 Shen Jean Lim
Shen Jean Lim Luke R. Thompson
Luke R. Thompson Kelly D. Goodwin
Kelly D. Goodwin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 09 January 2023
Sec. Deep-Sea Environments and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1067482
This article is part of the Research TopicBiodegradation of Emerging Marine ContaminantsView all 8 articles
The methane ice worm Sirsoe methanicola is the only marine polychaete species observed to colonize the methane hydrates of the Gulf of Mexico. Methane hydrates are ephemeral features of deep-sea cold seeps, and finding worm-colonized hydrates is rare; thus, little is known about these organisms. Recent metagenomic analysis predicted prokaryotic taxa and pathways from S. methanicola gut contents and worm fragments. Here, we increase the genetic information known about S. methanicola by assembling its nuclear rRNA genes (18S rRNA and 28S rRNA), mitochondrial genome (mitogenome), and other protein-coding genes from metagenomic data. Assembled 18S rRNA and 28S rRNA gene sequences of S. methanicola were near-identical to previously reported S. methanicola sequences. The 17,403-bp mitogenome of S. methanicola is the first mitogenome sequence of the family Hesionidae, consisting of 39.03% G+C content, 13 protein-coding genes, 24 tRNAs (including two split trnM genes), and 2 rRNA genes. Protein-coding genes in the S. methanicola metagenomes assigned to the phylum Annelida were involved in cell adhesion, signaling, ubiquitin system, metabolism, transport, and other processes. From the metagenomes, we also found 42 homologs of the cytochrome P450 (CYP) superfamily putatively involved in polycyclic aromatic hydrocarbon (PAH) metabolism. Our results encourage further studies into the genetic adaptations of S. methanicola to its methane hydrate habitat, especially in the context of deep-sea ecology and nutrient cycling.
Methane is a potent greenhouse gas, and methane hydrates represent one of the largest carbon reservoirs in the world; therefore, the dynamics of these deposits garner interest with regard to global carbon cycles, climate change, and as sources of alternative energy (National Energy Technology Laboratory, 2017). In marine cold seep locations, methane rises from the seafloor and may freeze into a crystalline clathrate structure under sufficiently low temperature and high pressure. These deposits are commonly known as methane hydrates (also called methane clathrates, gas hydrates, methane ice, hydromethane, or fire ice) (Kvenvolden, 1995; Tunnicliffe et al., 2003). A variety of species have been identified at cold seeps, such as chemosymbiotic bivalves, polychaetes, shrimps, amphipods, cnidarians, and sponges, but most invertebrates do not interact physically with gas hydrates (Desbruyères and Toulmond, 1998; Sibuet and Olu, 1998; Fisher et al., 2000; Tunnicliffe et al., 2003; Van Dover et al., 2003; Levin, 2005; Dubilier et al., 2008). A notable exception is the marine polychaete, Sirsoe methanicola (previously Hesiocaeca methanicola). First discovered in 1997 in the Green Canyon area of the Gulf of Mexico, USA, S. methanicola creates burrows in methane hydrates and inhabits them mostly with single occupancy (Desbruyères and Toulmond, 1998; Fisher et al., 2000). Members of the Sirsoe genus have also been found in deep-sea whale falls, vents, and seeps, but S. methanicola is the only known Sirsoe species to inhabit methane hydrates (Shimabukuro et al., 2019) and is the only macrofauna known to inhabit deposits in the Gulf of Mexico. Besides S. methanicola, a novel alvinocarid shrimp morphospecies has been found atop exposed methane hydrates in the Blake Ridge Diapir of the South Atlantic Bight (Van Dover et al., 2003).
Sirsoe methanicola could play important ecological roles in the methane hydrate habitat through bioturbation, methane release, and subsequent methane hydrate dissociation. Sirsoe methanicola is thought to introduce oxygen to a methane hydrate by generating water currents on the surface with its parapodia (Fisher et al., 2000). The resulting oxygen can support microaerophilic microbial growth and facilitate depression formation on the methane hydrate surface, leading to its subsequent dissociation (Fisher et al., 2000).
Sirsoe methanicola possesses a functional digestive system with a gut and appears to be a bacterivore feeding on a variety of bacteria from the surface of gas hydrates, although details of its life history or genetic capacities are not fully understood (Fisher et al., 2000; Becker et al., 2013). Methane hydrates provide a variety of potential substrates to support microbial life, including thermogenic methane, hydrogen sulfide, hydrocarbon gases (such as ethane, propane, isobutane, butane, and pentane), and carbon dioxide (Kvenvolden, 1995; Fisher et al., 2000; Lanoil et al., 2001; Joye et al., 2004; Mills et al., 2005). Despite the “methane” moniker of the ice worm habitat, our recent metagenomic analysis of S. methanicola gut contents and worm fragments revealed a paucity of reads assigned to aerobic or anaerobic methanotrophic taxa (Lim et al., 2022). Metagenomes associated with S. methanicola were instead dominated by Sulfurospirillum and included other prokaryotic taxa capable of nitrogen, sulfur, and carbon cycling (Lim et al., 2022).
From the S. methanicola metagenomes, we identified microbial genes involved in the degradation of hydrocarbon compounds, such as alkanes, benzoate, toluene, xylene, and phenol (Lim et al., 2022). Results were consistent with reports of low and high molecular weight hydrocarbons found in the methane hydrate where S. methanicola was initially collected (Fisher et al., 2000) and the emanation of a petroleum smell from the guts of S. methanicola individuals during dissection (Lim et al., 2022). The high number of metagenomic reads sequenced during the microbiome study (>2M paired-end reads per library) provided an opportunity to mine for host-related genes (Lim et al., 2022). Here we recovered and analyzed mitogenome, gene, and cytochrome P450 (CYP) enzyme superfamily annotations to explore the genetic features of this deep sea polychaete in relation to its unique deep-sea ecology.
Although little is known about S. methanicola, the shallow marine polychaete Capitella teleta has been well studied. Capitella teleta feeds on shallow marine sediments rich in organic matter such as fuel oil and other pollutants (Blake et al., 2009). Degradation of the polycyclic aromatic hydrocarbon (PAH) fluoranthene by C. teleta has been demonstrated, likely without aid from its gut microbiome (Forbes et al., 2001; Selck et al., 2003; Jang et al., 2020). Involvement of CYP was postulated because CYP-dependent activity and CYP expression in C. teleta increased with PAH exposure (Li et al., 2004; Dejong and Wilson, 2014). Sequencing has been described for the C. teleta genome (Simakov et al., 2013) with an annotated CYPome (Dejong and Wilson, 2014) and for the gut microbiome (Hochstein et al., 2019; Jang et al., 2020; Jang et al., 2021). Given the hydrocarbon rich habitat of S. methanicola (Fisher et al., 2000), we hypothesized that the S. methanicola genetic repertoire may include analogous hydrocarbon degradation capability.
Procedures for sample collection, processing, sequencing, and metagenomic analyses were previously documented (Lim et al., 2022), including diagrams that illustrated sample collection design. Details of bioinformatic methods were provided in the supplemental information. Methods are re-summarized here for the readers’ convenience, with additional details provided for analysis of eukaryotic sequences.
Live S. methanicola specimens were collected as part of the R/V Seward Johnson cruise SJ-2009-GOM, operated by Harbor Branch Oceanographic Institute, Fort Pierce, FL, USA. Using the manipulator arm of the crewed Johnson Sea-Link II, specimens were retrieved from a Gulf of Mexico methane hydrate in the Green Canyon area GC234 (27°44.7526’ N, 91°13.3168’ W) at a depth of 542.8 meters on October 3, 2009 at 10:31 am UTC during Dive #3751. All specimens were rinsed with 0.2-μm filtered seawater prior to aseptic dissection to expose the worm gut, and five out of the seven dissected specimens were spawned on the ship prior to dissection.
Gut contents were extracted from the seven specimens with a sterile syringe without removal of the gut itself. Samples were placed into microcentrifuge tubes, centrifuged at 13,200 rpm for 15 minutes on the ship, and the supernatant was removed. The remaining pellet was preserved in 95% ethanol and frozen at –80°C on the ship and upon returning to the laboratory. The sample used for Illumina HiSeq sequencing (Tube A) contained gut contents pooled from two unspawned worms. The sample used for Illumina MiSeq sequencing (Tube D) contained the gut contents of one spawned worm. Worm fragments contained various tissues (including heads and bristles, and guts) left over from the dissection and gut content extraction. These were pooled in a 4-ml sterile polycarbonate tube, covered with 95% ethanol, and stored at –20°C on the ship and at –80°C upon returning to the laboratory. The sample used for HiSeq sequencing (Tube Loc-2) contained the worm fragments pooled from all seven dissected worms.
DNA was extracted from all processed samples using the Qiagen DNeasy Tissue & Blood Kit (Valencia, CA, USA) on February 6, 2012 and quantified using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For Illumina HiSeq sequencing, 1 μg of DNA each from Tube A and Tube Loc-2 was sheared into 200–300-bp fragments using the Covaris S2 instrument (Woburn, MA, USA) to produce library G and library W, respectively. The fragments were end-repaired and used for library preparation using the TruSeq DNA Sample Preparation Kit (Illumina) with 12 cycles of amplification. From each library, 200–250-bp fragments were selected using gel size selection for paired-end sequencing at Scripps Research (formerly The Scripps Research Institute; La Jolla, CA, USA) on a single lane of the Illumina HiSeq 2000 (2 x 100 bp) platform. For Illumina MiSeq sequencing, the Nextera XT library preparation kit (Illumina) was used to prepare metagenomic library G-Mi from Tube D. This library was quantitated using the Qubit™ dsDNA assay (Life Technologies, Austin, TX, USA) and sequenced on the Illumina MiSeq (2 x 300 bp) platform at San Diego State University.
Reads from the three metagenomic libraries (W, G, and G-Mi) were assembled by various software to obtain full-length small subunit (SSU) rRNA sequences, whole metagenomes, and the worm mitogenome, as detailed below.
Reads from each library were assembled separately into full-length eukaryotic and prokaryotic SSU rRNA sequences using the default parameters of phyloFlash v3.4 (Gruber-Vodicka et al., 2020). Assembled sequences classified by phyloFlash as S. methanicola were searched against NCBI GenBank (Benson et al., 2018) via the NCBI blastn web interface (Johnson et al., 2008) to identify matching genes and sequences (Table 1).

Table 1 Taxonomic assignments to the polychaete S. methanicola based on nuclear 18S rRNA and 28S rRNA gene annotations recovered from the three metagenomic libraries.
Reads were trimmed at Q-score threshold of 30 using Trim Galore! V0.6.5 (https://github.com/FelixKrueger/TrimGalore), a wrapper tool around cutadapt (Martin, 2011), and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Read qualities pre- and post-trimming were assessed with FastQC v0.11.9. Each metagenomic library was individually assembled on the KBase server (Arkin et al., 2018) using MEGAHIT v1.2.9 (Li et al., 2016) with the “meta-large” preset option for large and complex assembly (kmin=21, kmax=99, and kstep=20). Reads from the three metagenomic libraries were not co-assembled because of computational resource limitations. The MiSeq-sequenced library G-Mi from the gut contents in Tube D was additionally assembled using the “mega-sensitive” option of MEGAHIT (kmin=21, kmax=255, and kstep=20), the default parameters of IDBA-UD v1.1.3 (Peng et al., 2012), and metaSPAdes v3.13.0 (k=21,33,55,77,99,127) (Nurk et al., 2017). For consistency with other HiSeq-sequenced metagenomes, the metagenome assembled from G-Mi with the “meta-large” preset option was used for 18S and 28S rRNA gene sequence retrieval and functional annotations.
