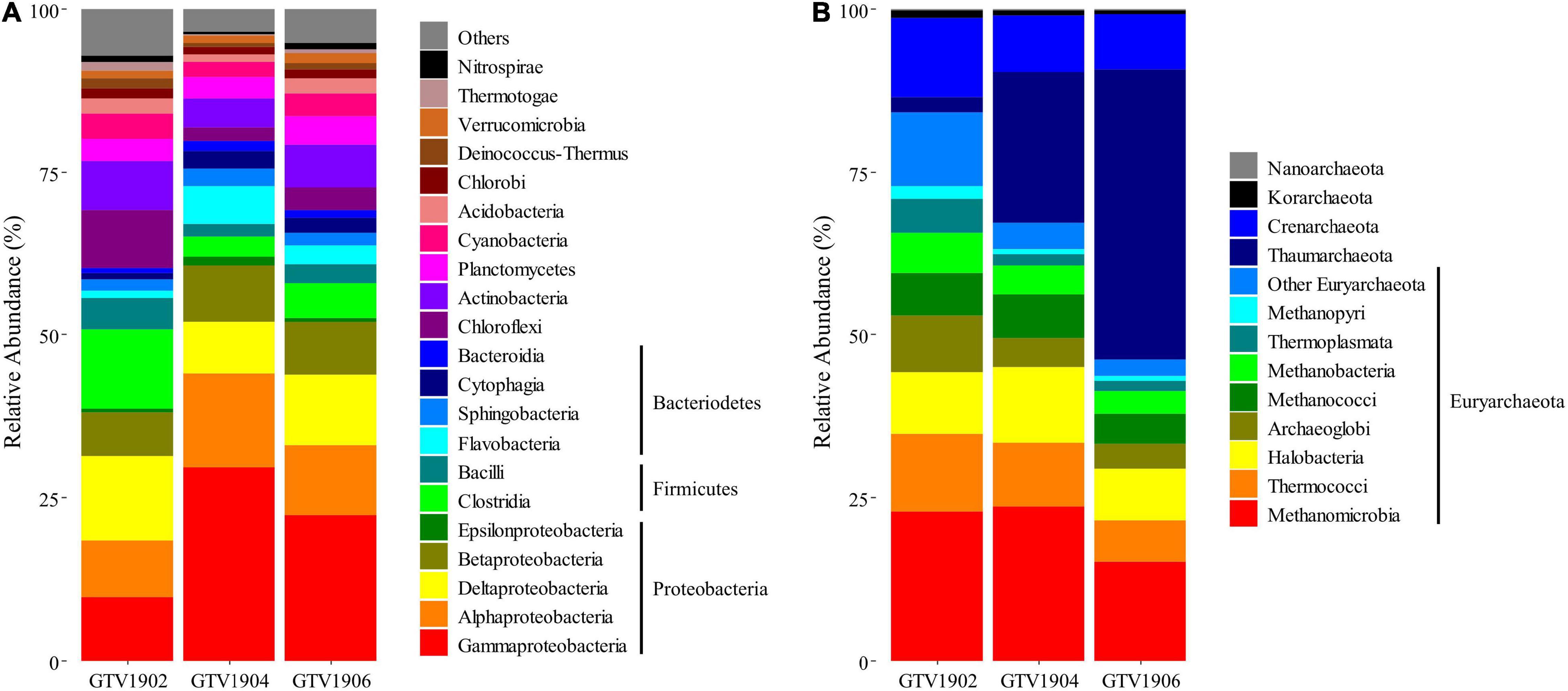
Figure 2. Taxonomic composition and relative abundance of (A) Bacteria and (B) Archaea in the three different metagenomes based on protein-coding genes using RefSeq database at class level. “Others” represent those accounting for <1% of the total hits in each sample and are shown in gray at the top of each bar.
A detailed taxonomic analysis of the archaeal representatives found in the different metagenomes is shown in Figure 2B. While GTV1902 and GTV1904 archaeal community was dominated by Euryarchaeota, the archaeal community of GTV1906 was equally dominated by Euryarchaeota and Thaumarchaeota. Methanomicrobia was the most prevalent class in GTV1902 and GTV1904 accounting for 22.9% and 23.7% of the total assigned sequences, respectively. Unclassified (derived from Thaumarchaeota) was the most abundant class accounting for 44.4% of the total assigned reads of GTV1906 and followed by Methanomicrobia (15.1%). GTV1902 exhibited a predominance of Aciduliprofundum while the thaumarchaeotal genera Nitrosopumilus dominated the archaeal community in both GTV1904 and GTV1906 (Supplementary Figure 4 and Supplementary Table 4).
Metagenome-Assembled Genomes
The GTV1902 assembly generated 61,732 contigs of sizes from 500 to 105,415 bp with an N50 of 12,795. The GTV1904 assembly generated 57,779 contigs of sizes from 500 to 99,270 base pairs with an N50 of 9,946 while 38,882 contigs of sizes from 500 to 39,457 base pairs with an N50 of 9,769 were retrieved from GTV1906 assembly (Supplementary Table 5). Metagenome binning yielded 20 medium- to high-quality MAGs on the established standards (Bowers et al., 2017) including 4 Archaea and 16 Bacteria (Supplementary Table 6). Taxonomic annotation based on the GTBD-Tk revealed that the 16 bacterial MAGs could be assigned to 8 phyla including the KSB1 (2 MAGs), Actinobacteriota (3), Bacteroidota (4), Chloroflexota (1), Gemmatimonadota (2), Nitrospirota (1), Planctomycetota (1), and Proteobacteria (2). In addition, we assembled four archaeal MAGs, which were affiliated to two phyla: Thermoplasmatota (3) and Asgardarchaeota (1) (Supplementary Table 6). Except for archaeal phylum Asgardarchaeota, similar taxa as the MAGs assembled here were observed in the 16S rRNA gene amplicon data.
Functional Composition
To determine the functional potential of the communities across the different metagenomes, we analyzed the relative abundance of key metabolic genes in the three CIR metagenomes. Biomass production in deep-sea hydrothermal vents is fueled by the oxidation of reduced inorganic compounds to fix inorganic carbon, therefore, key metabolic genes for carbon, sulfur, nitrogen, and methane metabolism were analyzed. The overall functional potential was similar across metagenomes with majority of the genes universally present across all samples and a few differences in relative abundances. The relative normalized abundances of the key genes involved in energy generation and biogeochemical cycling based on assembled contigs and unassembled raw reads are shown in Figure 3 and Supplementary Figure 5, respectively.

Figure 3. The relative abundance of key metabolic genes involved in energy-yielding processes and carbon fixation pathways in the different metagenome assemblies. Each bubble size represents the relative abundance of specific genes against the total number of KO hits in each assembly.
Carbon Fixation
Genes related to the key enzymes of Calvin–Benson–Bassham (CBB) cycle (rbcLS and prkB) were found in all metagenomic datasets (Figure 3 and Supplementary Table 7) however they were highly enriched in OVF (GTV1904 and GTV1906). Sequences were most related to gammaproteobacterial genera Nitrosococcus and Allochromatium, among others were observed (Supplementary Table 8). The three key genes of the reductive tricarboxylic acid (rTCA) cycle, ATP citrate lyase (acly), 2-oxoglutarate ferredoxin oxidoreductase (korABCD), and pyruvate ferredoxin oxidoreductase (porABCD) are higher in GTV1902 (Invent E) compared to GTV1904 and GTV1906. However, the marker gene for the complete rTCA cycle ATP citrate lyase (aclAB) was not found in GTV1902 (Figure 3 and Supplementary Table 7). The acly sequences found in the GTV1906 sites were closely affiliated with Chlorobi. In contrast sequences in GTV1904 share high similarities with those from Epsilonproteobacteria (Table 2 and Supplementary Table 8). Markers for the Wood-Ljungdhal (WL) (=reductive acetyl-CoA) pathway had a slightly higher abundance in GTV1906 (OVF) metagenome (Figure 3). These genes were closely related to Firmicutes and Deltaproteobacteria groups (Supplementary Table 8). The key enzymes for the 3-hydroxypropionate (3-HP) and 3-HP/4-hydroxybutyrate (3-HP/4-HB) cycle, 4-hydroxybutyryl-CoA dehydratase (abfD), propionyl-CoA synthetase (prpE), and malyl-CoA lyase (mcl), were also detected in low abundance, however, malonyl-coA reductase (mcr) was missing in all datasets (Figure 3 and Supplementary Table 7).
