 Rui Wang1,2†
Rui Wang1,2† Xuanzhao Li
Xuanzhao Li Jiwei Qi
Jiwei Qi- 1State Key Laboratory of Stem Cell and Reproductive Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
- 2Department of Reproductive Medicine, The First People’s Hospital of Yunnan Province, Kunming, China
- 3Key Laboratory of Animal Ecology and Conservation Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
- 4School of Life Sciences, Institute of Life Sciences and Green Development, Hebei University, Baoding, China
- 5Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, College of Life Sciences, Beijing Normal University, Beijing, China
One of the most striking exceptions to strict maternal inheritance of mitochondrial DNA (mtDNA) in the animal kingdom is a system called doubly uniparental inheritance (DUI), which exists in several bivalve species. DUI is characterized by the presence of two distinct sex-associated mitochondrial lineages: one transmitted through eggs (F-type mtDNA) and the other through sperm (M-type mtDNA). Presently, most known species exhibiting DUI belong to the freshwater bivalve order Unionoida. Other groups with species exhibiting DUI include the orders Mytiloida, Veneroida, and Nuculanoida. In Veneroida, the complete M-type mtDNA is available for two species. We report the presence of DUI in three species belonging to genus Macridiscus (Macridiscus melanaegis, Macridiscus multifarious, and Macridiscus semicancellata), in the order Veneroida, further obtaining their complete M-type mitogenomes. The M-type mitogenome sizes for M. melanaegis, M. multifarious, and M. semicancellata were 19,019 bp, 18,694 bp, and 18,726 bp, respectively, and the mean nucleotide difference between M-type and F-type mitogenomes was 21–23%. We compared the M-type and F-type mitogenomes and found that they show roughly the same genome features, except for gene order. In phylogenetic analyses of Veneroida, a “gender-joining” pattern was revealed within Macridiscus, similar to the pattern of “partial” Mytilus complex (except Unionida). This new insight provides novel evidence supporting the theory that Veneroida and Mytiloida have a more similar DUI pattern than Unionida. A large-scale rearrangement between the sex-linked mitogenomes of the three Macridiscus species was reported. From the observed rearrangement patterns, gene rearrangement between the two sex-linked mitogenomes could be explained by the tandem duplication and random loss (TDRL) model of dimer-mitogenome. This is the first report of heterogeneous genomes with two types of large-scale arrangements in the same organism, and may be contribute significantly to the study of mitochondrial recombination mechanisms.
Introduction
Doubly uniparental inheritance (DUI), one of the most striking exceptions to strict maternal inheritance of mitochondrial DNA (mtDNA) in the animal kingdom, has been observed in several bivalve species (Zouros et al., 1994a; Zouros et al., 1994b). DUI is characterized by the presence of two distinct sex-associated mitochondrial lineages, i.e., the female type (F-type mtDNA), which is transmitted through the eggs to all offspring, and the male type (M-type mtDNA), which is present in sperm and enters all eggs at the time of fertilization but is only retained in and transmitted through the male offspring. In adults, the F-type mtDNA predominates in all tissues of both sexes, except for male gonads, in which the M-type mtDNA predominates. However, a few exceptions, such as Ruditapes philippinarum, have been reported, in which the M-type mtDNA is largely predominant in male somatic tissues (Ghiselli et al., 2011). In addition, adult females are generally homoplasmic, while adult males are heteroplasmic (Zouros, 2012). The two sex-linked mtDNAs are therefore inherited separately, and as a result, they evolve independently and more rapidly than mtDNA in most other animals, with the M-type evolving faster than the F-type. Furthermore, a high level of sequence divergence between the two sex-linked mitogenomes has been observed, ranging from 10% to 50% (Breton et al., 2007; Passamonti and Ghiselli, 2009; Zouros, 2012).
To date, majority of species reported to exhibit DUI belong to the freshwater bivalve order Unionoida. Other species with DUI belong to the orders Mytiloida, Veneroida, and Nuculanoida. To date, few species in Veneroida have been reported to exhibit DUI, and the complete M-type mtDNA is available for only two species (Passamonti and Scali, 2001; Bettinazzi et al., 2016).
In this study, we investigated the presence of DUI in three species of genus Macridiscus, a taxon closely related to R. philippinarum. Genus Macridiscus belongs to the family Veneridae (Bivalvia: Veneroida), and includes three clam species with similar morphology. These commercially exploited shellfish are endemic to the western Pacific, extending from the coast of Japan to northwestern Australia (Kong et al., 2012; Ye et al., 2015). In our previous work, we sequenced the complete F-type mitogenomes of three Macridiscus species (Qi et al., 2019). Subsequently, we determined the presence of DUI in these three species, and sequenced their complete M-type mitogenomes.
Materials and methods
Sample collection and DNA extraction
The specimens of Macridiscus multifarius were sampled from natural beds in Haiyang (Shandong Province, China). Macridiscus melanaegis specimens were purchased from the Rongcheng farmers’ market (Shandong Province, China), and Macridiscus semicancellata specimens were purchased from the Zhoushan farmers’ market (Zhejiang Province, China). All specimens were dissected while alive, and gonadal extract was collected using a glass capillary and inspected microscopically, to confirm sexual maturity and to determine the sex. Adductor muscle samples were collected from 15 males of each species, and their sperms were stored in liquid nitrogen, following the procedure described in (Ghiselli et al., 2011). Precautions were taken to avoid contamination. Total genomic DNA was extracted from both the adductor muscle tissue and gonadal extract, using the CTAB and phenol/chloroform methods (Winnepenninckx et al., 1993). Three male individuals, one from each species (specimen HYM4 for M. multifarious, RCM3 for M. melanaegis, and ZSM9 for M. semicancellata) were selected for further sequencing, based on their total nucleic acid yield.
PCR amplification and sequencing
The three complete M-type mitogenomes were amplified in two large non-overlapping fragments, using long and accurate polymerase chain reaction (LA-PCR) (Cheng et al., 1994) paired with primer-walking sequencing. The gap at the primer position was then joined by redesigning the two pairs of primers, with same PCR conditions. The primer pairs used for amplification are shown in Table 1 and were designed based on our preliminary experiment. We first acquired a fragment of the partial rrnL gene sequence, using paired 16sar and 16sbr primers (Simon et al., 1994), from the male gonads of the three Macridiscus species. The results of sequencing, based on DNA extracted from the adductor muscle tissue, the partial rrnL gene exhibited a 21% difference from that of the F-type mitogenome. We then employed paired LCO1490 and HCO2198 primers (Folmer et al., 1994) in an attempt to obtain the M-type partial cox1 gene; however, a 23% sequence difference was observed between the M- and F-type sequences for M. multifarius. Based on this sequence and the three Macridiscus F-type mitogenomes obtained in our previous research, primers were designed to be paired with those of the rrnL gene from M. melanaegis and M. semicancellata. Finally, the designed primers were used to join the break between the cox1 and rrnL genes.

Table 1 LA-PCR Primers and conditions for amplifying the M-type mitogenomes of the three Macridiscus species.
LA-PCR amplifications were carried out in 25 µL reaction mixtures, containing 150 ng genomic DNA, 1 × Phanta® Max Buffer (Mg2+ plus), 200 µM of each dNTP, 0.2 μM of each primer, and 0.5 U Phanta® Max Super-Fidelity DNA Polymerase (Vazyme biotech, Nanjing, China). The thermal cycling conditions consisted of pre-denaturation at 95°C for 3 min, followed by 35 cycles of denaturation at 95°C for 15 s, annealing at the appropriate temperature for 15 s, and extension at 72°C for 1–7 min depending on the expected lengths of the PCR products, and a final extension at 72°C for 7 min (Table 1). PCR results were visualized via electrophoresis on 1% agarose gel stained with Ethidium bromide (EB), and then purified using the E.Z.N.A.® Gel Extraction Kit (Omega Bio-tek, Inc., Georgia, USA). PCR products were sequenced using the PCR primers of 5’ and 3’ ends, and then using primers designed according to the ends of the newly obtained sequences, until the sequences from both the directions met. All sequencing primers were designed to ensure an overlap of at least 100 bp between the abutting fragments to ensure proper sequence assembly. Sequencing was performed using the BigDye® Terminator Cycle Sequencing Kit (v3.1) on an ABI PRISM® 3730 Genetic Analyzer (both from Applied Biosystems, Foster City, CA, USA).
