 Hailong Ge1†
Hailong Ge1† Haoyu Zhang2†
Haoyu Zhang2† Lijun Yang1Haoyu Wang1Limei Tu1Zhuojin Jiang1Jing Zheng1Bolin Chen1Juan Chen3
Lijun Yang1Haoyu Wang1Limei Tu1Zhuojin Jiang1Jing Zheng1Bolin Chen1Juan Chen3 Yun Li1*
Yun Li1* Zhijian Wang1*
Zhijian Wang1*- 1Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), Key Laboratory of Aquatic Science of Chongqing, Southwest University, Chongqing, China
- 2Animal Disease Prevention and Food Safety Key Laboratory of Sichuan Province, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
- 3The Ministry of Education Key Laboratory of Laboratory Medical Diagnostics, The College of Laboratory Medicine, Chongqing Medical University, Chongqing, China
Background
Bigmouth buffalo (Ictiobus cyprinellus) belongs to the family Catostomidae. It is one of the largest freshwater fish endemics in North America, with a body length of more than 1.25 m and a body weight of more than 36 kg (Eddy and Underhill, 1974). Bigmouth buffalo is characterized by good meat quality and taste, a large size, rapid growth, strong disease resistance, relatively early maturity, and high reproductive ability. Due to its important economic value, it is highly valued among more than 30 species of Catostomidae that are abundant in America. Bigmouth buffalo has been a commercially important fish in the United States since the 19th century (Hoffbeck, 2001). In the 21st century, the value of its fisheries in the upper Mississippi River basin alone has exceeded US$1 million per year (Great Lakes Mississippi River Interbasin Study (GLMRIS), 2012). In 1971, the area of intensive bigmouth buffalo breeding in Arkansas, USA, reached 136 hm2. As an important economic fish, bigmouth buffalo has been successfully introduced for breeding and popularized in many countries. Bigmouth buffalo was introduced from the United States into Russia and Ukraine (from the former Soviet Union) in 1971, and it has since been widely farmed in reservoirs and ponds. In 1993 and 1994, China successively introduced bigmouth buffalo yolk sac fry and 3–5 cm fry from the United States in four batches, including more than 20,000 tails. After careful breeding and domestication, these bigmouth buffalo reached sexual maturity in 1996, and artificial induction and hatching success were achieved (Wang et al., 1997).
A 112-year-old bigmouth buffalo was found in 2019, making it the longest-lived species identified among the ~12,000 freshwater bony fish known globally (Lackmann et al., 2019). Recently published research shows that bigmouth buffalo exhibit negligible senescence in multiple physiological systems despite living for nearly a century (Sauer et al., 2021). At present, studies about bigmouth buffalo mainly focus on hybridization and breeding promotion (Jin et al., 2011; Chu, 2015), embryonic development (Osborn and Self, 1966), mitochondrial whole-genome analyses (Liu et al., 2015), environmental pollution stress (Wang et al., 1997; Doering et al., 2019; Zhang et al., 2020), drug tolerance (Qu et al., 2001), life history and ecology (Coulter et al., 2018; Wang et al., 2018; Keevin et al., 2019; Dba et al., 2021), and population activity (Moen, 1974; Enders et al., 2019). As the longest-lived freshwater bony fish identified to date, the research value of bigmouth buffalo is becoming increasingly evident, and related research will gradually intensify.
In this study, we applied long-read pacific bioscience (PacBio) isomer sequencing (ISO-seq) to produce the first full-length transcriptome assembly for bigmouth buffalo. The use of the PacBio sequencing platform, especially without reference genome sequence, is an ideal method for constructing reference transcriptome assembly (Dong et al., 2015; Kuo et al., 2017; Workman et al., 2018). We used the PacBio platform to process full-length mRNA of five major organs, consistent cycle sequencing (CCS). Since bigmouth buffalo lacks high-quality draft genome sequences, we believed that our data of high-quality transcriptome reference sequence could be helpful for transcriptome analysis in the future under a variety of conditions.