The 28S rRNA gene sequence from the ribosomal large subunit (LSU) of S. methanicola was retrieved by running blastn searches implemented in BLAST 2.10.1+ (Camacho et al., 2009). Published 28S rRNA gene sequence (NCBI accession: DQ442611) for S. methanicola (Ruta et al., 2007) were queried against the MEGAHIT-assembled metagenomes (Li et al., 2016) from the gut contents and worm fragments (Table 1).
Draft mitogenomes of S. methanicola were recovered by querying previously reported 16S rRNA gene sequence (NCBI accession: DQ442582) and cytochrome c oxidase subunit I (cox1) sequence (NCBI accession: DQ513295) for S. methanicola (Ruta et al., 2007) against the MEGAHIT-assembled metagenomes from the gut contents and worm fragments using the blastn function in the BLAST+ application (Camacho et al., 2009). Matching 18,000-bp and 16,108-bp contigs from the gut content and worm fragment metagenomes, respectively, were searched against NCBI GenBank using the web blastn interface (Johnson et al., 2008). These contigs were deduced to be S. methanicola mitogenomic sequences, based on matches to mitochondrial sequences from the genus Sirsoe and matches to mitogenomes of Polychaeta species.
Sequences from both S. methanicola draft mitogenomes were compared against each other using the web blastn interface and annotated with the MITOS web server (Bernt et al., 2013) and MitoZ v2.3 (Meng et al., 2019) based on the invertebrate mitochondrial code. The draft mitogenome assembled from the gut contents contained no missing genes, while the draft mitogenome from the worm fragments was missing 16 genes. The mitogenome assembled from the gut contents was retained for further annotation. Gene annotations produced by MITOS and MitoZ were manually reviewed and corrected by aligning the S. methanicola draft mitogenome with the Goniada japonica mitogenome (NC_026995/KP867019) (Chen et al., 2016), which was identified by MitoZ to be the most closely related to the S. methanicola draft genomes. Manual mitogenome annotation was aided by the blastn web interface with the Coding Sequences (CDS) feature display and performed according to tutorials published by NCBI (https://support.nlm.nih.gov/knowledgebase/article/KA-05223/en-us). Internal stop codons were identified in the sequences encoding cytochrome c oxidase subunit I (cox1) and NADH dehydrogenase subunit 2 (nd2). The internal stop codon in cox1 was due to a 497-bp insertion in the S. methanicola draft mitogenome and these bases were manually removed. The internal stop codon in NADH dehydrogenase subunit 2 (nd2) was due to an insertion causing a frameshift in the S. methanicola draft mitogenome. This frameshift was additionally verified by aligning the translated nucleotide sequence of nd2 in the S. methanicola draft genome with the protein and nucleotide sequences of nd2 in Hesionides sp. PA-2020 (MN855167/QHT64973) (Alves et al., 2020) using web blastx and tblastn searches against NCBI nr/nt. To correct the frameshift, a 100-bp gene region that was not homologous to Goniada japonica and Hesionides sp. PA-2020 nd2 sequences was removed from the S. methanicola draft mitogenome. The original draft mitogenomes recovered from the gut content (18,000 bp) and worm fragment (16,108 bp) metagenomes are provided in the Supplementary Data, and the manually annotated S. methanicola mitogenome is deposited to NCBI RefSeq with the accession number NC_064058.
MitoZ (Meng et al., 2019) was used to compute the G+C content (window size=50) and sequencing depth of each library (library G-Mi, library G, and library W) along the final representative S. methanicola mitogenome. The GenBank annotations, G+C content, and depth data files were used to visualize the S. methanicola genome with circos v0.69-8 (Krzywinski et al., 2009), based on the circos configuration file templates generated by MitoZ. MEGAX (Kumar et al., 2018) was used to calculate the relative synonymous codon usage (RSCU) in the S. methanicola mitogenome, which is the frequency of a codon divided by the average frequency of all synonymous codons for an amino acid (Sharp and Li, 1987). An RSCU value of 1 indicates no codon usage bias, while RSCU values above and below 1 represent positive and negative bias, respectively. Codons with RSCU values >1.6 were considered overrepresented, while codons with RSCU values <0.6 were considered underrepresented (Wong et al., 2010).
Nucleotide sequences of 16S rRNA, 18S rRNA, 28S rRNA, and cox1 genes assembled from S. methanicola (see subsections 2.3.1, 2.3.2.1, and 2.3.2.2) were concatenated and used for phylogenetic analysis. Comparisons utilized a subset of species selected from Table 1 of Rouse et al. (Rouse et al., 2018) that had reference sequences available for all four of the analyzed genes (16S rRNA, 18S rRNA, 28S rRNA, and cox1). These included 32 species from the family Hesionidae and the outgroup species Dsyponetus caecus from family Chrysopetalidae (Table 2). Sequences from the genus Sirsoe, including S. methanicola, S. dalailamai, S. munki, and S. sirikos, were part of a larger clade of the hesionid subfamily Psamathinae (Pleijel, 1998).
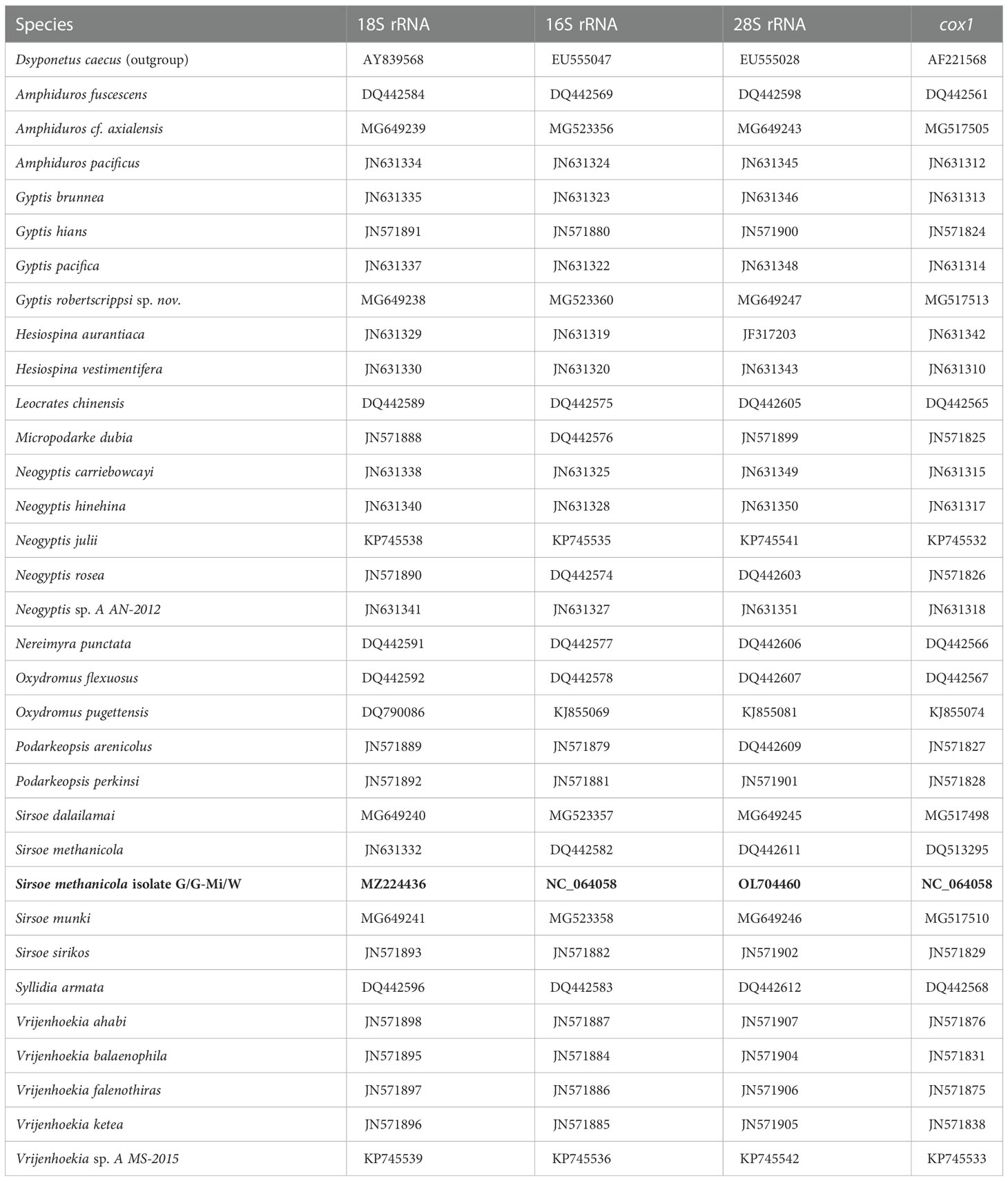
Table 2 GenBank accession numbers for Hesionidae and Chrysopetalidae (outgroup) species used for phylogenetic analysis. Sirsoe methanicola sequences assembled from this study are highlighted in bold.
In addition, amino acid sequences of protein-coding genes (PCGs) annotated in the assembled S. methanicola mitogenome (see subsection 2.3.2.2) were compared with published mitogenomes. No assembled Hesionidae mitogenomes were available; therefore, 35 mitogenomes from the order Phyllodocida (Table S1) were retrieved from NCBI’s Organelle Genome Resources (Sayers et al., 2021). Additionally, the S. methanicola mitogenome sequence was queried against NCBI GenBank (Benson et al., 2018) using the NCBI blastn web interface (Johnson et al., 2008) to identify two other Phyllodocida mitogenomes not listed in Organelle Genome Resources (Table S1). The mitogenome of Hydroides elegans from the polychaete order Sabellida, retrieved from Organelle Genome Resources, was used as the outgroup (Table S1). Phylogenetic analysis employed 12 of 13 PCGs annotated in these 37 Phyllodocida mitogenomes. The atp8 gene encoding ATP synthase F0 subunit 8 was excluded because it was absent in three mitogenomes (Table S1).
All sequences were downloaded from the NCBI database. Sequences for each gene were separately aligned with MAFFT v7.475 using the L-INS-I option recommended for <200 sequences, which is an iterative refinement method that produces accurate multiple sequence alignments with local pairwise alignment information (Katoh and Standley, 2013). Conserved blocks within each multiple sequence alignment were identified using gblocks v0.91b (Castresana, 2000). Using gblocks, conserved blocks for 16S rRNA, 18S rRNA, 28S rRNA, and cox1 were concatenated into a single nucleotide sequence alignment, while conserved blocks for the protein-coding genes in the mitogenomes were concatenated into a single amino acid sequence alignment.
MEGAX (Kumar et al., 2018) was used to identify the best model for each concatenated alignment. For the concatenated 16S rRNA+18S rRNA+28S rRNA+cox1 alignment, the best model was the General Time Reversible model (Nei and Kumar, 2000) with a discrete Gamma distribution that allows for evolutionary invariable sites (GTR+G+I). For the concatenated amino acid sequence alignment from mitogenomes, the best model was the General Reversible Mitochondria model (Adachi and Hasegawa, 1996) with Gamma distribution and frequencies (mtREV24+G+F). Phylogenetic trees for both concatenated alignments were constructed separately in MEGAX (Kumar et al., 2018) using the Maximum Likelihood method with 1,000 bootstrap replicates. The Maximum Composite Likelihood (MCL) or Jones-Taylor-Thornton model (Jones et al., 1992) was used to estimate a pairwise distance matrix for the nucleotide and protein sequence alignments, respectively. These matrices were used to search for initial trees heuristically using Neighbor-Join and BioNJ algorithms. Subsequently, a tree topology with the highest log likelihood value was predicted for each concatenated alignment.