Assembly, annotation, and sequence analyses of the mitogenomes
Overlapping fragments obtained after sequencing were assembled using the SeqMan Pro program (Lasergene version 7.1, DNASTAR, Madison, WI). The protein-coding genes (PCGs) and two rRNA genes were identified using combined analyses with the MITOS pipeline (http://mitos.bioinf.uni-leipzig.de) (Bernt et al., 2013c), applying the invertebrate mitochondrial genetic code. ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to locate the putative open reading frames (ORFs) for PCGs. BLASTx (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to compare the putative ORFs with the published nucleotide and amino acid sequences of related species, and T-Coffee (http://tcoffee.vital-it.ch/apps/tcoffee/index.html) (Di et al., 2011) was used for protein alignment using structural information and homology extension. The boundaries of each gene were determined using multiple alignments of known bivalve mitochondrial sequences. The tRNA genes were identified and their secondary structures were predicted using tRNAScan-SE v.2.0 (Lowe and Chan, 2016) and ARWEN 1.2 (Laslett and Canback, 2008). Nucleotide composition and codon usage were analyzed with MEGA 7.0 (Kumar et al., 2016). Strand asymmetry was measured using the formulas GC skew = (G - C)/(G + C) and AT skew = (A - T)/(A + T) (Perna and Kocher, 1995). The divergence of protein genes in synonymous (Ks) and non-synonymous (Ka) sites was calculated using DnaSP v5 (Librado and Rozas, 2009). Potential tandem repeats in the mitogenome sequences were searched for using Tandem Repeats Finder 4.0 (Benson, 1999). Mitogenome maps were drawn using GenomeVx online tool (Conant and Wolfe, 2008), followed by manual modification.
Phylogenetic and evolutionary rearrangement comparisons
To investigate the phylogenetic relationships between the species exhibiting DUI within the Veneridae family, we used the 23 mitogenomes currently available, including 15 F-type and two M-type mitogenomes of R. philippinarum and Meretrix lamarckii, respectively, obtained from GenBank (last accessed 30 January 2019), three F-type Macridiscus mitogenomes from our previous research, and the three M-type Macridiscus mitogenomes determined in the present study. Mactra chinensis and Coelomactra antiquata, belonging to superfamily Mactroidea, were used as outgroups (Supplementary Table 1). We used 12 PCGs for analyses, and the atp8 gene was excluded. The nucleotide sequences of the 12 mitochondrial PCGs were aligned using the MAFFT server (https://www.ebi.ac.uk/Tools/msa/mafft/) (Katoh and Toh, 2008) and poorly aligned regions were removed using Gblocks v.0.91b (Castresana, 2000), with options allowing gaps for all positions and less strict flanking positions. The 12 separate nucleotide sequence alignments were concatenated into one contig with 9,062 sites for each species using SequenceMatrix (Vaidya et al., 2011). The best-fitting partitioning scheme and molecular evolution models were estimated using PartitionFinder-2.1.1 (Lanfear et al., 2012), based on the Akaike Information Criterion corrected (AICc) and a greedy approach.
Phylogenetic relationships were established using the maximum likelihood (ML) and Bayesian inference (BI) methods. The ML analysis was performed using RaxML8 (Stamatakis, 2014), with the search strategy set to rapid bootstrapping and 1000 bootstrap replicates. ML bootstrap proportions greater than 70% were considered as a strong support for the resolved clades (Hillis and Bull, 1993; Wilcox et al., 2002). The BI analysis was performed using MrBayes 3.2.6 (Ronquist et al., 2012) with Metropolis coupled Markov Chain Monte Carlo (Metropolis coupled MCMC or MC3) algorithm. Three independent runs were executed, each including four chains of default heating values, and one million generations were run, sampling trees every 1000 generations, with the initial 25% being discarded as burn-in. Stationarity was reached when the average standard deviation of split frequencies was less than 0.01. Posterior probability support values higher than 0.95 were considered as a strong support (Huelsenbeck et al., 2004).
We mapped all known gene orders of Veneroidea on the resulting phylogeny, and then used CREx (Bernt et al., 2007) to determine the evolutionary scenario to have occurred between the breach. Finally, the different gene orders were searched for the conserved gene blocks identified by Bernt (Bernt et al., 2013a). We performed synteny alignment with Last version 984 (Kiełbasa et al., 2011) using the parameters ‘-m 2 -E 0.001 -l 30 -P 6’.
Results
Overall genomic features
The M-type mitogenomes of the three Macridiscus species sequenced were circular molecules. These mitogenomes for M. melanaegis, M. multifarius, and M. semicancellata measured 19,019 bp, 18,694 bp, and18,726 bp (GenBank accession no. OP187076, OP187077 and OP187078), respectively (Figure 1; Supplementary Table 2). Length differences were mostly due to the variation in size of the non-coding regions of the mitogenomes, which measured of 3,406 bp, 2,706 bp, and 2,864 bp, respectively. These M-type mitogenomes were composed of 36 genes: 12 PCGs (excluding the atp8 gene), two rRNA, and 22 tRNA genes. All genes were located on the same “+” strand, and the mitogenomes of the three Macridiscus species shared the same gene order.
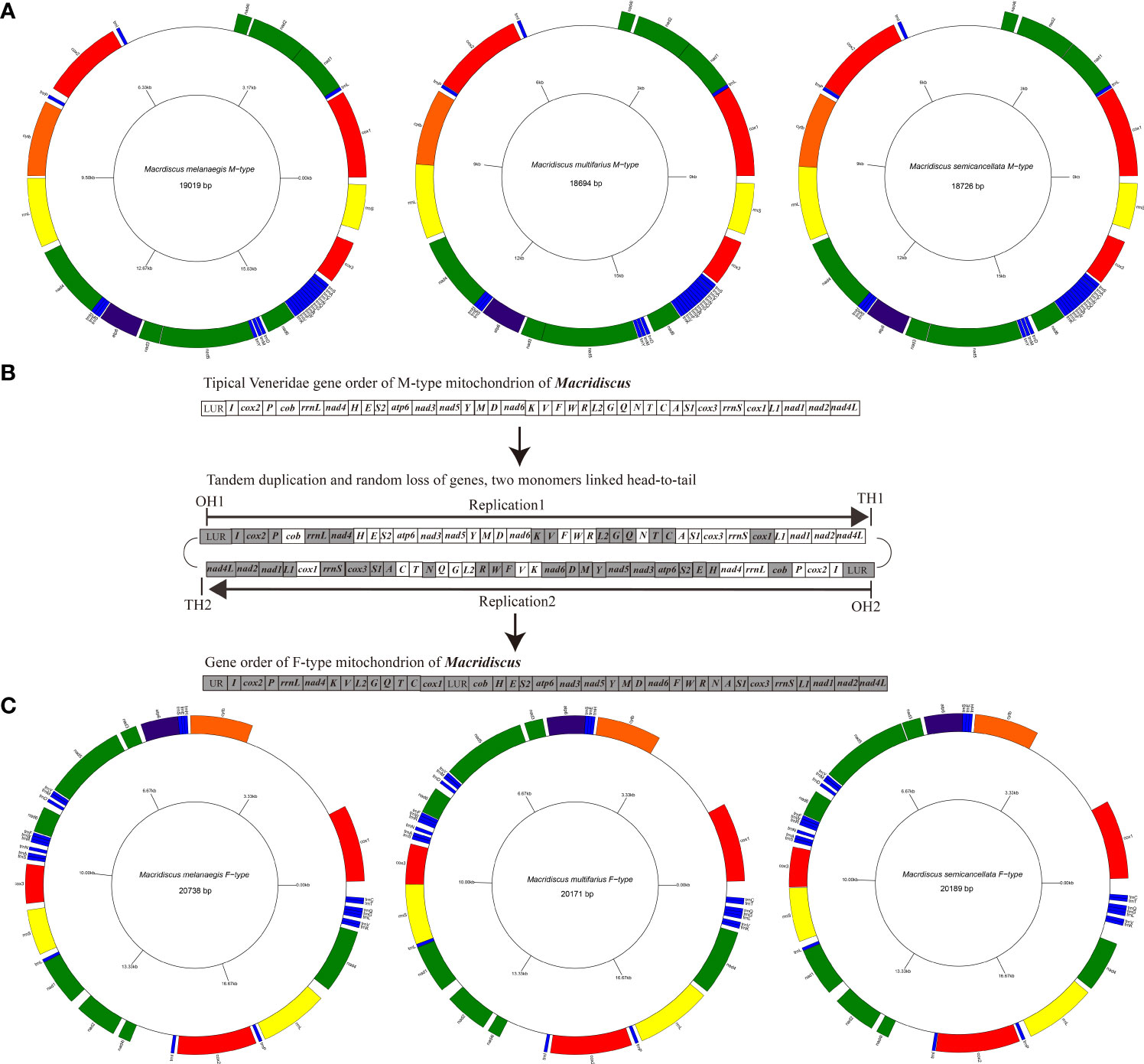
Figure 1 Inferred intermediate steps from the M-type gene order to F-type gene order of Macridiscus mitogenome. (A) M-type Mitochondrial gene maps of the three Macridiscus species. All the genes transcribed from the plus strand. Genes are mapped in anticlockwise starting from cox1. (B) Intermediate steps of Tandem duplication and random loss of genes. Protein-coding genes tRNA rRNA and LUR are indicated by boxes. OH and TH indicate the heavy-strand origin and termination of replication. The direction of transcription is shown by arrows. (C) F-type Mitochondrial gene maps of the three Macridiscus species.
