 Zhen Qin
Zhen Qin Qianqian Ge3†
Qianqian Ge3† Ping Liu
Ping Liu Jitao Li
Jitao Li- 1Key Laboratory of Sustainable Development of Marine Fisheries, Ministry of Agriculture and Rural Affairs, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
- 2National Experimental Teaching Demonstration Center of Aquatic Science, Shanghai Ocean University, Shanghai, China
- 3Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, China
Saline-alkaline waters are stressful environments where most aquatic animals can’t survive normally, and alkalinity is one of the key limited environmental factors. Due to strong adaptability to environment, the ridgetail white prawn Exopalaemon carinicauda is a potential good species suitable for large-scale culture in saline-alkaline waters. Exploring its alkaline adaptability mechanism will help to guide more marine crustaceans to saline-alkaline culture. In this study, an integrative analysis of the gill-specific transcriptome and proteome at 0, 12, and 36 h after alkalinity stress was performed to identify important regulators and pathways involved in alkalinity adaption of E. carinicauda. A total of 3,157 differentially expressed genes (DEGs) and 443 differentially expressed proteins (DEPs) were identified at 12 and 36 h compared with 0 h. Base on the transcriptome analysis, the Gene Ontology (GO) enriched terms were mainly related to ion transport, including “calcium-transporting ATPase activity,” “ATPase coupled ion transmembrane transporter activity,” “divalent inorganic cation transmembrane transporter activity,” etc., and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways mainly refer to the processes of endocrine system at both 12, and 36 h. Based on the proteomic analysis, KEGG pathways related to lipolysis and amino acids metabolism were significantly enriched at 12 h, and carbohydrate metabolism and immune response were significantly enriched at 36 h. There were significantly up-regulated expressions of ion transport related genes including aquaporin, carbonic anhydrase, ammonium transporter Rh type A-like, Na+/H+-exchanger, etc., as well as ion transport proteins including V-type proton ATPase 116 kDa subunit a-like isoform X1, sodium-potassium ATPase beta, vesicle associated membrane protein, etc. after alkalinity exposure, which indicating their important roles in response to alkalinity stress. The results of integrated analysis between proteome and transcriptome showed that up-regulated DEG/DEP (aldehyde dehydrogenase) was significantly enriched at 12 h and the up-regulated DEG/DEP (peptidylglycine alpha) was significantly enriched at 36 h, suggesting the two molecules may be critical in response to alkalinity change. This study reveals the first time-course, gill-specific, combined transcriptomic and proteomic profiling associated with alkalinity adaption of E. carinicauda and provides new insights into the mechanisms underlying the molecular response to alkalinity stress in shrimp.
Introduction
With the influence of climate, topography and other natural and human factors, the global soil and water salinization is increasing, which can be found in more than 100 countries on six continents (Chang and Liang, 2020). There are 244 million acres of saline-alkaline land and 114 million acres of low-lying saline-alkaline water area in China, which are widely distributed in 19 provinces, cities and autonomous regions in northwest, northeast and north China (Yao et al., 2010; Lin et al., 2013; Fan et al., 2021). Saline–alkaline waters are stressful environments in which only relatively few organisms are able to survive and breed, such as Gymnocypris przewalskii, Leuciscus waleckii, etc. (Wang et al., 2003; Dong et al., 2020; Li H. et al., 2020). Alkalinity is one of the key environmental factors in saline-alkaline water that can have direct effects on the physiological status of aquatic animals (Yao et al., 2010, 2012; González-Vera and Brown, 2017; Zhao et al., 2020). Maoxiao et al. (2018) increased saline–alkaline water from 1.22 to 45.00 mmol⋅L–1 and found that the survival rate of Sinonovacula constricta decreased significantly. When the carbonate alkalinity was 8.0 and pH was 8.2 for the long-term saline–alkaline toxicity test, S. constricta highly toxic death and growth inhibition occurred in the first month (Maoxiao et al., 2018). Through an experiment of sodium bicarbonate alkalinity stress, the gill filament of L. waleckii became fuller, the gill lamella became longer as well as the interval between lamellae became larger (Shan et al., 2020). When fish are exposed to water with high alkalinity, the surface of fish would secrete a lot of mucus, resulting in hemorrhoids bleeding and even death occur (Tiffany et al., 2009; Ouellet et al., 2013). Sun et al. (2020) reported that carbonate alkalinity can change the normal metabolism of crucian in terms of changing the osmotic pressure regulation capacity, antioxidant capacity, ammonia metabolism at low concentrations, and as the concentration of alkalinity increases, various metabolic processes are inhibited, causing chronic damage to the body (Sun et al., 2020). Therefore, exploring more species suitable for saline-alkaline aquaculture is an effective strategy for the development and utilization of saline-alkaline water.
As one of the important shrimp species, the ridgetail white prawn Exopalaemon carinicauda is widely distributed in the Yellow Sea and Bohai Sea, which occupies a key position in the field of shrimp mariculture in eastern China (Feng et al., 2019a). This species has many advantages such as rapid growth, good reproduction capabilities and strong disease resistance (Li et al., 2015). In addition, it adapts well to a wide variety of environmental conditions (salinity 3–30, pH 4.8–10.5, alkalinity: 1.4–10.0) (Ge et al., 2019). Recently, E. carinicauda has been successfully cultured and bred in the saline–alkaline ponds at Dongying City, Shandong province (approximate salinity 5–8, pH 8.5–9.5, carbonate alkalinity 1.4–8.0 mmol⋅L–1) and Cangzhou City, Hebei province (approximate salinity 10–25, pH 8.3–9.2, carbonate alkalinity 3.5–13.0 mmol⋅L–1), China, suggesting that it has a high tolerance to saline–alkaline stress (Ge et al., 2019). In view of these, E. carinicauda is a potential good species suitable for large-scale cultivation in saline-alkaline waters. Exploring its saline-alkaline adaptability mechanism will help to guide more marine economic crustaceans to saline-alkaline aquaculture.
At present, the studies on the mechanism of saline-alkaline tolerance mainly focus on the fish survived in this harsh environment. For example, Magadi tilapia (Alcolapia grahami), which inhabits in Magadi lake (pH = 10.0, salinity 18), increases tolerance to high pH in blood and tissues facilitated by high water alkalinity, furthermore it has developed unique strategies to overcome problems related to ammonia excretion. E.g., excretion of nitrogen mainly in form of urea instead of NH3/NH4+ (contrary to almost all other teleosts) (Pörtner et al., 2010). G. przewalskii have the capacity to tolerate combined high salinity and alkalinity, which is the core of the entire ecosystem in Qinghai Lake (salinity 13–15, alkalinity 26–32 mmol⋅L–1, pH 9.1–9.5) (Yao et al., 2016; Li H. et al., 2020). The previous experimental results showed that alkaline stress inhibited the ammonia excretion of the naked carp, and deal with environmental changes by antioxidant and phosphorylation stress response (Yao et al., 2016). Recently, studies on L. waleckii that inhabit Dari Lake (alkalinity 53.57 mmol⋅L–1, pH 8.25–9.6) have been performed, and findings revealed that there are substantial genome and expression changes that allow L. waleckii to cope with extreme alkaline environments (Chen et al., 2019; Dong et al., 2020), and unveiled significant expression differences in genes that encode important modulators of stress adaptation and tolerance, including carbonic anhydrases, heat shock proteins, superoxide dismutase, etc. (Xu et al., 2013; Lee et al., 2017). As for crustaceans, there are many researches on the alkalinity tolerance range and physiological change of crustacean, but few researches on the molecular mechanism of saline-alkaline tolerance. González-Vera and Brown (2017) found that alkalinity levels of 250 mg⋅L–1 as CaCO3 were lethal for Macrobrachium rosenbergii (González-Vera and Brown, 2017). Yang et al. (2004) conducted carbonate alkalinity toxicology experiments on juvenile Litopenaeus vannamei and found that the safe concentration (SC) was 2.90 mmol L–1 under the conditions of pH 7.50–8.72 and salinity 3.29–4.84 (Yang et al., 2004). By using suppressive subtractive hybridization, Yao et al. (2010, 2012) found that under high carbonate alkalinity stress, L. vannamei controlled homeostasis by increasing ion regulation, and the immune function was inhibited. Ge et al. (2019) found that three kinds of carbonic anhydrase (CA) were cloned in E. carinicauda, and CAc and CAg were proved to be important regulators under saline-alkaline stress. More information on the mechanisms of saline-alkaline tolerance in crustaceans needs to be uncovered.
Recently, RNA sequencing (RNA-seq) transcriptomics and Tandem Mass Tags (TMT) proteomics have been widely used to study gene and protein expression profiles for understanding biological processes of crustaceans in response to environmental stress (Buckley et al., 2016; Christie et al., 2017). Thus in this study, we employed RNA-Seq and TMT technics to reveal the transcriptome and proteomics differences of E. carinicauda at different alkalinity stress time. The genes, proteins and pathways responding to alkaline stress were analyzed comprehensively. The discoveries of this study will be contributed to illustrate the molecular mechanism of crustaceans adapting to saline-alkaline stress.
Materials and Methods
Sample Collection
The healthy adult E. carinicauda (body length 5.65 ± 0.5 cm, body weight 1.52 ± 0.38 g) were collected from the Haichen Aquaculture Co., Ltd., in Rizhao, China. All shrimps were acclimated in the normal sea water (salinity 30, pH = 8.2, carbonate alkalinity 1.0–2.0 mmol⋅L–1) for 2 weeks before the experiment. According to the 72 h half lethal concentration of carbonate alkalinity (LC50) 8.26 mmol⋅L–1 explored by our research group in the past, the experimental shrimps were subjected to acute carbonate alkalinity stress (Li M. D. et al., 2020). Before formal experiment, the salinity-alkalinity solution was firstly prepared using sodium bicarbonate (NaHCO3) power for precipitation at least 24 h, and the concentrations were expressed as 1 mmol⋅L–1. In formal experiment, we adjusted the water alkalinity by NaHCO3 saturated solution twice a day for maintaining the original concentrations at 8–9 mmol⋅L–1. We sampled the gills subject to treatment for 0, 12, and 36 h, and frozed them in liquid nitrogen as well as stored them at –80°C for extracting protein and RNA. Gills from three shrimp were pooled into one biological replicate and six biological replicates for each time point were prepared. Half of the samples were sequenced for transcriptome and half for proteome.
RNA Extraction, Sequencing, and de novo Assembly
The TRIzol® reagent (Invitrogen, Carlsbad, CA) was used to extract the total RNA from gill samples according to the manufacturer’s protocol and then detected by 2100 bioanalyzer (Agilent) and nd-2000 (nano drop technologies) to concentration and quality of total RNA. cDNA library was constructed for each of the three pooled samples and sequenced on an Illumina HiSeq Xten/NovaSeq6000 according to the manufacturer’s instructions at the GENEWIZ (Genewiz, Suzhou, China). Before assembly, the lower quality sequences and 5’ adaptor contaminants from the sequence raw reads were removed by data cleaning analysis. Subsequently, the high-quality reads were pooled together and assembled using Trinity package with default parameters to construct a pool of unique consensus sequences to serve as a reference sequence set. In this study, the original transcriptome data were stored in the National Center for Biotechnology Information (NCBI), with the accession number PRJNA756619.
Protein Extraction, Tandem Mass Tags Labeling, and LC-ESI-MS/MS Analysis
Fifty milligrams tissue was separated from each frozen specimen on dry ice. Subsequently, these tissues were used for proteomic analyses. The gill tissues samples were ground into a fine power in liquid nitrogen. Then, the power was suspended in lysis buffer [1% sodium deoxycholate (SDS), 8 M urea] for inhibiting protease activity. The mixture was submerged at 4°C for 30 min and immediately homogenized using an ultrasound for 2–3 cycles at a speed (40 kHz vibrational frequency and 40 W) at 60 s per cycle. Samples were centrifuged at 16,000 × g for 30 min at 4°C after ultrasonicated. The supernatant was collected for extracting the desired 2 mg ml–1 of protein into a clean Eppendorf tube, which followed by BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific, United States) in line with the instructions of manufacturer. Finally, these samples were storage at –80°C.
Sixty micrograms protein from each sample with the master mix were added to cold acetone (sixfold volumes of sample) at –20°C for 4 h. After centrifugation at 10,000 × g for 20 min at 4°C, the pellet was re-suspended with 100 μL 50 mM riethylammonium bicarbonate (TEAB) buffer. Subsequently, it was digested overnight at 37°C by adding 1.1 μg of sequencing grade modified porcine trypsin (Promega, V5113). TMT reagents were reconstituted in 40 μL anhydrous acetonitrile. Then the digested peptides were transferred to the TMT reagent vial, while incubated at room temperature for 1 h. In our study, nine samples of gills were harvested and labeled as (0 h)-1, 2, 3, (12 h)-1, 2, 3 and (36 h)-1, 2, 3. Finally, all samples were pooled, desalted and vacuum-dried.