Metagenomic reads obtained from S. methanicola gut contents (library G) and worm fragments (library W) were mapped separately to the genome of C. teleta (NCBI accession: GCA_000328365). Worm-related sequences were identified in these metagenomes by mapping trimmed reads using the default parameters of Bowtie2 v2.4.1 (Langmead and Salzberg, 2012) and SAMtools v1.10 (Li et al., 2009). Reads mapped to each C. teleta coding sequence were counted using HTSeq v0.12.4 (Anders et al., 2015). Each mapped C. teleta coding sequence was matched to the corresponding protein or nucleotide sequence in the S. methanicola metagenomes through blastp and tblastn searches performed using BLAST+ (Camacho et al., 2009), respectively. Mapping results of HiSeq-sequenced reads from libraries G and W were reported; results for MiSeq-sequenced reads from library G-Mi were not reported because of low coverage.
From the metagenomes assembled by MEGAHIT (Li et al., 2016), 346,292 nucleotide sequences that did not bin into bacterial metagenome-assembled genomes (MAGs) (Lim et al., 2022) were retrieved from the three libraries W, G, and G-Mi. These sequences were combined and annotated using the WebAUGUSTUS server (Hoff and Stanke, 2013). A training set containing the nucleotide and protein sequences in the genome of C. teleta (GCA_000328365) (Simakov et al., 2013) was submitted to WebAUGUSTUS to generate parameters for eukaryotic gene prediction in S. methanicola. From the C. teleta genomic and protein data, eukaryotic protein-coding genes in the S. methanicola metagenomes were predicted ab initio by WebAUGUSTUS using the default parameters (report genes on both strands; no alternative transcripts; and predict partial and complete genes). Assembled contigs >1,000 bp from the S. methanicola metagenomes were used for eukaryotic gene prediction. Since WebAUGUSTUS has an input limit of 250,000 sequences per prediction job, assembled S. methanicola nucleotide sequences from all metagenomes were split into two datasets containing 195,167 contigs that were ≥3,000 bp long and 151,125 contigs that were <3,000 bp.
Predicted protein sequences from the S. methanicola metagenomes were submitted to the ghostKOALA web server (Kanehisa et al., 2016) maintained by the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2020) for the assignment of taxonomy and KEGG Orthology (KO) terms. Protein sequences classified by ghostKOALA as Annelids were extracted based on similarities to sequences in the KEGG Genome database (Kanehisa et al., 2020). All KO terms classified as Annelids were submitted to the KEGG mapper server to identify complete pathway modules. Protein sequences in the complete pathway module M00141 (C1-unit interconversion, eukaryotes) were verified to be homologs of annelid sequences through web blastp searches against NCBI nr (Johnson et al., 2008).
We compared 84 CYP superfamily protein sequences described in C. teleta (Dejong and Wilson, 2014) with those predicted from the S. methanicola metagenomes to identify eukaryotic CYP homologs that may respond to or detoxify PAHs. Based on the HTSeq output, no reads were mapped to the P450 genomic regions. Cytochrome P450 protein sequences in S. methanicola were alternatively identified by clustering WebAUGUSTUS-predicted protein sequences in the Sirsoe methanicola metagenomes with C. teleta cytochrome P450 protein sequences sequentially at 100%, 90%, 80%, 70%, 60%, 50%, 40% and 30% (psi-cd-hit command) global identity thresholds using CD-HIT (Li and Godzik, 2006). Sirsoe methanicola protein sequences that clustered with C. teleta’s cytochrome P450 protein sequences were verified to be annelid CYP sequence homologs through web blastp searches against NCBI nr (Johnson et al., 2008). Sequence clusters were visualized with the igraph v1.2.6 R package (Csárdi and Nepusz, 2006).
A total of three metagenomic libraries were sequenced from S. methanicola specimens collected from a Gulf of Mexico methane hydrate located at GC234 (27°44.7526’ N, 91°13.3168’ W; Figure 1). Metagenomic sequencing provided the following numbers of paired-end reads: 1) 236.8M from gut contents pooled from two worms (HiSeq library G, Tube A); 2) 244.1M for non-axenic worm fragments containing gut tissues (HiSeq library W, Tube Loc-2); and 3) 1.3M for a gut content library sequenced with Illumina MiSeq (library G-Mi, Tube D). The text here details assignments associated with the host organism, S. methanicola. Analysis of prokaryotic diversity and function for this data set are provided under separate cover (Lim et al., 2022).

Figure 1 (A) Sirsoe methanicola individual viewed under a compound microscope (photo credit: R. Emlet/C. Young aboard R/V Seward Johnson; (B) S. methanicola individuals colonizing depressions on a methane hydrate (photo credit: SJ-2009-GOM-JSL2-3751-014, Johnson Sea Link II, Harbor Branch Oceanographic Institute); (C) Map showing location of the sampling site (generated from https://www.simplemappr.net. Accessed December 15, 2022).
Assembly by phyloFlash (Gruber-Vodicka et al., 2020) yielded 1,817-bp 18S rRNA gene sequences from library G and library G-Mi and a 1,781-bp 18S rRNA gene sequence from library W (Table 1). Based on web blastn alignments, these sequences were 100% identical to each other and to another 1,778-bp segment of the 18S rRNA gene sequence published for S. methanicola (NCBI accession: JN631332) (Pleijel et al., 2012) (Table 1). A previous 18S rRNA sequence (1,745 bp) obtained from a clone library using the S. methanicola samples collected in this study (Xin, 2013) showed 99% identity to JN631332.
All three metagenomes assembled with MEGAHIT (Li et al., 2016) identified 28S rRNA LSU gene sequences that were 782 bp in length and 100% identical to each other (Table 1). These sequences shared 99% global sequence identity with a 770-bp segment of the 28S rRNA gene sequence published for S. methanicola (NCBI accession: DQ442611) (Ruta et al., 2007), differing only by one gap at position 417 in the alignment with DQ442611 (Table 1).
From the metagenomes assembled with MEGAHIT (Li et al., 2016), we recovered draft mitogenomes of S. methanicola through sequence searches using the previously reported 16S rRNA gene sequence (NCBI accession: DQ442582) and cox1 sequence (NCBI accession: DQ513295) for S. methanicola (Ruta et al., 2007) as query. Searching against the gut content metagenome from the MiSeq-sequenced library G-Mi produced no hit. However, matching 16S rRNA and cox1 sequences were identified in a 18,000-bp contig assembled from the HiSeq-sequenced library G and another 16,108-bp contig assembled from the HiSeq-sequenced library W. Both contigs were deduced to be S. methanicola mitogenomic sequences, based on matches to mitochondrial sequences from the genus Sirsoe and matches to mitogenomes of Polychaeta species in NCBI GenBank (Benson et al., 2018). Based on MITOS (Bernt et al., 2013) and MitoZ (Meng et al., 2019) annotations, these draft mitogenomes were non-circular. The draft mitogenome assembled from the gut contents contained 38 genes with no missing genes, while the draft mitogenome assembled from the worm fragments contained only 22 genes and was missing 16 genes. Local alignment using the blastn web interface showed a shared region of 10,066 bp between both mitogenomes with 99% sequence identity and one gap. The mitogenome assembled from the gut contents was retained for downstream annotation and analysis to produce a representative mitogenome of S. methanicola (NCBI accessions: NC_064058/OM914591; Figure 2).
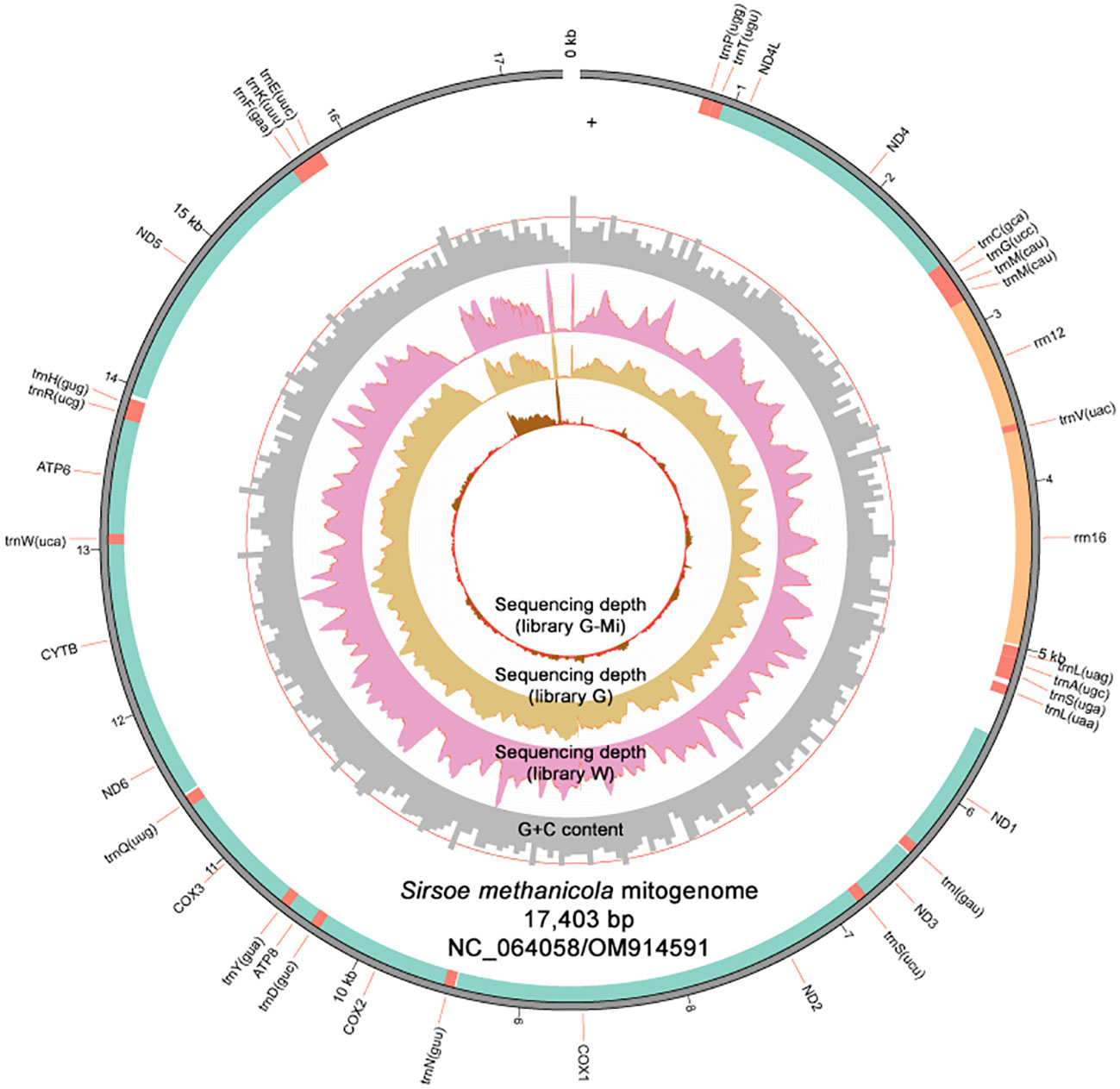
Figure 2 Map of the linear 17,403-bp S. methanicola mitogenome (NCBI accessions: NC_064058/OM91459) visualized by Circos implemented in MitoZ. The mitogenome includes protein-coding (green), rRNA (orange), and tRNA (red) gene regions. The red line on the G+C content track marks the G+C content threshold of 50%. The red lines on the depth distribution for library G-Mi indicate regions with sequencing depths lower than 20.
