
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 10 November 2021
Sec. Marine Evolutionary Biology, Biogeography and Species Diversity
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.754332
 Huan Wang1,2,3
Huan Wang1,2,3 Meng Wang1,2,3Shen Fan1,2,3Jie Lu1,2,3
Meng Wang1,2,3Shen Fan1,2,3Jie Lu1,2,3 Yi Lan4,5
Yi Lan4,5 Menggong Li6
Menggong Li6 Jiangyan Li7
Jiangyan Li7 Rulong Liu7
Rulong Liu7 Jin Sun6Jiasong Fang7Pei-Yuan Qian4,5Yu-Zhong Zhang1,2,3
Jin Sun6Jiasong Fang7Pei-Yuan Qian4,5Yu-Zhong Zhang1,2,3 Weipeng Zhang1,4*
Weipeng Zhang1,4*Culture enrichment was developed to discover the uncharted microbial species in the environmental microbiota. Yet this strategy has not been widely used to study microbes of deep-sea environments. Here, we report the cultivation and metagenomic analysis of oceanic sediment microbiota collected from 6,477 m deep in the Mariana Trench. The sediment samples were cultured anaerobically in the laboratory for 4 months, before being subjected to full-length 16S rRNA gene sequencing using the PacBio technique and metagenome sequencing using both the Illumina and Oxford Nanopore techniques. The 16S rRNA gene analyses revealed 437 operational taxonomic units specific to the cultured microbes, despite the lower diversity of the cultured microbiota in comparison with the original. Metagenome analyses revealed the prevalence of functions related to respiration, energy production, and stress response in the cultured microbes, suggesting these functions may contribute to microbial proliferation under laboratory conditions. Binning of the assembled metagenome contigs of the cultured microbiota generated four nearly complete genomes affiliated to yet unclassified species under the genera Alcanivorax, Idiomarina, Sulfitobacter, and Erythrobacter. Excepting Alcanivorax, the other three genera were almost undetectable in the original samples and largely enriched in the cultured samples. The four genomes possessed a variety of genes for carbohydrate utilization and nitrite reduction, pointing to an ability to respire diverse carbon sources using nitrite as the final electron acceptor. Taken together, the findings suggest that a combination of culture enrichment and long-read sequencing is an ideal way to mine novel microbial species in the hadal environment, particularly species that are rare in their native environmental niches, and thus expand our understanding of the hadal microbial diversity and function.
Due to the limitation of culturing conditions (Nichols et al., 2010; Zhang et al., 2018c) or the characteristics like slow growth (Vartoukian et al., 2010), most microbial species comprising environmental microbiota cannot be isolated (Deming and Baross, 2000). In order to discover and analyze the yet uncharted microbial species in environmental samples, culture enrichment can be carried out under laboratory conditions. Studies have proved that culture enrichment can improve the recovery rate of previously uncultured bacteria (Gich et al., 2012; Pham and Kim, 2012). Integrating laboratory culture enrichment and DNA sequencing techniques, culture-enriched metagenomics provides an ideal way to study environmental samples that have a limited microbial biomass or proportion. This approach has been recently used in the analyses of the microbiota in human cystic fibrosis sputum, which contains mostly human rather than microbial DNA, and has led to the identification of new species and an improvement in microbial genome assembly efficiency (Whelan et al., 2020). Culture-enriched metagenomics can be further improved along with the development of sequencing techniques. As yet, it has not been widely used in the study of marine microbiota, especially those within deep-sea environments, such as the hadal zone.
The areas at a water depth of between 6,000 and 11,000 m make up the hadal zone, which are the least explored domains in the ocean, and most are separated in trenches (Wolff, 1959; Jamieson et al., 2010). In recent years, the topic of hadal zone biology has increasingly become a research hotspot and many microbiologists have conducted explorations of the hadal microbial sphere. Based on these efforts, we already know that the hadal microbiota encompass heterotrophic microbes that apparently differ from the abyssal zone (Nunoura et al., 2015; Peoples et al., 2018; Du et al., 2021). The microbes in the hadal sediment are mainly composed of Proteobacteria (typically Gammaproteobacteria), Chloroflexi, Actinobacteria, and Planctomycetes (Chen et al., 2021; Fan et al., 2021), as well as Archaea such as Thaumarchaeota, Woesearchaeota, and Nanoarchaeaeota (Cui et al., 2019; Wang et al., 2021). Among these microbes, Proteobacteria and Thaumarchaeota dominate the shallower part of the hadal sediment, and the relative abundance of Chloroflexi, Woesearchaeota, and Marinimicrobia usually increases along the depth, which might be attributed to the concentration of oxygen and nitrate (Hiraoka et al., 2020). These microbes directly or indirectly participate in material transformation and play an important role in the biogeochemical cycle, such as anammox, denitrification, sulfate reduction, sulfate reduction, and sulfide oxidation (Xu et al., 2018; Zhang et al., 2018b; Gao et al., 2019; Xue et al., 2020).
In past decades, DNA cloning library and amplification of small subunit (SSU) rRNA genes are widely used to study the composition of hadal microbial community (Eloe et al., 2011; Ahmad et al., 2020; Schauberger et al., 2021). However, due to the problems relevant to sequencing depth and primer preference, microbial groups of low abundance are difficult to detect from original environmental samples. Moreover, despite the isolation and cultivation of a variety of hadal microbes (Zhao et al., 2020), such as Euzebyella marina (Liu et al., 2019) and Winogradskyella ouciana (He et al., 2021), in laboratory conditions, the full spectrum of laboratory culturable hadal microbes remains unestimated and the genomic characteristics of the microbes inhabiting the hadal zone await further study.
In the present study, we performed culture-enriched metagenomics on hadal sediment samples collected at a 6,477 m depth of the Mariana Trench. Hadal sediments were subjected to anaerobic culturing for 4 months, followed by 16S rRNA gene amplicon sequencing using the PacBio technique and metagenomics sequencing using both the Oxford Nanopore and Illumina techniques. Through these methods, we studied the diversity of microbes that can be cultured in laboratory conditions as well as their genomic characteristics.
Three sediment samples were collected from the surface seafloor in the Mariana Trench (10°813′N, 141°180′E) at 6,477 m on December 11, 2019. Each sample (25 cm3) was divided into two parts: one was used in culture enrichment while the other was stored at −80°C, from which three “cultured” replicates and three “original” replicates were generated. In organic-rich sediments, microbial respiration depletes oxygen and nitrate within a few centimeters below the sediment surface, and makes subsurface sediments entirely dominated by anaerobic carbon remineralization processes (Durbin and Teske, 2011). Hadal sediments are usually considered as anaerobic environments, which are inhabited by a number of anaerobes (Schauberger et al., 2021). Thus, we cultured the sampled hadal sediments in a flask with anaerobic headspace to mimic the in situ environment. The headspace air in the conical flask was replaced by a mixture of 90% N2, 5% H2, and 5% CO2 and the microbes were allowed to consume organic carbons contained in the sediments. The samples then were incubated in the dark at 16°C for 4 months.
DNA from the original and cultured samples was extracted with the Onestep-Lysis Bacteria DNA Kit (Nobelab Biotechnology, Beijing) and stored at −20°C. Full-length 16S rRNA genes from the original and enriched samples were amplified by PCR with barcode primers. We used an amplification mixture of a total volume of 50 μL containing 25 μL prime star Max premix (R045, Takara, Beijing), 2.5 μL 27F primers (5′-AGAGTTTGATCMTGGCTCAG-3′), 2.5 μL 1492R primers (5′-GGTTACCTTGTTACGACTT-3′), 1 μL DNA template, and 19 μL distilled H2O. The barcodes linked to 27F and 1492R sequences for the labeling of samples are given in Supplementary Table 1. The thermal conditions for PCR amplification included a denaturation step at 95°C for 5 min and 30 cycles of amplification (95°C for 30 s, 57°C for 30 s, and 72°C for 2 min), and a final extension step at 72°C for 5 min. PCR products were detected by electrophoresis with agarose gel of 2% concentration. According to the concentration of PCR products, the samples were mixed equally and then the PCR products were detected by 2% agarose gel electrophoresis. The target bands were recovered from the gel recovery kit provided by Qiagen company. The sequencing adaptors were connected to both ends of the amplified DNA fragment by DNA ligase. The DNA fragment was purified by AMPure PB magnetic beads to construct SMRT Bell library. The constructed library was quantified by Qubit concentration, the size of the inserted fragment was detected by Agilent 2100, and then sequenced on the PacBio RS II platform by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China), with more than 15,000 reads for each sample.
