 Michael T. Connelly
Michael T. Connelly Crystal J. McRae
Crystal J. McRae Pi-Jen Liu4
Pi-Jen Liu4 Cecily E. Martin
Cecily E. Martin Nikki Traylor-Knowles
Nikki Traylor-Knowles- 1Department of Marine Biology and Ecology, Rosenstiel School of Marine and Atmospheric Science, University of Miami, Coral Gables, FL, United States
- 2Department of Biological Sciences, Simon Fraser University, Burnaby, BC, Canada
- 3Department of Natural Resources and Environmental Studies, National Dong Hwa University, Hualien, Taiwan
- 4Graduate Institute of Marine Biology, National Dong Hwa University, Pingtung, Taiwan
- 5Department of Mechanical Engineering, Imperial College London, London, United Kingdom
Symbioses between eukaryotes and their associated microbial communities are fundamental processes that affect organisms’ ecology and evolution. A unique example of this is reef-building corals that maintain symbiotic associations with dinoflagellate algae (Symbiodiniaceae) and bacteria that affect coral health through various mechanisms. However, little is understood about how coral-associated bacteria communities affect holobiont heat tolerance. In this study, we investigated these interactions in four Pocillopora coral colonies belonging to three cryptic species by subjecting fragments to treatments with antibiotics intended to suppress the normal bacteria community, followed by acute heat stress. Separate treatments with only antibiotics or heat stress were conducted to compare the effects of individual stressors on holobiont transcriptome responses and microbiome shifts. Across all Pocillopora species examined, combined antibiotics and heat stress treatment significantly altered coral-associated bacteria communities and caused major changes in both coral and Cladocopium algal symbiont gene expression. Individually, heat stress impaired Pocillopora protein translation and activated DNA repair processes, while antibiotics treatments caused downregulation of Pocillopora amino acid and inorganic ion transport and metabolism genes and Cladocopium photosynthesis genes. Combined antibiotics-heat stress treatments caused synergistic effects on Pocillopora and Cladocopium gene expression including enhanced expression of oxidative stress response genes, programed cell death pathways and proteolytic enzymes that indicate an exacerbated response to heat stress following bacteria community suppression. Collectively, these results provide further evidence that corals and their Symbiodiniaceae and bacteria communities engage in highly coordinated metabolic interactions that are crucial for coral holobiont health, homeostasis, and heat tolerance.
Introduction
Microbial symbioses are fundamental to the structure and functioning of marine ecosystems, and influence marine organisms’ ecology, evolution, and stress responses (O’Brien et al., 2019; Wilkins et al., 2019; Apprill, 2020). Healthy reef-building corals (Order Scleractinia) maintain associations with endosymbiotic algae (Family Symbiodiniaceae) and bacteria, and are popular models for the study of complex microbial symbioses that underpin marine ecosystem processes (Blackall et al., 2015; Thompson et al., 2015).
Coral-associated bacteria communities exhibit patterns of phylosymbiosis and cophylogeny (Pollock et al., 2018) and demonstrate genomic signatures that are suggestive of highly interdependent symbiotic relationships between corals and bacteria (Robbins et al., 2019). Recent studies have also uncovered evidence for intimate associations between bacteria and Symbiodiniaceae, indicating that diverse multi-partner symbiotic interactions are present in coral holobionts (Motone et al., 2020; Maire et al., 2021). Certain coral-associated bacteria have been investigated for their roles in affecting corals’ responses to heat stress, with different bacteria taxa exerting either negative or positive effects on coral holobiont resistance and resilience (Tout et al., 2015; Zaneveld et al., 2016; Ziegler et al., 2017, 2019; Epstein et al., 2019; Avila-Magaña et al., 2021). Other environmental stressors such as nutrient enrichment can also alter coral bacteria communities (Pogoreutz et al., 2018; Ziegler et al., 2019), but the interactive effects of different stressors applied in combination and their effects on coral health are still the focus of much research (Maher et al., 2019).
In contrast to other environmental stressors, broad-spectrum antibiotics treatments have previously been used to directly suppress the coral-associated bacteria community and generate hypotheses about how bacteria might affect coral holobiont health and homeostasis (Hodgson, 1990; Gilbert et al., 2012; Sweet et al., 2014; Sweet and Bythell, 2015; Glasl et al., 2016). Altogether, these studies have documented negative effects of antibiotics on coral health and hypothesized that antibiotic suppression of the native microbiota enables the proliferation of potential pathogens. These results have led to concerns about antibiotics as environmental pollutants on coral reefs (Zhang R. et al., 2018, 2019), which merit special consideration as antibiotics treatments are now being broadly deployed as field interventions to combat stony coral tissue loss disease (SCTLD) in the Caribbean (Neely et al., 2020). Despite these novel applications and concerns over the use of antibiotics on coral reefs, the specific genes and pathways involved in coral holobiont responses to antibiotic suppression of the bacteria community have not been explored. The experimental study of these processes can inform our knowledge of risks that antibiotics pose to coral health in the field and can also improve our understanding of coral holobiont interactions under normal conditions.
The coral genus Pocillopora is among the most widespread and important reef-building coral taxa in the world, with a distribution from east Africa to western Central America. For this reason, Pocillopora corals have become popular experimental model species and Pocillopora holobiont responses to heat stress and other environmental stressors have been thoroughly studied (Traylor-Knowles et al., 2011; Mayfield et al., 2014; Vidal-Dupiol et al., 2014; Zhou et al., 2018; Poquita-Du et al., 2019; Li et al., 2020). These studies suggest that Pocillopora coral transcriptomes are generally more responsive to heat stress than their associated Symbiodiniaceae transcriptomes, and that expression of genes involved in metabolism, protein folding and immune pathways such as heat shock proteins (HSPs) and tumor necrosis factor receptors (TNFRs) are important components of the Pocillopora coral heat stress response (Zhou et al., 2017; Zhang Y. et al., 2018). Other studies have assessed Pocillopora-associated bacteria community shifts that occur during heat stress, nutrient enrichment, and treatment with putative beneficial microbes (Tout et al., 2015; Pogoreutz et al., 2018; Rosado et al., 2018; Maher et al., 2019; Ziegler et al., 2019; Li et al., 2020, 2021; Zhang Y. et al., 2021). However, despite the critical importance of coral-associated bacteria to holobiont health, no study has simultaneously assessed coral holobiont transcriptome and microbiome responses to acute heat stress following direct antibiotic suppression of the native bacteria community. These studies are needed to better understand the role of coral-associated bacteria in coral holobiont health and heat tolerance, and to inform ongoing efforts that seek to conserve and restore coral reef ecosystems by enhancing corals’ natural resilience through the application of beneficial microbes (Peixoto et al., 2021; Voolstra et al., 2021).
To investigate how antibiotics treatments suppress the native coral bacteria community and affect coral holobiont interactions and acute heat stress responses, we performed an experiment with four Pocillopora coral colonies (genotypes) collected from Kenting National Park in southern Taiwan. These four distinct genotypes belonged to three morphologically cryptic Pocillopora species: Pocillopora acuta (n = 2) and Pocillopora damicornis (n = 1) (Schmidt-Roach et al., 2014), and the undescribed species Pocillopora type 8a (n = 1) (Gélin et al., 2017; Johnston et al., 2017). In this experiment we examined the effects of antibiotics, acute heat stress, and combined antibiotics-heat stress treatments on coral host and Cladocopium symbiont gene expression through RNAseq and bacteria composition through 16S gene amplicon sequencing. Through this work, we identified genes involved in coral-bacteria interactions to gain a better understanding of how coral-associated bacteria communities affect coral holobiont stress responses. We found that there were differences between the four Pocillopora genotypes from distinct cryptic species and sites, as expected. However, we also found that there were shared responses across all genotypes and species examined. Most notably, for all genotypes we found that both antibiotics and combined antibiotics-heat stress treatments altered Pocillopora coral bacteria community composition and affected Pocillopora coral and Cladocopium symbiont gene expression. We hypothesize that the changes in bacteria community composition exacerbated the holobiont heat stress responses causing microbial dysbiosis and the activation of programed cell death pathways. These results have implications for understanding how multi-partner interactions affect coral holobiont health and homeostasis and highlight the utility of antibiotics treatments as an experimental tool to suppress and manipulate coral-associated bacteria communities.
Materials and Methods
Coral Study Site, Collection, Fragmentation, and Acclimation
Colonies of Pocillopora corals were collected from two reefs within Kenting National Park in southern Taiwan (Kenting National Park Permit #10621218300) Houwan (22° 2′29′′ N, 120°41′43′′ E) and Wanglitung (21°59′43′′ N, 120°42′22′′ E) (Supplementary Figure 1). Two Pocillopora colonies with morphological similarity to P. damicornis were collected at each site, Houwan and Wanglitung, and colonies (hereafter referred to as HW1, HW2, WT1, and WT2) were collected in July 2017 (Table 1). Colonies were acclimated in a flow-through system receiving ambient seawater under natural sunlight at the National Museum of Marine Biology and Aquarium (NMMBA), and then split into a minimum of 12 replicate 2 cm2 fragments per colony that were affixed to numbered plastic tags and allowed to recover for 3 weeks before the experiment.
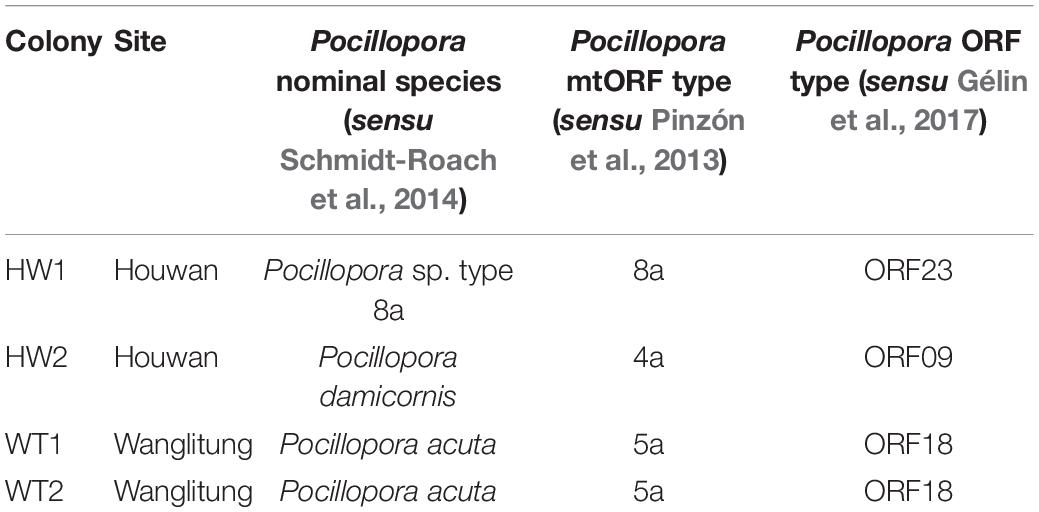
Table 1. Origin sites and species designations of the Pocillopora coral colonies used in the antibiotics and heat stress experiment.
Heat Stress, Antibiotics, and Antibiotics-Heat Stress Treatment Experiment
Three experimental treatments (heat stress, antibiotics, and antibiotics-heat stress) and a control were designed to examine the separate and interactive effects of acute heat stress and antibiotic bacteria suppression on the Pocillopora holobiont transcriptome and bacteria community composition (Figure 1). Triplicate fragments from each coral genotype were placed into individual autoclaved 400 mL glass jars filled with filtered seawater (1 μm) and randomly assigned to experimental or seawater control treatments (Figure 1). Jars were placed into two recirculating systems, one for the control and antibiotics treatment and another for the heat stress and antibiotics-heat stress treatments. Blue and white LED lights were operated on a 12 h:12 h light:dark cycle with even distribution across fragment jars. At 9:00 am in the morning of each day of the 3-day experiment, partial water changes were completed using 100 mL of filtered seawater (1 μm). The control system was maintained at 28°C for the full 72 h, to mimic the mean summer seawater temperature in Kenting National Park (Keshavmurthy et al., 2019).
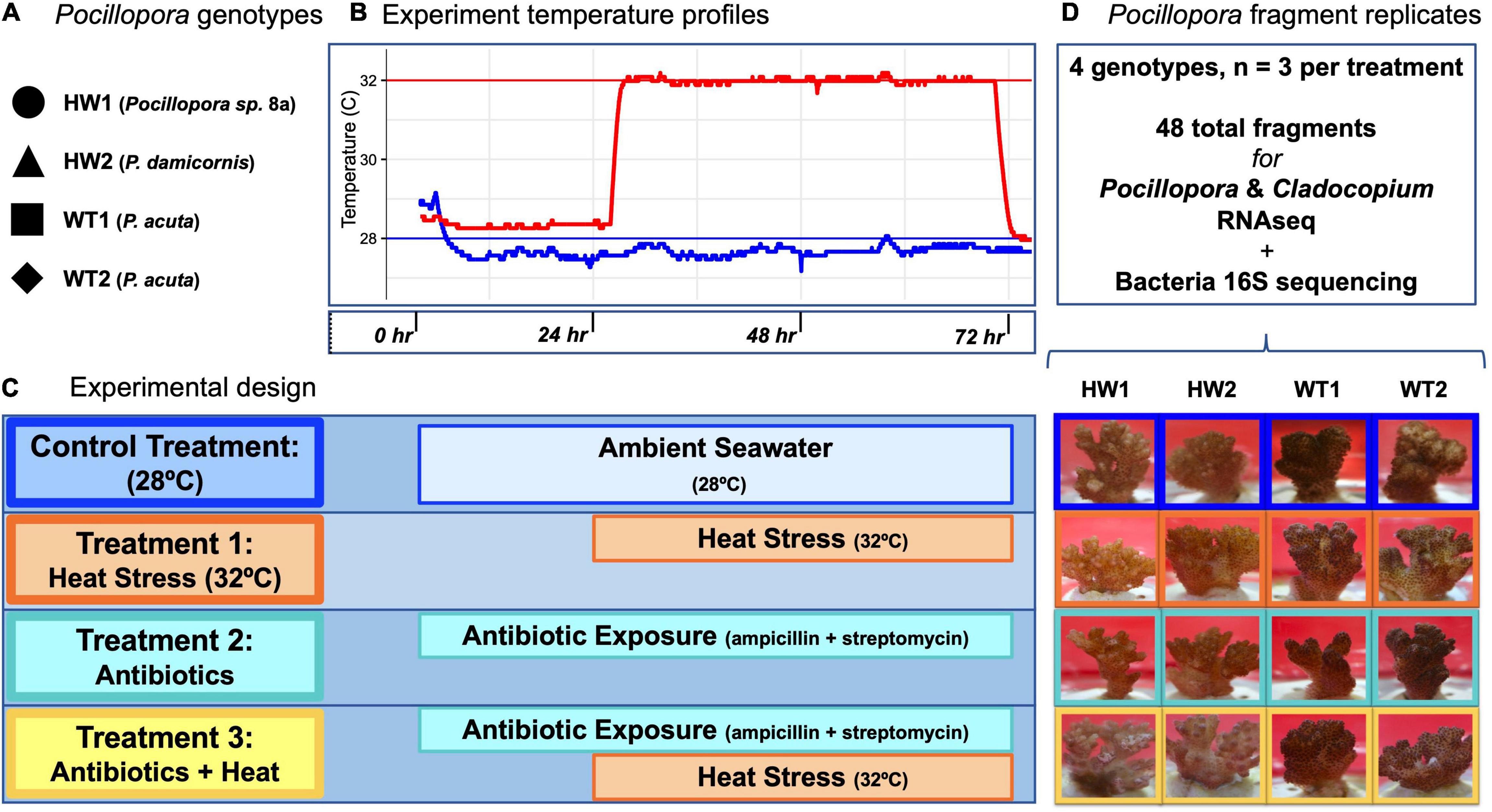
Figure 1. Experimental design for heat stress, antibiotics, and antibiotics-heat stress treatments. (A) Pocillopora genotypes and species identifications used in the experiment. (B) Experiment temperature profiles for control (blue) and heat stress (red) aquarium systems. (C) Experimental design indicating the intensity and duration of the control, heat stress, antibiotics, and antibiotics-heat stress treatments. (D) Fragment replicates used for downstream sequencing analyses and representative photographs of Pocillopora coral fragments from each genotype in each treatment at the time of sampling. All fragments in the control, heat stress, and antibiotics treatment appeared visually healthy, while two fragments in the antibiotics-heat stress treatment showed early signs of tissue loss.