The S. methanicola mitogenome was non-circular, consisting of 17,403 bp with 39.03% G+C content. Regions of low sequencing depths across all three libraries were mostly observed at the end of the mitogenome between 16 kb and 17.3 kb (Figure 2). The mitogenome contained 13 PCGs, 24 tRNAs (including two split trnM genes), and 2 rRNAs (12S rRNA and 16S rRNA). All genes were located on the positive strand of the mitogenome. The full-length 1,302-bp 16S rRNA gene was 99% identical to a 538-bp partial 16S rRNA gene sequence reported for S. methanicola (NCBI accession: DQ442582) (Ruta et al., 2007), differing by four nucleotides. The full-length 1,536-bp cox1 gene shared 99% identity with a 629-bp partial cox1 gene sequence (NCBI accession: DQ513295) (Pleijel et al., 2008) with two nucleotide mismatches.
Most PCGs identified in S. methanicola used AUG as the start codon, except for three that used AUC as an alternative start codon. Stop codons used in the S. methanicola mitogenome included the truncated U– stop codon in seven PCGs, UAA in four PCGs, and UAG in NADH dehydrogenase subunit 6 (nd6). Analysis of RSCU values of 64 codons showed positive bias for half of the codons (RSCU >1) and negative bias for the other half (Figure 3). UAA, the preferred stop codon over UAG, and amino acids with only two codons showed positive bias for one over the other (Figure 3). More than one codon was preferred for alanine, glycine, leucine, proline, serine, threonine, and valine (Figure 3). Among these, the UCU codon for serine was over-represented with RSCU >1.6 (Figure 3). Underrepresented codons with RSCU <0.6 included CUG for leucine, AUG for methionine, CCG for proline, ACG for threonine, GCG for alanine and AGU and AGG for serine (Figure 3).
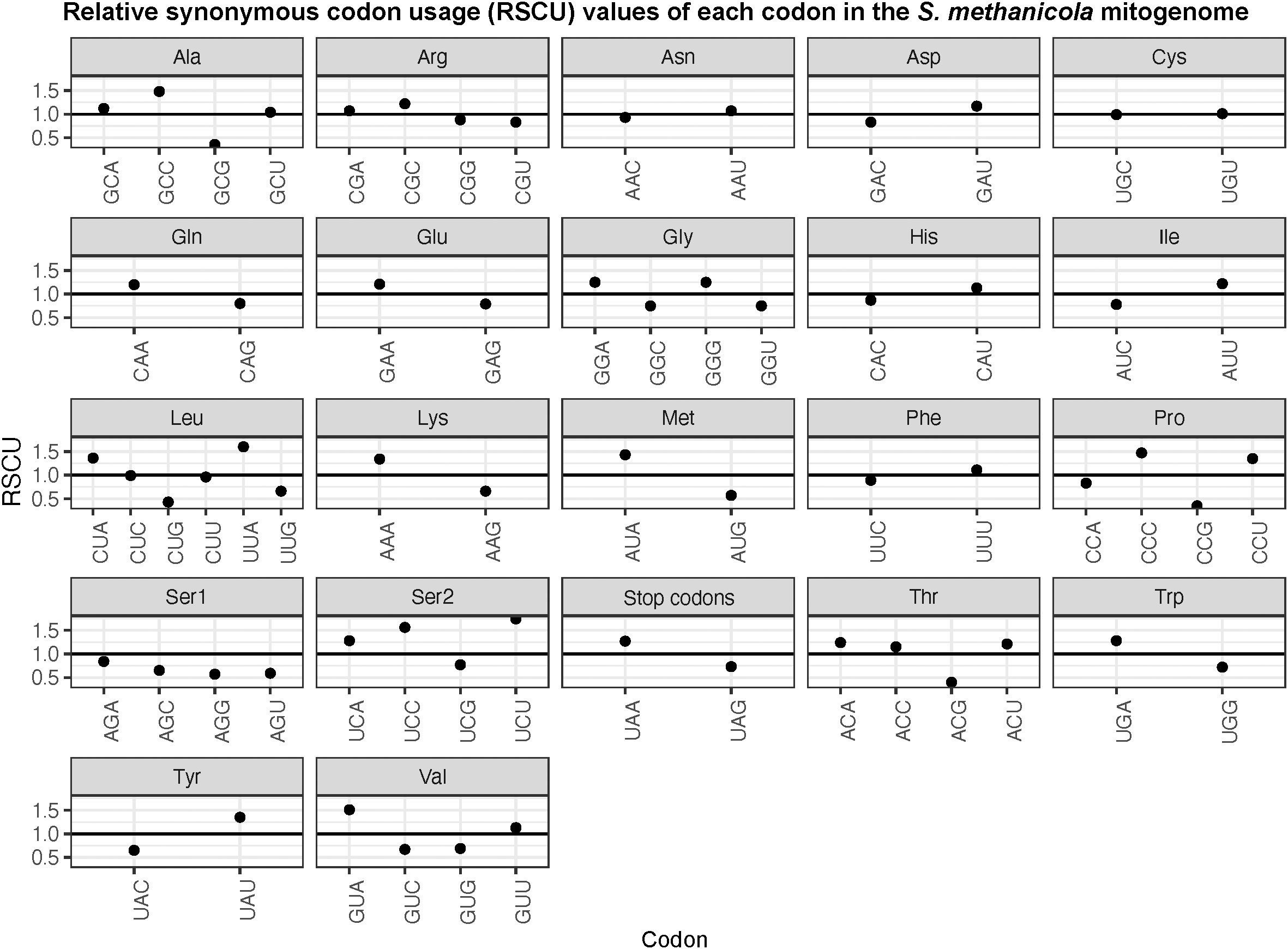
Figure 3 Relative synonymous codon usage (RSCU) values for each codon of each amino acid in the S. methanicola mitogenome. The black horizontal line on each plot marks the RSCU threshold of 1.
The phylogeny of concatenated 16S rRNA, 18S rRNA, 28S rRNA, and cox1 nucleotide gene sequences from the S. methanicola metagenome in relation to sequences available for the family Hesionidae (Figure 4) was consistent with the most recent published phylogeny (Rouse et al., 2018). Sirsoe methanicola sequences from this study were most closely related to previously published S. methanicola sequences (Ruta et al., 2007), and S. methanicola was most closely related to its sister species S. dalailamai (Rouse et al., 2018).
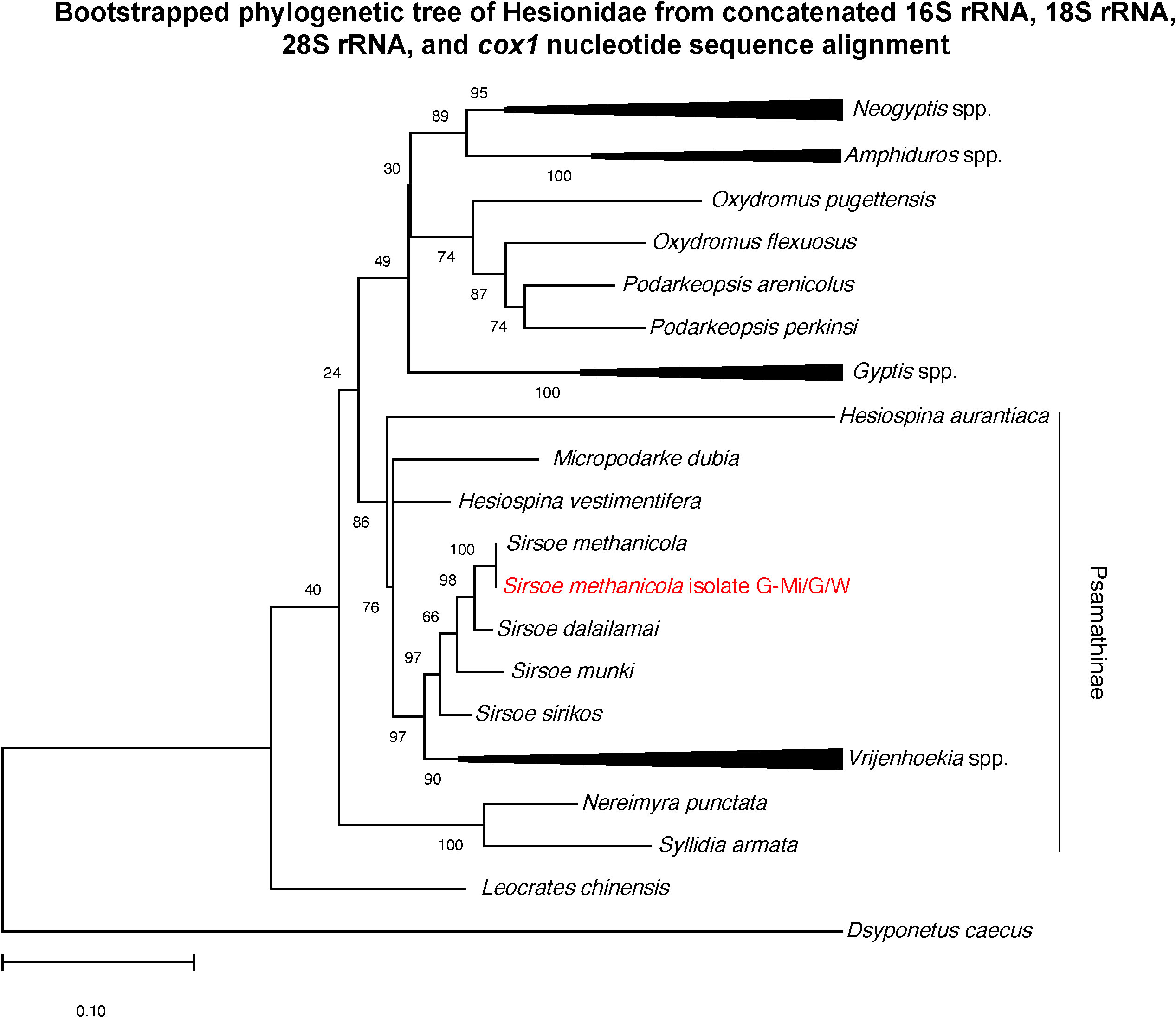
Figure 4 Bootstrapped maximum likelihood phylogenetic tree of concatenated 16S rRNA, 18S rRNA, 28S rRNA, and cox1 nucleotide sequences from S. methanicola metagenomic libraries G/W/G-Mi (red text) in relation to sequences from other species in the family Hesionidae. The tree was constructed by MEGAX from the concatenated 1,446-bp alignment from 33 specimens. The tree with the highest log likelihood (–10081.68) is shown, with branch lengths indicating the number of substitutions per site. Bootstrap values on tree nodes indicate the percentage of trees, based on 1,000 replicates, in which taxa from a node are clustered together. The outgroup species used was Dsyponetus caecus from the polychaete family Chrysopetalidae. Accession numbers of these sequences are provided in Table 2.
The phylogeny of amino acid sequences from the S. methanicola mitogenome in relation to 12 PCGs available for the order Phyllodocida showed the S. methanicola mitogenome to be most closely related to the mitogenome of Goniada japonica (Chen et al., 2016) from family Goniadidae (Figure 5), as predicted by MitoZ (Meng et al., 2019). Both mitogenomes were placed in a well-supported clade (98% bootstrap confidence) with the mitogenomes of Glycera capitata and Hemipodia simplex from the family Glyceridae (Figure 5). Of the 37 Phyllodocida mitogenomes, most (n=19) were from the family Nereididae (Figure 5). The mitogenomes of the Chrysopetalidae species Craseoschema thyasiricola and Chrysopetalum debile did not cluster together on the phylogenetic tree (Figure 5). The Chrysopetalum debile mitogenome clustered with mitogenomes from the Hesionidae-Goniadidae-Glyceridae clade with only 62% bootstrap confidence (Figure 5). Similar to previously reported phylogeny (Cejp et al., 2022), the Craseoschema thyasiricola mitogenome clustered with the mitogenome of Iheyomytilidicola lauensis from the family Nautiliniellidae with 100% bootstrap confidence (Figure 5). Both of these polychaetes are endosymbionts in deep-sea bivalves (Cejp et al., 2022).
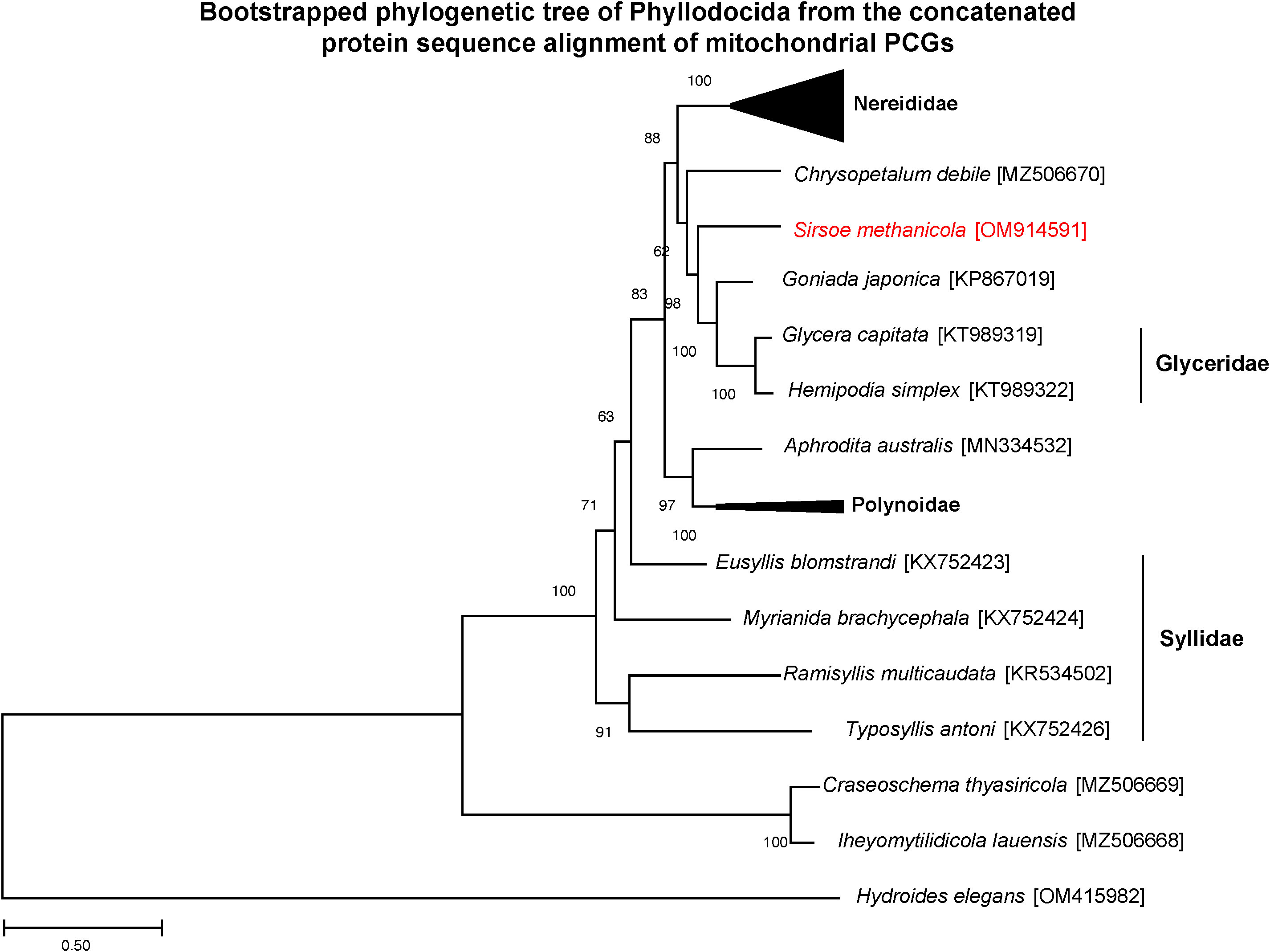
Figure 5 Bootstrapped maximum likelihood phylogenetic tree of concatenated protein sequences of 12 PCGs from the S. methanicola mitogenome (red text) in relation to mitogenomes available from the order Phyllodocida. The tree was constructed by MEGAX from the concatenated 2,648-aa alignment from 35 mitochondrial genomes. The tree with the highest log likelihood (–62222.03) is shown, with branch lengths indicating the number of substitutions per site. Bootstrap values on tree nodes indicate the percentage of trees, based on 100 replicates, in which taxa from a node are clustered together. The outgroup species used was Hydroides elegans from the polychaete order Sabellida. Accession numbers of all mitogenome sequences used are provided in Table S1.
Reads from two S. methanicola metagenomic libraries (libraries G and W) predominantly mapped to the 28S-5.8S-18S rRNA operon of C. teleta. Mapping was also observed to genes encoding structural components (actin and collagen alpha), signaling proteins (enterin neuropeptide, Fc-receptor like 1 homolog, phosphodiesterase 8B homolog, and ankyrin repeat domain-containing protein 26 homolog), RNA-directed DNA polymerase from transposon BS, cilia- and flagella-associated protein 20, putative glycosyltransferase, acidic repeat-containing protein, and hypothetical proteins (Figure 6).
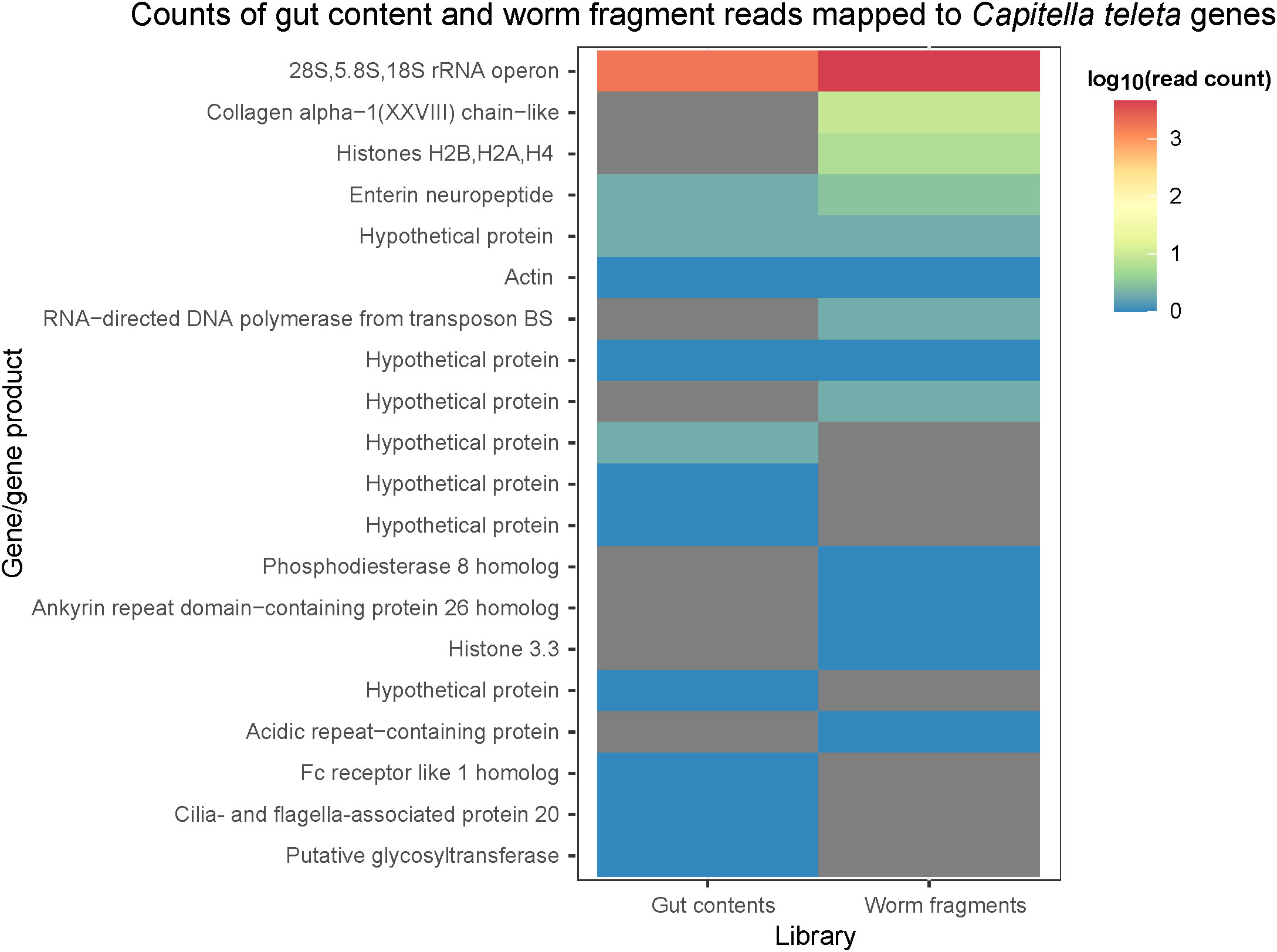
Figure 6 log10-transformed counts of reads from library G and library W (x-axis) mapped to genes/gene products in the Capitella teleta genome (y-axis), as quantified using HTSeq. Zero read counts are represented as grey cells.
Using the nucleotide and protein sequences in the C. teleta genome as training data, we predicted 79,493 eukaryotic protein-coding genes in the assembled S. methanicola metagenomes using WebAUGUSTUS (Hoff and Stanke, 2013). Of these protein sequences, ghostKOALA (Kanehisa et al., 2016) assigned taxonomy to 98% (77,530) and KO terms to 35% (28,270), based on mapping to complete genomes or functionally characterized individual protein sequences in the KEGG Genome database (Kanehisa et al., 2020). Protein sequences were mostly assigned to the KEGG-defined broad taxonomic groups “Animals” (77%; Table 3), with 2% (1,799) assigned to the phylum Annelida. All Annelid sequences were predicted based on mapping to sequences in the genome of the freshwater leech Helobdella robusta from class Clitellata (KEGG accession T0327 and NCBI accession GCF_000326865.1) (Simakov et al., 2013). Smaller numbers of protein sequences from the metagenomes were assigned to the groups “Bacteria”, “Plants”, “Fungi”, “Protists”, “Archaea” and “Viruses” (Table 3).
