Abstract
Sea urchin is an indicator of coastal environmental changes in the global warming era, and is also a model organism in developmental biology and evolution. Due to the depletion of wild resources, new aquaculture techniques for improving stocks have been well studied. The gut microbiome shapes various aspects of a host’s physiology. However, these microbiome structures and functions on sea urchins, particularly Mesocentrotus nudus and Strongylocentrotus intermedius which are important marine bioresources commonly found in Japan, have not been fully investigated yet. Using metagenomic approaches including meta16S and shotgun metagenome sequencings, the structures, functions, and dynamics of the gut microbiome of M. nudus and S. intermedius, related to both habitat environment and host growth, were studied. Firstly, a broad meta16S analysis revealed that at the family level, Psychromonadaceae and Flavobacteriaceae reads (38–71%) dominated in these sea urchins, which is a unique feature observed in species in Japan. Flavobacteriaceae reads were more abundant in individuals after rearing in an aquarium with circulating compared to one with running water. Campylobacteraceae and Vibrionaceae abundances increased in both kinds of laboratory-reared sea urchins in both types of experiments. 2-weeks feeding experiments of M. nudus and S. intermedius transplanted from the farm to laboratory revealed that these gut microbial structures were affected by diet rather than rearing environments and host species. Secondly, further meta16S analysis of microbial reads related to M. nudus growth revealed that at least four Amplicon Sequence Variant (ASV) affiliated to Saccharicrinis fermentans, which is known to be a nitrogen (N2) fixing bacterium, showed a significant positive correlation to the body weight and test diameter. Interestingly, gut microbiome comparisons using shotgun metagenome sequencing of individuals showing higher and lower growth rates revealed a significant abundance of “Nitrate and nitrite ammonification” genes in the higher-grown individuals under the circulating water rearing. These findings provide new insights on the structure-function relationship of sea urchin gut microbiomes beyond previously reported nitrogen fixation function in sea urchin in 1950s; we discovered a nitrate reduction function into ammonium for the growth promotion of sea urchin.
Introduction
Sea urchin is an important aquatic resource worldwide. However, its global production has been decreasing since the 1990s (Stefánsson et al., 2017). Strongylocentrotus intermedius and Mesocentrotus nudus are important fishery resources in many Asian countries. S. intermedius is found on the intertidal and subtidal rocky seabed in the northern region in the Pacific Ocean, the Sea of Japan, the Korean peninsula, northeastern China, Sakhalin, and Vladivostok (Agatsuma, 2013). M. nudus is found on the intertidal and subtidal seabed and is distributed from Dalian in China to Primorskyi Kray in Russia and Japan (Agatsuma, 2013; Takagi et al., 2019; Ding et al., 2020). As a result of the great efforts of pioneer biologists, seed production of both species has been established, currently on a practical level in Japan. Sea urchin has been used for over a hundred years as a model organism in developmental biology research (Wilson, 1895). Knowledge of sea urchins in the field of biology has expanded to include (1) the effects of toxic substances on their immune system, reproduction, and development (Nobre et al., 2015; Brown et al., 2020; Pikula et al., 2020; Rendell-Bhatti et al., 2021), (2) the gene expression involved in sea urchin fertilization and development stages (Li et al., 2020; Wessel et al., 2021; Cui et al., 2022), (3) the nervous system (Wood et al., 2018; Martín-Durán and Hejnol, 2021; Formery et al., 2021), and (4) sea urchin genomes (Sodergren et al., 2006; Kudtarkar and Cameron, 2017; Kinjo et al., 2018; Warner et al., 2021). Sea urchins have also been studied in various aspects related to the impact of current changing environments, such as ocean acidification and global warming to their development and growth (Dworjanyn and Byrne, 2018; García et al., 2018; Zhao et al., 2018; Houlihan et al., 2020).
Sea urchin has been studied for a long time as fisheries resources and model organisms of biology. However, there have been a few studies on general views of their gut microbes’ structure, function, and dynamics, particularly using individual-level metagenome approaches (Hakim et al., 2015, 2016, 2019, Yao et al., 2019; Faddetta et al., 2020; Miller et al., 2021). The abundances of reads assigned to phyla Bacteroidetes and Proteobacteria were observed in the gut of Tripneustes gratilla, and those assigned to phyla Fusobacteria and Proteobacteria in Diadema setosum and Stomopneustes variolaris (Yao et al., 2019), and the order Vibrionales was abundant in wild American green sea urchin (Hakim et al., 2016). Compared to the important roles of gut microbiota in various animals, including humans (Sharma et al., 2019; Youngblut et al., 2019; Fong et al., 2020; Fan and Pedersen, 2021; Morais et al., 2021; Tang et al., 2021), knowledge of gut microbes of sea urchin is behind that of other animals. Only the roles of nitrogen-fixing microbes in the gut of sea urchins have been discussed since the 1950s to answer why C/N rich diet provides nutrition to sea urchins (Mann, 1977).
In this study, we applied a non-destructive individual methodology (Yamazaki et al., 2016) to sea urchin species, S. intemedius and M. nudus, in Japan for the first time, (1) to examine the external factors, e.g., environments and diets, affecting the structure of the sea urchin gut microbiome, and (2) to explore the function of those gut microbiomes and specific microbes contributing to host growth by monitoring fecal microbiome changes. Here, we report new insights into the gut microbiome’s structure, function, and dynamics of two sea urchin species in Japan.
Materials and Methods
Samples Collection
M. nudus (n = 13) cultured at the Esan Seedling Center (ESC), Hakodate, and S. intermedius (n = 4) cultured at the Toi Seedling Center (TSC), Hakodate, in 2014 and 2015, were used in these individual microbiome studies. Both ESC and TSC were also designated to be Farm below. The procedure for rearing the two species of sea urchin larvae was carried out based on Sakai et al. (2003). Wild individuals of M. nudus (n = 4) and S. intermedius (n = 4) collected at Menagawa, Hakodate, in 2015 were also used as a comparison. We did not need a specific authorization to handle wild animals’ feces. The seawater samples taken from sea urchin rearing in both running and circulation-water system experiments were also used.
Rearing Experiments
Two types of rearing experiments were performed to understand (1) factors affecting the shape of the gut microbiome (Experiment 1; Exp. 1) and (2) microbiome functions affecting the host growth (Experiment 2; Exp. 2) (Supplementary Figure 1).
Experiment 1 (Exp. 1)
Nine individuals, including the two species M. nudus and S. intermedius were used for this experiment. Five specimens of M. nudus (test diameter 24.43 ± 6.69 mm, weight 8.67 ± 5.91 g) were collected at the farm ESC. They were fed with microalgae throughout the larval stage and subsequently a brown alga, Saccharina japonicus, at juvenile stage until collection time. Four S. intermedius (weight 2.88 ± 0.76 g) were obtained from the farm TSC, and were fed with microalgae until that collection. After feces collection on these farms, the sea urchins were transplanted to the laboratory of Hakodate Fisheries Experiment Station (HFES), which is equipped with a running-water rearing system and reared for 2-weeks. At the HFES laboratory, an ambient natural seawater temperature of approximately 15°C was used to feed sea urchins individually in a caged 2 L polyethylene terephthalate (PET) aquarium created with water exchange holes with full feeding of fresh S. japonicus thalli. After 2 weeks of rearing at the laboratory, feces samples were collected. Seawater samples from farm and laboratory aquarium were collected and used for microbiome analyses.
Experiment 2 (Exp. 2)
Eight specimens of M. nudus (test diameter 22.25 ± 4.47 mm, weight 4.77 ± 2.13 g) were taken from ESC, and they were transplanted to the laboratory of Hokkaido University, and fed with a fine slice of boiled frozen S. japonicus thalli. They were fed for 6 weeks in a circulating-water rearing system maintained at an optimal temperature of 18°C. Every day, half of the water was replaced with fresh artificial seawater. Artificial seawater (SEALIFE, Marine Tech, Japan) was used. Feces and seawater samples were collected at the farm ESC. They were also collected bi-weekly after start rearing in the laboratory.
Collection of Feces and Seawater
Feces and seawater samples for microbiome analyses were collected according to Yamazaki et al. (2016). In brief, feces collections on-site and at the farm HFES were performed inside an instant clean booth, illuminated by ultraviolet light for 15 min (GL-15, Panasonic, Japan). Sea urchin individuals were cleaned using filter-sterilized seawater and moved individually into sterile beakers with filter-sterilized seawater until feces were released. The filter-sterilized seawater was prepared as follows; the natural seawater taken from a coastal area at a depth of 3–4 m was transferred to pressure tanks and filtered through a 0.22 μm Sterivex filter (Sterivex-GV Sterile Vented Filter Unit 0.22 μm, EMD, Millipore, United States) under positive pressure using filtered (0.22 μm) high purity N2 gas. Feces was collected into 1.5 mL tubes using adopted 5 mL pipette tips. Feces collected on-site and at the farm HFES were immediately frozen in a sterilized 1.5 mL tube on dry ice, transferred to our lab, and kept at –80°C until the DNA extraction was performed. A total of 5 L of the seawater was collected and filtered through a 0.22 μm Sterivex filter. The Sterivex filter was filled with a SET buffer (sucrose 20%, EDTA 50 mM, Tris-HCl 50 mM) and rapidly frozen on dry ice, and kept at –80°C until the DNA extraction was performed.