For data acquisition, Proteome Discoverer (Thermo Fisher Scientific, Version 2.2) was used for generating raw data. ProteomeXchange dataset PXD028137 has been reserved by the PRIDE repository for a dataset. Labeled peptides were analyzed by online nano flow liquid chromatography tandem mass spectrometry performed on an 9RKFSG2_NCS-3500R system (Thermo Fisher Scientific, United States) connected to a Q Exactive Plus quadrupole orbitrap mass spectrometer (Thermo Fisher Scientific, United States) through a nanoelectrospray ion source. The Q Exactive Plus was operated in the data-dependent acquisition mode (DDA) to automatically switch between full scan MS and MS/MS acquisition. The survey of full scan MS spectra (m/z 350–1,300) was acquired in the Orbitrap with 70,000 resolutions. The automatic gain control (AGC) target at 3e6 and the maximum fill time was 20 ms. Then the top 20 most intense precursor ions were selected into collision cell for fragmentation by higher-energy collision dissociation (HCD). The MS/MS resolution was set at 35,000 (at m/z 100), the automatic gain control (AGC) target at 1e5, the maximum fill time at 50 ms, and dynamic exclusion was 18 seconds.
Data Analysis
In order to remove technical sequences, including adapter, poly-N and the quality of bases lower than 20, the pass filter data in the fastq format were processed by Trimmomatic (v0.30) to provide high-quality, clean data. Meanwhile, all the downstream analyses depended on high-quality clean data. Firstly, he trimmed reads were aligned to the databases [NR, Swiss prot, Pfam, COG, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG)]. It allows multiple alignments and up to two mismatches when mapping reads to the reference. Secondly, differentially expressed gene (DEG) was screened by the DESeq2 and Edger. FPKM (Fragments Per Kilobases per Millionreads) was used to normalized the data. After adjusting by BH for controlling the false discovery rate, the differential gene screening criteria was P < 0.05 and |log2FC| ≥ 1. Differentially expressed proteins (DEPs) were those exhibiting P < 0.05 and fold change (FC) > 1.20 or < 0.87 between groups. Finally, gene Set Enrichment Analysis (GSEA) was also used to detect the expression changes of the whole gene set. Annotation of all identified proteins was performed using GO1,2 and KEGG pathway.3
Real-Time Quantitative PCR Analysis
To validate the differential expression of mRNAs identified by high-throughput sequencing, 12 DEGs in transcriptome were randomly selected for Quantitative PCR (qPCR) analyses at 12 and 36 h, respectively. In addition, 8 transcripts were selected from consistent DEGs/DEPs to verify the expression trend of DEGs/DEPs identified after combined analysis. Primers of mRNAs used for the qPCR analysis are shown in the Supplementary Table 1. With the 18S rRNA as the internal reference, the primers were designed as follows: 5′-TATACGCTAGTGGAGCTGGAA-3′, and 3′-GGGGAGGTAGTGACGAAAAAT-5′. In short, qPCR was performed using the SYBR Green PCR Master Mix (Life Technologies, United States) according to the manufacture’s protocol in a 7,500 fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The three samples of transcriptome at each time point were used to perform the qPCR, and each biological repeat was performed technical replicates in triplicate. The relative expression of target genes was calculated using CT method (2–△△Ct method) (Tapia et al., 2017). The data were analyzed on the online Majorbio Cloud Platform (Shanghai, China).
Results
Transcriptome Analysis of Exopalaemon carinicauda in Response to Alkalinity Stress
A total of 24,112 transcripts/unigenes were generated from RNA-seq analysis in the gills of E. carinicauda after 0, 12, and 36 h alkalinity stress. A total of 3,157 DEGs were identified at the three time points. Compared with 0 h, 427 up- and 734 down-regulated genes at 12 h, 379 up- and 1,602 down-regulated genes at 36 h were, respectively, detected (Figure 1A). Overall, 1,161 and 1,981 DEGs were detected at 12 and 36 h compared with 0 h, respectively (Figure 1A). Obviously, alkalinity stress led to dramatic alteration of gene expression with time point. Venn diagram of the DEGs illustrated that 182 genes were shared in both 0 vs. 12 h and 0 vs. 36 h (Figure 1B). Hierarchical clustering results revealed that alkalinity stress response varied dramatically at the three time points (Figure 1C).
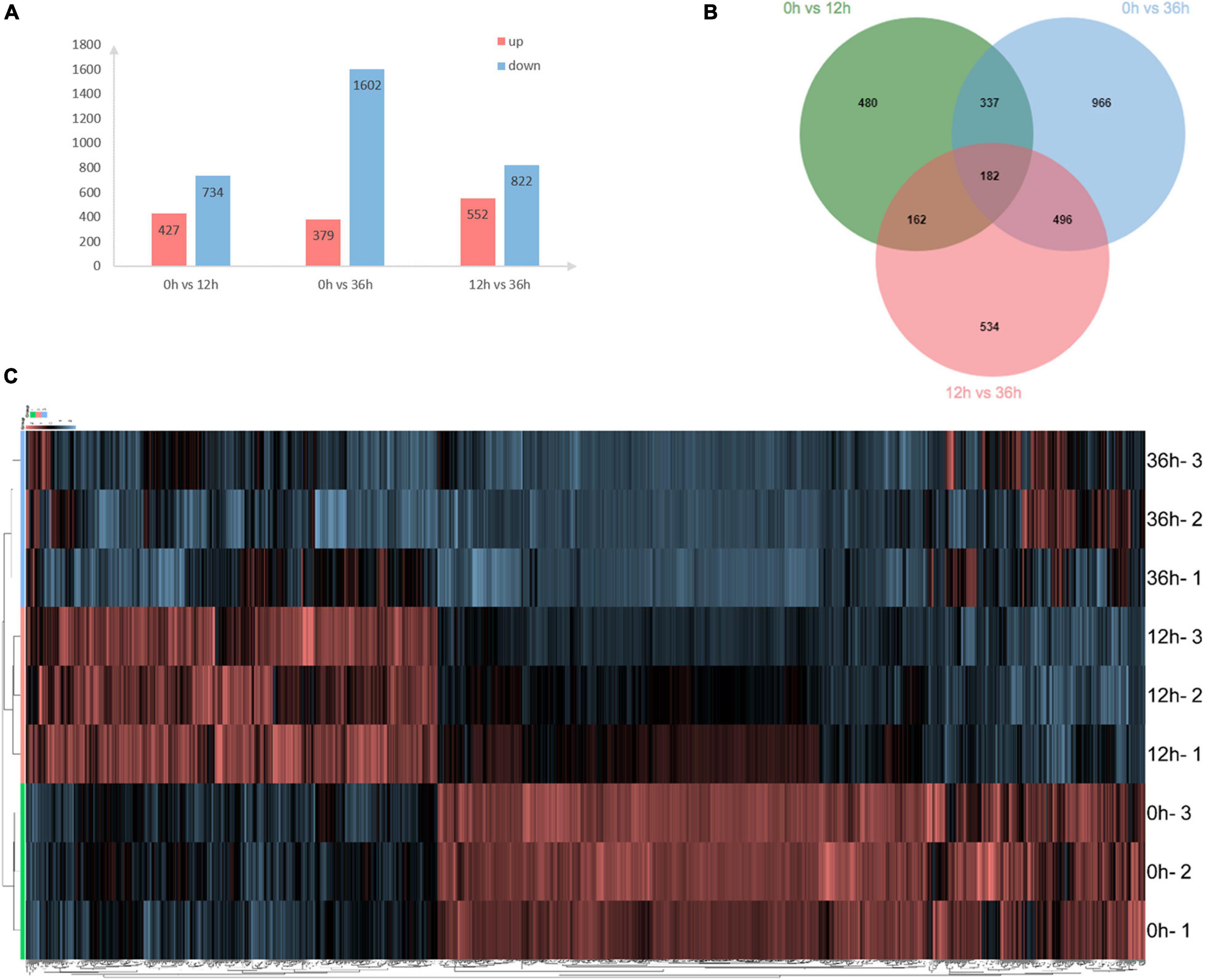
Figure 1. Distributionlocations of differentially expressed genes in the transcriptome. (A) Distribution of differentially expressed genes (DEGs) following different time effects in gills of E. carinicauda. (B) DEGs number and Venn diagram of overlap of the different groups. (C) Hierarchical cluster analysis of differentially expressed genes based on the data of log2FC.
GO annotation analysis showed that 43 GO terms (P < 0.05) in total were classified into 17, 14, and 11 sub-categories of three major categories: biological processes, cellular components and molecular function, respectively (Supplementary Figure 1). In 0 vs. 12 h, 7 of the top 20 GO enriched terms were related to ion transport, including “coupled to transmembrane movement of ions, phosphorylative mechanism,” “calcium-transporting ATPase activity,” “calcium ion transmembrane transporter activity,” “cation-transporting ATPase activity,” “ATPase coupled ion transmembrane transporter activity,” “divalent inorganic cation transmembrane transporter activity” (Figure 2A). In 0 vs. 36 h, 4 of the top 20 GO enriched terms were related to ion transport, including “calcium ion transmembrane transporter activity,” “active ion transmembrane transporter activity,” “metal ion transmembrane transporter activity,” and “divalent inorganic cation transmembrane transporter activity” (Figure 2B).
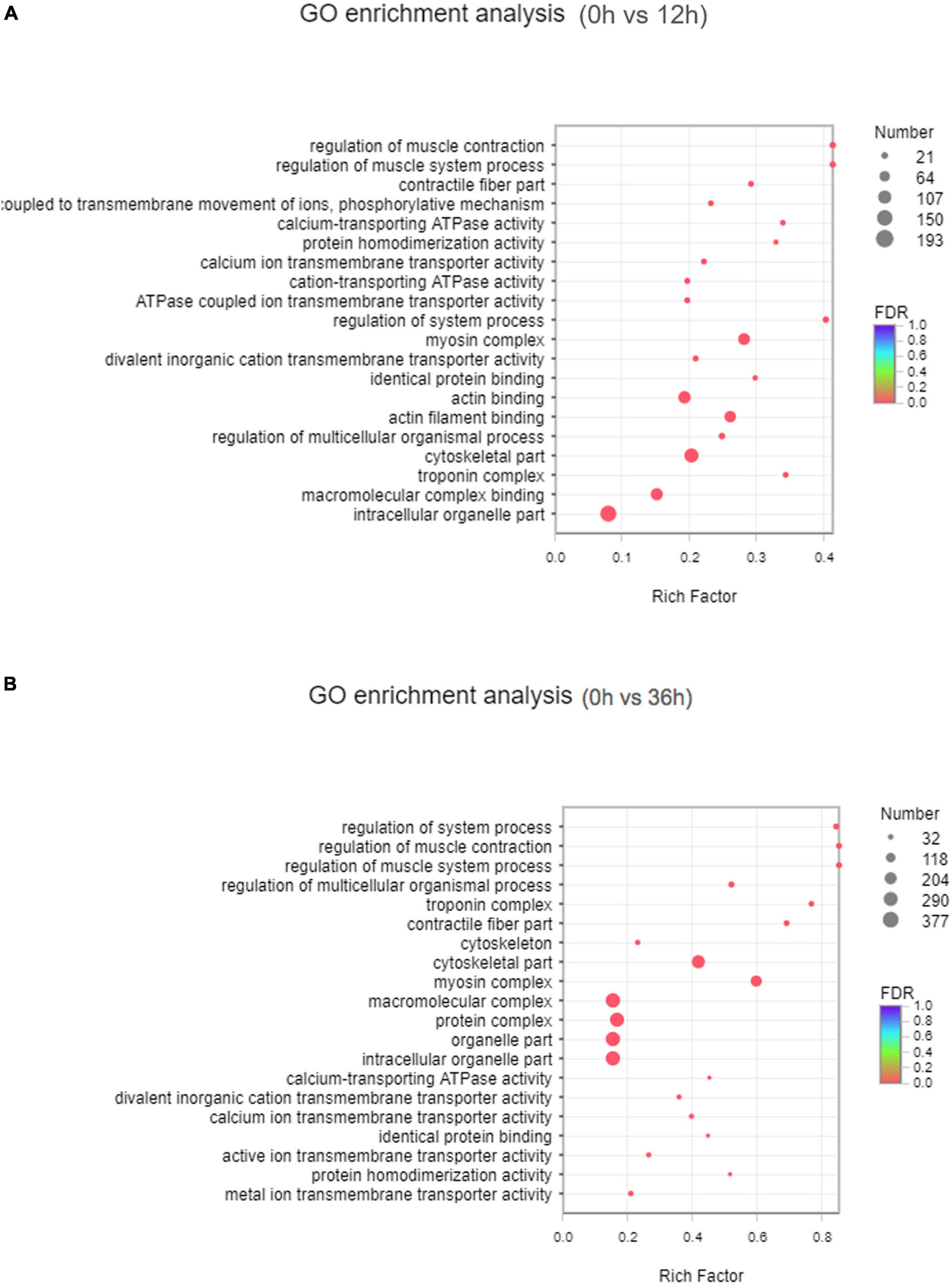
Figure 2. GO was significantly enriched in the transcriptome. (A) Analysis of the GO in 0 vs. 12 h group. (B) Analysis of the GO in 0 vs. 36 h group.
KEGG pathway analysis was performed to investigate pathways that were significantly altered following exposure to alkalinity stress. The 43 KEGG pathways can be divided into seven categories, including Metabolism (11), Genetic Information Processing (4), Environmental Information Processing (3), Cellular Processes (4), Organismal Systems (10), Human Diseases (11). The top 4 KEGG pathways with the most amount of annotation sequences were “signal transduction,” “endocrine system,” “cardiovascular disease,” and “circulatory system” (Supplementary Figure 1B). Besides, a comparative analysis of the KEGG pathways revealed that 36 pathways were shared in 0 vs. 12 h and 0 vs. 36 h. Among the first 20 pathways that are co enriched, we found that the top five of pathway enrichment as “Adrenergic signaling in cardiomyocytes,” “Cardiac muscle contraction,” “Thyroid hormone signaling pathway,” “cGMP-PKG signaling pathway,” and “Viral myocarditis” (Figure 3). In addition, compared with 12 h, there were more DEGs enriched in these five pathways at 36 h. It is worth noting that three of five pathways refer to the processes of endocrine system.