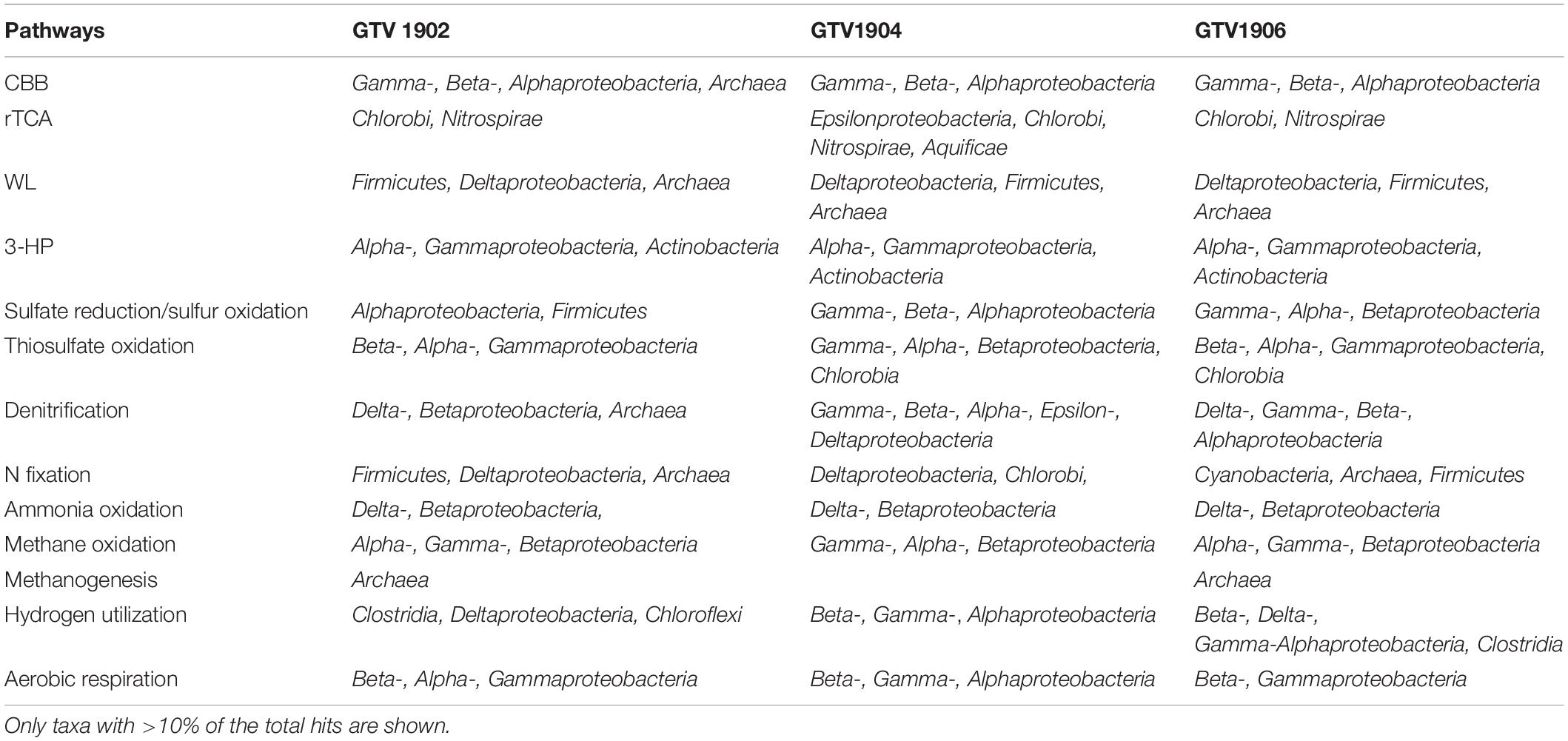
Table 2. Assignment of metabolic roles to major taxonomic groups based on detection of key genes-coding proteins in the metagenomes.
Sulfur Metabolism
The complete set of genes encoding the enzymes dissimilatory sulfite reductase (dsrAB), adenylylsulfate reductase (aprAB), and sulfate adenylyltransferase (sat) were enriched in GTV1904 (Figure 3). Majority of the dsrAB sequences in all the metagenomic datasets were taxonomically assigned to Gamma- (e.g., Allochromatium, Thioalkalivibrio, Alkalilimnicola), Alpha- (e.g., Rhodomicrobium, Magnetospirillum), Betaproteobacteria (e.g., Thiobacillus, Sideroxydans) as well as Chlorobi representatives, with the remainder being closely related to the reductive type of Deltaproteobacteria, Firmicutes, and Nitrospirae (Table 2 and Supplementary Table 9). Most aprAB (66.2%) hits were affiliated to Firmicutes and Deltaproteobacteria in GTV1902 metagenome while those from GTV1904 and GTV1906 were largely assigned to Beta-, and Gammaproteobacteria representing 88.9% and 81.9%, respectively.
Because the same set of genes are involved in both types of dissimilatory sulfur metabolism, the oxidative or reductive directionality of the pathway cannot easily be deduced from taxonomy and the presence of these genes alone. To determine whether the dsrAB genes identified were associated with sulfate reduction or sulfur oxidation, we inferred the taxonomy and catalytic type of the dsrAB based on their phylogenies. Sequence analysis of the dsrAB proteins assembled from the GTV1904 and GTV1906 metagenomes suggested that were of the oxidative type, which clustered with sulfur-oxidizing bacteria affiliated to Alpha- (e.g., Rhodospirillales) and Gammaproteobacteria such as Thiogranum, Thiotrichaceae, Acidithiobacillales, and other unclassified hydrothermal environmental sequences (Supplementary Figures 6, 7). Sulfate-reducing bacteria-type dsrAB sequences affiliated to Deltaproteobacteria (Desulfobulbaceae) were also detected. In contrast, the DsrAB protein tree demonstrated that the dsrAB genes assembled from GTV1902 were only of the reductive type affiliated with genes from uncultured sulfate-reducing bacterium (Supplementary Figures 6, 7).
Incomplete oxidation of sulfides can produce thiosulfate, which can be oxidized to sulfate through sulfur-oxidizing proteins (soxABXYZ). The key genes involved in sulfur-oxidizing (SOX) system were found in all the metagenomes with a higher relative abundance in GTV1906 whereas thiosulfate sulfurtransferase (tst) that oxidize thiosulfate to sulfite (SO32–) and sulfide:quinone oxidoreductase encoding gene (sqr) for periplasmic sulfide oxidation, similar abundances were identified across all metagenomes. Potential oxidation of reduced sulfur compounds via the SOX system was affiliated to the Alpha- and Gammaproteobacteria (Table 2 and Supplementary Table 9).
Nitrogen Metabolism
All the genes, including periplasmic nitrate reductase (napA), nitrate reductase (narGH), nitrite reductase (NO-forming) (nirKS), nitric oxide reductase (norBC), and nitrous oxide reductase (nosZ) encoding for complete denitrification pathway were identified in all three metagenomes, but with divergent abundances, being overrepresented in GTV1904 (Figure 3), with most of the sequences assigned to Gammaproteobacteria members (Table 2 and Supplementary Table 10). Dissimilatory nitrate reduction to ammonia via nitrite reductases (nirBD) were also detected and were found to be closely related to Beta- and Gammaproteobacteria groups. Nitrogenases (nifDHK), genes required for nitrogen fixation and ammonia monooxygenases (amoABC), and key enzymes in ammonia oxidation were absent across all samples; however, genes encoding for hydroxylamine oxidase (hao) mainly associated with Deltaproteobacteria were detected across all metagenomes (Figure 3 and Table 1). Members of the genera Anaeromyxobacter, Nitrosomonas, and Nitrosospira seemed to be the primary ammonia oxidizers in all metagenomes (Supplementary Table 10).