Microbial DNA Extraction
According to the manufacture’s protocol, the microbial DNA extraction of sea urchin feces was performed using the NucleoSpin Soil Kit (MACHEREY-NAGEL, Germany). According to the modified manufacture’s protocol, the microbial DNA extraction from seawater was performed using the NucleoSpin Tissue Kit (MACHEREY-NAGEL) (Yamazaki et al., 2016). We used 20% SDS and proteinase K (20 mg/mL) for pre-lysis instead of buffer T1 and proteinase K. We also used 1 mL buffer B3 instead of 200 μL.
Meta16S Sequencing and Downstream Analyses
PCR amplification and amplicon sequencing of the V1-V2 region on 16S rRNA gene were also performed according to Yamazaki et al. (2016) with minor modifications. Amplified PCR products using 27Fmod with barcode sequences and 338R primers were sequenced using MiSeq (Illumina, United States). Data was further qualified by the removal of reads with average quality values below 25. Filter-passed reads were filed as FASTA for downstream analysis after trimming off both primer sequences.
The sequence data of 16S rRNA genes was analyzed using the Quantitative Insights Into Microbial Ecology 2 (QIIME2) (Bolyen et al., 2019) version 2019.1. Sequence denoise, dereplication, and chimeras filtering were performed using the DADA2 pipeline (Callahan et al., 2016). During the “dada2” pipelines, the representative Amplicon Sequence Variant (ASV) sequences, consist of reads with 100% similarity, were constructed. ASVs were assigned taxonomic status through the “q2-feature-classifier” plugin (Bokulich et al., 2018) with the Greengeenes (version 13.8) database trained by Naïve Bayes methods. ASVs assigned to chloroplast or mitochondria were removed from the dataset through the “filter-features” plugin. Using this dataset, further analyses were conducted. For beta diversity, we performed weighted and unweighted UniFrac analysis (Lozupone et al., 2011) and visualized them in a PCoA plot based on a phylogenetic tree generated from the Greengenes database through the FastTree pipeline. To verify if specific ASVs abundance correlated to sea urchin body weight, we performed multiple Person correlations, and p-values were adjusted using the Holm (1979) method. We searched for highly (r > 0.6) and significantly (correlated p < 0.05) correlated ASVs against the bodyweight and test diameter of the sea urchins. ASVs that occurred in only one sample were removed from this analysis. Growth rate was calculated by Excel software. Meta16S sequences retrieved from the public database were also included in understanding the overview structure of the gut microbiome of sea urchin worldwide; SRP062365 (Hakim et al., 2015), SRP076869 (Hakim et al., 2016), and PRJNA504890 (Hakim et al., 2019).
Shotgun Metagenomic Sequencing and Analysis
Shotgun metagenome sequencing using the Illumina platform was performed on the high and low growth individuals based on their growth rate. Analysis of correlation coefficient used Excel software using the standardized number of leads and the body weight and test diameter of M. nudus was performed. The ASVs with an absolute correlation coefficient greater than 0.6 were defined as bacterial communities that correlated with sea urchin growth.
Individuals of M. nudus with high growth rates and low growth rates were selected, and the DNAs from fecal samples at both 4th and 6th weeks of rearing were metagenomically sequenced. The shotgun metagenome sequencing was performed on the HiSeq platform by Hokkaido System Science, Co., Ltd., Sapporo, Japan. The DNA quality and quantity were estimated using NanoDrop, Qubit Fluorometer, and Agilent 2200 TapeStatin System. Using TruSeq Nano DNA LT Sample Prep Kit, genomic DNA was fragment and insert DNA of 350 bp was selected and connected adaptor sequences. Sixty nanograms of each DNA sample were performed on the HiSeq platform. Sequences showing low fluorescence purity were removed.
The FASTQ format sequence data set was uploaded to MG-RAST server version 3.5. For QC, after dereplication was performed using the DRISEE method, low quality (25 < QV) sequences were removed with Dynamic Trim. The encoded gene region of protein/rRNA was predicted using reads that passed QC according to the “FragGeneScan” algorithm. The predicted proteins were clustered (similarity ≥ 90%) by BLAST analysis using UCLUST. Sequence assignment was performed in MG-RAST server under conditions excluding the following: expected value ≤ 1 × 10–5, minimum identity ≤ 60%, base pair ≤ 15, amino acid minimum alignment. Taxonomic annotation was performed using the NCBI database, and the functional annotation was performed using the SEED database (Overbeek et al., 2014). Post-assignment metagenomic sequence data was analyzed using MG-RAST tools and STAMP v2.0.9 software (Parks et al., 2014). To test if the abundance of each functional feature of the high and low growth sea urchin differed, Fisher’s exact test with Newcombe-Wilson confidence interval calculation method and Storey’s FDR for multiple test correction method was used with STAMP software. p < 0.05 was considered statistically significant. To visualize metagenomic results, profile bar plots and extended error bar plots were generated at Subsystem Levels 1 and 3. To plot data, results were filtered by q-value (< 0.05) and effect size (> 0.1 or > 0.2) (Yadav et al., 2015). To investigate the taxonomic affiliation of the significantly different functional features between the high and low growth sea urchin, we used the MG-RAST workbench tool implementing KEGG Orthology and GenBank database to annotate with functional features.
Results
Total Sequence Reads, Quality Trimming and Amplicon Sequence Variant Designation
Meta16S Sequence
In Exp. I, a total of 239,928 raw reads from 17 samples [M. nudus (n = 6), S. intermedius (n = 8) and rearing seawater (n = 3)] were produced (Supplementary Table 1). After QC, eukaryotic-derived reads were removed from these subjects to obtain 153,084 reads (131,198 reads and 21,886 reads from sea urchin and seawater samples, respectively). The total number of ASVs obtained from 17 samples was 1,250.
From eight wild sea urchin samples, we obtained a total of 276,858 raw reads (M. nudus n = 4, S. intermedius n = 4). After trimmed and removed eukaryotic reads, 96,332 qualified reads from the sea urchin samples (31,857 and 64,475 reads from M. nudus and S. intermedius) were produced. A total of 147,224, and 309 ASVs from M. nudus, S. intermedius, and wild animals have been observed (Supplementary Table 2).
In Exp. II, a total of 1,223,407 raw reads were obtained from a total of 36 samples [M. nudus samples (n = 32) and rearing seawater (n = 4)]. After QC, reads derived from eukaryotic microorganisms were removed, and 880,046 reads (775,774 reads and 104,272 reads, respectively, from sea urchin and seawater samples) were obtained. The total number of ASVs obtained from 36 samples were 1,613 (Supplementary Table 2).
Shotgun Metagenome Sequence
From the high growth individuals reared, 4,435,332 and 2,268,127 reads were obtained at the 4th and 6th weeks, respectively. After quality filtering, 3,848,669 reads (4th week) and 2,188,052 (6th week) were used for MG-RAST annotation, and a total of 3,329,288 and 1,707,418 reads were annotated from the high growth individuals reared for each sample. From the low growth individuals reared, 3,346,079 and 3,192,106 reads were obtained at the 4th and 6th weeks, respectively. After Quality filtering, the remaining 3,254,461 reads (4th week) and 3,117,838 (6th week) were used for MG-RAST annotation. A total of 2,560,781 and 2,306,340 were annotated from the low growth individuals at the 4th and 6th weeks, respectively (Supplementary Table 3).
Overall Structure of Gut Microbiome of Sea Urchins Mesocentrotus nudus and Strongylocentrotus intermedius in Japan
Compared to previously reported data for American green sea urchin (Lytechinus variegatus) (Hakim et al., 2015, 2016) and purple sea urchin (Strongylocentrotus purpuratus) (Hakim et al., 2019) gut microbiome, we did not find any apparent differences in average dominance of sea urchin microbiome in Japan from those of American species green and purple sea urchins at phylum-level, which consisted of more than 80% Proteobacteria and Bacteroidetes (Figure 1). However, relative abundances of Proteobacteria and Bacteroidetes fluctuated (more than 15% standard deviation) in relation to each other depend on rearing conditions those of Proteobacteria decreased in M. nudus under circulated water aquarium, and those of Bacteroidetes showed the opposite results (Figure 1A and Supplementary Figure 2).