Acute Heat Stress Treatment
The acute heat stress treatment was maintained for 48 h at 32°C (∼0.9 DHWs) after an initial 24 h at 28°C and a 90-min temperature ramp to 32°C. Temperatures in the control system and heat stress system were recorded at 1-min intervals with HOBO® Pendant data loggers (Onset, MA, United States) (Figure 1). This acute heat stress was done to contrast the short-term transcriptomic responses of healthy corals with normal bacteria communities against those with bacteria communities that had been previously suppressed by antibiotics treatment.
Antibiotics Treatment
The antibiotics treatment consisted of a combined exposure to 100 μg/mL ampicillin (Sigma-Aldrich A9393) and 100 μg/mL streptomycin sulfate (Sigma-Aldrich 6501), which were administered together to each jar as 400 μL of 100 mg/mL stock solution. These antibiotics were selected for their broad-spectrum antibacterial activity and different modes of action against bacterial cell wall cross-linking and protein synthesis, respectively. Previous studies that have used ampicillin and streptomycin indicate that the chosen concentrations of these antibiotics are non-toxic to the coral host or associated Symbiodiniaceae, and preliminary experiments supported this observation (Soffer et al., 2008; Gilbert et al., 2012; Sweet et al., 2014). Coral fragments were exposed to the antibiotic solution for a total of 72 h before fragment sampling and preservation. For daily partial water changes, 100 mL of antibiotic-treated seawater was used to maintain consistent levels of antibiotic exposure.
Antibiotics-Heat Stress Treatment
A combined antibiotics-heat stress treatment was conducted to parse apart the relative importance of stability within the coral-associated bacteria community toward the holobiont heat stress response. The antibiotics-heat stress treatment consisted of the same combined ampicillin and streptomycin treatment as in the individual antibiotics treatment. Corals were incubated in the antibiotic cocktail for 24 h at 28°C before exposure to acute heat stress at 32°C for 48 h (Figure 1), to allow 24 h for the antibiotic suppression of the coral bacteria community to occur before the start of the heat stress. For daily partial water changes, 100 mL of antibiotic-treated seawater was used to maintain consistent levels of antibiotic exposure for a total of 72 h through the entire experiment (Figure 1).
Collection and Preservation of Coral Tissue Samples
After the experiment was completed, fragments were photographed, and 1 cm2 coral fragments were cut using stainless steel bone cutters that were cleaned in 70% ethanol between samples (Supplementary Figure 4). These fragments were placed into tubes with 5 mL of RNA stabilization solution consisting of 70% w/v ammonium sulfate, 20 mM EDTA, and 25 mM sodium citrate adjusted to pH 5.2 with 1.0 M H2SO4. Fragments were incubated at room temperature for 30 min and frozen at −80°C. Frozen samples were then transported to the University of Miami Rosenstiel School of Marine and Atmospheric Science in Miami, FL, United States, in coolers on dry ice for further processing.
RNA Extraction, cDNA Library Preparation and Sequencing
Total RNA was isolated from the coral tissue using the Qiagen RNeasy Kit and eluted in 30 μL of RNase-free water. RNA concentration and purity were quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher). cDNA libraries were then created using the Illumina TruSeq RNA Library Prep Kit v2 with Poly-A selection according to the manufacturer’s protocol. cDNA concentrations were quantified using the Qubit fluorometer and Qubit dsDNA BR kit (Thermo Fisher Q32853). cDNA libraries were shipped overnight on dry ice to the University of Utah’s Huntsman Cancer Center for 50 bp single-end sequencing on the Illumina HiSeq 2000.
Pocillopora Mitochondrial Open Reading Frame Type Determination and Symbiodiniaceae Community Profiling
Pocillopora mitochondrial open reading frame (mtORF) consensus sequences were extracted for each genotype and used to identify the closest mtORF haplotype in GenBank as described previously (Connelly et al., 2020), and these mtORF haplotypes were used to assign species designations following the most recent taxonomic revision to the genus Pocillopora (Schmidt-Roach et al., 2014). The genus-level composition of the Symbiodiniaceae community was assessed by aligning non-coral reads against reference sequences of Symbiodiniaceae clade-delineating loci (ITS1, ITS2, and cp23S) with BWA (Li and Durbin, 2009) as in Ladner et al. (2012) to show that all colonies were dominated by Cladocopium goreauii (Supplementary Table 2).
Pocillopora and Cladocopium Dual RNAseq Data Analysis
Raw FASTQ files were checked for base pair quality using the FASTQC program1, then Illumina adapter sequences and reads with poor-quality bases were removed using Trimmomatic v0.36 (Bolger et al., 2014). Trimmed reads were aligned against the P. damicornis genome using the program STAR v2.5.3a in two-pass mode (Dobin et al., 2013; Cunning et al., 2018), and the remaining unaligned reads were then aligned against the C. goreauii genome (Liu et al., 2018). Unsorted BAM files containing the Pocillopora or Cladocopium-aligned transcripts were quantified at the gene level using the featureCounts command in the Subread program (Liao et al., 2014), with only transcripts that were assigned as accurately single-mapping being retained for gene expression analysis in R version 4.0.3 (R Core Team, 2020).
Pocillopora and Cladocopium gene counts were filtered to include only genes with counts ≥ 10 in at least 90% of samples, and counts were normalized using the variance-stabilizing transformation (vst) in the R package DESeq2 (Love et al., 2014). Principal coordinates analysis (PCoA) of Manhattan distances was used to visualize the overall transcriptome patterns of coral and symbiont normalized counts, and pairwise PERMANOVA tests in the R package vegan (Oksanen et al., 2019; Martinez Arbizu, 2020) were used to test for significant differences in transcriptome profiles between experimental treatments.
Statistical analysis of differential gene expression was conducted using generalized linear models implemented in DESeq2. Average log2 fold change (LFC) for each gene was estimated by comparing normalized gene expression counts between treatments, and significantly differentially expressed genes (DEGs) were identified based on a Benjamini–Hochberg false discovery rate-adjusted p-value cutoff of <0.05. The set properties of the DEGs lists produced by each experimental treatment contrasted against the control treatment were visualized using the R package VennDiagram (Chen and Boutros, 2011).
To identify modules of co-expressed Pocillopora and Cladocopium genes across all treatments, weighted gene co-expression network analysis (WGCNA) was performed. The R package WGCNA (Langfelder and Horvath, 2008) was used to construct a signed co-expression network using vst-normalized gene counts and the bi-weight mid-correlation statistic. The resulting adjacency matrix was transformed into connection strengths using a soft-thresholding power of 20 for Pocillopora and 5 for Cladocopium, based on a scale-free topology fit index. Modules were identified using a cut height of 0.99 on the topological overlap matrix and a minimum module size of 50 genes, and modules with >70.0% similar expression profiles were merged. The expression of each network module “eigengene” was correlated against a binary matrix containing categorical sample treatment information to identify modules with expression patterns linked to a given treatment.
Gene ontology (GO) and Eukaryotic Orthologous Group (KOG) class annotations were obtained for the P. damicornis and C. goreauii genome using the eggNOG-mapper online tool2 (Huerta-Cepas et al., 2017). Fisher’s exact tests for enrichment of GO terms and KOG categories associated with transcriptome changes among sets of differentially expressed genes and co-expression module genes were conducted using GO_MWU3 and the R package KOGMWU (Wright et al., 2015; Barfield et al., 2018; Matz, 2019).
Bacteria DNA Extraction and 16S rRNA Gene Amplicon Sequencing
Total DNA was extracted using the Qiagen Dneasy PowerSoil kit from all coral fragment samples in the experiment (Weber et al., 2017). PCR amplification of the V4 region of the bacterial 16S gene was completed with the PCR primers 515F and 806R with single-indexed Illumina adapters according to the Earth Microbiome Project protocols (Apprill et al., 2015). PCR amplicons were checked for quality and size distribution on a 1.5% TE-agarose gel, and amplicon DNA concentration was determined using a Qubit fluorometer and Qubit dsDNA BR kit (Thermo Fisher Q32853). 16S amplicons from all samples and controls were purified using AMPure XP beads (Beckman Coulter A63881) and 80.0% ethanol washes to remove contaminants, then final DNA amplicon products were normalized to 4 nM concentration and pooled before submission to the University of Miami Center for Genome Technology (CGT) for 300 bp paired-end sequencing on the Illumina MiSeq.
Bacteria Community Data Analysis
Raw 16S sequence reads were processed using the QIIME2 pipeline version 2020.2 (Bolyen et al., 2018). The QIIME2 plugin q2-demux was used to visualize read quality and the plugin DADA2 (Callahan et al., 2016) was used to remove primer sequences, trim poor-quality bases, dereplicate reads, identify chimeric sequences and merge paired-end reads. The plugin q2-feature-table (McDonald et al., 2012) was used to generate sequence summary statistics based upon sample metadata. Bacterial 16S rRNA sequences were taxonomically classified using the plugin q2-feature-classifier (Bokulich et al., 2018) and a Naïve Bayes Classifier previously trained on the SILVA 16S rRNA database (Quast et al., 2013) version 132 QIIME2 release based on the 515F/806RB primer pair4 at the 99.0% similarity amplicon sequence variant (ASV) level. The plugins q2-alignment (Katoh and Standley, 2013) and q2-phylogeny (Price et al., 2010) were used to create a phylogenetic tree for further downstream analyses, and the plugin q2-taxa was used to remove all mitochondria and chloroplast sequences from the dataset. The plugin q2-diversity was used to assess the core diversity metrics of the dataset and create sequence depth rarefaction plots in the QIIME2 environment. Finally, the output tables containing ASV counts, phylogenetic trees and sample metadata were exported from QIIME2 and imported into R for statistical analyses.
All bacteria community statistical analyses were conducted in R using the packages phyloseq (McMurdie and Holmes, 2013), CoDaSeq (Gloor and Reid, 2016), vegan (Oksanen et al., 2019), ggplot2 (Wickham, 2016), and the tidyverse (Wickham et al., 2019). ASVs with a mean count < 3 were filtered from the dataset and ASV abundances were adjusted with the package zCompositions before the centered log-ratio (clr) transformation was performed for compositional data analysis (Palarea-Albaladejo and Martín-Fernández, 2015; Gloor et al., 2017). Alpha-diversity metrics were calculated for the unfiltered dataset using the Chao1 richness, Simpson evenness and Shannon diversity indices, and non-parametric Kruskal–Wallis tests were used to test for significant differences between treatments, reef sites, and coral genotypes (Xia and Sun, 2017; Ziegler et al., 2019). Beta-diversity was calculated using Aitchison dissimilarity matrices and visualized using principal coordinates analysis (PCA) (Gloor et al., 2017). Tests for homogeneity of multivariate dispersions (PERMDISP) and permutational analysis of molecular variance (PERMANOVA) (Anderson, 2017) were used to test for differences in bacteria community beta-diversity between treatments, reef sites and coral genotypes. Tests for differential abundance of bacteria ASVs between experimental treatments and the control treatment were completed with the R package ALDEx2 (Fernandes et al., 2013, 2014).
Results
Pocillopora and Cladocopium Transcriptome Profiling Reveals Strong Species and Genotype Effects on Coral and Symbiont Gene Expression
RNAseq of 24 Pocillopora tissue samples yielded approximately 647.1 million raw reads ranging from 8.2 to 20.4 million reads per sample (Supplementary Table 1 and Supplementary Figure 5). Approximately 69.3% of total reads aligned to the P. damicornis genome while 16.1% of total reads aligned to the C. goreauii genome (Supplementary Figure 2). This was supported by preliminary alignments of non-coral reads to algal ITS2 reference sequences that suggested that the symbiont community was dominated by C. goreauii (Supplementary Table 2) as has been previously observed for Pocillopora corals at these sites (Keshavmurthy et al., 2014; Brener-Raffalli et al., 2018). The 14.6% of total reads that did not align to either reference were discarded from downstream analysis. Filtering only genes with counts ≥ 10 in at least 90% of samples left 12,867 Pocillopora genes and 9,818 Cladocopium genes remaining for downstream expression analyses.
Principal coordinates analysis visualization of vst-normalized gene counts indicated that Pocillopora genotype accounted for the most variance in overall Pocillopora and Cladocopium gene expression (Figures 2A,B). PCoA visualization showed the different genotypes clustering separately along PC1 with HW1 (Pocillopora sp. 8a) as the most dissimilar from the other three genotypes and HW2 (P. damicornis) in between HW1 and the WT1 and WT2 genotypes (P. acuta). PERMANOVA tests revealed significant effects of coral genotype (Pocillopora: F = 8.21, R2 = 0.36, p < 0.001; Cladocopium: F = 8.36, R2 = 0.37, p < 0.001) and experimental treatment (Pocillopora: F = 3.64, R2 = 0.20, p < 0.001; Cladocopium: F = 2.36, R2 = 0.14, p < 0.001) on overall transcriptome patterns.