Data Description
Sample Collection and RNA Preparation
The bigmouth buffalo (Ictiobus cyprinellus) used in this experiment was taken from the Key Laboratory of Freshwater Fish Resources and Reproductive Under the Breeding Environment of Development of the Ministry of Education, 1 year old, sex unknown, physical health, no disease, and no damage. In this study, the animal welfare protocols and experimental procedures applied were carried out in accordance with the recommendations of animal research ethics guidelines, and the program complied with the relevant ethical regulations of the Key Laboratory of Freshwater Fish Resources and Reproductive Development of the Ministry of Education. Samples of five organs, namely, gills, skin, brain, liver, and muscle, were frozen in liquid nitrogen before storage at −80°C immediately after dissection. We mixed frozen tissue samples of five organs together and extracted RNA from them.
Library Construction and SMRT Sequencing
The tissue was ground in TRIzol reagent (Life Technologies, Thermo Fisher Scientific, Shanghai, China) on dry ice to extract the total RNA, following the protocol provided by the manufacturer. Then, we used Agilent 2100 Bioanalyzer (Agilent, Beijing, China) to evaluate the RNA integrity. A NanoDrop microspectrophotometer (Thermo Fisher, Thermo Fisher Scientific, Shanghai, China) was used to determine the purity and concentration of RNA. mRNA was enriched using Oligo (dT) magnetic beads (NEB). We used Clontech SMARTer PCR cDNA Synthesis Kit (Takara Bio, Takara Biomedical Technology, Beijing, China) to reverse transcribe mRNA into cDNA. PCR cycle optimization was used to determine the optimal amplification cycle number for the downstream large-scale PCR. Then, the optimized cycle number was adopted to generate double-stranded cDNA. In addition, >5 kb size selection was performed using the BluePippin™ Size-Selection System, and the size-selected cDNA was mixed in equal amounts with the non-size-selected cDNA. Then, large-scale PCR was performed for subsequent construction of the SMRTbell library. Then, DNA damage repair, end repair, and ligation with sequencing adapters of the cDNAs were processed. The SMRT sequencing was performed on the PacBio Sequel II platform (PacBio) (Supplementary Figure 1A).
Generation of Full-Length Transcriptomes
The SMRT Link (version 9.0.0) pipeline was used to process the raw sequencing data. First of all, the CCS function was used to extract high-quality circular consensus sequences (CCS, HiFi reads) from the subread BAM file. The sequences containing structures of 5′ primers, 3′ primers, and polyA were considered as full-length sequences (full-length reads, FL reads). Full-length non-chimeric (FLNC) reads were formed by removing primers, barcodes, trimmed polyA tails, and concatemers of full passes and then used to generate complete isoforms by cluster. Similar FLNC reads were obtained using the cluster function to cluster the consistent sequences (which use minimap2 mapping to transcripts). Then, we used CD-HIT (version 4.6.7) to further correct the consistent sequences. Finally, BUSCO4 (https://busco.ezlab.org/) was used for assembly access. The high-quality isoforms according to these results (prediction accuracy ≥ 0.99) were used for our annotation of the transcriptome.
Functional Annotation of PacBio Isoforms
To annotate the isoforms, they were subjected to BLAST (National Center for Biotechnology Information, USA) (version 2.11.0) analysis compared with the NR database (http://www.ncbi.nlm.nih.gov), the SWISS-PROT protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg), and the Clusters of Orthologous Groups (COG)/EuKaryotic Orthologous Groups (KOG) database (http://www.ncbi.nlm.nih.gov/COG) with the BLASTx program. We applied a threshold of e-value > 1e−5 to obtain similar genes. The Gene Ontology (GO) (updated 5 January 2019) annotations were analyzed using Blast2GO software (version 5.2.0). The score of top 20 and the result of high-scoring segment pair (HSP) hits higher than 33 were obtained to conduct the Blast2GO (BioBam, Spain; Conesa et al., 2005) analysis. Then, WEGO software (https://wego.genomics.cn/images/gky400.pdf) (version 2.0) was used to perform the functional classification (Ye et al., 2006).
Predicting Gene Structure of Isoforms
For further information, we processed the predicted coding sequences (CDSs), alternative splicing (AS) isoforms, simple sequence repeats (SSRs), long non-coding RNAs (lncRNAs), and transcription factors (TFs). Open reading frames (ORFs) were detected by applying ANGEL software1 (version 2.4) to the isoform sequences to predict the coding sequence regions (Shimizu et al., 2006).