For metagenomic sequencing, equal amounts of DNA from the three cultured samples were pooled and subjected to Illumina NovaSeq 6000 sequencing (60 Gb data) and Oxford Nanopore sequencing (60 Gb data) on corresponding platforms by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). The detailed steps for Illumina NovaSeq 6000 sequencing: qualified DNA samples were randomly interrupted in about 350 bp by ultrasonic disruption device, and the whole Illumina library was prepared by end repair, adding poly A tail, adding adaptor, purification, and PCR amplification. After the construction of the library, Qubit2.0 was used for preliminary quantification. Then the library was diluted to 2 ng/μL, and Agilent 2100 was used to detect the insert size of the library. After the insert size met the expectations, the effective concentration of the Illumina library was accurately quantified by Q-PCR (effective concentration of the library >3 nM) to ensure the quality of the library. The detailed steps for Oxford Nanopore sequencing: DNA was interrupted by Megauptor, and the fragments of more than 10 kb were screened by Bluepinpin. After end repair and adding poly A tail and barcodes, then the samples with different barcodes were mixed according to the equal mole number, add adaptors, purify and use Qubit to detect concentration. The library was mixed with sequencing reagent for on-line sequencing.
After sequencing, 16S rRNA gene reads were extracted according to the barcodes, and the split reads were saved in bam format. Circular consensus sequencing (CCS) mode was used for sequence correction, with CCS = 3, minimum accuracy 0.99, and a length cutoff of between 1,340 and 1,640 bp, to produce fastq files. Primer sequences were removed with the software cutadapt (Martin, 2011), and chimeras were filtered out using VSEARCH (v2.13.3) (Rognes et al., 2016), resulting in clean reads. The six files containing clean reads were merged and clustered at a 97% similarity level to generate operational taxonomic units (OTUs) and build a representative sequence database. Taxonomic classification of the representative sequences was performed using RDP Classifier (v2.13) with default parameters (Wang et al., 2007). Finally, to avoid further sequencing errors, OTUs containing fewer than three reads were removed, and the relative abundance of the remaining OTUs was calculated based on read numbers across different samples. QIIME2 (Bolyen et al., 2019) was used to draw rarefaction curves and calculate alpha-diversities. For each sample, 50,000 reads were included for rarefaction calculation with intervals of 500 sequences, and 10 permutations were conducted.
The Illumina metagenome data in fastq format were subjected to quality control using the NGS QC Toolkit (v2.3.3) (Patel and Jain, 2012) to remove the primer/adaptor and low-quality sequences with the default cutoffs. he Nanopore metagenome data in fast5 format were transformed into fastq format with the software Guppy (Wick et al., 2019) and quality controlled using the software NanoPlot (v1.18.2) (De Coster et al., 2018). The Illumina and Oxford Nanopore data were cross-assembled using OPERA-MS (v0.9.0) software (Bertrand et al., 2019), and the quality of the assembled contigs was evaluated using QUAST (v4.6.3) (Mikheenko et al., 2016). Open reading frames (ORFs) were predicted with Prodigal (Hyatt et al., 2010) in the metagenome mode and annotated by searching against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using BLASTp (E-value < 1e−7). We conducted metagenomic binning using MaxBin (v2.2.7) (minimum contig length was 10,000) (Wu et al., 2016) and MetaBAT (v2.15) (Kang et al., 2019). CheckM (v1.1.2) (Parks et al., 2015) was employed to evaluate the completeness and contamination of the metagenome assembled genomes (MAGs). GTDB-Tk (v0.3.2) (Chaumeil et al., 2019) was used for taxonomic classification of the MAGs. We predicted the ORFs of the MAGs using Prodigal in single-genome mode and annotated them using the KEGG database (E-value < 1e−7). KEGG Mapper1 was used to reconstruct the gene metabolic pathways.
Six samples were used for 16S rRNA gene amplicon sequencing, including three samples without laboratory cultivation (designated “original samples”) and three samples that were subjected to anaerobic cultivation for 4 months (designated “cultured samples”). PacBio RS II was used to sequence the full-length 16S rRNA genes of the six samples to generate 18,253 (O1), 38,627 (O2), 57,375 (O3), 58,121 (C1), 41,855 (C2), and 48,415 (C3) clean reads (see Supplementary Table 2 for details). For alpha-diversity analyses, saturated rarefaction curves were obtained following the construction of four different metrics, abundance-based coverage estimator (ACE), Chao1, Shannon, and Simpson, indicating that the sequencing depth was sufficient to encompass the microbial diversity of these samples (Supplementary Figure 1). Moreover, according to the comparison of alpha-diversity levels, the diversity of the original samples was significantly higher than that of the cultured samples (Supplementary Figure 2).
After identifying OTUs at a 97% similarity level, the community structures of the six samples were profiled. The OTUs present in the original and cultured samples were summarized, which showed that the three original samples altogether possessed 1,059 OTUs, and the three cultured samples possessed 512 OTUs (Figure 1). Notably, 437 OTUs appeared only in the cultured samples and accounted for 85.35% of the cultured OTUs (Figure 1). The original samples contained 41 phyla, the majority of which comprised Proteobacteria (85.351–88.231%), Acidobacteria (1.816–2.961%), Chloroflexi (2.098–2.554%), and Planctomycetes (1.361–1.903%), while the cultured samples contained 10 phyla, including Proteobacteria (99.764–99.788%), Bacteroidetes (0.168–0.210%), Firmicutes (0.009–0.020%), Actinobacteria (0.007–0.010%), Fusobacteria (0–0.010%), Planctomycetes (0–0.004%), Chloroflexi (0–0.003%), Hydrogenedentes (0–0.003%), Gemmatimonadetes (0–0.002%), and Candidatus Saccharibacteria (0–0.002%) (Supplementary Figure 3). We identified 337 genera in the original samples (Figure 2), which predominantly included Shewanella (21.339–24.454%), Halomonas (13.845–20.131%), Marinobacter (10.223–14.719%), and Herbaspirillum (5.157–6.951%), while the enriched samples contained only 64 genera, the majority of which were Marinobacter (37.791–52.511%), Alteromonas (12.838–37.009%), Sulfitobacter (7.708–11.054%), and Idiomarina (7.551–10.514%). In total, 18 genera, including Reyranella, Salinimonas, Mangrovitalea, were unique to the cultured samples and accounted for 28.13% of all enriched genera (Supplementary Table 3). Student’s t-test showed that Marinobacter, Sulfitobacter, Idiomarina, Reyranella, and Erythrobacter were significantly (p-value < 0.01) enriched in the cultured samples (Figure 3).