Other Metabolic Pathways
Cytochrome c oxidase and cbb3-type cytochrome c oxidase complexes involved in aerobic respiration were predominately identified in OVF samples GTV1904 and GTV1904 (Figure 3). Both cytochrome c oxidase and cbb3-type cytochrome c oxidase genes majorly affiliated to aerobic proteobacterial lineages (Table 2 and Supplementary Table 11). GTV1904 showed a higher abundance of both hydrogenases (hyaAB) and [NiFe]-hydrogenases (hydAB) with the highest similarity to Proteobacteria and Firmicutes (Table 2 and Supplementary Table 12). Genes encoding enzymes involved in methane oxidation (pmoABC and mdh12) and marker genes for methanogenesis (metyl-CoM reductase; mcr) were absent in all metagenomes. However, traces of soluble methane monooxygenase (mmoX) were identified in GTV1904 and GTV1906 (Figure 3 and Supplementary Table 13).
Comparative Metagenomic Analysis
To explore the unique features of the CIR metagenomes, we compared our datasets to a collection of selected metagenomes obtained by shotgun sequencing from different ocean ridges (Supplementary Table 2). The double hierarchical clustering of the samples and KEGG pathways is shown in Figure 4. A cluster analysis of the KEGG level 3 pathways did group the samples into two clusters; the hydrothermally influenced sediments formed one group while the hydrothermal chimney samples clustered together and formed another group. The three CIR metagenomes (GTV1902, GTV1904, and GTV1906) clustered most closely with RB35 sample from MAR, and they formed a group with the sample MG35 from MAR and OCT chimney sample from the SWIR, whereas the rest of the chimney samples from SWIR (ToMo, BaC, PiMo, and Chan) grouped separately. A very similar grouping pattern was observed by clustering of samples based on the SEED subsystems and RefSeq databases (Supplementary Figures 8, 9).
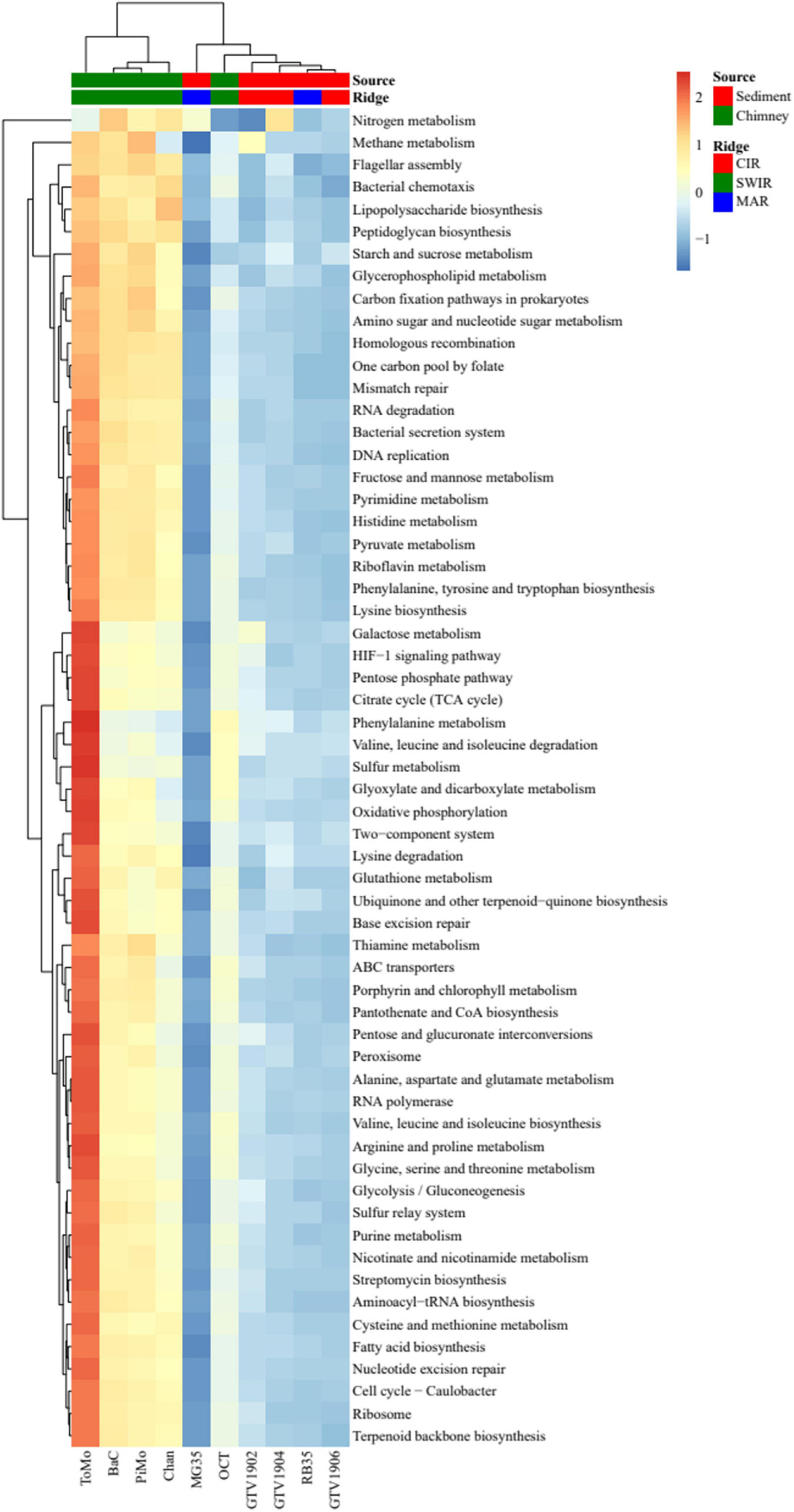
Figure 4. Two-way cluster analysis of samples and level 3 KEGG pathways based on the relative abundances of the KEGG-annotated sequences with hits in each category. The heatmap was clustered based on the Euclidean distance matrix. The shading is proportional to the normalized values calculated based on the functional hits of each category per sample. Only pathways with sum of hits >186,000 across the 10 metagenomes are shown.
The hydrothermally influenced or low activity field sediment samples (GTV1902, GTV1904, GTV1906, RB35, and MG35) were almost depleted in KEGG level 3 categories, while the chimney samples (ToMo, BaC, PiMo, Chan, and OCT) showed higher abundance in majority of the KEGG categories. Compared to other hydrothermally influenced sediments, GTV1902 and GTV1904 samples were more enriched with enzymes for methane and nitrogen metabolisms respectively. Further comparison of the taxonomic relative abundances of the ten metagenomes against the RefSeq database revealed that GTV1902 had a higher proportion of sequences related with Euryarchaeota and Korarchaeota, when compared with other metagenomes (Supplementary Figure 9).
Discussion
Microbial communities in deep-sea sediments play important roles in biogeochemical processes (Zeng et al., 2021). This study is the first to report the microbial community of deep-sea sediment from recently discovered hydrothermal vents in the CIR using shotgun metagenomic sequencing. To reveal the unique features of the CIR microbial community structures we conducted a comparative study with datasets from different hydrothermal systems, including five chimney metagenomes from Old City in SWIR (Lecoeuvre et al., 2021), and two sediment metagenomes from the Azores in the MAR (Cerqueira et al., 2018).
Microbial Community Composition
Taxonomic analyses revealed that the bacterial community was highly diverse. Proteobacteria (especially Gammaproteobacteria) was the most dominant followed by Firmicutes and Bacteroidetes. Although the three metagenomes shared several taxa, they differed with respect to overall phyla abundances (Figure 2). Sulfur-oxidizing genera Thioalkalivibrio, and ammonia-oxidizing bacteria Nitrosococcus were the most abundant genera within the Gammaproteobacteria. Previous investigation on the microbial diversity in the Indian Ocean Ridge (IOR) also revealed that Proteobacteria were the dominant bacterial phylum, but with contrasting proportions in its classes (Cao et al., 2014; Lecoeuvre et al., 2021). Serpentinite-hosted systems are enriched in hydrogen and methane as a result of seawater-rock reactions (Charlou et al., 2002; Proskurowski et al., 2008). The presence of H2 and metal oxides (mainly Fe and Mn) as alternative electron acceptors especially at OVF site (Kim et al., 2020) may have contributed to the abundance of Firmicutes present in the CIR samples. Among the phylum Firmicutes Clostridium and Bacillus were the predominant genera found in the three metagenomes. Representatives of Bacillus are known for their tolerance in extreme environments. Members of the genus Clostridium are strict anaerobes that use hydrogen as electron donor to fix CO2, autotrophically, through the acetyl-CoA pathway. In this study, a higher abundance of Firmicutes representatives in the metagenomes capable of H2-dependent carbon fixation indicates that the microbial communities depend on these hydrogen-utilizing microorganisms for biomass production. The presence of high concentrations of methane and sulfides in the OVF could explain the predominance of methane-oxidizing Gammaproteobacteria related to the genus Methylococcus. Indeed, several studies have shown that the predominance of Gammaproteobacteria is associated with serpentinite-hosted hydrothermal fields (Brazelton et al., 2006; Lecoeuvre et al., 2021). Members of Chloroflexi, Actinobacteria, and Planctomycetes among others which are generally recovered from deep-sea hydrothermal environments (Zeng et al., 2021) were also identified in the CIR metagenomes.