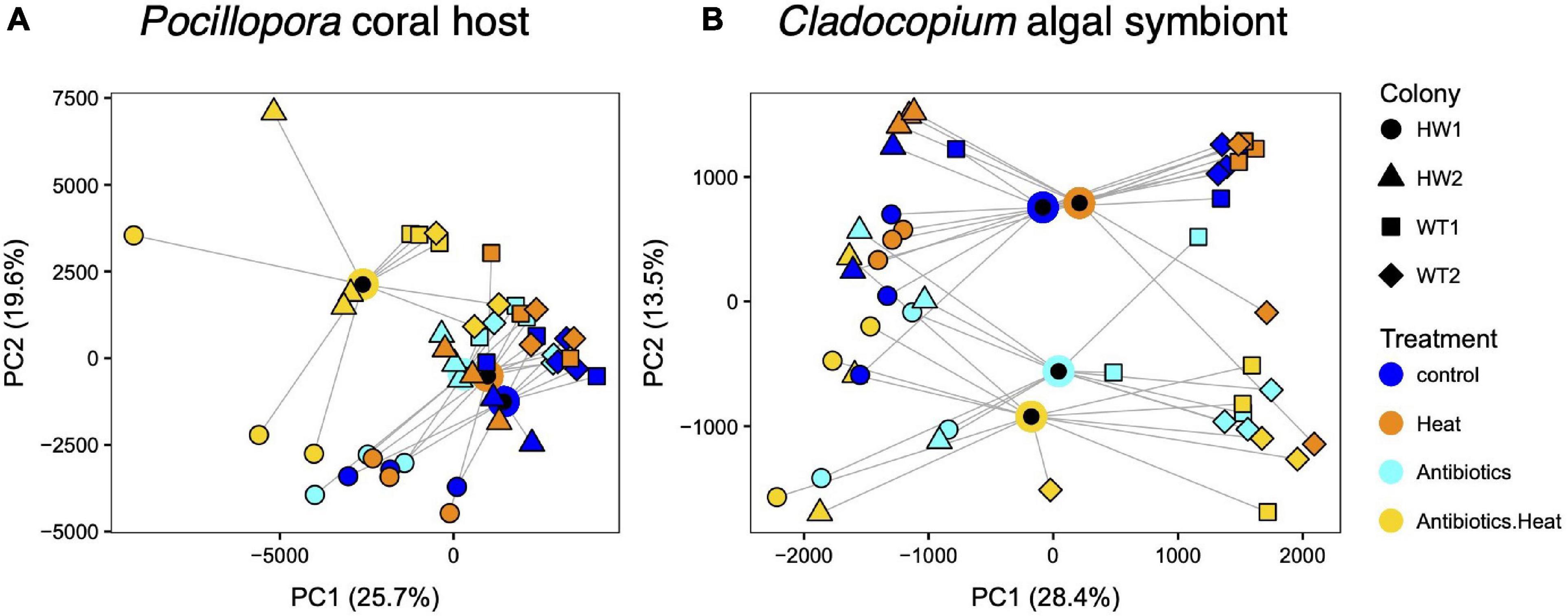
Figure 2. Principal coordinates analysis (PCoA) of transcriptome patterns in (A) Pocillopora corals and (B) Cladocopium algal symbionts reveals coral host cryptic species variation and experimental treatment effects, including a synergistic effect of combined antibiotics-heat treatment on coral host and algal symbiont gene expression. Point shape represents the sample colony (genotype) and color represents the treatment, with blue for the control treatment, orange for the heat treatment, cyan for the antibiotics treatment, and yellow for the combined treatment. Treatment centroids are plotted as black circles outlined in each treatment color with lines connected to each sample within the treatment.
Differential Gene Expression Analysis Reveals Effects of Acute Heat Stress, Antibiotics, and Antibiotics-Heat Stress Treatments
In addition to Pocillopora host genotypic effects, PCoA and PERMANOVA revealed a combined effect of the antibiotics-heat stress treatment on Pocillopora and Cladocopium gene expression. Samples in the antibiotics-heat stress treatment grouped separately from samples in the control treatment and the individual heat stress or antibiotics treatments in the PCoA plot (Figure 2). Furthermore, pairwise PERMANOVA tests between treatments only identified significant differences (p < 0.001) in Pocillopora transcriptome profiles between the antibiotics-heat treatment and the individual control, antibiotics, and heat stress treatments, which were not significantly different from each other (Supplementary Table 3).
This pattern was confirmed via the DESeq2 differential gene expression analysis that identified Pocillopora and Cladocopium DEGs between each of the heat stress, antibiotics, and antibiotics-heat stress treatments versus the control treatment for each genotype and all genotypes analyzed together (Table 1). The all-genotypes analysis detected 702 coral DEGs (185 up and 517 down) and 35 symbiont DEGs (11 up and 24 down) between the heat stress treatment and the control treatment, 730 coral DEGs (369 up and 361 down) and 1,614 symbiont DEGs (804 up and 810 down) between the antibiotics treatment and control treatment, and 6,118 coral DEGs (3,117 up and 3,001 down) and 2,936 symbiont DEGs (1,558 up and 1,378 down) between the antibiotics-heat stress treatment and the control treatment (Table 1 and Supplementary Table 3). Venn diagram analysis of the DEG set intersects revealed that many Pocillopora DEGs were uniquely differentially expressed in the antibiotics-heat stress treatment (5,044, 82.4%, Figure 3A), indicating a synergistic effect of antibiotics and heat stress on the Pocillopora transcriptome (Gunderson et al., 2016). This same pattern was also observed in the Cladocopium DEGs in the antibiotics-heat stress treatment (1,971, 67.1%, Figure 3B).
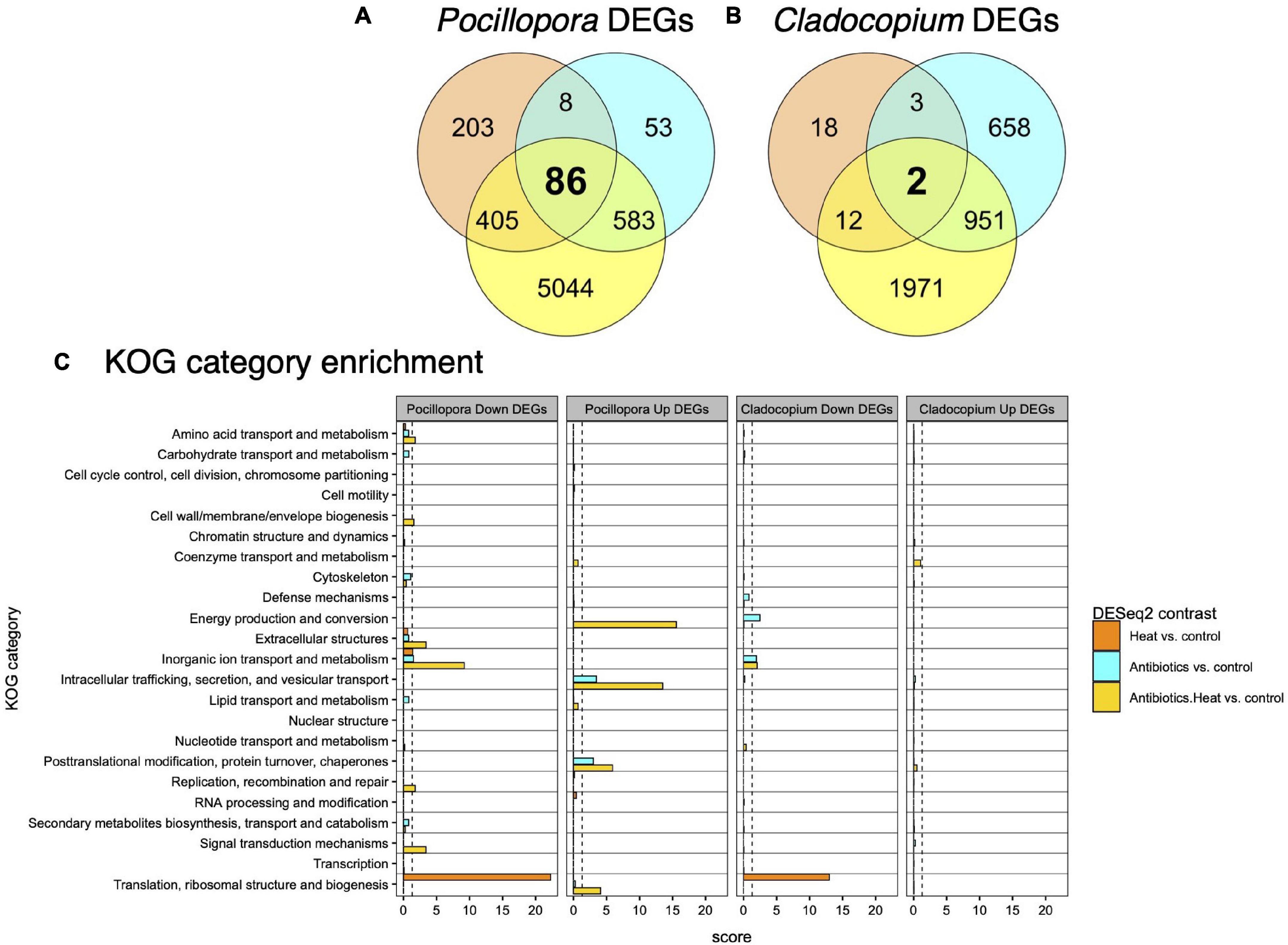
Figure 3. The overall summary of shared and unique DEGs detected in Pocillopora and Cladocopium. Venn diagrams of the number of shared and unique DEGs detected among experimental treatments contrasted with the control treatment for (A) Pocillopora corals and (B) Cladocopium algal symbionts. Thousands more DEGs are detected in the antibiotics and heat combined treatment than the individual antibiotics or heat stress treatments for both coral host and algal symbiont. (C) Barplots of enriched KOG categories for Pocillopora and Cladocopium upregulated and downregulated DEGs reveals that heat stress, antibiotics, and combined treatments affect different functional classes of genes. KOG categories are shown on the y-axis, and the x-axis depicts the enrichment score [–log10(padj)] for each KOG category within each set of upregulated or downregulated DEGs.
Eukaryotic Orthologous Group and Gene Ontology Enrichment Identifies Pocillopora and Cladocopium Biological Processes Affected by Heat Stress, Antibiotics, and Antibiotics-Heat Stress Treatments
Enrichment analysis of the Pocillopora and Cladocopium DEGs detected in treatment contrasts that had annotations to 23 non-overlapping KOG categories revealed distinct biological responses to the heat stress, antibiotics, and antibiotics-heat stress treatments (Figure 3C).
Pocillopora upregulated DEGs in the heat stress treatment were not significantly enriched (padj < 0.05) for any KOG categories or GO terms (Figure 3C). However, Pocillopora downregulated DEGs in the heat stress treatment were enriched for the KOG categories ‘translation, ribosomal structure and biogenesis’ and ‘inorganic ion transport and metabolism’ (Figure 3C) and this was supported by enriched biological process (BP) GO terms (n = 36, padj < 0.05), including ‘protein localization to endoplasmic reticulum,’ ‘cytoplasmic translation,’ and ‘ribosome biogenesis’ (Supplementary Table 4).
Pocillopora upregulated DEGs in the antibiotics treatment were enriched for the KOG categories ‘intracellular trafficking, secretion, and vesicular transport’ and ‘posttranslational modification, protein turnover, chaperones’ (Figure 3C), and were enriched for BP GO terms (n = 20, padj < 0.05) including ‘endoplasmic reticulum to Golgi vesicle-mediated transport,’ ‘response to topologically incorrect protein,’ ‘canonical Wnt signaling pathway,’ and ‘regulation of hydrolase activity’ (Supplementary Table 4). Pocillopora downregulated DEGs in the antibiotics treatment were enriched for the KOG categories ‘inorganic ion transport and metabolism’ and ‘cytoskeleton’ (Figure 3C), however no BP GO terms were significantly enriched at the padj < 0.05 level (Supplementary Table 4). Notable downregulated Pocillopora DEGs in the antibiotics treatments included five amino acid transporters (pdam_00001802, pdam_00001810, pdam_00011663, pdam_00012562, and pdam_00017019), one ammonium transporter (pdam_00014954), and several enzymes involved in amino acid biosynthetic pathways including glutamate carboxypeptidase 2 (pdam_00006146), adenosylhomocysteinase (pdam_00017020), S-adenosylmethionine synthase (pdam_00004044), and alanine aminotransferase 1 (pdam_00024643) (Supplementary Table 3).
Pocillopora upregulated DEGs in the antibiotics-heat stress treatment were enriched for the KOG categories ‘energy production and conversion,’ ‘intracellular trafficking, secretion and vesicular transport,’ ‘posttranslational modification, protein turnover, chaperones,’ and ‘translation, ribosomal structure and biogenesis’ (Figure 3C), as well as many BP GO terms (n = 92, padj < 0.05), among which the most significantly enriched were ‘cellular respiration,’ ‘electron transport chain,’ and ‘establishment of protein localization to mitochondrion.’ Other highly enriched terms included ‘antigen processing and presentation,’ ‘generation of precursor metabolites and energy’ ‘oxidation-reduction process,’ and ‘proteolysis’ (Supplementary Table 4).
Pocillopora downregulated DEGs in the antibiotics-heat stress treatment were enriched for the KOG categories ‘inorganic ion transport and metabolism,’ ‘extracellular structures,’ ‘signal transduction mechanisms,’ ‘amino acid transport and metabolism,’ ‘replication, recombination and repair,’ and ‘cell wall/membrane/envelope biogenesis’ (Figure 3C), and these processes were supported by the most highly enriched BP GO terms (n = 154, padj < 0.05) including ‘sodium ion transport,’ ‘metal ion transport,’ ‘microtubule organizing center organization,’ ‘response to wounding,’ and ‘cell–cell adhesion via plasma-membrane adhesion molecules’ (Supplementary Table 4).
The few Cladocopium upregulated DEGs in the heat stress treatment were not enriched for any KOG categories or BP GO terms, whereas Cladocopium downregulated DEGs in the heat stress treatment were enriched for the KOG category ‘translation, ribosomal structure and biogenesis’ and the BP GO terms (n = 4, padj < 0.05) ‘cytoplasmic translation,’ ‘amide biosynthetic process,’ ‘maturation of LSU-rRNA,’ and ‘ribosomal large subunit biogenesis’ (Supplementary Table 4).
