 José Martin Pujolar
José Martin Pujolar Morten T. Limborg
Morten T. Limborg Moritz Ehrlich
Moritz Ehrlich Cornelia Jaspers
Cornelia Jaspers- 1Centre for Gelatinous Plankton Ecology and Evolution, DTU Aqua - Technical University of Denmark, Lyngby, Denmark
- 2Center for Evolutionary Hologenomics, GLOBE Institute, University of Copenhagen, Copenhagen, Denmark
- 3Rosenstiel School of Marine and Atmospheric Science, University of Miami, Miami, FL, United States
High throughput low-density SNP arrays provide a cost-effective solution for population genetic studies and monitoring of genetic diversity as well as population structure commonly implemented in real time stock assessment of commercially important fish species. However, the application of high throughput SNP arrays for monitoring of invasive species has so far not been implemented. We developed a species-specific SNP array for the invasive comb jelly Mnemiopsis leidyi based on whole genome re-sequencing data. Initially, a total of 1,395 high quality SNPs were identified using stringent filtering criteria. From those, 192 assays were designed and validated, resulting in the final panel of 116 SNPs. Markers were diagnostic between the northern and southern M. leidyi lineages and highly polymorphic to distinguish populations. Despite using a reduced representation of the genome, our SNP panel yielded comparable results to using a whole genome re-sequencing approach (832,323 SNPs), recovering similar values of genetic differentiation between samples and detecting the same clustering groups when performing Structure analyses. The resource presented here provides a cost-effective, high throughput solution for population genetic studies, allowing to routinely genotype large number of individuals. Monitoring of genetic diversity and effective population size estimations in this highly invasive species will allow for the early detection of new introductions from distant source regions or hybridization events. Thereby, this SNP chip represents an important management tool in order to understand invasion dynamics and opens the door for implementing such methods for a wider range of non-indigenous invasive species.
Introduction
Ecosystems face a series of stressors related to anthropogenic activities. Unintentional translocation of species outside their natural dispersal ranges has attracted much attention in recent years (Chown et al., 2015; Seebens et al., 2017; Seebens et al., 2021) due to their profound impacts on ecosystem functioning (Thomaz et al., 2015). Impacts of invasive non-indigenous species include habitat destruction, introduction of diseases and outcompeting native species for food and other resources, which can lead to the decline and extinction of native species and re-structuring of ecosystems (Bax et al., 2003; Molnar et al., 2008). Especially marine systems face increased invasion rates, which is related to increased trade and aquaculture activities, facilitating the transport and thereby arrival of non-indigenous species into new environments (Hulme et al., 2008).
The comb jelly Mnemiopsis leidyi is a ctenophore native to the East coast of Americas (Costello et al., 2012). It is non-indigenous in large areas of western Eurasia (reviewed in Jaspers et al., 2018) and considered to be one of the top successful marine invasive species in the world (Lowe et al., 2000). Previous genetic studies identified two distinct native lineages in North America (Reusch et al., 2010; Bayha et al., 2015), a northern group occurring in New England and a southern group occurring in the Gulf of Mexico and Florida, based on data from mitochondrial and nuclear microsatellite markers. Using whole-genome re-sequencing data, Jaspers et al. (2021) reconstructed the demographic history of the recent invasions in Eurasia, tracing back the invasion of the Black Sea to the Gulf of Mexico population, the invasion of the North/Baltic Sea to the New England population and the invasion of the western Mediterranean to a secondary steppingstone invasion from the Black Sea. The species possesses some of the common characteristics of successful invaders: short generation time of two weeks (Baker and Reeve, 1974), high growth and reproduction rate with over 10,000 eggs per day (Jaspers et al., 2015), few natural enemies (predators, competitors, parasites and diseases), good dispersers, generalists with a broad diet (Costello et al., 2012) and a wide environmental tolerance (Purcell et al., 2001). Interestingly, low salinity (e.g. Baltic Sea) represents an invasion barrier for the northern invasive populations (Jaspers et al., 2011), while the southern population thrives in low saline environments such as the Caspian Sea and the Sea of Azov (Shiganova et al., 2001; Jaspers et al., 2018). This indicates that information about population origin is important for predicting range expansions due to changes in environmental reaction norms.
Irrespectively of international conventions to halt unintentional species translocations (e.g. IMO Ballast Water Management Convention www.imo.org), non-indigenous species are on a rise and projected to continue to increase in the future (Seebens et al., 2021). On a local scale, molecular tools have been implemented to detect non-indigenous species at an early stage (Dias et al., 2017) in order to allow for eradication actions to be effective (Hopkins et al., 2011). However, advances and cost reduction in sequencing technologies offer the opportunity to use genomic information to address invasion dynamics and recurrent introductions of model species (Jaspers et al., 2021), which can be used to approximate overall translocation activities from different source areas.
Single nucleotide polymorphisms (SNPs) have become the marker of choice in genetics research due to their high abundance and wide distribution in the genome, codominant inheritance, low mutation rate and relatively easy and cost-effective discovery and genotyping. Next-generation sequencing technologies enable the identification of hundreds of thousands of SNPs using either whole genome sequencing or reduced representation approaches (Baird et al., 2008; Davey et al., 2011). High throughput low-density SNP arrays provide a cost-effective solution for long-term genetic monitoring and have been established in commercially important species through the application of SNP chips (reviewed in LaFramboise, 2009). This approach not only allows for fast genotyping of a large number of individuals, but also the collection of genome-wide SNP data. Limitations include ascertainment bias (genotyping arrays contain biased sets of pre-ascertained SNPs), high dependence on DNA quality that might yield fewer reliable SNPs in poor-DNA quality samples and high development costs. However, although development is costly and time consuming, sequencing costs continue to fall rapidly and investment is long-term. SNP arrays have been applied in areas as diverse as human forensics and diagnostics, aquaculture, improvement of crops, selective breeding and conservation and management of fisheries. Commercially-available SNP arrays exist for a number of marine species including Atlantic salmon Salmo salar (Houston et al., 2014), rainbow trout Oncorhynchus mykiss (Palti et al., 2015), Atlantic cod Gadus morhua (Pocwierz-Kotus et al., 2015; Hemmer-Hansen et al., 2018), Pacific oyster Crassostrea gigas (synonym: Magallana gigas) and European oyster Ostrea edulis (Gutierrez et al., 2017), European sea bass Dicentrarchus labrax and gilthead seabream Sparus aurata (Penaloza et al., 2021), among others.
