 Hongbo Jiang
Hongbo Jiang Jie Bao
Jie Bao Jinghui Liu
Jinghui Liu Qijun Chen
Qijun Chen- Key Laboratory of Livestock Infectious Diseases in Northeast China, Ministry of Education, Key Laboratory of Zoonosis, Shenyang Agricultural University, Shenyang, China
Enterocytospora artemiae (EAM) mainly parasitizes the hepatopancreas of Palaemonetes sinensis. Serious infection leads to hepatopancreatic lesions, which greatly reduce the vitality of P. sinensis. Currently, EAM is detected via conventional PCR methods. However, conventional PCR has low sensitivity and cannot be used for accurate quantitative detection of EAM or its parasitic activity in host tissues. In this study, we designed a pair of specific primers based on the sequence of the ribosomal protein S9 gene (RPS9; GenBank accession number: MZ420734) to establish and optimize a SYBR Green I real-time fluorescent quantitative PCR detection method for EAM. Only EAM appeared as a bright and single target band, whereas other microorganisms did not, indicating that the primer for RPS9 had high specificity. This method displayed optimum amplification effects at an annealing temperature of 55°C, and the melting curve of the product produced a single peak. The established method showed a good linear relationship from 2.2 × 108 to 2.2 × 101 copies/μL. The relationship between the number of cycle thresholds (Ct) and the logarithm of the initial template amount (x) conformed to Ct = −3.281 log x + 36.543 (R2 = 0.998). Amplification efficiency was 101.737%, and the lower limit of detection sensitivity was 2.2 × 101 copies/μL. Good intra- and inter-group repeatability was observed within the linear range. The sensitivity of this method was more than 200 times higher than that of nested PCR. Thus, detection data obtained using this method may be useful as a technical reference for rapid and accurate identification of EAM infection and for the prevention and control of EAM during P. sinensis breeding.
Introduction
Microsporidia are a group of common fungal intracellular parasites consisting of over 200 genera and 1600 species (Stentiford et al., 2016). This group has a wide range of hosts, including almost all lower invertebrates, as well as higher vertebrates. Microsporidia are opportunistic pathogens, which easily infect patients receiving immunosuppressive therapy for congenital immune disorders (Pol et al., 1993; Didier and Weiss, 2011). Currently, 17 microsporidia species belonging to nine genera are known for their ability to infect humans (Pan et al., 2018). In addition to causing harm to human health, many microsporidia cause economic damage to important animal and plant industries (Kent and Speare, 2005; Higes et al., 2013). For example, the pepper disease of silkworms, which plagued the silkworm industry for more than two centuries, is caused by Nosema bombycis, a microsporidian (Nageswara Rao et al., 2004). Decapoda, an order of crustaceans that hosts microsporidia, has been studied for more than 100 years. As early as the end of the nineteenth century, French researchers reported that Thelohania infects Palaemon rectirostris, P. serratus, Crangon vulgaris, Procambarus clarkii, and Astacus fluviatilis as well as other shrimp (Stentiford and Dunn, 2014). Reportedly, the infection rate as well as the transmission range of Enterocytozoon hepatopenaei (EHP) in Litopenaeus vannamei has been increasing annually, making it one of three major pathogens endangering healthy L. vannamei cultures (Biju et al., 2016; Chaijarasphong et al., 2020).
Enterocytospora artemiae (EAM) was first discovered in Artemia in 2013. It was considered as an obligate parasite of Artemia that was confined to Europe and America (Rode et al., 2013). However, in 2020, it was found that this microsporidian had spread to Asia and was infecting the economically important Chinese grass shrimp, Palaemonetes sinensis (Jiang et al., 2020). EAM mainly parasitizes the hepatopancreas of P. sinensis (Jiang et al., 2020). Serious EAM infections lead to hepatopancreatic lesions and greatly reduce the vitality of P. sinensis. Although the mortality rate is not high, absorption of host nutrition by these microsporidia leads to increased breeding costs. Microsporidia are minute and do not cause obvious symptoms after infecting a host. Therefore, it is necessary to establish a rapid and accurate method to detect these parasites. Currently, only conventional PCR methods are used to detect EAM (Rode et al., 2013; Jiang et al., 2020). However, conventional PCR has low sensitivity and cannot be used for accurate, quantitative detection of EAM or its parasitic activity in host tissues. When the number of microsporidia infecting a host is low, observation via traditional staining followed by microscopy or electron microscopy becomes ineffective, preventing various species of microsporidia from being accurately determined. In contrast, real-time fluorescence quantitative PCR (qPCR), which is associated with advantages such as high specificity as well as high sensitivity, consumes less time and allows the copy number of the initial DNA template in the sample to be quantitated. The qPCR detection process is completed in a relatively closed and independent system, which prevents cross contamination of the sample and environment (Kotková et al., 2018; Li et al., 2019). In this study, we designed a nested PCR method, leading to the establishment of a qPCR protocol that detects EAM using SYBR Green I fluorescent dye. We compared the sensitivity and detection efficacy of conventional PCR, nested PCR, and qPCR to ensure rapid and accurate identification of EAM infections to control and prevent infection during P. sinensis breeding.
Materials and Methods
Sample Collection of Palaemonetes sinensis
P. sinensis was collected from farms 1, 2, and 3 in Panjin, Liaoning province. These shrimp were transported to Shenyang Agricultural University for temporary cultivation. During the temporary cultivation period, 30 shrimp were randomly selected from each farm to detect the prevalence via different methods. During sampling, the live shrimp were anesthetized on ice for 5 min, and the hepatopancreases were removed quickly and placed in a centrifuge tube containing 100% alcohol for further analysis.
DNA Extraction
DNA was extracted from the tissues of P. sinensis in strict accordance with the instructions of the TIANamp Marine Animals DNA Kit (Tiangen, Beijing). The extracted DNA was diluted to 50 ng/μL after determination of DNA concentration with an ultra-microspectrophotometer (K5500, Beijing Kaiao Technology Development Co., Ltd.), and then was stored at −20°C.
Conventional PCR Amplification and Nested PCR Primer Design
Previously reported universal 18S rDNA primers, V1F and 1492R, were used for PCR amplification (Rode et al., 2013). The V1F and 1492R sequences are listed in Table 1. The amplification procedure was as follows: denaturation at 94°C for 3 min; 35 cycles (denaturation at 94°C for 45 s, annealing at 45°C for 30 s, and extension at 72°C for 90 s); and a final extension at 72°C for 5 min. The PCR reaction system contained a total of 15 μL, with 13 μL of 2 × Taq Plus Master Mix, 0.5 μL of Primer-F, 0.5 μL of Primer-R, and 1 μL of DNA template.

Table 1. Primer sequences and PCR product size.
Primers, V1F, and 1492R were used for the first round of nested PCR amplification. The second-round primers, YW-F and YW-R, are shown in Table 1. The target segment was 322 bp. The amplification procedure was as follows: denaturation at 94°C for 3 min; 35 cycles (denaturation at 94°C for 30 s, annealing at 55°C for 45 s, and extension at 72°C for 45 s); and a final extension at 72°C for 5 min. The PCR reaction system was the same as in the first round of nested PCR amplification, and DNA samples were taken from the first round of PCR and diluted 1000 times.
Preparation of Plasmid Standard
Conventional PCR amplification: Using the EAM-positive samples as the template, the ribosomal protein S9 (RPS9) primers were used for amplification. The primers specific for RPS9 (GenBank accession number: MZ420734), RPS9-F, and RPS9-R, were designed using Primer 5.0 (Supplementary File 1). The RPS9-F and RPS9-R primer sequences are shown in Table 1. The PCR procedure was as follows: pre-denaturation at 94°C for 3 min; 35 cycles (denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s); and a final extension at 72°C for 5 min. The PCR reaction system was the same as that described for nested PCR.
Purification and recovery of DNA fragments: The PCR product was dispensed into the well of a 1.5% agarose gel, which was then transferred to an electrophoresis apparatus and run at 100 V for 38 min. After electrophoresis, a strong, positive single strip of 206 bp was rapidly excised from the gel, and the gel containing the target strip was placed in a centrifuge tube, following which DNA was recovered using a FastPure Gel DNA Extraction Mini Kit (Vazyme, Nanjing, China).
Vector recombination: A reaction solution (5 μL total volume) composed of pMD-19-T vector (1 μL), extracted DNA samples (1 μL), and ddH2O (3 μL) was prepared. Next, 5 μL of solution I was added to the prepared reaction solution and left to stand overnight at 4°C.
Strain screening: A mixture of vector solution and Escherichia coli DH5α competent cells was added to a 1.5 mL centrifuge tube, placed on ice for 30 min, heated at 42°C for 45 s, and placed on ice for 1 min. Subsequently, 890 μL of LB medium was added, and the mixture was shaken at 37°C for 60 min. Selection plates were made by adding 35 μL of X-gal and 5 μL of IPTG to LB agar medium containing ampicillin (AMP). Next, 100 μL of bacterial solution was spread on the selection plate, and the plate was placed in an incubator at 37°C overnight after sealing with parafilm. A single white colony was picked from the plate using a pipette tip and dissolved in 100 μL of ddH2O. One microliter of the plasmid DNA sample was verified via sequencing (Sangon, Shanghai, China). The remaining liquid was poured into a centrifuge tube containing 10 mL of AMP-LB liquid and placed in an orbital shaker for approximately 4 h until the liquid was turbid. Plasmids were extracted using a FastPure Plasmid Mini Kit (Vazyme, Nanjing, China).
Optimization of the Quantitative PCR Amplification Conditions
The initial concentration of the plasmid was 7.3 ng/μL. According to the formula copies/μL = initial concentration (ng/μL) × 6.02 × 1014/[(base number of vector fragment + base number of target gene) × 660], the copy number of the recombinant plasmid was 2.2 × 109 copies/μL. The recombinant plasmid was diluted using a 10-fold serial dilution with DEPC-treated water, and a 2.2 × 108–2.2 × 101 copies/μL plasmid standard was used as a template for qPCR amplification. The recombinant plasmid gradient was used as the standard, and the total reaction system volume was 20 μL. The PCR reaction system contained 10.0 μL of 2 × ChamQ Universal SYBR qPCR Master Mix, 0.4 μL of RPS9-F, 0.4 μL of RPS9-R, 8.2 μL of ddH2O, and 1 μL of recombinant plasmid. The blank group was treated with ddH2O instead of DNA samples. The amplification reaction was performed in the Applied Biosystems QuantStudio 3 (Thermo Fisher Scientific, Waltham, MA, United States). After pre-denaturation at 95°C for 30 s, 40 cycles of amplification at 95°C for 10 s, X°C for 30 s (X was the annealing temperature gradient and the set range was 50–65°C) and 72°C for 30 s were carried out to select the best annealing temperature. The data analysis was performed using QuantStudio Design and Analysis Software v1.4 (Applied Biosystems). The optimum annealing temperature was 55°C, and the reaction procedure was as follows: pre-denaturation at 95°C for 30 s and 40 cycles (95°C for 10 s; 55°C for 30 s; 72°C for 30 s).
Plotting of Quantitative PCR Standard Curve for Enterocytospora artemiae
The recombinant plasmid was diluted via a 10-fold serial dilution, which yielded a dilution series of 8 concentrations ranging from 2.2 × 108 to 2.2 × 101 copies/μL. The standard curve of the relationship between the logarithm of the initial quantity of the standard template (x) and the cycle threshold number (Ct) of the amplification products was established. The quality of the standard curve was determined by analyzing the correlation coefficient and amplification efficiency.
Sensitivity Comparison, Specificity Analysis, and Stability Evaluation of Quantitative PCR Primer RPS9
The sensitivity of qPCR and conventional PCR (RPS9), indicated by recombinant plasmids with a concentration of 2.2 × 108–2.2 × 101 copies/μL, and detection limits for qPCR and conventional PCR were analyzed by determining whether showing an S-shaped amplification curves and whether the target bands appeared separately. Furthermore, qPCR, nested PCR (V1F/1492R—YW-F/YW-R), and conventional PCR (V1F/1492R) were used to detect the 90 shrimp hepatopancreas samples simultaneously.
DNA from the hepatopancreases of healthy P. sinensis was used as the negative control, and microsporidia (EHP, Hepatospora eriocheir and Microsporidia sp.), viruses (white spot syndrome), bacteria (Pseudomonas putida and Vibrio parahaemolyticus), and fungi (Metschnikowia bicuspidata) were detected simultaneously. These pathogens were obtained from our laboratory. Specificity was analyzed using conventional PCR (RPS9) and qPCR.
To determine the stability of qPCR, serially diluted positive plasmid DNA (2.2 × 108–2.2 × 101 copies/μL) was used for qPCR templates, in which three replicates were set for each template concentration. Stability assessment consisted of two parts: intra-group repetition (in the same experiment) and inter-group repetition (in the same group). For intra-group repetition, the mean value, standard deviation, and coefficient of variation were calculated using the Ct value of three replicates (Liu et al., 2016). For inter-group repetition, the experiment was repeated thrice. The mean value, standard deviation, and coefficient of variation were calculated using the Ct value of three experiments (Ding et al., 2017).
Tissue Tropisms and Detection of Potential Enterocytospora artemiae Carriers by Quantitative PCR
Tissue tropisms: 10 shrimp were randomly selected from the temporary container for tissue tropism detection. The selected tissues included the hepatopancreas, gut, gill, muscle, and stomach.
Potential carriers: Biological and water samples were collected from the rice field of infected P. sinensis. The biological samples included fish, two Chanodichthys erythropterus and five Pseudorasbora parva; shrimp, five Macrobrachium nipponense and four Neocaridina denticulate; five cladocerans; and five copepods. The hepatopancreas tissues of fish and shrimp and the whole cladocerans and copepods were used for DNA extraction. The water samples from the rice field were filtered using a 0.22 μM filter membrane. The filter membrane was then washed with distilled water, the supernatant was discarded after centrifugation (8000 rpm, 10 min), and the precipitate was used to extract DNA. The extracted DNA was diluted to 50 ng/μL, and then was detected by qPCR and nested PCR. PCR amplification products of nested PCR positive samples were verified EAM via sequencing (Sangon, Shanghai, China).
Results
Specificity Analysis
The specificity of qPCR primers was analyzed by detecting other microsporidia, fungi, bacteria, and viruses. Only the hepatopancreatic DNA of EAM-infected shrimp appeared as a bright, single target band (Figure 1A), indicating that the primer RPS9 had high specificity. In addition, the qPCR amplification curve showed that only samples with positive EAM showed amplification curves, whereas no fluorescence signal was detected for non-target genes (Figure 1B), further indicating that the primer showed excellent specificity.
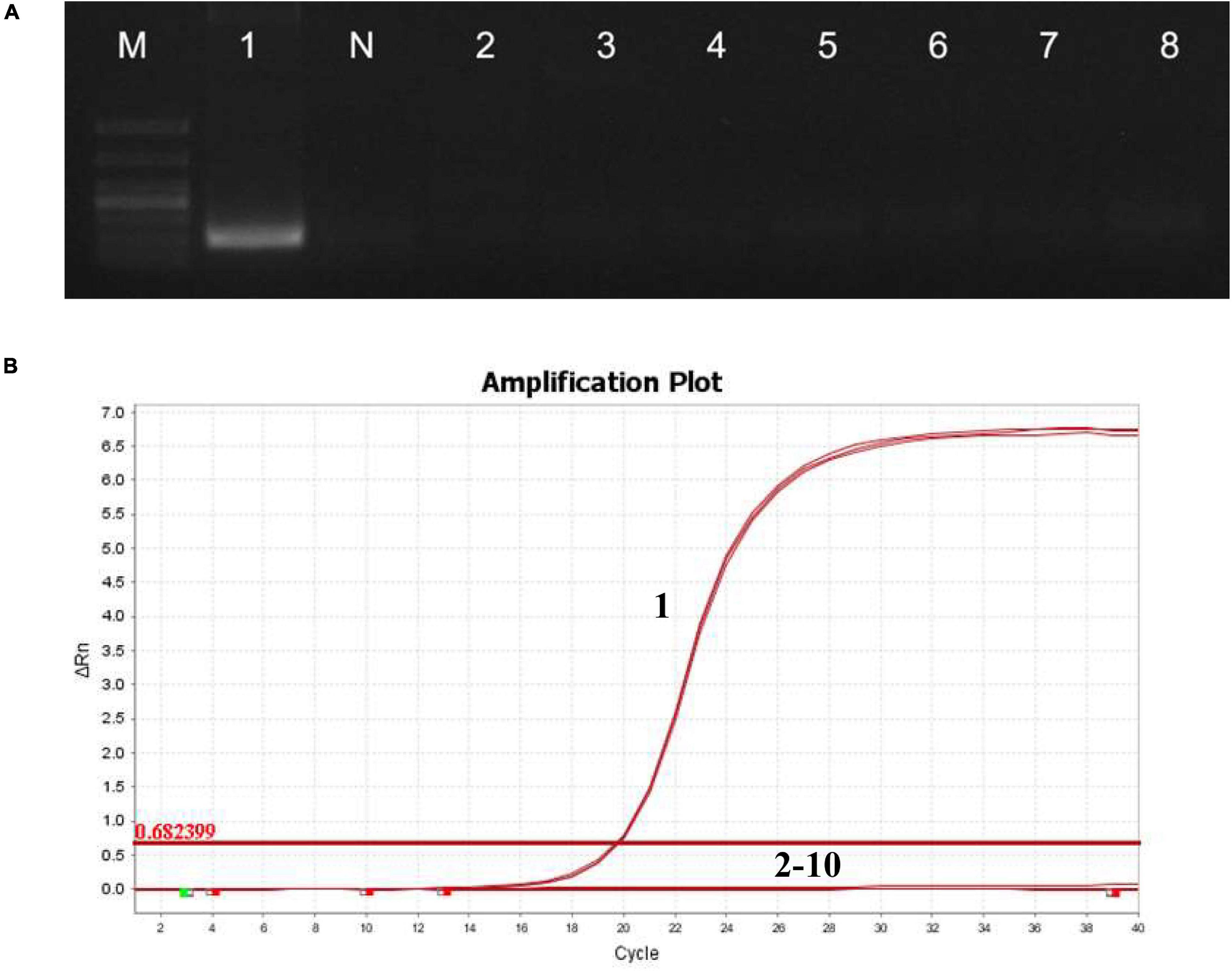
Figure 1. Specificity of detection of RPS9. (A) Electrophoresis of PCR products amplified using primer RPS9. M: DL1000; 1: EAM; N: healthy shrimp hepatopancreas DNA; 2: E. hepatopenaei; 3: H. eriocheir; 4: M. bicuspidata; 5: Microsporidia sp.; 6: P. putida; 7: V. parahaemolyticus; and 8: white spot syndrome virus. (B) Specificity of qPCR amplification curve of RPS9. 1: EAM; 2: E. hepatopenaei; 3: H. eriocheir; 4: M. bicuspidata; 5: Microsporidia sp.; 6: P. putida; 7: V. parahaemolyticus; 8: white spot syndrome virus; 9: healthy shrimp hepatopancreas DNA; and 10: water.
Construction of Quantitative PCR Standard Curve
The established qPCR system for EAM was used to amplify eight standard templates (2.2 × 108–2.2 × 101 copies/μL) with 10-fold gradient dilution, and the standard curve of the relationship between the threshold Ct value and the logarithm of the initial quantity of standard templates (x) was established. The qPCR results showed a strong fluorescence signal from 2.2 × 108 to 2.2 × 101 copies/μL (Figure 2A). The relationship was Ct = − 3.281 log x + 36.543 (Figure 2B). The correlation coefficient R2 was 0.998, and the amplification efficiency was 101.737%.
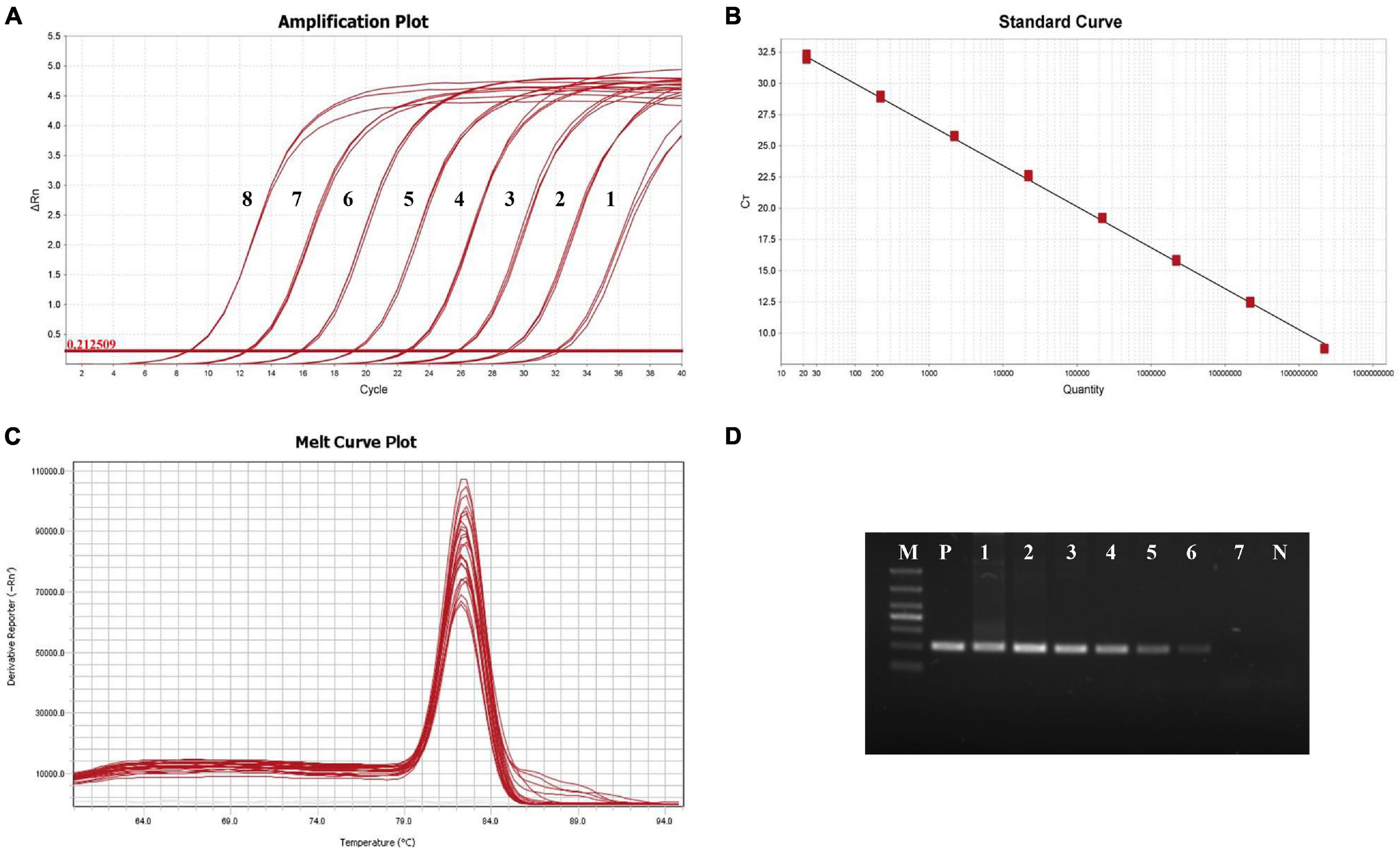
Figure 2. Amplification of the standard samples. (A) The amplification curve of RPS9-qPCR. 1: 2.2 × 101 copies/μL; 2: 2.2 × 102 copies/μL; 3: 2.2 × 103 copies/μL; 4: 2.2 × 104 copies/μL; 5: 2.2 × 105 copies/μL; 6: 2.2 × 106 copies/μL; 7: 2.2 × 107 copies/μL; and 8: 2.2 × 108 copies/μL. (B) Standard curve of qPCR-RPS9. (C) Melting curve of qPCR-RPS9. (D) Agarose electrophoresis of conventional PCR-RPS9. M: DL1000; P: EAM; N: Hepatopancreas DNA of healthy shrimp; 1: 2.2 × 107 copies/μL; 2: 2.2 × 106 copies/μL; 3: 2.2 × 105 copies/μL; 4: 2.2 × 104 copies/μL; 5: 2.2 × 103 copies/μL; 6: 2.2 × 102 copies/μL; and 7: 2.2 × 101 copies/μL.
Melting curve analysis (Figure 2C) showed that the curves of three parallel amplification products in each gradient group merged to produce a single melting peak at 82.4°C, with overlapping positions. This unique melting peak indicated that there was no non-specific amplification or primer dimer formation during the amplification process.
Stability Assessment
Six concentrations of plasmid (2.2 × 108–2.2 × 101 copies/μL) were selected, and a repeatability analysis was conducted using three parallel samples of the same gradient within the group. The coefficient of variation of Ct was much less than 1% (Table 2), indicating that intra-group repeatability was very good. All coefficients of variation between groups were less than 2% (Table 2), indicating that the method showed good repeatability, thereby ensuring the stability and reliability of the test results.
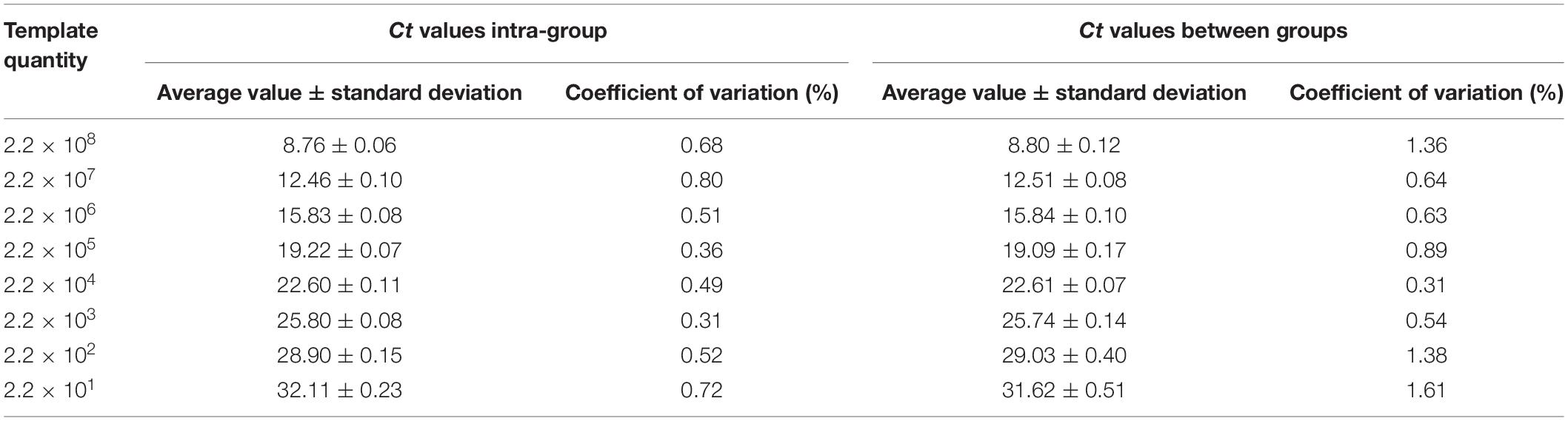
Table 2. Repeatability of detection of EAM by RPS9-qPCR.
Comparison of Sensitivity Between Quantitative PCR and Conventional PCR
Standard plasmid templates (2.2 × 108–2.2 × 101 copies/μL) were used for conventional PCR and qPCR. The amplified products of conventional PCR were analyzed by electrophoresis on a 1.5% agarose gel. The primer, RPS9, was detected by conventional PCR at 2.2 × 102 copies/μL, and at this concentration, the electrophoresis band was dim and almost invisible to the eye (Figure 2D).
With qPCR, the fluorescence signal could be detected from 2.2 × 101 to 2.2 × 108 copies/μL (Figure 2A). When the template concentration was 2.2 × 101 copies/μL, the cycle threshold was 32.11 and had good S-shaped amplification curves, indicating that the detection limit of qPCR was 2.2 × 101 copies/μL and that sensitivity was over 10 times that of conventional PCR.
Detection of Samples by Nested PCR and Quantitative PCR for Enterocytospora artemiae
Nested PCR showed that, of the 90 P. sinensis hepatopancreas samples, 56 were positive, indicating a detection rate of 62.22% (56/90). The ratio of PCR samples negative in the first round to PCR samples positive in the second round was 37/56 (66.07%), and the ratio of positives in both rounds was 19/56 (33.93%). The qPCR results showed that 73 shrimp samples were positive, with a detection rate of 81.11% (73/90). Ct was substituted in the established formula Ct = − 3.281 log x + 36.543. The E. artemiae content ranged from 4.4 × 101 to 6.3 × 107 copies/μL. EAM could be detected only when the level of 1.4 × 105 copies/μL was exceeded in the first round of PCR. When the copy number of EAM small subunit (SSU) rRNA was lower than 4.6 × 103 copies/μL, the results for nested PCR were negative, whereas the limit of qPCR for EAM was 2.2 × 101 copies/μL. This indicates that the sensitivity of qPCR for EAM was over 200 times higher than that of nested PCR for detecting P. sinensis in hepatopancreas samples.
Tissue Tropisms and Potential Carrier Detection by Quantitative PCR for Enterocytospora artemiae
The EAM load in the five tissues differed (Table 3). It was highest in the hepatopancreas, followed by the gut, gill, stomach, and muscle. However, there was no difference in the positive rate of EHP among the five tissues, all of which were 80%.

Table 3. Tissue tropisms of EAM in P. sinensis.
The water samples collected from the rice field of infected P. sinensis were all positive by qPCR detection. Some samples of fish and shrimp in the collected aquatic biological samples were positive by PCR detection, and the positive rate of cladocerans and copepods reached 80% (Table 4). However, C. erythropterus and M. nipponense was not detected by nested PCR (Table 4). Nested PCR positive samples were confirmed as EAM after sequencing and blast analysis, indicating that qPCR detection method had high sensitivity.
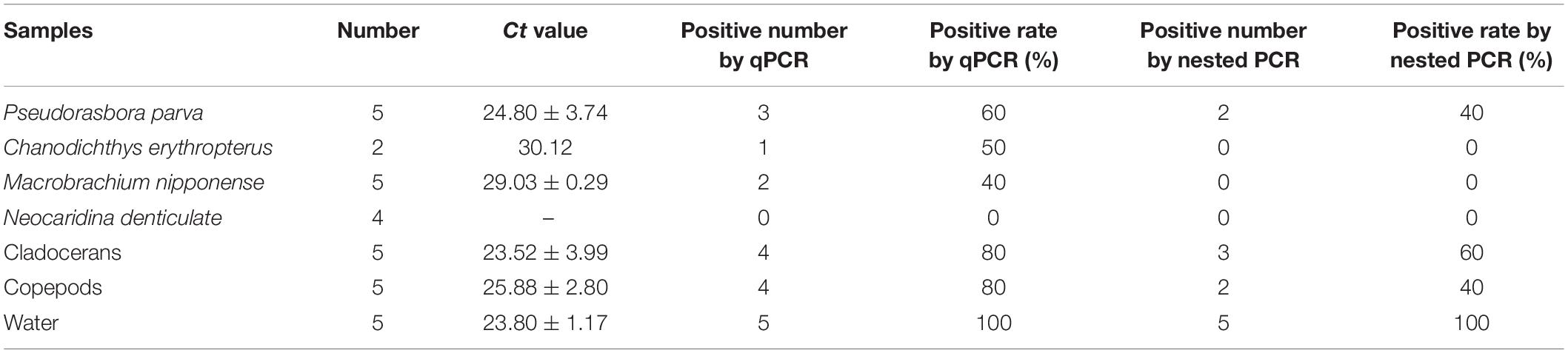
Table 4. Detection rate of EAM in samples from rice field of infected P. sinensis by qPCR and nested PCR.
Discussion
Rapid and accurate identification of pathogens is the basis of aquatic animal disease control. Microscopic examination is a rapid method. However, microsporidia can be observed under high magnification only when infection is severe. In addition, it is difficult to accurately identify microsporidia only by appearance due to inconsistencies during different developmental stages. Therefore, it is impossible to obtain accurate information regarding E. artemiae infection via microscopic examination. Based on this, highly sensitive molecular methods, such as PCR (Tang et al., 2015), nested PCR (Jaroenlak et al., 2016), qPCR (Liu et al., 2018; Piamsomboon et al., 2019), loop-mediated isothermal amplification (Suebsing et al., 2013; Cai et al., 2018), and recombinase polymerase amplification (Ma et al., 2021), are used instead of microscopic methods. Compared with conventional PCR, qPCR has the advantages of high sensitivity, good repeatability and quantification. Furthermore, qPCR reduces non-specific amplification and shortens detection time. Two common qPCR methods are the TaqMan probe and the SYBR Green I fluorescent dye method. Although the TaqMan probe method shows higher specificity, its cost of detection is also high, limiting its practical use. The SYBR Green I fluorescence method has the advantages of low cost and easy implementation and is more suitable for rapid detection. Notably, qPCR is often used in the detection of microsporidia and is considered to be more sensitive than conventional PCR (Naung et al., 2020). Wang et al. (2020) used qPCR to detect the polar tube protein 2 gene when quantitatively analyzing EHP in infected shrimp and reported that sensitivity was increased by at least two orders of magnitude compared with that of conventional PCR. Phelps and Goodwin (2007) used the same technology to detect Notemigonus crysoleucas and reported that the minimum gene copy number that could be detected in each reaction was as low as 10. Because the SSU rRNA gene of microsporidium is not a single-copy gene, ultrasonic treatment of spores can be used to extract DNA, which improves detection sensitivity, enabling 0.14 spores to be detected at the lowest level in each reaction (Phelps and Goodwin, 2007). In this study, the sensitivity of qPCR for detecting EAM was higher than that of conventional PCR and nested PCR. With real samples, nested PCR did not detect target DNA fragments below 4.6 × 103 copies/μL, and the sensitivity was only approximately 1/200 of that of qPCR for EAM. In contrast, in a batch of real samples, the detection rate of qPCR for EAM (81.11%) was significantly higher than that of nested PCR (62.22%). Meanwhile, the qPCR method was used to detect EAM tissue tropism and the carriers in the culture environment. The tissue tropism detection found that the content in the hepatopancreas was the highest, indicating that it was the main location of EAM, which was consistent with previous research (Rode et al., 2013; Jiang et al., 2020). At the same time, EAM was also detected in the gut, gill, stomach and muscle, and the same results have been obtained with EHP (Santhoshkumar et al., 2017; Cheng et al., 2018). Although EAM was detectable in these tissues in this study, whether EAM can infect the cells of these tissues needs to be further studied. Furthermore, the same infection rates were obtained in different tissues, which may be related to the high content of EAM in P. sinensis. Certainly, hepatopancreatic tissue was preferred because the load was the highest (Table 4). However, when hepatopancreas tissue is used for the determination of other indexes in research, tissues such as the gut can be considered as an alternative for detecting infection. Environmental detection found EAM in some biological samples, except for Neocaridina denticulata, and the detection rate in water was 100%. Sequencing showed that the positive sample of nested PCR was EAM, but the detection rate was significantly lower than qPCR, indicating that qPCR method has high sensitivity and can find carriers in the environment. This can provide guidance for transmission route research and prevent EAM infection.
Researchers have attempted to use qPCR to detect different microsporidia genes, such as those encoding the SSU rRNA, the polar tube protein, tubulin, and the spore wall protein (Jaroenlak et al., 2016; Han et al., 2018; Piamsomboon et al., 2019; Wang et al., 2020). Of these, the SSU rRNA gene carries the most commonly used detection target sequence. However, this gives rise to false positive results and shows high similarity with SSU rRNA genes of different species, leading to false detections (Lucchi et al., 2012; Tangprasittipap et al., 2013; Hitakarun et al., 2014). Sequence alignment showed that the EAM SSU rDNA gene (GenBank No. MT 645708) had 94% shared homology with the SSU rRNA gene of Globulispora mitoporans (GenBank No. KT762153), which shared more than 85% identity with five other species (Jiang et al., 2020). Therefore, we selected a specific target to modify the SYBR Green I qPCR detection method to detect microsporidia in P. sinensis. In this study, we established a qPCR method for the detection of EAM using RPS9 and established a minimum copy number of 22 copies/μL, indicating a highly sensitive diagnostic standard. RPS9 is widely distributed in yeast, bacteria, parasites, mammals, and humans. Studies have shown that RPS9 is a highly conserved gene, which plays a role in DNA repair, self-translation regulation, development regulation, malignant transformation of normal cells as well as in other undetected functions and is often used for pathogen detection (Lindström and Zhang, 2008; Lindström and Nistér, 2010; Araújo et al., 2012). In addition, whereas three species of microsporidia were detected using this study, no specific bands or interference reactions pertaining to other shrimp pathogens were found (Figure 2), indicating that the qPCR-RPS9 method showed high specificity.
Establishment of a qPCR method specifically for microsporidium not only enabled microsporidia to be detected sensitively and efficiently, but also allowed quantification. Analyzing microsporidium load is extremely important when conducting research on the prevention and control of microsporidia. Liu et al. (2016) reported that EHP load was negatively correlated with the growth of L. vannamei. When the relative copy number of EHP SSU rDNA in the hepatopancreas exceeded 103 copies/ng hepatopancreatic DNA, the growth of the shrimp was significantly slower, indicating that the effect of EHP on shrimp growth had reached a high-risk level (Liu et al., 2016). Thus, detection of microsporidium load enables better breeding strategies to be formulated. A serious microsporidium infection in P. sinensis causes body color turbidity and hepatopancreatic lesions, seriously reducing aquaculture yields (Jiang et al., 2020). Therefore, establishment of a rapid and quantitative detection method may help determine the severity of infection. In addition, this method may be useful for clarifying the relationship between EAM load and growth, leading to productive P. sinensis cultivation.
Conclusion
Serious EAM infection leads to hepatopancreatic lesions, which greatly reduce the vitality of P. sinensis. Conventional PCR has low sensitivity and cannot be used for accurate quantitative detection of EAM. In this study, a SYBR Green I real-time fluorescent quantitative PCR detection method based on the sequence of the ribosomal protein S9 gene was established and optimized. This method had high specificity and stability and was more than 200 times more sensitive than nested PCR. Thus, detection data obtained using this method may be useful as a technical reference for rapid and accurate identification of EAM infection and for the prevention and control of EAM during P. sinensis breeding.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Animal Experiments Ethics Committee of Shenyang Agricultural University.
Author Contributions
HJ, JB, and QC were involved in designing of the research and wrote the manuscript. HJ, JB, JL, YC, and SH performed the majority of the experiment, data processing, analysis, and interpretation. CF and XL assisted in sample collection and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Liaoning province Department of Education fund item (LSNQN202002), the China Agriculture Research System of MOF and MARA (CARS-48), the Liaoning Science and Technology Mission Project (2020JH 5/10400147 and 2020JH5/10400115), and the Liaoning Province Key R&D Planning Guidance Plan Project (2019JH8/102 00018).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.730569/full#supplementary-material
References
Araújo, H. F., Campos, P. C., Camargo, D. R., Pereira, F. N., Samuel, M. L., Oliveira, M. A., et al. (2012). Immune response and protective efficacy of S9 ribosomal protein of Streptococcus pneumoniae in a model of sepsis. Can. J. Microbiol. 58, 1055–1062. doi: 10.1139/w2012-083
Biju, N., Sathiyaraj, G., Raj, M., Shanmugam, V., Baskaran, B., Govindan, U., et al. (2016). High prevalence of Enterocytozoon hepatopenaei in shrimps Penaeus monodon and Litopenaeus vannamei sampled from slow growth ponds in India. Dis. Aquat. Organ. 120, 225–230. doi: 10.3354/dao03036
Cai, S. X., Kong, F. D., Xu, S. F., and Yao, C. L. (2018). Real-time loop-mediated isothermal amplification for rapid detection of Enterocytozoon hepatopenaei. PeerJ 6:e5993. doi: 10.7717/peerj.5993
Chaijarasphong, T., Munkongwongsiri, N., Stentiford, G. D., Aldama-Cano, D. J., Thansa, K., Flegel, T. W., et al. (2020). The shrimp microsporidian Enterocytozoon hepatopenaei (EHP): biology, pathology, diagnostics and control. J. Invertebr. Pathol. 2020:107458. doi: 10.1016/j.jip.2020.107458
Cheng, D. Y., Qiu, L., Song, Z. L., Wan, X. Y., Dong, X., Xie, G. S., et al. (2018). Differences between populations and tissues of Litopenaeus vannamei infected with Enterocytozoon hepatopenaei. Prog. Fishery Sci. 39, 83–92. doi: 10.19663/j.issn2095-9869.20170426001
Didier, E. S., and Weiss, L. M. (2011). Microsporidiosis: not just in AIDS patients. Curr. Opin. Infect. Dis. 24, 490–495. doi: 10.1097/QCO.0b013e32834aa152
Ding, Z. F., Chen, J. Q., Lin, J., Zhu, X. S., Xu, G. H., Wang, R. L., et al. (2017). Development of In situ hybridization and real-time PCR assays for the detection of Hepatospora eriocheir, a microsporidian pathogen in the Chinese mitten crab Eriocheir sinensis. J. Fish Dis. 40, 919–927. doi: 10.1111/jfd.12573
Han, J. E., Tang, K., and Hyung, K. J. (2018). The use of beta-tubulin gene for phylogenetic analysis of the microsporidian parasite Enterocytozoon hepatopenaei (EHP) and in the development of a nested PCR as its diagnostic tool. Aquaculture 495, 899–902. doi: 10.1016/j.aquaculture.2018.06.059
Higes, M., Meana, A., Bartolomé, C., Botías, C., and Martín-Hernández, R. (2013). Nosema ceranae (Microsporidia), a controversial 21st century honey bee pathogen. Environ. Microbiol. Rep. 5, 17–29. doi: 10.1111/1758-2229.12024
Hitakarun, A., Tan-ariya, P., Siripattanapipong, S., Mungthin, M., Piyaraj, P., Naaglor, T., et al. (2014). Comparison of PCR methods for detection of Leishmania siamensis infection. Parasit. Vectors 7:458. doi: 10.1186/s13071-014-0458-x
Jaroenlak, P., Sanguanrut, P., Williams, B. A., Stentiford, G. D., Flegel, T. W., Sritunyalucksana, K., et al. (2016). A nested PCR assay to avoid false positive detection of the microsporidian Enterocytozoon hepatopenaei (EHP) in environmental samples in shrimp farms. PLoS One 11:e0166320. doi: 10.1371/journal.pone.0166320
Jiang, H., Chen, Y., Bao, J., Li, X., Feng, C., Xing, Y., et al. (2020). Isolation of the parasite Enterocytospora artemiae from Chinese grass shrimp (Palaemonetes sinensis)-first report in Asia. Front. Cell. Infect. Microbiol. 10:580088. doi: 10.3389/fcimb.2020.580088
Kent, M. L., and Speare, D. J. (2005). Review of the sequential development of Loma salmonae (Microsporidia) based on experimental infections of rainbow trout (Oncorhynchus mykiss) and Chinook salmon (O. tshawytscha). Folia Parasitol. 52, 63–68. doi: 10.14411/fp.2005.009
Kotková, M., Sak, B., and Kváč, M. (2018). Differences in the intensity of infection caused by Encephalitozoon cuniculi genotype II and III - Comparison using quantitative real-time PCR. Exp. Parasitol. 192, 93–97. doi: 10.1016/j.exppara.2018.07.019
Li, P., Mi, R., Zhao, R., Li, X., Zhang, B., Yue, D., et al. (2019). Quantitative real-time PCR with high-throughput automatable DNA preparation for molecular screening of Nosema spp. in Antheraea pernyi. J. Invertebr. Pathol. 164, 16–22. doi: 10.1016/j.jip.2019.04.003
Lindström, M. S., and Nistér, M. (2010). Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. PLoS One 5:e9578. doi: 10.1371/journal.pone.0009578
Lindström, M. S., and Zhang, Y. (2008). Ribosomal protein S9 is a novel B23/NPM-binding protein required for normal cell proliferation. J. Biol. Chem. 283, 15568–15576. doi: 10.1074/jbc.M801151200
Liu, Y. M., Qiu, L., Sheng, A. Z., Wan, X. Y., Cheng, D. Y., and Huang, J. (2018). Quantitative detection method of Enterocytozoon hepatopenaei using TaqMan probe real-time PCR. J. Invertebr. Pathol. 151, 191–196. doi: 10.1016/j.jip.2017.12.006
Liu, Z., Zhang, Q. L., Wan, X. Y., Ma, F., and Huang, J. (2016). Development of real-time PCR assay for detecting microsporidian Enterocytozoon hepatopenaei and the application in shrimp samples with different growth rates. Prog. Fishery Sci. 37, 119–126. doi: 10.11758/yykxjz.20150512003
Lucchi, N. W., Poorak, M., Oberstaller, J., DeBarry, J., Srinivasamoorthy, G., Goldman, I., et al. (2012). A new single-step PCR assay for the detection of the zoonotic malaria parasite Plasmodium knowlesi. PLoS One 7:e31848. doi: 10.1371/journal.pone.0031848
Ma, C., Fan, S., Wang, Y., Yang, H., Qiao, Y., Jiang, G., et al. (2021). Rapid detection of Enterocytozoon hepatopenaei infection in shrimp with a real-time isothermal recombinase polymerase amplification assay. Front. Cell. Infect. Microbiol. 11:631960. doi: 10.3389/fcimb.2021.631960
Nageswara Rao, S., Muthulakshmi, M., Kanginakudru, S., and Nagaraju, J. (2004). Phylogenetic relationships of three new microsporidian isolates from the silkworm, Bombyx mori. J. Invertebr. Pathol. 86, 87–95. doi: 10.1016/j.jip.2004.05.004
Naung, M., Webster, T., Lloyd, R., Leaniz, C., and Consuegra, S. (2020). A novel qPCR assay for the rapid detection and quantification of the lumpfish (Cyclopterus lumpus) microsporidian parasite Nucleospora cyclopteri. Aquaculture 531:735779. doi: 10.1016/j.aquaculture.2020.735779
Pan, G., Bao, J., Ma, Z., Song, Y., Han, B., Ran, M., et al. (2018). Invertebrate host responses to microsporidia infections. Dev. Comp. Immunol. 83, 104–113. doi: 10.1016/j.dci.2018.02.004
Phelps, N. B., and Goodwin, A. E. (2007). Validation of a quantitative PCR diagnostic method for detection of the microsporidian Ovipleistophora ovariae in the cyprinid fish Notemigonus crysoleucas. Dis. Aquat. Organ. 76, 215–221. doi: 10.3354/dao076215
Piamsomboon, P., Choi, S. K., Hanggono, B., Nuraini, Y. L., Wati, F., Tang, K., et al. (2019). Quantification of Enterocytozoon hepatopenaei (EHP) in penaeid shrimps from Southeast Asia and Latin America using TaqMan probe-based quantitative PCR. Pathogens 8:233. doi: 10.3390/pathogens8040233
Pol, S., Romana, C. A., Richard, S., Amouyal, P., Desportes-Livage, I., Carnot, F., et al. (1993). Microsporidia infection in patients with the human immunodeficiency virus and unexplained cholangitis. N. Engl. J. Med. 328, 95–99. doi: 10.1056/NEJM199301143280204
Rode, N. O., Landes, J., Lievens, E. J., Flaven, E., Segard, A., Jabbour-Zahab, R., et al. (2013). Cytological, molecular and life cycle characterization of Anostracospora rigaudi n. g., n. sp. and Enterocytospora artemiae n. g., n. sp., two new microsporidian parasites infecting gut tissues of the brine shrimp Artemia. Parasitology 140, 1168–1185. doi: 10.1017/S0031182013000668
Santhoshkumar, S., Sivakumar, S., Vimal, S., Abdul Majeed, S., Taju, G., Haribabu, P., et al. (2017). Biochemical changes and tissue distribution of Enterocytozoon hepatopenaei (EHP) in naturally and experimentally EHP-infected whiteleg shrimp, Litopenaeus vannamei (Boone, 1931), in India. J. Fish Dis. 40, 529–539. doi: 10.1111/jfd.12530
Stentiford, G. D., Becnel, J. J., Weiss, L. M., Keeling, P. J., Didier, E. S., Williams, B. A. P., et al. (2016). Microsporidia - emergent pathogens in the global food chain. Trends Parasitol. 32, 336–348. doi: 10.1016/j.pt.2015.12.004
Stentiford, G. D., and Dunn, A. M. (2014). “Microsporidia in aquatic invertebrates,” in Microsporidia: Pathogens of Opportunity, eds L. M. Weiss and J. J. Becnel (Hoboken, NJ: John Wiley & Sons, Inc), 579–604. doi: 10.1002/9781118395264.ch23
Suebsing, R., Prombun, P., Srisala, J., and Kiatpathomchai, W. (2013). Loop-mediated isothermal amplification combined with colorimetric nanogold for detection of the microsporidian Enterocytozoon hepatopenaei in penaeid shrimp. J. Appl. Microbiol. 114, 1254–1263. doi: 10.1111/jam.12160
Tang, K. F., Pantoja, C. R., Redman, R. M., Han, J. E., Tran, L. H., and Lightner, D. V. (2015). Development of in situ hybridization and PCR assays for the detection of Enterocytozoon hepatopenaei (EHP), a microsporidian parasite infecting penaeid shrimp. J. Invertebr. Pathol. 130, 37–41. doi: 10.1016/j.jip.2015.06.009
Tangprasittipap, A., Srisala, J., Chouwdee, S., Somboon, M., Chuchird, N., Limsuwan, C., et al. (2013). The microsporidian Enterocytozoon hepatopenaei is not the cause of white feces syndrome in whiteleg shrimp Penaeus (Litopenaeus) vannamei. BMC Vet. Res. 9:139. doi: 10.1186/1746-6148-9-139
Keywords: Palaemonetes sinensis, Enterocytospora artemiae, qPCR detection method, RPS9, specific and quantitative detection
Citation: Jiang H, Bao J, Liu J, Chen Y, Feng C, Li X, Huang S and Chen Q (2021) Development of a Quantitative PCR Method for Specific and Quantitative Detection of Enterocytospora artemiae, a Microsporidian Parasite of Chinese Grass Shrimp (Palaemonetes sinensis). Front. Mar. Sci. 8:730569. doi: 10.3389/fmars.2021.730569
Received: 25 June 2021; Accepted: 09 September 2021;
Published: 28 September 2021.
Edited by:
Xiangli Tian, Ocean University of China, ChinaReviewed by:
Shihao Li, Institute of Oceanology, Chinese Academy of Sciences (CAS), ChinaHa Thanh Dong, Asian Institute of Technology, Thailand
Copyright © 2021 Jiang, Bao, Liu, Chen, Feng, Li, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qijun Chen, cWlqdW5jaGVuNzU5QHN5YXUuZWR1LmNu; c3lhdWNxakAxMjYuY29t
†These authors have contributed equally to this work