To analyze the AS events of the isoforms from our data, the COding GENome reconstruction Tool Cogent (version 3.3) was used for the purpose to divide the transcripts into gene families, which use k-mer similarity algorithm based on a De Bruijn graph and then reconstruct families to produce coding reference genome. Then, we further analyzed AS events using the SUPPA program2 (2.2) among the isoforms. The MIcroSAtellite server (http://pgrc.ipk-gatersleben.de/misa/) (MISA, version 1.0) was used for microsatellite annotation of the whole transcriptome. We used Primer 1.1.4 program to design primer pairs in the flanking regions of SSRs for subsequent validation from MISA results (Rozen and Skaletsky, 2000).
We used CNCI3 (version 2.0) and CPC4 (version 0.92r2) programs to access protein-coding potential of transcripts without annotations for potential long non-coding RNAs CPC reference database use UniProt sequences in SWISS-PROT database. LncRNA analysis used full-length transcripts apart from the four major databases. Results predicted by both software programs were considered to represent “non-coding” sequences and were taken as the final lncRNA results. We used Infernal (version 1.1.2) software (http://eddylab.org/infernal/) for sequence alignment (Nawrocki and Eddy, 2013). LncRNAs were classified according to sequence conservation and secondary structures. The animal protein-coding sequences of isoforms were aligned by using hmmscan (version 3.1b2) (http://hmmer.org/download.html) against TFdb5 (version 2.0) to predict TF families (Zhang et al., 2015).
Software Parameters
(1) SMRT Link: version 9.0.0, ccs–min-length 50 –max-length 15,000 –min-passes 1 –min-snr 2.5 –min-rq 0.8 (min_predicted_accuracy) cluster: –use-qvs, other parameters are set as default. (2) cd-hit-est: version 4.6.7, -c 0.99 -T 6 -G 0 -aL 0.90 -AL 100 -aS 0.99 -AS 30. (3) Blast2GO: version 2.3.5, default parameters. (4) WEGO: version 2.0, default parameters. (5) ANGEL: version 2.4, default parameters. (6) Cogent: version 3.3, default parameters. (7) SUPPA: version 2.2, default parameters. (8) MIcroSAtellite: version 1.0, definition: unit_size, min_repeats 2-6 3-5 4-4 5-4 6-4. (9) Interruptions: max_difference_between_2_SSRs 100. (10) Primer: version 1.1.4, default parameters. (11) CNCI: version 2.0, default parameters. (12) Cpc: version 0.92r2, default parameters. (13) Infernal: version 1.1.2, default parameters.
Results
Quality Control of the Full-Length Transcriptomes
We obtained a total of 38,548,713 reads (76.18 Gb nucleotides) (Supplementary Figure 2A), with 2,121 bp of average length and 2,581 bp of N50 length. Then the data were deposited in the NCBI SRA database with project number PRJNA718003. All the subreads were further analyzed into CCS. A total of 1,063,453 CCSs were generated with an average length of 2,805 bp. FLNC reads used Minimap2 error correction to polish the third-generation sequence data into clusters. We generated 57,678 high-quality isoforms (HQ isoforms, prediction accuracy ≥ 0.99) for further analysis, and 1,415 low-quality sequences (low-quality isoforms, LQ isoforms, prediction accuracy <0.99) were obtained but not used in subsequent analysis. Later, we used CD-HIT-EST to process FLNC reads into clusters to remove redundant sequences, which was defined as the sequence similarity >99%. Finally, 54,319 full-length non-redundant transcripts were generated, with the features, i.e., 14,591 bp of maximum length and 3,308 bp of N50 sequence length (Supplementary Figure 2D). After the SMRT sequencing pipeline processing, we used BUSCO6 to evaluate the sequence completeness, and the result shows that the number of non-redundant sequence we generated was significantly reduced compared with the previous data (Supplementary Figure 2E).
Prediction of Coding Sequences and Functional Annotation
Full-length non-redundant transcripts were aligned against four databases using BLASTx. A total of 52,443 (96.54%) transcripts were annotated, and 40,067 (73.76%) transcripts were annotated in all four databases. The prediction and functional annotation of the encoded transcripts resulted in the annotation of 52,415 (96.49%), 52,182 (96.07%), 48,376 (89.06%), and 40,150 (73.92%) transcripts in the non-redundant (NR) database, KEGG database, SWISS-PROT, and KOG database, respectively (Figure 1A), as full-length transcripts.