Euryarchaeota, Thaumarchaeota, and Crenarchaeota were the dominant archaeal phyla identified in the three metagenomes studied. Previous studies have shown that these are known to be the three major archaeal phyla found in deep-sea hydrothermal vents in the chimney fluids from SWIR (Lecoeuvre et al., 2021) as well as in hydrothermal sediments from the MAR (Cerqueira et al., 2018). Similar to a recent metagenomic study in serpentinite-hosted Old City hydrothermal field in SWIR (Lecoeuvre et al., 2021), Euryarchaeota was the most frequently observed archaeal phyla in the three metagenomes. Specifically, majority of the Euryarchaeota sequences in the GTV1902 were assigned to Aciduliprofundum a thermoacidophilic iron and sulfur reducing heterotroph originally isolated from deep-sea hydrothermal vents (Reysenbach et al., 2006). On the other hand, potential methanogenic Archaea (i.e., Methanosarcina) were predominant in GTV1904 and GTV1906. The presence of these methanogenic Archaea in methane-rich sediments from OVF suggests that the high concentration of methane at this site may be accumulated as a result of both biotic and abiotic processes.
In addition, although in low abundances Korarchaeota and the Nanoarchaeota represented by the genera “Candidatus Korarchaeum” and Nanoarchaeum respectively were also observed in our metagenomes. Several 16S rRNA gene studies have reported Korarchaeota to be found in extreme terrestrial and marine environments including hot springs (Barns et al., 1994), coastal thermal areas (Yan et al., 2018), as well as hydrothermal systems (Takai and Sako, 1999; Teske et al., 2002; Schrenk et al., 2003). However, “Cand. Korarchaeum” has been reported in terrestrial hot springs (Elkins et al., 2008) and more recently in a shallow hydrothermal vent in Eolian Islands (Gugliandolo and Maugeri, 2019). To the best of our knowledge, this is the first metagenome survey to observe “Cand. Korarchaeum” in deep-sea hydrothermal vents. The finding of Cand. Korarchaeum in hydrothermally influenced deep-sea sediments in CIR suggests that Korarchaeum are globally distributed in extreme environments.
Carbon Metabolism
Carbon fixation by deep-sea hydrothermal vents organisms has been reported to occur through several metabolic pathways such as CBB cycle, rTCA cycle, 3-HP, 3-HP/4-HB, WP pathway, and dicarboxylate/4-HB cycle (Nakagawa and Takai, 2008; Minic and Thongbam, 2011). In this study key genes for CBB cycle were affiliated primarily with sulfur-oxidizing and nitrifying Gammaproteobacteria as well as Euryarchaeota members (Supplementary Table 8) similar to what has been reported from the metagenomic studies in the SWIR (Cao et al., 2014; Lecoeuvre et al., 2021) and other deep-sea vent systems (Elsaied et al., 2007; Nakagawa and Takai, 2008; Cerqueira et al., 2018).
The rTCA cycle in deep-sea hydrothermal systems is preferred by Epsilonproteobacteria, Aquificae, and Nitrospirae (Nakagawa and Takai, 2008; Minic and Thongbam, 2011; Cerqueira et al., 2018; Zeng et al., 2021) which were also observed in our datasets. However, compared to the above-mentioned groups, majority of the acl sequences a key enzyme in the rTCA cycle were affiliated to the phylum Chlorobi represented by Chlorobium, Chlorobaculum, and Pelodictyon (Supplementary Table 8). The rTCA is ubiquitous among the Chlorobiaceace family (Imhoff, 2014; Ward and Shih, 2021) whose members have been identified in deep-sea black smoker along the East Pacific Rise (Beatty et al., 2005). The predominance of Chlorobi acl indicates that the rTCA in the metagenomes is supported by geothermal light which provides a selective advantage for phototrophs over chemoautotrophs. Like other studies in deep-sea sediments (Cerqueira et al., 2018), some key enzymes in the 3-HP pathway were missing in our metagenomes suggesting that the 3-HP pathway was not an important carbon fixation pathway in the hydrothermally influenced sediments in OVF and Invent E sites. Therefore, carbon fixation is thought to occur either via CBB, and/or the rTCA in the OVF and Invent E metagenomes.
Sulfur Metabolism
The accumulation of metal oxides and sulfides in OVF (Kim et al., 2020) make OVF site as a great habitant for sulfur-oxidizing microorganisms. Sulfur-oxidation in deep-sea hydrothermal vents occurs via the reverse sulfate reduction and SOX pathways. Majority of the key genes in sulfur cycle observed in the OVF metagenomes were affiliated to families Ectothiorhodospiraceae and Chromatiaceae within the class Gammaproteobacteria. Sulfur-chemolithoautotrophs such as Thiomicrospira and Thioalkalivibrio and photolithotrophic Allochromatium were the dominant sulfur-oxidizing genera which were consistent with the findings from previous studies in other deep-sea vent systems (Nakagawa and Takai, 2008; Yamamoto and Takai, 2011; Cao et al., 2014; Cerqueira et al., 2018).
Taxonomic annotation and analysis of the rTCA pathway revealed several sequence hits related to sulfur-oxidizing Epsilonproteobacteria representatives of the genera Sulfurimonas and Sulfurovum in the OVF sediments. Interestingly, sulfur oxidation via the SOX complex was dominated by the sulfur-oxidizing Gammaproteobacteria (e.g., Thiomicrospira and Thioalkalivibrio genera), although the Epsilonproteobacteria are known to use the SOX pathway for oxidation of reduced sulfur compounds in deep-sea environments (Yamamoto and Takai, 2011). This result is consistent with the predominance of Gammaproteobacteria and low proportion of Epsilonproteobacteria observed in sediments. Epsilonproteobacteria are known to dominate less oxygenated/high sulfide hydrothermal fluids and chimneys (Hou et al., 2020) while the Gammaproteobacteria require presence of high reduced compounds and oxygen concentrations. The level of hydrothermal activity together with the degree of mixing of diffuse flow fluids with surrounding seawater in OVF possibly favors Gammaproteobacteria over Epsilonproteobacteria. This indicates that oxidation of reduced sulfur compounds in OVF occurs in multiple niches via both the reverse sulfate reduction and SOX pathway similar to what was observed in hydrothermally influenced Rainbow sediments in the MAR (Cerqueira et al., 2018) and consistent with other hydrothermal systems (Hügler et al., 2010).
The presence of key metabolic genes for sulfur utilization affiliated with Alphaproteobacteria and Bacteroidetes groups suggests the role of these taxa in the sulfur cycle. Sulfur-oxidizing Alphaproteobacteria and Bacteroidetes are widely distributed in marine environments including deep-sea hydrothermal vents (Sylvan et al., 2013; Stokke et al., 2015). The sulfur-oxidizing members of the Rhodobacterales within Alphaproteobacteria are enriched in all samples and these were also observed in Old City hydrothermal vent in SWIR (Lecoeuvre et al., 2021). An amplicon survey in inactive sulfide chimneys in Lau Basin also reports the dominance of sulfur-oxidizing Alphaproteobacteria and Bacteroidetes linages outside the chimneys (Sylvan et al., 2013).