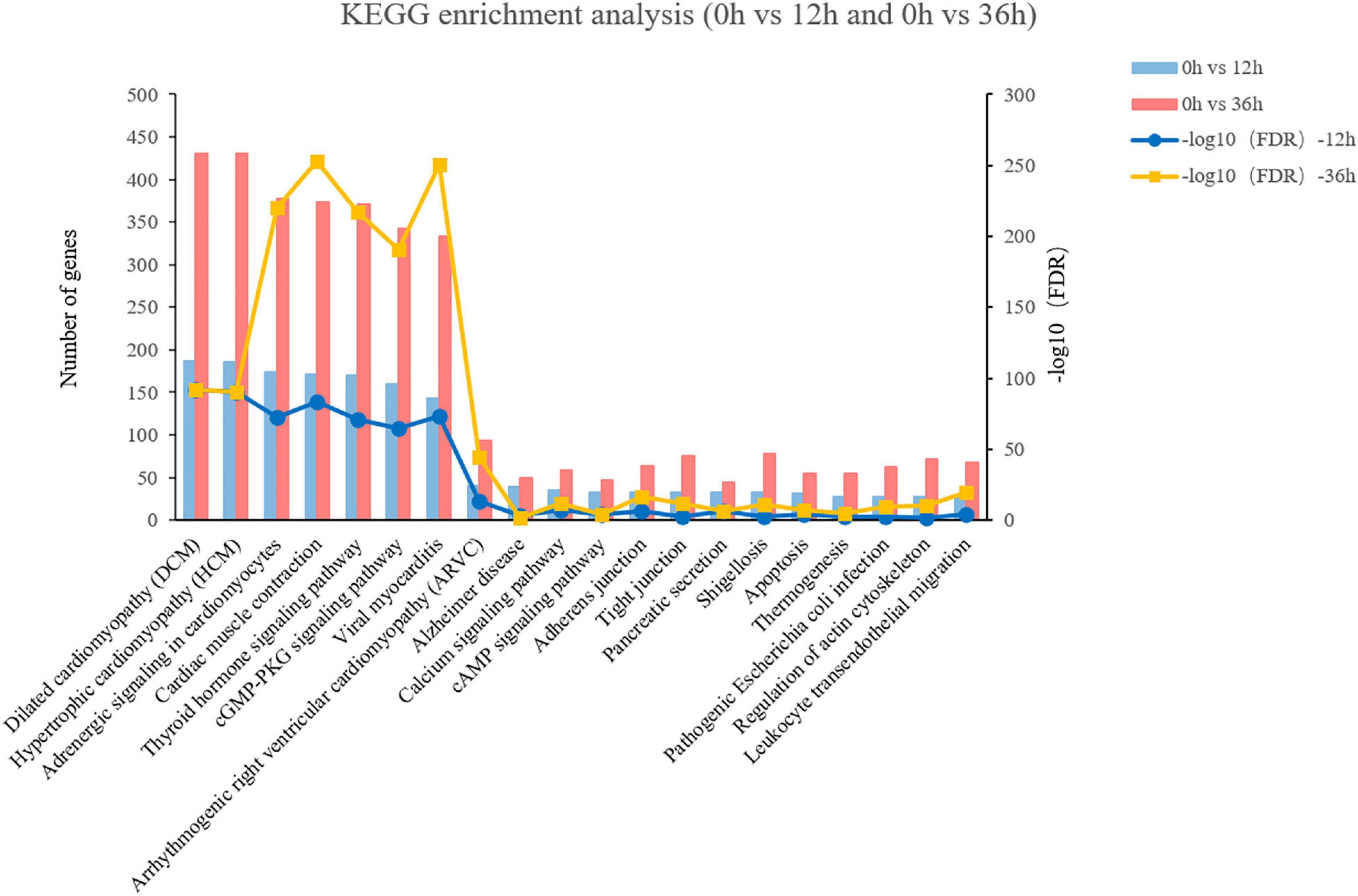
Figure 3. The top 20 of the most enriched KEGG pathways of 0 vs. 12 h and 0 vs. 36 h in the transcriptome.
In addition, some important ion transport related genes have been identified in DEGs, including sarco/endoplasmic reticulum calcium ATPase isoform, glycosyl phosphatidylinositol linked carbonic anhydrase, sodium chloride cotransporter, sodium and chloride dependent GABA transporter, glycosyl phosphatidylinositol linked carbonic anhydrase, aquaporin, Na+/K+-ATPase, sodium calcium exchanger. Some ion transport related genes were significantly different in 0 vs. 12 h and 0 vs. 36 h, such as aquaporin, proton channel, glycosyl-phosphatidylinositol-linked carbonic anhydrase, ammonium transporter Rh type A-like, etc. were up-regulated in 0 vs. 12 h (Figure 4A). Besides, gene related to sodium/hydrogen exchanger, calcium/calmodulin-dependent protein kinase, sodium-dependent multivitamin transporter, pre-mRNA-splicing factor ATP-dependent RNA helicase PRP1 were up-regulated in 0 vs. 36 h (Figure 4B). Thus it can be seen that the DEGs are mainly related to inorganic ion transport at early time point (12 h), and then related to the transport of organic macromolecules at later time point (36 h).
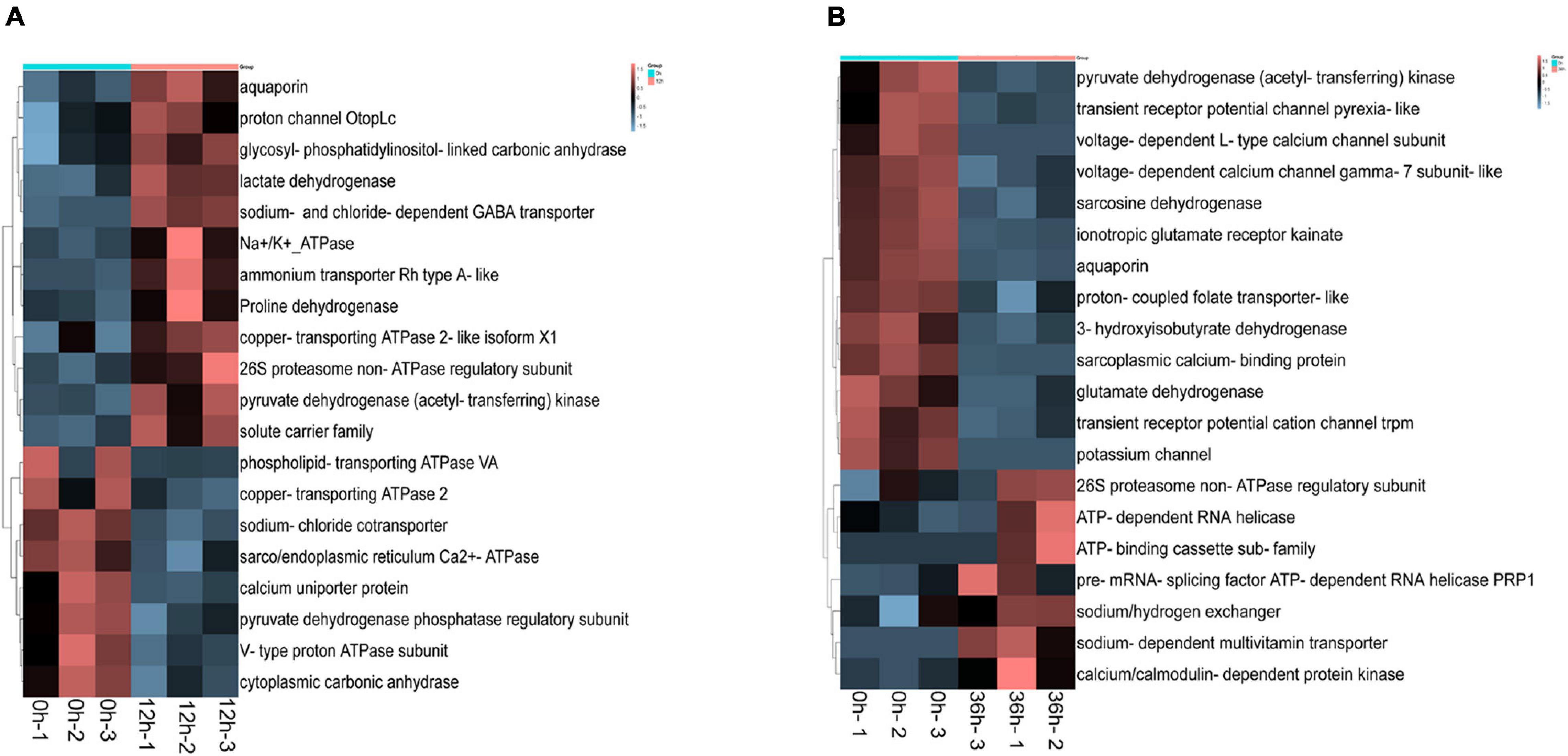
Figure 4. Differentially expressed genes (DEGs) heat map of transcriptome associated with alkalinity stress. (A) Heat map of DEGs in 0 vs. 12 h. (B) Heat map of DEGs in 0 vs. 36 h.
Proteomic Analysis of Alkalinity Stress
To further understand the effects of alkalinity stress on E. carinicauda, label-free LC-MS/MS analysis was used to evaluate the expression at three time points. TMT analysis of the gills proteome in E. carinicauda showed 58,224 queries in the database (a total of 429,067 spectra) and resulted in 3,334 protein hits. A total of 443 DEPs were identified at the three time points. Compared with 0 h, 239 DEPs were identified at 12 h, with 164 (68.6%) up- and 75 (31.4%) down-regulated proteins (Figure 5A). Two hundred thirty-six DEPs were identified at 36 h, with 136 (57.6%) up- and 100 (42.4%) down-regulated proteins (Figure 5B). Similarly, the protein expression at 36 h showed variations compared with 12 h (Figure 5C). Compared with 12 h, 85 DEPs were identified, with 47 (55.3%) up- and 38 (44.7%) down-regulated proteins at 36 h. In proteomic data, there were more up-regulated proteins than down-regulated proteins in protein expression, which is different from the result that down-regulated genes account for the majority in transcriptome data. Here, 85 proteins (37 up- and 48 down-regulated) were significantly expressed (P < 0.05) at both 12 and 36 h (Figure 5D).
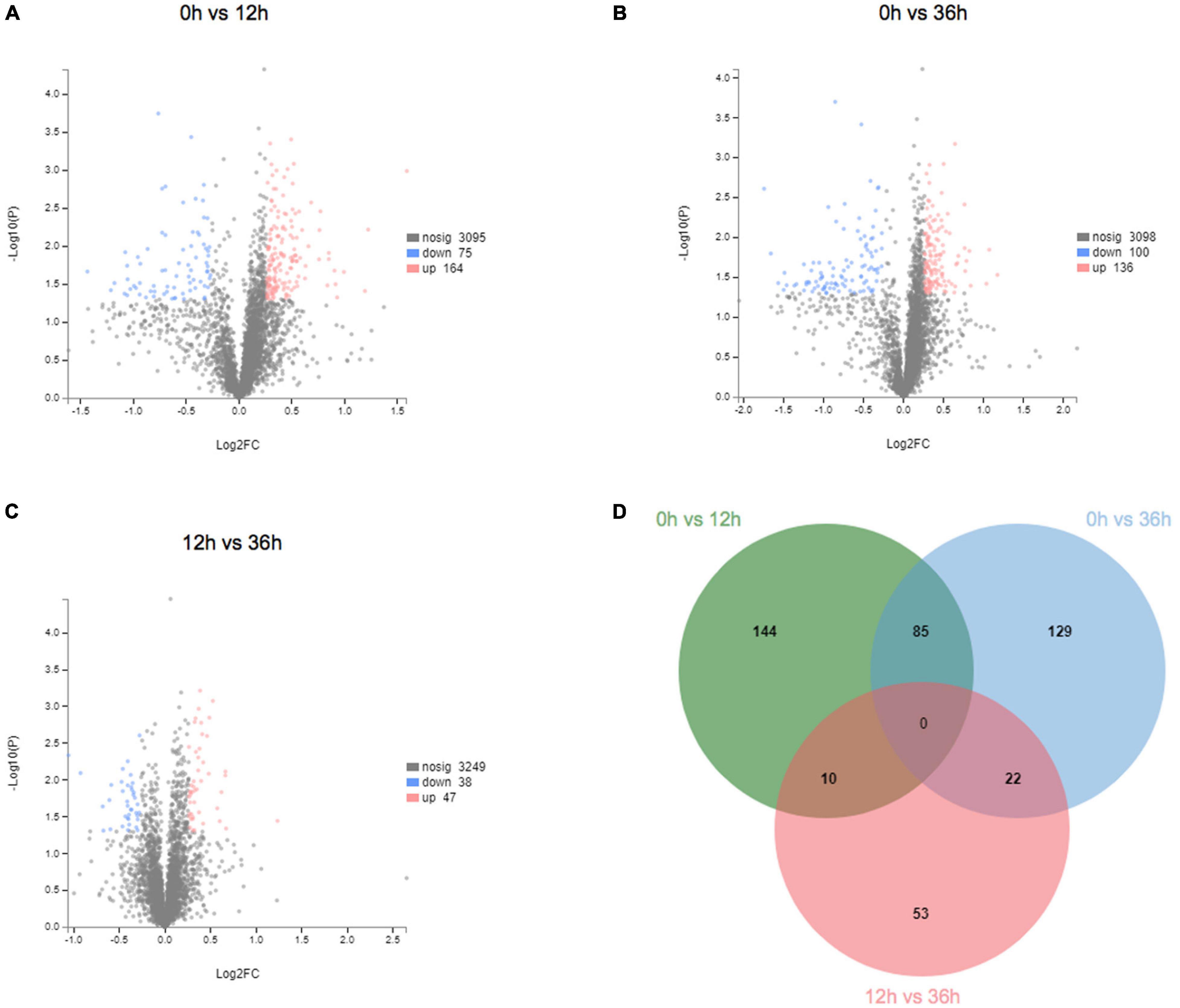
Figure 5. Expression and trend of differentially expressed proteins in proteome. (A) Volcano plot of 0 vs. 12 h. (B) Volcano plot of 0 vs. 36 h. (C) Volcano plot of 12 vs. 36 h. (D) Venn diagram of differentially expressed proteins between the different groups.