Nucleotide composition is presented in Table 2. The A+T content of the three M-type mitogenomes were 66.88%, 66.48%, and 67.20%, respectively. All nucleotide compositions were strongly skewed, away from C and in favor of G (GC-skew = 0.32 – 0.35), and from A in favor of T (AT-skew = -0.18 – -0.16).
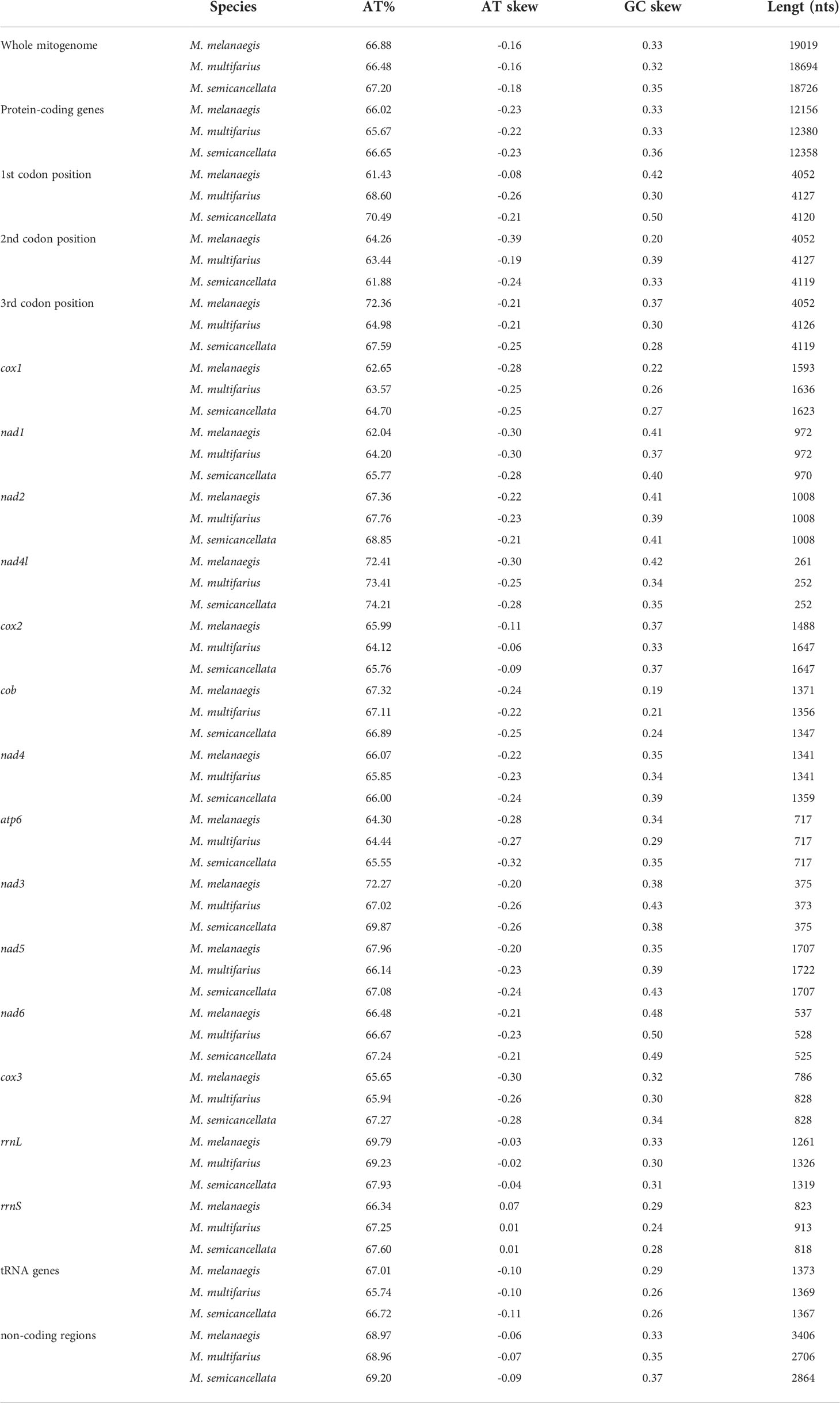
Table 2 AT content, AT and GC skew of the M-type mitogenomes of the three Macridiscus species, The order of genes is consistent with mitogenomes.
Protein coding genes
The 12 PCGs identified in the three M-type mitogenomes were analyzed, and the absence of the atp8 gene was consistent with the condition observed in some other bivalve species (Yu et al., 2008; Breton et al., 2010; Plazzi et al., 2011; Plazzi et al., 2016). Recently, Plazzi et al. (2016) reported the atp8 gene in 71 out of the 100 bivalve species analyzed for their mitogenomes. However, we did not find the atp8 gene by annotation, possibly because this gene has a short but variable length, and shows high variation in its amino acid composition (Gissi et al., 2008).
Four different start codons were found in the PCGs of the three M-type mitogenomes: ATA (12 times in total in the three mitogenomes), ATT (12), ATG (7), and GTG (5). Except the two common stop codons, i.e., TAG (13) and TAA (20), we observed truncated stop codons T- (thymine) in cox1 and nad3 genes of M. multifarius, and in nad1 gene of M. semicancellata. The truncated T would be completed (UAA) by the addition of 3’ A residues to the mRNA (Schuster and Stern, 2009; Temperley et al., 2010; Wen et al., 2017). All these codons are typical to the invertebrate mtDNA (Wolstenholme, 1992).
The PCGs of the mitogenomes contained 4,052 codons in M. melanaegis, 4,125 codons in M. multifarious, and 4,119 codons in M. semicancellata. The codon usage in the three M-type mitogenomes (Supplementary Table 3) was similar to that in other Veneridae species (Bettinazzi et al., 2016). In all three mitogenomes, the most frequent codon was UUU (311, 323, and 331 hits, respectively), and the least common codon was CGC (3, 6, and 0 hits, respectively). The most common amino acid in the three mitogenomes was leucine (582, 588, and 584 hits respectively), while arginine was the rarest (66, 78, and 72 hits, respectively).
The non-synonymous (Ka) and synonymous (Ks) substitution rates reflect the evolutionary dynamics of protein-coding sequences across closely related species. The Ka/Ks ratios of the 12 common PCGs of all pairs of the three M-type mitogenomes were less than 1, ranging from 0.019 to 0.278 (Supplementary Table 4; Supplementary Figure 1), indicating that these genes were under negative or purifying selection.
rRNAs and tRNAs
Standard rRNAs were identified via comparison with other bivalve rRNA genes; rrnS was located between cox3 and cox1, while rrnL was located between cob and nad4. The predicted secondary structures of these mitogenome rRNAs are given in Supplementary Figure 2.
The three M-type mitogenomes presented a typical complete set of 22 tRNAs, i.e., two tRNAs each for serine and leucine, and one tRNA for each of the other 18 amino acids (Supplementary Figure 3). The tRNAs ranged in size from 55 to 67 bp, and most of them could be folded into the canonical clover-leaf secondary structure, except those for serine, which were missing the “DHU” arm, thus being simplified down to several base pairs.
Non-coding regions
In M. melanaegis, M. multifarius, and M. semicancellata, the non-coding sequences represented 17.91% (~3,406 bp), 14.48% (~2,706 bp), and 15.29% (~2,864 bp) of their mitogenomes, respectively. The A + T content of the non-coding regions in the three mitogenomes was 68.97%, 68.96%, and 69.20%, respectively, with negative AT-skew (-0.09 – -0.06) and positive GC-skew (0.33 – 0.37).