We observed differences in gene abundances as well as differences in microbial community abundance suggesting that Invent E was distinct from the OVF. OVF sediments (GTV1904 and GTV1906) were dominated by Gammaproteobacteria while Deltaproteobacteria were predominant in GTV1902. The predominance of Deltaproteobacteria and Firmicutes in GTV1902 (Figure 2) suggests dissimilatory sulfate-reduction dominates in Invent E site. However, it should be noted that dissimilatory sulfite reductase (dsrAB) responsible for oxidation of sulfur to sulfite, adenylylsulfate reductase (aprAB) were nearly absent in GTV1902 metagenome. A similar set of dsr genes is found in both sulfur-reducing microorganism and sulfur-oxidizing microorganisms (Müller et al., 2015). Recent findings of Thorup et al. (2017), show that Deltaproteobacteria possess the capabilities to perform dissimilatory sulfate reduction and oxidation by the same reversible enzymes, sat, apr, and dsr. Based on phylogeny of the dsrAB genes assembled from GTV1902, the identified sequences were of the reductive type affiliated with uncultured sulfate-reducing bacterium. This affiliation is strongly suggestive that the Deltaproteobacteria identified in GTV1902 perform dissimilatory sulfate reduction. Within OVF, many sulfur-oxidizing bacteria were found, such as Ectothiorhodospiraceae, and Thiotrichaceae families, but also some sulfate-reducing bacteria such as Desulfobulbaceae. Analysis of dsrAB genes also indicated that sulfate reduction by Deltaproteobacteria appeared to be occurring exclusively in GTV1902 while sulfur oxidation by the Gammaproteobacteria was occurring at OVF. This result is also consistent with the finding of higher total sulfur concentrations in OVF (GTV1904 and GTV1906) samples compared to GTV1902 (Invent E) which is most likely controlling the community structure of total bacteria and sulfur-cycling bacteria.
Nitrogen Metabolism
Nitrate and nitrite are important electron acceptors for oxidation of reduced sulfur compounds although they are usually depleted in serpentinite-hosted systems (Nakagawa and Takai, 2008; Zeng et al., 2021). Analysis of key genes involved in the nitrogen cycle identified genes involved in the dissimilatory nitrate reduction, and denitrification, across all three metagenomes which were consistent with the diverse taxonomic abundance of nitrogen metabolizing taxa in the three metagenomes including Proteobacteria, Firmicutes, Chlorobi, Cyanobacteria, and Archaea among others. Denitrification pathway was the most prevalent among metagenomes affiliated with primarily the Delta- and Gammaproteobacteria. Similar observations of denitrification-related taxa have been documented in the SWIR (Cao et al., 2014; Lecoeuvre et al., 2021). These findings indicate that sulfur-oxidation/sulfate reduction coupled with denitrification are important energy-generating pathways fueling the microbial communities at the OVF and Invent E sites. Although marker genes amoABC for ammonia oxidation were not detected in three metagenomes, ammonia-oxidizing members in the Betaproteobacteria such as Nitrosomonas and Nitrosospira are potential ammonia oxidizers identified in all three metagenomes consistent with what was reported previously from hydrothermally influenced sediments from Rainbow vent in MAR (Cerqueira et al., 2018) but different from SWIR Old City chimneys where Thaumarchaeota was reported to be involved in ammonia oxidation (Lecoeuvre et al., 2021).
Other Metabolic Pathways
Methane concentrations produced by serpentinization reactions in OVF site were recorded up to 52.5 nM (Kim et al., 2020), therefore, methane oxidation is expected in the OVF sediments. The pmmo catalyzes the production of methanol, which is further oxidized to formaldehyde by a periplasmic methanol dehydrogenase. However, pmmo/mmo key enzymes in the methanotrophy are missing in all datasets with a very low abundance of mmoX observed exclusively in OVF. Potential methane-oxidizing Methylococcales (Gammaproteobacteria) which have been reported in several serpentinite-hosted hydrothermal systems including Old City in the SWIR (Brazelton et al., 2006; Cerqueira et al., 2018; Lecoeuvre et al., 2021) were observed in the studied sediments. The presence of methanotrophic Methylococcus in our datasets suggests a possibility that methane oxidation exists in OVF sediments although it might not be an important energy-yielding metabolic pathway.
Interestingly, the key genes for methanogenesis were missing in all metagenomes, however methanogenic Archaea dominated by Methanosarcinales were observed and being almost exclusive to GTV902 (Supplementary Table 13). Previous studies have also reported similar methanogenic Archaea in several deep-sea hydrothermal vents, such as presence of Methanosarcinales in Old City in SWIR (Lecoeuvre et al., 2021), Lost City (MAR) chimneys being dominated by Methanosarcinales (Brazelton et al., 2006), as well as the predominance of Methanococcales in CIR Kairei field (Takai et al., 2004). Although we recognize that no significant conclusions can be made due to lack of replicate samples, we speculate that Invent E site is highly enriched in hydrogen that supports the high abundance of methanogenic Archaea observed in this site. Nevertheless, much work remains to be conducted within each of these vent fields for a clear understanding of the resident microbial communities and their metabolic potentials.
Apart from the metabolic pathways discussed above, the geochemistry of the vents especially OVF offers preferable conditions for aerobic respiration hydrogen metabolism. Although our study didn’t focus on these pathways, it was evident through taxonomic characterization which reveals presence of genera such as Geobacter, Thiobacillus, Anaeromyxobacter, Acidithiobacillus, Sideroxydans, and Pseudomonas among others capable of hydrogen utilization and aerobic respiration. Majority of the hydrogenases identified in the OVF site were closely related to Beta-, Gamma-, and Alphaproteobacteria while Clostridia, Deltaproteobacteria, and Chloroflexi dominated in the Invent E site (Supplementary Table 12). Chemosynthesis in deep-sea hydrothermal environments may be coupled to both aerobic and anaerobic respiration. Genes encoding cytochrome c oxidase and cbb3-type cytochrome c oxidase involved in aerobic respiration were identified across metagenomes (Figure 3) suggesting that chemosynthesis in OVF and Invent E depends on oxygen as a terminal electron acceptor other than nitrate, sulfate, etc. Both cytochrome c oxidase and cbb3-type cytochrome c oxidase genes majorly affiliated to aerobic proteobacterial linages (Table 2 and Supplementary Table 11). Thus, oxygen derived from the constant mixing of diffuse fluids with the surrounding seawater is thought to be a key electron acceptor allowing sulfur-oxidizing Proteobacteria to thrive in the OVF and Invent E sites.
Metagenome Comparison From Other Hydrothermal Systems
Currently, no metagenomes from hydrothermal vent field in the CIR are available for comparison. Within the SWIR, only two metagenome studies have been reported, one from chimney sulfides (Cao et al., 2014) and more recently from Old City hydrothermal system (Lecoeuvre et al., 2021). Unfortunately, only the metagenomes from Old City were used for comparison due to the unavailability of the sequencing data of sulfides metagenomes in NCBI’s database. Metagenome comparisons showed that the Old City chimney metagenomes clustered together except sample OCT, which may be attributed to the fact that OCT chimney had a greater influence on seawater intrusions than in other chimney samples (Lecoeuvre et al., 2021). In addition, metagenomes from hydrothermal vent chimneys (ToMo, BaC, PiMo, Chan, and OCT) were enriched in metabolic functions suggesting that hydrothermal microbial communities have evolved extensive metabolisms as an adapting strategy in response to the steep chemical and thermal gradients observed at vent chimneys.
Rainbow metagenome (RB35 collected 100 m from the active vent) did not cluster together with its counterpart Menez Gwen (MG35 collected near the chimney) from the MAR but rather closely clustered next to our metagenomes from the CIR. The difference in the microbial community structure of the two metagenomes from MAR is also consistent with the one that has been reported before (Cerqueira et al., 2018). The physicochemical conditions of the Rainbow vent and OVF are strikingly different (for example, the diffuse flow of OVF is weak with low temperature, metal-poor, and methane-rich (Kim et al., 2020), whereas Rainbow is characterized with very high rates of fluid flow ∼360°C and high metal contents (Cerqueira et al., 2018), however the sediment samples studied both represent low hydrothermal activity samples that are continuously exposed to the surrounding sea water as a result these prevailing environmental conditions have a significant influence on the microbial community structure and distribution of functional gene categories. This result indicates the stratification in hydrothermally influenced and hydrothermal microbial communities.