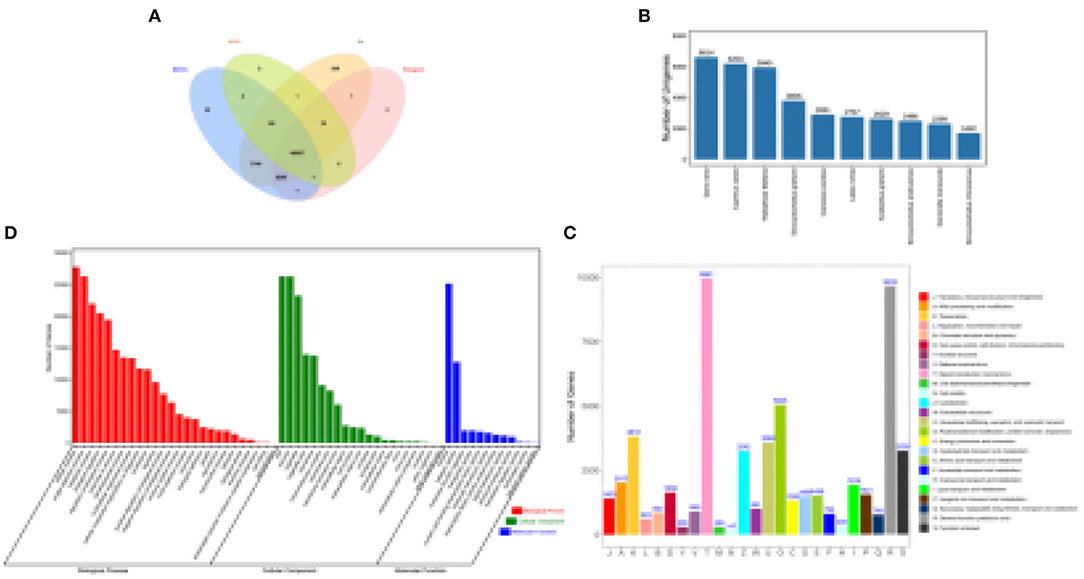
Figure 1. Characterization of functional annotation. (A) Overlapping of the different results annotated by four databases. (B) Protein taxonomic distribution annotated by non-redundant protein database. (C) Two levels of GO functional annotations of full-length transcripts. (D) Functional classification annotated in the EuKaryotic Orthologous Groups (KOG) database.
The majority of annotation result is Cyprinidae family in the NR annotation results, where the most abundant species was Danio rerio (6,614, 12.62%), and the other species in the top 10 results were Cyprinus carpio (6,204, 11.84%), Triplophysa tibetana (5,945, 11.34%), Sinocyclocheilus graham (3,806, 7.26%), Carassius auratus (2,891, 5.52%), Labeo rohita (2,757, 5.26%), Anabarilius grahami (2,629, 5.02%), Sinocyclocheilus anshuiensis (2,486, 4.74%), Danionella translucida (2,299, 4.39%), and Sinocyclocheilus rhinocerous (1,692, 3.23%) (Figure 1B).
In the KOG annotations, 40,150 (73.92%) transcripts were divided into 25 subcategories, with the highest percentage being involved in signal transduction mechanisms (Figure 1D). Regarding functional annotations, 34,958 (64.36%) transcripts were classified into 59 subcategories in the GO database. Among these subcategories, those receiving the most abundant annotations under the biological process category (54.89%) were cellular processes (6.68%), single-tissue processes (6.33%), metabolic processes (5.28%), and biological regulation (4.93%). Among cellular components (33.37%), the GO terms showing the highest abundance were cells (6.33%), cell components (6.32%), and organelles (5.59%). The molecular function (11.45%) GO term with the highest abundance was binding (6.15%) (Figure 1C).
The KEGG results annotated from 52,182 transcripts (96.07%) showed 357 pathways in total, including 6 primary categories with 45 secondary subcategories. And the “metabolism” pathway occupies a major position (Table 1).