The largest non-coding region was located between nad4l and trnI, with a length variation of 1,813–2,180 bp. Three tandem repeats were found in the longest non-coding regions (Supplementary Figure 4). The first motif consisted of nearly 4.2 identical copies of an 11 bp unit located at the 5,142-5,187 bp position in M. multifarious. The second and third were both located at the 5,137-5,237 bp position in M. semicancellata, one had 9.2 copies of an 11 bp unit, and the other had 3 copies of a 10 bp unit, and the two tandem repeats exhibited an overlap of 4 bp.
Phylogenetic analysis and rearrangement
ML and BI trees, based on the sequences of 12 concatenated PCGs (excluding atp8), belonging to 23 mitogenomes currently available, were constructed (Figure 2). The complete dataset was composed of 9,062 sites. Partition Finder suggested the partition of PCGs in two clusters, and the optimal method was GTR+I+G for cox1, cox3, cob, nad1, nad2, nad3, nad4, nad5, nad6, and atp6 genes, and GTR+G for cox2 and nad4l genes. The congruent topologies and bootstrap proportion for ML, and posterior probability for BI were found to be strongly supporting in all nodes. Veneridae was separated into Tapetinae and Meretricinae as two monophyletic clades. The three Macridiscus species were in the sister taxon of the DUI species R. philippinarum, and their M-type and F-type genomes were separated from each other, with a “gender-joining” phylogenetic pattern (Zouros, 2012).
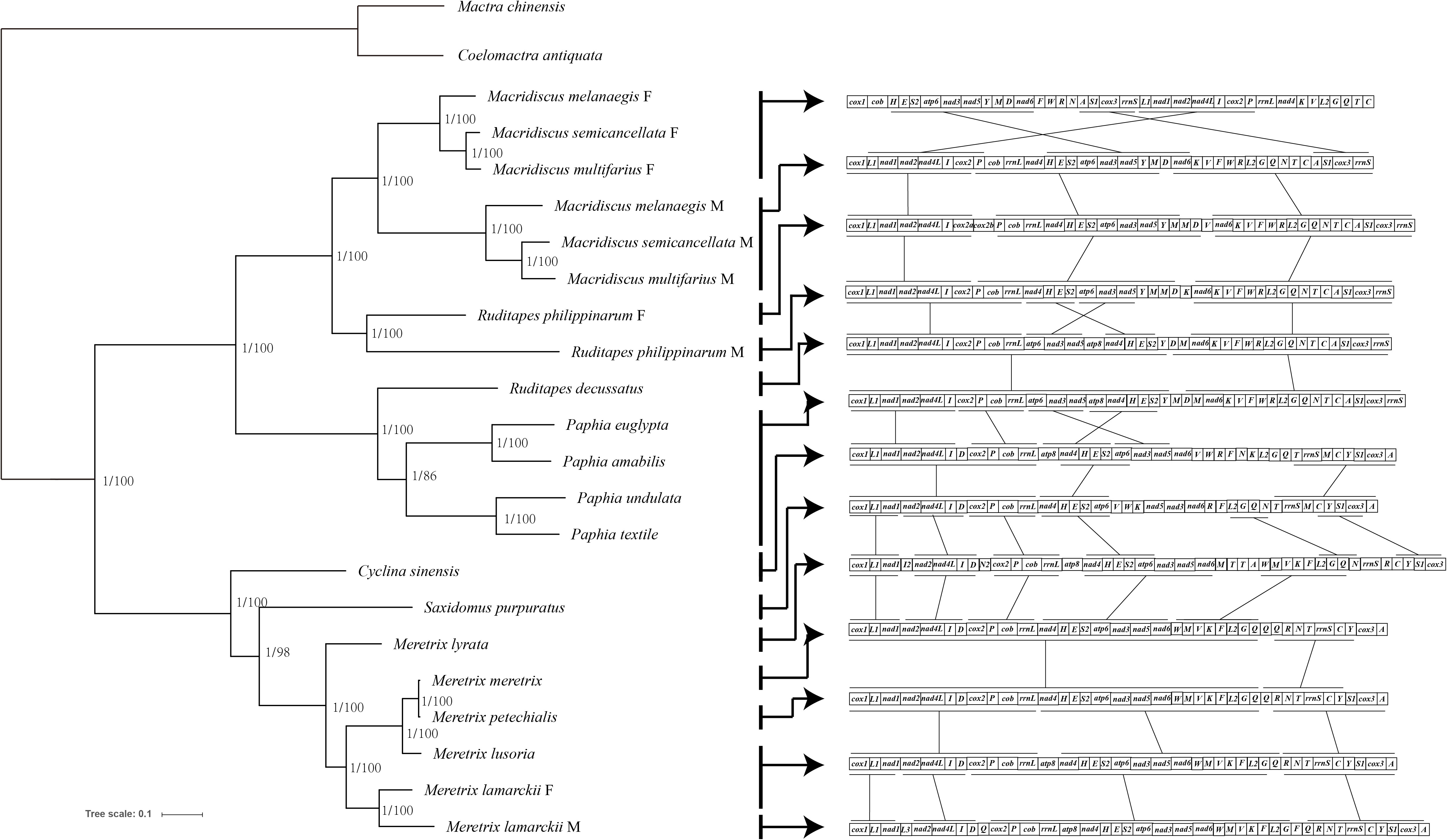
Figure 2 Phylogeny of the Family Veneridae based on nucleotide sequences of the 12 PCGs and rearrangement of mtDNA. The topology is ML tree. Numbers in front of the nodes are Bayesian posterior probabilities and back of the nodes are ML bootstrap proportions, right side ligature is conservative gene block.
The gene order of the three M-type mitogenomes was identical, and almost consistent with the F-type R. philippinarum (except duplication and deletion of cox2, trnV, and trnM genes). The M-type Macridiscus mitogenomes shared three gene blocks with the F-type R. philippinarum, i.e., cox1 – trnL1 – nad1 – nad2 – nad4l – trnI – cox2, trnP – cob – rrnL – nad4 – trnH – trnE – trnS2 – atp6 – nad3 – nad5 – trnY – trnM, and nad6 – trnK – trnV – trnF – trnW – trnR – trnL2 – trnG – trnQ – trnN – trnT – trnC – trnA – trnS1 – cox3 – rrnS (Figure 2). However, the gene order of the three M-type Macridiscus mitogenomes was completely different from that of the F-type mitogenomes.
Discussion
Comparison of the DUI genome features of the two sex-linked genomes
The F-type mitogenomes of the three Macridiscus species have been reported in our previous study (Qi et al., 2019). In the present study, we focused on comparing the three pairs of sex-linked mitogenomes of these species. The intraspecific mean nucleotide divergence between M-type and F-type mitogenomes of the three Macridiscus species was around 21 – 23%. Among Veneridae species, this level of divergence is more than that observed in M. lamarckii, and roughly equal to that in R. philippinarum (Passamonti and Scali, 2001; Bettinazzi et al., 2016). In terms of genome length, the F-type mitogenome was longer than the M-type. This size variation was mainly due to the two long unassigned regions in the F-type mitogenomes, totaling 5,053 bp in M. melanaegis, 4,202 bp in M. multifarius, and 4,606 bp in M. semicancellata (location between cob and cox1, and between trnI and nad4l), while the M-type mitogenome included only one such region, of 2,180 bp, 1,813 bp, and 1,867 bp, respectively (between trnI and nad4l).
Both types of mitogenomes exhibited roughly the same genome composition, nucleotide composition, PCG length, and type of start, stop and PCG codons. However, the M-type mitogenome has higher non-synonymous (Ka) and synonymous (Ks) substitution rates than the F-type mitogenome (Supplementary Figures 1, 5), i.e., the M-type mitogenome experiences a more relaxed selection pressure than the F-type mitogenome (Ort and Pogson, 2007; Śmietanka et al., 2009). This may be because F-type mitochondria are the energizing organelles of all tissues of female individuals and somatic cells of male individuals, while M-type mitochondria are only present in the germ cell line of male individuals, and play an important role only at a certain stage of gametogenesis, which allows the frequency of amino acid substitutions in M-type mitochondrial genome PCGs to increase (Obata et al., 2011). In addition, compared to the F-type, the nad4l gene in the M-type mitogenome has a very high non-synonymous mutation rate, and its Ka/Ks value is significantly higher than other genes, which can be quite low, and even zero, in the F-type mitogenome (Qi et al., 2019). We speculate that it might be due to gene order rearrangement within the two sex-linked genomes and transposition of the control region caused by the TDRL process; one of the Long Unassigned Region (LUR) might undergo loss of function or become a pseudogene (detailed in following sections), resulting in a transfer of position of the heavy chain replication origin. The nad4l gene is found immediately upstream of the LUR region in the M-type mitogenome, while the F-type LUR is located between cox1 and cob genes, far from nad4l. In the classical mitochondrial gene replication theory, according to the strand displacement model, genes that are close to the origin of replication are exposed to a single-stranded state for a longer period of time than other genes, and therefore undergo accelerated mutation due to longer chemical oxidative damage (Clayton, 1991; Ren et al., 2010; Yuan et al., 2012). Studies have shown that oxidative damage is significantly higher in the sperm than in the egg (Stewart et al., 1996). This pattern was also observed in the mitochondrial genomes of six Arcidae species (Sun et al., 2016).