FIGURE 1
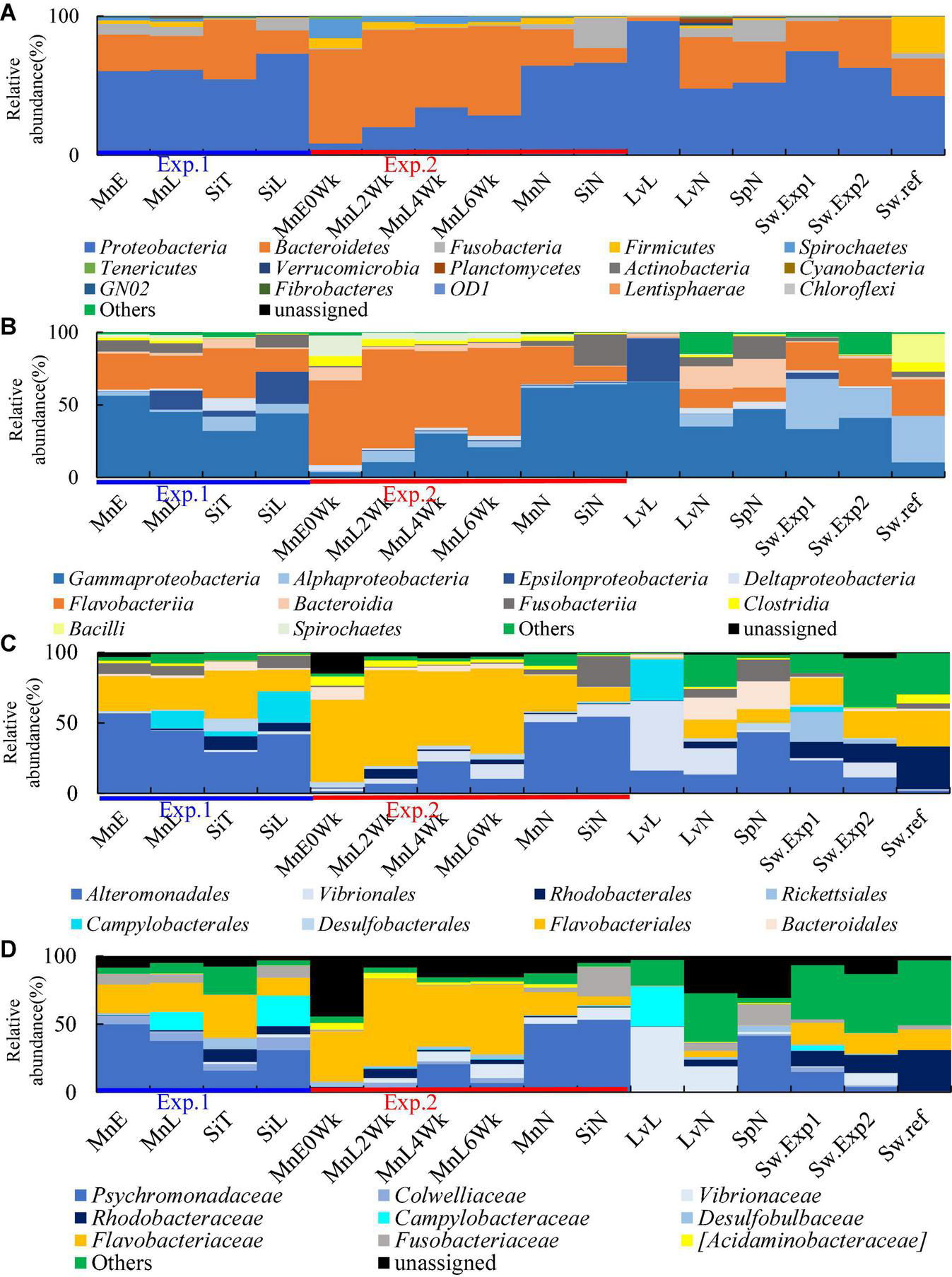
Average microbial community structure of sea urchin gut microbiome. (A) Phylum (B) Class. (C) Order. (D) Family. Mn, M. nudus; Si, S. intermedius; Lv, L. variegatus; Sp, S. purpuratus; E, the Esan Seedling Center; T, the Toi Seedling Center; N, Natural; L, Laboratory; 0Wk, Zero week; 2Wk, two week; 4Wk, four week; 6Wk, six week; Sw.Exp1, Seawater Experiment 1; Sw.Exp2, Seawater Exp. 2; Sw.ref, Seawater reference.
Key members of each sea urchin microbiome could be more clearly defined below class level at the family level (Figures 1B–D, 2 and Supplementary Figures 3, 4). Families Psychromonadaceae and Flavobacteriaceae dominated 38–71% on sea urchins in Japan under all rearing conditions. Meanwhile, these two families dominated between 60 and 67% in wild sea urchins, whereas in seawater around 15–31%. The species of S. purpuratus and L. variegatus were dominated by 41% and less than 5.1% of Psychromonadaceae and Flavobacteriaceae, respectively. Only Psychromonadaceae was present in wild S. purpuratus. The relative abundances of families Campylobacteraceae and Vibrionaceae were affected by rearing conditions. Campylobacteraceae abundances increased in laboratory aquarium conditions in two species of sea urchins in Japan and L. variegatus. Vibrionaceae abundances increased in M. nudus in the circulated water aquarium and L. variegatus in the laboratory aquarium. Abundance of Rhodobacteraceae (around 5–10%) was rather high in S. intermedius both in wild and laboratory reared individuals. Fusobacteriaceae was found in small quantities and was detected 2– 20% in all sea urchin species and under all rearing conditions (Figure 1D).
FIGURE 2
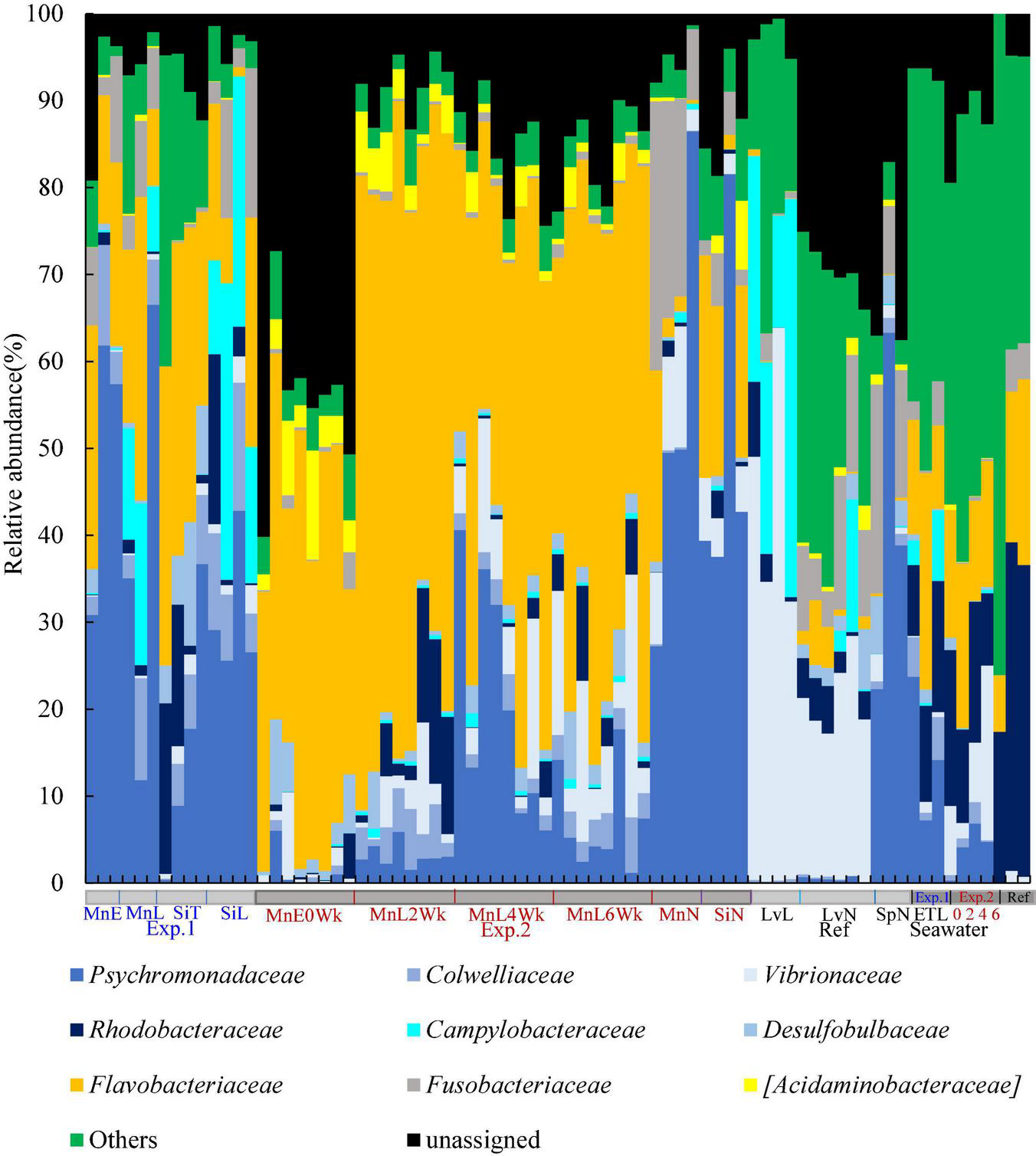
Family-level individual microbial community structure of sea urchin gut microbiome. Mn, M. nudus; Si, S. intermedius; Lv, L. variegatus; Sp, S. purpuratus; E, the Esan Seedling Center; T, the Toi Seedling Center; N, Natural; L, Laboratory; 0Wk, Zero week; 2Wk, two week; 4Wk, four week; 6Wk, six week; Sw.Exp1, Seawater Experiment 1; Sw.Exp2, Seawater Exp. 2; Sw.ref, Seawater reference.