Conclusion
In this study, using a metagenomic approach, we report for the first time the microbial ecology of the recently discovered hydrothermal vents (OVF and Invent E) located along the slow-spreading CIR. The microbial community structures and metabolic properties across metagenomes were largely similar although dominant taxa and functional potential vary between vent fields. The oxidation and reduction of sulfur may be important energy metabolism pathways in the two sampling sites. We illustrated these key metabolic pathways fueling microbial communities at the OVF and Invent E sites (Figure 5). The Gammaproteobacteria are the major primary producers at OVF site fixing CO2 via CBB cycle and retrieving energy from oxidation of reduced mineral sulfides through reverse sulfate reduction pathway or SOX complex leading to the oxidation of sulfide to sulfate. Deltaproteobacteria lineages known to be involved in sulfate reduction characterized Invent E. We speculate that the Deltaproteobacteria and Firmicutes utilize the sulfates from the seawater, fixing CO2 through the WL cycle contributing to the production hydrogen sulfide in the hydrothermal fluids. In addition, hydrogen from serpentinization processes supplies metabolic energy for methanogenic Archaea. Oxygen and nitrates from the surrounding seawater are possible electron acceptors for sulfate-reducing and sulfur-oxidizing bacteria.
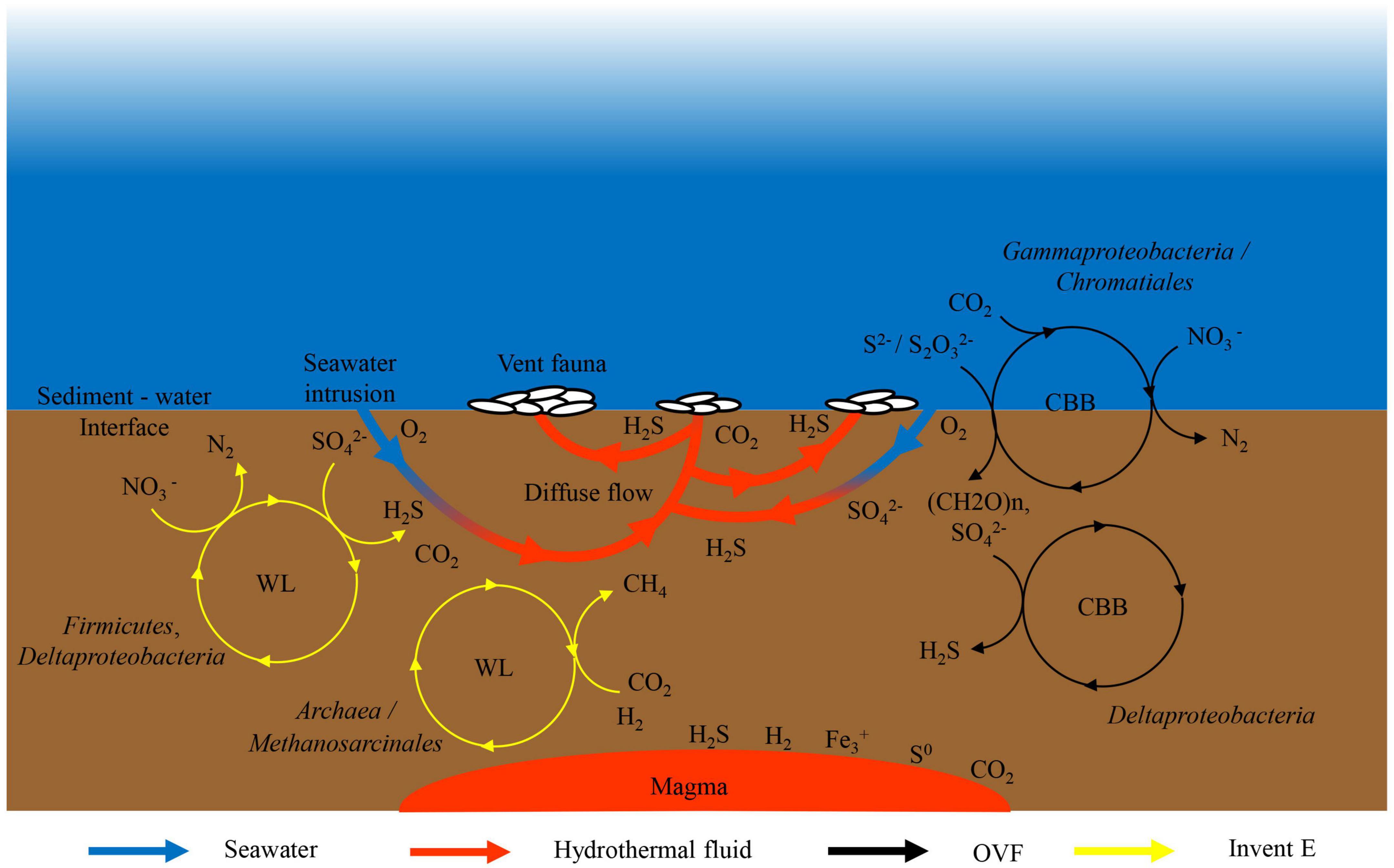
Figure 5. Schematic illustration of key microbial metabolisms of carbon, sulfur, methane, and nitrogen in the diffuse flow hydrothermal vents at OVF and Invent E sites. SO42–, sulfate; S0, sulfur; S2O32–, thiosulfate; S2–, sulfide; H2S, hydrogen sulfide; NO3–, nitrate; N2, nitrogen; H2, hydrogen; CH4, methane; CO2, carbon dioxide; O2, oxygen.
Although our study is limited by lack of replicate samples and sufficient environmental data, our results indicate that OVF and Invent E may exhibit diverging geochemistry reflected in the sediment microbial ecology. Autotrophic Gammaproteobacteria known to be involved in oxidation of reduced sulfur compounds, using oxygen or nitrate as electron acceptors to fix carbon via CBB characterized OVF. The microbial lineages and functional profiles of sediments from diffuse-flow OVF are similar to hydrothermally influenced sediments from highly active vents. This study provides a baseline of the microbial ecology associated with deep-sea sediments from low activity hydrothermal vents in the CIR. Further sampling and investigations are required to broadly determine the microbial metabolic activities and specific contributions of the above-described metabolic pathways in slow to intermediate spreading hydrothermal systems.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm. nih.gov/, PRJNA751690 and http://metagenomics.anl.gov/, mgm4898492.3, mgm4898484.3, and mgm4898483.3.
Author Contributions
TN conducted the experiments and data analyses and prepared the manuscript. YK collected the sediment samples. M-JP assisted in data analyses and graphics. DL contributed metal concentration data. J-HL conceived the study. YK and KK supervised the study. All authors reviewed and approved the final manuscript.
Funding
This research was funded by the Ministry of Oceans and Fisheries Republic of Korea, as part of the project titled “Understanding the deep-sea biosphere on seafloor hydrothermal vents in the Indian Ridge” (Grant No. 20170411).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the crew members and participants on R/V ISABU cruise during the 2019 Indian Ocean Expedition for their assistance in collecting the samples. We would also like to thank Jonguk Kim from Deep-Sea and Seabed Mineral Resources Research Center, KIOST, who provided us with the original map of the study area.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.784807/full#supplementary-material
Footnote
References
Barns, S. M., Fundyga, R. E., Jeffries, M. W., and Pace, N. R. (1994). Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. U.S.A. 91, 1609–1613. doi: 10.1073/pnas.91.5.1609
Beatty, J. T., Overmann, J., Lince, M. T., Manske, A. K., Lang, A. S., Blankenship, R. E., et al. (2005). An obligately photosynthetic bacterial anaerobe from a deep-sea hydrothermal vent. Proc. Natl. Acad. Sci. U.S.A. 102, 9306–9310. doi: 10.1073/pnas.0503674102
Beaulieu, S. E., and Szafrański, K. M. (2020). InterRidge Global Database of Active Submarine Hydrothermal Vent Fields Version 3.4. PANGAEA. doi: 10.1594/PANGAEA.917894
Bowers, R., Kyrpides, N., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Brazelton, W. J., Schrenk, M. O., Kelley, D. S., and Baross, J. A. (2006). Methane- and sulfur-metabolizing microbial communities dominate the Lost City hydrothermal field ecosystem. Appl. Environ. Microbiol. 72, 6257–6270. doi: 10.1128/AEM.00574-06
Cao, H., Wang, Y., Lee, O. O., Zeng, X., Shao, Z., and Qian, P. Y. (2014). Microbial sulfur cycle in two hydrothermal chimneys on the Southwest Indian Ridge. mBio 5:e00980-13. doi: 10.1128/mBio.00980-13
Cerqueira, T., Barroso, C., Froufe, H., Egas, C., and Bettencourt, R. (2018). Metagenomic signatures of microbial Communities in deep-sea hydrothermal sediments of Azores vent fields. Microb. Ecol. 76, 387–403. doi: 10.1007/s00248-018-1144-x
Charlou, J., Donval, J., Fouquet, Y., Jean-Baptiste, P., and Holm, N. (2002). Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field (36 14’ N, MAR). Chem. Geol. 191, 345–359. doi: 10.1016/S0009-2541(02)00134-1
Dick, G. J. (2019). The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat. Rev. Microbiol. 17, 271–283. doi: 10.1038/s41579-019-0160-2
Ding, J., Zhang, Y., Wang, H., Jian, H., Leng, H., and Xiao, X. (2017). Microbial community structure of deep-sea hydrothermal vents on the ultraslow Spreading Southwest Indian Ridge. Front. Microbiol. 8:1012. doi: 10.3389/fmicb.2017.01012
Elkins, J. G., Podar, M., Graham, D. E., Makarova, K. S., Wolf, Y., Randau, L., et al. (2008). A korarchaeal genome reveals insights into the evolution of the Archaea. Proc. Natl. Acad. Sci. U.S.A. 105, 8102–8107. doi: 10.1073/pnas.0801980105
Elsaied, H. E., Kimura, H., and Naganuma, T. (2007). Composition of archaeal, bacterial, and eukaryal RuBisCO genotypes in three Western Pacific arc hydrothermal vent systems. Extremophiles 11, 191–202. doi: 10.1007/s00792-006-0025-2
Fisher, C. R., Takai, K., and Le Bris, N. (2007). Hydrothermal vent ecosystems. Oceanography 20, 14–23. doi: 10.5670/oceanog.2007.75
Francheteau, J., Needham, H., Choukroune, P., Juteau, T., Seguret, M., Ballard, R. D., et al. (1979). Massive deep-sea sulphide ore deposits discovered on the East Pacific Rise. Nature 277, 523–528.