Cladocopium upregulated DEGs in the antibiotics treatment were also not enriched for any KOG categories or BP GO terms. Cladocopium downregulated DEGs in the antibiotics treatment were enriched for the KOG categories ‘energy production and conversion’ and ‘inorganic ion transport and metabolism’ and BP GO terms (n = 4, padj < 0.05) ‘cytoplasmic translational elongation,’ ‘photosynthesis,’ ‘cellular response to hydrogen peroxide,’ and ‘cellular response to toxic substance.’ Among these Cladocopium downregulated DEGs was a chloroplastic aminomutase (SymbC1.scaffold4424.2), a chloroplastic delta-aminolevulinic acid dehydratase (SymbC1.scaffold548.4), six aminotransferases (SymbC1.scaffold31.2, SymbC1.scaffold2551.4, SymbC1.scaffold 2895.2, SymbC1.scaffold2898.1, SymbC1.scaffold2207.2, and SymbC1.scaffold1758.2), an amino acid permease (Symb C1.scaffold7668.1), a chloroplastic glutamate synthase (Sym bC1.scaffold1423.11), a type-3 glutamine synthase (SymbC1. scaffold371.1), a mitochondrial basic amino acid transporter (SymbC1.scaffold1251.1), and a fumarate reductase enzyme (SymbC1.scaffold10507.1).
Cladocopium upregulated DEGs in the antibiotics-heat stress treatment were not enriched for any KOG categories or BP GO terms, whereas Cladocopium downregulated DEGs in the antibiotics-heat stress treatment were enriched for the KOG category ‘inorganic ion transport and metabolism,’ and no BP GO terms (Supplementary Table 4).
WCGNA Reveals Modules of Co-expressed Immunity, Symbiosis, and Stress Response Genes Correlated to Experimental Treatments
WGCNA based on vst-transformed counts identified 23 Pocillopora and 13 Cladocopium co-expressed gene modules that were labeled according to module size along a standard sequence of colors within the WGCNA R package (Figure 4). Of these, 12 Pocillopora modules and 8 Cladocopium modules displayed module eigengene expression that was significantly correlated to one or more treatments (p < 0.05).
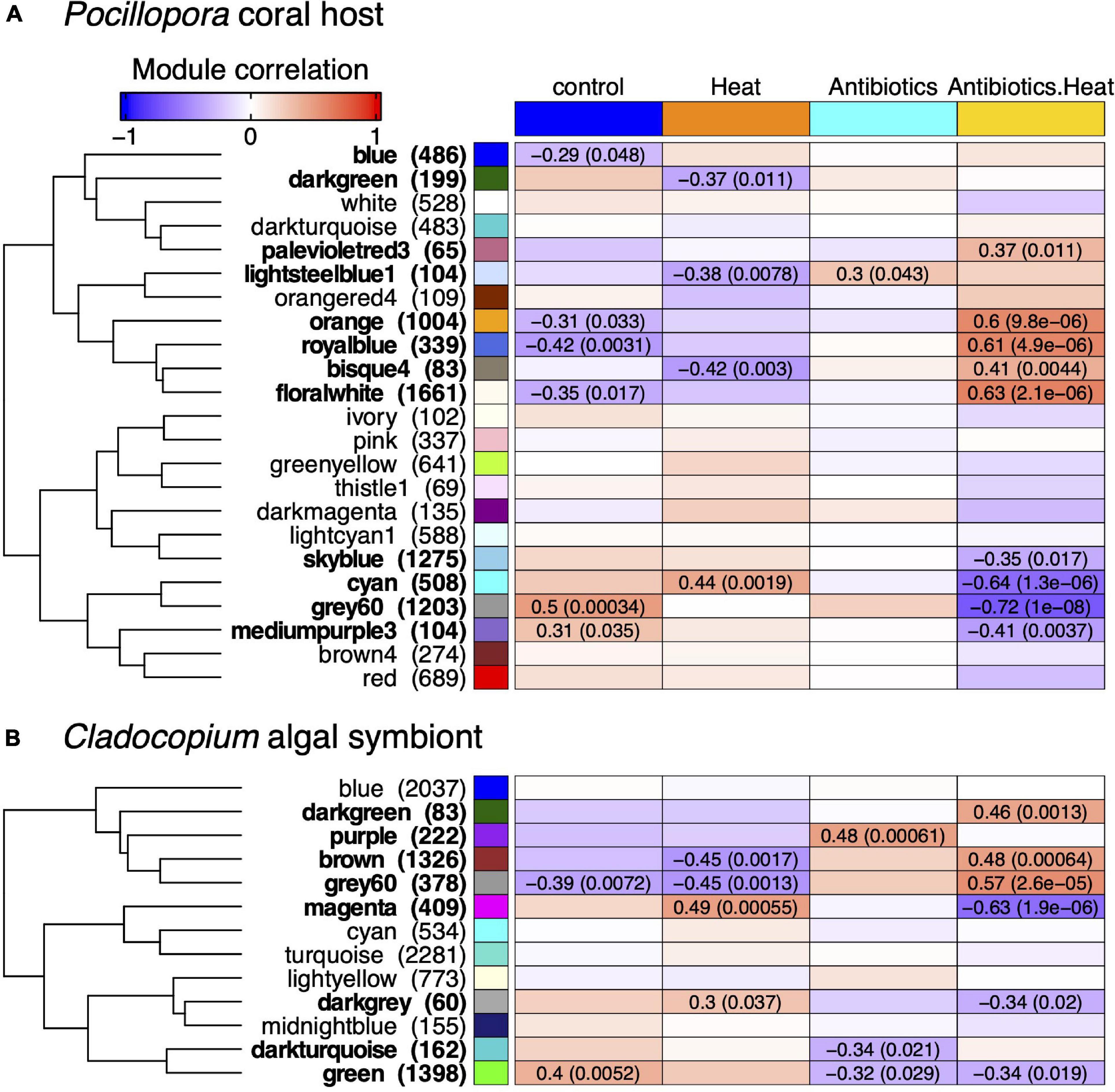
Figure 4. WGCNA reveals (A) Pocillopora and (B) Cladocopium co-expressed gene modules correlated to experimental treatments. The heatmap color represents the correlation coefficient of module eigengene expression to each treatment ranging from blue for negative correlation (–1) to red for positive correlation (+1), and the Pearson correlation coefficient and p-value is listed in each cell with a significant correlation (p < 0.05). The eigengene hierarchical clustering dendrogram is depicted to the left of the heatmap, and the number of genes in each module is listed in parentheses next to the module label. There are 12 Pocillopora and 8 Cladocopium modules that are significantly correlated to one or more treatments.
For Pocillopora, there were 2 positively (‘grey60’ and ‘mediumpurple3’) and 4 negatively (‘royalblue,’ ‘floralwhite,’ ‘orange,’ and ‘blue’) correlated modules to the control treatment, 1 positively (‘cyan’) and 3 negatively (‘bisque4,’ ‘lightsteelblue1,’ and ‘darkgreen’) correlated modules to the heat stress treatment, 1 module positively correlated to the antibiotics treatment (‘lightsteelblue1’), and 5 positively (‘floralwhite,’ ‘royalblue,’ ‘orange,’ ‘bisque4,’ and ‘palevioletred3’) and 4 negatively (‘grey60,’ ‘cyan,’ ‘mediumpurple3,’ and ‘skyblue’) correlated modules to the antibiotics-heat stress treatment (Figure 4A).
For Cladocopium, there was 1 positively (‘green’) and 1 negatively (‘grey60’) correlated modules to the control treatment, 2 positively (‘magenta’ and ‘darkgrey’) and 2 negatively (‘grey60’ and ‘brown’) correlated modules to the heat stress treatment, 1 positively (‘purple’) and 2 negatively (‘darkturquoise’ and ‘green’) correlated modules to the antibiotics treatment, and 3 positively (‘grey60,’ ‘brown,’ and ‘darkgreen’) and 3 negatively (‘magenta,’ ‘darkgrey,’ and ‘green’) correlated modules the antibiotics-heat stress treatment (Figure 4B).
Among all the significantly correlated Pocillopora modules, the ‘lightsteelblue1’ module (n = 104 genes) was the only module positively correlated to the antibiotics treatment and was also negatively correlated to the heat stress treatment. This module was enriched for the KOG category ‘signal transduction mechanisms’ and BP GO terms (n = 38, padj < 0.05) related to immune processes such as ‘activation of immune response,’ ‘leukocyte activation,’ and ‘response to bacterium’ (Supplementary Tables 5, 6). The module hub gene was an endoribonuclease (ZC3H12B, pdam_00012426) (Table 2) that may function as an RNase involved in immunomodulation via mRNA decay, and other co-expressed genes included the immune-related transcription factors ETS-related transcription factor Elf-4 (ELF4, pdam_00016137), ETS domain-containing protein Elk-1 (ELK1, pdam_00003296), baculoviral IAP repeat-containing protein 2 (BIRC2, pdam_00007333), and neural proliferation differentiation and control protein 1 (NPDC1, pdam_00019344).
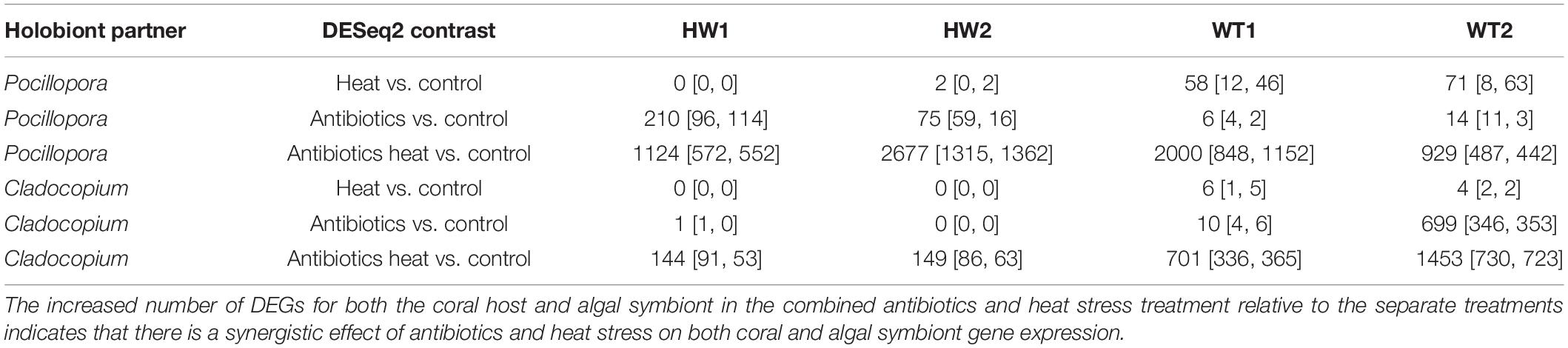
Table 2. Number of Pocillopora and Cladocopium DEGs (# of genes DESeq2 padj < 0.05 [up, down]) detected in contrasts between each experimental treatment and the control treatment for each genotype and all genotypes together.
The ‘floralwhite’ module was the largest module (n = 1,661 genes) and was negatively correlated to the control treatment and positively correlated to the antibiotic-heat treatment (Figure 4A). This module was highly enriched for the KOG categories ‘energy production and conversion,’ ‘intracellular trafficking, secretion, and vesicular transport’, ‘posttranslational modification, protein turnover, chaperones’ and ‘translation, ribosomal structure and biogenesis,’ as well as the BP GO terms (n = 131, padj < 0.05) ‘generation of precursor metabolites and energy,’ ‘protein polyubiquitination,’ and ‘proteasome assembly’ (Supplementary Tables 5, 6). The ‘floralwhite’ module hub gene was a proteolytic subunit of the mitochondrial ATP-dependent Clp protease complex (CLPP, pdam_00002241) (Table 2), and other co-expressed genes within the ‘floralwhite’ module were involved in immune-related processes including the master immune regulator NF-κB (pdam_00003205), tumor necrosis factor receptor-associated factor 6 (TRAF6, pdam_00017873), and mannan-binding lectin-associated serine protease 1 (MASP1, pdam_00000447). Additional upregulated and co-expressed genes were involved in pro-apoptosis pathways and protein degradation such as apoptosis regulator BAX (pdam_00019537), programed cell death 6-interacting protein (pcdip6, pdam_00015525), matrix metalloproteinase 25 (MMP25, pdam_00023163), and multiple 26S proteasome subunits.
The ‘orange’ module (n = 1,004 genes) was also negatively correlated to control treatment and positively correlated to antibiotics-heat stress treatment (Figure 4A). The ‘orange’ module was enriched for the KOG category ‘cytoskeleton,’ and the most highly enriched BP GO terms (n = 74, padj < 0.05) included ‘response to oxidative stress,’ ‘cellular amino acid catabolic process,’ and ‘response to temperature stimulus’ (Supplementary Tables 5, 6). Although the module hub gene (pdam_00024070) lacked annotation, other highly connected and upregulated genes were involved in the oxidative stress response, such as catalase (pdam_00001882), peroxidase (pdam_00000351), peroxiredoxin-6 (pdam_00009060) and apoptosis-inducing factor 2 (AIFM2, pdam_00001271), which is involved in directly triggering programed cell death pathways during oxidative stress.
Conversely, the ‘grey60’ (n = 1,203 genes) and ‘mediumpurple3’ (n = 104 genes) modules were both positively correlated to the control treatment and negatively correlated to the antibiotics-heat treatment. The ‘grey60’ module was enriched for the KOG categories ‘extracellular structures,’ ‘inorganic ion transport and metabolism,’ ‘signal transduction mechanisms,’ and ‘transcription’ as well as many BP GO terms (n = 305, padj < 0.05) related to cellular housekeeping functions and homeostasis maintenance (Supplementary Tables 5, 6). The ‘mediumpurple3’ module was enriched for the KOG category ‘signal transduction mechanisms’ and several BP GO terms (n = 25, padj < 0.05) involved in epithelial immune processes such as ‘regulation of antibacterial peptide biosynthetic process,’ ‘positive regulation of humoral immune response,’ ‘cell–cell adhesion,’ and ‘columnar/cuboidal epithelial cell development.’ The module hub gene was a fibroblast growth factor receptor (FGFR, pdam_00020781), and there were numerous other tyrosine kinase receptors within the module (Supplementary Tables 5, 6).
The ‘cyan’ module (n = 508 genes) was positively correlated to the control treatment and heat stress treatment and negatively correlated the antibiotics-heat treatment, and was enriched for the KOG categories ‘cell cycle control, cell division, and chromosome partitioning,’ ‘chromatin structure and dynamics,’ ‘cytoskeleton,’ ‘nucleotide transport and metabolism,’ ‘replication, recombination, and repair,’ and ‘secondary metabolites biosynthesis, transport, and catabolism,’ and the top enriched BP GO terms (n = 209, padj < 0.05) included numerous DNA damage response and repair processes (Supplementary Tables 5, 6).
For Cladocopium, only the ‘purple’ module (n = 222 genes) was positively correlated with antibiotics treatment, however, no KOG categories or BP GO terms were significantly enriched (Figure 4B). The module hub gene was a 26S proteasome regulatory subunit (SymbC1.scaffold7524.1) (Table 2), and other genes in the module that were also upregulated in the antibiotics treatment included a D-amino acid dehydrogenase small subunit protein (SymbC1.scaffold805.5), an isoleucine-tRNA ligase (SymbC1.scaffold2776.4), a vacuolar amino acid transporter associated with ATP-mediated isoleucine uptake (SymbC1.scaffold2075.3), and two sodium-dependent amino acid transporters (SymbC1.scaffold8224.5 and SymbC1.scaffold8948.3).
The largest significant Cladocopium module was the ‘green’ module (n = 1,398 genes), which was positively correlated to the control treatment and negatively correlated to both the antibiotics and antibiotics-heat stress treatment, and was enriched for the KOG category ‘energy production and conversion’ and BP GO terms ‘photosynthesis’ and ‘nucleoside monophosphate biosynthetic process.’ The ‘darkturquoise’ module (n = 162 genes) was also negatively correlated to the antibiotics treatment and had a chloroplastic hub gene of fucoxanthin-chlorophyll binding protein E (SymbC1.scaffold3718.1), an element of the light-harvesting complex associated with photosystem II.
Both the Cladocopium ‘magenta’ (n = 409 genes) and ‘darkgrey’ (n = 60 genes) modules were positively correlated to the heat stress treatment and negatively correlated to the antibiotics-heat stress treatment (Figure 4B). The ‘magenta’ module was enriched for the KOG category ‘signal transduction mechanisms,’ however, no BP GO terms were enriched. Conversely, the ‘brown’ (n = 1,326 genes) and ‘grey60’ (n = 378 genes) modules were negatively correlated to the heat stress treatment and positively correlated to the antibiotics-heat stress treatment. The ‘brown’ module was enriched for the KOG category ‘translation, ribosomal structure and biogenesis’ and BP GO terms ‘cytoplasmic translation,’ ‘membrane docking,’ ‘ncRNA metabolic process,’ and ‘RNA/ncRNA processing,’ and its hub gene was autophagy-related protein 9 (ATG9, SymbC1.scaffold517.1), a protein involved in autophagic vesicle formation. Genes related to ncRNA metabolism and processing included piRNA biogenesis protein Exd1 (SymbC1.scaffold5866.5), an Elav-like RNA-binding protein associated with miRNA processing (SymbC1.scaffold526.10), and an insulin-like mRNA-binding protein capable of shielding target transcripts from microRNA-mediated degradation (SymbC1.scaffold2518.1).
Antibiotics Alters Pocillopora-Associated Bacteria Communities
Sequencing of bacteria 16S rRNA gene V4 region on the Illumina MiSeq at the University of Miami CGT was completed in April 2019. A total of 3,716,629 demultiplexed reads were obtained from all 48 samples, with an average sequence length of 300 bp (Supplementary Figure 3). After read denoising, classification against the SILVA database and filtering of mitochondria and chloroplast-derived sequences, 470 coral-associated ASVs were detected representing 17 phyla, 31 classes, 84 orders, and 135 bacterial and archaeal families. The most abundant bacterial phyla were Proteobacteria, Bacteroidetes, and Cyanobacteria, and the most abundant families within the phylum Proteobacteria were Endozoicomonadaceae, Rhodobacteraceae, and Alteromonadaceae (Figure 5B). Filtering out low-abundance taxa with mean counts < 3 left 36 high-abundance ASVs remaining for compositional data analysis of bacteria community alpha and beta-diversity. Antibiotics treatments caused significant changes in coral-associated bacteria community beta-diversity according to PCA visualization of center-log ratio transformed abundances, as PC1 separated samples in both antibiotics treatments from the control and heat stress treatment samples and explained 24.8% of the variance (Figure 5A). PC2 explained 20.8% of the variance and partially separated the P. acuta genotypes (WT1 and WT2) from the P. damicornis genotypes (HW1 and HW2), in part due to a higher relative abundance of Endozoicomonas bacteria (Figures 5A,B).
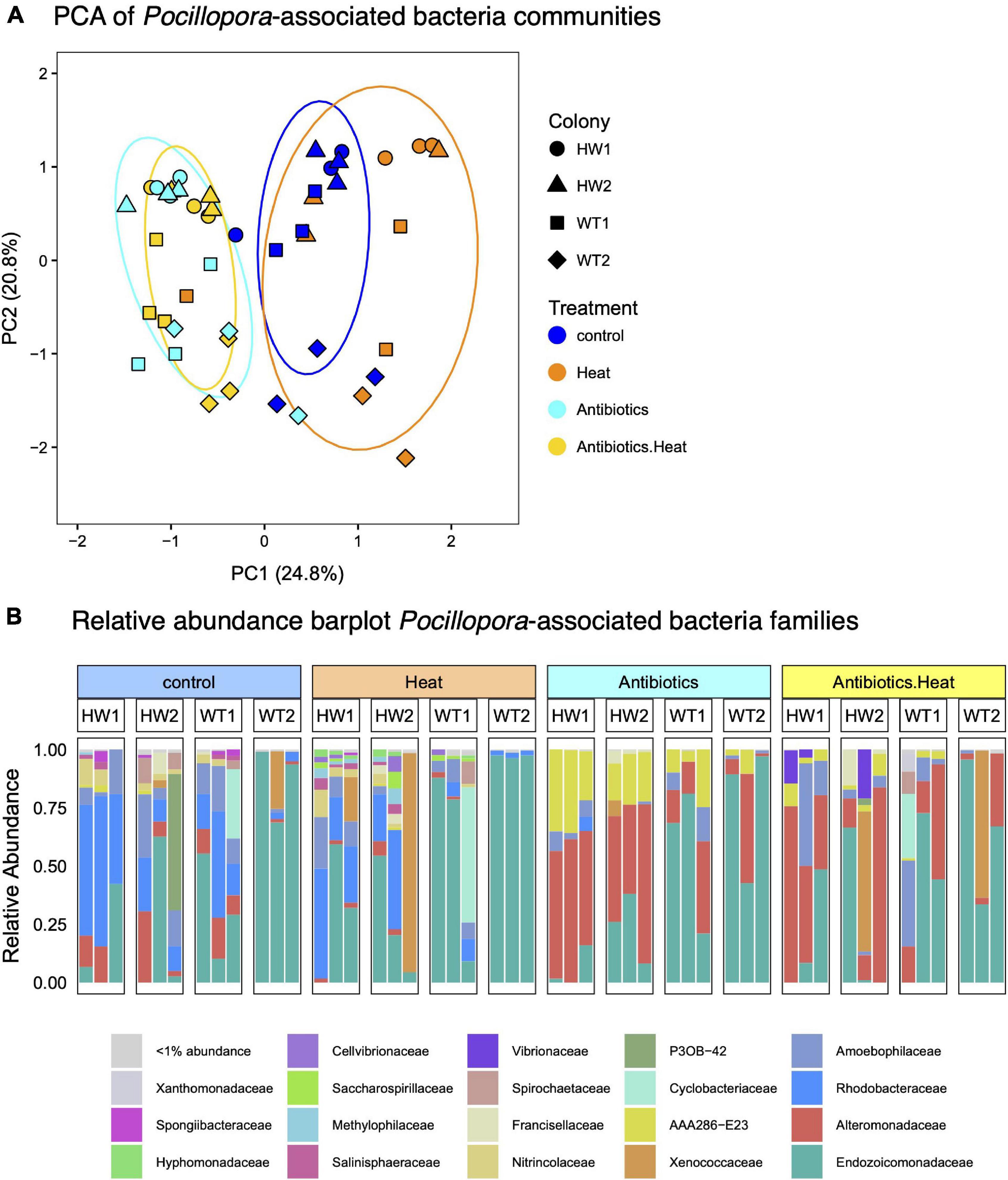
Figure 5. Bacteria 16S sequencing demonstrates that antibiotics treatments alter coral-associated bacteria communities, and that certain coral-associated bacteria families exhibit differential susceptibility to antibiotics treatment. (A) Principal components analysis (PCA) plot of coral-associated bacteria communities reveals that antibiotics-treated samples (yellow and cyan) cluster separately from heat stress and control treatment samples (blue and orange). Point shape represents the sample colony (genotype) and color represents the treatment, with blue for the control treatment, orange for the heat treatment, cyan for the antibiotics treatment, and yellow for the combined treatment. Colored ellipses represent the 95% confidence interval. (B) Barplot of the relative abundance of bacteria families in each treatment reveals significant declines in the abundance of coral-associated Rhodobacteraceae bacteria (royal blue) and increases in Alteromonadaceae bacteria (red) in antibiotics-treated corals, while Endozoicomonadaceae bacteria (dark green) tend to dominate both WT genotypes and are not eliminated by antibiotics treatment.
Non-parametric Kruskal–Wallis tests detected significant differences in bacteria community alpha-diversity metrics between experimental treatments (Chao1, Kruskal–Wallis chi-squared = 8.62, p = 0.035), with lower alpha-diversity observed in antibiotics-treated samples. PERMDISP tests revealed inhomogeneity of dispersions between treatments (F = 3.44, p = 0.027), and PERMANOVA tests also revealed significant differences between treatment centroids (F = 5.28, R2 = 0.26, p < 0.001), indicating differences in bacteria community composition in antibiotics-treated samples.
Specifically, antibiotics treatments reduced the relative abundance of bacteria in the family Rhodobacteraceae while bacteria in the family Alteromonadaceae persisted and increased in relative abundance (Figure 5B and Supplementary Figure 4). Bacteria in the genus Endozoicomonas were also observed to persist through antibiotics treatment but did not increase in relative abundance (Figure 5B and Supplementary Figure 4).
Furthermore, paired ALDEx2 tests that contrasted each experimental treatment with the control identified specific differentially abundant ASVs. Three ASVs in the family Rhodobacteraceae and one ASV in the genus Neptuniibacter (family Nitrincolaceae) were significantly reduced in both antibiotics and antibiotics-heat stress treatment samples (Supplementary Figure 5). Two ASVs in the family Alteromonadaceae had increased relative abundance in the antibiotics treatment and antibiotics-heat stress treatment (Supplementary Figure 5). BLASTn searches of these ASVs against the NCBI 16S rRNA database uncovered 100% sequence similarity (e-values < 3e–123) to Alteromonas aestuariivivens, an ampicillin-resistant bacterium isolated from a South Korea tidal flat (Park et al., 2017) and Aestuariibacter aggregatus, another ampicillin-resistant bacterium isolated from Yellow Sea (Wang et al., 2010).
Discussion
On coral reefs, antibiotics are increasingly being considered for use in coral disease intervention strategies and as environmental pollutants that are potentially damaging to coral health (Zhang R. et al., 2018; Zhang R. et al., 2019; Neely et al., 2020; Walker et al., 2021). Our experiment reveals that antibiotic suppression of coral-associated bacteria communities produces dramatic changes in Pocillopora coral and Cladocopium symbiont gene expression and bacteria community composition, resulting in microbial dysbiosis and diminished holobiont heat tolerance. These findings corroborate results from previous studies that documented negative effects of antibiotics on coral health (Gilbert et al., 2012; Glasl et al., 2016), and provide new insights into the complex multi-partner interactions that occur within Pocillopora coral holobionts (Brener-Raffalli et al., 2018; Pogoreutz et al., 2018; Geissler et al., 2021; Haydon et al., 2021; Li et al., 2021).
Antibiotics Treatment Disrupts Expression of Genes Involved in Coral Holobiont Metabolic Interactions
Pocillopora and Cladocopium transcriptome responses to antibiotics treatment revealed changes in intracellular trafficking and vesicular transport and inorganic ion transport and metabolism genes and upregulation of immune-related genes in the Pocillopora ‘lightsteelblue1’ module. These changes suggest that disruptions of the coral-associated bacteria community negatively affect coral holobiont metabolic exchanges and inter-partner signaling, resulting in immune inflammation and microbiome dysregulation (Figures 4, 5 and Supplementary Table 3). This transcriptomic evidence also supports the notion that these holobiont metabolic interactions entail the exchanges of amino acids, inorganic ions, and other secondary metabolites among the Pocillopora coral host, its Cladocopium algal symbionts and specific bacteria within the coral holobiont (Matthews et al., 2020; Peixoto et al., 2021). Previous studies have identified diazotrophic bacteria as a source of fixed nitrogen to coral holobionts (Olson et al., 2009; Benavides et al., 2017; Lesser et al., 2018; Rädecker et al., 2021), and have implicated coral-associated bacteria in dimethylsulfoniopropionate (DMSP) metabolism (Raina et al., 2010; Tandon et al., 2020) and the production of siderophores capable of alleviating iron and trace metal limitation (Hopkinson and Morel, 2009; Schalk et al., 2011; Amin et al., 2017; Reich et al., 2020; Peixoto et al., 2021). Importantly, while transcriptome read alignments suggest these Pocillopora genotypes were Cladocopium-dominated, direct measurement of Symbiodiniaceae community composition via ITS2 amplicon sequencing (Boulotte et al., 2016; Hume et al., 2019) in future studies would be preferred for assessing interactions of different Symbiodiniaceae genera with associated bacteria.
While the magnitude of Pocillopora gene expression changes were similar for the acute heat stress treatment and antibiotics treatment relative to the control treatment, Cladocopium gene expression had more DEGs in response to antibiotics treatment than to the heat stress treatment (Figures 2B, 3B). This difference was driven in part by the downregulation of genes involved in energy production and photosynthesis in the Cladocopium ‘green’ and ‘darkturquoise’ modules that were negatively correlated to antibiotics treatment, such as the light-harvesting fucoxanthin-chlorophyll A–C binding proteins E and F, a caroteno-chlorophyll A-C-binding protein and several photosystem I proteins (Figures 3C, 4B and Supplementary Table 4).
We hypothesize that the downregulation of Cladocopium photosynthesis and metabolism genes in the antibiotics treatment may be related to disruptions in nitrogen cycling and amino acid exchanges between the Pocillopora coral, Cladocopium symbionts and members of the associated bacteria community. Five Pocillopora amino acid transporters were downregulated in the antibiotics treatment (Supplementary Table 3), however, for Cladocopium three amino acid transporters and an isoleucine tRNA ligase were upregulated in the ‘purple’ module, and other genes involved in amino acid metabolism were downregulated in the ‘green’ and ‘darkturquoise’ modules. The role of amino acids in generating energy via the TCA cycle (Akram, 2014) or alternatively as metabolites in chlorophyll biosynthesis (Vavilin and Vermaas, 2002) could potentially play a role in Cladocopium photosynthetic impairment. This second hypothesis is consistent with observations of decreased chlorophyll a in nitrogen-starved C. goreauii (Zhou et al., 2021) and stable photochemical efficiency and dramatically increased growth rates in Breviolium minutum cultures supplemented with casein amino acids (Kirk et al., 2020). Although the Pocillopora and Cladocopium genomes possess pathways for synthesizing most amino acids and heterotrophic feeding may account for substantial input of essential amino acids to the coral holobiont (Fox et al., 2019; Ferrier-Pagès et al., 2021), these observations suggest that certain coral-associated and/or Symbiodiniaceae-associated bacteria may provide in hospite Symbiodiniaceae with additional nitrogenous compounds that benefit holobiont metabolism (Benavides et al., 2017; Geissler et al., 2021; Glaze et al., 2021; Rädecker et al., 2021). While more experimental evidence is required to validate this conclusion, Cladocopium symbionts were recently shown to associate with diverse bacteria located within algae cells and on external cell surfaces, which supports the idea that Symbiodiniaceae-bacteria metabolic exchanges may be more common than previously recognized (Camp et al., 2020; Matthews et al., 2020; Maire et al., 2021).