The gut microbiome of S. intermedius fed microalgae on the farm resembled those of M. nudus fed kelp thallus both in the farm and laboratory (Figures 1, 3). In detail, dominant microbial taxa (>5%) of the gut microbiome of S. intermedius in the farm, laboratory, and in wild conditions consisted of Psychromonadaceae (16, 31, and 53%, respectively), Flavobacteriaceae (32, 13, and 7%), Rhodobacteraceae (10, 6, and 1%), and Colwelliaceae (5, 9, and 0.2%). Fusobacteriaceae, Campylobacteraceae, and Vibrionaceae fluctuated more in the S. intermedius gut microbiome. Similarly, those of the gut microbiome of M. nudus mainly consisted of Psychromonadaceae (38–50%), Flavobacteriaceae (17–21%), and Fusobacteriaceae (4–8%). As mentioned above, Psychromonadaceae/Flavobacteriaceae/Vibrionaceae ratio dynamically fluctuated in M. nudus in a recirculating-water system; 1:41:0.4 at the start of rearing were 1:7.3:1.4 at the end of 6th week rearing.
FIGURE 3
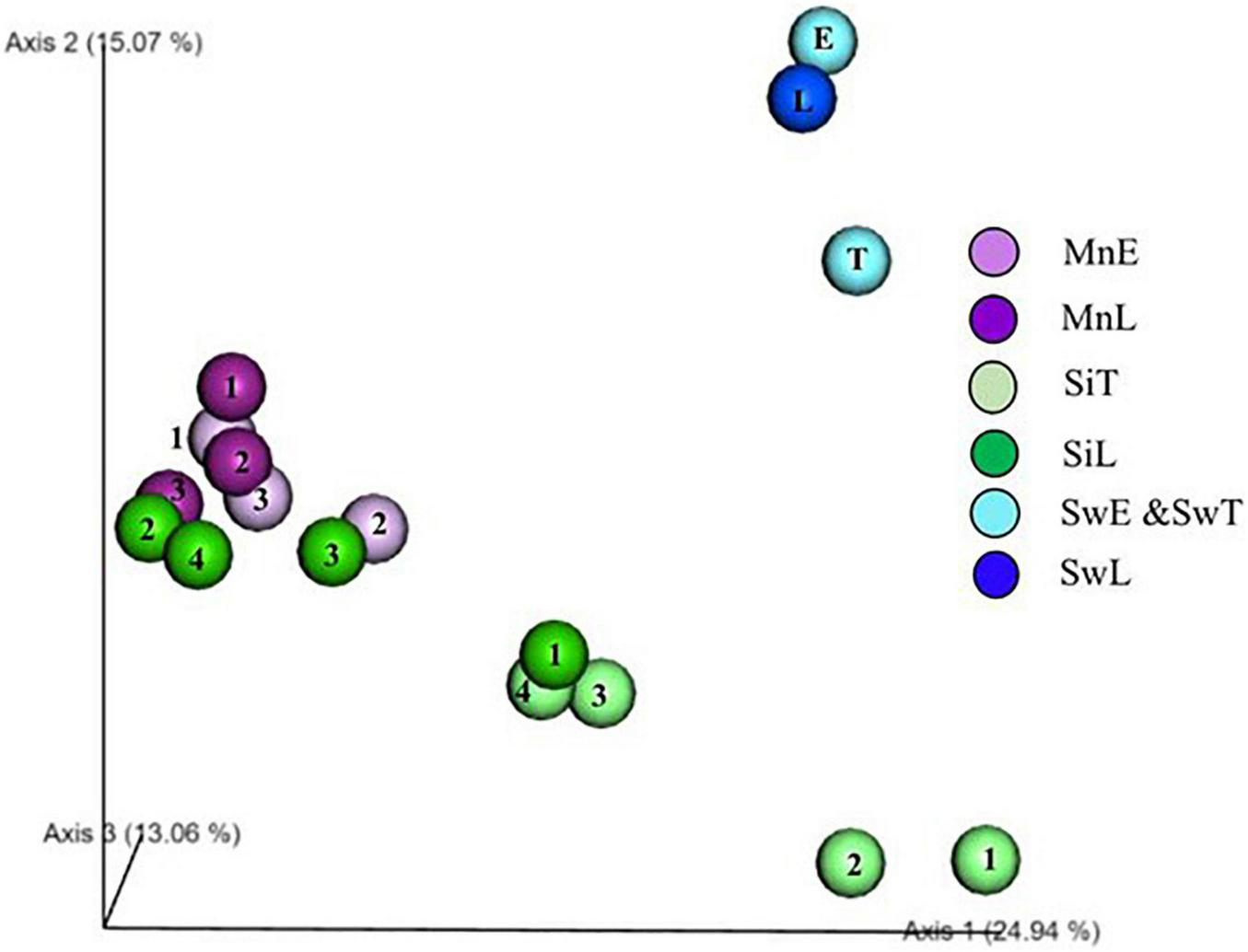
PCoA plot of sea urchin microbiome. Unweighted Unifrac 3D PCoA plot. The numbers (1, 2, 3, 4) in symbol indicates individual number of sea urchin samples. Mn, M. nudus; Si, S. intermedius; E, the Esan Seedling Center; T, the Toi Seedling Center; L, Laboratory; Sw, Seawater.
Factors Affecting Sea Urchin Gut Microbiome
Two sea urchins of different species were transplanted to a laboratory aquarium from farms to assess what factors, e.g., diets, rearing environments, and host species affect the shaping of the gut microbiomes of sea urchin in Exp. 1. PCoA plot based on unweighted UniFrac revealed a grouping of both gut microbiomes of M. nudus in farm and laboratory after 2 week feeding trials. Three of four individual gut microbiomes in the laboratory reared S. intermedius were shifted to the group of M. nudus, and the other one was likely to be closer to those of M. nudus (Figure 3).
Amplicon Sequence Variants Correlated to Hosts’ Body Weight and Shell Length
A total of seven ASVs were found in the gut microbiota of sea urchins reared in a circulating aquarium in Exp. 2, showing a positive correlation with host test diameter and body weight (Figure 4 and Table 1). The five ASVs identified correlated with the body weight of sea urchin (r > 0.6) with statistical significance (p < 0.05) (Figures 4A,B,D, and Table 1). Three of the five ASVs (BW1, BW2, and BW4) with a host weight gain and correlation coefficient of 0.6 or higher and a significant correlation coefficient (p < 0.05) were closest to Saccharicrinis fermentans sequence with 91.2–91.5% similarities (Table 1). The other two ASVs (BW3 and BW5) had the sequences closet to Kiritimatiellaeota bacterium S-5,007 (89.7% similarity) and Saccharicrinis carchari (88.2% similarity), respectively. Moreover, two ASVs correlated with test diameter (DM1, ASV had positive and DM2, ASV negative correlation) (r > 0.6, r < –0.6) with statistical significance (p < 0.05). One ASV (DM1) showed a positive correlation had the sequence closest to S. fermentans sequences (91.5% similarity) (Table 1). One ASV (DM2) showed a negative correlation with the sequences closest to Flaviramulus ichthrvoenteri sequence (88.6% similarity).