Here we present the first implementation of a high throughput SNP array specifically-designed for monitoring an invasive non-indigenous species of no commercial value. We developed and validated a low-density SNP array for M. leidyi that will allow routine collection of genome-wide SNP data on a large number of individuals and populations, hence providing a cost-effective and fast method for genetic monitoring of this highly invasive species. The array will allow monitoring of changes in genetic diversity and genetic structure in invasive M. leidyi populations, the detection of admixed individuals and the arrival of new genotypes. This knowledge is crucial in order to better understand on-going population dynamics of invasive species and inform about current or re-current introduction events in marine systems.
Materials and methods
SNP calling and filtering
SNP assay design was conducted using a total of 72 whole-genome re-sequenced individuals from Jaspers et al. (2021). Read quality was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx-toolkit). Adapter sequences were soft-clipped (i.e., base quality set to 2) and low-quality bases removed using Trimmomatic v0.36 (Bolger et al., 2014). Subsequently, paired and unpaired reads were mapped to the M. leidyi reference genome using the mem algorithm in BWA v0.7.15 (Li and Durbin, 2009). A trimmed version of the genome was used containing only scaffolds >10 Kb and excluding mitochondrial sequences, for a total of 1,254 scaffolds. Sequence alignments were pre-processed using Picard v2.6.0 (Broad Institute 2018 https://broadinstitute.github.io/picard/) which included removing duplicate reads arising during library amplification.
Variant calling was performed using the Genome Analysis Toolkit GATK v3.8 (McKenna et al., 2010) with the following parameters: variant quality score > 30, variant quality by depth > 7, Phred-scaled p-value using Fisher’s test to detect strand bias < 32, SOR (symmetric odds ratio) > 2, RMS mapping quality > 58, z-score from the Wilcoxon rank sum test of Alt vs. Ref read position bias between -1.2 and 1.75, z-score from the Wilcoxon rank sum test of Alt vs. Ref read mapping qualities > -0.05 and distance from scaffold ends > 1Kb. Only biallelic SNPs genotyped in all individuals (100% call rate) and with a minor allele frequency (MAF) > 0.05 were retained.
SNP low-density array design and scoring
Final SNP selection to design a low-density array was based on the following criteria: first, the array should include at least 33% diagnostic SNPs between the northern and the southern M. leidyi groups inferred from genetic data (Jaspers et al., 2021). To do so, we calculated allele frequencies between the pooled northern populations (Woods Hole = native, Rhode Island, NE USA and Sylt = invasive, Germany, North Sea, N Europe) and the pooled southern populations (Miami = native, S USA, Varna = invasive, Bulgaria, Black Sea and Villefranche-sur-Mer = invasive, France, Mediterranean Sea) and noted those SNPs with significant allelic differences in a contingency chi-square test (p < 0.05). Ranking of SNPs did not differ if based ranking on i) FST or ii) differences in allele frequencies. Second, select SNPs with the highest MAF, which are considered to be the most informative for monitoring allele frequency changes over time and for distinguishing among populations within groups (e.g., Woods Hole vs. Sylt). Third, all selected SNPs were distributed across different scaffolds in the M. leidyi genome to guarantee a good representation of the genome, or in the cases when they were in the same scaffold, they were at least 10kb apart to ensure they were unlinked and segregating randomly. Moreover, linkage disequilibrium was tested for all SNP pairs using the web-based tool LDlink (Machiela and Chanock, 2015). Finally, the last SNP selection criterion required at least 500 bp flanking sequence both upstream and downstream each target SNP in order to allow for optimal primer design. SNPs meeting all criteria were considered as diagnostic for the purpose of the study and submitted to the D3 Assay Design (https://d3.fluidigm.com) that creates primers for SNP panels for use on the Fluidigm genomics system.
SNPs were genotyped on 96.96 Dynamic Arrays (Fluidigm Corporation, San Francisco) using the Fluidigm EP1 instrumentation. The Fluidigm system uses nano-fluidic circuitry to genotype simultaneously 96 individual samples with up to 96 assays, for a total of 9,216 parallel reactions (Seeb et al., 2009). The Fluidigm SNP Genotyping Analysis software was used to call genotypes and compile data. Each assay was assessed individually on the basis of plot quality (samples were expected to group in non-dispersed compact clusters according to genotype) and match between observed and expected clustering patterns. Genotyped individuals included a subset of 46 re-sequenced individuals from Jaspers et al. (2021) from which genotypes were known: 6 individuals from Miami (native, Florida, S USA), 11 from Sylt (invasive, Germany, North Sea, N Europe), 12 from Varna (invasive, Bulgaria, Black Sea), 5 from Villefranche-sur-Mer (invasive, France, Mediterranean Sea, S Europe) and 12 from Woods Hole (native, Rhode Island, NE USA). Each individual was included in the array twice for validation.