Twenty-four GO terms were significant, including 10 molecular function, 3 cellular component, and 11 biological process sub-categories (Supplementary Figure 2). Based on the DEPs data in 0 vs. 12 h, GO enrichment analysis was conducted and the most significantly enriched GO terms are shown in Figure 6A, only “retrograde transport, endosome to Golgi” had a significant difference (P < 0.001). Based on the DEPs data in 0 vs. 36 h, “actomyosin structure organization,” “myofibril assembly,” “cellular component assembly involved in morphogenesis,” “myofilament,” “striated muscle myosin thick filament,” “myosin filament,” “M band,” and “myosin complex” were the most dominant GO terms (P < 0.001).
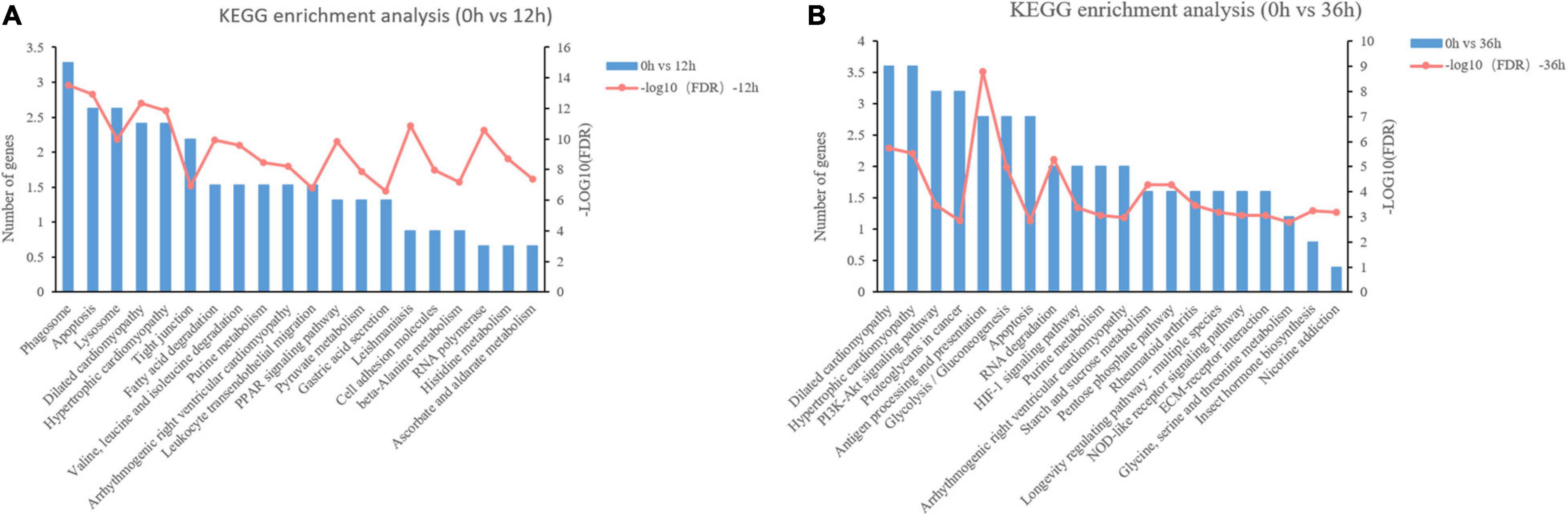
Figure 6. KEGG enrichment analysis of differentially expressed proteins (DEPs) in proteome. (A) KEGG pathway enrichment analysis of DEPs in 0 vs. 12 h. (B) KEGG pathway enrichment analysis of DEPs in 0 vs. 36 h.
In addition to the GO term analysis, KEGG pathway analysis was also conducted to assign the DEPs to six categories. The number of secondary category of KEGG is the same as that in the transcriptome. The annotation results of KEGG pathway are similar to those of transcriptome. Obviously, the signal transduction pathway in environmental information processing and the transport and catabolism pathway in cellular processes annotated the most proteins (Supplementary Figure 2B). KEGG-enrichment analyses were conducted on the DEGs, and the top 20 significant KEGG pathways in 0 vs. 12 h and 0 vs. 36 h are shown in Figure 6. Among the KEGG pathways in 0 vs. 12 h, lipolysis and amino acids metabolism (Fatty acid degradation, PPAR signaling pathway, Valine, leucine and isoleucine degradation, Histidine metabolism, etc.) and immune system (“Phagosome,” “Apoptosis,” “Dilated cardiomyopathy,” “Hypertrophic cardiomyopathy,” “Leishmaniasis,” “Lysosome,” etc.) related pathways were identified to be enriched (Figure 6A). Among the KEGG pathways in 0 vs. 36 h, carbohydrate metabolism (“Glycolysis/Gluconeogenesis,” “Starch and sucrose metabolism,” “Pentose phosphate pathway”) and immune system (Antigen processing and presentation, Hypertrophic cardiomyopathy, Dilated cardiomyopathy) were significantly enriched (P < 0.01) (Figure 6B).
We compared the DEPs among gill tissue at different time points under the same alkalinity stress. The comparison of 0 vs. 12 h and 0 vs. 36 h rendered the resemble number of DEPs. And the up-regulated DEPs are more than the down-regulated ones. Based on the criteria that P < 0.01, some proteins that may play important roles in alkalinity stress were listed in Figure 7. These DEPs were considered as relating to alkalinity stress, we mainly pay close attention to these DEPs in up-regulated. In the present study, sodium-potassium ATPase, sodium- and chloride-dependent GABA transporter, vacuolar protein sorting-associated protein and chloride intracellular channel were up-regulated in 0 vs. 12 h. Meanwhile, alkalinity stress related signaling pathways as hemocyanin, cathepsin, and V-type proton ATPase 116 kDa subunit were significantly up-regulated in 0 vs. 36 h.
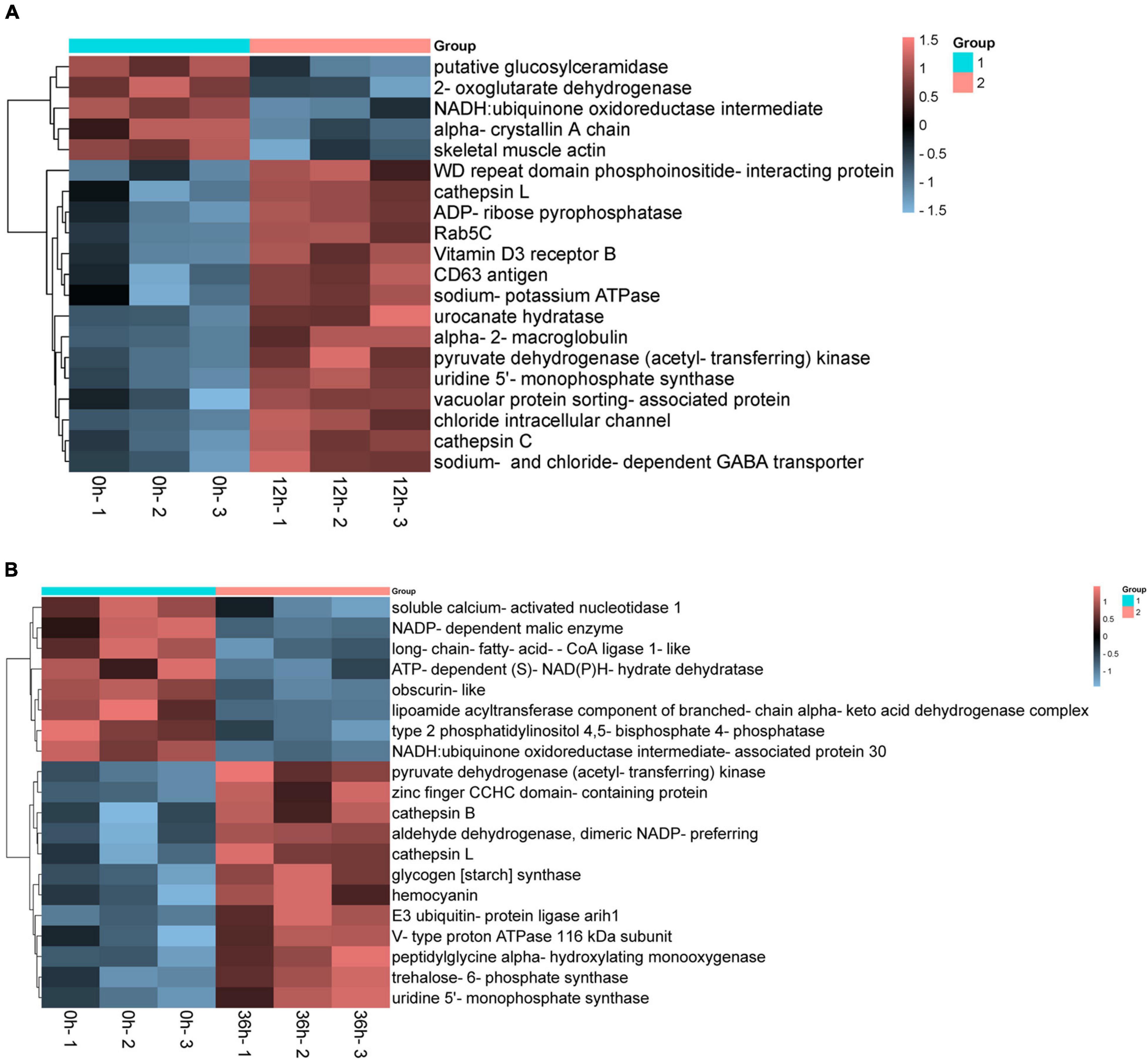
Figure 7. Cluster heat maps of differentially expressed proteins (DEPs) associated with alkalinity stress in the proteome. (A) Hierarchical cluster analysis of saline-alkaline related DEPs in 0 vs. 12 h. (B) Hierarchical cluster analysis of saline-alkaline related in 0 vs. 36 h based on the data of p < 0.01.
Integrated Analysis Between Proteome and Transcriptome
In total, 18,170 mRNAs and 6,159 proteins were quantified. Analysis for relationship between gene and protein resulted in the identification of 2,825 mRNAs to the corresponding proteins. Accession numbers with coverage in both protein and mRNA data sets were identified to accurately compare protein and mRNA regulation. DEGs and DEPs relationship analysis of 0 vs. 12 h and 0 vs. 36 h based on the data of log ratio fold change. Consistent and differential expression analysis of transcriptome and proteome was carried out to study the relationship between DEGs and DEPs of E. carinicauda under alkalinity stress. This study identified only 21 and 60 significantly regulated matching DEGs/DEPs in 0 vs. 12 h and 0 vs. 36 h comparisons, respectively, which indicated that gill protein and mRNA expression had a moderate level of overall correlation (Figures 8, 9). In 0 vs. 12 h, 3 DEGs/DEPs were consistent up-regulated, 15 DEGs/DEPs were consistent down-regulated and 3 DEGs/DEPs were opposite expression (Figures 8, 9A). In 0 vs. 36 h, 3 DEGs/DEPs were up-regulated, 52 DEGs/DEPs were down-regulated and 5 DEGs/DEPs were opposite expression (Figures 8, 9B). The entries which were significantly regulated at both mRNA and protein levels represent clear coordinated and essentially linear gene expression although the 0 vs. 12 h was higher than 0 vs. 36 h (compare A and B in Supplementary Figure 3). A correlation analysis of these data sets identified most of them were respectively changed with the same trend (Supplementary Figure 3).