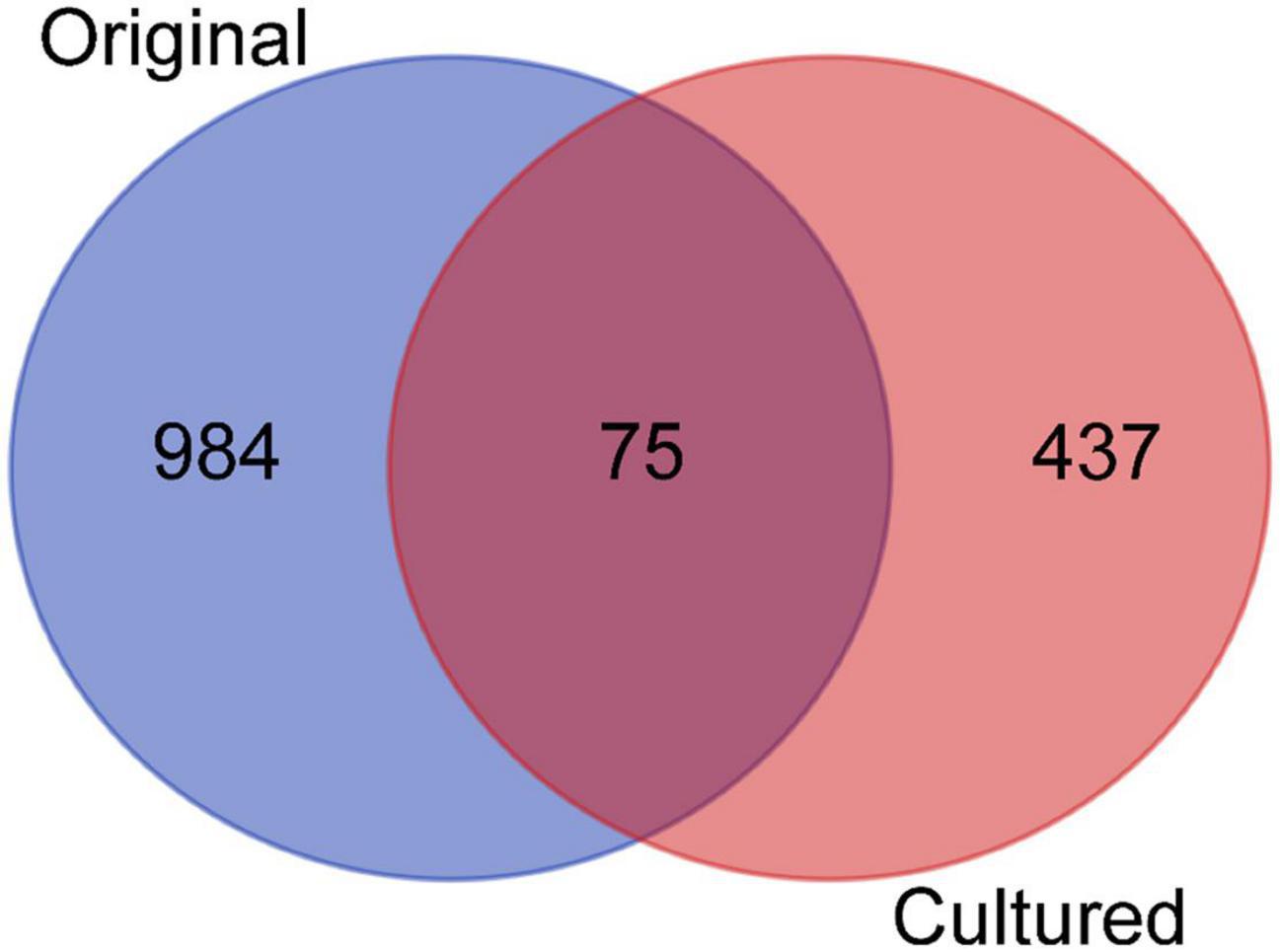
Figure 1. Venn diagram showing the distribution of operational taxonomic units (OTUs) across the original and cultured samples. The three replicates of the original and the cultured samples were combined, respectively, and compared.
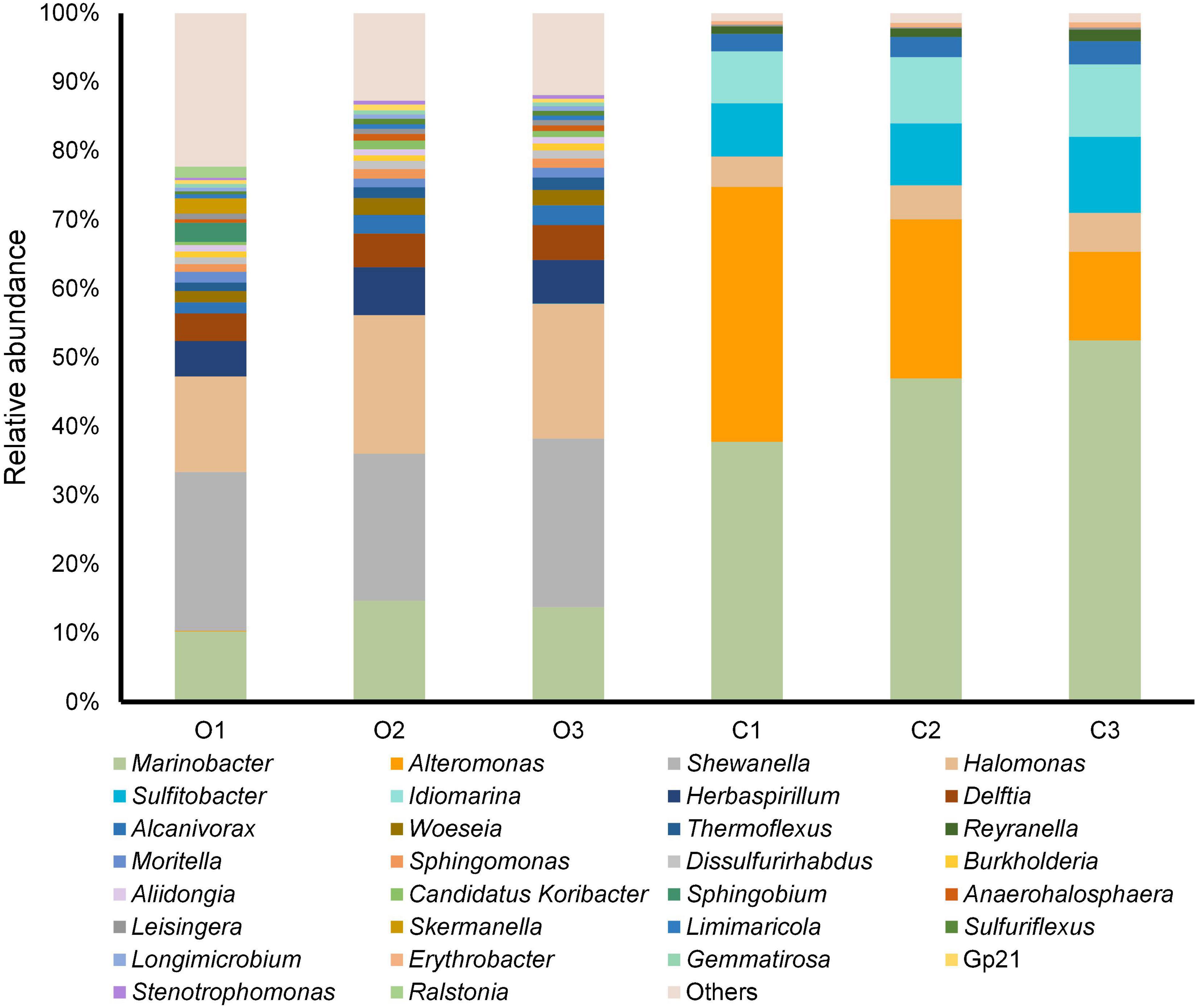
Figure 2. Genus-level taxonomic structure of the original and cultured sediment samples. The top 30 abundant genera in terms of maximum relative abundance are shown. C1–3 represent the three cultured samples, and O1–3 represent the three original samples without laboratory cultivation.