Data analysis
Genetic diversity indices including observed and expected heterozygosities for each sampling location and the fixation index (FST) between population pairs in accordance with Weir and Cockerham (1984) were calculated using vcftools v0.1.14 (Danecek et al., 2011). Standardized pairwise FST values were used to conduct a Principal Component Analysis (PCA) in order to visualize population structure using smartPCA from the Eigensoft package (Patterson et al., 2006). Dedicated PCAs for the northern and southern population only were also constructed. Population structure was also explored using the Bayesian assignment approach implemented in STRUCTURE (Pritchard et al., 2000), a clustering algorithm which infers the most likely number of groups (K) in the data. The analysis was performed with K = 1–5, assuming an admixture model, correlated allele frequencies and without population priors. A burn-in of 100,000 steps followed by one million additional Markov Chain Monte Carlo iterations were performed. For each K, 10 independent runs were conducted to check for consistency. The most likely K was inferred using the method of Evanno et al. (2005), which measures the highest increase of the ad hoc statistic ΔK based on the rate of change in the log probability of data between successive K values. Finally, in order the assess the power and accuracy of the SNP panel, reduced data from Jaspers et al. (2021) using 832,323 SNPs was re-analyzed using the same subset of 46 individuals genotyped with the SNP panel, including new FST calculations as well as new PCA and STRUCTURE analyses.
Before the analysis, the power of the selected SNP markers to discriminate between the northern and southern lineages and identify hybrids was assessed using recom-sim (https://github.com/salanova-elliott/recom-sim), a program for generating simulated hybrids from population samples. Based on the observed allelic frequencies at the selected SNPs in the northern and southern lineage baseline populations (Woods Whole and Miami, respectively), the program generated 100 random genotypes of pure northern lineage, 100 of pure southern lineage and 100 of F1 hybrid. All simulated individuals were then blindly reassigned to their most probable category using STRUCTURE (Pritchard et al., 2000) and NEWHYBRIDS (Anderson and Thompson, 2002). The software NEWHYBRIDS uses a Bayesian approach to identify hybrids by computing the posterior probability of each individual to belong to a pure parental class or be hybrid. Both STRUCTURE and NEWHYBRIDS were run for 100,000 iterations in the burn-in followed by one million Markov Chain Monte Carlo iterations.
Results
SNP design and validation
A total of 1,395 high quality SNPs were identified after an initial dataset of 7.3 million SNPs based on whole genome re-sequencing of Mnemiopsis leidyi from native and invasive distribution ranges. The initial dataset was strictly filtered in order to increase the success of assay design, so that all SNPs presented 2 alleles, had a MAF > 0.05 and a 100% call rate. After ranking diagnostic SNPs by MAF and removing those SNPs without at least 500 bp flanking sequence, a total of 412 candidate SNP sequences were submitted to Fluidigm for assay design. A final number of 192 assays passed the in silico primer design.
Validation runs for all 192 assays run on a Fluidigm machine, showed the following results: 149 successful assays (77.6%) with a perfect match between observed and expected genotypes called in Jaspers et al. (2021) from whole genome re-sequencing data, 40 failed assays (20.8%) with all individuals grouped in a single cluster, and 3 mis-matched assays (1.6%) in which quality was good but the number of observed clusters did not match expectations. At all assays, duplicate samples of the same individual showed a 100% match with each other.
Successful assays were ranked according to most informative for the purpose of the study, including diagnostic SNPs between the northern and southern lineages and SNPs with the highest MAF. The best 116 assays were selected for the final SNP panel (see Table S1).
Power of the SNP panel
In order to test the power of the SNP panel, parental and hybrid simulated individuals were reassigned blindly. Using STRUCTURE, admixture proportions ranged from 0.00-0.02 in the southern lineage to 0.93-1.00 in the northern lineage (Figure S1). All hybrids showed intermediate admixture proportions (0.37-0.63). Using NEWHYBRIDS, a 100% correct assignment was made for all individuals, including all simulated hybrids. This suggests that our SNP panel has enough discriminatory power to identify northern and southern lineage individuals and their respective hybrids.
Genetic diversity
Average observed heterozygosity was 0.23 ± 0.10 with similar values found for the northern (0.26 ± 0.18) and southern (0.20 ± 0.21) groups. The northern group included 26 SNPs with MAF > 0.25, 35 SNPs with MAF 0.16-0.25 and 28 SNPs with MAF 0.06-0.15. The southern group included 41 SNPs with MAF > 0.25, 8 SNPs with MAF 0.16-0.25 and 11 SNPs with MAF 0.06-0.15. SNPs with high MAF > 0.25 were present in all populations (Miami: 27; Varna: 30; Villefranche-sur-Mer: 37; Woods Hole: 27; Sylt: 25), as well as moderately high MAF 0.16-0.25 (Miami: 11; Varna: 16; Villefranche-sur-Mer: 12; Woods Hole: 31; Sylt: 41).
Genetic differentiation
A total of 75 SNPs were diagnostic between the northern/southern group and at 29 of those SNPs the most common allele was different in the northern and southern groups. Subsequently, genetic differentiation between pooled northern vs. pooled southern populations was high (FST = 0.18) and pair-wise comparison between northern and southern populations ranged between FST = 0.15-0.23. Within groups, the lowest genetic differentiation was found between the native and invasive northern populations (Woods Hole vs. Sylt, FST = 0.03). A higher differentiation was found between the native and invasive southern populations (Miami vs. Varna, FST = 0.09; Miami vs. Villefranche-sur-Mer, FST = 0.11), while a fixation index of FST = 0.05 was found between the two southern invasive populations, Varna and Villefranche-sur-Mer (Table 1).