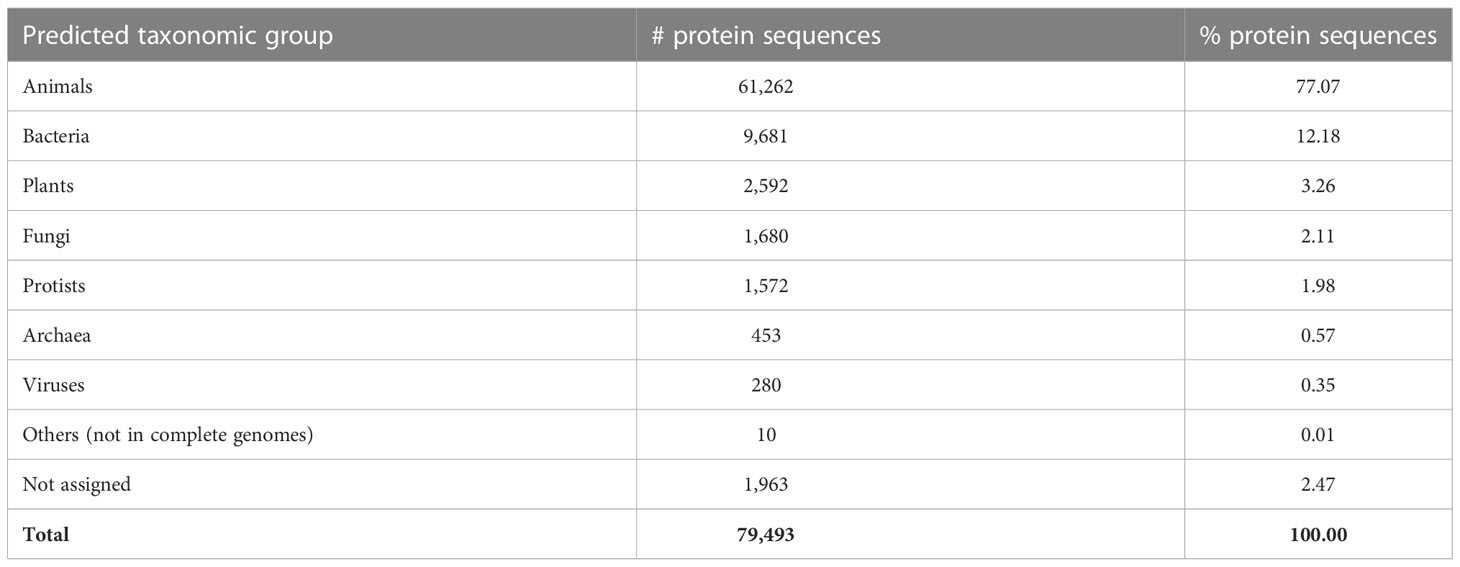
Table 3 Broad taxonomic classification of protein sequences annotated from the combined S. methanicola metagenomes by ghostKOALA, based on mapping to complete genomes or functionally characterized individual protein sequences in the KEGG Genome database.
Of the Annelid sequences, the most abundant KEGG Orthology term mapped to H. robusta from S. methanicola metagenomes was innexin (Table 4). Other abundant KEGG Orthology terms were involved in cell adhesion, signaling, the ubiquitin system, metabolism, transport, and other processes (Table 4). KEGG mapper analysis mapping all Annelid KO terms to KEGG pathway modules revealed one complete pathway module associated with eukaryotic C1-unit interconversion (M00141). This module comprised two genes assigned to K00600 (glycine hydroxymethyltransferase) and two genes assigned to K00288 (methylenetetrahydrofolate dehydrogenase (NADP+)/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase).
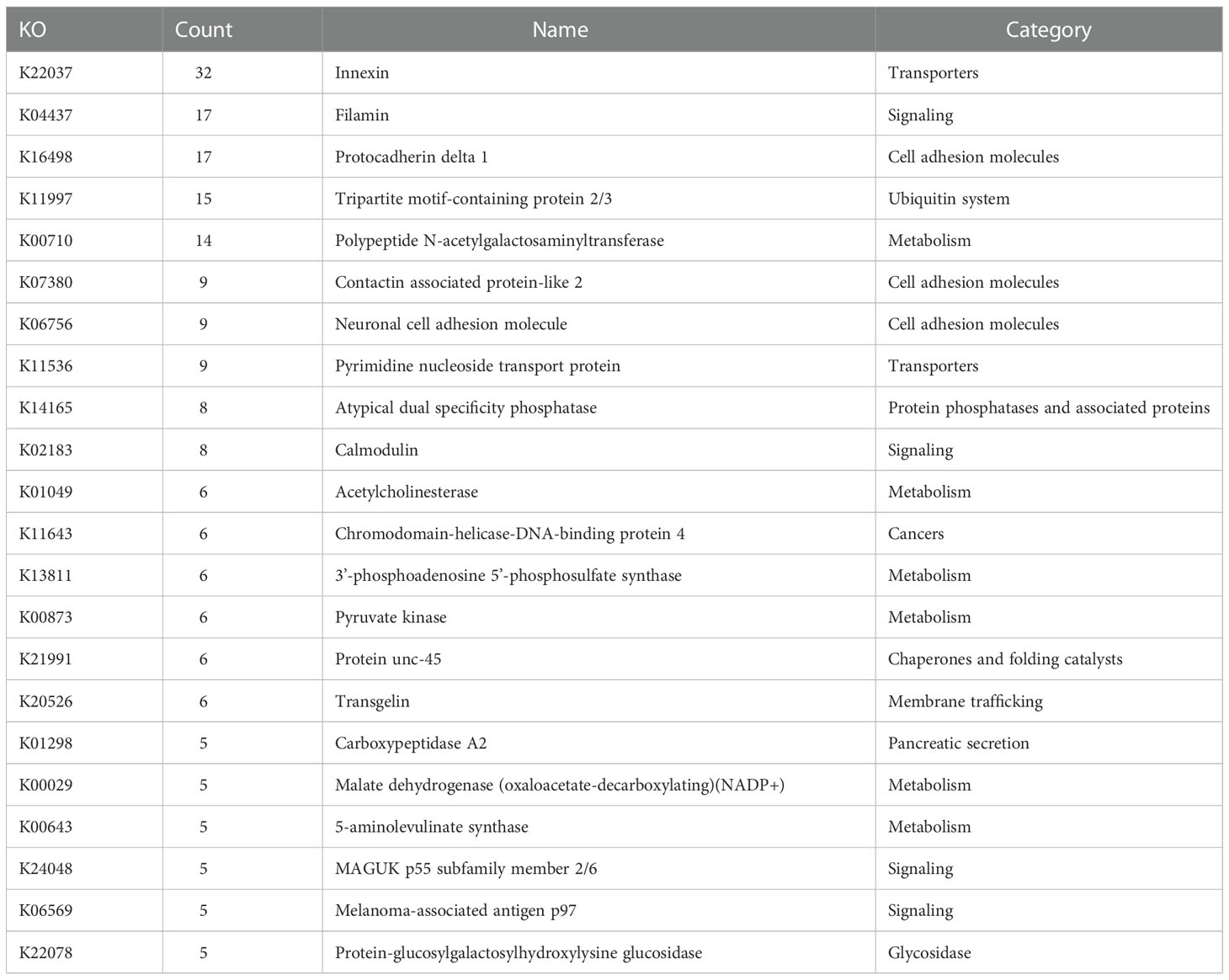
Table 4 Most abundant KEGG Orthology (KO) terms mapped by ghostKOALA from the S. methanicola metagenomes to Annelid sequences in the KEGG Genome database.
Using reference C. teleta CYP genomic annotations (Dejong and Wilson, 2014) to identify potential CYP sequences in S. methanicola that may respond to or detoxify PAHs, we identified 42 predicted protein sequences from the S. methanicola metagenomes that were homologous (30% to 79% identical) to 37 cytochrome P450 sequences in C. teleta (Figure 7 and Table S2). In C. teleta, expression of both CYP331A1 and CYP4AT1 was shown to increase with exposure to PAHs (Li et al., 2004). From the S. methanicola metagenomes, we identified a 177-aa protein sequence sharing ~38% local sequence identity and ~55% local sequence similarity to CYP331A1. This sequence was part of a 6,485-bp contig encoding only one protein product. The sequence also shared 49% identity to an unnamed protein product of the polychaete Owenia fusiformis (NCBI accession: CAH1774988), as well as 50% identity and 69% similarity to CYP 3A29-like sequences from the brachiopod Linguna found inhabiting an intertidal zone in Kasari Bay, Japan (NCBI accessions: XP_023933140, XP_013408119, XP_013408125, XP_013408132, XP_013408139, XP_013408146, XP_013408154). We also identified another 63-aa protein sequence with ~37% local sequence identity and ~62% local sequence similarity to CYP4AT1. This sequence was part of a 5,913-bp contig encoding only one protein product. The sequence was 56% identical and 76% similar to a hypothetical protein predicted in the Helobdella robusta genome (Simakov et al., 2013), and 48% identical and 82% similar to an unnamed protein product of Owenia fusiformis.
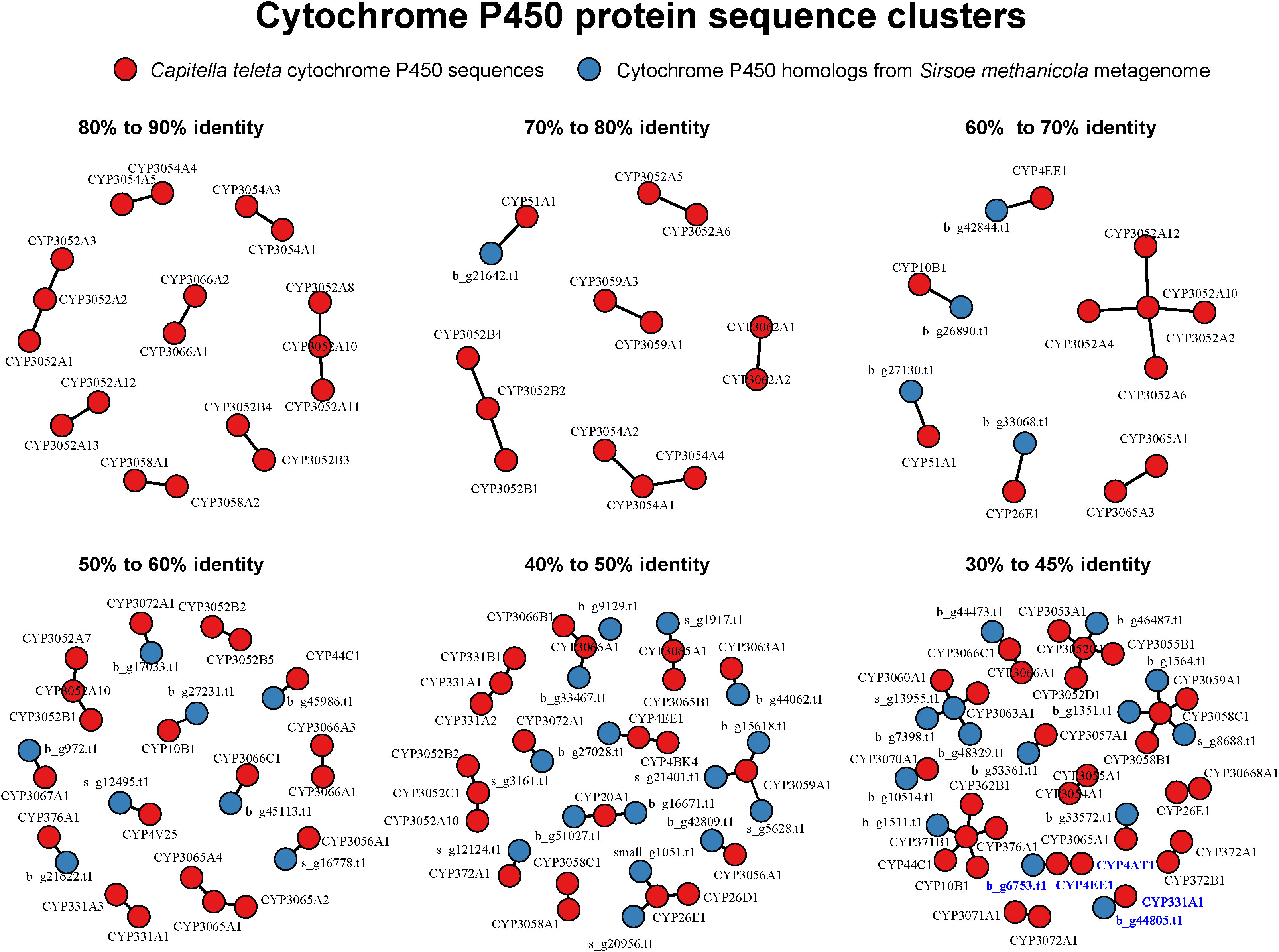
Figure 7 Clusters of cytochrome P450 (CYP) protein sequences from Capitella teleta compared to those identified in the S. methanicola metagenomes, visualized using the igraph R package. Each node represents a protein sequence, and connected nodes represent protein sequences sharing the specified range of global % sequence identity. Protein sequences in the S. methanicola metagenomes in contigs ≥3,000 bp contain the prefix b_, while those in contigs <3,000 bp contain the prefix s_. Clusters containing protein sequences similar to CYP331A1 and CYP4AT1 in C. teleta are highlighted in blue text. Protein sequences of CYP homologs recovered from the S. methanicola metagenomes are listed in Table S2.