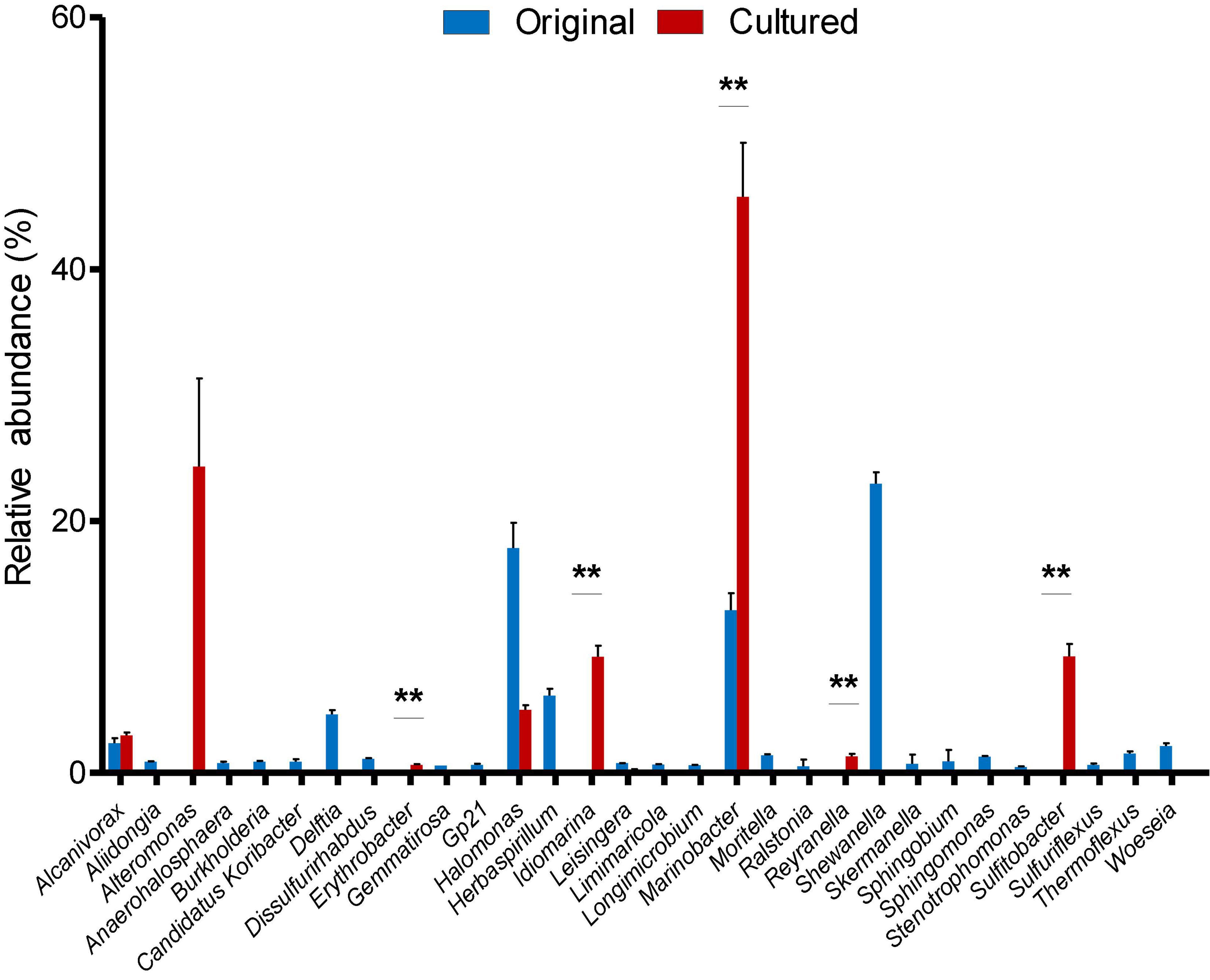
Figure 3. Relative abundance of the 30 most abundant genera in the original and cultured sediment samples. Mean values and standard errors are shown. “**” represents p-value < 0.01 produced by two-tailed Student’s t-test.
Despite efforts to optimize the technique (e.g., using different DNA extraction kits), we were unable to extract enough DNA from the original sediment samples (total DNA yield <50 μg) to perform metagenomic sequencing. By contrast, we were able to conduct both Illumina and Oxford Nanopore sequencing on the DNA extracted from the cultured samples (the total DNA yield from one sample was >1,000 μg, and the three cultured samples were pooled before sequencing) to generate 43.72 Gb of metagenome data with Illumina and 48.03 Gb data with Oxford Nanopore. All of the Illumina reads have a read length of 150 bp, and the N50 of the Oxford Nanopore reads was 2,685 bp with a maximum length of 114,365 bp (detailed information is given in Supplementary Table 4). The Illumina and Oxford Nanopore reads were assembled together, resulting in a total of 1,460 contigs, 283 of which were longer than 50 Kbp. The N50 was 241,431 bp (Supplementary Table 5), suggesting that the metagenomic data were well assembled.
To study the functional structure of the cultured sediment microbiota, 94,485 ORFs were predicted from the assembled contigs. After searching against the KEGG database, 64,826 ORFs could be annotated, and the 40 most abundant (in terms of the number of ORFs) KEGG genes are shown in Figure 4. These KEGG-annotated genes included quinone reductases (K00540), iron complex outer membrane receptor protein (K02014), methyl-accepting chemotaxis proteins (K03406), and diverse transposases and recombinases (K07497, K07483, K07484, K04763, K07481, and K07493).
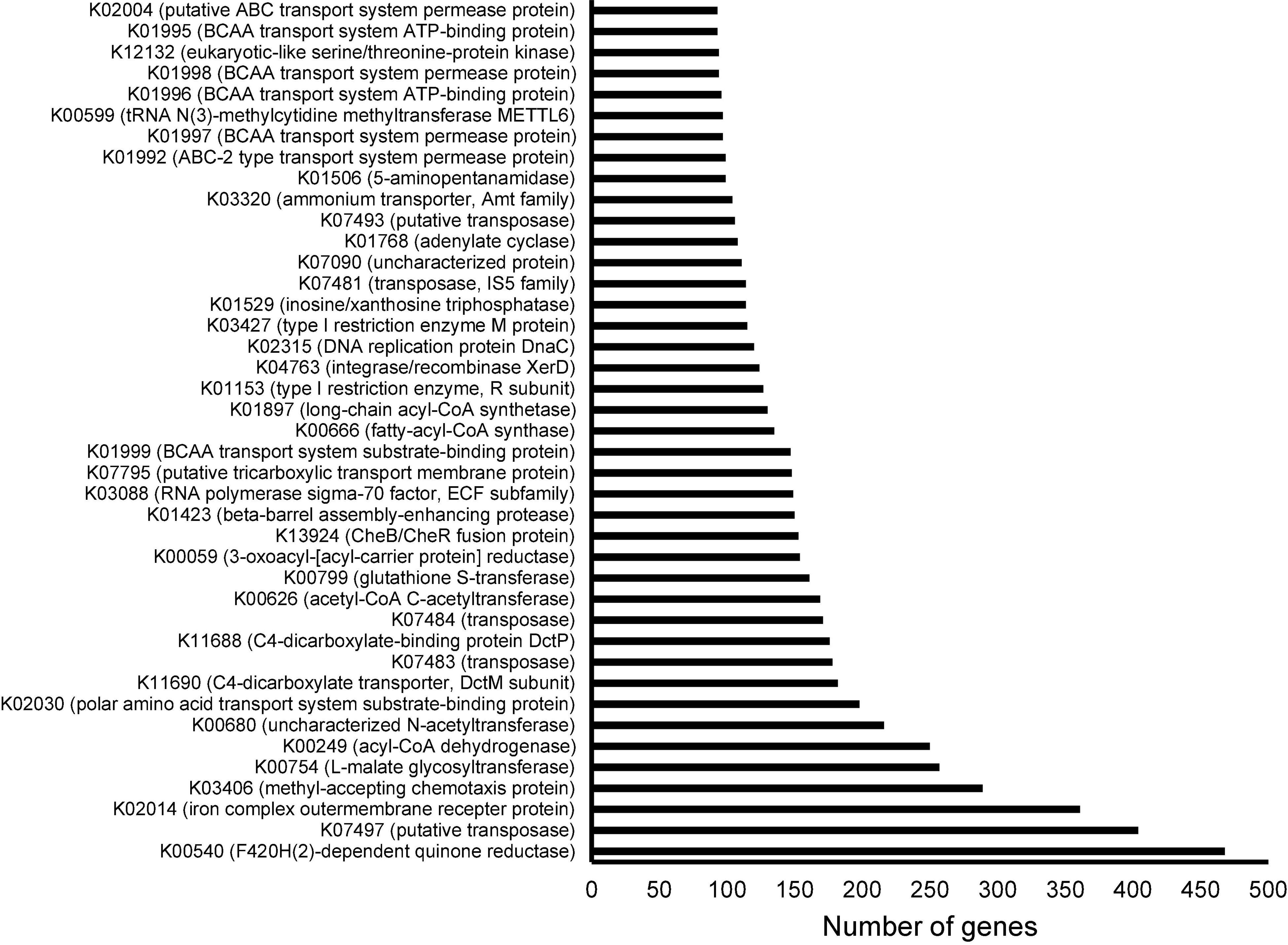
Figure 4. Annotation of open reading frames (ORFs) derived from the metagenome of the cultured sediment samples based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The three cultured sediment samples were pooled for both Illumina and Oxford Nanopore sequencing followed by assembly and ORF prediction. The top 40 KEGGs in terms of number of corresponding ORFs are shown.