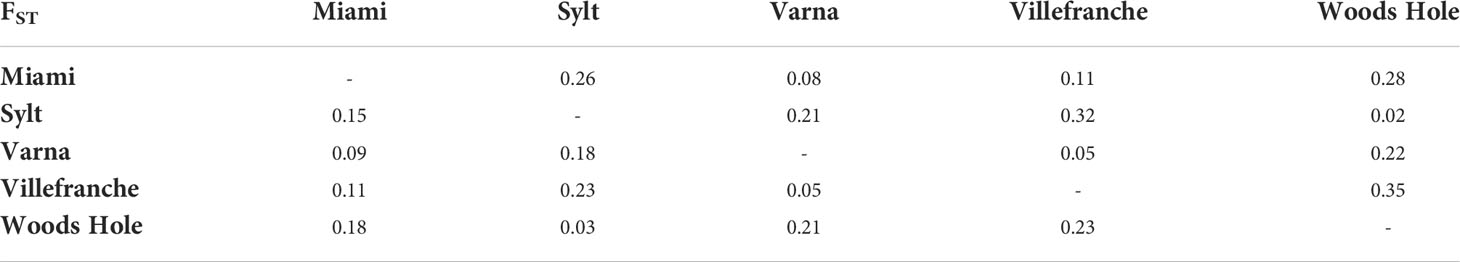
Table 1 Pairwise genetic differentiation (FST) between M. leidyi sampling locations at 116 SNPs (present study; lower diagonal) and at 832,323 SNPs (re-analysis of whole genome data using the same individuals; upper diagonal).
Principal component analysis (PCA) including all individuals supports the values of genetic differentiation found among samples, with the first component explaining 16.2% of the total variance and clearly separating the northern vs. southern populations, while the second component (2.3%) separated the southern native vs. southern invasive populations (Figure 1). Dedicated PCAs for the northern and southern populations could on the whole separate Woods Hole vs. Sylt and Varna vs. Villefranche-sur-Mer except for a few mixed individuals (Figure 1). A more robust separation of the populations could be observed using Structure, which suggested a scenario of two clusters (K = 2), grouping both northern vs. all southern populations, as the most likely (Figure 2). A K = 3 scenario separated the native southern population (Miami) vs. the invasive southern populations (Varna and Villefranche-sur-Mer). A K = 4 scenario separated the native (Woods Hole) vs. invasive (Sylt) northern populations, although with a large number of mixed individuals. Finally, a K = 5 scenario separated the two southern invasive populations (Varna vs. Villefranche-sur-Mer).

Figure 1 Principal Component Analysis (PCA) comparing results of Mnemiopsis leidyi SNP sets from whole genome re-sequencing at 832,323 SNPs (left) and newly developed SNP Chip at 116 SNPs (right) for all native (Miami: red; Woods Hole: blue, USA) and invasive (Sylt: purple, GER; Varna: green, BG; Villefranche-sur-Mer: orange, FR) populations (top panel A, B), northern populations only (mid panel, C, D) and southern populations only (bottom panel E, F).
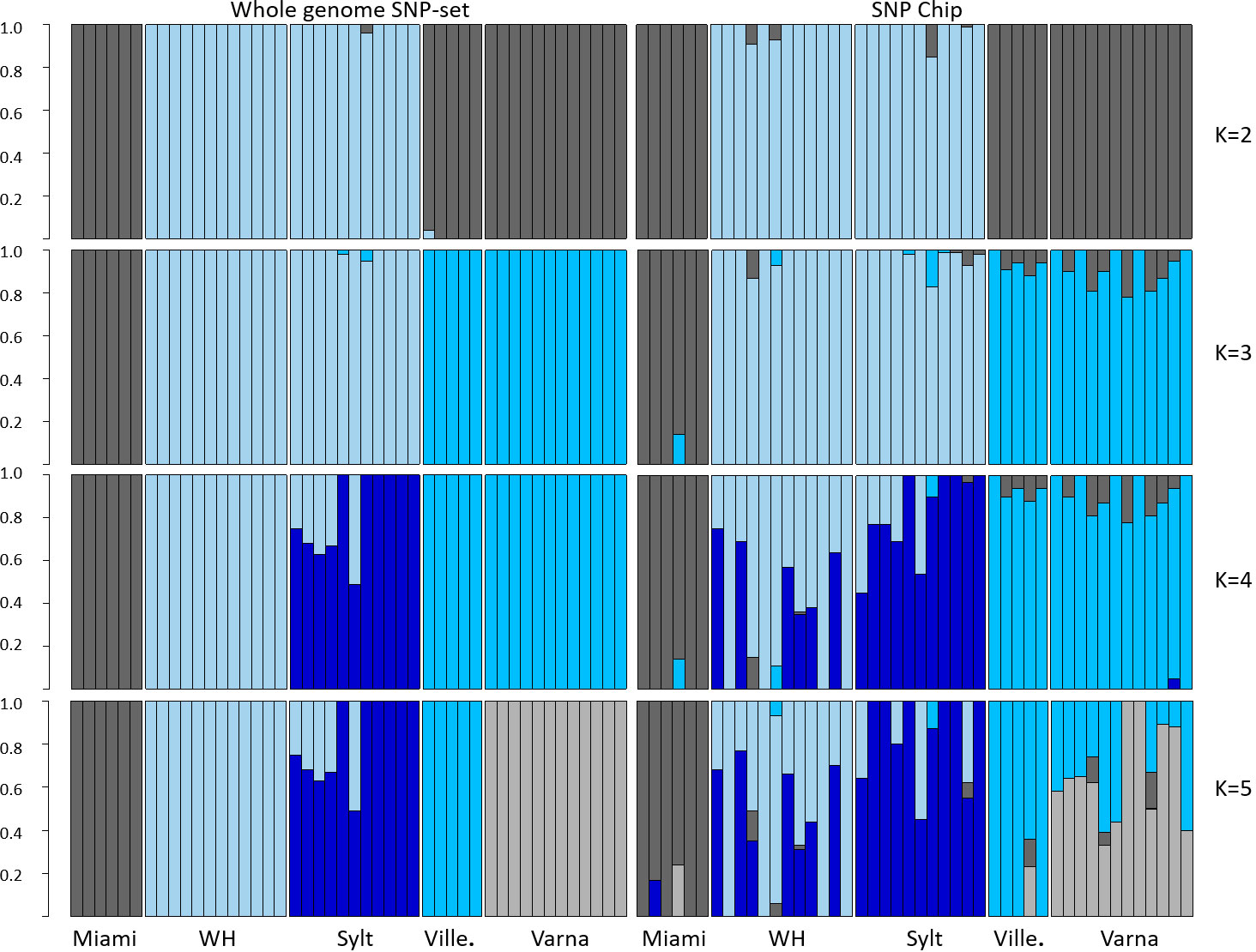
Figure 2 Structure plots comparing results of Mnemiopsis leidyi SNP sets from re-analysed whole genome re-sequencing at 832,323 SNPs (left) and newly developed SNP Chip at 116 SNPs (right) of the ctenophore for all native (Miami and Woods Hole – WH, USA) and invasive (Sylt, GER; Villefranche-sur-Mer – Ville., FR; Varna, BG) populations for the most likely scenario at K = 2 and alternative scenarios at K = 3–5.
