 Laura Geissler1†
Laura Geissler1† Valentine Meunier2†
Valentine Meunier2† Nils Rädecker1,3
Nils Rädecker1,3 Gabriela Perna1
Gabriela Perna1 Riccardo Rodolfo-Metalpa2
Riccardo Rodolfo-Metalpa2 Fanny Houlbrèque2
Fanny Houlbrèque2 Christian R. Voolstra1*
Christian R. Voolstra1*- 1Department of Biology, University of Konstanz, Konstanz, Germany
- 2ENTROPIE UMR 9220 (CNRS, IRD, UR, UNC, IFREMER) Institut de Recherche pour le Développement, Nouméa, New Caledonia
- 3Laboratory for Biological Geochemistry, School of Architecture, Civil and Environmental Engineering, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
The ecological success of corals depends on their association with microalgae and a diverse bacterial assemblage. Ocean acidification (OA), among other stressors, threatens to impair host-microbial metabolic interactions that underlie coral holobiont functioning. Volcanic CO2 seeps offer a unique opportunity to study the effects of OA in natural reef settings and provide insight into the long-term adaptations under a low pH environment. Here we compared nitrogen-fixing bacteria (diazotrophs) associated with four coral species (Pocillopora damicornis, Galaxea fascicularis, Acropora secale, and Porites rus) collected from CO2 seeps at Tutum Bay (Papua New Guinea) with those from a nearby ambient CO2 site using nifH amplicon sequencing to characterize the effects of seawater pH on bacterial communities and nitrogen cycling. Diazotroph communities were of generally low diversity across all coral species and for both sampling sites. Out of a total of 25 identified diazotroph taxa, 14 were associated with P. damicornis, of which 9 were shared across coral species. None of the diazotroph taxa, however, were consistently found across all coral species or across all samples within a species pointing to a high degree of diazotroph community variability. Rather, the majority of sampled colonies were dominated by one or two diazotroph taxa of high relative abundance. Pocillopora damicornis and Galaxea fascicularis that were sampled in both environments showed contrasting community assemblages between sites. In P. damicornis, Gammaproteobacteria and Cyanobacteria were prevalent under ambient pCO2, while a single member of the family Rhodobacteraceae was present at high relative abundance at the high pCO2 site. Conversely, in G. fascicularis diazotroph communities were indifferent between both sites. Diazotroph community changes in response to OA seem thus variable within as well as between host species, potentially arguing for haphazard diazotroph community assembly. This warrants further research into the underlying factors structuring diazotroph community assemblages and their functional role in the coral holobiont.
Introduction
Corals are adapted to the oligotrophic conditions of the tropical ocean via symbiotic associations with a variety of microbes, which extend their metabolic repertoire to support the efficient uptake and recycling of limiting nutrients (Bang et al., 2018; Robbins et al., 2019; Pogoreutz et al., 2020). The resulting metaorganism is commonly referred to as the coral holobiont (Knowlton and Rohwer, 2003; Jaspers et al., 2019). Besides endosymbiotic dinoflagellate algae of the family Symbiodiniaceae that transfer photosynthetically fixed carbon to other holobiont constituents in exchange for shelter and inorganic nutrients (Muscatine and Porter, 1977; LaJeunesse et al., 2018), associated microbes include bacteria, archaea, fungi, and viruses that all contribute to coral holobiont physiology and stress resilience (Bourne et al., 2016; Peixoto et al., 2021; Voolstra et al., 2021). Although corals can cover almost up to 100% of their carbon demand via nutrient exchange with their endosymbiotic algae (Falkowski et al., 1984; Muscatine, 1990), biomass synthesis requires the supplementation of translocated carbon with other nutrients (e.g., nitrogen). Consequently, productivity and growth of coral holobionts is argued to be limited by bioavailable nitrogen (Falkowski et al., 1993). Although corals can acquire bioavailable nitrogen through heterotrophic feeding (Grottoli et al., 2006; Houlbreque and Ferrier-Pages, 2009; Meunier et al., 2021a) and the uptake of dissolved organic nitrogen from the surrounding water (Grover et al., 2008; Tilstra et al., 2019; El-Khaled et al., 2020), they also rely on nitrogen-fixing bacteria and archaea (Rädecker et al., 2015). These so-called diazotrophs are capable of reducing N2 to ammonia (NH3) via the nitrogenase enzyme and supposedly comprise a significant source of fixed nitrogen to coral holobiont members (Fiore et al., 2010; Lema et al., 2012, 2014; Pogoreutz et al., 2017b). Typically, amplicon sequencing of the nifH gene encoding for a conserved subunit of the nitrogenase enzyme is used for the characterization of diazotroph communities associated with corals (Gaby and Buckley, 2012; Lema et al., 2012; Lesser et al., 2018; Bednarz et al., 2019). These studies indicate that diazotroph communities vary widely between locations and coral host species. Alphaproteobacteria, especially of the orders Rhizobiales and Rhodobacterales, as well as Gammaproteobacteria and Cyanobacteria are commonly found to comprise the largest part of these diazotroph communities in tropical and temperate corals (Lesser et al., 2004, 2018; Lema et al., 2012; Bednarz et al., 2019).
Given that the coral holobiont typically exists in a nitrogen-limited state (Rädecker et al., 2015, 2021), diazotrophy may help corals adjust to differences in environmental nitrogen availability (Rädecker et al., 2015; Benavides et al., 2017). In support of this, changes in environmental conditions are often met with changes in the composition of microbial communities associated with corals (Jessen et al., 2013; Roder et al., 2015; Ziegler et al., 2019). Shifts in microbial community composition and abundance supposedly change the metabolic repertoire to support the coral holobiont to adapt to changing environmental conditions (Reshef et al., 2006; Ziegler et al., 2017; Voolstra and Ziegler, 2020). Given the potential importance of diazotrophs, changes in diazotroph activity, abundance, and community composition could directly affect the response of the coral holobiont to environmental change (Santos et al., 2014; Pogoreutz et al., 2017a). In this light, ocean acidification (OA), the reduction of the pH in the Earth’s oceans due to increasing absorption of CO2 from the atmosphere, poses a direct threat to calcifying organisms such as corals (Orr et al., 2005; O’Brien et al., 2016). Reduced carbonate ion concentrations as a result of OA increase energy requirements of calcification in the coral holobiont (Cohen and Holcomb, 2009; Crook et al., 2013; Strahl et al., 2015). The resulting reduction in energy availability in the coral holobiont could thus directly affect the symbiotic interactions within the coral holobiont (McCulloch et al., 2017; D’Olivo et al., 2019).
Currently, our knowledge regarding the effects of OA on nitrogen fixation in coral holobionts is limited. Rädecker et al. (2014) found that gross nitrogen fixation decreases in Seriatopora hystrix coral holobionts when exposed to OA conditions for short periods of time. These findings, however, cannot account for any long-term environmental adaptation of coral holobionts under such conditions, including potential shifts in diazotroph community composition. Volcanic CO2 seeps offer a unique opportunity to study the long-term effects of OA on coral holobionts. Local pCO2 at some of these sites resemble levels that are expected for global oceans in the coming century (Caldeira, 2005). Coral communities at these low pH seeps show a reduced diversity with massive corals like Porites spp. being favored over more structurally complex (e.g., branching or foliose growth forms) species (Fabricius et al., 2011). Corals at these sites have been reported to have higher net photosynthesis rates (Biscéré et al., 2019), suggesting a stimulation of Symbiodiniaceae photosynthetic activity due to enhanced CO2 availability (Rädecker et al., 2017). In addition, the bacterial microbiomes of some coral species were less diverse (Morrow et al., 2015). However, a more recent study showed that massive Porites species showed almost no difference in their associated bacterial communities under high pCO2 (O’Brien et al., 2018).
In this study, we compared the diazotroph communities of four species of hermatypic corals using a nifH-based amplicon sequencing approach to characterize changes in abundance, diversity, and microbiome composition of samples from ambient and high pCO2 volcanic seep sites in Papua New Guinea. After stringent selection of functional nifH phylotypes and samples, we show that in at least one of the species, Pocillopora damicornis, different phylogenetic groups of nitrogen-fixing bacteria are present between ambient and high pCO2 sites, suggesting separate functional roles in the coral holobiont across host species.
Materials and Methods
Sample Collection
Sample collection took place during the boreal spring, between May 26th and June 7th 2018, aboard the R/V Alis, as described in Meunier et al. (2021b). At a hydrothermal vent area at Tutum Bay (Ambitle Island, New Ireland Province, Papua New Guinea), samples were collected from one site exposed to high pCO2 (4°3′44.82′′S 153°34′56.09′′E) and one reference (control) site with ambient pCO2 (4°4′0.60′′S 153°34′41.43′′E) located ca. 1.8 km south of the main vent site (Biscéré et al., 2019; Pichler et al., 2019). Collections took place at the same depths (between 3–8 m) for both sampling sites. Coral fragments were cut with a bone cutter while SCUBA diving and transported in individual zip-lock bags to the laboratory within 30 min of collection. We collected a total of 24 samples from 4 coral species at the high pCO2 site: 7 samples of Acropora secale, 5 of Galaxea fascicularis, 7 of Pocillopora damicornis, and 5 of Porites rus. Similarly, we sampled 27 colonies from 4 coral species at the control site: 10 fragments of A. secale, 5 of G. fascicularis, 9 of P. damicornis, and 3 of P. rus. Thus, a total of 51 samples were collected from both sites. Samples were immersed in DNA/RNA Shield (Zymo, Freiburg, Germany) and shipped on dry ice for DNA extraction, nifH amplification, and sequencing. The P. damicornis samples are the same as those analyzed in Meunier et al. (2021b).