FIGURE 4
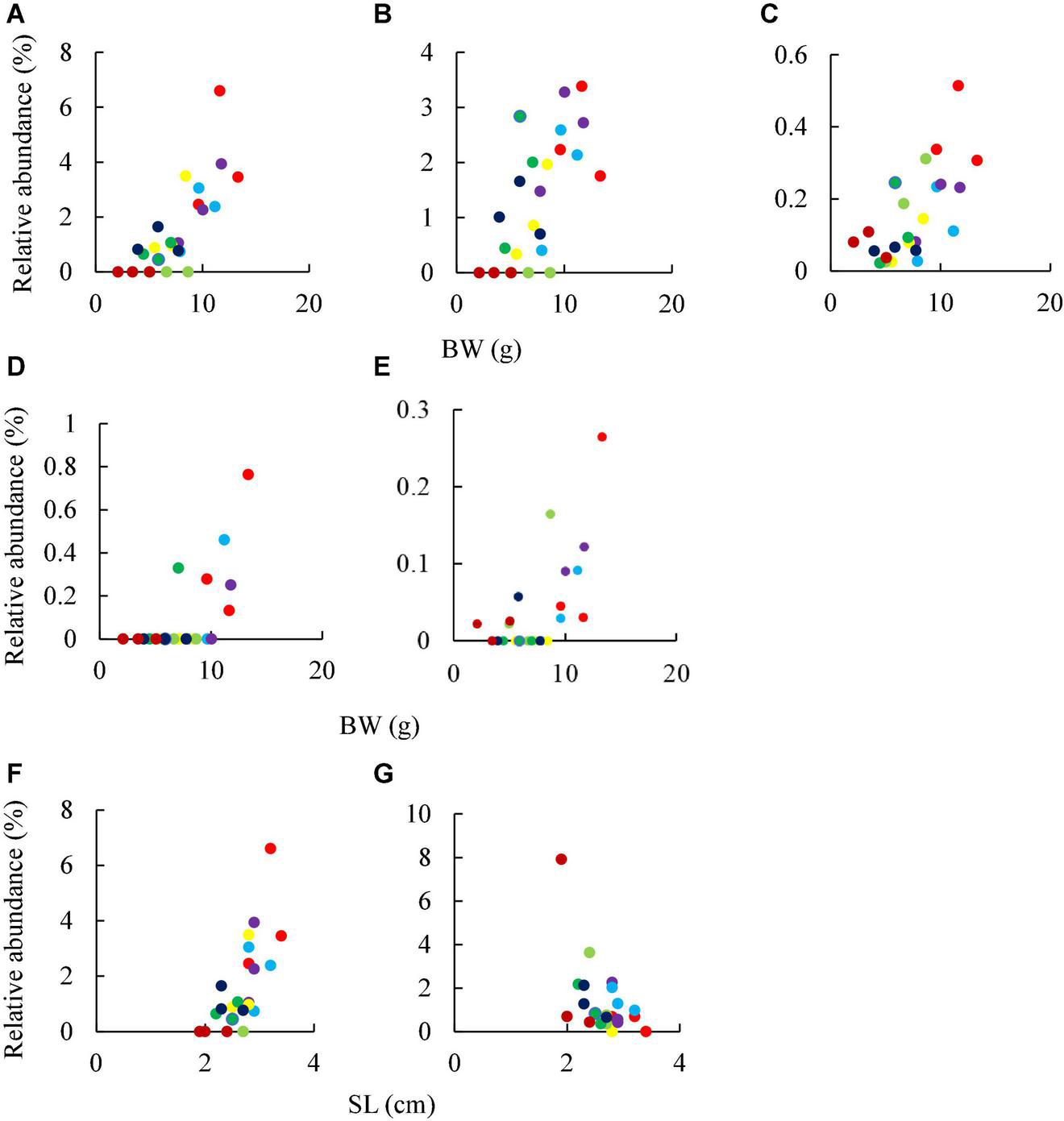
Correlation between ASV abundance and host growth in Exp. 2. Same colors showed the data collected from same samples number at different time point. BW, hosts’ bodyweight; SL, hosts’ diameter. (A) BW1, (B) BW2, (C) BW3, (D) BW4, (E) BW5, (F) DM1, and (G) DM2, see Table 1 for details. Sample numbers:  1,
1,  2,
2,  3,
3,  4,
4,  5,
5,  6,
6,  7, and
7, and  8.
8.
TABLE 1
| ASV ID | ID | Closest taxa | Identity (%) of the 16S rRNA gene sequences | Correlation | p-value |
| 7ad7a993096d762eea0f11b6e9b12697 | BW1 | Saccharicrinis fermentans | 91.5 | 0.768726 | 1.14E–05 |
| 45adbed4e7caeb4864617a35df730e45 | BW2 | Saccharicrinis fermentans | 91.2 | 0.668635 | 0.000355 |
| 0acddd2c18bc6b015bf0f83dab2bb20b | BW3 | Kiritimatiellaeota bacterium S-5,007 | 89.7 | 0.66822 | 0.000359 |
| 05b07909be6b43512e1d49e488c795df | BW4 | Saccharicrinis fermentans | 91.2 | 0.643274 | 0.000697 |
| c7395fe383e772bb5df6a28ae6e900b1 | BW5 | Saccharicrinis carchari | 88.2 | 0.641202 | 0.000734 |
| 7ad7a993096d762eea0f11b6e9b12697 | DM1 | Saccharicrinis fermentans | 91.5 | 0.628922 | 0.000995 |
| 1292c35cfbc3711fd999120ffa6f9a69 | DM2 | Flaviramulus ichthyoenteri | 88.6 | –0.68511 | 0.000221 |
ASVs correlated to growth of the sea urchin M. nudus (Exp. 2).
Functional Analysis of Sea Urchin Gut Microbiome
Shotgun metagenome analysis of gene function subsystem level 1 (Supplementary Figure 5) and subsystem level 3 (Figure 5) between each functional genes showed significantly different proportion at both 4th and 6th week. The comparison of functional genes between bacteria with higher and lower growth rates found abundance differences in more than 34 functional genes (Supplementary Table 4). The two highest genes occupied more than 15% on the relative proportion on functional genes level were a nitrite reductase [NAD(P)H] large subunit (EC 1.7.1.4) and periplasmic nitrate reductase precursor (EC 1.7.99.4), that affiliated to “nitrate and nitrite ammonification.” We observed a rising trend in bacterial abundance between the 4th and 6th weeks (Figure 5), which correlates with the existence of potential genes involved in the growth of the sea urchin M. nudus. These potential genes were identified as belonging to the order Vibrionales.
FIGURE 5
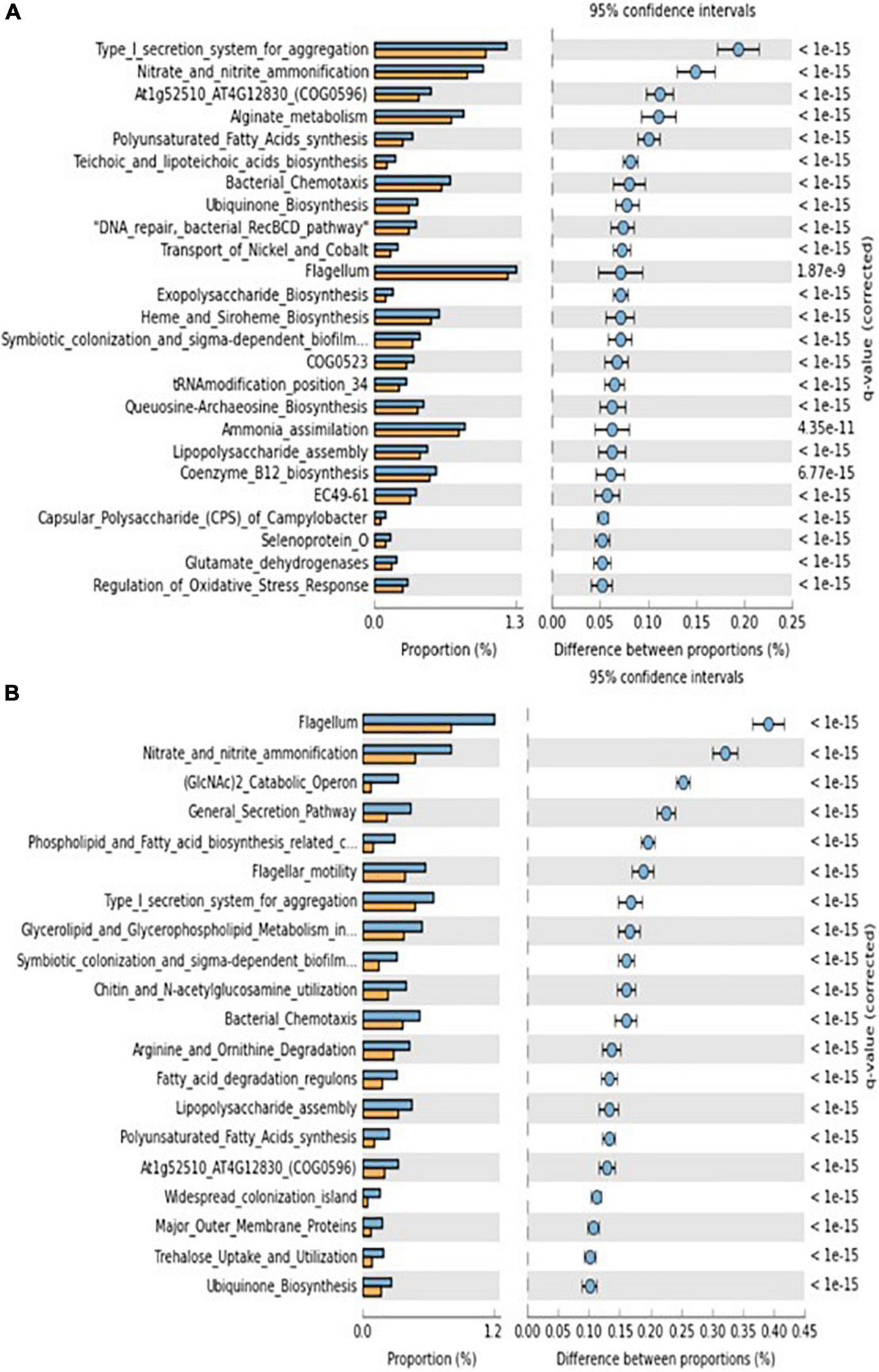
Metagenome gene functions (subsystem level 3) significantly abundant at individuals showing higher lower growth rates. (A) 4 weeks and (B) 6 weeks. Blue and orange bars indicate higher and lower grown sea urchin individuals, respectively.
Discussion
Landmark findings on the reproductive physiology, development, and body structure of sea urchins are available (Ernst, 1997; Sodergren et al., 2006; Lawrence, 2013), however, knowledge on the structure and function of the gut microbiome, which is likely to influence host physiology and development, is extremely limited. N2-fixation (Guerinot and Patriquin, 1981) and amino acid supply (Fong and Mann, 1980) reported in 1980s were the most recent representative findings on the functional aspects of the sea urchin microbiomes using classical feeding experiments. Gut microbial community studies of sea urchin have recently re-emerged using next-generation sequencing technology in contributing to accumulated knowledge; (1) the microbiome of purple sea urchin in European Paracentrotus lividus differs in each part of the gut tract (Meziti et al., 2007), (2) the gut microbiota of the American species of green sea urchin L. variegatus may complement host metabolism (Hakim et al., 2016), (3) the gut microbiome of the European species of purple sea urchin P. lividus inhibits the growth of microbes introduced from the environment and may play an important role in gut microbiota homeostasis (Laport et al., 2018), (4) the diversity and function of the gut microbiome of the American species of purple sea urchin may be reduced due to the increase in seawater temperature associated with global warming (Brothers et al., 2018), (5) geographical factors may have a more significant influence than diet on the formation of the symbiotic microbiome of larvae of the humpback sea urchin (Carrier et al., 2019), and 6) the possibility that sea urchin in the family Shizadteridae continue to have a symbiotic relationship with common gut bacteria even before species divergence (Ziegler et al., 2020). However, our understanding of the structure and function of the gut microbiota in sea urchin in Japan, such as M. nudus and S. intermedius, is behind other sea urchin species. It is necessary to expand our knowledge for further innovation of effective seed production technology for these sea urchin species in Japan. Even in marine invertebrates, fine-scale gut microbiome studies using individual microbiome approaches, which can monitor microbiome changes structurally and functionally without sacrificing individual animals, have revealed unexpected contributions of the gut microbiome to host growth and physiology of sea cucumber, which belong in the same phylum as sea urchin (Yamazaki et al., 2016). Therefore, for the first time we applied these individual microbiome approaches to expand our knowledge in finding new associations between the sea urchin host and its gut microbiomes.
The increase of Vibrionaceae and Campylobacteriaceae, in more detail, genus Vibrio and Arcobacter, respectively, was observed in both M. nudus and S. intermedius after rearing in running water more than 2 weeks. Increases of these taxa have been reported in studies using American green sea urchin (Hakim et al., 2015, 2016). An increase of more than 20% in Vibrionaceae and Campylobacteriaceae observed in those reared sea urchin indicates that both bacterial families may be defined as a fluctuated community in the gut microbiome of sea urchin (Hakim et al., 2015, 2016).
By comparing our findings to those of other researchers (Hakim et al., 2015, 2016; Yao et al., 2019), it was determined that seawaters had a negligible effect on the gut microbiome of sea urchins. When two distinct species of sea urchins were transferred from the farm to the laboratory settings, comparable microbiome changes were found in the sea urchin fed with the kelp S. japonicus thalli. Our research discovered that diet has a significant effect on the gut microbiota of sea urchin species. It is reasonable to conclude that the rearing environment of sea urchin and host species have minimal impact on gut microbiomes. Our study established that diet significantly influences the gut microbiota of sea urchin species. The similar phenomenon was seen in a variety of species, including humans, indicating that gut microbiota shaping mechanisms are evolutionarily conserved (e.g., Wilson et al., 2020).
Five ASVs showed strong positive correlations with body weight or test diameter in the sea urchin M. nudus. Among these ASVs, a bacterium similar to S. fermentans was affiliated. This bacterium has been reported to be capable of fermenting amygdalin, glucose, mannitol, and sucrose (Yang et al., 2014). Since the kelp, fed in these studies, contained relatively high amounts of mannitol, it may contribute to the growth of the host through fermentation of the diet ingredients. In addition, S. fermentans is also known to possess a N2-fixing ability (Inoue et al., 2015). Guerinot and Patriquin (1981) reported the possibility that N2-fixing bacteria exist in the digestive tract of sea urchins S. droebachiensis and play a role in supplying nitrogen sources to the host. Therefore, it is possible that Saccharicrinis- like bacteria also contribute to the growth of sea urchins M. nudus and S. intermedius through their N2-fixing ability.
The number of functional genes involved in “nitrate and nitrite ammonification” was significantly higher in high-growth individuals at subsystem level 3 when sea urchin reared in a circulating aquarium. “Nitrate and nitrite ammonification” is also called dissimilatory nitrate reduction to ammonification (DNRA), a reaction in which nitrate is converted to ammonium under anaerobic conditions. The genes nrfE, nrfF, and nrfG are required to activate cytochrome c nitrite reductase, NrfA, in Escherichia coli (Eaves et al., 1998). One of the functions of NrfA is to produce ammonium in the periplasm, which is transported to the cytoplasm where it is converted to glutamine by an amino acid, for assimilation (Einsle, 2011). The utilization of ammonium produced by bacteria has been reported mainly in plants. In addition to this, recent studies on nitrogen dynamics in the digestive tract of aquatic insects have shown that there may be a pathway by which gut microorganisms take up nitrate-nitrogen from the host digestive tract and convert it to ammonium through DNRA for utilization by the host (Ayayee et al., 2019). Nitrate accumulation is commonly observed in a closed aquarium system, and the concentration reaches to 10 time higher than those determined in the ocean (e.g., Kim et al., 2017). Unfortunately, we did not determine the nitrate concentrations in this study, nitrate accumulation in the circulating aquarium might trigger the dynamics of DNRA by the gut microbiome of sea urchin. We might discover a veiled nitrogen metabolism through the gut microbiome of sea urchin, which could contribute host growths under those aquaculture conditions. Fong and Mann (1980) suggested the existence of gut microorganisms that supply amino acids to sea urchins. Although many studies have shown that N2-fixing bacteria exist in the gut microbiota of sea urchin and that they may play a role in supplying nitrogen source to the host, few studies have addressed the possibility that other nitrogen cycling pathways may play a role in supplying nitrogen source to sea urchin host. The presence of DNRA-related respiratory cytochrome c nitrite reductase has been reported in several organisms, including E. coli and Vibrio fischeri, and it is likely that Vibrionales, which was found to be a candidate for the nrfEFG gene, also possesses it. Vibrionales, which were found as a candidate with the nrfEFG gene, are also likely to have the gene. In the future, it will be necessary to isolate gut microorganisms and clarify their nitrogen metabolisms that contribute to the nitrogen supply to the host, with a view to their application as probiotics in fisheries.
Conclusion
Overall, Psychromonadaceae and Flavobacteriaceae dominated in gut microbiomes of two species of sea urchins in Japan, M. nudus and S. intermedius. Diets rather than rearing environments and host species is the most potent factor structuring gut microbiomes of the sea urchins. S. fermentans was identified as ASV showing a positive correlation with host shell length and body weight in M. nudus, suggesting a link in the symbiotic association through nitrogen fixation. Vibrionales possessing genes responsible for nitrate and nitrite ammonification pathway also correlated to the growth of sea urchin species in Japan. Our study showed M. nudus and S. intermedius could be excellent candidates for studying marine invertebrates’ structure and function relationships. Further studies in the utilization of gut microbiomes of sea urchin could allow us to develop sustainable technology of fisheries resources in a changing world.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The 16S rRNA and metagenome data sets generated in this study were deposited in DDBJ/GenBank/ENA database under BioProject accession number PRJDB12341.