Re-analysis of whole genome SNP data
Re-analysis of the same 46 individuals included in our study, genotyped at 832,323 SNPs from Jaspers et al. (2021), showed high genetic differentiation between the northern and southern populations (FST = 0.18), ranging from 0.22 to 0.35. Genetic differentiation was also high among southern populations (FST = 0.05-0.11), with the lowest FST value found between Woods Hole and Sylt (FST = 0.02) (Table 1), with similar results obtained from less stringent filtered dataset including 1.6 Mio. SNPs. Accordingly, PCA analysis supported the separation of the northern and southern populations, with the first component explaining 10.1% of the variance (Figure 1). The northern native and invasive population clustered together, as did the two southern invasive populations, but were clearly separated when using dedicated PCAs (Figure 1). Structure analysis suggested a scenario of K = 2 clusters corresponding to all northern vs. all southern populations, as most likely (Figure 2). A K = 3 scenario separated the southern native vs. southern invasive populations, a K = 4 scenario separated the northern native vs. northern invasive population, while a K = 5 scenario separated the two southern invasive populations.
Discussion
A SNP array for one of the world’s top invasive species
Single nucleotide polymorphism (SNP) arrays provide a cost-effective solution for genetic studies and have been extensively applied since the advent of the genomic era, first in human studies and later in both model and non-model organisms (LaFramboise, 2009). A plethora of SNP arrays have been developed for commercially-important aquaculture species, enabling the implementation of genomic selection to improve profitability and sustainability programs including Atlantic salmon, rainbow trout, Atlantic cod, European sea bass and gilthead sea bream (Boudry et al., 2021). However, the application of high throughput SNP arrays towards the monitoring of invasive species has so far not been implemented for non-commercial species yet.
We present the development and validation of the first high-throughput low-density SNP array specifically-designed to monitor one of the top invasive species in the world (Lowe et al., 2000). The comb jelly Mnemiopsis leidyi, originating from the West coast of North America, has invaded western Eurasia in recent years, including the Black Sea, the Mediterranean Sea and the North Sea. The SNP panel consists of a total of 116 fully-informative SNPs, of which the top-ranked 96 SNPs can potentially be implemented in a Fluidigm plate for routinely scoring a large number of individuals and populations, hence providing a cost-effective and fast method for genetic monitoring of M. leidyi. Using a similar approach, Karlsson et al. (2011) developed a SNP chip constituted by 60 diagnostic SNPs (from a pool of 7K SNPs) capable of identifying Atlantic salmon individuals as being wild or farmed. Ferchaud et al. (2014) generated a low-density SNP array to distinguish between freshwater and marine populations of threespine sticklebacks Gasterosteus aculeatus, encompassing markers located in regions presumably under selection along with neutral markers.
Is our SNP panel as good as using whole-genome data?
Comparison of genetic differentiation and population structure analyses clearly indicates that the SNP chip presented in this study leads to similar results as utilizing whole genome re-sequencing data. In detail, the presented panel of 116 SNPs could clearly distinguish the two genetic lineages of M. leidyi, the northern group corresponding to the native population in New England and the invasive population in northern Europe and the southern group corresponding to the native population in Gulf of Mexico/Florida and the invasive population in the Black Sea, Caspian Sea and Mediterranean Sea (Jaspers et al., 2021). A total of 75 SNP markers were diagnostic between the two groups. Accordingly, genetic differentiation between the two lineages was high (FST = 0.18) and explorative analyses (PCA, STRUCTURE) clearly separated the two groups. Besides being able to distinguish between the northern and southern groups, the SNPs included in our array are characterized by presenting high genetic variability and being highly informative, which is important for future monitoring and traceability studies. In this sense, all populations had at least 25 highly polymorphic SNPs and between 38-66 SNPs with a MAF > 0.15.
Despite being a reduced representation of the genome, fixation indices (FST) calculated with the SNP panel at 116 SNPs were similar to the ones obtained when re-analyzing data from Jaspers et al. (2021) at 832,323 SNPs, with both datasets showing similar FST values. Similar values were also obtained when comparing the two northern populations, Woods Hole and Sylt (FST = 0.03 in our study and FST = 0.02 in re-analysed data from Jaspers et al., 2021) and when comparing the southern populations, with nearly identical values found in both studies. Structure analysis also yielded similar results and the groupings suggested at K = 2, K = 3, K = 4 and K = 5 were the same either using our SNP panel or the 832,323 SNPs, re-analysed from Jaspers et al. (2021), with first a partitioning of northern vs. southern lineages, then a split of southern native vs. southern invasive populations, then a split of northern native vs. invasive populations and finally the split between the two southern invasive populations. While the proportion of admixed individuals was higher using our SNP panel, the number and assignment of groups was identical. Overall, the reduced SNP dataset was able showcase the known relationship between the M. leidyi groups (northern vs. southern) but also within groups (native vs. invasive). Both when using the whole-genome dataset and when using our SNP chip data the invasive (Sylt) and native (Woods Hole) northern populations were difficult to distinguish since genetic differentiation across samples is low. While the SNP panel is a reduced representation of the genome, our analyses show that the SNP panel is powerful enough to effectively conduct monitoring and traceability studies in M. leidyi.