ORF genes with different endogenization
It is interesting to note that several species with DUI appear to possess lineage-specific mitochondrial ORFs, one in the F-type, and one in the M-type. According to in silico predictions, such ORFs are correlated with DUI emergence and establishment, due to their involvement in sex determination, reproduction, embryo development, and spermatogenesis (Breton et al., 2011a; Breton et al., 2011b; Milani et al., 2016). The ORFs have an ancient viral origin and are non-homologous to other known proteins. Moreover, they might possess the ability to invade the germ line, producing a selfish element (Milani et al., 2015; Mitchell et al., 2016). Although such ORFs share some common features, their alignments are possible only among sequences of closely related species (Milani et al., 2013). However, on comparison with the related R. philippinarum, a well-studied species (Milani et al., 2014a), it was revealed that the ORFs are positioned between nad4l and nad2 of R. philippinarum (523 bp long) (Milani et al., 2013), while there is no non-coding region of more than 71 bp in the same position in Macridiscus species. Furthermore, we search homologous regions of the R. philippinarum ORF sequences in all mitochondrial non-coding regions of Macridiscus. We found a 126 – 150 bp long candidate for the female supernumerary ORF (FORF) gene in the three F-type Macridiscus mitogenomes, between nad4l and trnI genes, and a 279 – 351 bp long candidate for the male supernumerary ORF (MORF) gene in the M-type mitogenome, at the same position (alignment of protein using structural information and homology extension is shown in Supplementary Figure 6; location and sequence information is detailed in Supplementary Table 5). Furthermore, The position is consistent with the position found by studies on Unionids and Mytilus species which at the beginning of LUR (Breton et al., 2011b; Milani et al., 2013). However, the FORF and MORF of Macridiscus are non-homologous with other known ORFs of Unionids and Mytilus species, even for closely related species, such as R. philippinarum and M. lamarckii. Based on the hypothetical model proposed by Milani et al. (2016), we speculate that this might be due to several independent endogenization events. The untestable homology does not appear to be a result of rapid evolution, but it might be due the fact that the ORFs of R. philippinarum and Macridiscus have been derived from the endogenization of viral sequences after species differentiation.
Phylogenetic analyses and new clues for the DUI pattern of Veneridae
A robust phylogenetic framework is necessary to elucidate the origin, function, and molecular evolutionary consequences of DUI. To determine the DUI pattern between two sex-linked mitogenomes of Macridiscus, and to compare with other DUI species, ML and BI trees, based on nucleotide sequences of 12 concatenated PCGs, belonging to 23 sequences available from NCBI, were constructed (Figure 2). The three M-type Macridiscus mitogenomes clustered together as a monophyletic group, separate from another monophyletic group composed of the three F-type Macridiscus mitogenomes. Macridiscus belongs to the order Veneroida, however, other species of Veneroida, Mytiloida, Nuculanoida, and members of Autobranchia, excluding the Mytilus species complex, exhibit a “taxon-joining” pattern, in which the F-type and M-type mitogenomes sequences cluster together in a monophyletic group. However, Macridiscus species exhibited a “gender-joining” phylogenetic pattern, i.e., the F-type and M-type mitogenomes formed two separate clusters, which was similar to order Unionoida, except that the split did not date back to 200 million years, as in Unionids (Doucet-Beaupré et al., 2010). The split between the F-type and M-type Macridiscus mitogenomes is more recent, occurring after the split in the two sex-linked mitogenomes of R. philippinarum. This suggests that the Macridiscus DUI pattern is more like a “partial” complex of the DUI pattern of genus Mytilus. The DUI pattern is mixed-type for the four Mytilus species, i.e., it is “gender-joining” for Mytilus edulis and Mytilus trossulus, while Mytilus galloprovincialis occurs as a sister group of the F-type of Mytilus californianus, but separate from the M-type of M. californianus (Zouros, 2012; Gusman et al., 2016). This new insight, i.e., the “gender-joining” phylogenetic pattern found in the order Veneroida, indicates that Veneroida and Mytiloida both exhibit “taxon-joining” and “gender-joining” patterns, while the Unionoida only has the “gender-joining” pattern, and the two sex-linked lineages have higher divergence. We identified the Mytilus species complex, which is not the only known case of a gender-joining pattern among Autobranchia, and provide new evidence for the dichotomy separating the DUI species into two taxa, i.e., Unionidae and Mytilidae + Veneridae (Bettinazzi et al., 2016).
If we consider the hypothesis of a single origin of DUI, an explanation for the “gender-joining” pattern observed in Macridiscus would be that the last masculinization event occurred in the common ancestor of the three species, followed by species differentiation. Such masculinization events have also been observed in Mytilus (Stewart et al., 2009; Zouros, 2012). The underlying mechanism is that F-type obtains sperm transmission elements from the male control region by recombination, thereby invading the M-type mtDNA transmitted via sperm, and resetting the nucleotide divergence between F-type and M-type mtDNA to zero, following which the M-type mtDNA accumulates more mutations than the F-type, because of relaxed selective constraints, and the M-type “selfish element” is active only if this function cannot be performed by the F-type mtDNA (Ort and Pogson, 2007; Śmietanka et al., 2009; Obata et al., 2011). According to the hypothesis of multiple origin of DUI, developed through in silico analyses (Milani et al., 2013; Milani et al., 2014b; Milani et al., 2016). The three Macridiscus lineages likely experienced a unique viral independent endogenization event, or different mechanisms, that modified SMI to DUI, because the ORF gene is non-homologous, even to closely-related R. philippinarum and M. lamarckii. However, the debate about the theory of origin of DUI, i.e., single or multiple origin, remains unresolved.
A TDRL model of the dimer-mitogenome rearrangement mechanism
The species in the phylum Mollusca are extraordinarily variable in the arrangement of genes in their mitogenomes (Boore and Brown, 1994; Boore and Brown, 1998). Bivalves display an even higher degree of gene rearrangements in their mitogenomes, compared to other Mollusca species (Serb and Lydeard, 2003). However, such a large-scale rearrangement, as observed between the sex-linked mitogenomes of the three Macridiscus species, occurs less frequently (Figure 2). It is difficult, therefore, to propose a mechanism to account for the observed differences in the mitogenome structure. Mitochondrial gene rearrangements are usually explained by the recombination model (Lunt and Hyman, 1997) or the TDRL model (Boore, 2000). The TDRL model is widely accepted, and requires only indirect evidences, such as pseudogenes and residues of tandemly duplicated sequences, as an intermediate supporting step (Xia et al., 2016). According to this model, the newly synthesized mitogenome contains duplicated genes, one of which is subsequently and randomly lost due to errors, such as imprecise terminations, strand slippages, and/or mispairings, in mtDNA replication (Macey et al., 1997; Macey et al., 1998). To compare the gene order in family Veneridae, and to determine the evolution scenario between the taxa, the CREx tool was used. The gene order of F-type Macridiscus mitogenomes was very different from the others, and the M-type was almost consistent with the F-type R. philippinarum mitogenome and was more like the ancestral Veneridae order. CREx employs the TDRL model (tandem duplication and random loss model) of dimer-mitogenome to explain the evolutionary scenario between two sex-linked mitogenomes.