Antibiotics Treatment Alters Pocillopora-Associated Bacteria Communities
Treatments of Pocillopora corals with the broad-spectrum antibiotics ampicillin and streptomycin caused significant changes in coral-associated bacteria community composition (Figure 5A), as antibiotics treatment substantially reduced the relative abundance of bacteria in the family Rhodobacteraceae while bacteria in the family Alteromonadaceae persisted and increased in relative abundance (Figure 5B).
Bacteria community analysis of these Pocillopora corals from southern Taiwan revealed similar results to previous studies demonstrating that P. acuta corals in Taiwan and elsewhere in the Indo-Pacific have high relative abundances of Endozoicomonas bacteria (Brener-Raffalli et al., 2018; Epstein et al., 2019; Connelly et al., 2020), however, further studies on how variation in coral host species, site effects and environmental factors affect Pocillopora corals’ associations with Endozoicomonas bacteria are needed to corroborate this trend. Furthermore, these results indicate that Endozoicomonas-dominated microbiomes may be more resistant to experimental antibiotic perturbation (Figure 5B and Supplementary Figure 4). The persistence of Endozoicomonas bacteria throughout multiple stressors suggests that these bacteria are either resistant to environmental perturbations or perhaps are effectively shielded from external stressors because of their location as cellular aggregates within Pocillopora coral tissues (Work and Aeby, 2014; Neave et al., 2017; Pogoreutz et al., 2018).
Furthermore, it appears that other bacteria that persist through antibiotics treatment, such as the bacteria ASVs similar to the ampicillin-resistant Alteromonas spp. and A. aggregatus, might act as opportunistic pathogens that activate coral immune responses and cause microbial dysbiosis during antibiotics-heat stress treatment (Wang et al., 2010; Park et al., 2017; Zaneveld et al., 2017). More in-depth research on the taxonomic diversity, localization, and metabolic functions of Pocillopora coral-associated bacteria communities is necessary to fully understand how interactions with specific bacteria influence coral holobiont health, and these results highlight the potential of antibiotics treatments as an experimental tool to manipulate these associations.
Antibiotics-Heat Stress Treatment Causes Microbial Dysbiosis and Exacerbates Pocillopora and Cladocopium Transcriptome Stress Responses
Despite the strong influence of coral genotype on both Pocillopora and Cladocopium gene expression, differential gene expression and gene co-expression network analyses revealed distinct effects of the separate acute heat stress and antibiotics treatments as well as a strong combined effect of the antibiotics-heat treatment (Figure 2 and Table 1). Samples in the antibiotics-heat stress treatment clustered separately from the heat stress, antibiotics, and control treatment samples in PCoA, and thousands more DEGs were detected in the antibiotics-heat treatment than either individual heat stress or antibiotics treatment (Table 1 and Figures 2, 3). This dramatic transcriptional activation of Pocillopora and Cladocopium energy conversion and stress response genes suggest that in the antibiotics-heat stress treatment, bacteria community disruption negatively impacts Pocillopora coral holobiont metabolism and immune homeostasis such that opportunistic microbes flourish and the corals’ ability to collectively mount an efficient heat stress response and defend against infection is diminished (Figures 3C, 5B). This is supported by functional enrichment analyses of Pocillopora DEGs and the co-expressed ‘floralwhite,’ ‘orange,’ ‘grey60,’ ‘mediumpurple3,’ and ‘cyan’ modules that indicate Pocillopora corals’ responses to the antibiotics-heat stress treatment involved increases in energy production and conversion, immune inflammation, proteolysis and apoptosis at the expense of diminished holobiont metabolism, epithelial immune integrity, ion transport, inter-partner signal transduction, and DNA maintenance (Figure 3C). This suggests that in the antibiotics-heat treatment, oxidative damage signals together with a dysregulated bacteria community triggers programed cell death pathways through the activation of tumor necrosis factor and caspase pathways, and proteolytic effectors such as MMPs and the 26S proteasome (Wecker et al., 2018; Chuang, 2020; Jiang et al., 2020). Indeed, many of these energy conversion, oxidative stress response and proteolysis genes were co-expressed within the Pocillopora ‘floralwhite’ and ‘orange’ modules and the Cladocopium ‘brown’ module, making the hub genes of each of these modules appealing targets for future functional characterization, pharmacological inhibition and gene knockdown/knockout studies (Table 3) (Cleves et al., 2020a,b; Jiang et al., 2020).
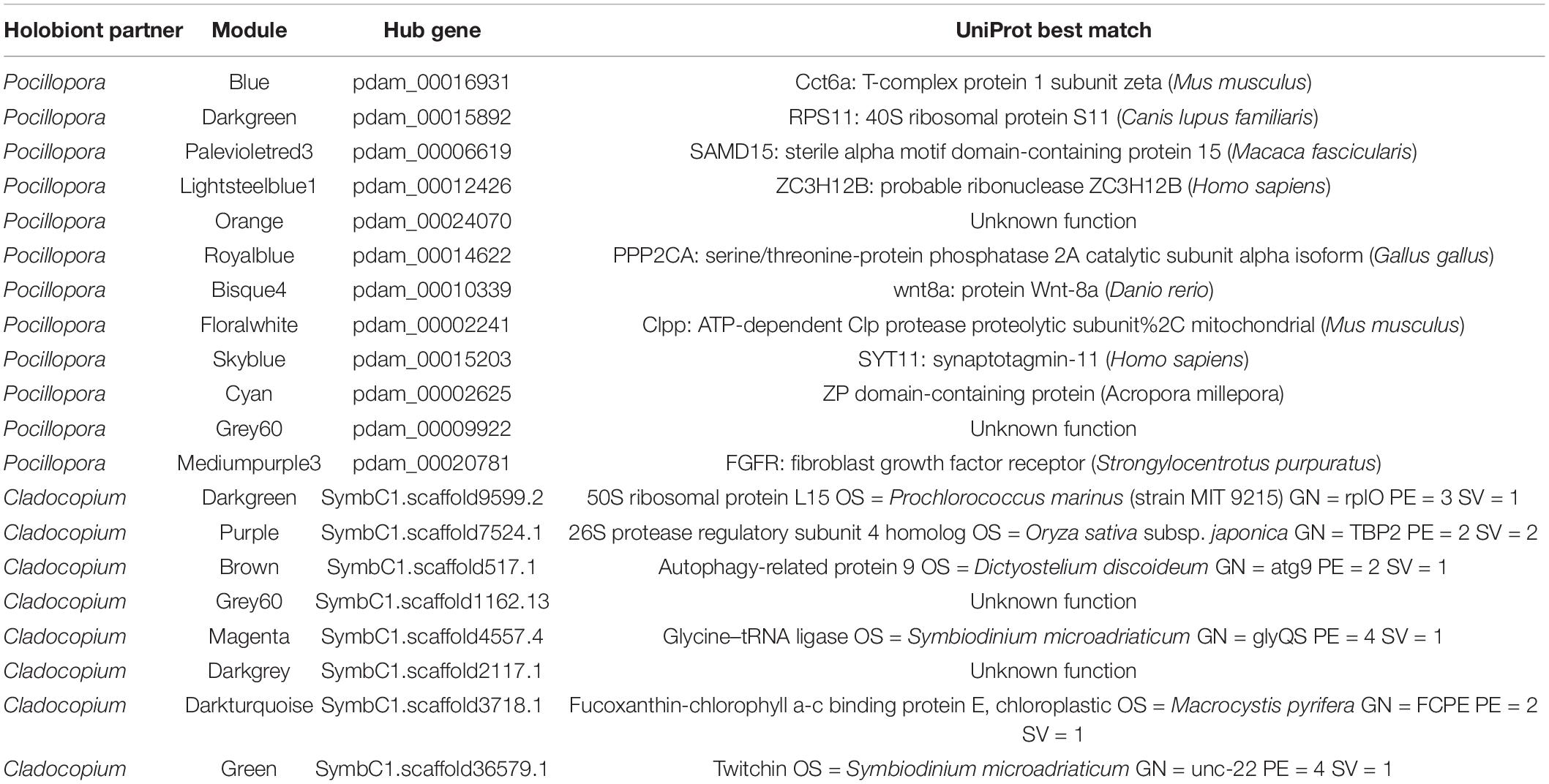
Table 3. Hub genes of Pocillopora and Cladocopium WGCNA modules with significant correlations to experimental treatments.
Implications for Coral Conservation and Disease Intervention Strategies
The results on the effect of antibiotic suppression of coral bacteria community and host-microbe interactions lead us to warn coral reef conservation practitioners to be cautious in considering field-based applications of antibiotics treatments as potential disease therapies, as there may be numerous unintended consequences for coral health. Current mitigation strategies being used throughout the Caribbean for treating stony coral tissue loss disease (SCTLD) involve treatments with antibiotics-infused epoxies (Voss et al., 2019; Walker and Pitts, 2019; Neely et al., 2020; Shilling, 2020). Our results suggest that ampicillin and streptomycin based antibiotic treatments do not non-selectively eliminate pathogenic bacteria, and more investigation into the efficacy and safety of these approaches should be conducted.
Future laboratory work should attempt to carefully manipulate associations between different coral genotypes and microbial symbiotic partners (including Symbiodiniaceae and bacteria) to move toward the generation of gnotobiotic (i.e., known-microbiome) corals for use in functional experiments. This has been shown to be feasible in cnidarian model systems such as Hydra, Nematostella, and Aiptasia (Murillo-Rincon et al., 2017; Domin et al., 2018; Medrano et al., 2019; Costa et al., 2021; Dungan et al., 2021), but is only beginning to be explored in reef-building corals and other cnidarians (Lin et al., 2019; Cunning and Baker, 2020; Weiland-Bräuer et al., 2020). Experiments that apply a combination of antibiotics treatments followed by targeted probiotic delivery or “microbiome recovery” treatments will yield insights into whether disturbed coral-associated bacteria communities and host stress response phenotypes can be “rescued” by the re-introduction of one or several beneficial microbes (Peixoto et al., 2017, 2021; Rosado et al., 2018; Damjanovic et al., 2019; Assis et al., 2020; Santoro et al., 2021). Future work would also benefit from the inclusion of baseline samples and additional time points to track the temporal changes in bacteria community composition and transcriptome responses across coral colonies.
Collectively, the results of this experiment highlight the vital importance of coral-associated bacteria communities to coral holobiont metabolism, immunity, and acute heat stress responses, and provide a basis for future mechanistic studies of how symbiotic interactions between multiple partners shape coral holobiont health, evolution, and ecological resilience (Engelberts et al., 2021; Voolstra et al., 2021).
Data Availability Statement
The mRNA and 16S sequencing datasets generated for this study can be found in the NCBI Sequence Read Archive under accession PRJNA587509. All scripts used in the RNAseq and 16S data analysis are available on GitHub: https://github.com/michaeltconnelly/EAPSI_Pocillopora_AxH.
Author Contributions
MC and NT-K designed the research. MC, CJM, and P-JL conducted the experiments. MC and CEM analyzed the data. MC, CEM, and NT-K wrote the manuscript. All authors participated in the editing of the manuscript.
Funding
This work was supported by the National Science Foundation East Asia and Pacific Summer Institute (EAPSI) Program Award #1713962 and the Taiwan Ministry of Science and Technology (MOST) S.I.T. program 2017 awarded to MC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the scientists and research support staff at the National Museum of Marine Biology and Aquarium, especially Chih-Jui (Ray) Tan for field assistance and site environmental data. Additional thanks to Anderson Mayfield for sharing laboratory space at the NMMBA, and to Ross Cunning and Stephanie Rosales for bioinformatics assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.814124/full#supplementary-material
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://eggnog-mapper.embl.de/
- ^ https://github.com/z0on/GO_MWU
- ^ https://data.qiime2.org/2020.2/common/silva-132-99-515-806-nb-classifier.qza
References
Akram, M. (2014). Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 68, 475–478. doi: 10.1007/s12013-013-9750-1
Amin, S. A., Green, D. H., Hart, M. C., Kupper, F. C., Sunda, W. G., and Carrano, C. J. (2017). Photolysis of ion – siderophore chelates promotes bacteria – algal mutualism Photolysis of iron – siderophore chelates promotes bacterial – algal mutualism. Environ. Sci. 106, 2–7.