Author contributions
MY, JY, SM, and YS did molecular biology experiments and data analysis. MY, JY, and YS collected samples. JY, SM, and TS conceived of the study and coordinated the experiments and data analysis. AH, MY, SM, and TS mainly wrote this manuscript. All authors read and approved the final manuscript.
Funding
The studies were supported by KAKEN 19K22262.
Acknowledgments
We thank Toi Seedling Center and Esan Seedling Center for sample collection. We also thank to Ms. Kodama, Mr. Y. Nomura, and Dr. Y. Yamazaki, Hokkaido University, for technical assistant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.802754/full#supplementary-material
References
1
Agatsuma Y. (2013). “Hemicentrotus pulcherrimus, Pseudocentrotus depressus, and Heliocidaris crassispina,” in Sea Urchins: Biology and Ecology, ed.LawrenceJ. M. (San Diego, CA: Academic Press), 461–473. 10.1016/b978-0-12-396491-5.00030-7
2
Ayayee P. Bhattacharyya S. Arnold T. Werne J. Leff L. (2019). Experimental Investigation of Potential Biological Nitrogen Provisioning by Freshwater Insect Gut Microbiomes Using 15N Isotope Analysis. Available online at: https://www.preprints.org/manuscript/201908.0034/v1(accessed August 5, 2019).
3
Bokulich N. A. Kaehler B. D. Rideout J. R. Dillon M. Bolyen E. Knight R. et al (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin.Microbiome6:90. 10.1186/s40168-018-0470-z
4
Bolyen E. Rideout J. R. Dillon M. R. Bokulich N. A. Abnet C. C. Al-Ghalith G. A. et al (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2.Nat. Biotechnol.37852–857. 10.1038/s41587-019-0209-9
5
Brothers C. J. Van Der Pol W. J. Morrow C. D. Hakim J. A. Koo H. McClintock J. B. (2018). Ocean warming alters predicted microbiome functionality in a common sea urchin.Proc. R. Soc. B Biol. Sci.285:20180340. 10.1098/rspb.2018.0340
6
Brown K. E. King C. K. Harrison P. L. (2020). Impacts of petroleum fuels on fertilization and development of the Antarctic sea urchin Sterechinus neumayeri.Environ. Toxicol. Chem.39:2527–2539. 10.1002/etc.4878
7
Callahan B. J. McMurdie P. J. Rosen M. J. Han A. W. Johnson A. J. A. Holmes S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data.Nat. Methods13581–583. 10.1038/nmeth.3869
8
Carrier T. J. Dupont S. Reitzel A. M. (2019). Geographic location and food availability offer differing levels of influence on the bacterial communities associated with larval sea urchins.FEMS Microbiol. Ecol.95:fiz103. 10.1093/femsec/fiz103
9
Cui Z. Zhang J. Sun Z. Liu B. Han Y. Zhao C. et al (2022). Testis-specific expression pattern of dmrt1 and its putative regulatory region in the sea urchin (Mesocentrotus nudus).Comp. Biochem. Physiol. B Biochem. Mol. Biol.257:110668. 10.1016/j.cbpb.2021.110668
10
Ding J. Zheng D. Sun J. Hu F. Yu Y. Zhao C. et al (2020). Effects of water temperature on survival, behaviors and growth of the sea urchin Mesocentrotus nudus: new insights into the stock enhancement.Aquaculture519301–308. 10.1016/j.aquaculture.2019.734873
11
Dworjanyn S. A. Byrne M. (2018). Impacts of ocean acidification on sea urchin growth across the juvenile to mature adult life-stage transition is mitigated by warming.Proc. R. Soc. B Biol. Sci.285:20172684. 10.1098/rspb.2017.2684
12
Eaves D. J. Grove J. Staudenmann W. James P. Poole R. K. White S. A. et al (1998). Involvement of products of the nrfEFG genes in the covalent attachment of haem c to a novel cysteine–lysine motif in the cytochrome c552 nitrite reductase from Escherichia coli.Mol. Microbiol.28205–216. 10.1046/j.1365-2958.1998.00792.x
13
Einsle O. (2011). Structure and function of formate-dependent cytochrome c nitrite reductase, NrfA.Methods Enzymol.496399–422. 10.1016/B978-0-12-386489-5.00016-6
14
Ernst S. G. (1997). A century of sea urchin development.Am. Zool.37250–259. 10.1093/icb/37.3.250
15
Faddetta T. Ardizzone F. Faillaci F. Reina C. Palazzotto E. Strati F. et al (2020). Composition and geographic variation of the bacterial microbiota associated with the coelomic fluid of the sea urchin Paracentrotus lividus.Sci. Rep.10:21443. 10.1038/s41598-020-78534-5
16
Fan Y. Pedersen O. (2021). Gut microbiota in human metabolic health and disease.Nat. Rev. Microbiol.1955–71. 10.1038/s41579-020-0433-9
17
Fong W. Li Q. Yu J. (2020). Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer.Oncogene394925–4943. 10.1038/s41388-020-1341-1
18
Fong W. Mann K. H. (1980). Role of gut flora in the transfer of amino acids through a marine food chain.Can. J. Fish. Aquat. Sci.37:e100092. 10.1139/f80-009
19
Formery L. Orange F. Formery A. Yaguchi S. Lowe C. J. Schubert M. et al (2021). Neural anatomy of echinoid early juveniles and comparison of nervous system organization in echinoderms.J. Comp. Neurol.5291135–1156. 10.1002/cne.25012
20
García E. Hernández J. C. Clemente S. (2018). Robustness of larval development of intertidal sea urchin species to simulated ocean warming and acidification.Mar. Environ. Res.13935–45. 10.1016/j.marenvres.2018.04.011
21
Guerinot M. L. Patriquin D. G. (1981). The association of N2-fixing bacteria with sea urchins.Mar. Biol.62197–207. 10.1007/BF00388183
22
Hakim J. A. Koo H. Dennis L. N. Kumar R. Ptacek T. Morrow C. D. et al (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus.Front. Microbiol.6:1047. 10.3389/fmicb.2015.01047
23
Hakim J. A. Koo H. Kumar R. Lefkowitz E. J. Morrow C. D. Powell M. L. et al (2016). The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles.FEMS Microbiol. Ecol.92:fiw146. 10.1093/femsec/fiw146
24
Hakim J. A. Schram J. B. Galloway A. W. Morrow C. D. Crowley M. R. Watts S. A. et al (2019). The purple sea urchin Strongylocentrotus purpuratus demonstrates a compartmentalization of gut bacterial microbiota, predictive functional attributes, and taxonomic co-occurrence.Microorganisms7:35. 10.3390/microorganisms7020035
25
Holm S. (1979). A simple sequentially rejective multiple test procedure.Scand. J. Stat.665–70.
26
Houlihan E. P. Espinel-Velasco N. Cornwall C. E. Pilditch C. A. Lamare M. D. (2020). Diffusive boundary layers and ocean acidification: implications for sea urchin settlement and growth.Front. Mar. Sci.7:972. 10.3389/fmars.2020.577562
27
Inoue J. I. Oshima K. Suda W. Sakamoto M. Iino T. Noda S. et al (2015). Distribution and evolution of nitrogen fixation genes in the phylum Bacteroidetes.Microbes Environ.3044–50. 10.1264/jsme2.ME14142
28
Kim Y. Bonn W. V. Aw T. G. Rose J. B. (2017). Aquarium viromes: viromes of human-managed aquatic systems.Front. Microbiol.8:1231. 10.3389/fmicb.2017.01231
29
Kinjo S. Kiyomoto M. Yamamoto T. Ikeo K. Yaguchi S. (2018). HpBase: a genome database of a sea urchin, Hemicentrotus pulcherrimus.Dev. Growth Differ.60174–182. 10.1111/dgd.12429
30
Kudtarkar P. Cameron R. A. (2017). Echinobase: an expanding resource for echinoderm genomic information.Database2017:bax074. 10.1093/database/bax074
31
Laport M. S. Bauwens M. Collard M. George I. (2018). Phylogeny and antagonistic activities of culturable bacteria associated with the gut microbiota of the sea urchin (Paracentrotus lividus).Curr. Microbiol.75359–367. 10.1007/s00284-017-1389-5
32
Lawrence J. M. (2013). Sea Urchins: Biology and Ecology.San Diego, CA: Academic Press.