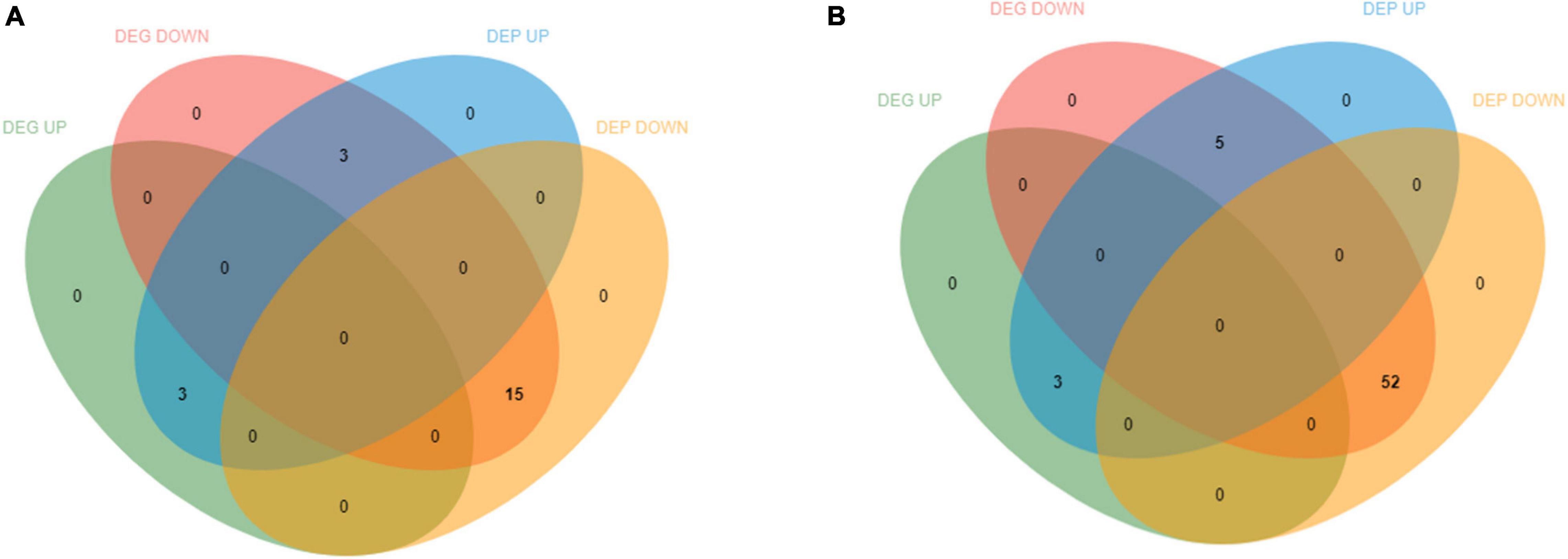
Figure 8. Differentially expressed genes (DEGs) and differentially expressed proteins (DEPs) distribution in transcriptome and proteome association analysis. (A) The differences between 0 vs. 12 h correlated data were up-regulated and down regulated DEGs and DEPs. (B) The differences between 0 vs. 36 h correlated data were up-regulated and down regulated DEGs and DEPs.
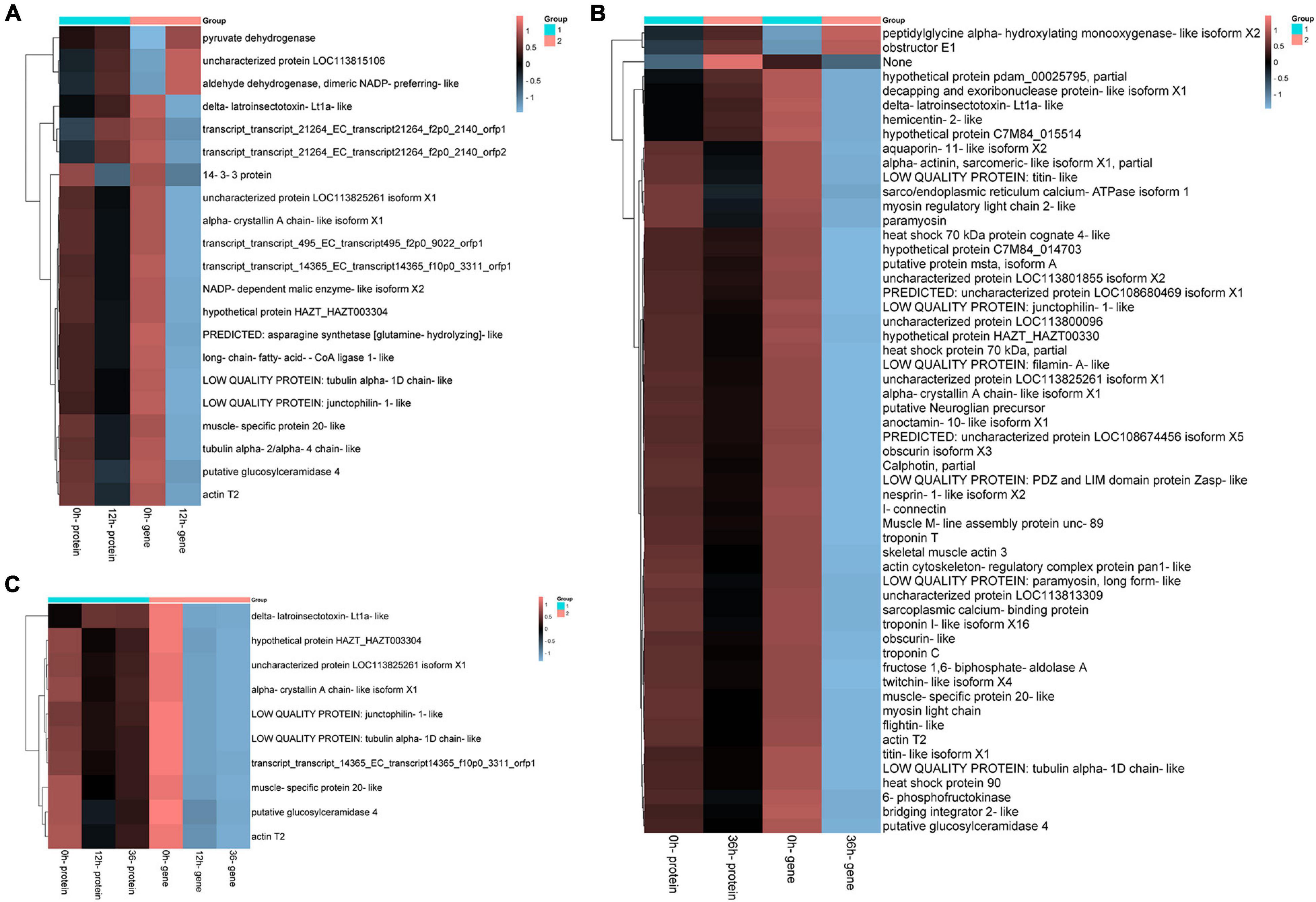
Figure 9. Significantly enriched differentially expressed genes (DEGs)/differentially expressed proteins (DEPs) in both transcriptome and proteome. (A) Hierarchical cluster analysis of 21 DEGs in 0 vs. 12 h group. (B) Hierarchical cluster analysis of 60 differentially expressed genes in 0 vs. 36 h group based on the data of log ratio fold change. (C) Hierarchical cluster analysis of 10 pairs of protein gene pairs in both groups.
Consistently up-regulated DEGs/DEPs in 0 vs. 12 h mainly included aldehyde dehydrogenase (ALDH), pyruvate dehydrogenase and cell adhesion, and consistently down-regulated DEGs/DEPs in 0 vs. 12 h were primarily concerned with metabolism, especially “Pyruvate metabolism,” “Alanine, aspartate and glutamate metabolism,” “Thiamine metabolism,” and “Sphingolipid metabolism” (Figure 9A). The consistently up-regulated DEGs/DEPs in 0 vs. 36 h were mainly involved in peptidylglycine alpha (PPG), obstructor E1 and “no description,” while the consistently down-regulated DEGs/DEPs in 0 vs. 36 h included Calphotin, heat shock protein, troponin and aquaporin (Figure 9B). However, there were a few genes, including Intermediate filament protein, delta-latroinsectotoxin, GTP binding, Chitin binding, hemicentin, and decapping and exoribonuclease protein were regulated with the opposite trend at the mRNA and protein levels (Figures10A,B). Correlation analyses of DEGs/DEPs between 0 vs. 12 h and 0 vs. 36 h showed 10 same DEGs/DEPs, but there were no groups related to ion transporters/channels (Figure 10C).
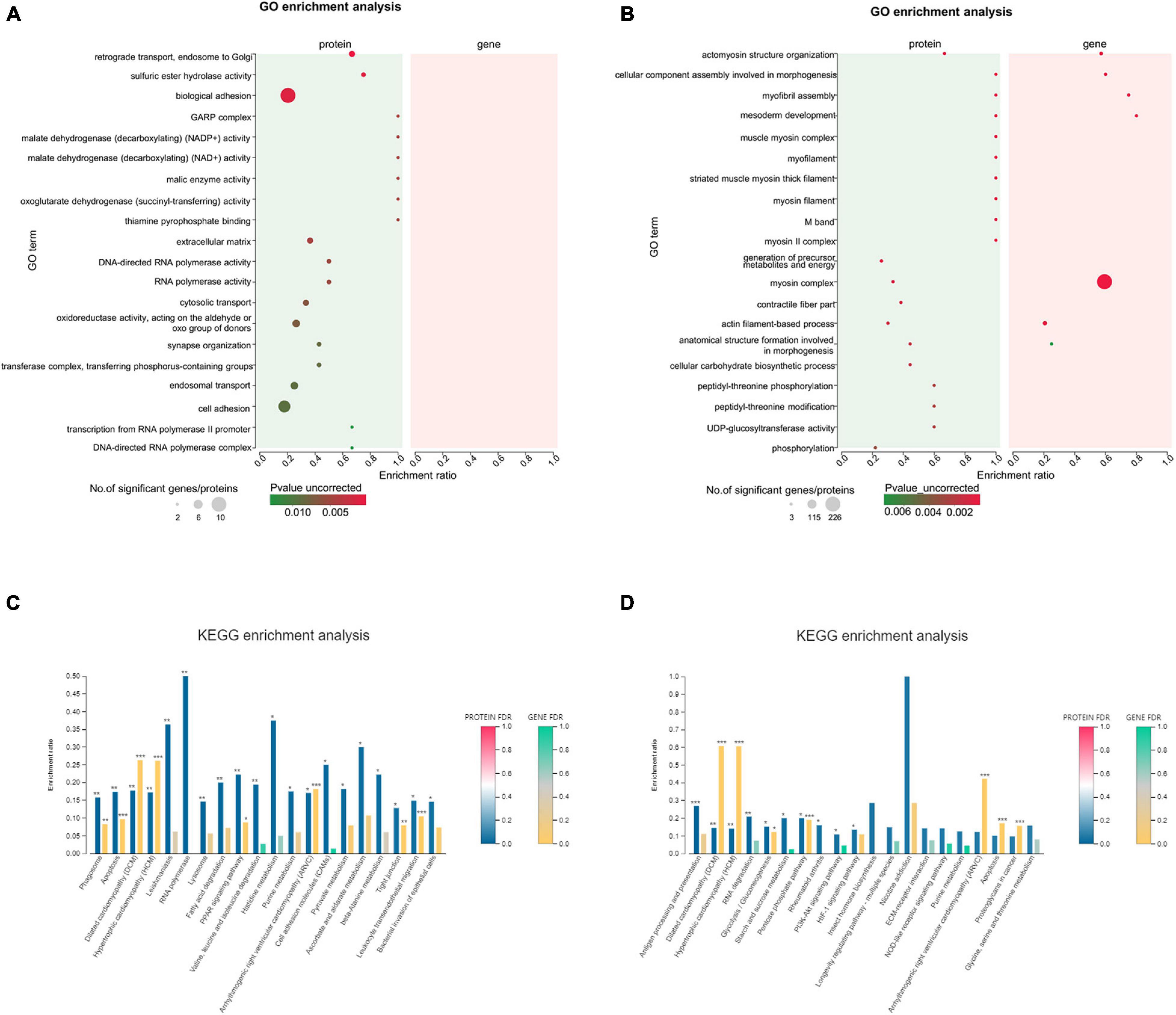
Figure 10. The top 20 GO terms and KEGG pathways significantly enriched in differentially expressed genes (DEGs)/differentially expressed proteins (DEPs). (A,B) Integrated GO term enrichment analysis of DEGs and DEPs for the intracellular protein fraction. (C,D) Integrated KEGG pathway enrichment analysis of DEGs and DEPs for the intracellular protein fraction. *FDR < 0.05; **FDR < 0.01; ***FDR < 0.001.
The 247 GO terms were enriched in 0 vs. 12 h. Disappointingly, the top 20 GO terms were not matched DEGs/DEPs (Figure 10A). Meanwhile, 315 GO terms were significantly enriched in 0 vs. 36 h, and the only highly enriched GO term was myosin complex in the top 20 GO terms (Figure 10B). KEGG analyses were performed for two groups, as well as lists the top 20 of significantly regulated proteins and mRNAs (Figures 10C,D). We found that the up-regulated DEG/DEP-ALDH in 0 vs. 12 h were identified (Figure 10C), which was also enriched in the Histidine metabolism pathway. In addition, the up-regulated DEG/DEP-PPG in 0 vs. 36 h, which was also enriched in the PI3K-Akt signaling pathway (Figure 10D). Highly enriched KEGG pathways for significantly up-regulated DEGs/DEPs, apart from signal transduction already mentioned, also involved immune system including “Antigen processing and presentation” and “NOD-like receptor signaling pathway”.
Quantitative Verification of Differentially Expressed Genes Related to Alkalinity Response
To verify the accuracy of the RNA-seq data, 12 transcripts were randomly selected from transcriptome data for qPCR (Figures 11A,B). The expression changes generally agreed with those from the RNA sequencing analysis, showing that the RNA-Seq data were reliable for reflecting the response of E. carinicauda to salinity-alkalinity stress. To better understand the regulation of expression of transcription associated proteins, eight transcripts were selected from consistent DEGs/DEPs in 0 vs. 12 h and 0 vs. 36 h for qPCR (Figures 11C,D). The results of qPCR were compared with the RNA-Seq analysis results. qPCR showed that the candidate genes have the same tendency, which further verified the reliability of the multi-omics analysis results.