Genetic monitoring to tackle biological invasions
Besides traceability, a highly polymorphic SNP panel is also important for monitoring, which allows to follow the changes in genetic diversity and genetic composition of the populations over time. Spatiotemporal SNP analyses have been used to monitor changes in genetic variability and effective population sizes in many species, in particular commercial fish such as Atlantic cod Gadus morhua (Therkildsen et al., 2013a; Therkildsen et al., 2013b), Atlantic herring Clupea harengus (Martinez-Barrio et al., 2016) and brown trout Salmo trutta (Saint-Pé et al., 2019; Bekkevold et al., 2020; Bekkevold et al., 2021). The study of Jaspers et al. (2021) showed how M. leidyi invasions in the North Sea area are ongoing, which is explained by the high connectivity that characterizes northern European ports (Jaspers et al., 2018) and the extensive trade activity between the NE coast of the USA and northern Europe. Hence it is crucial to monitor the development of M. leidyi populations in the North Sea/Baltic sea region, which can be easily done with the SNP array presented here. By analyzing new spatial samples we could detect new invasions and trace the origin of the invaders, while by analyzing temporal samples we can monitor populations and changes in genetic composition, genetic diversity and estimated effective population sizes to help evaluate the invasive potential of newly emerging populations. This includes potential exchange of individuals across regions, such as introductions of individuals from the low saline Black Sea into Northern Europe. As low salinity is currently setting an invasion barrier to the range expansion of M. leidyi in Northern Europe (Jaspers et al., 2011; Jaspers et al., 2018), the introduction of low saline thriving individuals could lead to a further range expansion of M. leidyi in Northern Europe.
The high throughput low-density SNP array presented here represents a cost-effective solution for population genetic studies and monitoring of genetic diversity and provides an important toolbox of M. leidyi genetic resources. While alternative resequencing methods have decreasing costs to date, sequencing is still expensive, requires high quality DNA and therefore hampers whole genome methods to be implemented in monitoring programs scoring routinely large number of individuals. It also opens the door for implementing such methods for a wider range of problematic non-indigenous species. Monitoring of genetic diversity in highly invasive species allows for the early detection of new introductions from distant source regions or hybridization events, which represent an important management tool in order to understand invasion dynamics. Detecting both the frequency and the origin of invasion events is essential for developing targeted management actions to improve the characterization of risk areas prone to recurrent invasions as well as to detect and potentially halt species introductions in general.
Conclusion
By using the comb jelly M. leidyi as a model organism, we outline that genomic, high throughput methods can cost effectively be integrated in monitoring activities to improve the characterization of areas prone to re-current species introductions. Thereby this method provides the opportunity to generate the critical temporal context required to assess the current invasion risk of areas for informing risk assessment and decision making by policy makers and the scientific community.
Data availability statement
All data are available on github (https://github.com/martipujolar) and dryad (https://datadryad.org/stash/dataset/doi:10.5061/dryad.18931zd11). Research described in the publication complies with all relevant national laws, implementing the Nagoya Protocol – for reference to permits see original whole genome re-sequencing data published in Jaspers et al. (2021).
Author contributions
JP: Data generation, analyses, writing final draft. ML: Conceptualization, methodology, writing and commenting. ME: Conceptualization, methodology, writing and commenting. CJ: Conceptualization, methodology, data generation, analyses, writing final draft. All authors contributed to the article and approved the submitted version.
Funding
This study was financed by VILLUM FONDEN, Denmark (Grant ID 25512 to CJ), the Danish Council for Independent Research and the European Commission—Marie-Curie Program with the DFF-MOBILEX mobility Grant No. DFF-1325-00102B (to CJ) and by Cluster of Excellence 80 ‘The Future Ocean’, which is funded within the framework of the Excellence Initiative by the German Research Council (DFG) on behalf of the German federal and state governments (Grant ID CP1539 to PI: CJ).