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gamo, T., Chiba, H., Yamanaka, T., Okudaira, T., Hashimoto, J., Tsuchida, S., et al. (2001). Chemical characteristics of newly discovered black smoker fluids and associated hydrothermal plumes at the Rodriguez Triple Junction, Central Indian Ridge. Earth Planet. Sci. Lett. 193, 371–379. doi: 10.1016/S0012-821x(01)00511-8
Gomez-Alvarez, V., Teal, T. K., and Schmidt, T. M. (2009). Systematic artifacts in metagenomes from complex microbial communities. ISME J. 3, 1314–1317. doi: 10.1038/ismej.2009.72
Gugliandolo, C., and Maugeri, T. L. (2019). Phylogenetic diversity of Archaea in shallow hydrothermal vents of Eolian Islands, Italy. Diversity 11:156. doi: 10.3390/d11090156
Halbach, P., Blum, N., Münch, U., Plüger, W., Garbe-Schönberg, D., and Zimmer, M. (1998). Formation and decay of a modern massive sulfide deposit in the Indian Ocean. Miner. Deposita 33, 302–309. doi: 10.1007/s001260050149
Han, Y., Gonnella, G., Adam, N., Schippers, A., Burkhardt, L., Kurtz, S., et al. (2018). Hydrothermal chimneys host habitat-specific microbial communities: analogues for studying the possible impact of mining seafloor massive sulfide deposits. Sci. Rep. 8:10386. doi: 10.1038/s41598-018-28613-5
Hoek, J., Banta, A., Hubler, F., and Reysenbach, A. L. (2003). Microbial diversity of a sulphide spire located in the Edmond deep-sea hydrothermal vent field on the Central Indian Ridge. Geobiology 1, 119–127. doi: 10.1046/j.1472-4669.2003.00015.x
Hou, J., Sievert, S. M., Wang, Y., Seewald, J. S., Natarajan, V. P., Wang, F., et al. (2020). Microbial succession during the transition from active to inactive stages of deep-sea hydrothermal vent sulfide chimneys. Microbiome 8:102. doi: 10.1186/s40168-020-00851-8
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernandez-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Hügler, M., Gärtner, A., and Imhoff, J. F. (2010). Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. 73, 526–537. doi: 10.1111/j.1574-6941.2010.00919.x
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Imhoff, J. F. (2014). “The family chlorobiaceae,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer Berlin Heidelberg), 501–514. doi: 10.1007/978-3-642-38954-2_142
Jiang, H., Lei, R., Ding, S. W., and Zhu, S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. doi: 10.1186/1471-2105-15-182
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2007). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359
Kawagucci, S., Miyazaki, J., Noguchi, T., Okamura, K., Shibuya, T., Watsuji, T., et al. (2016). Fluid chemistry in the Solitaire and Dodo hydrothermal fields of the Central Indian Ridge. Geofluids 16, 988–1005. doi: 10.1111/gfl.12201
Keegan, K. P., Trimble, W. L., Wilkening, J., Wilke, A., Harrison, T., D’Souza, M., et al. (2012). A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput. Biol. 8:e1002541. doi: 10.1371/journal.pcbi.1002541
Kent, W. J. (2002). BLAT – the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kieser, S., Brown, J., Zdobnov, E. M., Trajkovski, M., and McCue, L. A. (2020). ATLAS: a Snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinformatics 21:257. doi: 10.1186/s12859-020-03585-4
Kim, J., Son, S. K., Kim, D., Pak, S. J., Yu, O. H., Walker, S. L., et al. (2020). Discovery of active hydrothermal vent fields along the Central Indian Ridge, 8-12 degrees S. Geochem. Geophys. Geosyst. 21:e2020GC009058. doi: 10.1029/2020GC009058
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lecoeuvre, A., Menez, B., Cannat, M., Chavagnac, V., and Gerard, E. (2021). Microbial ecology of the newly discovered serpentinite-hosted Old City hydrothermal field (southwest Indian ridge). ISME J. 15, 818–832. doi: 10.1038/s41396-020-00816-7
Liao, S., Tao, C., Li, H., Barriga, F. J. A. S., Liang, J., Yang, W., et al. (2018). Bulk geochemistry, sulfur isotope characteristics of the Yuhuang-1 hydrothermal field on the ultraslow-spreading Southwest Indian Ridge. Ore Geol. Rev. 96, 13–27. doi: 10.1016/j.oregeorev.2018.04.007
Liu, C., Cui, Y., Li, X., and Yao, M. (2021). microeco: an R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 97:fiaa255. doi: 10.1093/femsec/fiaa255
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, F., Bagchi, S., Chaterji, S., Gerlach, W., Grama, A., Harrison, T., et al. (2019). MG-RAST version 4-lessons learned from a decade of low-budget ultra-high-throughput metagenome analysis. Brief. Bioinform. 20, 1151–1159. doi: 10.1093/bib/bbx105
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Minic, Z., and Thongbam, P. D. (2011). The biological deep sea hydrothermal vent as a model to study carbondioxide capturing enzymes. Mar. Drugs 9, 719–738. doi: 10.3390/md9050719
Müller, A. L., Kjeldsen, K. U., Rattei, T., Pester, M., and Loy, A. (2015). Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 9, 1152–1165. doi: 10.1038/ismej.2014.208
Münch, U., Lalou, C., Halbach, P., and Fujimoto, H. (2001). Relict hydrothermal events along the super-slow Southwest Indian spreading ridge near 63° 56’ E—mineralogy, chemistry and chronology of sulfide samples. Chem. Geol. 177, 341–349. doi: 10.1016/S0009-2541(00)00418-6
Nakagawa, S., and Takai, K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65, 1–14. doi: 10.1111/j.1574-6941.2008.00502.x
Nakamura, K., and Takai, K. (2014). Theoretical constraints of physical and chemical properties of hydrothermal fluids on variations in chemolithotrophic microbial communities in seafloor hydrothermal systems. Prog. Earth Planet. Sci. 1:5. doi: 10.1186/2197-4284-1-5
Nakamura, K., and Takai, K. (2015). “Indian Ocean hydrothermal systems: seafloor hydrothermal activities, physical and chemical characteristics of hydrothermal fluids, and vent-associated biological communities,” in Subseafloor Biosphere Linked to Hydrothermal Systems: TAIGA Concept, eds J.-I. Ishibashi, K. Okino, and M. Sunamura (Tokyo: Springer Japan), 147–161. doi: 10.1007/978-4-431-54865-2_12
Nakamura, K., Watanabe, H., Miyazaki, J., Takai, K., Kawagucci, S., Noguchi, T., et al. (2012). Discovery of new hydrothermal activity and chemosynthetic fauna on the Central Indian Ridge at 18–20 S. PLoS One 7:e32965. doi: 10.1371/journal.pone.0032965
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., McVeigh, D. H. R., Rajput, B., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H.-Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Pak, S. J., Moon, J. W., Kim, J., Chandler, M. T., Kim, H. S., Son, J., et al. (2017). Widespread tectonic extension at the Central Indian Ridge between 8S and 18S. Gondwana Res. 45, 163–179. doi: 10.1016/j.gr.2016.12.015
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Proskurowski, G., Lilley, M. D., Seewald, J. S., Fruh-Green, G. L., Olson, E. J., Lupton, J. E., et al. (2008). Abiogenic hydrocarbon production at Lost City hydrothermal field. Science 319, 604–607. doi: 10.1126/science.1151194
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2021). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reysenbach, A.-L., Liu, Y., Banta, A. B., Beveridge, T. J., Kirshtein, J. D., Schouten, S., et al. (2006). A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature 442, 444–447. doi: 10.1038/nature04921
Rho, M., Tang, H., and Ye, Y. (2010). FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191. doi: 10.1093/nar/gkq747
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schrenk, M. O., Kelley, D. S., Delaney, J. R., and Baross, J. A. (2003). Incidence and diversity of microorganisms within the walls of an active deep-sea sulfide chimney. Appl. Environ. Microbiol. 69, 3580–3592. doi: 10.1128/Aem.69.6.3580-3592.2003
Sieber, C. M. K., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843. doi: 10.1038/s41564-018-0171-1
Son, J., Pak, S. J., Kim, J., Baker, E. T., You, O. R., Son, S. K., et al. (2014). Tectonic and magmatic control of hydrothermal activity along the slow-spreading Central Indian Ridge, 8 degrees S-17 degrees S. Geochem. Geophys. Geosyst. 15, 2011–2020. doi: 10.1002/2013gc005206
Steinegger, M., and Soding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Stokke, R., Dahle, H., Roalkvam, I., Wissuwa, J., Daae, F. L., Tooming-Klunderud, A., et al. (2015). Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ. Microbiol. 17, 4063–4077. doi: 10.1111/1462-2920.12970
Sylvan, J. B., Sia, T. Y., Haddad, A. G., Briscoe, L. J., Toner, B. M., Girguis, P. R., et al. (2013). Low temperature geomicrobiology follows host rock composition along a geochemical gradient in Lau basin. Front. Microbiol. 4:61. doi: 10.3389/fmicb.2013.00061
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T., and Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS One 9:e105592. doi: 10.1371/journal.pone.0105592
Takai, K., and Sako, Y. (1999). A molecular view of archaeal diversity in marine and terrestrial hot water environments. FEMS Microbiol. Ecol. 28, 177–188. doi: 10.1016/S0168-6496(98)00103-2
Takai, K., Gamo, T., Tsunogai, U., Nakayama, N., Hirayama, H., Nealson, K. H., et al. (2004). Geochemical and microbiological evidence for a hydrogen-based, hyperthermophilic subsurface lithoautotrophic microbial ecosystem (HyperSLiME) beneath an active deep-sea hydrothermal field. Extremophiles 8, 269–282. doi: 10.1007/s00792-004-0386-3
Tao, C. H., Lin, J., Guo, S. Q., Chen, Y. J., Wu, G. H., Han, X. Q., et al. (2012). First active hydrothermal vents on an ultraslow-spreading center: Southwest Indian Ridge. Geology 40, 47–50. doi: 10.1130/G32389.1
Teske, A., Hinrichs, K. U., Edgcomb, V., de Vera Gomez, A., Kysela, D., Sylva, S. P., et al. (2002). Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl. Environ. Microbiol. 68, 1994–2007. doi: 10.1128/AEM.68.4.1994-2007.2002
Thorup, C., Schramm, A., Findlay, A. J., Finster, K. W., and Schreiber, L. (2017). Disguised as a sulfate reducer: growth of the deltaproteobacterium Desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. mBio 8:e00671-17. doi: 10.1128/mBio.00671-17
Van Dover, C. L., Humphris, S. E., Fornari, D., Cavanaugh, C. M., Collier, R., Goffredi, S. K., et al. (2001). Biogeography and ecological setting of Indian Ocean hydrothermal vents. Science 294, 818–823. doi: 10.1126/science.1064574
Wang, Y., Han, X., Petersen, S., Frische, M., Qiu, Z., Li, H., et al. (2017). Mineralogy and trace element geochemistry of sulfide minerals from the Wocan Hydrothermal Field on the slow-spreading Carlsberg Ridge, Indian Ocean. Ore Geol. Rev. 84, 1–19. doi: 10.1016/j.oregeorev.2016.12.020
Wang, Y., Han, X., Zhou, Y., Qiu, Z., Yu, X., Petersen, S., et al. (2020). The Daxi Vent Field: an active mafic-hosted hydrothermal system at a non-transform offset on the slow-spreading Carlsberg Ridge, 6° 48’ N. Ore Geol. Rev. 129:103888. doi: 10.1016/j.oregeorev.2020.103888
Ward, L. M., and Shih, P. M. (2021). Phototrophy and carbon fixation in Chlorobi postdate the rise of oxygen. bioRxiv [preprint]. doi: 10.1101/2021.01.22.427768
Wickham, H., Chang, W., Henry, L., Pedersen, T. L., Takahashi, K., et al. (2016). ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. R Package Version. 3.3.5.
Wilke, A., Harrison, T., Wilkening, J., Field, D., Glass, E. M., Kyrpides, N., et al. (2012). The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13:141. doi: 10.1186/1471-2105-13-141
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Yamamoto, M., and Takai, K. (2011). Sulfur metabolisms in Epsilon- and Gamma-Proteobacteria in deep-sea hydrothermal fields. Front. Microbiol. 2:192. doi: 10.3389/fmicb.2011.00192
Yan, L. J., Yu, D., Hui, N., Naanuri, E., Viggor, S., Gafarov, A., et al. (2018). Distribution of archaeal communities along the coast of the Gulf of Finland and their response to oil contamination. Front. Microbiol. 9:15. doi: 10.3389/fmicb.2018.00015
Yang, W., Tao, C., Li, H., Liang, J., Liao, S., Long, J., et al. (2017). 230Th/238 U dating of hydrothermal sulfides from Duanqiao hydrothermal field, Southwest Indian Ridge. Mar. Geophys. Res 38, 71–83. doi: 10.1007/s11001-016-9279-y
Yang, Z., Xiao, X., and Zhang, Y. (2020). Microbial diversity of sediments from an inactive hydrothermal vent field, Southwest Indian Ridge. Mar. Life Sci. Technol. 2, 73–86. doi: 10.1007/s42995-019-00007-0
Zeng, X., Alain, K., and Shao, Z. (2021). Microorganisms from deep-sea hydrothermal vents. Mar. Life Sci. Technol. 3, 204–230. doi: 10.1007/s42995-020-00086-4
Zhou, Y. D., Zhang, D. S., Zhang, R. Y., Liu, Z. S., Tao, C. H., Lu, B., et al. (2018). Characterization of vent fauna at three hydrothermal vent fields on the Southwest Indian Ridge: implications for biogeography and interannual dynamics on ultraslow-spreading ridges. Deep Sea Res. Part I 137, 1–12. doi: 10.1016/j.dsr.2018.05.001
Keywords: slow to intermediate spreading, hydrothermal vent, microbial community, metagenome, Central Indian Ridge (CIR)
Citation: Namirimu T, Kim YJ, Park M-J, Lim D, Lee J-H and Kwon KK (2022) Microbial Community Structure and Functional Potential of Deep-Sea Sediments on Low Activity Hydrothermal Area in the Central Indian Ridge. Front. Mar. Sci. 9:784807. doi: 10.3389/fmars.2022.784807
Received: 28 September 2021; Accepted: 17 January 2022;
Published: 17 February 2022.
Edited by:
Pei-Yuan Qian, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaReviewed by:
Man Kit Cheung, The Chinese University of Hong Kong, ChinaZongze Shao, Third Institute of Oceanography, China
Copyright © 2022 Namirimu, Kim, Park, Lim, Lee and Kwon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kae Kyoung Kwon, a2trd29uQGtpb3N0LmFjLmty