We further assumed that the two processes leading to the M-type to F-type gene arrangements in Macridiscus are as follows (Figures 1B, 3). The first step would be a tandem duplication of the entire mitogenome, resulting in a dimeric molecule, with the two monomers linked head-to-tail (Figures 1B, 3). The genes and coding regions of the dimeric mtDNA are assumed to have retained their functions at this time, so that transcription could be initiated normally at the promoters (both L-strand and H-strand). Unlike vertebrates, the invertebrates do not have a “WANCY” region to indicate the origin of L-strand (Sahyoun et al., 2014), and hence, we cannot determine the location of promoters on the L-strand. Subsequently, the functionality of transcriptional promoters in one of the non-coding regions would have been lost or severely impaired, due to mutation or fragment loss. The sets of genes under the control of the disabled promoter would become pseudogenes, and the sequences containing them would be free to mutate, and to disappear from the genome. Due to the accumulation of deleterious mutations in one of the copies of duplicated genes, the selection pressure on the size of the mitogenomic DNA would rapidly remove the non-functional pseudogenes (Bernt et al., 2013b). In the end, the genes transcribed would be clustered together as a new order. Alternatively, in the first step, transcription would be initiated normally at the promoters on both strands. Subsequently, when the replication reached the transcriptional terminator of the non-coding regions, whose functionality was lost or severely impaired due to mutation or fragment loss, the tandem duplication of the entire mitogenome till the second functional normal terminator resulted in a single stranded molecule with the two monomers linked head-to-tail (Figures 1B and 3). The sets of genes under the control of the terminator would be duplicated and be free to mutate and disappear from the genome for the aforementioned reason. In the end, the genes transcribed would be clustered together as a new order.
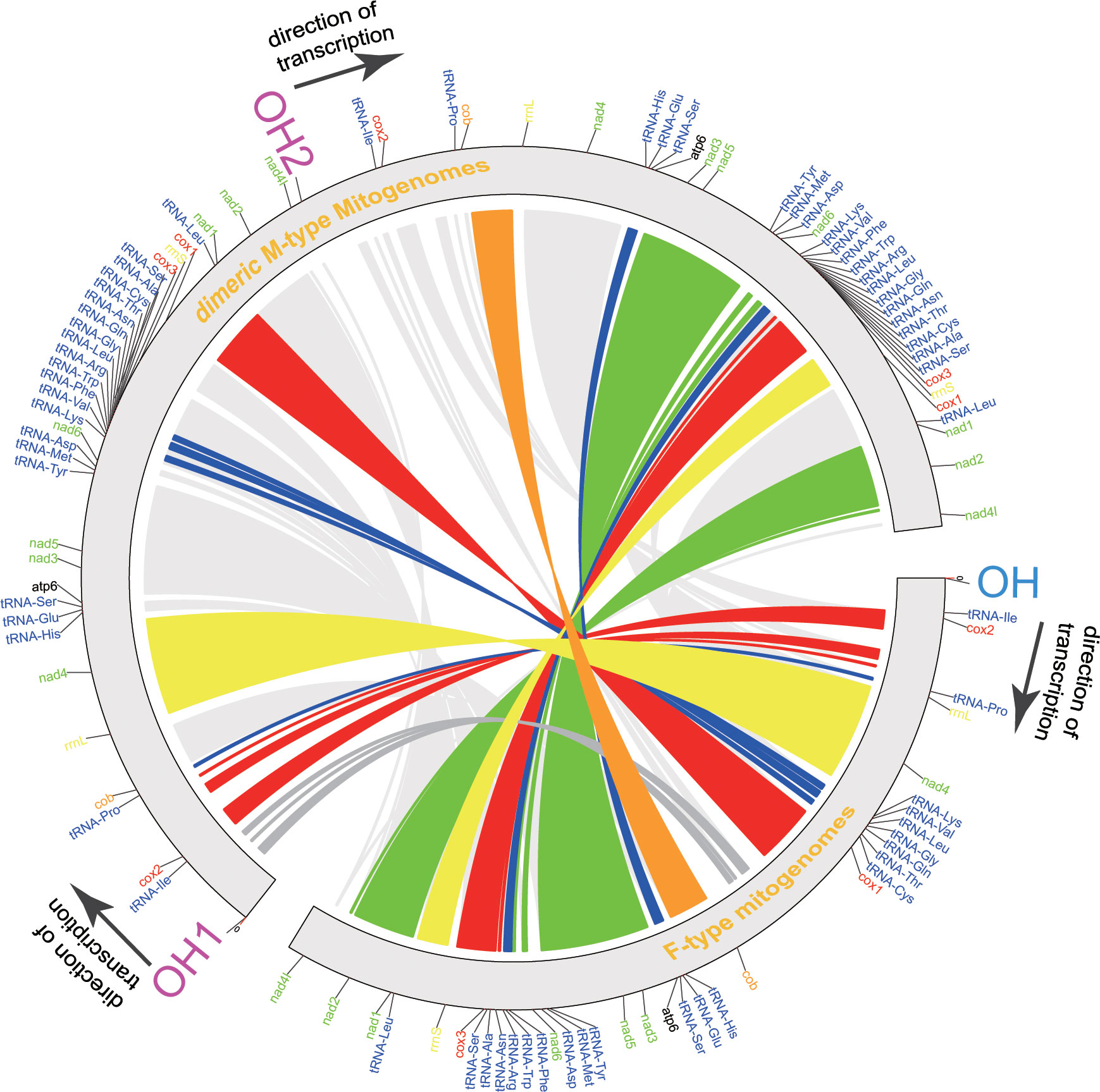
Figure 3 Circos diagrams synteny of dimeric M-type and F-type Macridiscus mitochondrion.
These theories are supported by the presence of two large non-coding regions in the F-type mitogenome, of which the one located between nad4l and trnI is the residue of tandemly duplicated sequences for an intermediate step. Despite the duplication of the complete genome, dimeric mitogenomes are not common in mitogenomic replication, however, such dimeric mitogenome molecules have been reported in many animals, such as millipedes (Lavrov et al., 2002), isopods (Neuroni et al., 1999), and flatfishes (Shi et al., 2013; Shi et al., 2015). Such a structure is not unimaginable and can be seen as duplication of a very large fragment.
Reuse potential
In Veneroida, R. philippinarum was the first species reported to exhibit DUI, and studies on R. philippinarum have resolved the origin and mechanism of DUI to a great extent (Ghiselli et al., 2011; Milani et al., 2014a; Guerra et al., 2016). Subsequently, the acquisition of the M-type M. lamarckii mtDNA made this species the only mitogenomic experimental counterpart of R. philippinarum (Bettinazzi et al., 2016). Therefore, our study has increased the total number of known sex-linked mitogenomes of species with DUI in Veneroida to five. Genus Macridiscus presents the second case of a “gender-joining” pattern of phylogenetic reconstruction among Autobranchia, with the Mytilus species complex being the other. This may suggest that Veneroida and Mytiloida may have some similarities in the mechanism of DUI. Further research on ORFs and masculinization events of Macridiscus are required to determine the origin of DUI, i.e., single or multiple origin, and to understand the evolution and molecular machinery of DUI.
The mitochondrial rearrangement event between the two sex-linked mitogenomes of Macridiscus species may not be related to the DUI system itself, but rather to the stochastic processes. However, due to the characteristics of the DUI system, it was first discovered in the animal kingdom that two extensive rearranged mitochondria exist in the same individual. This may contribute significantly to the study of mtDNA recombination, because there are two heteroplasmic and significantly different gene orders of mitochondria in close enough physical proximity to allow a crossover event. This makes the detection of mitochondrial recombination events very convenient and allows for easy understanding of the basic issues related to recombination (White et al., 2008; Zouros, 2012).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by Committee for Animal Experiments of the Institute of Zoology, CAS.
Author contributions
JQ supervised the study and managed the project. RW and JQ collected the samples in-field. RW and XL performed the genome sequencing, assembly, annotation, genetic data analyses, and writing of the draft manuscript. JQ, RW, and XL contributed to the taxonomy and phylogeny issues. RW, XL, and JQ finished the final manuscript with contributions from all other authors. All authors contributed to the article and approved the submitted version.
Funding
The grants supporting this research were received from the National Key R&D Program of China (2021YFC2700300) and the National Natural Science Foundation of China (31771633).