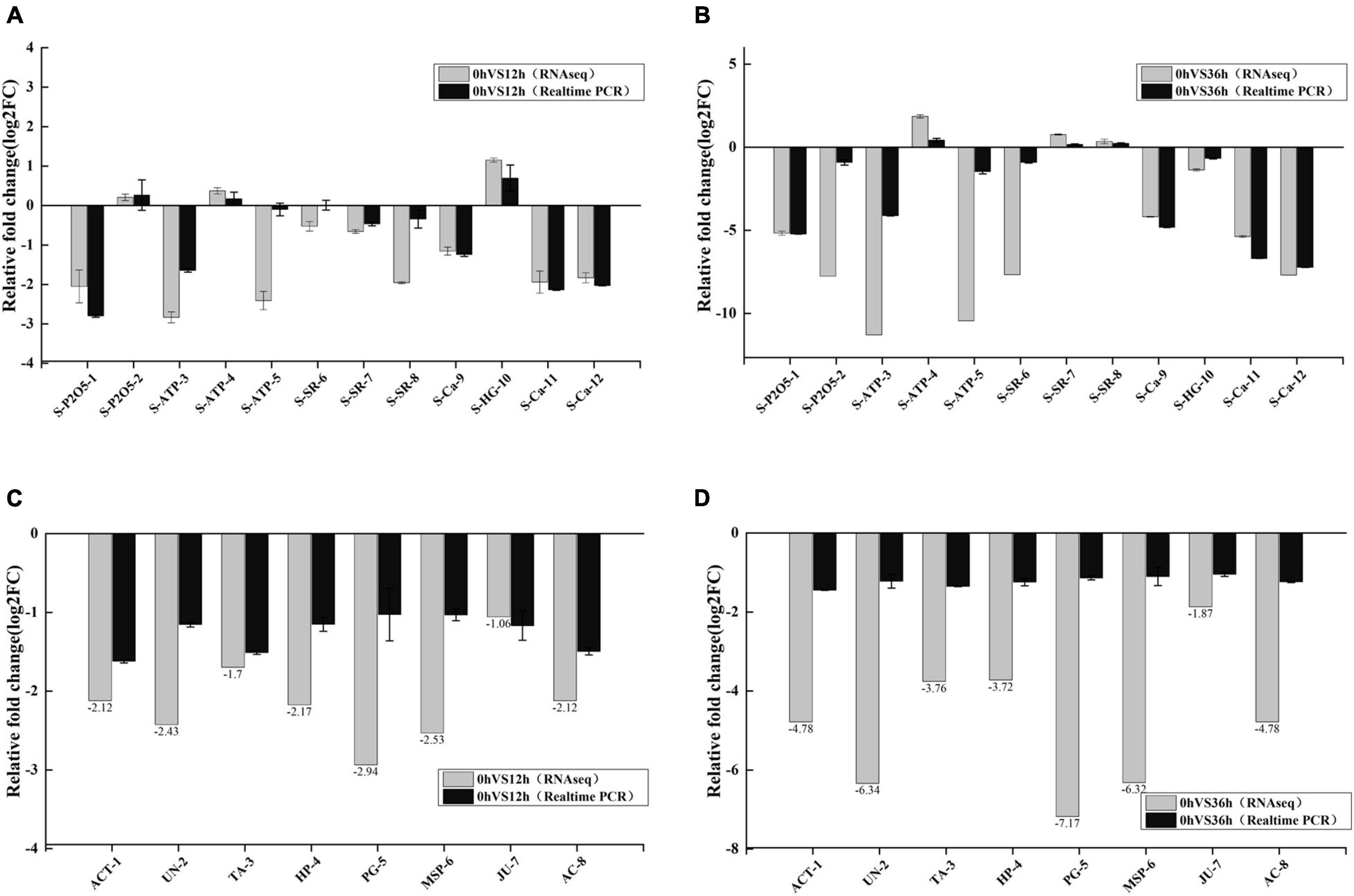
Figure 11. The results were verified by quantitative PCR (qPCR). (A,B) Relative fold change of different mRNAs between qPCR and mRNA-sequencing results in the transcriptome. Relative expression levels from the RNA-seq results were calculated as log2FC values. (C,D) qPCR validation of co-existing differentially expressed genes (DEGs)/differentially expressed proteins (DEPs) in 0 vs. 12 h and 0 vs. 36 h groups. Relative expression levels from the RNA-seq results were calculated as log2FC values.
Discussion
Saline-alkaline water aquaculture has become one of the effective ways to use saline-alkaline water resources. Environmental factors including salinity, alkalinity and pH cause stress to the aquatic animals by interfering with physiological homeostasis. As a result, only a few species can survive under the natural conditions of saline-alkaline water. Therefore, it is important for the aquaculture to study the molecular adaptation mechanism in aquatic animals over the course of adapting to saline-alkaline water. Using transcriptomic and proteomic data analysis, we sought to determine the molecular events under alkalinity stress.
In the present study, 3,157 DEGs was identified by transcriptomic analysis in alkalinity stress. GO and KEGG analysis were conducted to identify genes and pathways that mainly related to ion transport and metabolism. GO analysis results indicated that GO enriched terms were mainly related to ion transport, including “calcium-transporting ATPase activity,” “ATPase coupled ion transmembrane transporter activity,” “divalent inorganic cation transmembrane transporter activity,” etc., which indicated that the ion-regulated function of lots of DEGs. Accordingly, numerous ion transport enzymes and ion transporters associated with osmoregulation were identified after alkalinity stress. KEGG pathways related to the processes of endocrine system were extensively enriched at both 0 vs. 12 h and 0 vs. 36 h, such as “Adrenergic signaling in cardiomyocytes,” “Thyroid hormone signaling pathway,” “cGMP-PKG signaling pathway.” In addition, the energy metabolic pathways in the 0 vs. 36 h group were unexpectedly rich, which indicated that the alkalinity adaptability of E. carinicauda not just depend on osmoregulatory capacity but also depend on metabolic reorganization to make related energetic support. “Adrenergic signaling in cardiomyocytes,” “Thyroid hormone signaling pathway,” “cGMP-PKG signaling pathway” play vital roles in lipid, carbohydrate and protein metabolism (Fabbri and Moon, 2016; Guo et al., 2018; Si et al., 2019). More importantly, at some segments, some ion transport enzymes activity of the gills and the density of epithelium of cells riched in mitochondria can be significantly raised by hormones during the environmental acclimation (Marshall et al., 2000). As stated above, endocrine system related signaling pathways are likely to be critical because their stimulation on basal metabolic rate and energy supply. In addition, ion transport related genes, such as aquaporin, carbonic anhydrase, ammonium transporter Rh have been reported in many studies. Aquaporins are a class of water channel proteins that specifically transport water, glycerol, ammonia, and urea across cell membranes and affect different osmoregulation in many physiological functions and cellular processes (Lema et al., 2018; Cao and Shi, 2019). Ma et al. (2020) found that the mRNA expression levels of aquaporins were significantly up-regulated after salinity stress in Trachidermus fasciatus. In our results, aquaporin content increased significantly after 12 h of alkalinity stress. We speculate that this alleviated alkalinity stress through this water-specific channel from the gills in order to balance osmotic pressure in vivo, leading to a more active osmoregulatory activity in this treatment. Interestingly, down-regulated that occurred in the aquaporin in the 36 h-treatment in the gills were accompanied by an increase in the time. Such an effect does come in agreement with previous studies that claim a limited osmoregulatory role of this gene in the gills (Kokou et al., 2019). Carbonic anhydrase (CA) is considered to be one of the central molecular components of the systemic process of ion transport and osmoregulation (Han et al., 2018). Ali et al. (2015) suggested that carbonic anhydrase is involved in the response to changes in pH and in systemic acid–base balance in freshwater crayfish. Ammonium transporter Rh involved in the ammonia excretory process have been identified in crustaceans (Si et al., 2018). It has been reported that the expression of Rh protein increased significantly in the gill of Eriocheir sinensis, Portunus trituberculatus, and L. vannamei after abiotic stress, and ammonia excretion was inhibited after Rh protein knockdown in Caenorhabditis elegans and P. trituberculatus (Martin et al., 2011; Ren et al., 2015; Aida et al., 2016; Si et al., 2018). The Na+/H+ exchanger in gill cells creates an electrochemical gradient for regulation osmotic pressure, which can help aquatic organisms balance the changes of osmotic pressure in vivo (Castaldo et al., 2020). The potential promotion of ammonia secretion also in Na+/H+ exchanger (Fehsenfeld and Wood, 2018). In our analysis, we found upregulation of expression of the Na+/H+-exchanger at 36 h which agrees with values previously reported for Sydney rock oysters (Parker et al., 2017). This might be due to a high salinity-alkalinity, potentially resulting in upregulation of apical acid extruding mechanisms in order to maintain intracellular pH in the gill epithelium. Combined with the results of this study, it can be speculated that Rh was a regulated promote ion transport in white shrimp after salinity-alkalinity stress. According to the above, alkalinity stress can cause a large number of ion transport processes in the shrimp, such as bicarbonate ion, ammonium ion and hydrogen ion.
Based on the proteomic analysis, 443 DEPs were identified after alkalinity stress. In the transcriptome, the results of GO enrichment and KEGG enrichment in 0 vs. 12 h and 0 vs. 36 h are similar, but there are significant differences in the proteome. In general, signal transduction pathways such as ion transport were significantly enriched in the proteome in the early stage of alkalinity stress. In addition, it also contained some immune and metabolic pathways. However, the transport routes of organic macromolecules were significantly enriched at 36 h. Similar results have been obtained in previous studies exploring environmental stress (exposure) in shrimps and crabs, in which the metabolic and immune enzyme activities is altered in proportion to stress duration (Duan et al., 2019; Zhang et al., 2021). After 36 h of alkalinity stress, the enriched energy metabolism process increased significantly. In the process of translation from mRNA to protein, it will undergo multiple modifications to lead to the non-linear relationship between them, including the stabilization and degradation of mRNA (Christian and Ivan, 2019), as well as the modification of protein translational initiation, elongation, and termination (Tahmasebi et al., 2018). In agreement with this notion, a substantial percentage of proteins were associated with lipolysis and amino acids metabolism at 12 h, while most pathways were related to carbohydrate metabolism and immune system at 36 h. We speculated that the gill cells of shrimp were engaged in adjusting the osmotic pressure balance by increasing free amino acid content at the early stage of alkalinity stress, but at the later stage, gill cells tended to acclimate the environmental stress. In addition, ion transport related genes, such as V-type proton ATPase 116 kDa subunit a-like isoform X1, sodium-potassium ATPase beta and chloride intracellular channel were also identified in proteome. As a pH stress response enzyme, V-ATPase can adapt to environmental changes by changing its structure, state and quantity under stress conditions, and effectively reduce the damage of stress to organisms. Wang et al. (2012) found that under pH 9.3 stress, the gene expression of V-ATPase in L. vannamei increased at 3 h, which was 10 times higher than that of the control, and then down-regulated. This is similar to the results of high pH stress in Cherax quadricarinatus (Ali et al., 2017). A significant upregulation of V-type proton ATPase 116 kDa subunit a-like isoform X1 was observed in the present study, which was in agreement with our hypothesis that a high carbonate alkalinity leads to the increase of pH in water, and high pH further promotes the regulation of hydrogen ion in order to maintain ionic balance. Vesicle associated membrane protein is capable of regulated exocytosis and play an important role of maintaining homeostasis, cell growth and development, and polarity (Brumshtein et al., 2014; Singh et al., 2018). Furthermore, a micro tubule dependent ammonia excretion mechanism with vesicle associated membrane protein was verified in P. trituberculatus (Ren et al., 2015). Similarly, the protein expression of vesicle associated membrane protein was also increased in this study. Acting as membrane localized protein, sodium-potassium ATPase (NKA) is responsible for establishing electrochemical gradients by providing the driving force (Li et al., 2021). The accumulation of NKA was indicative of increased need for ATP in order to maintain the acid-base balance when exposed to alkalinity stress.