Acknowledgments
We thank Prof. T. B. H. Reusch for constructive discussions and input during the initial SNP chip design and Dr. Corinna Breusing for discussions. We thank Dorte Meldrup for running the SNP Chip and laboratory work at DTU Aqua in Silkeborg.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1019001/full#supplementary-material
References
Anderson E. C., Thompson E. A. (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160, 1217–1229. doi: 10.1093/genetics/160.3.1217
Baird N. A., Etter P. D., Atwood T. S., Currey M. C., Shiver A. L., Lewis Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequence RAD markers. PloS One 3 (10), e3376. doi: 10.1371/journal.pone.0003376
Baker L. D., Reeve M. R. (1974). Laboratory culture of the lobate ctenophore Mnemiopsis leidyi with notes on feeding and fecundity. Mar. Biol. 26, 57–62. doi: 10.1007/BF00389086
Bax N., Williamson A., Aguero M., Gonzalez E., Geeves W. (2003). Marine invasive alien species: A threat to global diversity. Mar. Policy 27, 313–323. doi: 10.1016/S0308-597X(03)00041-1
Bayha K. M., Chang M. H., Mariani C. L., Richardson J. L., Edwards D. L., DeBoer, et al. (2015). Worldwide phylogeography of the invasive ctenophore Mnemiopsis leidyi based on nuclear and mitochondrial DNA data. Biol. Invasions 17, 827–850. doi: 10.1007/s10530-014-0770-6
Bekkevold D., Höjesjö J., Nielsen E. E., Aldvén D., Als T. D., Sodeland M., et al. (2020). Northern European Salmo trutta populations are genetically divergent across geographical regions and environmental gradients. Evol. Appl. 13 (2), 400–416. doi: 10.1111/eva.12877
Bekkevold D., Piper A., Campbell R., Rippon P., Wright R. W., Crundwell C., et al. (2021). Genetic stock identification of sea trout (Salmo trutta) along the British North Sea coast shows prevalent distance migration. ICES J. Mar. Sci. 78 (3), 952–966. doi: 10.1093/icesjms/fsaa240
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Boudry P., Allal F., Aslam M. L., Bargelloni L., Bean T. P., Brard-Fudulea S., et al. (2021). Current status and potential of genomic selection to improve selective breeding in the main aquaculture species of international council for the exploration of the Sea (ICES) member countries. Aquac. Rep. 20, 100700. doi: 10.1016/j.aqrep.2021.100700
Chown S. L., Hodgins K. A., Griffin P. C., Oakeshott J. G., Byrne M., Hoffmann A. A. (2015). Biological invasions, climate change and genomics. Evol. Appl. 8 (1), 23–46. doi: 10.1111/eva.12234
Costello J. H., Bayha K. M., Mianzan H. W., Shiganova T. A., Purcell J. E. (2012). Transitions of Mnemiopsis leidyi from a native to an exotic species: A review. Hydrobiologia 690, 21–46. doi: 10.1007/s10750-012-1037-9
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi: 10.1093/bioinformatics/btr330
Davey J. W., Hohenlohe P. A., Etter P. D., Boone J. Q., Catchen J. M., Blaxter M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510. doi: 10.1038/nrg3012
Dias P. J., Fotedar S., Muenoz J., Hewitt M. J., Lukehurst S., Hourston M., et al. (2017). Establishment of a taxonomic and molecular reference collection to support the identification of species regulated by the western Australian prevention list for introduced marine pests. Manage. Biol. Invasions 8 (2), 215–225. doi: 10.3391/mbi.2017.8.2.09
Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14 (8), 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Ferchaud A. L., Pedersen S. H., Bekkevold D., Jian J., Niu Y., Hansen H. H., et al. (2014). A low-density SNP array for analyzing differential selection in freshwater and marine populations of threespine stickleback (Gasterosteus aculeatus). BMC Genomics 15, 867. doi: 10.1186/1471-2164-15-867
Gutierrez A. P., Turner F., Gharbi K., Talbot R., Lowe N. R., Penaloza C., et al. (2017). Development of a medium density combined-species SNP array for pacific and european oysters (Crassostrea gigas and Ostrea edulis). G3-Genes Genom. Genet. 7 (7), 2209–2218. doi: 10.1534/g3.117.041780
Hemmer-Hansen J., Hussy K., Baktoft H., Huwer B., Bekkevold D., Haslob H., et al. (2018). Genetic analyses reveal complex dynamics within a marine fish management area. Evol. Appl. 12 (4), 830–844. doi: 10.1111/eva.12760
Hopkins G. A., Forrest B. M., Jiang W., Gardner J. P. A. (2011). Successful eradication of a non-indigenous marine bivalve from a subtidal soft-sediment environment. J. Appl. Ecol. 48 (2), 424–431. doi: 10.1111/j.1365-2664.2010.01941.x
Houston R. D., Taggart J. B., Cezard T., Bekaert M., Lowe N. R., Downing A., et al. (2014). Development and validation of a high density SNP genotyping array for Atlantic salmon (Salmo salar). BMC Genomics 15, 90. doi: 10.1186/1471-2164-15-90
Hulme P. E., Bacher S., Kenis M., Klotz S., Kuhn I., Minchin D., et al. (2008). Grasping at the routes of biological invasions: A framework for integrating pathways into policy. J. Appl. Ecol. 45 (2), 303–414. doi: 10.1111/j.1365-2664.2007.01442.x
Jaspers C., Costello J. H., Colin S. P. (2015). Carbon content of Mnemiopsis leidyi eggs and specific egg production rates in northern Europe. J. Plankton Res. 37 (1), 11–15. doi: 10.1093/plankt/fbu102
Jaspers C., Ehrlich M., Pujolar J. M., Kunzel S., Bayer T., Limborg M. T., et al. (2021). Invasion genomics uncover contrasting scenarios of genetic diversity in a widespread marine invader. Proc. Natl. Acad. Sci. U.S.A. 118 (51), e2116211118. doi: 10.1073/pnas.2116211118
Jaspers C., Huwer B., Antajan E., Hosia A., Hinrichsen H. H., Biastoch A., et al. (2018). Ocean current connectivity propelling the secondary spread of a marine invasive comb jelly across western Eurasia. Glob. Ecol. Biogeogr. 27, 814–827. doi: 10.1111/geb.12742
Jaspers C., Møller L. F., Kiørboe T. (2011). Salinity gradient of the Baltic Sea limits the reproduction and population expansion of the newly invaded comb jelly Mnemiopsis leidyi. PloS One 6, e24065. doi: 10.1371/journal.pone.0024065
Karlsson S., Moen T., Lien S., Glover K. A., Hindar K. (2011). Generic genetic differences between farmed and wild Atlantic salmon identified from a 7K SNP chip. Mol. Ecol. Resour. 11 (s1), 247–153. doi: 10.1111/j.1755-0998.2010.02959.x
LaFramboise T. (2009). Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Res. 37 (13), 4181–4193. doi: 10.1093/nar/gkp552
Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Lowe S., Browne M., Boudjelas S., De Poorter M. (2000). 100 of the world’s worst invasive alien species: A selection from the global invasive species database (Auckland, New Zealand: Hollands Printing Ltd), 12.