To further study the function of the cultured microbiota, genome binning was performed on the assembled Illumina and Oxford Nanopore metagenomic data. Four high-quality MAGs affiliated to the genera Alcanivorax, Idiomarina, Sulfitobacter, and Erythrobacter were obtained (detailed information is given in Table 1). Excepting Alcanivorax, the other three genera were almost undetectable in the original samples and largely enriched in the cultured samples (Figure 3), and thus genomic analysis of these MAGs can be an approach to explain the mechanisms of proliferation of certain taxa during the culture enrichment. In addition to these four MAGs, the MAGs from Marinobacter might be more related to the diversity, because this genus showed the highest relative abundance in the cultured samples. However, binning of Marinobacter genomes failed, likely due to the existence of very close relatives in the cultured sample. The completeness of all the obtained MAGs was above 97%, and contamination was lower than 3%. Moreover, the number of contigs contained in each MAG was less than 10. Based on house-keeping gene classification using the GTDB-TK tool, none of the four MAGs could be classified to the species level, and they were designated Alcanivorax sp. HT1, Idiomarina sp. HT2, Sulfitobacter sp. HT3, and Erythrobacter sp. HT4. ORF prediction revealed 3,681, 2,819, 3,632, and 2,984 ORFs for the four MAGs (HT1–4), of which 2,643, 2,183, 2,722, and 2,139, respectively, could be annotated by KEGG.
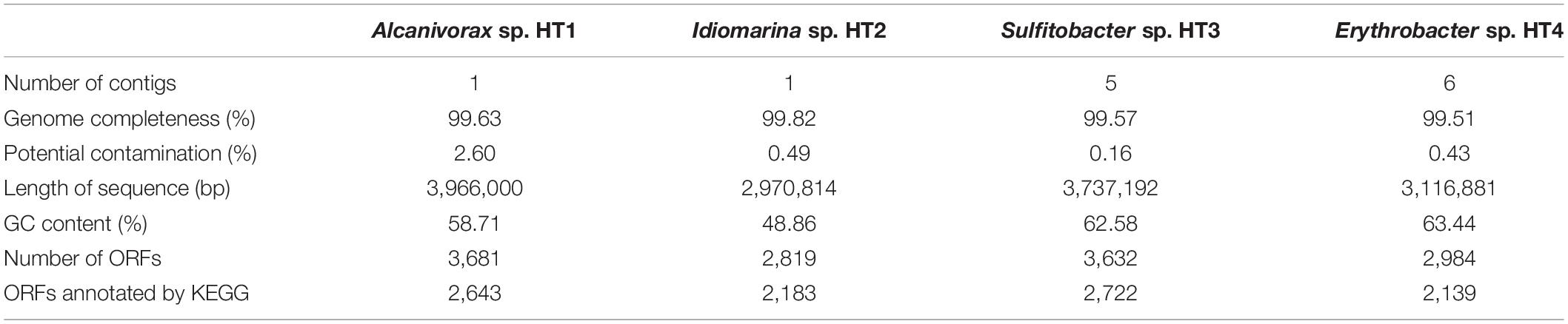
Table 1. Information on the four metagenome assembled genomes (MAGs) recovered from the metagenome of cultured sediment samples.
The metabolic pathways of the four MAGs were reconstructed using KEGG Mapper (Figure 5). The complete tricarboxylic acid cycle pathway and 5-phosphoribosyl-1-pyrophosphate biosynthetic pathway were identified in all four strains. The complete pentose phosphate (PP) pathway was present in Sulfitobacter sp. HT3, while the other three strains lacked the oxidative phase of the pathway. The complete Entner–Doudoroff (ED) pathway was present in Idiomarina sp. HT2, Sulfitobacter sp. HT3, and Erythrobacter sp. HT4. The complete Embden–Meyerhof–Parnas (EMP) pathway was detected in Idiomarina sp. HT2, while Alcanivorax sp. HT1, Sulfitobacter sp. HT3, and Erythrobacter sp. HT4 lacked key enzymes. With the exception of Sulfitobacter sp. HT3, all the MAGs included the complete glyoxylate cycle pathway. The ethylmalonyl and propanoyl-CoA metabolic pathways were detected in Sulfitobacter sp. HT3 and Erythrobacter sp. HT4. Moreover, nucleotide sugars and UDP-N-acetyl-D-glucosamine biosynthetic pathways were detected in Idiomarina sp. HT2, Sulfitobacter sp. HT3, and Erythrobacter sp. HT4. The glycogen biosynthetic pathway was detected in Sulfitobacter sp. HT3, while C5 and C10–C20 isoprenoid and terpenoid backbone biosynthetic pathways were found in Idiomarina sp. HT2 and Erythrobacter sp. HT4. Alcanivorax sp. HT1 and Erythrobacter sp. HT4 had complete sulfate assimilation pathways, evidence of sulfur metabolism. The sulfate assimilation pathway of Idiomarina sp. HT2 and Sulfitobacter sp. HT3 lacked the enzyme that reduces ammonium persulfate to 3′-phosphoadenosine-5′-phosphosulfate. In addition, Sulfitobacter sp. HT3 had genes for the sox sulfur oxidation system. In terms of nitrogen metabolism, the genomes of Alcanivorax sp. HT1 and Erythrobacter sp. HT4 had the assimilatory nitrate reductase gene (nasA), Idiomarina sp. HT2 and Sulfitobacter sp. HT3 had the nitrite reductase genes (nirKS), while Alcanivorax sp. HT1 and Erythrobacter sp. HT4 had the nitrite reductase genes (nirBD). Thus, the cultured hadal microbes may employ diverse pathways for central carbon metabolism and denitrification might be related to their anaerobic respiration.