In our analysis, we found a weak but not insignificant correlation between expression patterns of molecular phenotype of mRNA and protein. As demonstrated by many studies, systematic comparisons between transcriptome and proteome level responses have revealed generally poor correlation between mRNA and protein regulation in invertebrates (Schenk et al., 2019; Yang et al., 2020; Root et al., 2021). It has been reported that this may be due to different regulatory mechanisms involved in gene expression and protein synthesis, regulatory factors and technical limitations in the methods used (Lundberg et al., 2010; Haider and Pal, 2013; Tavares et al., 2018). Among physiological mechanisms with low correlations between transcriptome and proteome, post-transcriptional mechanisms such as translation, protein degradation and modification have been described (Ahn et al., 2017; Tavares et al., 2018). Of which, most of these proteins were respectively changed with the same trend in the combined analysis of DEGs and DEPs in this study. This indicating that the expression alterations of these proteins are mainly controlled by transcriptional changes (Ding et al., 2019). Nevertheless, actually the transcripts are not always consistent with the proteins which are the final products. A few genes were regulated with the opposite trend at the mRNA and protein levels. This may be due to the transport of proteins between cells and tissues, indicating the involvement of post-transcriptional regulation for these genes/proteins (Li and Wang, 2021). To be specific, as we all know, such inconsistency is possibly caused by the posttranscriptional and translational processing which regulates the protein location, quantity, as well as efficiency in the cell (Du et al., 2021). Besides, accumulating evidence has shown the involvement of miRNAs, methylation and alternative splicing in environment stress (Cao et al., 2019; Feng et al., 2019b; Ng et al., 2020). Even so, there was still several consistently up-regulated DEGs/DEPs significantly enriched, such as aldehyde dehydrogenase (ALDH) at 12 h and peptidylglycine (PPG) alpha at 36 h. ALDHs are a superfamily of NADP+-dependent enzymes that various aldehydes to the corresponding carboxylic acids using the coenzyme NAD+ or NADP+ (Perozich et al., 1999; Fong et al., 2003). The ALDH is involved in the histidine metabolism, and the main metabolic pathways in organisms are deamination by histidine deaminase, histamine formation by decarboxylase and amino transfer reaction (Fong et al., 2003). An increase in blood HCO3– concentration leads to an increase in pH after the aquatic animals enter high alkalinity water. Due to the decrease of H+ in the blood, the balance of H+ + NH3 ⇌ NH4+ disorder, and ammonia mainly exists in the form of NH3. At the same time, H+ in the environment is too low to form NH4+ with NH3 transported outside the body, which inhibits the process of ammonia transport and eventually leads to the accumulation of NH3 in aquatic organism. This study points that ALDH may promote the transfer of amino groups from in vivo to in vitro. The up-regulated expression of ALDH in this paper confirms this statement. PPG is another up-related DEG/DEP in our study. It not only participates in signal transduction, but also interferes with the immune system. PPG is a key enzyme in the bioactivation of neuropeptides, which control intracellular transport, targeting, and enzyme activity (Donlon and Ryan, 2019; Back et al., 2020). These two DEGs/DEPs are up-regulation in the proteome and transcriptome in this study. We speculate that ALDH and PPG play a key role in alkalinity stress.
In summary, comparative transcriptomic and proteomic analysis of E. carinicauda in response to alkalinity stress at 0, 12, and 36 h revealed molecular adaption mechanisms. In transcriptome, DEGs were extensively enriched into GO terms related to ion transport, and KEGG pathways related to the processes of endocrine system in both 0 vs. 12 h and 0 vs. 36 h. In proteome, DEPs were highly enriched into KEGG pathways related to lipolysis and amino acids metabolism in 0 vs. 12 h, and carbohydrate metabolism and immune system in 0 vs. 36 h. Upregulated expressions of genes or proteins related to ion transport were identified by DEGs and DEPs after alkalinity stress. Although there was poor correlation between mRNA and protein regulation, there was still several consistently up-regulated DEGs/DEPs significantly enriched, including aldehyde dehydrogenase and peptidylglycine alpha, which may play the key role in alkalinity adaption. This study may provide valuable information on the adaption mechanism of the shrimp gill under alkalinity stress.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Center for Biotechnology Information (NCBI) with the accession number PRJNA756619 and the proteomic data was reserved in the PRIDE repository with the accession number PXD028137.
Ethics Statement
The animal study was reviewed and approved by the Animal Ethics Committee of Yellow Sea Research Institute, CAFS. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
JTL and QG conceived and designed the project. ZQ and QG wrote the manuscript. ZQ and JW measured and collected the data. ZQ and ML collected the samples and carried out the analysis. PL, JL, and JTL supervised, wrote, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Key R&D Program of China (2019YFD0900404-03), the National Natural Science Foundation of China (32072974 and 31702319), the China Agriculture Research System of MOF and MARA (CARS-48) and the Central Public-interest Scientific Institution Basal Research Fund, CAFS (2020TD46).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.759923/full#supplementary-material
Supplementary Figure 1 | GO and KEGG annotation results of the transcriptome. (A) GO annotation of differentially expressed genes (DEGs). (B) KEGG annotation of DEGs.
Supplementary Figure 2 | GO and KEGG annotation results of the proteome. (A) GO annotation of differentially expressed proteins (DEPs). (B) KEGG annotation of DEPs.
Supplementary Figure 3 | Transcriptome and proteome expression correlation scatter diagram. (A) Expression correlation scatter plot of 0 vs. 12 h. (B) Expression correlation scatter plot of 0 vs. 36 h.
Footnotes
References
Ahn, S. J., Gu, T., Koh, J., and Rice, K. C. (2017). Remodeling of the Streptococcus mutans proteome in response to LrgAB and external stresses. Sci. Rep. 7:14063. doi: 10.1038/s41598-017-14324-w
Aida, A., Michael, J. O. D., Jay, K., Sheena, B., Jason, R. T., David, M., et al. (2016). Ammonia excretion in Caenorhabditis elegans: physiological and molecular characterization of the rhr-2 knock-out mutant. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 195, 46–54. doi: 10.1016/j.cbpa.2016.02.003
Ali, M. Y., Pavasovic, A., Mather, P. B., and Prentis, P. J. (2015). Transcriptome analysis and characterisation of gill-expressed carbonic anhydrase and other key osmoregulatory genes in freshwater crayfish Cherax quadricarinatus. Data Brief 5, 187–193. doi: 10.1016/j.dib.2015.08.018
Ali, M., Pavasovic, A., Mather, P., and Prentis, P. (2017). Expression patterns of two carbonic anhydrase genes, Na+/K+-ATPase and V-type H+-ATPase, in the freshwater crayfish, Cherax quadricarinatus, exposed to low pH and high pH. Aust. J. Zool. 65, 50–59. doi: 10.1071/ZO16048
Back, N., Luxmi, R., Powers, K. G., Mains, R. E., and Eipper, B. A. (2020). Peptidylglycine alpha-amidating monooxygenase is required for atrial secretory granule formation. Proc. Natl. Acad. Sci. U.S.A. 117, 17820–17831. doi: 10.1073/pnas.2004410117
Brumshtein, B., Esswein, S. R., Landau, M., Ryan, C. M., Whitelegge, J. P., Phillips, M. L., et al. (2014). Formation of amyloid fibers by monomeric light chain variable domains. J. Biol. Chem. 289, 27513–27525. doi: 10.1074/jbc.M114.585638
Buckley, S. J., Fitzgibbon, Q. P., Smith, G. G., and Ventura, T. (2016). In silico prediction of the G-protein coupled receptors expressed during the metamorphic molt of Sagmariasus verreauxi (Crustacea: Decapoda) by mining transcriptomic data: RNA-seq to repertoire. Gen. Comp. Endocrinol. 228, 111–127. doi: 10.1016/j.ygcen.2016.02.001
Cao, J., and Shi, F. (2019). Comparative analysis of the aquaporin Gene Family in 12 Fish Species. Animals 9:233. doi: 10.3390/ani9050233
Cao, M., Wang, D. M., Kong, F. N., Wang, J. H., Xu, K. P., and Mao, Y. X. (2019). A genome-wide identification of osmotic stress-responsive MicroRNAs in Pyropia haitanensis (Bangiales, Rhodophyta). Front. Mar. Sci. 6:766. doi: 10.3389/fmars.2019.00766
Castaldo, G., Pillet, M., Slootmaekers, B., Bervoets, L., Town, R. M., Blust, R., et al. (2020). Investigating the effects of a sub-lethal metal mixture of Cu, Zn and Cd on bioaccumulation and ionoregulation in common carp, Cyprinus carpio. Aquat. Toxicol. 218:105363. doi: 10.1016/j.aquatox.2019.105363
Chang, Y. M., and Liang, L. Q. (2020). Advances of research of physiological and molecular mechanisms related to alkali-saline adaptation for fish species inhabiting alkali-saline water. J. Fish. China 45, 798–812. doi: 10.11964/jfc.20190311681
Chen, B. H., Xu, J., Cui, J., Pu, F., Peng, W. Z., Chen, L., et al. (2019). Transcriptional differences provide insight into environmental acclimatization in wild amur ide (Leuciscus waleckii) during spawning migration from alkalized lake to freshwater river. Genomics 111, 267–276. doi: 10.1016/j.ygeno.2018.11.007
Christian, P., and Ivan, D. (2019). Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822. doi: 10.1126/science.aax3769
Christie, A. E., Roncalli, V., Cieslak, M. C., Pascual, M. G., Yu, A., Lameyer, T. J., et al. (2017). Prediction of a neuropeptidome for the eyestalk ganglia of the lobster Homarus americanus using a tissue-specific de novo assembled transcriptome. Gen. Comp. Endocrinol. 243, 96–119. doi: 10.1016/j.ygcen.2016.11.001
Ding, Z. H., Fu, L. L., Tie, W. W., Yan, Y., Wu, C. L., Hu, W., et al. (2019). Extensive post-transcriptional regulation revealed by transcriptomic and proteomic integrative analysis in cassava under drought. J. Agric. Food Chem. 67, 3521–3534. doi: 10.1021/acs.jafc.9b00014
Dong, C. J., Duan, X. D., Younis, L. M., Zhang, M., Ma, X., Chen, B. H., et al. (2020). Mitogenomic perspectives on the adaptation to extreme alkaline environment of amur ide (Leuciscus waleckii). Mar. Biotechnol. 22, 220–232. doi: 10.1007/s10126-020-09946-7
Donlon, J., and Ryan, P. (2019). Peptidylglycine monooxygenase activity of monomeric species of growth hormone. Heliyon 5:e02436. doi: 10.1016/j.heliyon.2019.e02436
Du, C. X., Li, H., Liu, C., and Fan, H. F. (2021). Understanding of the postgerminative development response to salinity and drought stresses in cucumber seeds by integrated proteomics and transcriptomics analysis. J. Proteomics 232, 104062. doi: 10.1016/j.jprot.2020.104062
Duan, Y. F., Wang, Y., Liu, Q. S., Zhang, J. S., and Xiong, D. L. (2019). Changes in the intestine barrier function of Litopenaeus vannamei in response to pH stress. Fish Shellfish Immunol. 88, 142–149. doi: 10.1016/j.fsi.2019.02.047
Fabbri, E., and Moon, T. W. (2016). Adrenergic signaling in teleost fish liver, a challenging path. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 199, 74–86. doi: 10.1016/j.cbpb.2015.10.002
Fan, Z., Wu, D., Zhang, Y. Y., Li, J. N., Xu, Q. Y., and Wang, L. S. (2021). Carbonate alkalinity and dietary protein levels affected growth performance, intestinal immune responses and intestinal micromicroflora in Songpu mirror carp (Cyprinus carpio Songpu). Aquaculture 545:737135. doi: 10.1016/j.aquaculture.2021.737135
Fehsenfeld, S., and Wood, C. M. (2018). Section-specific expression of acid-base and ammonia transporters in the kidney tubules of the goldfish Carassius auratus and their responses to feeding. Am. J. Physiol. Renal Physiol. 315, F1565–F1582. doi: 10.1152/ajprenal.00510.2017
Feng, Y. Y., Zhai, Q. Q., Wang, J. J., Li, J. T., and Li, J. (2019a). Comparison of florfenicol pharmacokinetics in Exopalaemon carinicauda at different temperatures and administration routes. J. Vet. Pharmacol. Ther. 42, 230–238. doi: 10.1111/jvp.12734
Feng, Y. Y., Zhang, D. N., Lv, J. J., Gao, B. Q., Li, J., and Liu, P. (2019b). Identification of SNP markers correlated with the tolerance of low-salinity challenge in swimming crab (Portunus trituberculatus). Acta Oceanol. Sin. 38, 41–47. doi: 10.1007/s13131-019-1428-0