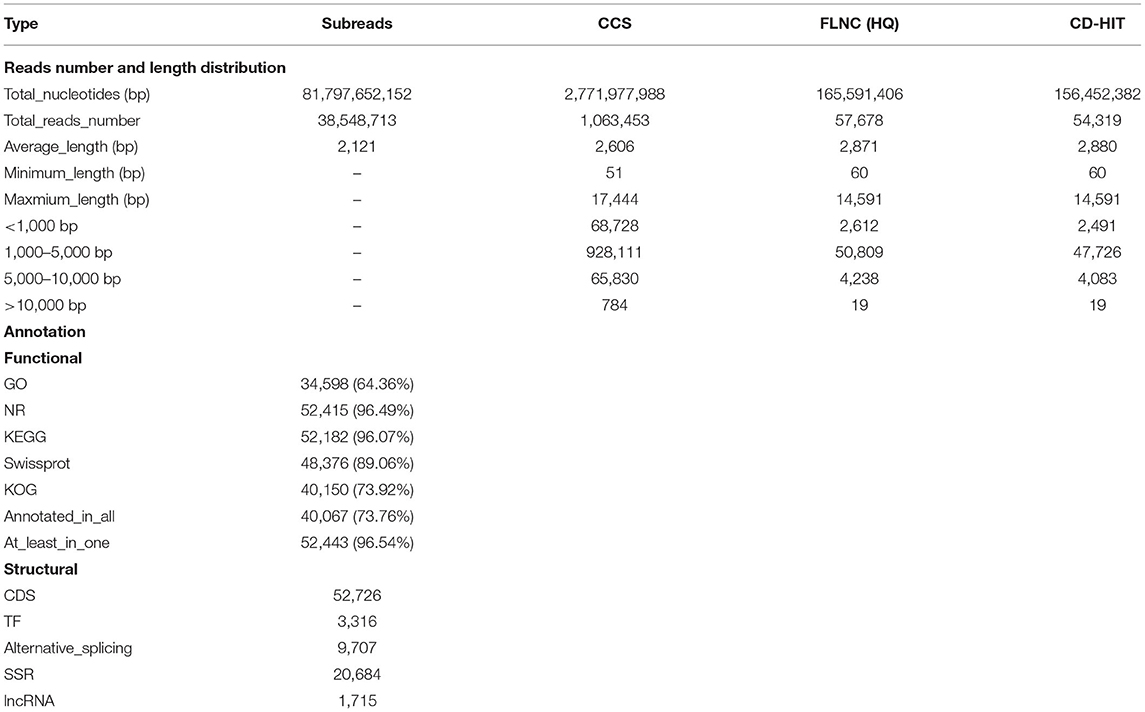
Table 1. Reads and annotation statistics for the ISO-seq transcripts.
Gene Structure Predictions
Protein-encoding transcripts were identified from the mRNA transcripts used for the ANGEL prediction of CDSs. A total of 52,726 (97.07%) transcripts were predicted (Supplementary Figure 3E and Table 1). Most of these CDSs were shorter than 2,500 bp. A total of 766 domains from a total of 25,024 isoforms were aligned via protein domain prediction.
RNA AS is a widespread biological phenomenon. It occurs after mRNA transcription before the formation of template DNA, and these events helped single protein-coding genes to form multiple proteins and increased biodiversity. We obtained 9,707 AS events at last (Supplementary Figure 3A), due to the lack of the reference genome of bigmouth buffalo, only 1,892 events were classified into 7 types of AS events (Supplementary Figure 3B) and the most identified event was reserved introns (RIs, 1,382).
We used high-quality isoforms, promoted genetic research to study genetic diversity, and analyzed SSRs identified in the ISO-seq library. For complex SSRs, a total of 20,684 SSRs were detected from 13,272 transcripts (Supplementary Figure 3C). The results showed that the dinucleotide repeats were the prominent part, with the number of 9,471 (65.14%), and most of them showed 4–7 duplicates. In the total of our results, the higher annotation number were dinucleotide repeats with 8–11 duplicates and trinucleotide repeats with 4–7 duplicates (Supplementary Figure 3D).
Long non-coding RNA was also analyzed using our data, which possess the feature of polyA ends. Since the bigmouth buffalo reference genome was not available, the exons of lncRNA had not been evaluated, but the figure shows the different annotation results from two software (Supplementary Figure 3F). We used Hmm search to predict the TFs from all single protein-coding genes, and we obtained 3,316 TF transcripts. The first four common families were the zf-C2H2 (748), bHLH (271), TF_bZIP (255), and homeobox (253) families (Supplementary Figure 3G). The results may facilitate the further elucidation of transcriptional regulation.