Anderson, M. J. (2017). “Permutational multivariate analysis of variance (PERMANOVA),” in Wiley StatsRef Stat. Ref. Online, eds N. Balakrishnan, T. Colton, B. Everitt, W. Piegorsch, F. Ruggeri, and J. L. Teugels (Hoboken, NJ: John Wiley & Sons), 1–15. doi: 10.1002/9781118445112.stat07841
Apprill, A. (2020). The role of symbioses in the adaptation and stress responses of marine organisms. Ann. Rev. Mar. Sci. 12, 291–314. doi: 10.1146/annurev-marine-010419-010641
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. doi: 10.3354/ame01753
Assis, J. M., Villela, H., Abreu, F., Duarte, G., Roj, L., and Peixoto, R. (2020). Delivering beneficial microorganisms for corals (BMCs): rotifers as carriers of coral probiotics. Front. Microbiol. 11:608506. doi: 10.3389/fmicb.2020.608506
Avila-Magaña, V., Kamel, B., DeSalvo, M., Gómez-Campo, K., Enríquez, S., Kitano, H., et al. (2021). Elucidating gene expression adaptation of phylogenetically divergent coral holobionts under heat stress. Nat. Commun. 12:5731. doi: 10.1038/s41467-021-25950-4
Barfield, S. J., Aglyamova, G. V., Bay, L. K., and Matz, M. V. (2018). Contrasting effects of Symbiodinium identity on coral host transcriptional profiles across latitudes. Mol. Ecol. 27, 3103–3115. doi: 10.1111/mec.14774
Benavides, M., Bednarz, V. N., and Ferrier-Pagès, C. (2017). Diazotrophs: overlooked key players within the coral symbiosis and tropical reef ecosystems? Front. Mar. Sci. 4:10. doi: 10.3389/fmars.2017.00010
Blackall, L. L., Wilson, B., and Van Oppen, M. J. H. (2015). Coral-the world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347. doi: 10.1111/mec.13400
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17. doi: 10.1186/s40168-018-0470-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170a
Bolyen, E., Dillon, M., Bokulich, N. A., Abnet, C., Al-Ghalith, G., Alexander, H., et al. (2018). QIIME 2: reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6:e27295v2. doi: 10.7287/peerj.preprints.27295
Boulotte, N. M., Dalton, S. J., Carroll, A. G., Harrison, P. L., Putnam, H. M., Peplow, L. M., et al. (2016). Exploring the Symbiodinium rare biosphere provides evidence for symbiont switching in reef-building corals. ISME J. 10, 2693–2701. doi: 10.1038/ismej.2016.54
Brener-Raffalli, K., Clerissi, C., Vidal-Dupiol, J., Adjeroud, M., Bonhomme, F., Pratlong, M., et al. (2018). Thermal regime and host clade, rather than geography, drive Symbiodinium and bacterial assemblages in the Scleractinian coral Pocillopora damicornis sensu lato. Microbiome 6:39. doi: 10.1186/s40168-018-0423-6
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Camp, E. F., Kahlke, T., Nitschke, M. R., Varkey, D., Fisher, N. L., Fujise, L., et al. (2020). Revealing changes in the microbiome of Symbiodiniaceae under thermal stress. Environ. Microbiol. 22, 1294–1309. doi: 10.1111/1462-2920.14935
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.2307/2689606
Chuang, P. (2020). Signaling pathways in the coral polyp bail-out response. Coral Reefs 39, 1535–1548. doi: 10.1007/s00338-020-01983-x
Cleves, P. A., Shumaker, A., Lee, J. M., Putnam, H. M., and Bhattacharya, D. (2020a). Unknown to known: advancing knowledge of coral gene function. Trends Genet. 36, 93–104. doi: 10.1016/j.tig.2019.11.001
Cleves, P. A., Tinoco, A., Bradford, J., Perrin, D., Bay, L. K., and Pringle, J. (2020b). Reduced heat tolerance in a coral carrying CRISPR-induced mutations in the gene for a heat-shock transcription factor. Proc. Natl. Acad. Sci. U.S.A. 117, 28899–28905. doi: 10.1073/pnas.1920779117
Connelly, M. T., McRae, C. J., Liu, P.-J., and Traylor-Knowles, N. (2020). Lipopolysaccharide treatment stimulates Pocillopora coral genotype-specific immune responses but does not alter coral-associated bacteria communities. Dev. Comp. Immunol. 109:103717. doi: 10.1016/j.dci.2020.103717
Costa, R. M., Cárdenas, A., Loussert-fonta, C., Toullec, G., Meibom, A., Voolstra, C. R., et al. (2021). Surface topography, bacterial carrying capacity, and the prospect of microbiome manipulation in the sea anemone coral model Aiptasia. Front. Microbiol. 12:637834. doi: 10.3389/fmicb.2021.637834
Cunning, R., and Baker, A. C. (2020). Thermotolerant coral symbionts modulate heat stress-responsive genes in their hosts. Mol. Ecol. 29, 2940–2950. doi: 10.1111/mec.15526
Cunning, R., Bay, R. A., Gillette, P., Baker, A. C., and Traylor-Knowles, N. (2018). Comparative analysis of the Pocillopora damicornis genome highlights role of immune system in coral evolution. Sci. Rep. 8:16134. doi: 10.1038/s41598-018-34459-8
Damjanovic, K., van Oppen, M. J. H., Menéndez, P., and Blackall, L. L. (2019). Experimental inoculation of coral recruits with marine bacteria indicates scope for microbiome manipulation in Acropora tenuis and Platygyra daedalea. Front. Microbiol. 10:1702. doi: 10.3389/fmicb.2019.01702
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Domin, H., Zurita-Gutiérrez, Y. H., Scotti, M., Buttlar, J., Humeida, U. H., and Fraune, S. (2018). Predicted bacterial interactions affect in vivo microbial colonization dynamics in Nematostella. Front. Microbiol. 9:728. doi: 10.3389/fmicb.2018.00728
Dungan, A. M., van Oppen, M. J., and Blackall, L. L. (2021). Short-term exposure to sterile seawater reduces bacterial community diversity in the sea anemone, Exaiptasia diaphana. Front. Mar. Sci. 7:599314. doi: 10.3389/fmars.2020.599314
Engelberts, J. P., Robbins, S. J., Damjanovic, K., and Webster, N. S. (2021). Integrating novel tools to elucidate the metabolic basis of microbial symbiosis in reef holobionts. Mar. Biol. 168, 1–19. doi: 10.1007/s00227-021-03952-6
Epstein, H. E., Torda, G., Munday, P. L., and van Oppen, M. J. H. (2019). Parental and early life stage environments drive establishment of bacterial and dinoflagellate communities in a common coral. ISME J. 13, 1635–1638. doi: 10.1038/s41396-019-0358-3
Fernandes, A. D., Macklaim, J. M., Linn, T. G., Reid, G., and Gloor, G. B. (2013). ANOVA-Like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS One 8:e67019. doi: 10.1371/journal.pone.0067019
Fernandes, A. D., Reid, J. N. S., Macklaim, J. M., McMurrough, T. A., Edgell, D. R., and Gloor, G. B. (2014). Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 1–13. doi: 10.1186/2049-2618-2-15
Ferrier-Pagès, C., Martinez, S., Grover, R., Cybulski, J., Shemesh, E., and Tchernov, D. (2021). Tracing the trophic plasticity of the coral–dinoflagellate symbiosis using amino acid compound-specific stable isotope analysis. Microorganisms 9:182. doi: 10.3390/microorganisms9010182
Fox, M. D., Elliott Smith, E. A., Smith, J. E., and Newsome, S. D. (2019). Trophic plasticity in a common reef-building coral: insights from δ13C analysis of essential amino acids. Funct. Ecol. 33, 2203–2214. doi: 10.1111/1365-2435.13441
Geissler, L., Meunier, V., Rädecker, N., Perna, G., Rodolfo-Metalpa, R., Houlbrèque, F., et al. (2021). Highly variable and non-complex diazotroph communities in corals from ambient and high CO2 environments. Front. Mar. Sci. 8:754682. doi: 10.3389/fmars.2021.754682
Gélin, P., Postaire, B., Fauvelot, C., and Magalon, H. (2017). Reevaluating species number, distribution and endemism of the coral genus Pocillopora Lamarck, 1816 using species delimitation methods and microsatellites. Mol. Phylogenet. Evol. 109, 430–446. doi: 10.1016/j.ympev.2017.01.018
Gilbert, J. A., Hill, R., Doblin, M. A., and Ralph, P. J. (2012). Microbial consortia increase thermal tolerance of corals. Mar. Biol. 159, 1763–1771. doi: 10.1007/s00227-012-1967-9
Glasl, B., Herndl, G. J., and Frade, P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. doi: 10.1038/ismej.2016.9
Glaze, T. D., Erler, D. V., and Siljanen, H. M. P. (2021). Microbially facilitated nitrogen cycling in tropical corals. ISME J. 16, 68–77. doi: 10.1038/s41396-021-01038-1
Gloor, G. B., and Reid, G. (2016). Compositional analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can. J. Microbiol 703, 692–703.
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V., and Egozcue, J. J. (2017). Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8:2224. doi: 10.3389/fmicb.2017.02224
Gunderson, A. R., Armstrong, E. J., and Stillman, J. H. (2016). Multiple stressors in a changing world: the need for an improved perspective on physiological responses to the dynamic marine environment. Ann. Rev. Mar. Sci. 8, 357–378. doi: 10.1146/annurev-marine-122414-033953
Haydon, T. D., Seymour, J. R., Raina, J. B., Edmondson, J., Siboni, N., Matthews, J. L., et al. (2021). Rapid shifts in bacterial communities and homogeneity of symbiodiniaceae in colonies of Pocillopora acuta transplanted between reef and mangrove environments. Front. Microbiol. 12:756091. doi: 10.3389/fmicb.2021.756091
Hopkinson, B. M., and Morel, F. M. M. (2009). The role of siderophores in iron acquisition by photosynthetic marine microorganisms. BioMetals 22, 659–669. doi: 10.1007/s10534-009-9235-2
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hume, B. C. C., Smith, E. G., Ziegler, M., Hugh, J., Burt, M. W. J. A., and Voolstra, C. R. (2019). SymPortal: a novel analytical framework and platform for coral algal symbiont next – generation sequencing ITS2 profiling. Mol. Ecol. Resour. 19, 1063–1080. doi: 10.1111/1755-0998.13004
Jiang, S., Zhou, Z., Sun, Y., Zhang, T., and Sun, L. (2020). Coral gasdermin triggers pyroptosis. Sci. Immunol. 5:eabd2591. doi: 10.1126/SCIIMMUNOL.ABD2591
Johnston, E. C., Forsman, Z. H., Flot, J.-F., Schmidt-Roach, S. Pinzón, J. H., Knapp, I. S. S., et al. (2017). A genomic glance through the fog of plasticity and diversification in Pocillopora.Sci. Rep. 7:5991. doi: 10.1038/s41598-017-06085-3
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keshavmurthy, S., Kuo, C., Huang, Y., and Carballo-Bolaños, R. (2019). Coral reef resilience in taiwan: lessons from long- term ecological research on the coral reefs of Kenting national park (Taiwan). J. Mar. Sci. Eng. 7:388. doi: 10.3390/jmse7110388
Keshavmurthy, S., Meng, P. J., Wang, J. T., Kuo, C. Y., Yang, S. Y., Hsu, C. M., et al. (2014). Can resistant coral-Symbiodinium associations enable coral communities to survive climate change? A study of a site exposed to long-termhot water input. PeerJ 2:e327. doi: 10.7717/peerj.327
Kirk, A. L., Clowez, S., Lin, F., Grossman, A. R., and Xiang, T. (2020). Transcriptome reprogramming of symbiodiniaceae Breviolum minutum in response to casein amino acids supplementation. Front. Physiol. 11:574654. doi: 10.3389/fphys.2020.574654
Ladner, J. T., Barshis, D. J., and Palumbi, S. R. (2012). Protein evolution in two co-occurring types of Symbiodinium: an exploration into the genetic basis of thermal tolerance in Symbiodinium clade D. BMC Evol. Biol. 12:217. doi: 10.1186/1471-2148-12-217
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lesser, M. P., Morrow, K. M., Pankey, S. M., and Noonan, S. H. C. (2018). Diazotroph diversity and nitrogen fixation in the coral Stylophora pistillata from the Great Barrier Reef. ISME J. 12, 813–824. doi: 10.1038/s41396-017-0008-6
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, J., Long, L., Zou, Y., and Zhang, S. (2020). Microbial community and transcriptional responses to increased temperatures in coral Pocillopora damicornis holobiont. Soc. Appl. Microbiol. 23, 826–843. doi: 10.1111/1462-2920.15168
Li, J., Long, L., Zou, Y., and Zhang, S. (2021). Microbial community and transcriptional responses to increased temperatures in coral Pocillopora damicornis holobiont. Environ. Microbiol. 23, 826–843.
Liao, Y., Smyth, G. K., and Shi, W. (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lin, M. F., Takahashi, S., Forêt, S., Davy, S. K., and Miller, D. J. (2019). Transcriptomic analyses highlight the likely metabolic consequences of colonization of a cnidarian host by native or non-native Symbiodinium species. Biol. Open 8, 1–11. doi: 10.1242/bio.038281
Liu, H., Stephens, T. G., González-Pech, R. A., Beltran, V. H., Lapeyre, B., Bongaerts, P., et al. (2018). Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 1:95. doi: 10.1038/s42003-018-0098-3
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Maher, R. L., Rice, M. M., McMinds, R., and Burkepile, D. E. (2019). Multiple stressors interact primarily through antagonism to drive changes in the coral microbiome. Sci. Rep. 9:6834. doi: 10.1038/s41598-019-43274-8
Maire, J., Girvan, S. K., Barkla, S. E., David, A. P., Blackall, L. L., and Van Oppen, M. J. H. (2021). Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 15, 2028–2042. doi: 10.1038/s41396-021-00902-4
Martinez Arbizu, P. (2020). pairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. R Package Version 0.4.
Matthews, J. L., Raina, J. B., Kahlke, T., Seymour, J. R., van Oppen, M. J. H., and Suggett, D. J. (2020). Symbiodiniaceae-bacteria interactions: rethinking metabolite exchange in reef-building corals as multi-partner metabolic networks. Environ. Microbiol. 22, 1675–1687. doi: 10.1111/1462-2920.14918
Matz, M. V. (2019). KOGMWU: Functional Summary and Meta-Analysis of Gene Expression Data. R Package Version 1.2. Available online at: https://cran.r-project.org/web/packages/KOGMWU/index.html (accessed February 19, 2019).
Mayfield, A. B., Wang, Y. B., Chen, C.-S., Lin, C. Y., and Chen, S. H. (2014). Compartment-specific transcriptomics in a reef-building coral exposed to elevated temperatures. Mol. Ecol. 23, 5816–5830. doi: 10.1111/mec.12982
McDonald, D., Clemente, J. C., Kuczynski, J., Rideout, J. R., Stombaugh, J., Wendel, D., et al. (2012). The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience 464, 1–6. doi: 10.1186/2047-217X-1-7
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Medrano, E., Merselis, D. G., Bellantuono, A. J., and Rodriguez-Lanetty, M. (2019). Proteomic Basis of symbiosis: a heterologous partner fails to duplicate homologous success in a novel cnidarian- Symbiodiniaceae mutualism. Front. Microbiol. 10:1153. doi: 10.3389/FMICB.2019.01153
Motone, K., Takagi, T., Aburaya, S., Miura, N., Aoki, W., and Ueda, M. (2020). A zeaxanthin-producing bacterium isolated from the algal phycosphere protects coral endosymbionts from environmental stress. MBio 11:e01019-19.
Murillo-Rincon, A. P., Klimovich, A., Pemöller, E., Taubenheim, J., Mortzfeld, B., Augustin, R., et al. (2017). Spontaneous body contractions are modulated by the microbiome of Hydra. Sci. Rep. 7:15937. doi: 10.1038/s41598-017-16191-x
Neave, M. J., Rachmawati, R., Xun, L., Michell, C. T., Bourne, D. G., Apprill, A., et al. (2017). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 11, 186–200. doi: 10.1038/ismej.2016.95
Neely, K. L., Macaulay, K. A., Hower, E. K., and Dobler, M. A. (2020). Effectiveness of topical antibiotics in treating corals affected by Stony Coral Tissue Loss Disease. PeerJ 8:e9289. doi: 10.7717/peerj.9289
O’Brien, P. A., Webster, N. S., Miller, D. J., and Bourne, D. G. (2019). Host-Microbe coevolution: applying evidence from model systems to complex marine invertebrate holobionts. MBio 10:e02241-18. doi: 10.1128/mBio.02241-18
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). vegan: Community Ecology Package.