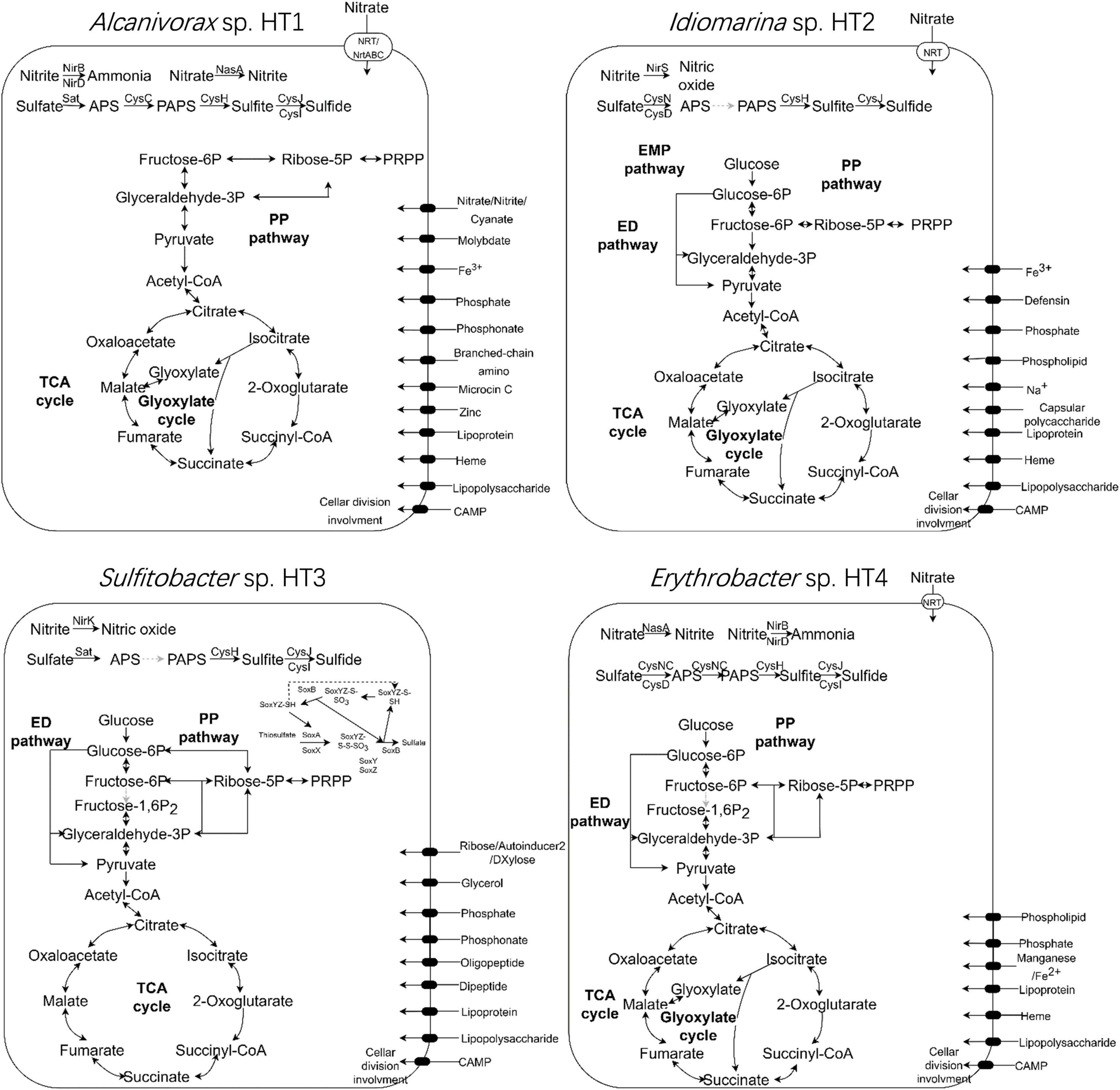
Figure 5. Metabolic pathways in the four genomes (Alcanivorax sp. HT1, Idiomarina sp. HT2, Sulfitobacter sp. HT3, and Erythrobacter sp. HT4) recovered from the metagenome of the cultured sediment samples. TCA cycle, tricarboxylic acid cycle; PP pathway, pentose phosphate pathway; ED pathway, Entner–Doudoroff pathway; EMP pathway, Embden–Meyerhof–Parnas pathway; PRPP, phosphoribosyl pyrophosphate; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; APS, adenosine-5′-phosphosulfate.
By integrating culture enrichment and 16S rRNA gene amplicon sequencing, we showed that about 7% of the collected hadal sediment microbiota can be cultured in the laboratory at sea-level pressure. The culturable taxa were confined to Proteobacteria, especially Gammaproteobacteria. Importantly, however, culture enrichment identified more than 85% OTUs that were not detected by direct sequencing. Moreover, culture enrichment increased the biomass of the sediment microbiota and facilitated the conduction of long-read sequencing for the assembly and analyses of nearly complete microbial genomes.
So far, a number of studies have investigated the microbiota of the hadal zone and showed that the hadal sediment and seawater are predominantly composed of Gammaproteobacteria (Zhang et al., 2018b; Hiraoka et al., 2020). Consistent with this, the results from the present study revealed that the sediment samples collected from 6,477 m depth of the Challenger Deep comprised mostly Marinobacter, and Halomonas, but it was surprising that several high-pressure-resistant bacteria, such as Psychromonas, were not detected. Members of Psychromonas have been frequently reported in previous hadal studies (Zhang et al., 2018a; Cui et al., 2019) and were demonstrated to be high-pressure-resistant copiotrophic members (Nogi et al., 2002). Regarding the difference between the original and cultured hadal taxa, we found that members of Marinobacter, Idiomarina, Sulfitobacter, Reyranella, and Erythrobacter were enriched in the cultured microbiota, suggesting that they can proliferate under laboratory conditions. Members of Marinobacter have been isolated from a variety of deep-sea environments, including cold seeps (Sun et al., 2020), hydrothermal vents (Zhou et al., 2020), and trenches (Cao et al., 2019; Ahmad et al., 2020). Halomonas strains have also been isolated from hadal trenches (Yan et al., 2020). Contrastingly, no strains of Idiomarina, Sulfitobacter, Reyranella, or Erythrobacter have yet been cultured from hadal environments, although researchers using several culture-independent methods (e.g., single-cell genomics) have documented the presence of Erythrobacter and Sulfitobacter in hadal trenches (Nunoura et al., 2016; Chen et al., 2020). Taken together, these results suggest that our culture enrichment conditions have expanded the diversity of the culturable hadal microbes; certain species could not be isolated, but they can be maintained in laboratory conditions within a community.
Our metagenome analyses based on the cross-assembly of the Illumina and Oxford Nanopore data uncovered the functional profile of the culturable hadal sediment microbiota in addition to its taxonomic diversity. The prevalence of quinone reductases (K00540) suggests that genes of this family are employed for anaerobic respiration and energy metabolism; quinone reductases are also employed by microbes to protecting them against oxidative stress and bactericidal agents (Gurumurthy et al., 2013; Cassagnes et al., 2018). The prevalence of iron complex outer membrane receptor protein (K02014) may be related to bacterial iron uptake (Locher et al., 1998). Methyl-accepting chemotaxis proteins (K03406) could be involved in bacterial chemotactic responses and niche adaptations for sensing changes in the environmental conditions by regulating gene transcription, allowing them to adapt to hadal environments and proliferate in laboratory conditions (Miller et al., 2009). Notably, the prevalence of a variety of transposases and recombinases (K07497, K07483, K07484, K04763, K07481, and K07493) implies that gene exchange would be a common event among the cultured sediment microbes. Transposases are important in gain-of-function processes and are likely to function in the adaptions to environmental change (Casacuberta and González, 2013). Together, these functions are likely to contribute to the success of the cultured microbes in terms of survival in the hadal environment and proliferation in the laboratory condition.
Analyses of the four nearly complete MAGs further revealed the functional diversity of the hadal environment microbes, especially the rare species, as three of the four MAGs were almost undetectable in the original samples. Analyses of these MAGs also shed light on the characteristics of culturable hadal microbes. First, the cultured microbes are probably copiotrophic bacteria, which can utilize a variety of organic carbons to proliferate. Based on the type of transporters present in the MAGs, the species utilize carbon sources such as polysaccharides and oligopeptides. Previous studies have demonstrated that polysaccharides recycled at the ocean bottom can remain there for a long time and are important carbon sources for the hadal microbiota (Nunoura et al., 2015; Luo et al., 2017). Evidence that protein degradation provides important carbon sources, indicated by the abundance of dipeptide and oligopeptide transporters, has been also documented in previous studies on deep-sea microbes (Zhang et al., 2015; Zeng et al., 2020). Excepting the Sulfitobacter sp. HT3, all the MAGs contained glyoxylate cycle genes, indicating that the bacteria can use acetate to grow. Regarding central carbon metabolism, the presence of the complete ED pathway and nitrite reductases in most MAGs suggests the microbes are probably facultative. All but Alcanivorax sp. HT1 had all ED pathway genes, and the complete EMP pathway was only present in Idiomarina sp. HT2. The EMP pathway generates more ATP than the ED pathway, and use of the EMP pathway is ubiquitous in terrestrial microorganisms (Flamholz et al., 2013); by contrast, most marine bacteria strains examined seem to rely on the ED pathway for glucose catabolism (Klingner et al., 2015). Thus, the cultured microbes in the present study may employ the ED pathway to convert glucose to pyruvate when oxygen is unavailable and use nitrite as an electron acceptor for respiration and energy production. Moreover, the metabolic features of Idiomarina sp. HT2 and Erythrobacter sp. HT4 are consistent with their niche adaptation. In a recent study (Poff et al., 2021), Idiomarina was demonstrated to be a particle-associated taxa that is associated with particles sinking from surface ocean to deep sea, and Erythrobacter is related to the “summer export pulse” events, which are defined as the periods when particulate carbon flux at 4,000 m exceeds the mean annual flux by 150%. Thus, members of Idiomarina and Erythrobacter are likely to be pelagic marine bacteria that can survive and be found in deep oceans, and their association with particles imply the ability of polysaccharide consumption. Members of Sulfitobacter, belonging to the well-known Roseobacter clade, are widely distributed in global oceans, from surface to deep sea, and they often displayed many typical traits of generalists, such as enrichment of gene transfer agents, sulfate and nitrate reductases, and genes for utilizing compounds with various carbon atoms (Newton et al., 2010). Thus, Sulfitobacter may have the ability to survive in various environments and proliferate when the external condition is optimal.