33
Li A. Espinoza J. Hamdoun A. (2020). Inhibitory effects of neurotoxin β-N-methylamino-L-alanine on fertilization and early development of the sea urchin Lytechinus pictus.Aquat. Toxicol.221:105425. 10.1016/j.aquatox.2020.105425
34
Lozupone C. Lladser M. E. Knights D. Stombaugh J. Knight R. (2011). UniFrac: an effective distance metric for microbial community comparison.ISME J.5169–172. 10.1038/ismej.2010.133
35
Mann K. H. (1977). Destruction of kelp-beds by sea-urchins: a cyclical phenomenon or irreversible degradation?Helgoländer Meeresunters30455–467. 10.1007/BF02207854
36
Martín-Durán J. M. Hejnol A. (2021). A developmental perspective on the evolution of the nervous system.Dev. Biol.475181–192. 10.1016/j.ydbio.2019.10.003
37
Meziti A. Kormas K. A. Pancucci-Papadopoulou M. A. Thessalou-Legaki M. (2007). Bacterial phylotypes associated with the digestive tract of the sea urchin Paracentrotus lividus and the ascidian Microcosmus sp.Russ. J. Mar. Biol.3384–91. 10.1134/S1063074007020022
38
Miller P. M. Lamy T. Page H. M. Miller R. J. (2021). Sea urchin microbiomes vary with habitat and resource availability.Limnol. Oceanogr. Lett.6119–126. 10.1002/lol2.10189
39
Morais L. H. Schreiber H. L. Mazmanian S. K. (2021). The gut microbiota–brain axis in behaviour and brain disorders.Nat. Rev. Microbiol.19241–255. 10.1038/s41579-020-00460-0
40
Nobre C. R. Santana M. F. M. Maluf A. Cortez F. S. Cesar A. Pereira C. D. S. et al (2015). Assessment of microplastic toxicity to embryonic development of the sea urchin Lytechinus variegatus (Echinodermata: Echinoidea).Mar. Pollut. Bull.9299–104. 10.1016/j.marpolbul.2014.12.050
41
Overbeek R. Olson R. Pusch G. D. Olsen G. J. Davis J. J. Disz T. et al (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST).Nucleic Acids Res.42D206–D214. 10.1093/nar/gkt1226
42
Parks D. H. Tyson G. W. Hugenholtz P. Beiko R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles.Bioinformatics303123–3124. 10.1093/bioinformatics/btu494
43
Pikula K. Zakharenko A. Chaika V. Em I. Nikitina A. Avtomonov E. et al (2020). Toxicity of carbon, silicon, and metal-based nanoparticles to sea urchin Strongylocentrotus intermedius.Nanomaterials10:1825. 10.3390/nano10091825
44
Rendell-Bhatti F. Paganos P. Pouch A. Mitchell C. D’Aniello S. Godley B. J. et al (2021). Developmental toxicity of plastic leachates on the sea urchin Paracentrotus lividus.Environ. Pollut.269:115744. 10.1016/j.envpol.2020.115744
45
Sakai Y. Tajima K. I. Agatsuma Y. (2003). “Mass production of seed of the Japanese edible sea urchins Strongylocentrotus intermedius and Strongylocentrotus nudus,” in Proceedings of the International Conference on Sea-Urchin Fisheries and Aquaculture, eds. J. M. Lawrence and O. Guzmán (Lancaster, PA: DEStech Publication), 289–298.
46
Sharma S. Awasthi A. Singh S. (2019). Altered gut microbiota and intestinal permeability in Parkinson’s disease: pathological highlight to management.Neurosci. Lett.712:134516. 10.1016/j.neulet.2019.134516
47
Sodergren E. Weinstock G. M. Davidson E. H. Cameron R. A. Gibbs R. A. Angerer R. C. et al (2006). The genome of the sea urchin Strongylocentrotus purpuratus.Science314941–952. 10.1126/science.1133609
48
Stefánsson G. Kristinsson H. Ziemer N. Hannon C. James P. (2017). Markets for sea urchins: A Review of Global Supply and Markets.Reykjavik: Matís.
49
Takagi S. Murata Y. Inomata E. Aoki M. N. Agatsuma Y. (2019). Production of high quality gonads in the sea urchin Mesocentrotus nudus (A. Agassiz, 1864) from a barren by feeding on the kelp Saccharina japonica at the late sporophyte stage.J. Appl. Phycol.314037–4048. 10.1007/s10811-019-01895-6
50
Tang W. Meng Z. Li N. Liu Y. Li L. Chen D. et al (2021). Roles of gut microbiota in the regulation of hippocampal plasticity, inflammation, and hippocampus-dependent behaviors.Front. Cell. Infect. Microbiol.10:876. 10.3389/fcimb.2020.611014
51
Warner J. F. Lord J. W. Schreiter S. A. Nesbit K. T. Hamdoun A. Lyons D. C. (2021). Chromosomal-level genome assembly of the painted sea urchin Lytechinus pictus: a genetically enabled model system for cell biology and embryonic development.Genome Biol. Evol.13:evab061. 10.1093/gbe/evab061
52
Wessel G. M. Wada Y. Yajima M. Kiyomoto M. (2021). Bindin is essential for fertilization in the sea urchin.Proc. Natl. Acad. Sci. U.S.A.118:e2109636118. 10.1073/pnas.2109636118
53
Wilson A. S. Koller K. R. Ramaboli M. C. Nesengani L. T. Ocvirk S. Chen C. et al (2020). Diet and the human gut microbiome: an international review.Dig. Dis. Sci.65723–740.
54
Wilson E. B. (1895). Archoplasm, centrosome and chromatin in the sea-urchin egg.J. Morphol.11443–478.
55
Wood N. J. Mattiello T. Rowe M. L. Ward L. Perillo M. Arnone M. I. et al (2018). Neuropeptidergic systems in pluteus larvae of the sea urchin Strongylocentrotus purpuratus: neurochemical complexity in a “simple” nervous system.Front. Endocrinol.9:628. 10.3389/fendo.2018.00628
56
Yadav V. Varshney P. Sultana S. Yadav J. Saini N. (2015). Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation.BMC Cancer15:581. 10.1186/s12885-015-1560-y
57
Yamazaki Y. Meirelles P. M. Mino S. Suda W. Oshima K. Hattori M. et al (2016). Individual Apostichopus japonicus fecal microbiome reveals a link with polyhydroxybutyrate producers in host growth gaps.Sci. Rep.6:21631. 10.1038/srep21631
58
Yang S. H. Seo H. S. Woo J. H. Oh H. M. Jang H. Lee J. H. et al (2014). Carboxylicivirga gen. nov. in the family Marinilabiliaceae with two novel species, Carboxylicivirga mesophila sp. nov. and Carboxylicivirga taeanensis sp. nov., and reclassification of Cytophaga fermentans as Saccharicrinis fermentans gen. nov., comb. nov.Int. J. Syst. Evol. Microbiol.641351–1358. 10.1099/ijs.0.053462-0
59
Yao Q. Yu K. Liang J. Wang Y. Hu B. Huang X. et al (2019). The composition, diversity and predictive metabolic profiles of bacteria associated with the gut digesta of five sea urchins in Luhuitou fringing reef (northern South China Sea).Front. Microbiol.10:1168. 10.3389/fmicb.2019.01168
60
Youngblut N. D. Reischer G. H. Walters W. Schuster N. Walzer C. Stalder G. et al (2019). Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades.Nat. Commun.10:2200.
61
Zhao C. Zhang L. Shi D. Ding J. Yin D. Sun J. et al (2018). Transgenerational effects of ocean warming on the sea urchin Strongylocentrotus intermedius.Ecotoxicol. Environ. Saf.151212–219. 10.1016/j.ecoenv.2018.01.014
62
Ziegler A. Gilligan A. M. Dillon J. G. Pernet B. (2020). Schizasterid heart urchins host microorganisms in a digestive symbiosis of mesozoic origin.Front. Microbiol.11:1697. 10.3389/fmicb.2020.01697
Summary
Keywords
sea urchin, gut, microbiome, Mesocentrotus nudus , Strongylocentrotus intermedius
Citation
Haditomo AHC, Yonezawa M, Yu J, Mino S, Sakai Y and Sawabe T (2021) The Structure and Function of Gut Microbiomes of Two Species of Sea Urchins, Mesocentrotus nudus and Strongylocentrotus intermedius, in Japan. Front. Mar. Sci. 8:802754. doi: 10.3389/fmars.2021.802754
Received
27 October 2021
Accepted
25 November 2021
Published
22 December 2021
Volume
8 - 2021
Edited by
Fabiano Thompson, Federal University of Rio de Janeiro, Brazil
Reviewed by
Reiji Tanaka, Mie University, Japan; Adam Michael Reitzel, University of North Carolina at Charlotte, United States
Updates
Copyright
© 2021 Haditomo, Yonezawa, Yu, Mino, Sakai and Sawabe.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoo Sawabe, sawabe@fish.hokudai.ac.jp
This article was submitted to Microbial Symbioses, a section of the journal Frontiers in Marine Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.