DNA Collection, nifH Amplification, and Sequencing
DNA was isolated from 51 coral fragments and 1 negative control (null input sample: to assess for DNA extraction kit contamination) using the Qiagen DNeasy 96 Blood & Tissue kit (Qiagen, Hilden, Germany) with slight modifications to the manufacturer’s instructions. Briefly, coral samples were fragmented with a bone cutter and transferred to Eppendorf tubes with 400 μl of ATL buffer and 20 μl of protease. Samples where then incubated in a thermal mixer (Eppendorf, Hamburg, Germany) for 2 h at 56°C and 600 rpm. Afterward, for each sample, 200 μl of the lysate were transferred into microtubes. To aid the disintegration of coral mucus, tubes were incubated at 56°C for 1 h. DNA extraction then proceeded according to the manufacturer’s instructions. DNA concentrations were quantified using Qubit and the dsDNA High Sensitivity Assay Kit (Invitrogen, Carlsbad, United States). To amplify the nifH gene, the IGK3 (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCIWTHTAYGGIAARGGIGGIATHGGIAA-3′) and the DVV (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGATIGCRAAICCICCRCAIACIACRTC-3′) primers were used (Illumina adaptor overhangs underlined) (Ando et al., 2005). Triplicate PCRs were performed according to Lesser et al. (2018) using the Qiagen Multiplex PCR kit, with a final primer concentration of 1.2 μM in a final reaction volume of 10 μl. Thermal cycler conditions for the nifH PCR amplifications were as follows: 95°C for 15 min, 40 cycles of 95°C for 45 s, 57°C for 45 s, and 72°C for 60 s, followed by a final extension step of 72°C at 10 min. A total of 5 μl of the PCR reactions were run on a 1% agarose gel to confirm successful amplification. Triplicates for each sample were pooled, and samples were purified using Agencourt AMPure beads (Agencourt Bioscience Corporation, Beverly, MA, United States). Samples were then indexed and Illumina sequencing adaptors were added using the Nextera XT Index Kit v2. Successful addition of indexes was confirmed by comparing the length of the initial PCR product to the corresponding indexed sample on a 1% agarose gel. Samples were then cleaned and normalized to 1 ng/μl using the SequalPrep Normalization Plate Kit (Invitrogen, Carlsbad, United States). All samples were pooled in a microtube (6 μl per sample) and concentrated using a CentriVap Benchtop Vacuum Concentrator (Labconco, Kansas City, MO, United States). Next, the quality of the library was assessed using the Agilent High Sensitivity DNA Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, United States). Quantification was done using Qubit (Qubit dsDNA High Sensitivity Assay Kit, Invitrogen, Carlsbad, United States). Sequencing was performed at 6 pM with 20% phiX on the Illumina MiSeq platform at 2 × 301 bp paired-end V3 chemistry according to the manufacturer’s specifications. Sequencing data determined in this study are available at NCBI BioProject PRJNA668126 at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA668126.
nifH Amplicon Sequencing Data Analysis Pipeline
Quality control of MiSeq nifH amplicon paired reads was done using the TaxADivA (TAXonomy Assignment and DIVersity Assessment) perl wrapper script (Gaby et al., 2018), as detailed in Lesser et al. (2018) with the following settings: -y -k -r 26 -l 29 –keepc4 -g 500. Subsequent to TaxADivA-based read merging, quality control and filtering, samples with <500 sequences were removed from subsequent analyses (Lesser et al., 2018). The remaining 41 samples (including the negative control) were combined into one fasta file and minimum entropy decomposition (MED) was employed at default settings to cluster reads into oligotypes in an unsupervised fashion (Eren et al., 2015). Representative sequences of the resultant 197 final MED nodes were translated to the protein level using FrameBot (Wang et al., 2013). As a reference dataset, the “nifh_prot_ref.fasta” file included with the FrameBot program was used. To separate nifH-like genes belonging to clusters IV and V, from true functional nifH genes belonging to clusters I-III (Raymond et al., 2004), we employed a three-criteria model sensu (Angel et al., 2018). First, after alignment in MAFFT (Katoh et al., 2002), representative sequences for each final MED node were analyzed using a Classification and Regression Tree (CART) model (Frank et al., 2016). Second, we constructed an Evolutionary Placement Algorithm (EPA) Tree (Berger et al., 2011) using the corresponding RaxML implementation (Stamatakis, 2014). Minimum entropy decomposition node representatives were aligned with a reference dataset from the NifMAP pipeline (Angel et al., 2018) that included representatives of all nifH-like gene clusters (“cluster.rep.nifH-chL-bchX.0.9_noDot.fasta”). Query sequences within this combined reference alignment were then placed on the reference tree (“RAxML_bipartitions.MultipleOriginal.tree” also from the NifMAP pipeline). Third, using node representative sequences as queries, a BLASTP (Camacho et al., 2009) search was conducted against the nifH gene database taken from the Zehr lab website (1 accessed October 2nd 2020). In addition, we looked for matches in the NCBI non-redundant (nr) protein database (excluding uncultured/environmental sample sequences and automatically generated model annotations) to further confirm the results of the initial BLASTP search. In case of mismatching results but similar percentages of protein identity between the two queries, the result to the curated Zehr nifH database was preferred. In order to determine the cluster designation for the representative sequences of all final MED nodes, a node was considered to be a true nifH-representative (i.e., belonging to clusters I-III) if at least two of the three criteria produced consistent results.
In order to obtain putative species-level annotations and to collapse the MED count matrix for downstream analyses, an OTU (operational taxonomic unit) clustering at the nucleotide level was conducted via CD-HIT-EST (Fu et al., 2012). The clustering cutoff was set to 91.9%, corresponding to the species-level cutoff previously reported (Gaby et al., 2018). Reference sequences of final MED nodes that only differed by their length, i.e., by means of additional or missing nucleotides at the 5′ or 3′ of the sequence, were combined in this clustering step, which led to the removal of 42 redundant MED nodes from an initial total of 197 MED nodes. Lastly, all samples that contained <100 sequences of cluster I-III nifH representatives were removed from subsequent analyses, retaining 25 of the 40 coral samples and 1 negative control. A maximum likelihood phylogenetic tree of all final OTUs was constructed using RAxML from 20 starting trees using the PROTGAMMAAUTO method to determine an optimal substitution model (LG + G in this case). Additionally, 1,000 bootstrap trees were calculated to support the bipartitions of the best ML tree. Trees were visualized using the R package ggtree (Yu et al., 2017).
Statistical analyses within and between sites and species as well as plotting were done in R using the packages phyloseq, ape, and vegan (R Development Core Team, 2015). Pairwise PERMANOVA tests within and between coral host species for a given sampling site were done using the custom R function “pairwise_adonis.R”2 based on a Bray-Curtis dissimilarity matrix. Comparisons of Shannon diversity, inverse Simpson’s Index, and the Chao1 estimator between the two sampling sites over all species were based on Wilcoxon rank sum tests. In addition, we conducted Wilcoxon rank sum tests on those coral host species with samples available from both sites (i.e., P. damicornis and G. fascicularis).
To determine OTUs that were differentially abundant between the control and seep site, linear discriminant analysis (LDA) was conducted in the LEfSe implementation on the Galaxy web application of the Huttenhower lab (3accessed October 2nd 2020). LEfSe statistical tests for differential abundance were carried out with alpha values for factorial Kruskal-Wallis and subsequent pairwise Wilcoxon tests considered significant at ≤0.05. For the LDA analysis, a threshold score of ≥2.0 for OTUs was considered indicative for a significant difference in the relative abundance between sites. This analysis was done across samples from all species, for samples of P. damicornis, and for samples of G. fascicularis.
Taxon-Specific nifH Quantitative PCR
Patterns of relative abundance based on nifH amplicon sequencing data were confirmed using qPCR (quantitative polymerase chain reaction). For this, OTU_31 was chosen due to its pervasive presence among P. damicornis samples. A specific primer pair for OTU_31 was designed based on its MED representative sequence using Primer-BLAST (Ye et al., 2012). The OTU_31 primer pair (forward: 5′-TTGCGATCCCAAAGCTGACT-3′; reverse: 5′-GCCGGACTCAACACAGAGAA-3′) amplifies a 157 bp long region of the nifH amplicon determined from the MiSeq sequencing. Primer products were checked on a 1.2% agarose gel for single bands after PCR amplification using the following protocol: 90°C for 10 min, followed by 40 cycles of 95°C, 57°C, and 72°C for 30 s each, and a final extension step of 72°C for 10 min. Upon confirmation of a single amplification product using gel electrophoresis, amplicon gel slices from different samples were cut out, and DNA was extracted and purified using the QIAquick Gel Extraction Kit (Qiagen, Germany) following the manufacturer’s protocol. The purified product was Sanger-sequenced to confirm that the amplified sequence was identical to the OTU_31 representative sequence (Supplementary Figure 1). The 16S rRNA gene amplicon primers 784F and 1061R (Andersson et al., 2008; Bayer et al., 2013) were used to produce a standard for relative abundance calculation. To confirm that primer amplification efficiency for the nifH and 16S primer pairs were similar and close to 100%, a 10-fold serial dilution of DNA from a sample with a known presence of OTU_31 was prepared for the determination of qPCR standard curves (Supplementary Figure 2). All qPCR reactions were run on a qTOWER3 84 using the innuMIX qPCR DSGreen Standard master mix (both Analytik Jena GmbH, Germany) with final primer concentrations of 0.5 μM in a reaction volume of 10 μl with a cycling protocol of 95°C for 10 min, 50 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s, followed by 72°C for 10 min, and a subsequent melting curve to assess uniformity of amplification and to confirm the absence of primer dimers. The qPCR samples were run in triplicate in addition to a no-template and an extraction kit negative control for both primer pairs, respectively. Fold change differences were calculated using the ΔΔCT method. For this, the mean ΔCT (CTnifH – CT16S) across all samples was subtracted from the ΔCTs of individuals samples and the calculated ΔΔCTs were expressed as relative fold changes of nifH abundance using 2–ΔΔCT. Pearson’s correlation coefficient was calculated to confirm that qPCR and nifH amplicon sequencing showed similar distribution patterns of OTU_31 in all tested samples.
Results
Overview of nifH Sequencing Effort and Filtering
We collected 51 samples (plus 1 negative control, see Methods) from four different coral species: 7 and 10 samples of Pocillopora damicornis, 5 and 5 samples of Galaxea fascicularis, 7 and 9 samples of Acropora secale, and 5 and 3 samples of Porites rus from the high and control CO2 site, respectively. For each species, samples were collected from a high carbon dioxide volcanic seep site with a partial pressure pCO2 = 1179 μatm (pHT 7.73) and a nearby control site with an ambient pCO2 = 436 μatm (pHT 8.12). After amplifying nifH-like genes using the IKG3 and DVV primer pair and MiSeq sequencing, a total of 7,143,933 read pairs (on average 137,383.3 read pairs/sample) were available for analysis. After quality filtering and read pair merging, 10 samples were excluded due to low sequence counts. With the remaining 41 samples, 6,240,993 sequences were assessed using MED. Post-MED, a total of 4,531,377 sequences remained, of which 637,413 (14.1%) were annotated as functional nitrogenase genes of nifH clusters I-III, while 3,893,964 (85.9%) were identified as belonging to cluster IV and V nitrogenase gene homolog representatives. On average, coral samples were represented by 110,521 sequences after MED. Of the 40 coral host samples that were considered during MED, 15 were discarded due to having <100 sequences annotated to nifH clusters I–III. Consequently, 25 samples remained for downstream analysis: 4 A. secale, 11 P. damicornis, 7 G. fascicularis, 3 P. rus (Table 1). These 25 samples comprised 2,662,462 sequences in total. On average 23.5% of sequences from all samples could be assigned to nifH clusters I-III. Although there were no statistically significant differences in the ratio of functional nitrogenase to nifH homolog annotated sequences between species (ANOVA, p = 0.08), P. damicornis samples showed the highest presence of cluster I–III to cluster IV–V sequences (84% of assesses sequenced) (Figure 1). In addition, samples from the control site had higher relative numbers of cluster I–III sequences (33% of assessed sequences) in comparison to the seeps site (15% of assessed sequences, t test, p = 0.1). The negative control (no input DNA extraction) comprised 6,984 sequences after MED and showed a higher relative number of functional nitrogenase sequences, with 92.7% of retained sequences annotated as true nifH (Table 1 and Supplementary Table 1).
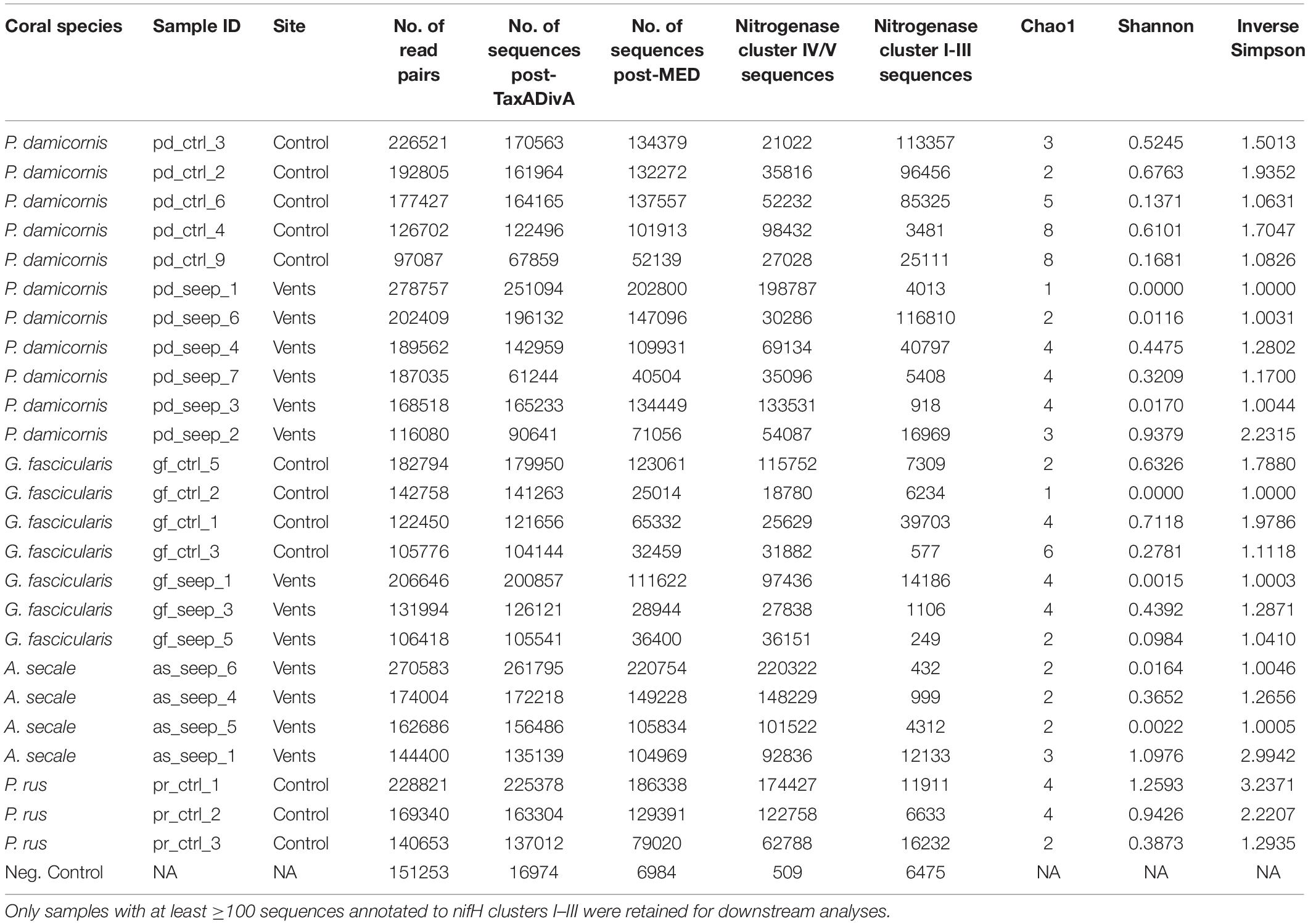
Table 1. nifH sequencing overview and alpha diversity measures of 25 coral samples from a high pCO2 (seep) and an ambient pCO2 (control) site and 1 negative control, retained after sequence processing and nifH gene filtering.
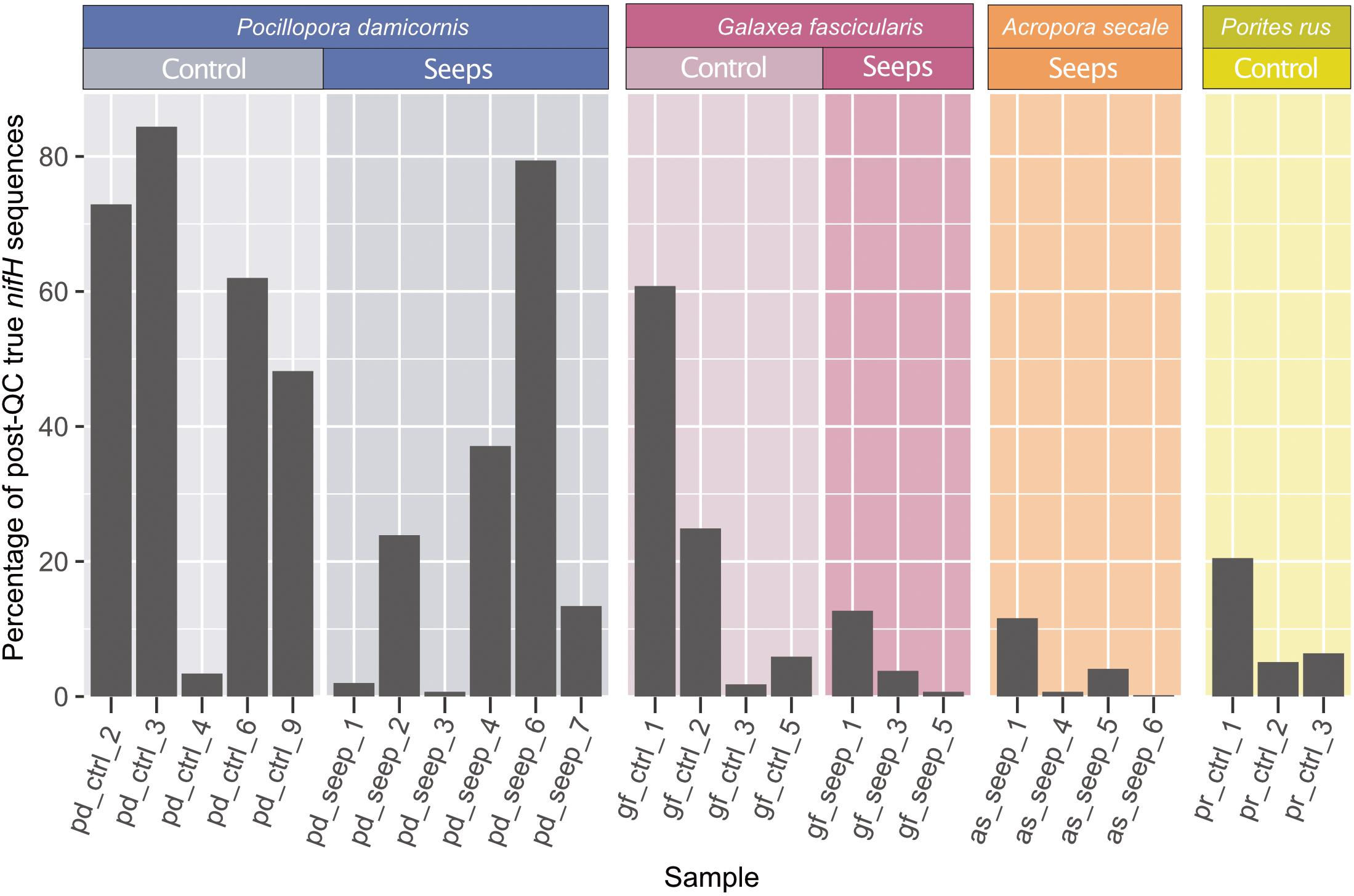
Figure 1. nifH sequencing overview. Percentage of retained sequences after quality control and minimum entropy decomposition (MED) that were annotated as true nitrogenase genes belonging to nifH Clusters I–III (black) relative to the total amount of post-QC retained sequences. Only the 25 samples with ≥100 true nifH sequences are shown and were kept for all subsequent analyses.
Phylogenetic nifH Analysis
Clustering of the 197 determined MED nodes into OTUs using a 91.9% sequence identity threshold in CD-HIT-EST resulted in 88 OTUs, of which 61 were designated as cluster IV nifH homologs or cluster V bacteriochlorophyll synthesis genes and not considered further (Supplementary Data 1). A maximum likelihood phylogenetic analysis confirmed that all cluster IV/V nifH homolog OTUs were monophyletic and highly conserved (Figure 2). The retained 27 OTUs were assigned to nifH cluster I–III. Of these, 25 were cluster I Mo-Fe nitrogenase representatives assigned as follows: Gammaproteobacteria (9 OTUs), Alphaproteobacteria (8 OTUs), Cyanobacteria (6 OTUs), and Epsilonbacteraeota (2 OTUs) (Waite et al., 2017) (Figure 2 and Supplementary Data 1). The majority of Gammaproteobacteria OTUs were identified as Alteromonadales, some closely matched Pseudomonadales and Vibrionales (Figure 2). All Alphaproteobacteria were annotated as Rhodobacterales according to BLAST in discordance with the Zehr database that annotated all OTUs except OTU_46 as Sphingomonadales. Notably, two OTUs belonging to the order of Rhizobiales were detected in the negative extraction (OTU_70: 65.52% of sequences in negative control, OTU_77: 27.19% of sequences in negative control; Supplementary Data 1), thus considered contaminants (red font annotation in Figure 2), and removed, leaving 23 cluster I OTUs for downstream analyses. Only one OTU (OTU_21) was assigned to nifH cluster II (anaerobe Mo-Fe nitrogenases), a bacterium of the class Opitutae. Likewise, only one OTU (OTU_13) was assigned to Cluster III (Mo-independent nitrogenases), a bacterium from the group Bacteroidetes/Chlorobi.
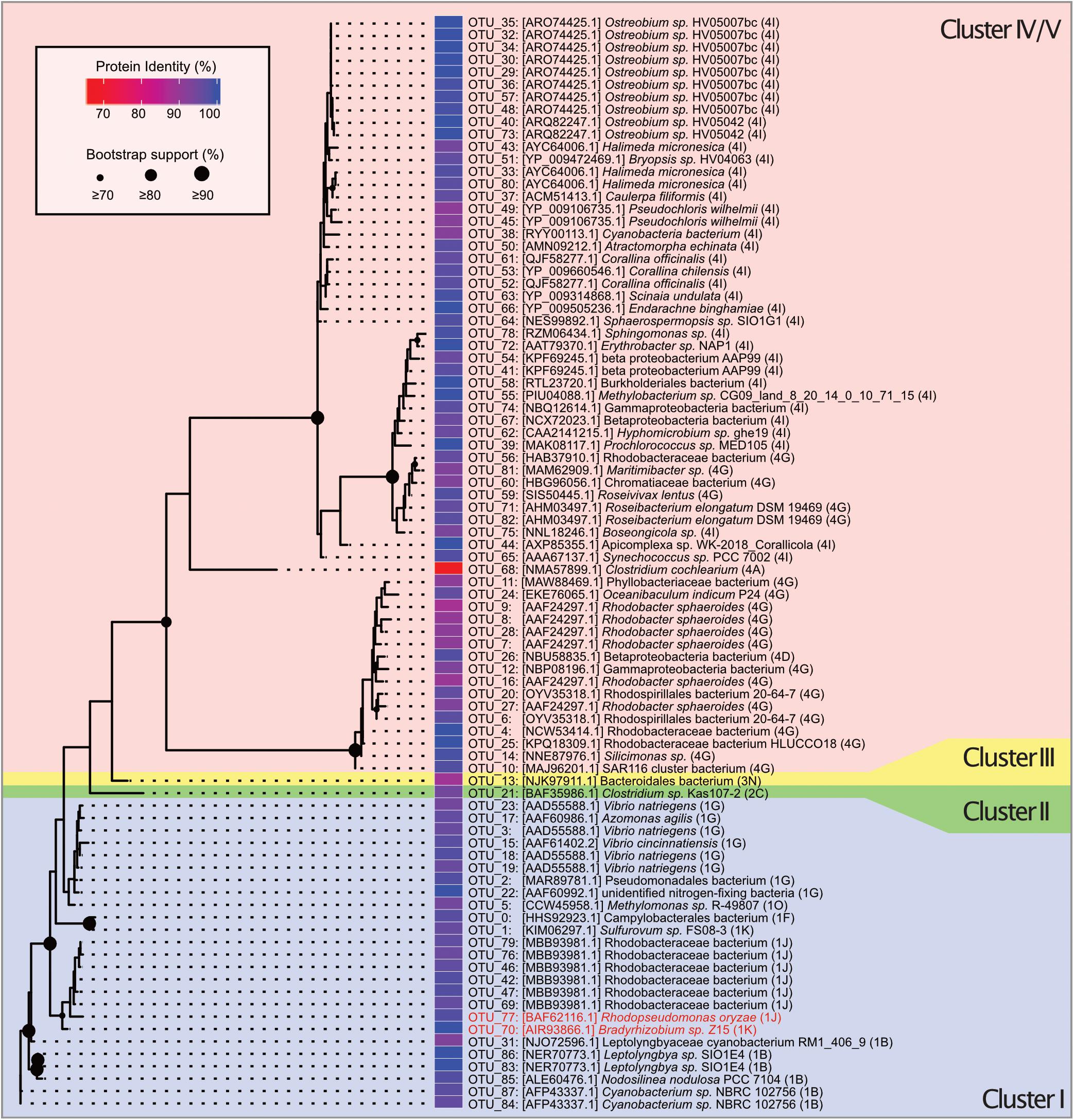
Figure 2. Unrooted maximum likelihood tree of 88 OTUs belonging to nifH gene clusters I–V constructed from 200 ML starting trees. The nifH cluster identities follow the classification and regression tree model shown in parentheses and with different background colors. Names and sequence IDs are best hits from BLASTP searches against NCBI’s non-redundant protein database. Colored cells show percent identity of the closest protein match to the OTU representative sequence. Circles on nodes indicate bootstrap support (1,000 bootstraps). Taxa identified as putative contaminants due to their presence in the negative extraction are marked in red annotation.
Diazotroph Community Composition of Coral Hosts From High pCO2 and Ambient pCO2 Sites
Based on the retained 25 OTUs, the highest number of distinct diazotrophs was found among samples of P. damicornis (14 OTUs), followed by G. fascicularis (12 OTUs), P. rus (8 OTUs), and A. secale (5 OTUs). Generally, there was little overlap in diazotroph community composition between coral species (Figure 3A). For each coral species, at least 25% of the OTUs were not observed in any other species, and none of the 25 OTUs was present in all four host species. Apart from the differences between coral species, diazotroph assemblage between colonies of the same species was also largely variant (Figure 3B). We could not detect any OTU that was consistently present across all samples of a given coral species, site, or host species-site combination with one exception. OTU_05 (Gammaproteobacteria) was found in all but one control site sample of G. fascicularis (Figure 3C). Further, we found three OTUs that were shared between three coral host species: OTU_31 (Cyanobacteria) was present in P. damicornis, G. fascicularis, and A. secale, while OTU_18 and OTU_23 (both Gammaproteobacteria) were present in P. damicornis, G. fascicularis, and P. rus. Thus, we found little similarity in diazotroph abundance between coral species as well as between colonies of a given coral species. A principal coordinate analysis of the Bray-Curtis dissimilarity matrix of relative OTU abundances between samples did not reveal a pattern of distinction between the seep and control site (Figure 3B; PERMANOVA: F = 0.88, p = 0.53). Rather, coral host species explained more of the variation between samples than site (coral species R2 = 0.22; site R2 = 0.03), with G. fascicularis and P. rus samples clustering closer together. On the other hand, samples of P. damicornis and A. secale appeared more spread out on the ordination. PERMANOVA analysis further supported this observation (F = 1.93, p = 0.0041) and pairwise comparisons indicated that the difference stems from the difference between G. fascicularis and P. damicornis samples (Supplementary Table 2; F = 2.84, FDR-adjusted p = 0.03) as well as between G. fascicularis and A. secale (F = 2.38, FDR-adjusted p = 0.03). There was no difference between sets of samples when site and coral host species were considered as factors (F = 0.75, p = 0.70).
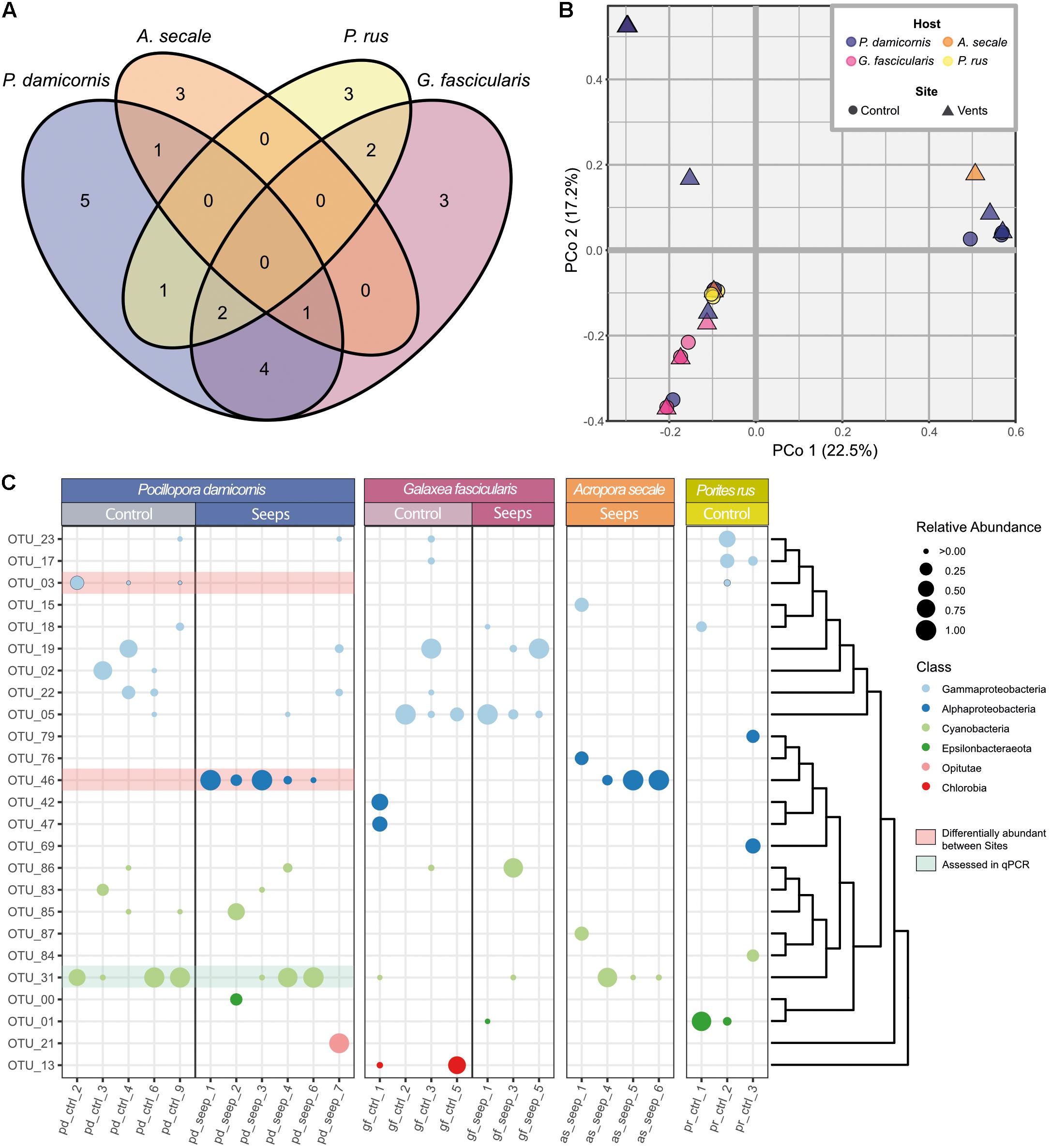
Figure 3. Diazotroph community composition of coral hosts from a high pCO2 and an ambient pCO2 site (A) Venn diagram showing number of observed OTUs and their distribution across the four examined coral species. (B) Principal coordinate analysis of Bray-Curtis distance matrix for all 25 coral samples with ≥100 sequences from the 25 cluster I-III nifH representative OTUs. First two principal coordinates are shown, explained variance in parentheses. (C) Relative abundances of OTUs per sample. OTUs are sorted according to their phylogenetic relationship as indicated by the dendrogram to the right. Class level phylogenetic association is shown in different dot colors. Red highlighted OTUs are differentially abundant between P. damicornis samples from the control and seeps site according to LEfSe analyses. OTU_31 (highlighted in green) was used for relative gene copy abundance estimates using qPCR.
Coral samples were associated with an average of only 3.08 ± 0.26 OTUs. In general, each sample was dominated by one or two highly abundant OTUs (Figure 3C). The OTU with the highest relative abundance in every sample made up on average 82.22 ± 4.02% of nifH sequences. Alpha diversity was generally very low among all samples (Table 1). There were no differences between samples or species from the control and seep site when looking at Shannon diversity (Wilcoxon = 45.5, p = 0.081), Inverse Simpson’s index (Wilcoxon = 44.5, p = 0.073), and Chao1 estimator (Wilcoxon = 54.5, p = 0.195) for all host species (Supplementary Figure 3), or when only samples of P. damicornis (Wilcoxon, all p > 0.05) or G. fascicularis (Wilcoxon, all p > 0.05), the two coral species with samples from both sites, were considered (Supplementary Table 3).
Testing for differential relative abundance of OTUs (LefSe analysis) between the high pCO2 and ambient site identified no OTUs that were indicative for either the seep or control site across all four coral host species. When only P. damicornis samples were considered, however, there were clear differences in the distribution of diazotroph taxa between samples from both sites. At the class level, Gammaproteobacteria were more prevalent at the ambient site, while Alphaproteobacteria, specifically OTU_46 (closest matching to family Rhodobacteraceae), were more prevalent at the high pCO2 site (LDA effect size = 5.41, p = 0.013). For control samples, OTU_03 (family Vibrionaceae) was identified as differentially abundant (LDA effect size = 5.13, p = 0.036), with Gammaproteobacteria as a whole being discriminant (LDA effect size = 5.40, p = 0.015). By contrast, in G. fascicularis, none of the OTUs exhibited a significant differential abundance between the high pCO2 and control site.
Confirmation of Amplicon Sequencing Results Using Taxon-Specific Quantitative PCR
We validated the nifH amplicon sequencing data using qPCR to assess and rule out potential biases stemming from differences in sequence amplification (see Figure 1) and specificity of degenerate primers (see Table 1) (Gaby and Buckley, 2012; Angel et al., 2018; Lesser et al., 2018; Tilstra et al., 2019). We chose OTU_31 as a representative because it was broadly and abundantly present among samples of P. damicornis (Figure 3C). Based on a qPCR standard curve, the specific primer pair for OTU_31 showed high efficiency (104.84%), comparable to the primer pair used for 16S rRNA gene amplification that was used as a reference (100.92%). Sanger sequencing of the single PCR product obtained confirmed that the amplicon is identical to the representative sequence of OTU_31 (Supplementary Figure 1). We assayed all samples of P. damicornis using the OTU_31 specific primer pair and the 16S rRNA gene primer pair (see Methods). The comparison of fold change values relative to 16S and the relative abundance in the nifH sequencing dataset showed high correlation between qPCR and amplicon sequences (Figure 4) with a Pearson correlation coefficient of r = 0.87 (p < 0.001).
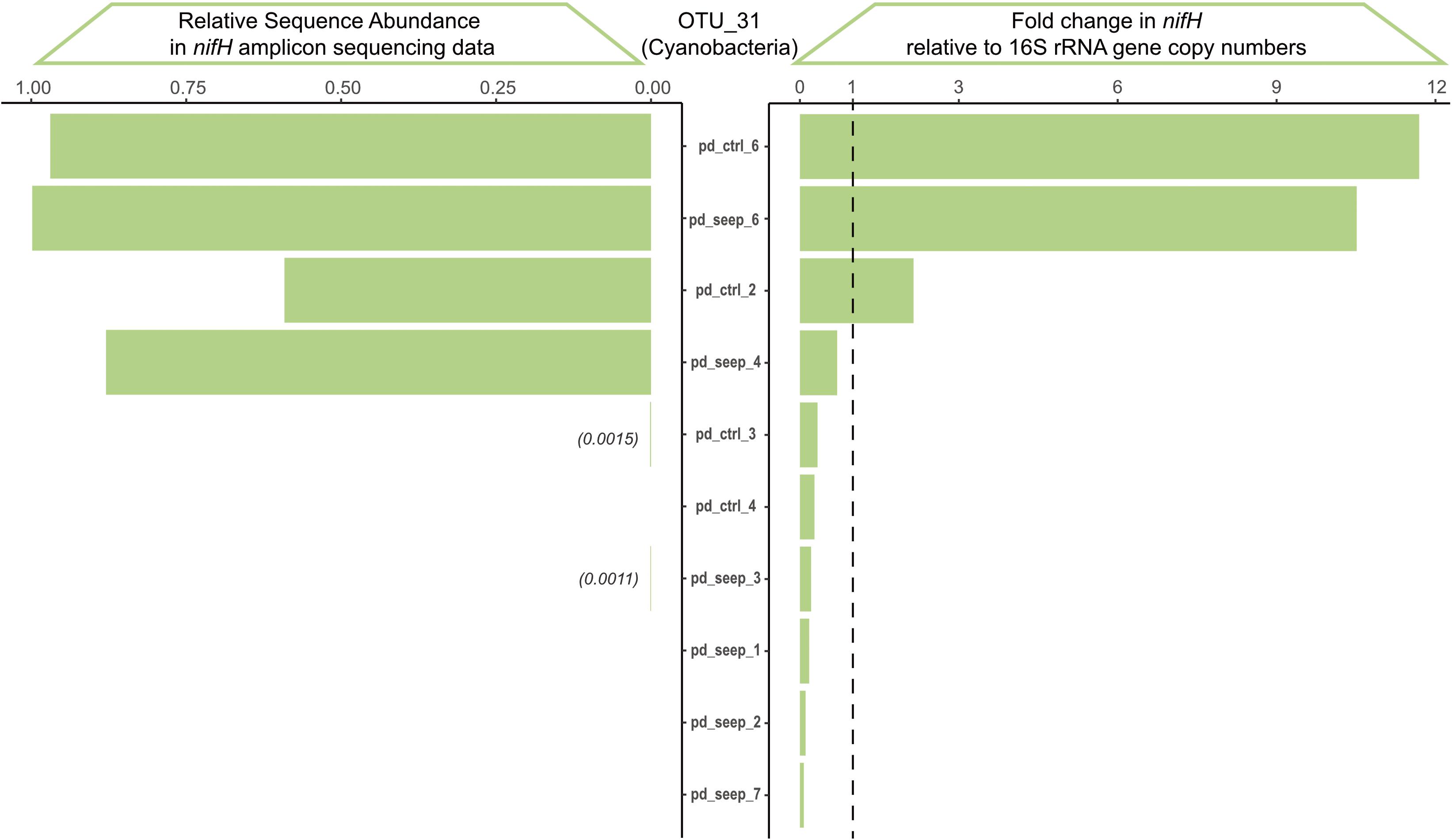
Figure 4. Validation of nifH amplicon sequencing results via qPCR based on OTU_31. OTU_31 annotates as a Cyanobacterium with broad prevalence across P. damicornis samples. The qPCR fold-change in nifH gene copy numbers was calculated relative to the 16S rRNA amplicon and shows high correlation to the relative sequence abundance determined from the nifH amplicon sequencing dataset across the 11 samples of P. damicornis (Pearson correlation coefficient r = 0.87, p = 4*10– 4).
Discussion
The functioning of the coral-algae symbiosis depends on the low, yet stable availability of nitrogen in the holobiont (Rädecker et al., 2021). The impacts of anthropogenic climate change and ocean acidification on corals will thus, in part, depend on the effect of these stressors on nitrogen cycling in the holobiont (Rädecker et al., 2015). The present study examined diazotroph, i.e., nitrogen-fixing communities, of four coral species from an ambient and a high pCO2 site in Papua New Guinea using amplicon sequencing of the nifH subunit of the nitrogenase gene to disentangle how host-identity and environmental conditions shape diazotroph assemblages in stony corals.
High False Positive Rates in nifH Sequencing Pose Unique Challenges When Analyzing Diazotroph Assemblages
To analyze nifH amplicon sequencing data, we employed a pipeline based on (Lesser et al., 2018) and modified using methods from Angel et al. (2018) to confidently assign sequences to nifH clusters. Amplification of the nifH gene is commonly achieved through the use of highly degenerate primers at high concentrations using a high number of PCR cycles. The consequential amplification and sequencing of homologous sequences that do not represent a functional nitrogenase subunit is inevitable. Thus, it is important for downstream analyses to identify and remove such false positives. Employment of three different methods to identify such sequences of nifH cluster IV and V (nifH homologs without known function and bacteriochlorophyll synthesis genes) (Raymond et al., 2004), we found that only 14.1% of sequences could be considered a functional nitrogenase gene in the present dataset. This is in line with what (Lesser et al., 2018) and (Angel et al., 2018) observed using the same primers. Notably, we found that the amount of functional nifH sequences was highly variable across individual samples, which varied widely in their overall read depths as well as the read depths of cluster I-III nifH sequences.
After removal of all cluster IV/V homologs of nifH from the initial 88 OTUs, 27 of them were identified as belonging to true, functional nitrogenase genes. Most of these nitrogenase sequences belonged to cluster I Mo-Fe nitrogenases, while cluster II and III were only represented by one OTU each. These cluster I-III nifH OTUs covered a wide range of phylogenetic backgrounds, with the commonly found groups of diazotroph associates of corals being represented by Cyanobacteria as well as Alpha- and Gamma proteobacteria. Notably, samples in this study contained two cluster I nifH OTUs belonging to the Rhizobiales order, closely related to Bradyrhizobium sp. and Rhodopseudomonas oryzae, respectively. Rhizobiales are commonly reported in diazotroph assemblages of corals (Santos et al., 2014; Lesser et al., 2018; Bednarz et al., 2019), sometimes comprising a large relative abundance of up to 70% (Lema et al., 2012). In this study, two Rhizobiales OTUs of the family Bradyrhizobiaceae were identified across samples, but they also amplified in the negative DNA extraction control (no sample input) where they were the most abundant. This finding cautions the high abundance and importance of Rhizobiales in coral diazotroph communities proposed previously and highlights the importance of implementing negative controls, as noted previously (Laurence et al., 2014; Salter et al., 2014; Weyrich et al., 2019). Following the above, we excluded these two OTUs (OTU_70, OTU_77) from consideration in downstream analysis.
Coral-Associated Diazotroph Communities Are Simple and Non-Conserved Within and Between Host Species
Previous studies suggest a mutualistic relationship between corals and their associated nitrogen-fixing bacteria that could provide adaptive potential to the holobiont under a changing environment (Lesser et al., 2007; Cardini et al., 2014; Benavides et al., 2016; Voolstra and Ziegler, 2020). The functional importance of diazotrophs could thus affect the coral holobiont in two opposing ways: (i) diazotroph community assemblages are flexible, thereby allowing to dynamically change in response to changing environmental conditions (Roder et al., 2015; Ziegler et al., 2017; Voolstra and Ziegler, 2020), or (ii) the dependence of the holobiont on fixed nitrogen results in a near obligate association with a low level of flexibility under changing environmental conditions.
We observed that most coral samples harbored only two to five distinct diazotrophs, and each sample was dominated by one or two taxa. The overall validity of the patterns observed in the amplicon sequencing results was confirmed by the high correlation between qPCR-based cycle threshold (CT) fold-change difference and relative abundance in the amplicon sequencing data of OTU_31. Thus, considering that we identified on average only one or two OTUs of high relative abundance in most samples, coral-associated diazotroph communities may be even less diverse than reported in previous studies (Lema et al., 2012, 2014; Lesser et al., 2018; Bednarz et al., 2019), at least for the coral species in this study.
Considering all samples analyzed, coral host species were predominantly associated with Alpha- and Gammaproteobacteria, as well as Cyanobacteria (Figure 3C). This is largely in line with previous studies of nifH-based analyses in corals that found pre-dominance of Alpha-, Delta-, Gammaproteobacteria (Zhang et al., 2016; Lesser et al., 2018), Alpha-, Delta-, Gammaproteobacteria as well as Cyanobacteria and Bacteroidetes (Bednarz et al., 2019), or Alphaproteobacteria (majoritively Rhizobiales, but see above) (Lema et al., 2014). Notably, Deltaproteobacteria, although found in other studies (Zhang et al., 2016; Bednarz et al., 2019), were not found in samples from this study.
In P. damicornis, diazotroph communities exhibited prevalent association with Gammaproteobacteria at the control site in comparison to samples from the high pCO2 site that were predominantly associated with an Alphaproteobacterium of the Rhodobacteraceae family (OTU_46, Figure 3C). This resembles findings by Yang et al. (2015) that observed a shifting dominance of Alpha- to Gammaproteobacteria in P. damicornis in a coast to open sea gradient. Bacteria of the family Rhodobacteraceae are commonly associated with stress, injury, or disease in corals (Sekar et al., 2008; Sunagawa et al., 2009; Roder et al., 2014a,b).
The absence of diazotroph diversity or conserved coral species-specific assemblages are striking and warrant further investigation regarding the underlying reasons and the broad validity of these observations. From the current data, it seems that diazotrophs may be opportunistically acquired (i.e., diazotroph assemblage is haphazard) from the surrounding environment, based on their functional contribution. As such, it may be unlikely to find a pattern of phylosymbiosis and cophylogeny as demonstrated previously for coral-associated bacteria (Pollock et al., 2018). This may provide one explanation for the de facto absence of conserved diazotroph community patterns. Considering the notion that corals fix on average less nitrogen than other benthic reef organisms (Cardini et al., 2014), diazotrophy of coral-associated microbes may not be as essential for holobiont functioning as previously thought.
Although volcanic CO2 seeps offer a unique opportunity to examine the consequences of OA in a natural setting, they offer only a steady-state view of microbiome composition under high pCO2 conditions and do not allow teasing apart the adaptive potential underlying microbiome assemblage differences. Further studies will be required to complement nifH sequencing results with physiological measurements to disentangle the contribution and implications of diazotroph microbiome composition for holobiont functioning under ambient and high pCO2 conditions. Ideally, isolating and culturing approaches of differentially abundant OTUs combined with genome sequencing will clarify a potential functional transition from heterotrophic Gammaproteobacteria to autotrophic Alphaproteobacteria under OA, as observed in P. damicornis. In addition, in situ transplantation experiments of coral fragments between high and ambient pCO2 sites offer a promising approach to study diazotroph assemblage dynamics.
Data Availability Statement
All sequencing data determined in this study are available at NCBI under BioProject PRJNA668126 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA668126).
Author Contributions
RR-M, FH, and CRV conceived and designed the study. LG, VM, NR, and GP generated data. LG, VM, and CRV analyzed data. RR-M, FH, and CRV contributed tools, reagents, and supervision. LG, NR, and CRV wrote the manuscript. LG, VM, NR, GP, RR-M, FH, and CRV revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded to RR-M by the French National Research Agency (ANR, project CARIOCA no. ANR15CE02-0006- 01, 2015), by Fonds Pacifique (project AMBITLE no. 1598, 2016), by the Flotte Océanographique Française for using the research vessel Alis and to FH by LabEx-Corail (FLAMENCO project). VM was the beneficiary of a Ph.D. grant from LabEx-Corail (MACADAM project).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to kindly thank the KAUST BioScience Core Laboratory (BCL) for assistance with sequencing. We are grateful to the local populations for access to their reef, and to the National Research Institute, the Milne Bay Provincial Research Committee, the New Ireland Provincial Administration, and the Conservation and Environment Protection Authority of Papua New Guinea and the University of Papua New Guinea for permits. Thanks to the crew of the R/V Alis and of the diving boat Chertan.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.754682/full#supplementary-material
Footnotes
- ^ https://www.jzehrlab.com/nifh
- ^ https://github.com/pmartinezarbizu/pairwiseAdonis
- ^ https://huttenhower.sph.harvard.edu/galaxy/
References
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. doi: 10.1371/journal.pone.0002836
Ando, S., Goto, M., Meunchang, S., Thongra-ar, P., Fujiwara, T., Hayashi, H., et al. (2005). Detection of nifH sequences in sugarcane (Saccharum officinarum L.) and pineapple (Ananas comosus [L.] Merr.). Soil Sci. Plant Nutr. 51, 303–308. doi: 10.1111/j.1747-0765.2005.tb00034.x
Angel, R., Nepel, M., Panhölzl, C., Schmidt, H., Herbold, C. W., Eichorst, S. A., et al. (2018). Evaluation of primers targeting the diazotroph functional gene and development of NifMAP – a bioinformatics pipeline for analyzing nifH amplicon data. Front. Microbiol. 9:703. doi: 10.3389/fmicb.2018.00703
Bang, C., Dagan, T., Deines, P., Dubilier, N., Duschl, W. J., Fraune, S., et al. (2018). Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology 127, 1–19. doi: 10.1016/j.zool.2018.02.004
Bayer, T., Neave, M. J., Alsheikh-Hussain, A., Aranda, M., Yum, L. K., Mincer, T., et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl. Environ. Microbiol. 79, 4759–4762. doi: 10.1128/AEM.00695-13
Bednarz, V. N., van de Water, J. A. J. M., Rabouille, S., Maguer, J.-F., Grover, R., and Ferrier-Pagès, C. (2019). Diazotrophic community and associated dinitrogen fixation within the temperate coral Oculina patagonica. Environ. Microbiol. 21, 480–495. doi: 10.1111/1462-2920.14480
Benavides, M., Bednarz, V. N., and Ferrier-Pagès, C. (2017). Diazotrophs: overlooked key players within the coral symbiosis and tropical reef ecosystems? Front. Mar. Sci. 4:10. doi: 10.3389/fmars.2017.00010
Benavides, M., Houlbrèque, F., Camps, M., Lorrain, A., Grosso, O., and Bonnet, S. (2016). Diazotrophs: a non-negligible source of nitrogen for the tropical coral Stylophora pistillata. J. Exp. Biol. 219, 2608–2612. doi: 10.1242/jeb.139451
Berger, S. A., Krompass, D., and Stamatakis, A. (2011). Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol. 60, 291–302. doi: 10.1093/sysbio/syr010
Biscéré, T., Zampighi, M., Lorrain, A., Jurriaans, S., Foggo, A., Houlbrèque, F., et al. (2019). High p CO 2 promotes coral primary production. Biol. Lett. 15:20180777. doi: 10.1098/rsbl.2018.0777
Bourne, D. G., Morrow, K. M., and Webster, N. S. (2016). Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Caldeira, K. (2005). Ocean model predictions of chemistry changes from carbon dioxide emissions to the atmosphere and ocean. J. Geophys. Res. 110:C09S04. doi: 10.1029/2004jc002671
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cardini, U., Bednarz, V. N., Foster, R. A., and Wild, C. (2014). Benthic N2 fixation in coral reefs and the potential effects of human-induced environmental change. Ecol. Evol. 4, 1706–1727. doi: 10.1002/ece3.1050
Cohen, A. L., and Holcomb, M. (2009). Why corals care about ocean acidification: uncovering the mechanism. Oceanography 22, 118–127.
Crook, E. D., Cohen, A. L., Rebolledo-Vieyra, M., Hernandez, L., and Paytan, A. (2013). Reduced calcification and lack of acclimatization by coral colonies growing in areas of persistent natural acidification. Proc. Natl. Acad. Sci. U.S.A. 110, 11044–11049. doi: 10.1073/pnas.1301589110
D’Olivo, J. P., Georgiou, L., Falter, J., DeCarlo, T. M., Irigoien, X., Voolstra, C. R., et al. (2019). Long-term impacts of the 1997–1998 bleaching event on the growth and resilience of massive porites corals from the central Red Sea. Geochem. Geophys. Geosyst. 20, 2936–2954. doi: 10.1029/2019GC008312
El-Khaled, Y. C., Roth, F., Tilstra, A., Rädecker, N., Karcher, D. B., Kürten, B., et al. (2020). In situ eutrophication stimulates dinitrogen fixation, denitrification, and productivity in Red Sea coral reefs. Mar. Ecol. Prog. Ser. 645, 55–66. doi: 10.3354/meps13352
Eren, A. M., Morrison, H. G., Lescault, P. J., Reveillaud, J., Vineis, J. H., and Sogin, M. L. (2015). Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 9, 968–979. doi: 10.1038/ismej.2014.195
Fabricius, K. E., Langdon, C., Uthicke, S., Humphrey, C., Noonan, S., De’ath, G., et al. (2011). Losers and winners in coral reefs acclimatized to elevated carbon dioxide concentrations. Nat. Clim. Chang. 1, 165–169. doi: 10.1038/nclimate1122
Falkowski, P. G., Dubinsky, Z., Muscatine, L., and McCloskey, L. (1993). Population control in symbiotic corals. Bioscience 43, 606–611. doi: 10.2307/1312147
Falkowski, P. G., Dubinsky, Z., Muscatine, L., and Porter, J. W. (1984). Light and the bioenergetics of a symbiotic coral. Bioscience 34, 705–709. doi: 10.2307/1309663
Fiore, C. L., Jarett, J. K., Olson, N. D., and Lesser, M. P. (2010). Nitrogen fixation and nitrogen transformations in marine symbioses. Trends Microbiol. 18, 455–463. doi: 10.1016/j.tim.2010.07.001
Frank, I. E., Turk-Kubo, K. A., and Zehr, J. P. (2016). Rapid annotation of nifH gene sequences using classification and regression trees facilitates environmental functional gene analysis. Environ. Microbiol. Rep. 8, 905–916. doi: 10.1111/1758-2229.12455
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gaby, J. C., and Buckley, D. H. (2012). A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. doi: 10.1371/journal.pone.0042149
Gaby, J. C., Rishishwar, L., Valderrama-Aguirre, L. C., Green, S. J., Valderrama-Aguirre, A., Jordan, I. K., et al. (2018). Diazotroph community characterization via a high-throughput nifH amplicon sequencing and analysis pipeline. Appl. Environ. Microbiol. 84, e01512–e01517. doi: 10.1128/AEM.01512-17
Grottoli, A. G., Rodrigues, L. J., and Palardy, J. E. (2006). Heterotrophic plasticity and resilience in bleached corals. Nature 440, 1186–1189. doi: 10.1038/nature04565
Grover, R., Maguer, J.-F., Allemand, D., and Ferrier-Pagès, C. (2008). Uptake of dissolved free amino acids by the scleractinian coral Stylophora pistillata. J. Exp. Biol. 211, 860–865. doi: 10.1242/jeb.012807
Houlbreque, F., and Ferrier-Pages, C. (2009). Heterotrophy in tropical Scleractinian corals. Biol. Rev. Camb. Philos. Soc. 84, 1–17. doi: 10.1111/j.1469-185x.2008.00058.x
Jaspers, C., Fraune, S., Arnold, A. E., Miller, D. J., Bosch, T. C. G., Voolstra, C. R., et al. (2019). Resolving structure and function of metaorganisms through a holistic framework combining reductionist and integrative approaches. Zoology 133, 81–87. doi: 10.1016/j.zool.2019.02.007
Jessen, C., Villa Lizcano, J. F., Bayer, T., Roder, C., Aranda, M., Wild, C., et al. (2013). In-situ effects of eutrophication and overfishing on physiology and bacterial diversity of the red sea coral Acropora hemprichii. PLoS One 8:e62091. doi: 10.1371/journal.pone.0062091
Katoh, K., Misawa, K., Kuma, K.-I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Knowlton, N., and Rohwer, F. (2003). Multispecies microbial mutualisms on coral reefs: the host as a habitat. Am. Nat. 162, S51–S62. doi: 10.1086/378684
LaJeunesse, T. C., Parkinson, J. E., Gabrielson, P. W., Jeong, H. J., Reimer, J. D., Voolstra, C. R., et al. (2018). Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral Endosymbionts. Curr. Biol. 28, 2570–2580. doi: 10.1016/j.cub.2018.07.008
Laurence, M., Hatzis, C., and Brash, D. E. (2014). Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS One 9:e97876. doi: 10.1371/journal.pone.0097876
Lema, K. A., Willis, B. L., and Bourne, D. G. (2012). Corals form characteristic associations with symbiotic nitrogen-fixing bacteria. Appl. Environ. Microbiol. 78, 3136–3144. doi: 10.1128/AEM.07800-11
Lema, K. A., Willis, B. L., and Bourne, D. G. (2014). Amplicon pyrosequencing reveals spatial and temporal consistency in diazotroph assemblages of the Acropora milleporamicrobiome. Environ. Microbiol. 16, 3345–3359. doi: 10.1111/1462-2920.12366
Lesser, M. P., Bythell, J. C., Gates, R. D., Johnstone, R. W., and Hoegh-Guldberg, O. (2007). Are infectious diseases really killing corals? Alternative interpretations of the experimental and ecological data. J. Exp. Mar. Bio. Ecol. 346, 36–44. doi: 10.1016/j.jembe.2007.02.015
Lesser, M. P., Mazel, C. H., Gorbunov, M. Y., and Falkowski, P. G. (2004). Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305, 997–1000. doi: 10.1126/science.1099128
Lesser, M. P., Morrow, K. M., Pankey, S. M., and Noonan, S. H. C. (2018). Diazotroph diversity and nitrogen fixation in the coral Stylophora pistillata from the Great Barrier Reef. ISME J. 12, 813–824. doi: 10.1038/s41396-017-0008-6
McCulloch, M. T., D’Olivo, J. P., Falter, J., Holcomb, M., and Trotter, J. A. (2017). Coral calcification in a changing world and the interactive dynamics of pH and DIC upregulation. Nat. Commun. 8:15686. doi: 10.1038/ncomms15686
Meunier, V., Bonnet, S., Benavides, M., Ravache, A., Grosso, O., Lambert, C., et al. (2021a). Diazotroph-derived nitrogen assimilation strategies differ by scleractinian coral species. Front. Mar. Sci. 8:692248. doi: 10.3389/fmars.2021.692248
Meunier, V., Geissler, L., Bonnet, S., Rädecker, N., Perna, G., Grosso, O., et al. (2021b). Microbes support enhanced nitrogen requirements of coral holobionts in a high CO2 environment. Mol. Ecol. doi: 10.1111/mec.16163 [Epub ahead of print].
Morrow, K. M., Bourne, D. G., Humphrey, C., Botté, E. S., Laffy, P., Zaneveld, J., et al. (2015). Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J. 9, 894–908. doi: 10.1038/ismej.2014.188
Muscatine, L. (1990). The role of symbiotic algae in carbon and energy flux in reef corals. Coral Reefs 25, 75–87.
Muscatine, L., and Porter, J. W. (1977). Reef corals: mutualistic symbioses adapted to nutrient-poor environments. Bioscience 27, 454–460. doi: 10.2307/1297526
O’Brien, P. A., Morrow, K. M., Willis, B. L., and Bourne, D. G. (2016). Implications of ocean acidification for marine microorganisms from the free-living to the host-associated. Front. Mar. Sci. 3:47. doi: 10.3389/fmars.2016.00047
O’Brien, P. A., Smith, H. A., Fallon, S., Fabricius, K., Willis, B. L., Morrow, K. M., et al. (2018). Elevated CO2 has little influence on the bacterial communities associated with the ph-tolerant coral, massive porites spp. Front. Microbiol. 9:2621. doi: 10.3389/fmicb.2018.02621
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., et al. (2005). Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms. Nature 437, 681–686. doi: 10.1038/nature04095
Peixoto, R. S., Sweet, M., Villela, H. D. M., Cardoso, P., Thomas, T., Voolstra, C. R., et al. (2021). Coral probiotics: premise, promise, prospects. Annu. Rev. Anim. Biosci. 9, 265–288. doi: 10.1146/annurev-animal-090120-115444
Pichler, T., Biscéré, T., Kinch, J., Zampighi, M., Houlbrèque, F., and Rodolfo-Metalpa, R. (2019). Suitability of the shallow water hydrothermal system at Ambitle Island (Papua New Guinea) to study the effect of high pCO2 on coral reefs. Mar. Pollut. Bull. 138, 148–158. doi: 10.1016/j.marpolbul.2018.11.003
Pogoreutz, C., Rädecker, N., Cárdenas, A., Gärdes, A., Wild, C., and Voolstra, C. R. (2017b). Nitrogen fixation aligns with nifH abundance and expression in two coral trophic functional groups. Front. Microbiol. 8:1187. doi: 10.3389/fmicb.2017.01187
Pogoreutz, C., Rädecker, N., Cárdenas, A., Gärdes, A., Voolstra, C. R., and Wild, C. (2017a). Sugar enrichment provides evidence for a role of nitrogen fixation in coral bleaching. Glob. Chang. Biol. 23, 3838–3848. doi: 10.1111/gcb.13695
Pogoreutz, C., Voolstra, C. R., Rädecker, N., Weis, V., Cardenas, A., and Raina, J.-B. (2020). “The coral holobiont highlights the dependence of cnidarian animal hosts on their associated microbes,” in Cellular Dialogues in the Holobiont, eds T. C. G. Bosch and M. G. Hadfield (Boca Raton, FL: CRC Press), 91–118.
Pollock, F. J., McMinds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921. doi: 10.1038/s41467-018-07275-x
R Development Core Team, (2015). A Language and Environment for Statistical Computing. 2008. Vienna: R Development Core Team.
Rädecker, N., Meyer, F. W., Bednarz, V. N., Cardini, U., and Wild, C. (2014). Ocean acidification rapidly reduces dinitrogen fixation associated with the hermatypic coral Seriatopora hystrix. Mar. Ecol. Prog. Ser. 511, 297–302. doi: 10.3354/meps10912
Rädecker, N., Pogoreutz, C., Gegner, H. M., Cárdenas, A., Roth, F., Bougoure, J., et al. (2021). Heat stress destabilizes symbiotic nutrient cycling in corals. Proc. Natl. Acad. Sci. U.S.A 118:e2022653118. doi: 10.1073/pnas.2022653118
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J., and Wild, C. (2015). Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497. doi: 10.1016/j.tim.2015.03.008
Rädecker, N., Pogoreutz, C., Wild, C., and Voolstra, C. R. (2017). Stimulated respiration and net photosynthesis in Cassiopeia sp. during glucose enrichment suggests in hospite CO2 limitation of algal endosymbionts. Front. Mar. Sci. 4:267. doi: 10.3389/fmars.2017.00267
Raymond, J., Siefert, J. L., Staples, C. R., and Blankenship, R. E. (2004). The natural history of nitrogen fixation. Mol. Biol. Evol. 21, 541–554. doi: 10.1093/molbev/msh047
Reshef, L., Koren, O., Loya, Y., Zilber-Rosenberg, I., and Rosenberg, E. (2006). The coral probiotic hypothesis. Environ. Microbiol. 8, 2068–2073. doi: 10.1111/j.1462-2920.2006.01148.x
Robbins, S. J., Singleton, C. M., Chan, C. X., Messer, L. F., Geers, A. U., Ying, H., et al. (2019). A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 4, 2090–2100. doi: 10.1038/s41564-019-0532-4
Roder, C., Arif, C., Bayer, T., Aranda, M., Daniels, C., Shibl, A., et al. (2014a). Bacterial profiling of white plague disease in a comparative coral species framework. ISME J. 8, 31–39. doi: 10.1038/ismej.2013.127
Roder, C., Arif, C., Daniels, C., Weil, E., and Voolstra, C. R. (2014b). Bacterial profiling of white plague disease across corals and oceans indicates a conserved and distinct disease microbiome. Mol. Ecol. 23, 965–974. doi: 10.1111/mec.12638
Roder, C., Bayer, T., Aranda, M., Kruse, M., and Voolstra, C. R. (2015). Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 24, 3501–3511. doi: 10.1111/mec.13251
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Santos, H. F., Carmo, F. L., Duarte, G., Dini-Andreote, F., Castro, C. B., Rosado, A. S., et al. (2014). Climate change affects key nitrogen-fixing bacterial populations on coral reefs. ISME J. 8, 2272–2279. doi: 10.1038/ismej.2014.70
Sekar, R., Kaczmarsky, L. T., and Richardson, L. L. (2008). Microbial community composition of black band disease on the coral host Siderastrea siderea from three regions of the wider Caribbean. Mar. Ecol. Prog. Ser. 362, 85–98. doi: 10.3354/meps07496
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Strahl, J., Stolz, I., Uthicke, S., Vogel, N., Noonan, S. H. C., and Fabricius, K. E. (2015). Physiological and ecological performance differs in four coral taxa at a volcanic carbon dioxide seep. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 184, 179–186. doi: 10.1016/j.cbpa.2015.02.018
Sunagawa, S., DeSantis, T. Z., Piceno, Y. M., Brodie, E. L., DeSalvo, M. K., Voolstra, C. R., et al. (2009). Bacterial diversity and white plague disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J. 3, 512–521. doi: 10.1038/ismej.2008.131
Tilstra, A., El-Khaled, Y. C., Roth, F., Rädecker, N., Pogoreutz, C., Voolstra, C. R., et al. (2019). Denitrification aligns with N2 fixation in Red Sea Corals. Sci. Rep. 9:19460. doi: 10.1038/s41598-019-55408-z
Voolstra, C. R., Suggett, D. J., Peixoto, R., Parkinson, J. E., Quigley, K., Silveira, C., et al. (2021). Extending the adaptive capacity of corals to survive climate change. Nat. Rev. Earth Environ. doi: 10.1038/s43017-021-00214-3 [Epub ahead of print].
Voolstra, C. R., and Ziegler, M. (2020). Adapting with microbial help: microbiome flexibility facilitates rapid responses to environmental change. Bioessays 42:e2000004. doi: 10.1002/bies.202000004
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Wang, Q., Quensen, J. F. III, Fish, J. A., Lee, T. K., Sun, Y., Tiedje, J. M., et al. (2013). Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. MBio 4, e592–e513. doi: 10.1128/mBio.00592-13
Weyrich, L. S., Farrer, A. G., Eisenhofer, R., Arriola, L. A., Young, J., Selway, C. A., et al. (2019). Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996. doi: 10.1111/1755-0998.13011
Yang, Q., Dong, J., Zhang, Y., Ling, J., Wang, D., Wu, M., et al. (2015). Diversity analysis of diazotrophs associated with corals from Xisha and Sanya, South China Sea. Aquat. Ecosyst. Health Manag. 18, 433–442. doi: 10.1080/14634988.2015.1092850
Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., and Madden, T. L. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. doi: 10.1186/1471-2105-13-134
Yu, G., Smith, D. K., Zhu, H., Guan, Y., and Lam, T. T.-Y. (2017). Ggtree : an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210x.12628
Zhang, Y., Yang, Q., Ling, J., Van Nostrand, J. D., Shi, Z., Zhou, J., et al. (2016). The shifts of Diazotrophic communities in spring and summer associated with Coral Galaxea astreata, Pavona decussata, and Porites lutea. Front. Microbiol. 7:1870. doi: 10.3389/fmicb.2016.01870
Ziegler, M., Grupstra, C. G. B., Barreto, M. M., Eaton, M., BaOmar, J., Zubier, K., et al. (2019). Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10:3092. doi: 10.1038/s41467-019-10969-5
Keywords: coral holobiont, diazotrophy, nitrogen fixation, climate change, ocean acidification, nifH, next-generation sequencing
Citation: Geissler L, Meunier V, Rädecker N, Perna G, Rodolfo-Metalpa R, Houlbrèque F and Voolstra CR (2021) Highly Variable and Non-complex Diazotroph Communities in Corals From Ambient and High CO2 Environments. Front. Mar. Sci. 8:754682. doi: 10.3389/fmars.2021.754682
Received: 06 August 2021; Accepted: 24 September 2021;
Published: 28 October 2021.
Edited by:
Aldo Cróquer, The Nature Conservancy, Dominican RepublicReviewed by:
Julie L. Meyer, University of Florida, United StatesDaniel Pech, The South Border College (ECOSUR), Mexico
Copyright © 2021 Geissler, Meunier, Rädecker, Perna, Rodolfo-Metalpa, Houlbrèque and Voolstra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian R. Voolstra, Y2hyaXN0aWFuLnZvb2xzdHJhQHVuaS1rb25zdGFuei5kZQ==
†These authors have contributed equally to this work