Reuse Potential
We reported for the first time the full-length transcriptome of bigmouth buffalo obtained using the PacBio SMRT platform. Our data of assembly and annotation results based on the ISO-seq technology could be helpful for further relative research on this species and could be of great benefit to future transcriptome analysis and genome sequencing of the bigmouth buffalo and its relatives.
Data Availability Statement
The raw sequencing data and files from the gene abundance analysis conducted in this study were deposited in the NCBI Sequence Read Archive (SRA) with accession number PRJNA718003.
Ethics Statement
The animal study was reviewed and approved by the Key Laboratory of Freshwater Fish Resources and Reproductive Development of the Ministry of Education.
Author Contributions
HG and HZ conceived and designed the experiment. ZJ, JZ, and BC raised fish. HG, LY, and LT dissected and collected fish tissue samples. HZ and HW completed the bioinformatics analysis. HG and JC provided tables and figures. HG and HZ drafted the manuscript. ZW and YL revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Financial Program of Ministry of Agriculture and Rural Affairs of China (Grant No. YYJZHC201921301350063) and the National Special Research Fund for Non-Profit Sector (Agriculture) (201203086).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.736188/full#supplementary-material
Footnotes
1. ^ANGEL software: References: ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA.
2. ^Leveraging transcript quantification for fast computation of alternative splicing profiles.
3. ^Utilizing sequence intrinsic composition to classify protein- coding and long non-coding transcripts.
4. ^CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine.
5. ^Huazhong University of Science and Technology. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors.
6. ^BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Download: https://busco.ezlab.org/.
References
Chu, X. M. (2015). Study on breeding adaptability of Big-mouth Buffalo in northern China. Modern Rural Sci. Techno. 2015:43.
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Coulter, A. A., Swanson, H. K., and Goforth, R. R. (2018). Seasonal variation in resource overlap of invasive and native fishes revealed by stable isotopes. Biol. Invasions 21, 315–321. doi: 10.1007/s10530-018-1832-y
Dba, B., Mct, A., Jtl, B., Afc, B., and Mdd, C. (2021). Complex to simple: fish growth along the illinois river network. Ecol. Complex. 45:100891. doi: 10.1016/j.ecocom.2020.100891
Doering, J. A., Villeneuve, D. L., Fay, K. A., Randolph, E. C., Jensen, K. M., Kahl, M. D., et al. (2019). Differential sensitivity to in vitro inhibition of cytochrome P450 aromatase (CYP19) activity among 18 freshwater fishes. Toxicol. Sci. 170, 394–403. doi: 10.1093/toxsci/kfz115
Dong, L., Liu, H., Zhang, J., Yang, S., Kong, G., Chu, J. S. C., et al. (2015). Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics 16:1039. doi: 10.1186/s12864-015-2257-y
Eddy, S., and Underhill, J. C. (1974). Northern Fishes 3rd edn. Minneapolis, MN: Univ. of Minnesota Press.
Enders, E. C., Charles, C., Watkinson, D. A., Kovachik, C., Leroux, D. R., Hansen, H., et al. (2019). analysing habitat connectivity and home ranges of bigmouth buffalo and channel catfish using a large-scale acoustic receiver network. Sustainability 11:3051. doi: 10.3390/su11113051
Great Lakes Mississippi River Interbasin Study (GLMRIS) (2012). Commercial Fisheries Baseline Economic Assessment - U.S. Waters of the Great Lakes, Upper Mississippi River, and Ohio River Basins, U.S. Army Corps of Engineers.
Hoffbeck, S. R. (2001). Without Careful Consideration Why Carp Swim in Minnesota's Waters. Minnesota Historical Society, Minnesota History.
Jin, W., Shi, D., Jianxin, Y., Yongping, G., Li, Y., Cijun, Z., et al. (2011). Induction of gynogenetic diploid by heterologous spermatozoa. Tianjin Fish. 2011, 23–31.