Fong, W. P., Cheng, C. H. K., and Tang, W. K. (2003). Purification and characterization of two distinct aldehyde-oxidizing enzymes from the liver of black seabream. Chem. Biol. Interact. 143–144, 159–167. doi: 10.1016/s0009-2797(02)00200-4
Ge, Q. Q., Li, J. T., Wang, J. J., Li, Z. D., and Li, J. (2019). Characterization, functional analysis, and expression levels of three carbonic anhydrases in response to pH and saline-alkaline stresses in the ridgetail white prawn Exopalaemon carinicauda. Cell Stress Chaperon. 24, 503–515. doi: 10.1007/s12192-019-00987-z
González-Vera, C., and Brown, J. H. (2017). Effects of alkalinity and total hardness on growth and survival of postlarvae freshwater prawns, Macrobrachium rosenbergii (De Man 1879). Aquaculture 473, 521–527. doi: 10.1016/j.aquaculture.2017.03.016
Guo, B. Y., Tang, Z. R., Wu, C. W., Xu, K. D., and Qi, P. Z. (2018). Transcriptomic analysis reveal an efficient osmoregulatory system in Siberian sturgeon Acipenser baeri in response to salinity stress. Sci. Rep. 8:14353. doi: 10.1038/s41598-018-32771-x
Haider, S., and Pal, R. (2013). Integrated analysis of transcriptomic and proteomic data. Curr. Genomics 14, 91–110. doi: 10.2174/1389202911314020003
Han, S. Y., Wang, B. J., Liu, M., Wang, M. Q., Jiang, K. Y., Liu, X. W., et al. (2018). Adaptation of the white shrimp Litopenaeus vannamei to gradual changes to a low-pH environment. Exontox. Environ. Safe 149, 203–210. doi: 10.1016/j.ecoenv.2017.11.052
Kokou, F., Con, P., Barki, A., Nitzan, T., Slosman, T., Mizrahi, I., et al. (2019). Short- and long-term low-salinity acclimation effects on the branchial and intestinal gene expression in the European seabass (Dicentrarchus labrax). Comp. Biochem. Physiol. A Mol. Integr. Physiol. 231, 11–18. doi: 10.1016/j.cbpa.2019.01.018
Lee, S. Y., Bang, I.-C., and Nam, Y. K. (2017). Complete mitochondrial genome of albino cyprinid loach, Misgurnus anguillicaudatus (Cypriniformes: Cobitidae). Conserv. Genet. Resour. 10, 507–510. doi: 10.1007/s12686-017-0861-7
Lema, S. C., Carvalho, P. G., Egelston, J. N., Kelly, J. T., and McCormick, S. D. (2018). Dynamics of gene expression responses for ion transport proteins and aquaporins in the gill of a euryhaline pupfish during freshwater and high-salinity acclimation. Physiol. Biochem. Zool. 91, 1148–1171. doi: 10.1086/700432
Li, H., Lai, Q. F., Yao, Z. L., Liu, Y. M., Gao, P. C., Zhou, K., et al. (2020). Ammonia excretion and blood gas variation in naked carp (Gymnocypris przewalskii) exposed to acute hypoxia and high alkalinity. Fish Physiol. Biochem. 46, 1981–1990. doi: 10.1007/s10695-020-00850-4
Li, J. T., Ma, P., Liu, P., Chen, P., and Li, J. (2015). The roles of Na+/K+-ATPase alpha-subunit gene from the ridgetail white prawn Exopalaemon carinicauda in response to salinity stresses. Fish Shellfish Immunol. 42, 264–271. doi: 10.1016/j.fsi.2014.10.043
Li, M. D., Li, J. T., Shi, K. P., He, Y. Y., Gao, B. Q., Liu, P., et al. (2020). Estimation of heritability and genetic correlation of saline-alkali tolerance in Exopalaemon carinicauda. Prog. Fish. Sci. 42, 120–126. doi: 10.19663/j.issn2095-9869.20200221001
Li, Y. L., and Wang, W. X. (2021). Integrated transcriptomics and proteomics revealed the distinct toxicological effects of multi-metal contamination on oysters. Environ. Pollut. 284:117533. doi: 10.1016/j.envpol.2021.117533
Li, Z. X., Tang, X. Q., Li, J., and He, Y. Y. (2021). Comparative proteomic and transcriptomic analysis reveals high pH-induced expression signatures of Chinese shrimp Fenneropenaeus chinensis. Funct. Integr. Genomics 21, 299–311. doi: 10.1007/s10142-021-00779-8
Lin, T. T., Lai, Q. F., Yao, Z. L., Lu, J. X., Zhou, K., and Wang, H. (2013). Combined effects of carbonate alkalinity and pH on survival, growth and haemocyte parameters of the Venus clam Cyclina sinensis. Fish Shellfish Immunol. 35, 525–531. doi: 10.1016/j.fsi.2013.05.006
Lundberg, E., Fagerberg, L., Klevebring, D., Matic, I., Geiger, T., Cox, J., et al. (2010). Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6:450. doi: 10.1038/msb.2010.106
Ma, Q., Liu, X. F., Li, A., Liu, S. F., and Zhuang, Z. M. (2020). Effects of osmotic stress on the expression profiling of aquaporin genes in the roughskin sculpin (Trachidermus fasciatus). Acta Oceanol. Sin. 39, 19–25. doi: 10.1007/s13131-020-1594-0
Maoxiao, P., Bo, Y., Xiaojun, L., Donghong, N., Tianyi, L., Zhiguo, D., et al. (2018). Effects of alkalinity and pH on survival, growth, and enzyme activities in juveniles of the razor clam, Sinonovacula constricta. Front. Physiol. 9:552. doi: 10.3389/fphys.2018.00552
Marshall, W., Daborn, K., Doyle, T., McCormick, S., and Singer, T. D. (2000). Regulation of ion transport by chloride cells of euryhaline teleost gill epithelium: hormones, tonicity, and cytoskeleton. Comp. Biochem. Physiol. A 126:101. doi: 10.1016/S1095-6433(00)80200-7
Martin, M., Fehsenfeld, S., Sourial, M. M., and Weihrauch, D. (2011). Effects of high environmental ammonia on branchial ammonia excretion rates and tissue Rh-protein mRNA expression levels in seawater acclimated Dungeness crab Metacarcinus magister. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 160, 267–277. doi: 10.1016/j.cbpa.2011.06.012
Ng, H. M., Ho, J. C. H., Nong, W., Hui, J. H. L., Lai, K. P., and Wong, C. K. C. (2020). Genome-wide analysis of MicroRNA-messenger RNA interactome in ex-vivo gill filaments, Anguilla japonica. BMC Genomics 21:208. doi: 10.1186/s12864-020-6630-0
Ouellet, J. D., Dubé, M. G., and Niyogi, S. (2013). Influence of elevated alkalinity and natural organic matter (NOM) on tissue-specific metal accumulation and reproductive performance in fathead minnows during chronic, multi-trophic exposures to a metal mine effluent. Ecotoxicol. Environ. Saf. 95, 104–112. doi: 10.1016/j.ecoenv.2013.05.027
Parker, L. M., O’connor, W. A., Byrne, M., Coleman, R. A., Virtue, P., Dove, M., et al. (2017). Adult exposure to ocean acidification is maladaptive for larvae of the Sydney rock oyster Saccostrea glomerata in the presence of multiple stressors. Biol. Lett. 13:20160798. doi: 10.1098/rsbl.2016.0798
Perozich, J., Nicholas, H., Wang, B. C., Lindahl, R., and Hempel, J. (1999). Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 8, 137–146. doi: 10.1110/ps.8.1.137
Pörtner, H. O., Schulte, P. M., Wood, C. M., and Schiemer, F. (2010). Niche dimensions in fishes: an integrative view. Physiol. Biochem. Zool. 83, 808–826. doi: 10.1086/655977
Ren, Q., Pan, L., Zhao, Q., and Si, L. (2015). Ammonia and urea excretion in the swimming crab Portunus trituberculatus exposed to elevated ambient ammonia-N. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 187, 48–54. doi: 10.1016/j.cbpa.2015.04.013
Root, L., Campo, A., Macniven, L., Con, P., Cnaani, A., and Kultz, D. (2021). Nonlinear effects of environmental salinity on the gill transcriptome versus proteome of Oreochromis niloticus modulate epithelial cell turnover. Genomics 113, 3235–3249. doi: 10.1016/j.ygeno.2021.07.016
Schenk, S., Bannister, S. C., Sedlazeck, F. J., Anrather, D., Minh, B. Q., Bileck, A., et al. (2019). Combined transcriptome and proteome profiling reveals specific molecular brain signatures for sex, maturation and circalunar clock phase. Elife 8:e41556. doi: 10.7554/eLife.41556
Shan, G., Yu-Mei, C., Xue-Fei, Z., Bo, S., Li-Ming, Z., Li-Qun, L., et al. (2020). The effect of different bicarbonate alkalinity on the gill structure of amur ide (leuciscus waleckii). Acta Hydrobiol. Sin. 44, 736–743. doi: 10.7541/2020.088
Si, L. G., Pan, L. Q., Wang, H. D., and Zhang, X. (2018). Identification of the role of Rh protein in ammonia excretion of the swimming crab Portunus trituberculatus. J. Exp. Biol. 221:jeb184655. doi: 10.1242/jeb.184655
Si, L. G., Pan, L. Q., Zhang, X., Wang, H. D., and Wei, C. (2019). Evidence that dopamine is involved in neuroendocrine regulation, gill intracellular signaling pathways and ion regulation in Litopenaeus vannamei. J. Exp. Biol. 222:jeb204073. doi: 10.1242/jeb.204073
Singh, B., Khurana, P., Khurana, J. P., and Singh, P. (2018). Gene encoding vesicle-associated membrane protein-associated protein from Triticum aestivum (TaVAP) confers tolerance to drought stress. Cell Stress Chaperones 23, 411–428. doi: 10.1007/s12192-017-0854-1
Sun, Y. C., Han, S. C., Yao, M. Z., Liu, H. B., and Wang, Y. M. (2020). Exploring the metabolic biomarkers and pathway changes in crucian under carbonate alkalinity exposure using high-throughput metabolomics analysis based on UPLC-ESI-QTOF-MS. RSC Adv. 10, 1552–1571. doi: 10.1039/c9ra08090b
Tahmasebi, S., Khoutorsky, A., Mathews, M. B., and Sonenberg, N. (2018). Translation deregulation in human disease (Review). Nat. Rev. Mol. Cell Biol. 19, 791–807. doi: 10.1038/s41580-018-0034-x
Tapia, D., Eissler, Y., Espinoza, J. C., and Kuznar, J. (2017). Inter-laboratory ring trial to evaluate real-time reverse transcription polymerase chain reaction methods used for detection of infectious pancreatic necrosis virus in Chile. Electron J. Biotechnol. 28, 20–26. doi: 10.1016/j.ejbt.2017.05.008
Tavares, G. C., Carvalho, A. F., Pereira, F. L., Rezende, C. P., Azevedo, V. A. C., Leal, C. A. G., et al. (2018). Transcriptome and proteome of fish-pathogenic Streptococcus agalactiae are modulated by temperature. Front. Microbiol. 9:2639. doi: 10.3389/fmicb.2018.02639
Tiffany, L. L., David, H. B., Jenifer, K. M., and Nathaniel, L. S. (2009). Effects of water hardness, alkalinity, and dissolved organic carbon on the toxicity of copper to the lateral line of developing fish. Environ. Toxicol. Chem. 28, 1455–1461. doi: 10.1897/08-283.1
Wang, H., Lai, Q. F., Fang, W. H., and Wang, J. (2003). Study on Aquaculture in Different Type of Saline-Alkali Water. Beijing: Ocean Press, 116–120.
Wang, L., Wang, W. N., Liu, Y., Cai, D. X., Li, J. Z., and Wang, A. L. (2012). Two types of ATPases from the pacific white shrimp, Litopenaeus vannamei in response to environmental stress. Mol. Biol. Rep. 39, 6427–6438. doi: 10.1007/s11033-012-1461-y
Xu, J., Ji, P. F., Wang, B. S., Zhao, L., Wang, J., Zhao, Z. X., et al. (2013). Transcriptome sequencing and analysis of wild Amur Ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS One 8:e59703. doi: 10.1371/journal.pone.0059703
Yang, F. Y., Sun, L. M., and Yang, X. Q. (2004). Toxicity of carbonate alkalinity to Penaeus vannamei juveniles. J. Fish. China 23, 3–6. doi: 10.3969/j.issn.1003-1111.2004.09.002
Yang, M. Y., Wang, Z. C., Wang, R., Zhang, X. M., Li, M. Y., Xin, J. J., et al. (2020). Transcriptomic and proteomic analyses of the mechanisms of overwintering diapause in soybean pod borer (Leguminivora glycinivorella). Pest Manag. Sci. 76, 4248–4257. doi: 10.1002/ps.5989
Yao, Z. L., Guo, W., Lai, Q. F., Shi, J. Q., Zhou, K., Qi, H., et al. (2016). Gymnocypris przewalskii decreases cytosolic carbonic anhydrase expression to compensate for respiratory alkalosis and osmoregulation in the saline-alkaline lake Qinghai. J. Comp. Physiol. B 186, 83–95. doi: 10.1007/s00360-015-0939-z
Yao, Z. L., Lai, Q. F., Zhou, K., Rizalita, R. E., and Wang, H. (2010). Developmental biology of medaka fish (Oryzias latipes) exposed to alkalinity stress. J. Appl. Ichthyol. 26, 397–402. doi: 10.1111/j.1439-0426.2009.01360.x
Yao, Z. L., Wang, H., Chen, L., Zhou, K., Ying, C. Q., and Lai, Q. F. (2012). Transcriptomic profiles of Japanese medaka (Oryzias latipes) in response to alkalinity stress. Genet. Mol. Res. 11, 2200–2246. doi: 10.4238/2012.June.15.2
Zhang, Y., Sun, K., Li, Z. Y., Chai, X. X., Fu, X. Y., Kholodkevich, S., et al. (2021). Effescts of acute diclofenac exposure on intestinal histology, antioxidant defense, and microbiota in freshwater crayfish (Procambarus clarkii). Chemosphere 263:128130. doi: 10.1016/j.chemosphere.2020.128130
Keywords: Exopalaemon carinicauda, alkalinity stress, transcriptomic, proteomic, adaption mechanism
Citation: Qin Z, Ge Q, Wang J, Li M, Liu P, Li J and Li J (2021) Comparative Transcriptomic and Proteomic Analysis of Exopalaemon carinicauda in Response to Alkalinity Stress. Front. Mar. Sci. 8:759923. doi: 10.3389/fmars.2021.759923
Received: 17 August 2021; Accepted: 27 September 2021;
Published: 15 October 2021.
Edited by:
Alaa El-Din Hamid Sayed, Assiut University, EgyptReviewed by:
Ting Chen, South China Sea Institute of Oceanology, Chinese Academy of Sciences (CAS), ChinaHongyu Ma, Shantou University, China
Copyright © 2021 Qin, Ge, Wang, Li, Liu, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jitao Li, bGlqdEB5c2ZyaS5hYy5jbg==
†These authors have contributed equally to this work