Acknowledgments
We express sincere thanks to Professors Jie Han and Jie Zhang for their long-term encouragement and support. We would like to especially thank Professor Ming Li for his technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1016779/full#supplementary-material
References
Śmietanka B., Burzyński A., Wenne R. (2009). Molecular population genetics of male and female mitochondrial genomes in European mussels mytilus. Mar. Biol. 156 (5), 913–925. doi: 10.1007/s00227-009-1137-x
Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bernt M., Bleidorn C., Braband A., Dambach J., Donath A., Fritzsch G., et al. (2013a). A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenet. Evol. 69 (2), 352–364. doi: 10.1016/j.ympev.2013.05.002
Bernt M., Braband A., Schierwater B., Stadler P. F. (2013b). Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 69 (2), 328–338. doi: 10.1016/j.ympev.2012.10.020
Bernt M., Donath A., Juhling F., Externbrink F., Florentz C., Fritzsch G., et al. (2013c). MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69 (2), 313–319. doi: 10.1016/j.ympev.2012.08.023
Bernt M., Merkle D., Ramsch K., Fritzsch G., Perseke M., Bernhard D., et al. (2007). CREx: inferring genomic rearrangements based on common intervals. Bioinformatics 23 (21), 2957–2958. doi: 10.1093/bioinformatics/btm468
Bettinazzi S., Plazzi F., Passamonti M. (2016). The complete female- and Male-transmitted mitochondrial genome of meretrix lamarckii. PloS One 11 (4), e0153631. doi: 10.1371/journal.pone.0153631
Boore J. L. (2000). The Duplication/Random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals Comparative Genomics. Computational Biology, vol 1. Springer, Dordrecht.
Boore J. L., Brown W. M. (1994). Mitochondrial genomes and the phylogeny of mollusks. Nautilus 108, 61–78.
Boore J. L., Brown W. M. (1998). Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 8 (6), 668–674. doi: 10.1016/S0959-437X(98)80035-X
Breton S., Beaupré H. D., Stewart D. T., Hoeh W. R., Blier P. U. (2007). The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23 (9), 465–474. doi: 10.1016/j.tig.2007.05.011
Breton S., Ghiselli F., Passamonti M., Milani L., Stewart D. T., Hoeh W. R. (2011a). Evidence for a fourteenth mtDNA-encoded protein in the female-transmitted mtDNA of marine mussels (Bivalvia: Mytilidae). PloS One 6 (4), e19365. doi: 10.1371/journal.pone.0019365
Breton S., Stewart D. T., Hoeh W. R. (2010). Characterization of a mitochondrial ORF from the gender-associated mtDNAs of mytilus spp. (Bivalvia: Mytilidae): Identification of the “missing” ATPase 8 gene. Mar. Genomics 3 (1), 11–18. doi: 10.1016/j.margen.2010.01.001
Breton S., Stewart D. T., Shepardson S., Trdan R. J., Bogan A. E., Chapman E. G., et al. (2011b). Novel protein genes in animal mtDNA: a new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol. Biol. Evol. 28 (5), 1645–1659. doi: 10.1093/molbev/msq345
Castresana J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17 (4), 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cheng S., Chang S. Y., Gravitt P., Respess R. (1994). Long pcr. Nature 369 (6482), 684–685. doi: 10.1038/369684a0
Clayton D. A. (1991). Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 7 (7), 453–478. doi: 10.1146/annurev.cb.07.110191.002321
Conant G. C., Wolfe K. H. (2008). GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24 (6), 861–862. doi: 10.1093/bioinformatics/btm598
Di T. P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J. M., et al. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39 (Web Server issue), 13–17. doi: 10.1093/nar/gkr245
Doucet-Beaupré H., Breton S., Chapman E. G., Blier P. U., Bogan A. E., Stewart D. T., et al. (2010). Mitochondrial phylogenomics of the bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol. Biol. 10 (1), 1–19. doi: 10.1186/1471-2148-10-50
Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. (1994). DNA Primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3 (5), 294.
Ghiselli F., Milani L., Passamonti M. (2011). Strict sex-specific mtDNA segregation in the germ line of the DUI species venerupis philippinarum (Bivalvia: Veneridae). Mol. Biol. Evol. 28 (2), 949–961. doi: 10.1093/molbev/msq271
Gissi C., Iannelli F., Pesole G. (2008). Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 101 (4), 301–320. doi: 10.1038/hdy.2008.62
Guerra D., Ghiselli F., Milani L., Breton S., Passamonti M. (2016). Early replication dynamics of sex-linked mitochondrial DNAs in the doubly uniparental inheritance species ruditapes philippinarum (Bivalvia veneridae). Hered. (Edinb) 116 (3), 324–332. doi: 10.1038/hdy.2015.105
Gusman A., Lecomte S., Stewart D. T., Passamonti M., Breton S. (2016). Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 4, e2760. doi: 10.7717/peerj.2760
Hillis D. M., Bull J. J. (1993). An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. System. Biol. 42 (2), 182–192. doi: 10.1093/sysbio/42.2.182
Huelsenbeck J. P., Rannala B., Buckley T. (2004). Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. System. Biol. 53 (6), 904–913. doi: 10.1080/10635150490522629
Katoh K., Toh H. (2008). Recent developments in the MAFFT multiple sequence alignment program. Briefings Bioinf. 9 (4), 286. doi: 10.1093/bib/bbn013
Kiełbasa S. M., Wan R., Sato K., Horton P., Frith M. C. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21 (3), 487–493. doi: 10.1101/gr.113985.110
Kong L., Matsukuma A., Hayashi I., Takada Y., Li Q. (2012). Taxonomy of macridiscus species (Bivalvia : Veneridae) from the western pacific: insight based on molecular evidence, with description of a new species. J. Molluscan Stud. 78 (1), 1–11. doi: 10.1093/mollus/eyr024
Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lanfear R., Calcott B., Ho S. Y., Guindon S. (2012). Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29 (6), 1695–1701. doi: 10.1093/molbev/mss020
Laslett D., Canback B. (2008). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24 (2), 172–175. doi: 10.1093/bioinformatics/btm573
Lavrov D. V., Boore J. L., Brown W. M. (2002). Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 19 (2), 163–169. doi: 10.1093/oxfordjournals.molbev.a004068
Librado P., Rozas J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Lowe T. M., Chan P. P. (2016). tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44 (Web Server issue), W54–W57. doi: 10.1093/nar/gkw413
Lunt D. H., Hyman B. C. (1997). Animal mitochondrial DNA recombination. Nature 387 (6630), 247. doi: 10.1038/387247a0
Macey J. R., Larson A., Ananjeva N. B., Fang Z., Papenfuss T. J. (1997). Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 14 (1), 91. doi: 10.1093/oxfordjournals.molbev.a025706
Macey J. R., Nd S. J., Larson A., Papenfuss T. J. (1998). Tandem duplication via light-strand synthesis may provide a precursor for mitochondrial genomic rearrangement. Mol. Biol. Evol. 15 (1), 71–75. doi: 10.1093/oxfordjournals.molbev.a025849
Milani L., Ghiselli F., Guerra D., Breton S., Passamonti M. (2013). A comparative analysis of mitochondrial ORFans: new clues on their origin and role in species with doubly uniparental inheritance of mitochondria. Genome Biol. Evol. 5 (7), 1408–1434. doi: 10.1093/gbe/evt101
Milani L., Ghiselli F., Iannello M., Passamonti M. (2014a). Evidence for somatic transcription of male-transmitted mitochondrial genome in the DUI species ruditapes philippinarum (Bivalvia: Veneridae). Curr. Genet. 60 (3), 163–173. doi: 10.1007/s00294-014-0420-7
Milani L., Ghiselli F., Maurizii M. G., Nuzhdin S. V., Passamonti M. (2014b). Paternally transmitted mitochondria express a new gene of potential viral origin. Genome Biol. Evol. 6 (2), 391–405. doi: 10.1093/gbe/evu021
Milani L., Ghiselli F., Passamonti M. (2016). Mitochondrial selfish elements and the evolution of biological novelties. Curr. Zool. 62 (6), 687–697. doi: 10.1093/cz/zow044
Milani L., Ghiselli F., Pecci A., Maurizii M. G., Passamonti M. (2015). The expression of a novel mitochondrially-encoded gene in gonadic precursors may drive paternal inheritance of mitochondria. PloS One 10 (9), e0137468. doi: 10.1371/journal.pone.0137468
Mitchell A., Guerra D., Stewart D., Breton S. (2016). In silico analyses of mitochondrial ORFans in freshwater mussels (Bivalvia: Unionoida) provide a framework for future studies of their origin and function. BMC Genomics 17, 597. doi: 10.1186/s12864-016-2986-6
Neuroni A. C., Fraefel M., Riedl R. (1999). Organization of the large mitochondrial genome in the isopod armadillidium vulgare. Genetics 151 (1), 203–210. doi: 10.1093/genetics/151.1.203
Obata M., Sano N., Komaru A. (2011). Different transcriptional ratios of male and female transmitted mitochondrial DNA and tissue-specific expression patterns in the blue mussel, mytilus galloprovincialis. Dev. Growth Differ. 53 (7), 878–886. doi: 10.1111/j.1440-169X.2011.01294.x
Ort B. S., Pogson G. H. (2007). Molecular population genetics of the Male and female mitochondrial DNA molecules of the California Sea mussel, mytilus californianus. Genetics 177 (2), 1087. doi: 10.1534/genetics.107.072934
Passamonti M., Ghiselli F. (2009). Doubly uniparental inheritance: Two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 28 (2), 79–89. doi: 10.1089/dna.2008.0807
Passamonti M., Scali V. (2001). Gender-associated mitochondrial DNA heteroplasmy in the venerid clam tapes philippinarum (Mollusca bivalvia).