The culture enrichment work was carried out during the COVID-19 outbreak, which hindered our utilization of high-pressure culture devices in our key laboratory to mimic the in situ conditions of the hadal environment. However, based on the current results, we propose that coupling laboratory enrichment with long-read sequencing is an ideal way to obtain high-quality microbial genomes from hadal microbiota. The taxonomic and functional diversity of the hadal microbiota could be expanded through further culture enrichment efforts.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA743701.
YL, JLi, RL, JS, JF, P-YQ, Y-ZZ, and WZ contributed to conception and design of the study. HW, MW, SF, JLu, and ML performed the experiments, and processed and analyzed the data. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by funding from the National Key Research and Development Program of China (2018YFC0310600 and 2018YFC1406700), the Fundamental Research Funds of Ocean University of China (842041010 and 841912035), the National Science Foundation of China (31630012, U1706207, 91851205 and 31870052), the Major Scientific and Technological Innovation Project of Shandong Province (2019JZZY010817), the Taishan Scholars Program of Shandong Province (tspd20181203), and the Fundamental Research Funds for the Central Universities (201961018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.754332/full#supplementary-material
Ahmad, W., Zheng, Y., Li, Y., Sun, W., Hu, Y., He, X., et al. (2020). Marinobacter salinexigens sp. nov., a marine bacterium isolated from hadal seawater of the Mariana Trench. Int. J. Syst. Evol. Microbiol. 70, 3794–3800. doi: 10.1099/ijsem.0.004236
Bertrand, D., Shaw, J., Kalathiyappan, M., Ng, A. H. Q., Kumar, M. S., Li, C., et al. (2019). Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 37, 937–944. doi: 10.1038/s41587-019-0191-2
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Cao, J., Liu, P., Liu, R., Su, H., Wei, Y., Liu, R., et al. (2019). Marinobacter profundi sp. nov., a slightly halophilic bacterium isolated from a deep-sea sediment sample of the New Britain Trench. Antonie Van Leeuwenhoek 112, 425–434. doi: 10.1007/s10482-018-1176-8
Casacuberta, E., and González, J. (2013). The impact of transposable elements in environmental adaptation. Mol. Ecol. 22, 1503–1517. doi: 10.1111/mec.12170
Cassagnes, L. E., Chhour, M., Pério, P., Sudor, J., Gayon, R., Ferry, G., et al. (2018). Oxidative stress and neurodegeneration: the possible contribution of quinone reductase 2. Free Radic. Biol. Med. 120, 56–61. doi: 10.1016/j.freeradbiomed.2018.03.002
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen, M., Song, Y., Feng, X., Tang, K., Jiao, N., Tian, J., et al. (2020). Genomic characteristics and potential metabolic adaptations of hadal trench Roseobacter and Alteromonas bacteria based on single-cell genomics analyses. Front. Microbiol. 11:1739. doi: 10.3389/fmicb.2020.01739
Chen, P., Zhou, H., Huang, Y., Xie, Z., Zhang, M., Wei, Y., et al. (2021). Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep. Genome Biol. 22:207. doi: 10.1186/s13059-021-02408-w
Cui, G., Li, J., Gao, Z., and Wang, Y. (2019). Spatial variations of microbial communities in abyssal and hadal sediments across the Challenger Deep. PeerJ 7:e6961. doi: 10.7717/peerj.6961
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Deming, J. W., and Baross, J. A. (2000). “Extreme deep-Sea environments,” in Nonculturable Microorganisms in the Environment, eds R. R. Colwell and D. J. Grimes (Boston, FL: Springer), 147–149.
Du, M., Peng, X., Zhang, H., Ye, C., Dasgupta, S., Li, J., et al. (2021). Geology, environment, and life in the deepest part of the world’s oceans. Innovation 2:100109. doi: 10.1016/j.xinn.2021.100109
Durbin, A. M., and Teske, A. (2011). Microbial diversity and stratification of South Pacific abyssal marine sediments. Environ. Microbiol. 13, 3219–3234. doi: 10.1111/j.1462-2920.2011.02544.x
Eloe, E. A., Shulse, C. N., Fadrosh, D. W., Williamson, S. J., Allen, E. E., and Bartlett, D. H. (2011). Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 3, 449–458. doi: 10.1111/j.1758-2229.2010.00223.x
Fan, S., Wang, M., Ding, W., Li, Y. X., Zhang, Y. Z., and Zhang, W. (2021). Scientific and technological progress in the microbial exploration of the hadal zone. Mar. Life Sci. Technol. 1–11. doi: 10.1007/s42995-021-00110-1
Flamholz, A., Noor, E., Bar-Even, A., Liebermeister, W., and Milo, R. (2013). Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc. Natl. Acad. Sci. U.S.A. 110, 10039–10044. doi: 10.1073/pnas.1215283110
Gao, Z. M., Huang, J. M., Cui, G. J., Li, W. L., Li, J., Wei, Z. F., et al. (2019). In situ meta-omic insights into the community compositions and ecological roles of hadal microbes in the Mariana Trench. Environ. Microbiol. 21, 4092–4108. doi: 10.1111/1462-2920.14759
Gich, F., Janys, M. A., König, M., and Overmann, J. (2012). Enrichment of previously uncultured bacteria from natural complex communities by adhesion to solid surfaces. Environ. Microbiol. 14, 2984–2997. doi: 10.1111/j.1462-2920.2012.02868.x
Gurumurthy, M., Rao, M., Mukherjee, T., Rao, S. P., Boshoff, H. I., Dick, T., et al. (2013). A novel F(420)-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol. Microbiol. 87, 744–755. doi: 10.1111/mmi.12127
He, X., Liu, R., Liang, J., Li, Y., Zhao, X., Ran, L., et al. (2021). Winogradskyella ouciana sp. nov., isolated from the hadal seawater of the Mariana Trench. Int. J. Syst. Evol. Microbiol. 71, doi: 10.1099/ijsem.0.004687
Hiraoka, S., Hirai, M., Matsui, Y., Makabe, A., Minegishi, H., Tsuda, M., et al. (2020). Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J. 14, 740–756. doi: 10.1038/s41396-019-0564-z
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jamieson, A. J., Fujii, T., Mayor, D. J., Solan, M., and Priede, I. G. (2010). Hadal trenches: the ecology of the deepest places on Earth. Trends Ecol. Evol. 25, 190–197. doi: 10.1016/j.tree.2009.09.009
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359
Klingner, A., Bartsch, A., Dogs, M., Wagner-Döbler, I., Jahn, D., Simon, M., et al. (2015). Large-Scale 13C flux profiling reveals conservation of the Entner-Doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl. Environ. Microbiol. 81, 2408–2422. doi: 10.1128/AEM.03157-14
Liu, J., Xue, C. X., Sun, H., Zheng, Y., Meng, Z., and Zhang, X. H. (2019). Carbohydrate catabolic capability of a Flavobacteriia bacterium isolated from hadal water. Syst. Appl. Microbiol. 42, 263–274. doi: 10.1016/j.syapm.2019.01.002
Locher, K. P., Rees, B., Koebnik, R., Mitschler, A., Moulinier, L., Rosenbusch, J. P., et al. (1998). Transmembrane signaling across the ligand-gated FhuA receptor: crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell 95, 771–778. doi: 10.1016/s0092-8674(00)81700-6
Luo, M., Gieskes, J., Chen, L., Shi, X., and Chen, D. (2017). Provenances, distribution, and accumulation of organic matter in the southern Mariana Trench rim and slope: implication for carbon cycle and burial in hadal trenches. Mar. Geol. 386, 98–106. doi: 10.1016/j.margeo.2017.02.012
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mikheenko, A., Valin, G., Prjibelski, A., Saveliev, V., and Gurevich, A. (2016). Icarus: visualizer for de novo assembly evaluation. Bioinformatics 32, 3321–3323. doi: 10.1093/bioinformatics/btw379
Miller, L. D., Russell, M. H., and Alexandre, G. (2009). “Diversity in bacterial chemotactic responses and niche adaptation,” in Advances in Applied Microbiology, eds A. L. Laskin, S. Sariaslani, and G. Gadd (Amsterdam: Elsevier Inc), 53–75.