Olson, N. D., Ainsworth, T. D., Gates, R. D., and Takabayashi, M. (2009). Diazotrophic bacteria associated with Hawaiian Montipora corals: diversity and abundance in correlation with symbiotic dinoflagellates. J. Exp. Mar. Biol. Ecol. 371, 140–146. doi: 10.1016/j.jembe.2009.01.012
Palarea-Albaladejo, J., and Martín-Fernández, J. A. (2015). ZCompositions – R package for multivariate imputation of left-censored data under a compositional approach. Chemom. Intell. Lab. Syst. 143, 85–96. doi: 10.1016/j.chemolab.2015.02.019
Park, S., Choi, S. J., Park, J. M., and Yoon, J. H. (2017). Alteromonas aestuariivivens sp. nov., isolated from a tidal flat. Int. J. Syst. Evol. Microbiol. 67, 2791–2797. doi: 10.1099/ijsem.0.002023
Peixoto, R. S., Rosado, P. M., Leite, D. C., de, A., Rosado, A. S., and Bourne, D. G. (2017). Beneficial microorganisms for corals (BMC): proposed mechanisms for coral health and resilience. Front. Microbiol. 8:341. doi: 10.3389/fmicb.2017.00341
Peixoto, R. S., Sweet, M., Villela, H. D. M., Cardoso, P. M., Thomas, T., Voolstra, C. R., et al. (2021). Coral probiotics: premise, promise, prospects. Annu. Rev. Anim. Biosci. 9, 265–288. doi: 10.1146/annurev-animal-090120-115444
Pinzón, J. H., Sampayo, E., Cox, E., Chauka, L. J., Chen, C. A., Voolstra, C. R., et al. (2013). Blind to morphology: genetics identifies several widespread ecologically common species and few endemics among Indo-Pacific cauliflower corals (Pocillopora, Scleractinia). J. Biogeogr. 40, 1595–1608. doi: 10.1111/jbi.12110
Pogoreutz, C., Rädecker, N., Cárdenas, A., Gärdes, A., Wild, C., and Voolstra, C. R. (2018). Dominance of Endozoicomonas bacteria throughout coral bleaching and mortality suggests structural inflexibility of the Pocillopora verrucosa microbiome. Ecol. Evol. 8, 2240–2252. doi: 10.1002/ece3.3830
Pollock, F. J., McMinds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921. doi: 10.1038/s41467-018-07275-x
Poquita-Du, R. C., Huang, D., Chou, L. M., and Mrinalini</snm>, and Todd, P. A. (2019). Short term exposure to heat and sediment triggers changes in coral gene expression and photo-physiological performance. Front. Mar. Sci. 6:121. doi: 10.3389/fmars.2019.00121
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rädecker, N., Pogoreutz, C., Gegner, H. M. Cárdenas, A., Perna, G. Geißler, L., et al. (2021). Heat stress reduces the contribution of diazotrophs to coral holobiont nitrogen cycling. ISME J. doi: 10.1038/s41396-021-01158-8
Raina, J. B., Dinsdale, E. A., Willis, B. L., and Bourne, D. G. (2010). Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 18, 101–108. doi: 10.1016/j.tim.2009.12.002
Reich, H. G., Rodriguez, I. B., LaJeunesse, T. C., and Ho, T. Y. (2020). Endosymbiotic dinoflagellates pump iron: differences in iron and other trace metal needs among the Symbiodiniaceae. Coral Reefs 39, 915–927. doi: 10.1007/s00338-020-01911-z
Robbins, S. J., Singleton, C. M., Chan, C. X., Messer, L. F., Geers, A. U., Ying, H., et al. (2019). A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 4, 2090–2100. doi: 10.1038/s41564-019-0532-4
Rosado, P. M., Leite, D. C. A., Duarte, G. A. S., Chaloub, R. M., Jospin, G., Nunes da Rocha, U., et al. (2018). Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation. ISME J. 13, 921–936. doi: 10.1038/s41396-018-0323-6
Santoro, E. P., Borges, R. M., Espinoza, J. L., Freire, M., Messias, C. S. M. A., Villela, H. D. M., et al. (2021). Coral microbiome manipulation elicits metabolic and genetic restructuring to mitigate heat stress and evade mortality. Sci. Adv. 7, 19–21. doi: 10.1126/sciadv.abg3088
Schalk, I. J., Hannauer, M., and Braud, A. (2011). New roles for bacterial siderophores in metal transport and tolerance. Environ. Microbiol. 13, 2844–2854. doi: 10.1111/j.1462-2920.2011.02556.x
Schmidt-Roach, S., Miller, K. J., Lundgren, P., and Andreakis, N. (2014). With eyes wide open: a revision of species within and closely related to the Pocillopora damicornis species complex (Scleractinia; Pocilloporidae) using morphology and genetics. Zool. J. Linn. Soc. 170, 1–33. doi: 10.1111/zoj.12092
Shilling, E. N. (2020). Stony Coral Tissue Loss Disease Intervention Strategies for Montastrea cavernosa. Master’s thesis. Ann Arbor, MI: ProQuest.
Soffer, N., Gibbs, P. D. L., and Baker, A. C. (2008). “Practical applications of contaminant-free Symbiodinium cultures grown on solid media,” in Proceedings of the 11th International Coral Reef Symposium, Ft. Lauderdale, FL, 159–163.
Sweet, M. J., Croquer, A., and Bythell, J. C. (2014). Experimental antibiotic treatment identifies potential pathogens of white band disease in the endangered Caribbean coral Acropora cervicornis. Proc. R. Soc. B Biol. Sci. 281, 20140094–20140094. doi: 10.1098/rspb.2014.0094
Sweet, M., and Bythell, J. C. (2015). White syndrome in Acropora muricata: nonspecific bacterial infection and ciliate histophagy. Mol. Ecol. 24, 1150–1159. doi: 10.1111/mec.13097
Tandon, K., Lu, C.-Y., Chiang, P.-W., Wada, N., Yang, S.-H., Chan, Y.-F., et al. (2020). Comparative genomics: dominant coral-bacterium Endozoicomonas acroporae metabolizes dimethylsulfoniopropionate (DMSP). ISME J. 14, 1290–1303. doi: 10.1038/s41396-020-0610-x
Thompson, J. R., Rivera, H. E., Closek, C. J., and Medina, M. (2015). Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4:176. doi: 10.3389/fcimb.2014.00176
Tout, J., Siboni, N., Messer, L. F., Garren, M., Stocker, R., Webster, N. S., et al. (2015). Increased seawater temperature increases the abundance and alters the structure of natural Vibrio populations associated with the coral Pocillopora damicornis. Front. Microbiol. 6:432. doi: 10.3389/fmicb.2015.00432
Traylor-Knowles, N., Granger, B. R., Lubinski, T. J., Parikh, J. R., Garamszegi, S., Xia, Y., et al. (2011). Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 12:585. doi: 10.1186/1471-2164-12-585
Vavilin, D. V., and Vermaas, W. F. J. (2002). Regulation of the tetrapyrrole biosynthetic pathway leading to heme and chlorophyll in plants and cyanobacteria. Physiol. Plant. 115, 9–24. doi: 10.1034/j.1399-3054.2002.1150102.x
Vidal-Dupiol, J., Dheilly, N. M., Rondon, R., Grunau, C., Cosseau, C., Smith, K. M., et al. (2014). Thermal stress triggers broad Pocillopora damicornis transcriptomic remodeling, while Vibrio coralliilyticus infection induces a more targeted immuno-suppression response. PLoS One 9:e107672. doi: 10.1371/journal.pone.0107672
Voolstra, C. R., Suggett, D. J., Peixoto, R. S., Parkinson, J. E., Quigley, K. M., Silveira, C. B., et al. (2021). Extending the natural adaptive capacity of coral holobionts. Nat. Rev. Earth Environ. 2, 747–762. doi: 10.1038/s43017-021-00214-3
Voss, J. D., Shilling, E. N., and Combs, I. R. (2019). Intervention and Fate Tracking for Corals Affected by Stony Coral Tissue Loss Disase in the Northern Florida Reef Tract. Miami, FL: Florida DEP, 1–23.
Walker, B. K., Turner, N. R., Noren, H. K. G., Buckley, S. F., and Pitts, K. A. (2021). Optimizing Stony Coral Tissue Loss Disease (SCTLD) intervention treatments on Montastraea cavernosa in an endemic zone. Front. Mar. Sci. 8:666224. doi: 10.3389/fmars.2021.666224
Walker, B., and Pitts, K. (2019). SE FL Reef-Building-Coral Response to Amoxicillin Intervention and Broader-Scale Coral Disease Intervention. Miami, FL: Florida DEP, 12.
Wang, Y., Wang, H., Liu, J., Lai, Q., Shao, Z., Austin, B., et al. (2010). Aestuariibacter aggregatus sp. nov., a moderately halophilic bacterium isolated from seawater of the Yellow Sea. FEMS Microbiol. Lett. 309, 48–54. doi: 10.1111/j.1574-6968.2010.02011.x
Weber, L., DeForce, E., and Apprill, A. (2017). Optimization of DNA extraction for advancing coral microbiota investigations. Microbiome 5:18. doi: 10.1186/s40168-017-0229-y
Wecker, P., Lecellier, G., Guibert, I., Zhou, Y., Bonnard, I., and Berteaux-Lecellier, V. (2018). Exposure to the environmentally-persistent insecticide chlordecone induces detoxification genes and causes polyp bail-out in the coral P. damicornis. Chemosphere 195, 190–200. doi: 10.1016/j.chemosphere.2017.12.048
Weiland-Bräuer, N., Pinnow, N., Langfeldt, D., Roik, A., Güllert, S., Chibani, C. M., et al. (2020). The native microbiome is crucial for offspring generation and fitness of Aurelia aurita. MBio 11:e02336-20. doi: 10.1128/mBio.02336-20
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag New York.
Wickham, H., Averick, M., Bryan, J., Chang, W., McGowan, L. D., François, R., et al. (2019). Welcome to the {tidyverse}. J. Open Source Softw. 4:1686. doi: 10.21105/joss.01686
Wilkins, L. G. E., Leray, M., Dea, A. O., Yuen, B., Peixoto, R., Pereira, T. J., et al. (2019). Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17:e3000533. doi: 10.1371/journal.pbio.3000533
Work, T. M., and Aeby, G. S. (2014). Microbial aggregates within tissues infect a diversity of corals throughout the Indo-Pacific. Mar. Ecol. Prog. Ser. 500, 1–9. doi: 10.3354/meps10698
Wright, R. M., Aglyamova, G. V., Meyer, E., and Matz, M. V. (2015). Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genomics 16:371. doi: 10.1186/s12864-015-1540-2
Xia, Y., and Sun, J. (2017). Hypothesis testing and statistical analysis of microbiome. Genes Dis. 4, 138–148. doi: 10.1016/j.gendis.2017.06.001
Zaneveld, J. R., Burkepile, D. E., Shantz, A. A., Pritchard, C. E., McMinds, R., Payet, J. P., et al. (2016). Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 7:11833. doi: 10.1038/ncomms11833
Zaneveld, J. R., McMinds, R., and Thurber, R. V. (2017). Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2:17121. doi: 10.1038/nmicrobiol.2017.121
Zhang, R., Yu, K., Li, A., Wang, Y., and Huang, X. (2019). Antibiotics in corals of the South China Sea: occurrence, distribution, bioaccumulation, and considerable role of coral mucus. Environ. Pollut. 250, 503–510. doi: 10.1016/j.envpol.2019.04.036
Zhang, R., Zhang, R., Yu, K., Wang, Y., Huang, X., Pei, J., et al. (2018). Occurrence, sources and transport of antibiotics in the surface water of coral reef regions in the South China Sea: potential risk to coral growth. Environ. Pollut. 232, 450–457. doi: 10.1016/j.envpol.2017.09.064
Zhang, Y., Yang, Q., Ling, J., Long, L., Huang, H., Yin, J., et al. (2021). Shifting the microbiome of a coral holobiont and improving host physiology by inoculation with a potentially beneficial bacterial consortium. BMC Microbiol. 21:130. doi: 10.1186/s12866-021-02167-5
Zhang, Y., Zhou, Z., Wang, L., and Huang, B. (2018). Transcriptome, expression, and activity analyses reveal a vital heat shock protein 70 in the stress response of stony coral Pocillopora damicornis. Cell Stress Chaperones 23, 711–721. doi: 10.1007/s12192-018-0883-4
Zhou, Z., Yu, X., Tang, J., Wu, Y., Wang, L., and Huang, B. (2018). Systemic response of the stony coral Pocillopora damicornis against acute cadmium stress. Aquat. Toxicol. 194, 132–139. doi: 10.1016/j.aquatox.2017.11.013
Zhou, Z., Zhang, G., Chen, G., Ni, X., Guo, L., Yu, X., et al. (2017). Elevated ammonium reduces the negative effect of heat stress on the stony coral Pocillopora damicornis. Mar. Pollut. Bull. 118, 319–327. doi: 10.1016/j.marpolbul.2017.03.018
Zhou, Z., Zhang, K., Wang, L., Su, Y., Wang, J., Song, T., et al. (2021). Nitrogen availability improves the physiological resilience of coral endosymbiont Cladocopium goreaui to high temperature. J. Phycol. 57, 1187–1198. doi: 10.1111/jpy.13156
Ziegler, M., Grupstra, C. G. B., Barreto, M. M., Eaton, M., BaOmar, J., Zubier, K., et al. (2019). Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10:3092. doi: 10.1038/s41467-019-10969-5
Keywords: coral, holobiont, transcriptome, microbiome, antibiotics, bacteria, dysbiosis
Citation: Connelly MT, McRae CJ, Liu P-J, Martin CE and Traylor-Knowles N (2022) Antibiotics Alter Pocillopora Coral-Symbiodiniaceae-Bacteria Interactions and Cause Microbial Dysbiosis During Heat Stress. Front. Mar. Sci. 8:814124. doi: 10.3389/fmars.2021.814124
Received: 12 November 2021; Accepted: 22 December 2021;
Published: 13 January 2022.
Edited by:
Kefu Yu, Guangxi University, ChinaReviewed by:
Xiaopeng Yu, Guangxi University, ChinaDanwei Huang, National University of Singapore, Singapore
Copyright © 2022 Connelly, McRae, Liu, Martin and Traylor-Knowles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikki Traylor-Knowles, bnRyYXlsb3Jrbm93bGVzQHJzbWFzLm1pYW1pLmVkdQ==
†Present address: Michael T. Connelly, Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution, Washington, DC, United States