Perna N. T., Kocher T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41 (3), 353–358. doi: 10.1007/bf01215182
Plazzi F., Ceregato A., Taviani M., Passamonti M. (2011). A molecular phylogeny of bivalve mollusks: ancient radiations and divergences as revealed by mitochondrial genes. PloS One 6 (11), e27147. doi: 10.1371/journal.pone.0027147
Plazzi F., Puccio G., Passamonti M. (2016). Comparative Large-scale mitogenomics evidences clade-specific evolutionary trends in mitochondrial DNAs of bivalvia. Genome Biol. Evol. 8 (8), 2544–2564. doi: 10.1093/gbe/evw187
Qi J.-W., Shi L.-Y., Lin Y.-Q., Han J. (2019). The complete mitochondrial genomes of three species of macridiscus dall 1902 (Bivalvia: Veneroidea: Veneridae) and their phylogenetic implications. J. Molluscan Stud. 85, 271–279. doi: 10.1093/mollus/eyz009
Ren J., Shen X., Jiang F., Liu B. (2010). The mitochondrial genomes of two scallops, argopecten irradians and chlamys farreri (Mollusca: Bivalvia): The most highly rearranged gene order in the family pectinidae. J. Mol. Evol. 70 (1), 57–68. doi: 10.1007/s00239-009-9308-4
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a Large model space. System. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Sahyoun A. H., Bernt M., Stadler P. F., Tout K. (2014). GC skew and mitochondrial origins of replication. Mitochondrion 17, 56–66. doi: 10.1016/j.mito.2014.05.009
Schuster G., Stern D. (2009). RNA Polyadenylation and decay in mitochondria and chloroplasts. Prog. Mol. Biol. Trans. Sci. 85, 393. doi: 10.1016/S0079-6603(08)00810-6
Serb J. M., Lydeard C. (2003). Complete mtDNA sequence of the north American freshwater mussel, Lampsilis ornata (Unionidae): An examination of the evolution and phylogenetic utility of mitochondrial genome organization in bivalvia (Mollusca). Mol. Biol. Evol. 20 (11), 1854–1866. doi: 10.1093/molbev/msg218
Shi W., Dong X. L., Wang Z. M., Miao X. G., Wang S. Y., Kong X. Y. (2013). Complete mitogenome sequences of four flatfishes (Pleuronectiformes) reveal a novel gene arrangement of l-strand coding genes. BMC Evol. Biol. 13, 173. doi: 10.1186/1471-2148-13-173
Shi W., Gong L., Wang S. Y., Miao X. G., Kong X. Y. (2015). Tandem duplication and random loss for mitogenome rearrangement in symphurus (Teleost: Pleuronectiformes). BMC Genomics 16, 355. doi: 10.1186/s12864-015-1581-6
Simon C., Frati F., Beckenbach A., Crespi B., Liu H., Flook P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. America 87 (6), 651–701. doi: 10.1093/aesa/87.6.651
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stewart D. T., Breton S., Blier P. U., Hoeh W. R. (2009). Masculinization events and doubly uniparental inheritance of mitochondrial DNA: A model for understanding the evolutionary dynamics of gender-associated mtDNA in mussels In: Pontarotti P. (eds) Evolutionary Biology. Springer, Berlin, Heidelberg. 163–173. doi: 10.1007/978-3-642-00952-5_9
Stewart D. T., Kenchington E. R., Singh R. K., Zouros E. (1996). Degree of selective constraint as an explanation of the different rates of evolution of gender-specific mitochondrial DNA lineages in the mussel mytilus. Genetics 143 (3), 1349–1357. doi: 10.1093/genetics/143.3.1349
Sun S., Li Q., Kong L., Yu H. (2016). Complete mitochondrial genomes of trisidos kiyoni and potiarca pilula: Varied mitochondrial genome size and highly rearranged gene order in arcidae. Sci. Rep. 6, 33794. doi: 10.1038/srep33794
Temperley R. J., Wydro M., Lightowlers R. N., Chrzanowskalightowlers Z. M. (2010). Human mitochondrial mRNAs–like members of all families, similar but different. Biochim. Et Biophys. Acta 1797 (6-7), 1081. doi: 10.1016/j.bbabio.2010.02.036
Vaidya G., Lohman D. J., Meier R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics the Int. J. Willi Hennig Soc. 27 (2), 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Wen H. B., Cao Z. M., Hua D., Xu P., Ma X. Y., Jin W., et al. (2017). The complete maternally and paternally inherited mitochondrial genomes of a freshwater mussel potamilus alatus (Bivalvia: Unionidae). PloS One 12 (1), e0169749. doi: 10.1371/journal.pone.0169749
White D. J., Wolff J. N., Pierson M., Gemmell N. J. (2008). Revealing the hidden complexities of mtDNA inheritance. Mol. Ecol. 17 (23), 4925–4942. doi: 10.1111/j.1365-294X.2008.03982.x
Wilcox T. P., Zwickl D. J., Heath T. A., Hillis D. M. (2002). Phylogenetic relationships of the dwarf boas and a comparison of Bayesian and bootstrap measures of phylogenetic support. Mol. Phylogenet. Evol. 25 (2), 361–371. doi: 10.1016/S1055-7903(02)00244-0
Winnepenninckx B., Backeljau T., Dewachter R. (1993). Extraction of high-Molecular-Weight DNA from mollusks. Trends Genet. 9 (12), 407–407. doi: 10.1016/0168-9525(93)90102-n
Wolstenholme D. R. (1992). Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141 (6), 173. doi: 10.1016/S0074-7696(08)62066-5
Xia Y., Zheng Y., Murphy R. W., Zeng X. (2016). Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in quasipaa boulengeri. BMC Genomics 17 (1), 965. doi: 10.1186/s12864-016-3309-7
Ye Y. Y., Wu C. W., Li J. J. (2015). Genetic population structure of Macridiscus multifarius (Mollusca: Bivalvia) on the basis of mitochondrial markers: strong population structure in a species with a short planktonic larval stage. PloS One 10 (12), e0146260. doi: 10.1371/journal.pone.0146260
Yuan Y., Li Q., Yu H., Kong L. (2012). The complete mitochondrial genomes of six heterodont bivalves (Tellinoidea and solenoidea): variable gene arrangements and phylogenetic implications. PloS One 7 (2), e32353. doi: 10.1371/journal.pone.0032353
Yu Z., Wei Z., Kong X., Shi W. (2008). Complete mitochondrial DNA sequence of oyster Crassostrea hongkongensis-a case of “Tandem duplication-random loss” for genome rearrangement in Crassostrea? BMC Genomics 9 (1), 477–489. doi: 10.1186/1471-2164-9-477
Zouros E. (2012). Biparental inheritance through uniparental transmission: The doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40 (1), 1–31. doi: 10.1007/s11692-012-9195-2
Zouros E., Ball A. O., Saavedra C., Freeman K. R. (1994a). Mitochondrial DNA inheritance. Nature 368 (6474), 818. doi: 10.1038/368818a0
Keywords: Macridiscus, DUI, paternally inheritance, mitochondrial genomes, TDRL rearrangement
Citation: Wang R, Li X and Qi J (2022) The complete paternally inherited mitochondrial genomes of three clam species in genus Macridiscus (Bivalvia: Veneridae): A TDRL model of dimer-mitogenome rearrangement of doubly uniparental inheritance. Front. Mar. Sci. 9:1016779. doi: 10.3389/fmars.2022.1016779
Received: 11 August 2022; Accepted: 30 August 2022;
Published: 20 September 2022.
Edited by:
Marco Casu, University of Sassari, ItalyReviewed by:
Khaled Mohammed-Geba, University of Maryland, College Park, United StatesMarco Passamonti, University of Bologna, Italy
Copyright © 2022 Wang, Li and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiwei Qi, cWlqaXdlaUBpb3ouYWMuY24=
†These authors have contributed equally to this work and share first authorship