CYP sequences are considered to be the same family and subfamily if they share 40% and 55% identity, respectively, according to the CYP nomenclature committee (Nelson et al., 1996). Based on these criteria, both homologs from the S. methanicola metagenomes did not belong to the CYP331 and CYP4 families. The protein sequence similar to CYP331A may belong to the CYP3 family, while the other protein sequence similar to CYP4AT1 was too short (66a) for CYP family assignment.
The genomes of deep sea invertebrates are poorly represented in genetic databases (Taylor and Roterman, 2017). Here, we advance knowledge concerning the genetic repertoire of a rarely studied bristle worm that can be found inhabiting Gulf of Mexico methane hydrates (Fisher et al., 2000; Becker et al., 2013). This polychaete has previously been barcoded using 16S rRNA, 18S rRNA, 28S rRNA, and cox1 genes (Ruta et al., 2007). In this study, 18S rRNA and 28S rRNA genes, the mitogenome, and certain protein-coding genes were assembled using shotgun metagenomic sequencing of S. methanicola gut contents and worm body fragments. The resulting nuclear 18S rRNA and 28S rRNA (Table 1) and mitochondrial 16S rRNA and cox1 gene sequences were 99% to 100% identical to marker gene sequences previously published for S. methanicola (Ruta et al., 2007; Pleijel et al., 2008). The phylogeny inferred from the concatenated alignment of these genes (Figure 4) was consistent with the most recent phylogeny available for Hesionidae, which clustered S. methanicola with S. dalailamai (Rouse et al., 2018) and placed all Sirsoe species within the hesionid group Psamathinae (Pleijel, 1998).
The S. methanicola mitogenome reported here is the first from the family Hesionidae (Figure 2). Further polymerase chain reaction (PCR) attempts are needed to finish the S. methanicola mitogenome, particularly to verify regions with missing sequences, low coverage, and manually corrected annotations. The mitogenome of S. methanicola was found most closely related to the mitogenome of Goniada japonica (Chen et al., 2016) from the family Goniadidae, and these were clustered with the mitogenomes of Glycera capitata and Hemipodia simplex from the family Glyceridae (Figure 5). Among these S. methanicola relatives, only the bristle worm G. capitata has been reported in the northern Gulf of Mexico at shallow (12–18 m) depth (Fauchald et al., 2009). Other Goniadidae and Glyceridae species have been found in shallow sediments (Fauchald et al., 2009) and deep-sea oil platform sediments (Granadosbarba and Solisweiss, 1997) of the Gulf of Mexico.
The S. methanicola mitogenome shared similar features with other reported annelid mitogenomes, including the common usage of AUG as the start codon, as well as the frequent occurrence of truncated U– stop codons which may be completed by alternative polyadenylation (Chen et al., 2016; Cejp et al., 2022). Additionally, UAA and UAG stop codons found in the S. methanicola mitogenome are the most common stop codons in polychaete mitogenomes species (Cejp et al., 2022). We also identified several codon biases in the S. methanicola mitogenome. Previous mitogenomic analysis of the polychaete family Chrysopetalidae revealed relaxed selection, particularly in the cytochrome c oxidase subunit III (cox3) gene, in deep-sea compared to shallow-water species (Cejp et al., 2022). Similar codon usage comparisons within the family Hesionidae could reveal habitat-specific adaptations; however in-depth mitogenome analysis of S. methanicola was hampered by the paucity of mitogenomes taxonomically or ecologically related to the species.
This study provides the first functional profile for S. methanicola. Annotations of the metagenomic data included genes for putative cell adhesion, signaling, ubiquitin system, metabolism, and transport, as well as genes homologous to innexins and the CYP superfamily (Figure 6, Table 4, and Figure 7). Innexins form gap junctions between neurons and are potentially useful for studying annelid phylogeny (Kandarian et al., 2012; Hughes, 2014). Using reference sequences from C. teleta, we predicted 42 CYP protein sequences in S. methanicola (Figure 7 and Table S2). Based on sequence identities, none of these sequences were assigned to the same family or subfamily as CYP331A1 and CYP4AT1, whose expression was shown to increase with PAH exposure in C. teleta (Li et al., 2004). Although we hypothesized that the worm may detoxify or consume organic compounds from the environment or its gut (Lim et al., 2022), further studies are required to validate the expression and functions of the cytochrome P450 protein sequences in S. methanicola.
This study provides the first mitogenome, protein-coding gene, and CYP enzyme superfamily annotations for S. methanicola living on the surface of a methane hydrate in the Gulf of Mexico. Future sampling will improve the genetic annotations of this poorly understood polychaete species, which has proven difficult to locate in the deep sea. Previous studies on C. teleta polychaetes harboring gut microbes had revealed important host-microbiome interface properties relevant to the cycling of environmental compounds (Dejong and Wilson, 2014; Jang et al., 2020; Jang et al., 2021). Our results encourage further comparative studies on the genomic and microbiome adaptations of this deep-sea worm to its unique habitat and how these adaptations contribute to the ecology and nutrient cycling of methane hydrates.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
SL conducted bioinformatics analyses and wrote the manuscript, under the guidance of LT. KG conceived this project and led manuscript editing. All authors edited the manuscript and approved the submission.
This research was carried out [in part] under the auspices of the Cooperative Institute for Marine and Atmospheric Studies (CIMAS), a Cooperative Institute of the University of Miami and the National Oceanic and Atmospheric Administration (cooperative agreement # NA20OAR4320472), with support from the NOAA Office of Oceanic and Atmospheric Research’s ‘Omics Initiative. LT was supported by award NA21OAR4320190 to the Northern Gulf Institute from NOAA’s Office of Oceanic and Atmospheric Research, U.S. Department of Commerce. Sample collection was supported in part by NSF OCE-0527139. MiSeq sequencing was performed through NSF DUE-1323809 that supported a Sequencing Technology Education using Microbial Metagenomes (STEMM) workshop at San Diego State University organized by Dr. Elizabeth Dinsdale. NIH grant 1R21-ES024105 supported preliminary computational analysis.
We are indebted to Dr. Terry Gaasterland for orchestrating the HiSeq sequencing and Dr. Craig M. Young for organizing the cruise that provided the methane ice worm samples. We thank Wei Xin of Smith College and Lee Edsall and Nick Smith of the University of California, San Diego (UCSD) for early data analysis efforts. We thank Dr. Michelle Wood (Co-PI on NSF OCE-0527139) for inviting KG on the cruise and for promoting metagenomic research, including this methane ice worm study. We acknowledge Dr. Richard Emlet at University of Oregon for the skilled dissection that provided the Tube A sample. Nucleic acid processing and HiSeq 2000 sequencing efforts were aided by Steve Head and Terry Gelbart at the Scripps Research Institute.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1067482/full#supplementary-material
Adachi J., Hasegawa M. (1996). Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol. 42 (4), 459–468. doi: 10.1007/BF02498640
Alves P. R., Halanych K. M., Santos C. S. G. (2020). The phylogeny of nereididae (Annelida) based on mitochondrial genomes. Zool. Scripta 49 (3), 366–378. doi: 10.1111/zsc.12413
Anders S., Pyl P. T., Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31 (2), 166–169. doi: 10.1093/bioinformatics/btu638
Arkin A. P., Cottingham R. W., Henry C. S., Harris N. L., Stevens R. L., Maslov S., et al. (2018). KBase: The united states department of energy systems biology knowledgebase. Nat. Biotechnol. 36 (7), 566–569. doi: 10.1038/nbt.4163
Becker E. L., Cordes E. E., Macko S. A., Lee R. W., Fisher C. R. (2013). Using stable isotope compositions of animal tissues to infer trophic interactions in gulf of Mexico lower slope seep communities. PloS One 8 (12), e74459. doi: 10.1371/journal.pone.0074459
Benson D. A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Ostell J., Pruitt K. D., et al. (2018). GenBank. Nucleic Acids Res. 46 (D1), D41–D47. doi: 10.1093/nar/gkx1094
Bernt M., Donath A., Jühling F., Externbrink F., Florentz C., Fritzsch G., et al. (2013). MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69 (2), 313–319. doi: 10.1016/j.ympev.2012.08.023
Blake J. A., Grassle J. P., Eckelbarger K. J. (2009). Capitella teleta, a new species designation for the opportunistic and experimental Capitella sp. I, with a review of the literature for confirmed records. Zoosymposia 2 (1), 25–53. doi: 10.11646/zoosymposia.2.1.6
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Castresana J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17 (4), 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cejp B., Ravara A., Aguado M. T. (2022). First mitochondrial genomes of chrysopetalidae (Annelida) from shallow-water and deep-sea chemosynthetic environments. Gene 815, 146159. doi: 10.1016/j.gene.2021.146159
Chen X., Li M., Liu H., Li B., Guo L., Meng Z., et al. (2016). The complete mitochondrial genome of the polychaete, Goniada japonica (Phyllodocida, goniadidae). Mitochondrial. DNA A DNA Mapp Seq Anal. 27 (4), 2850–2851. doi: 10.3109/19401736.2015.1053124
Csárdi G., Nepusz T. (2006). The igraph software package for complex network research. InterJournal Complex Syst. 1695, 1–9.
Dejong C. A., Wilson J. Y. (2014). The cytochrome P450 superfamily complement (CYPome) in the annelid Capitella teleta. PloS One 9 (11), e107728. doi: 10.1371/journal.pone.0107728
Desbruyères D., Toulmond A. (1998). A new species of hesionid worm, Hesiocaeca methanicola sp. nov. (Polychaeta: Hesionidae), living in ice-like methane hydrates in the deep gulf of Mexico. Cah. Biol. Mar. 39, 93–98. doi: 10.21411/CBM.A.BA5D76AF
Dubilier N., Bergin C., Lott C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat.Rev.Microbiol 6 (10), 725–740. doi: 10.1038/nrmicro1992
Fauchald K., Granados-Barba A., Solis-Weiss V. (2009). “Polychaeta (Annelida) of the gulf of Mexico,” in Gulf of Mexico origin, waters, and biota: Biodiversity. Eds. Felder D. L., Camp D. K. (College Station, USA: Texas A&M University Press), 751–788.