Newton, R. J., Griffin, L. E., Bowles, K. M., Meile, C., Gifford, S., Givens, C. E., et al. (2010). Genome characteristics of a generalist marine bacterial lineage. ISME J. 4, 784–798. doi: 10.1038/ismej.2009.150
Nichols, D., Cahoon, N., Trakhtenberg, E. M., Pham, L., Mehta, A., Belanger, A., et al. (2010). Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 76, 2445–2450. doi: 10.1128/AEM.01754-09
Nogi, Y., Kato, C., and Horikoshi, K. (2002). Psychromonas kaikoae sp. nov., a novel piezophilic bacterium from the deepest cold-seep sediments in the Japan Trench. Int. J. Syst. Evol. Microbiol. 52, 1527–1532. doi: 10.1099/00207713-52-5-1527
Nunoura, T., Hirai, M., Yoshida-Takashima, Y., Nishizawa, M., Kawagucci, S., Yokokawa, T., et al. (2016). Distribution and niche separation of planktonic microbial communities in the water columns from the surface to the hadal waters of the Japan Trench under the Eutrophic Ocean. Front. Microbiol. 7:1261. doi: 10.3389/fmicb.2016.01261
Nunoura, T., Takaki, Y., Hirai, M., Shimamura, S., Makabe, A., Koide, O., et al. (2015). Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on Earth. Proc. Natl. Acad. Sci. U.S.A. 112, 1230–1236. doi: 10.1073/pnas.1421816112
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Peoples, L. M., Donaldson, S., Osuntokun, O., Xia, Q., Nelson, A., Blanton, J., et al. (2018). Vertically distinct microbial communities in the Mariana and Kermadec trenches. PLoS One 13:e0195102. doi: 10.1371/journal.pone.0195102
Pham, V. H., and Kim, J. (2012). Cultivation of unculturable soil bacteria. Trends Biotechnol. 30, 475–484. doi: 10.1016/j.tibtech.2012.05.007
Poff, K. E., Leu, A. O., Eppley, J. M., Karl, D. M., and DeLong, E. F. (2021). Microbial dynamics of elevated carbon flux in the open ocean’s abyss. Proc. Natl. Acad. Sci. U.S.A. 118:e2018269118. doi: 10.1073/pnas.2018269118
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Schauberger, C., Glud, R. N., Hausmann, B., Trouche, B., Maignien, L., Poulain, J., et al. (2021). Microbial community structure in hadal sediments: high similarity along trench axes and strong changes along redox gradients. ISME J. 1–13. doi: 10.1038/s41396-021-01021-w
Sun, Q. L., Sun, Y. L., Sun, Y. Y., Luan, Z. D., and Lian, C. (2020). Marinobacter fonticola sp. nov., isolated from deep sea cold seep sediment. Int. J. Syst. Evol. Microbiol. 70, 1172–1177. doi: 10.1099/ijsem.0.003895
Vartoukian, S. R., Palmer, R. M., and Wade, W. G. (2010). Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiol. Lett. 309, 1–7. doi: 10.1111/j.1574-6968.2010.02000.x
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, Z., Wang, L., Liu, R., Li, Z., Wu, J., Wei, X., et al. (2021). Community structure and activity potentials of archaeal communities in hadal sediments of the Mariana and Mussau trenches. Mar. Life Sci. Technol. doi: 10.1007/s42995-021-00105-y
Whelan, F. J., Waddell, B., Syed, S. A., Shekarriz, S., Rabin, H. R., Parkins, M. D., et al. (2020). Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota. Nat. Microbiol. 5, 379–390. doi: 10.1038/s41564-019-0643-y
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20:129. doi: 10.1186/s13059-019-1727-y
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xu, Z., Wang, M., Wu, W., Li, Y., Liu, Q., Han, Y., et al. (2018). Vertical distribution of microbial eukaryotes from surface to the hadal zone of the Mariana Trench. Front. Microbiol. 9:2023. doi: 10.3389/fmicb.2018.02023
Xue, C. X., Liu, J., Lea-Smith, D. J., Rowley, G., Lin, H., Zheng, Y., et al. (2020). Insights into the vertical stratification of microbial ecological roles across the deepest seawater column on Earth. Microorganisms 8:1039. doi: 10.3390/microorganisms8091309
Yan, F., Fang, J., Cao, J., Wei, Y., Liu, R., Wang, L., et al. (2020). Halomonas piezotolerans sp. nov., a multiple-stress-tolerant bacterium isolated from a deep-sea sediment sample of the New Britain Trench. Int. J. Syst. Evol. Microbiol. 70, 2560–2568. doi: 10.1099/ijsem.0.004069
Zeng, X., Zhang, X., and Shao, Z. (2020). Metabolic adaptation to sulfur of hyperthermophilic Palaeococcus pacificus DY20341 from deep-sea hydrothermal sediments. Int. J. Mol. Sci. 21:368. doi: 10.3390/ijms21010368
Zhang, C. L., Xie, W., Martin-Cuadrado, A. B., and Rodriguez-Valera, F. (2015). Marine group II archaea, potentially important players in the global ocean carbon cycle. Front. Microbiol. 6:1108. doi: 10.3389/fmicb.2015.01108
Zhang, W., Tian, R. M., Sun, J., Bougouffa, S., Ding, W., Cai, L., et al. (2018a). Genome reduction in Psychromonas species within the gut of an Amphipod from the Ocean’s deepest point. MSystems 3;e00009–e18. doi: 10.1128/mSystems.00009-18
Zhang, X., Xu, W., Liu, Y., Cai, M., Luo, Z., and Li, M. (2018b). Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap Trench. Front. Microbiol. 9:2402. doi: 10.3389/fmicb.2018.02402
Zhang, Z., Wu, Y., and Zhang, X. H. (2018c). Cultivation of microbes from the deep-sea environments. Deep Sea Res. 2 Top. Stud. Oceanogr. 155, 34–43. doi: 10.1016/j.dsr2.2017.07.008
Zhao, X., Liu, J., Zhou, S., Zheng, Y., Wu, Y., Kogure, K., et al. (2020). Diversity of culturable heterotrophic bacteria from the Mariana Trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2, 181–193. doi: 10.1007/s42995-020-00027-1
Keywords: marine microbiota, hadal sediment, culture-enriched metagenomics, Oxford Nanopore sequencing, genome binning
Citation: Wang H, Wang M, Fan S, Lu J, Lan Y, Li M, Li J, Liu R, Sun J, Fang J, Qian P-Y, Zhang Y-Z and Zhang W (2021) Culture Enrichment Combined With Long-Read Sequencing Facilitates Genomic Understanding of Hadal Sediment Microbes. Front. Mar. Sci. 8:754332. doi: 10.3389/fmars.2021.754332
Received: 06 August 2021; Accepted: 19 October 2021;
Published: 10 November 2021.
Edited by:
XueWei Xu, Second Institute of Oceanography, Ministry of Natural Resources, ChinaReviewed by:
Iftikhar Ahmed, National Agricultural Research Center, PakistanCopyright © 2021 Wang, Wang, Fan, Lu, Lan, Li, Li, Liu, Sun, Fang, Qian, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weipeng Zhang, emhhbmd3ZWlwZW5nQG91Yy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.