Machiela M. J., Chanock S. J. (2015). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31 (21), 3555–3557. doi: 10.1093/bioinformatics/btv402
Martinez-Barrio A., Lamichhaney S., Fan G., Rafati N., Pettersson M., Zhang H., et al. (2016). The genetic basis for ecological adaptation of the Atlantic herring revealed by genome sequencing. eLife 5, e12081. doi: 10.7554/eLife.12081
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A. M., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Molnar J. L., Gamboa R. L., Revenga C., Spalding M. D. (2008). Assessing the global threat of invasive species to marine biodiversity. Front. Ecol. Environ. 6 (9), 485–492. doi: 10.1890/070064
Palti Y., Gao G., Liu S., Kent M. P., Lien S., Miller, et al. (2015). The development and characterization of a 57K single nucleotide polymorphism array for rainbow trout. Mol. Ecol. Resour. 15 (3), 662–672. doi: 10.1111/1755-0998.12337
Patterson N., Price A. L., Reich D. (2006). Population structure and eigenanalysis. PloS Genet. 2, e190. doi: 10.1371/journal.pgen.0020190
Penaloza C., Manousaki T., Franch R., Tsakogiannis A., Sonesson A., Aslam M. L., et al. (2021). Development and testing of a combined species SNP array for the European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata). Genomics 113 (4), 2096–2107. doi: 10.1016/j.ygeno.2021.04.038
Pocwierz-Kotus A., Kijewska A., Petereit C., Bernas R., Wiecaszek B., Arnyasi M., et al. (2015). Genetic differentiation of brackish water populations of cod Gadus morhua in the southern Baltic, inferred from genotyping using SNP-arrays. Mar. Genomics 19, 17–22. doi: 10.1016/j.margen.2014.05.010
Pritchard J. K., Stephens M., Donelly. P. (2000). Inference of population structure using multilocus genotype data. Genetics 155 (2), 945–959. doi: 10.1093/genetics/155.2.945
Purcell J. E., Shiganova T. A., Decker M. B., Houde E. D. (2001). The ctenophore Mnemiopsis in native and exotic habitats: U.S. estuaries versus the black Sea basin. Hydrobiologia 451, 145–176. doi: 10.1023/A:1011826618539
Reusch T. B. H., Bolte S., Sparwel M., Moss A. G., Javidpour J. (2010). Microsatellites reveal origin and genetic diversity of Eurasian invasions by one of the world’s most notorious marine invader, Mnemiopsis leidyi. Mol. Ecol. 19 (13), 2690–2699. doi: 10.1111/j.1365-294X.2010.04701.x
Saint-Pé K., Leitwein M., Tissot L., Poulet N., Guinand B., Berrebi P., et al. (2019). Development of a large SNPs resource and a low-density SNP array for brown trout (Salmo trutta) population genetics. BMC Genomics 20, 582. doi: 10.1186/s12864-019-5958-9
Seebens H., Bacher S., Blackburn T. M., Capinha C., Dawson W., Dullinger S., et al. (2021). Projecting the continental accumulation of alien species through to 2050. Glob. Change Biol. 27 (5), 970–982. doi: 10.1111/gcb.15333
Seebens H., Blackburn T. M., Dyer E. E., Genovesi P., Hulme P. E., Jeschke J. M., et al. (2017). No saturation in the accumulation of alien species worldwide. Nat. Commun. 8, 14435. doi: 10.1038/ncomms14435
Seeb J. E., Pascal C. E., Ramakrishnan R., Seeb L. W. (2009). SNP genotyping by the 5’-nuclease reaction: Advances in high-throughput genotyping with nonmodel organisms. Methods Mol. Biol. 578, 277–292. doi: 10.1007/978-1-60327-411-1_18
Shiganova T., Mirzoyan Z., Studenikina E., Volovik S., Siokou-Frangou I., Zervoudaki S., et al. (2001). Population development of the invader ctenophore Mnemiopsis leidyi, in the black Sea and in other seas of the Mediterranean basin. Mar. Biol. 139, 431–445. doi: 10.1007/s002270100554
Therkildsen N. O., Hemmer-Hansen J., Als T. D., Swain D. P., Morgan M. J., Trippel E. A., et al. (2013a). Microevolution in time and space: SNP analysis of historical DNA reveals dynamic signatures of selection in Atlantic cod. Mol. Ecol. 22 (9), 2424–2440. doi: 10.1111/mec.12260
Therkildsen N. O., Hemmer-Hansen J., Hedeholm R. B., Wisz M. S., Pampoulie C., Meldrup D., et al. (2013b). Spatiotemporal SNP analysis reveals pronounced biocomplexity at the northern range margin of Atlantic cod Gadus morhua. Evol. Appl. 6 (4), 690–705. doi: 10.1111/eva.12055
Thomaz S. M., Kovalenko K. E., Havel J. E., Kats L. B. (2015). Aquatic invasive species: General trends in the literature and introduction to the special issue. Hydrobiologia 746, 1–12. doi: 10.1007/s10750-014-2150-8
Keywords: jellyfish, non-indigenous species (NIS), molecular diversity, introduction events, management tool
Citation: Pujolar JM, Limborg MT, Ehrlich M and Jaspers C (2022) High throughput SNP chip as cost effective new monitoring tool for assessing invasion dynamics in the comb jelly Mnemiopsis leidyi. Front. Mar. Sci. 9:1019001. doi: 10.3389/fmars.2022.1019001
Received: 14 August 2022; Accepted: 26 September 2022;
Published: 13 October 2022.
Edited by:
Lorenzo Zane, University of Padua, ItalyReviewed by:
Yulong Li, Institute of Oceanology (CAS), ChinaSimo Njabulo Maduna, Norwegian Institute of Bioeconomy Research (NIBIO), Norway
Alberto Pallavicini, University of Trieste, Italy
Copyright © 2022 Pujolar, Limborg, Ehrlich and Jaspers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Martin Pujolar, am1hcHVAYXF1YS5kdHUuZGs=; Cornelia Jaspers, Y29qYUBhcXVhLmR0dS5kaw==