Fisher C. R., MacDonald I. R., Sassen R., Young C. M., Macko S. A., Hourdez S., et al. (2000). Methane ice worms: Hesiocaeca methanicola colonizing fossil fuel reserves. Naturwissenschaften 87 (4), 184–187. doi: 10.1007/s001140050700
Forbes V. E., Andreassen M. S. H., Christensen L. (2001). Metabolism of the polycyclic aromatic hydrocarbon fluoranthene by the polychaete Capitella capitata species I. Environ. Toxicol. Chem. 20 (5), 1012–1021. doi: 10.1002/etc.5620200511
Granadosbarba A., Solisweiss V. (1997). The polychaetous annelids from oil platforms areas in the southeastern gulf of Mexico: Phyllodocidae, glyceridae, goniadidae, hesionidae, and pilargidae, with description of Ophioglycera lyra, a new species, and comments on Goniada distorta Moore and Scoloplos texana maciolek & Holland. Proc. Biol. Soc. Washington 110 (3), 457–470.
Gruber-Vodicka H. R., Seah B. K. B., Pruesse E. (2020). phyloFlash: rapid small-subunit rRNA profiling and targeted assembly from metagenomes. mSystems 5 (5), e00920–e00920. doi: 10.1128/mSystems.00920-20
Hochstein R., Zhang Q., Sadowsky M. J., Forbes V. E. (2019). The deposit feeder Capitella teleta has a unique and relatively complex microbiome likely supporting its ability to degrade pollutants. Sci. Total Environ. 670, 547–554. doi: 10.1016/j.scitotenv.2019.03.255
Hoff K. J., Stanke M. (2013). WebAUGUSTUS–a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41 (Web Server issue), W123–W128. doi: 10.1093/nar/gkt418
Hughes A. L. (2014). Evolutionary diversification of insect innexins. J. Insect Sci. 14, 1–5. doi: 10.1093/jisesa/ieu083
Jang J., Forbes V. E., Sadowsky M. J. (2020). Lack of evidence for the role of gut microbiota in PAH biodegradation by the polychaete Capitella teleta. Sci. Total Environ. 725, 138356. doi: 10.1016/j.scitotenv.2020.138356
Jang J., Hochstein R., Forbes V. E., Sadowsky M. J. (2021). Bioturbation by the marine polychaete Capitella teleta alters the sediment microbial community by ingestion and defecation of sediment particles. Sci. Total Environ. 752, 142239. doi: 10.1016/j.scitotenv.2020.142239
Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36 (Web Server issue), W5–W9. doi: 10.1093/nar/gkn201
Jones D. T., Taylor W. R., Thornton J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8 (3), 275–282. doi: 10.1093/bioinformatics/8.3.275
Joye S. B., Boetius A., Orcutt B. N., Montoya J. P., Schulz H. N., Erickson M. J., et al. (2004). The anaerobic oxidation of methane and sulfate reduction in sediments from gulf of Mexico cold seeps. Chem. Geol. 205 (3), 219–238. doi: 10.1016/j.chemgeo.2003.12.019
Kandarian B., Sethi J., Wu A., Baker M., Yazdani N., Kym E., et al. (2012). The medicinal leech genome encodes 21 innexin genes: different combinations are expressed by identified central neurons. Dev. Genes Evol. 222 (1), 29–44. doi: 10.1007/s00427-011-0387-z
Kanehisa M., Furumichi M., Sato Y., Ishiguro-Watanabe M., Tanabe M. (2020). KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 49 (D1), D545–D551. doi: 10.1093/nar/gkaa970
Kanehisa M., Sato Y., Morishima K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428 (4), 726–731. doi: 10.1016/j.jmb.2015.11.006
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
Kvenvolden K. A. (1995). A review of the geochemistry of methane in natural gas hydrate. Organic Geochem. 23 (11), 997–1008. doi: 10.1016/0146-6380(96)00002-2
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Lanoil B. D., Sassen R., La Duc M. T., Sweet S. T., Nealson K. H. (2001). Bacteria and archaea physically associated with gulf of Mexico gas hydrates. Appl. Environ. Microbiol. 67 (11), 5143–5153. doi: 10.1128/AEM.67.11.5143-5153.2001
Levin L. A. (2005). “Ecology of cold seep sediments: interactions of fauna with flow, chemistry,” in Oceanography and marine biology. Eds. Gibson R. N., Atkinson R. J. A., Gordon J. D. M. (Boca Raton, FL, USA: Taylor & Francis), 1–46.
Li B., Bisgaard H. C., Forbes V. E. (2004). Identification and expression of two novel cytochrome P450 genes, belonging to CYP4 and a new CYP331 family, in the polychaete Capitella capitata sp.I. Biochem. Biophys. Res. Commun. 325 (2), 510–517. doi: 10.1016/j.bbrc.2004.10.066
Li W., Godzik A. (2006). CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 (13), 1658–1659. doi: 10.1093/bioinformatics/btl158
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi: 10.1093/bioinformatics/btp352
Li D., Luo R., Liu C. M., Leung C. M., Ting H. F., Sadakane K., et al. (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Lim S. J., Thompson L. R., Young C. M., Gaasterland T., Goodwin K. D. (2022). Dominance of Sulfurospirillum in metagenomes associated with the methane ice worm (Sirsoe methanicola). Appl. Environ. Microbiol. 88 (15), e0029022. doi: 10.1128/aem.00290-22
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Meng G., Li Y., Yang C., Liu S. (2019). MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47 (11), e63. doi: 10.1093/nar/gkz173
Mills H. J., Martinez R. J., Story S., Sobecky P. A. (2005). Characterization of microbial community structure in gulf of Mexico gas hydrates: comparative analysis of DNA- and RNA-derived clone libraries. Appl. Environ. Microbiol. 71 (6), 3235–3247. doi: 10.1128/AEM.71.6.3235-3247.2005
National Energy Technology Laboratory (2017). Methane hydrate science and technology: a 2017 update. (U.S. Department of Energy). Available at: https://www.netl.doe.gov/sites/default/files/netl-file/2017-Methane-Hydrate-Primer%5B1%5D.pdf.
Nelson D. R., Koymans L., Kamataki T., Stegeman J. J., Feyereisen R., Waxman D. J., et al. (1996). P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6 (1), 1–42. doi: 10.1097/00008571-199602000-00002
Nurk S., Meleshko D., Korobeynikov A., Pevzner P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27 (5), 824–834. doi: 10.1101/gr.213959.116
Peng Y., Leung H. C., Yiu S. M., Chin F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28 (11), 1420–1428. doi: 10.1093/bioinformatics/bts174
Pleijel F. (1998). Phylogeny and classification of hesionidae (Polychaeta). Zool. Scripta 27 (2), 89–163. doi: 10.1111/j.1463-6409.1998.tb00433.x
Pleijel F., Rouse G. W., Ruta C., Wiklund H., Nygren A. (2008). Vrijenhoekia balaenophila, a new hesionid polychaete from a whale fall off California. Zool. J. Linn. Soc. 152 (4), 625–634. doi: 10.1111/j.1096-3642.2007.00360.x
Pleijel F., Rouse G. W., Sundkvist T., Nygren A. (2012). A partial revision of Gyptis (Gyptini, ophiodrominae, hesionidae, aciculata, Annelida), with descriptions of a new tribe, a new genus and five new species. Zool. J. Linn. Soc. 165 (3), 471–494. doi: 10.1111/j.1096-3642.2012.00819.x
Rouse G. W., Carvajal J. I., Pleijel F. (2018). Phylogeny of hesionidae (Aciculata, Annelida), with four new species from deep-sea eastern pacific methane seeps, and resolution of the affinity of Hesiolyra. Invertebrate Systemat. 32 (5), 1050–1068. doi: 10.1071/IS17092
Ruta C., Nygren A., Rousset V., Sundberg P., Tillier A., Wiklund H., et al. (2007). Phylogeny of Hesionidae (Aciculata, polychaeta), assessed from morphology, 18S rDNA, 28S rDNA, 16S rDNA and COI. Zool. Scripta 36 (1), 99–107. doi: 10.1111/j.1463-6409.2006.00255.x
Sayers E. W., Beck J., Bolton E. E., Bourexis D., Brister J. R., Canese K., et al. (2021). Database resources of the national center for biotechnology information. Nucleic Acids Res. 49 (D1), D10–D17. doi: 10.1093/nar/gkaa892
Selck H., Palmqvist A., Forbes V. E. (2003). Biotransformation of dissolved and sediment-bound fluoranthene in the polychaete, Capitella sp. I. Environ. Toxicol. Chem. 22 (10), 2364–2374. doi: 10.1897/02-272
Sharp P. M., Li W. H. (1987). The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15 (3), 1281–1295. doi: 10.1093/nar/15.3.1281
Shimabukuro M., Carrerette O., Alfaro-Lucas J. M., Rizzo A. E., Halanych K. M., Sumida P. Y. G. (2019). Diversity, distribution and phylogeny of hesionidae (Annelida) colonizing whale falls: new species of Sirsoe and connections between ocean basins. Front. Mar. Sci. 6 (478). doi: 10.3389/fmars.2019.00478
Sibuet M., Olu K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. Part II: Topical Stud. Oceanogr. 45 (1), 517–567. doi: 10.1016/S0967-0645(97)00074-X
Simakov O., Marletaz F., Cho S.-J., Edsinger-Gonzales E., Havlak P., Hellsten U., et al. (2013). Insights into bilaterian evolution from three spiralian genomes. Nature 493 (7433), 526–531. doi: 10.1038/nature11696
Taylor M. L., Roterman C. N. (2017). Invertebrate population genetics across earth's largest habitat: the deep-sea floor. Mol. Ecol. 26 (19), 4872–4896. doi: 10.1111/mec.14237
Tunnicliffe V., Juniper S. K., Sibuet M. (2003). “Reducing environments of the deep-sea floor,” in Ecosystems of the world. Ed. Tyler P. A. (Amsterdam: The Netherlands: Elsevier Science), 81–110.
Van Dover C. L., Aharon P., Bernhard J. M., Caylor E., Doerries M., Flickinger W., et al. (2003). Blake Ridge methane seeps: characterization of a soft-sediment, chemosynthetically based ecosystem. Deep Sea Res. Part I: Oceanogr. Res. Pap. 50 (2), 281–300. doi: 10.1016/S0967-0637(02)00162-0
Wong E. H., Smith D. K., Rabadan R., Peiris M., Poon L. L. (2010). Codon usage bias and the evolution of influenza a viruses. codon usage biases of influenza virus. BMC Evol. Biol. 10, 253. doi: 10.1186/1471-2148-10-253
Keywords: deep-sea, Gulf of Mexico, methane hydrate, polychaete, worm, mitogenome
Citation: Lim SJ, Thompson LR and Goodwin KD (2023) Genetic features of the marine polychaete Sirsoe methanicola from metagenomic data. Front. Mar. Sci. 9:1067482. doi: 10.3389/fmars.2022.1067482
Received: 11 October 2022; Accepted: 19 December 2022;
Published: 09 January 2023.
Edited by:
Rachel Przeslawski, New South Wales Department of Primary Industries, AustraliaReviewed by:
Wang Minxiao, Institute of Oceanology (CAS), ChinaCopyright © 2023 Lim, Thompson and Goodwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly D. Goodwin, a2VsbHkuZ29vZHdpbkBub2FhLmdvdg==; Shen Jean Lim, anNsaW1AZy5jbGVtc29uLmVkdQ==
†Present address: Shen Jean Lim, College of Marine Science, University of South Florida, St. Petersburg, FL, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.