Keevin, T. M., Adams, S. R., Killgore, K. J., and Schaeffer, D. J. (2019). Effects of pressure changes induced by commercial navigation traffic on mortality of fish early life stages. J. Appl. Ichthyol. 35, 1105–1110. doi: 10.1111/jai.13953
Kuo, R. I., Tseng, E., Eory, L., Paton, I. R., Archibald, A. L., and Burt, D. W. (2017). Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics 18:323. doi: 10.1186/s12864-017-3691-9
Lackmann, A. R., Andrews, A. H., Butler, M. G., and Clark, M. E. (2019). Bigmouth Buffalo Ictiobus cyprinellus sets freshwater teleost record as improved age analysis reveals centenarian longevity. Commun. Biol. 2, 1–14. doi: 10.1038/s42003-019-0452-0
Liu, X., Du, H., Wu, C., Xiao, K., Qu, H., Zhang, X., et al. (2015). Complete mitochondrial genome of the Ictiobus cyprinellus (Actinopterygii: Ostariophysi). Mitochondrial DNA 27, 3242–3243. doi: 10.3109/19401736.2015.1007365
Moen, T. E. (1974). Population Trends, Growth, and Movement of Bigmouth Buffalo, Ictiobus Cyprinellus, in Lake Oahe, 1963-70. U.S. Fish and Wildlife Service (USFWS). Available online at: https://pubs.er.usgs.gov/publication/tp78
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Osborn, R. S., and Self, J. T. (1966). Observations on the spawning ecology of buffalos (Ictiobus bubalus and I. cyprinellus) in relation to parasitism. Proc. Okla. Acad. Sci. 46, 54–57.
Qu, C. Y., Hui, Y., and Ma, X. T. (2001). An acute toxic test of 6 kinds of drugs on the bigmouth buffalo fish ictiobus cyprinellus valenciennes. Res. Fish. 4, 42–43.
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 132, 365–386. doi: 10.1385/1-59259-192-2:365
Sauer, D. J., Heidinger, B. J., Kittilson, J. D., Lackmann, A. R., and Clark, M. E. (2021). No evidence of physiological declines with age in an extremely long-lived fish. Sci. Rep. 11:9065. doi: 10.1038/s41598-021-88626-5
Shimizu, K., Adachi, J., and Muraoka, Y. (2006). Angle: a sequencing errors resistant program for predicting protein coding regions in unfinished CDNA. J. Bioinform. Comput. Biol. 4, 649–664. doi: 10.1142/S0219720006002260
Wang, J., Chapman, D., Xu, J., Wang, Y., and Gu, B. (2018). Isotope niche dimension and trophic overlap between bigheaded carps and native filter-feeding fish in the lower Missouri River, USA. PLoS ONE 13:e0197584. doi: 10.1371/journal.pone.0197584
Wang, J., Hu, S., and Huang, Z. (1997). Experimental study on oxygen consumption rate and asphyxiation point of mullet larvae in America. Freshwater Fish. 1997, 7–9.
Workman, R. E., Myrka, A. M., Wong, G. W., Tseng, E., Welch, K. C., and Timp, W. (2018). Single-molecule, full-length transcript sequencing provides insight into the extreme metabolism of the ruby-throated hummingbird Archilochus colubris. GigaScience 7:giy009. doi: 10.1093/gigascience/giy009
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34, W293–W297. doi: 10.1093/nar/gkl031
Zhang, F., Man, Y. B., Mo, W. Y., and Wong, M. H. (2020). Application of spirulina in aquaculture: a review on wastewater treatment and fish growth. Rev. Aquacult. 12:12341. doi: 10.1111/raq.12341
Keywords: bigmouth buffalo, Ictiobus cyprinellus, full-length transcriptome, PacBio sequencing, ISO-seq
Citation: Ge H, Zhang H, Yang L, Wang H, Tu L, Jiang Z, Zheng J, Chen B, Chen J, Li Y and Wang Z (2021) Full-Length Transcriptome Sequencing From the Longest-Lived Freshwater Bony Fish of the World: Bigmouth Buffalo (Ictiobus Cyprinellus). Front. Mar. Sci. 8:736188. doi: 10.3389/fmars.2021.736188
Received: 04 July 2021; Accepted: 09 August 2021;
Published: 17 September 2021.
Edited by:
Jin Liu, Peking University, ChinaReviewed by:
Qiong Shi, Beijing Genomics Institute (BGI), ChinaHao Song, Institute of Oceanology, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Ge, Zhang, Yang, Wang, Tu, Jiang, Zheng, Chen, Chen, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Li, YXF1YXRpY3NAc3d1LmVkdS5jbg==; eXVubGljbkAxMjYuY29t; Zhijian Wang, d2FuZ3pqQHN3dS5lZHUuY24=; d2FuZ3pqMTk2OUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship