 Yong Wang
Yong Wang Wenli Li1
Wenli Li1 Qingmei Li
Qingmei Li Yingli Zhou
Yingli Zhou Dong Feng
Dong Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 21 July 2021
Sec. Marine Molecular Biology and Ecology
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.698945
Carbonates are globally distributed particularly around deep-sea cold seeps. The embedded microbes are fossil records of the past bioprocess but metagenomes of the carbonates have not been fully studied. In this study, we report microbial community structures and genomes of dominant species in cold-seep carbonates from the South China Sea (SCS) and Gulf of Mexico (GoM). The carbonates contained both anaerobic microbes represented by methane oxidizing archaea (ANME) and aerobic ammonia-oxidizing archaea (AOA). The samples from GoM were mostly composed of small microbial groups, indicating heavy degradation of the fossil microbes. The composition of the carbonate communities differed from that of cold seep sediments, suggesting alteration of cold-seep microbial structures during formation and weathering of carbonates. Extraction of 18S rRNA genes from metagenomic reads revealed prevalence of fungal species in the carbonates of the GoM. Genome binning resulted in 10 genomes for dominant prokaryotic species. The ANME genomes showed a short genetic distance to the relatives from the current cold seep sediments; the AOA genomes were affiliated with alpha ecotype dominating deep-sea sediments. Our study reports the genomes in ancient carbonates and sheds lights on microbial role in formation and bioweathering of carbonates.
Along continental coastal slopes, there are cold seeps on the seafloor (Boetius and Wenzhofer, 2013). Carbonate is formed in the deep-sea cold seeps to trap CO2 into minerals, which is one of important carbon capture processes on Earth. In cold seeps, anaerobic methane oxidizing archaea (ANME) consume methane to generate CO2 and then secret HCO3– (Reitner et al., 2005; Timmers et al., 2017), which is then precipitated with calcium and other metal ions. Syntrophic partners of ANME-1, ANME-2, and ANME-3 are sulfur reducing bacteria (SRB) that provide electron acceptors for the anaerobic oxidation of methane (AOM) (Niemann et al., 2006; Skarke et al., 2014; Li et al., 2020). An archaeal family Candidatus Methanoperedenaceae (known as ANME-2d) may reduce nitrate, nitrite and metal oxides for independent AOM (Niemann et al., 2006). As carbonate deposits are being formed, solution, particles and microbes in the cold seep sediments were embedded into the minerals. Therefore, carbonates can probably serve as an important reservoir of the past geological settings and fossil microbes. In the Black Sea, ANME and SRB were firstly revealed by microscopy (Reitner et al., 2005). Highly diverse microbial communities of deep-sea cold seep carbonates from mud volcano in Mediterranean Sea and cold seep in the Okinawa Though were then revealed by 16S rRNA gene cloning and GeoChip (Heijs et al., 2006; Li et al., 2018). ANME plus other archaeal groups unrelated to methane metabolism were also identified in the carbonates (Heijs et al., 2006). Aside from the anaerobes, there are aerobic microbial groups such as methanotrophic bacteria (Birgel et al., 2011), suggesting that the conditions and positions for the carbonate formation are variable. The precipitation of carbonate may therefore occur in both oxic and anoxic layers of a cold seep as a result of microbial consumption of methane occurring at different oxygen levels (Cavagna et al., 1999). On the other hand, carbonate deposit might be dissolved by sulfur oxidizing bacteria (SOB) (Leprich et al., 2021). This will release the previously buried microbes into the pore water of the cold seep and marine bottom waters, providing additional nutrient source and biodiversity for cold seep ecosystem. Discharging CO2 into deep-sea water with the dissolvement of carbonates can probably affect the global climate ultimately. However, the SOB have not been detected in carbonate deposits yet. At present, using metagenomic methods for microbial study on cold seep carbonates is still sparse. The genomes of fossil microbes and the community structures in cold seep carbonates at different ages have not been investigated. Whether the fossil ANME and SRB genomes resembled those of the relatives in the current cold seep remains a question.
In the South China Sea (SCS) and Gulf of Mexico (GoM), cold seeps were discovered at numerous sites (Roberts et al., 2010; Feng et al., 2018). Microbial communities and metagenomes have been studied to demonstrate the role of microbes in element cycling and subsequent formation of cold seep ecosystem (Skarke et al., 2014; Li et al., 2020). Biomarkers derived from ANME and bacteria have been examined and mineral components were used to indicate methane flux rate of cold seeps in the past (Bian et al., 2013; Feng and Chen, 2015; Ge et al., 2015). At present, the microbial communities in the cold seeps of the two areas have not been reported and there are not metagenomics data for better elucidation of microbial functions. In this study, we selected four sites in the SCS and GoM to compare their fossil microbes embedded in the carbonates in terms of community structure, diversity and phylogenomic divergence. The age of these carbonate samples had been estimated to be 1.2–46 ka (Birgel et al., 2011; Bian et al., 2013; Feng and Chen, 2015), which allowed to evaluate the impact of lasting time on the preservation of fossil microbes. Our results revealed the presence of a wide spectrum of microbes in the carbonates. The genomes of the dominant species showed a short genetic distance with their current relatives, which further highlights microbial role in carbonate precipitation process and age-dependent deterioration of fossil microbes.
During R/V “Dayangyihao” in June 2013, Jiaolong submersible collected carbonate samples from site F of the northern SCS at depth of 1,120–1,143 m. The carbonate sample collection from the SCS was also described elsewhere (Feng and Chen, 2015) (Supplementary Table 1). Carbonate samples were also taken from the northern GoM (Figure 1). GC140 from the Green Canyon (hereafter MEX1) was taken by remotely operated vehicle (ROV) Johnson-Sea-Link dive 2591 at the Green Canyon at depth of 260 m in 1989 (Bian et al., 2013). AC645 (hereafter MEX2) was obtained from the Alaminos Canyon by Alvin dive4197 at depth of 2,200 m (Birgel et al., 2011). The carbonate samples were cut into smaller ones (Figure 1). About 10 g inner parts of the carbonates were digested with 2.5 U DNaseI in 1 L buffered ddH2O for 30 min in 25°C. The carbonates were then cleaned with distilled water under ultrasonication for three times, each lasting for 30 min, followed by an ultrasonication treatment in 70% ethanol for 30 min. The clean carbonates were dissolved in 1% acetate solution. Triplicates were performed for each of the samples.

Figure 1. Carbonate samples and sampling sites. Carbonate samples for this study were collected from the Gulf of Mexico and the South China Sea (A), represented by SCS1 (B) and MEX2 (C). Details of the samples are shown in Supplementary Table 1.
The carbonate clays after dissolution were used for DNA extraction using a PowerMax Soil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, United States) following the manufacturer’s instruction. The 16S rRNA gene amplicons of the samples were obtained by a PCR reaction using universal primers 341F (5′-CCTAYGGGRBGCASCAG-3′) and 802R (5′-TACNVGGGTATCTAATCC-3′) that target V3-V4 region of 16S rRNA genes (Wang and Qian, 2009). A set of 8-nucleotide barcodes was added to the primers to distinguish the samples (Supplementary Table 2). The PCR was executed as below: denaturation firstly at 98°C for 10 s; 30 cycles including denaturation at 98°C for 10 s, annealing at 50°C for 15 s, and extension at 72°C for 30 s; and with a final extension at 72°C for 5 min. The 16S rRNA gene amplicons were prepared for Illumina sequencing by TruSeq® Nano DNA LT Kit (Illumina, San Diego, CA, United States). The amplicon libraries were sequenced with Illumina MiSeq platform (2 × 300 bp). Raw reads were separated for different samples using the barcode information and were then filtered by NGS QC Toolkit (v2.3.3) (Patel and Jain, 2012) with default parameters (base quality <20 for >70 per cent of a given read). The paired reads were merged with PEAR (v0.9.5) (Zhang et al., 2014). The resulted fragments were processed with QIIME2 pipeline (Caporaso et al., 2010b). The fragments with similarity >97% were firstly clustered into OTUs by UCLUST (Edgar, 2010). The longest fragment in each OTU was selected as the representative read and then used for alignment with the references in the Silva database (version 132) by PyNAST (Caporaso et al., 2010a). Chimeric reads were removed based on the result of ChimeraSlayer (Haas et al., 2011). Taxa assigned to mitochondria, chloroplast, and eukaryotes were deleted. Taxonomy assignment of representative reads was finally conducted by online silvangs1. Correlation analysis of genera was carried out by SparCC method (100 permutation).
The genomic DNA was sheared randomly to ∼350 bp by ultrasonication. DNA libraries were prepared by TruSeq® Nano DNA LT Kit (Illumina, San Diego, CA, United States) and sequenced on an Illumina NovaSeq6000 platform (2 × 150 bp). Quality control of raw reads was initiated by removing reads containing adaptor and the bases in low quality (<20) at both ends were also trimmed using Fastp (v0.20.0) (Chen et al., 2018) with -f 27 -t 36 -F 27 -T 73 -w 16 -c -q 20 -u 20 -g -W 5 -3 -l 50. FastUniq (v1.1) (Xu et al., 2012) was used for removal of duplicated reads with default parameters. The remaining data were searched using Blastn (Altschul et al., 1990) with e-value of 1e-10 against a database containing genomes for the species potentially detected in reagents (Salter et al., 2014). The mapped contaminant reads were removed. The high-quality paired reads of the same samples were co-assembled into scaffolds by SPAdes (v3.6.2) (Nurk et al., 2013) with parameters (–careful -k 21, 33, 55, 77, 99, 127).
MetaWRAP (v1.2) (Uritskiy et al., 2018) was used to perform genome binning by invoking metaBAT2 (Kang et al., 2019), CONCOCT (Alneberg et al., 2014) and MAXBin2 (Wu et al., 2016), and refined by Bin_refinement module for consolidation, integration and optimization with parameter of over 50% completeness rate and below 10% contamination rate with an evaluation of the genomes by CheckM (v1.0.12) (Parks et al., 2015). Deduplication was performed using dRep software (Olm et al., 2017) by initially clustering the genomes with 97% average nucleotide identity (ANI) value. Taxonomic classification of the genomes was performed using GTDB-tk (Chaumeil et al., 2019). Relative evolutionary divergence (RED) of the MAGs was calculated in genome taxonomy database (GTDB) (Parks et al., 2018). Prodigal (v2.6.2) (Hyatt et al., 2010) was applied for prediction of coding sequences (CDSs) and proteins in the MAGs. The proteins were annotated against KEGG Ortholog database through BlastKOALA (Kanehisa et al., 2016).
The 16S and 18S rRNA genes in metagenomics reads (16S and 18S miTags) were predicted using rRNA_HMM (Huang et al., 2009). The reads were assigned to OTUs at 97% similarity level using QIIME v1.9.1 (Kuczynski et al., 2011). Taxonomic classification of the representative 16S miTags in the OTUs was performed using vsearch (Rognes et al., 2016) in reference to SILVA132 (Quast et al., 2013) database. For eukaryotic classification with the same pipeline, those 18S miTags assigned to Metazoa were not taken into account.
The genomes of the closest relatives of the MAGs were downloaded from the NCBI. 43 commonly conserved proteins of MAGs and reference genomes were predicted using CheckM (v1.0.12) (Parks et al., 2015). The conserved proteins of genomes were aligned using hmmerAlign.py (integrated in CheckM). The alignment of 43 concatenated conserved proteins was used for construction of a phylogenomic tree with RAxML (Stamatakis, 2014) (v.8.2.12; -# 1000; -m PROTGAMMALG) after optimization with trimAl (Capella-Gutierrez et al., 2009) (v.1.4). 1,000 replicates were performed for bootstrap calculation. All 16S miTag sequences were aligned with MAFFT (Katoh and Standley, 2013) (v.7.407) and optimized with trimAl as well. RAxML (v.8.2.12; GTRCAT model) was used to build a 16S rRNA phylogenetic tree with bootstrap values based on 1,000 replicates. All the visualization of the phylogenetic tree was carried out by iTOL (Letunic and Bork, 2007).
The carbonate samples from four sites of the north SCS and GoM (Figure 1A and Supplementary Table 1) were cement-like minerals composed of nodules, bivalve shells and tiny pores (Figures 1B,C). Previous studies have provided mineral components, age estimate and isotopes of the samples (Birgel et al., 2011; Bian et al., 2013; Feng and Chen, 2015). For the SCS samples, their age ranged between 6.4 and 11 ka; for the GoM samples, the age is largely higher than 10 ka (Birgel et al., 2011; Bian et al., 2013; Feng and Chen, 2015). Triplicates of DNA extraction were executed for different samples from each site. DNA was successfully extracted for 16S rRNA gene amplification and metagenomics work for 10 of 12 carbonate triplicates.
Using Illumina sequencing, we obtained 66,695 16S rRNA amplicon reads for ten samples with a minimum of 2,627 reads (Supplementary Table 2). All the rarefaction curves of the observed OTUs reach a plateau (Supplementary Figure 1), which indicates that the sequencing depth is sufficient to cover the whole microbial communities. A sample from Green Canyon of GoM (MEX1-R1) is associated with the highest number of OTUs (n = 203) and Shannon index of 7.47 (Supplementary Table 2). The carbonates were dominated by Firmicutes, Proteobacteria, and Euryarchaeota (Supplementary Figure 2). However, the microbial communities differed among the samples to some extent. All the carbonate samples contained ANME species such as ANME-1a and ANME-2a-c, which were dominant Archaea in the samples (SCS1: 14.37%; SCS2: 7.39%; and MEX2: 4.87%) except MEX1 (Figure 2). This suggests that the carbonates were formed during methane seeping of the sites. As syntrophic partners of ANME, SRB in comparative percentage were only identified in SCS1-R1 and SCS1-R2. Moreover, ANME-2d that does not rely on SRB was not present in our samples. It is therefore likely that other carbonate samples (such as SCS2) that lacked SRB were formed above sulfur-methane transition zone by other microbes or abiological process (Boetius and Wenzhofer, 2013). The ANME archaea in the carbonates have likely been transferred to near-surface oxic sediment by seeping solution. This was indicated in 6–8 cm layer (JLS_3) of Jiaolong cold seep, where abundant ANME1 archaea were not associated with SRB in comparative amount (Wu et al., 2018). Paucity of syntrophic SRB was also observed in most of in ANME-enriched carbonates formed along coastal slopes of the North America (Leprich et al., 2021). Interestingly, the high relative abundance of ANME archaea in SCS samples was not associated with a comparatively high content of carbonate content in the samples. The previous geochemical analyses showed a carbonate content ranging from 37 to 67% for the SCS samples, much lower than that of 93–96% for the MEX2 sample (Birgel et al., 2011; Feng and Chen, 2015). Therefore, other environmental factors and microbial players might impact on the carbonate content in cold seep.
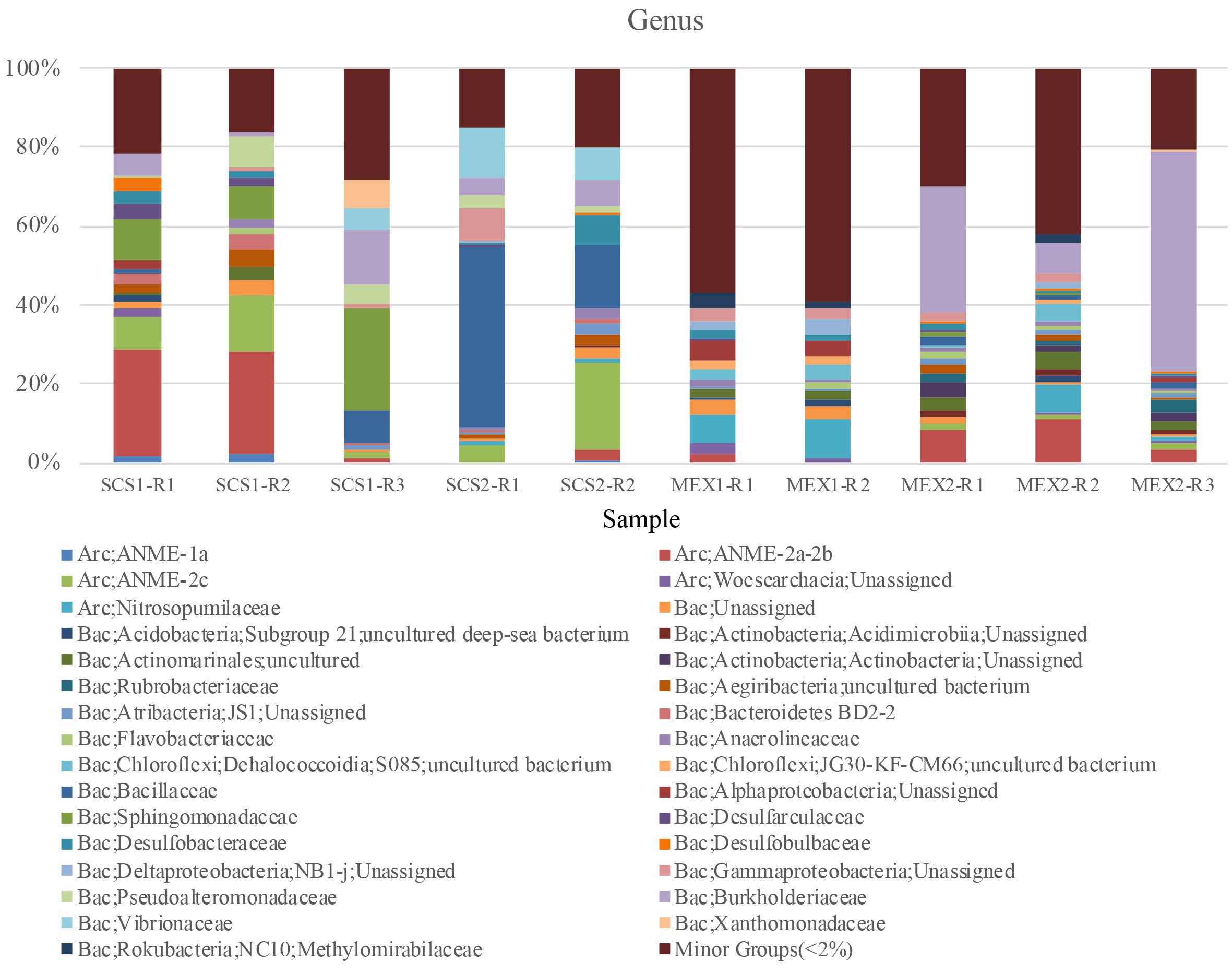
Figure 2. Microbial communities of carbonates. The samples were carbonates obtained from the South China Sea (SCS) and Gulf of Mexico (GoM). The community structures at genus level were based on analysis of 16S rRNA gene amplicons. The genera shown in this figure were at least >2% in two samples. Otherwise the genera will be combined into minor groups. The sample IDs are referred to Supplementary Table 2. R1–R3 in sample IDs denote replicates of PCR products.
Ammonia-oxidizing archaea (AOA) represented by Nitrosopumilaceae were prevalent in MEX1, while they were hardly detected in the SCS1 and SCS2 samples. Since AOA were present in oxic subsurface sediment layers and marine waters (Wang et al., 2019), carbonate vents were probably formed in anoxic layer and later extruded into shallower oxic sediment layers in the MEX sampling sites. In addition, respiration of heterotrophic microbes in the oxic layer can also generate CO2 for formation of AOA-containing carbonates. For the replicates of SCS1, the third sample SCS1-R3 was largely occupied by Bacillaceae and Sphingomonadaceae, suggesting discrepancy between the samples from the same site in the composition of microbial communities. Bacillaceae represented by Bacillus was also ranked as the first and second most abundant genera in SCS2-R1 and SCS2-R2 samples, respectively. In a previous report, several Bacillus strains were experimentally confirmed to be able to dissolve carbonates (Subrahmanyam et al., 2012). Overall, the samples from the GoM were more diversified than those from the SCS as the minor groups occupying <2% of the community were totaled to be more than half of the communities in the GoM samples (Figure 2). The loss of dominant microbial groups in the MEX2 suggests natural degradation of the cemented biomass under the long-term preservation. This sample had been estimated to be formed about 10 ka ago (Birgel et al., 2011). We cannot preclude the possibility that some microbes in the communities might dissolve the carbonate to take the fossil microbes as nutrient. In this study, we have removed the surface carbonate, and therefore most of carbonate-dissolving microbes could not be examined herein.
Correlation analysis revealed relationships between the major microbial groups in the SCS and MEX carbonates (Supplementary Figure 3). High betweenness centrality was found for Nitrosococcaceae and Nitrospinaceae which are known ammonia-oxidizing bacteria (AOB) and nitrite-oxidizing bacteria (NOB), respectively. This seems to be evidence of a microbial network centralized on aerobic nitrogen cycle in the carbonates. Nitrosococcaceae was also found to be a central node of the network between microbial habitants in Karst caves (Zhu et al., 2019), suggesting an unknown role of nitrifying microbes in carbonate formation. The nodes for the AOB and NOB were also negatively related to the ANME species, which agrees with their prevalence in oxic and anoxic conditions, respectively.
Next, we compared the microbial communities of cold seep sediments (JLS) and the carbonates of site F, along with those of MEX1 and MEX2, using principal coordinate analysis (Figure 3). F1 and F2 explain 53% of the differences between the samples. The cold seep sediments were obtained from an active seeping vent. From the surface JLS_1 to bottom JLS_7, four layers of the 14-cm core were studied in our previous work (Wu et al., 2018), demonstrating a transition of SRB to SOB at about 8 cm below surface. In this study, the cold seep sediments were obviously separated from the carbonates, although all were from site F of SCS. This suggests that carbonates cemented a slightly different group of microbes, compared with the cold seep sediments, or the communities have been shifted during carbonate weathering.
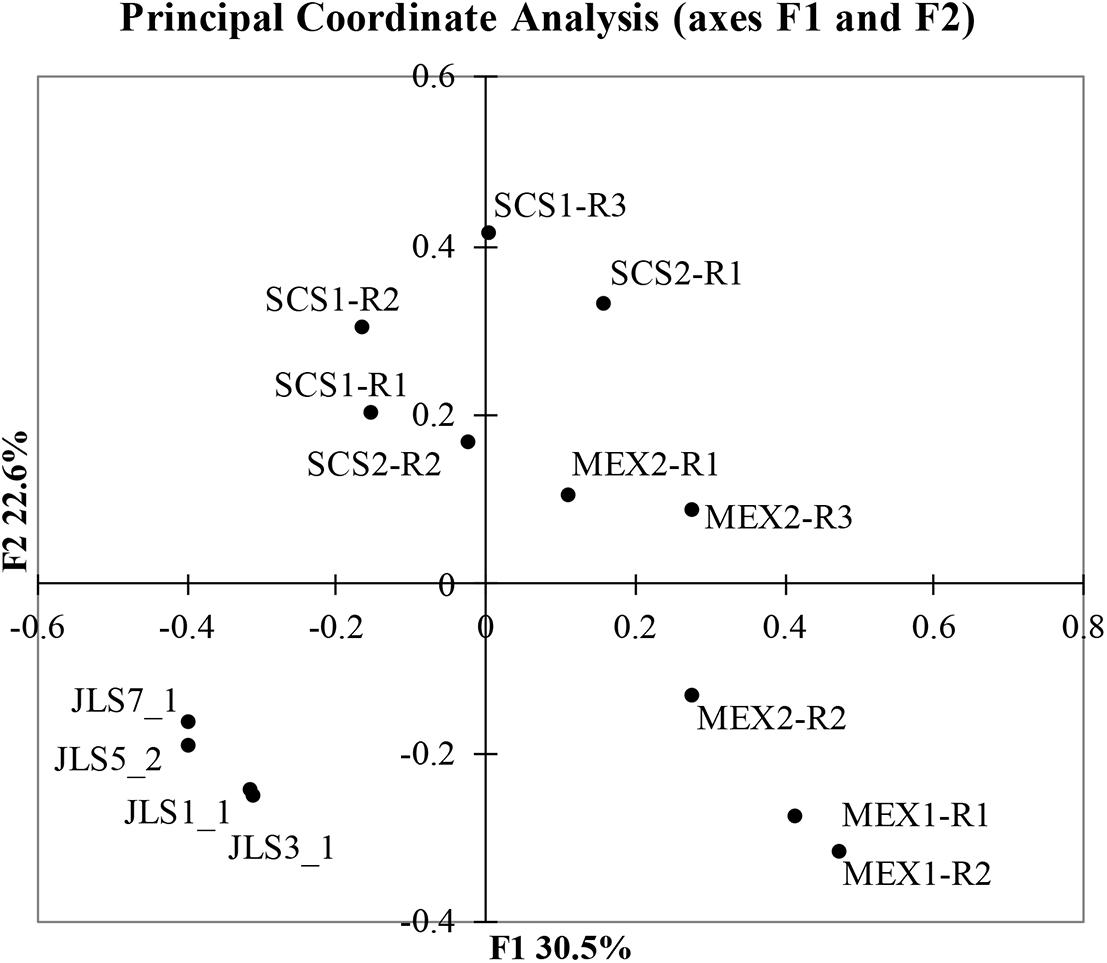
Figure 3. Principle coordinate analysis of microbial communities at genus level. The Bray-Curtis dissimilarity was calculated using community structures at genus level and was then used for the principle coordinate analysis.
We obtained a total of 49.2 Gbp metagenome data and 328 million Illumina reads for carbonate samples from the four sites. After quality control, 36.6 Gbp metagenomic data were retained for subsequent work. A total of 104,252 16S miTags were retrieved from the metagenomic reads for detection of prokaryotic community. The primer-independent 16S miTags allow for unbiased detection of microbial communities. Proteobacteria, Firmicutes and Euryarchaeota were still abundantly present in the metagenomes (Supplementary Figure 3). In addition, there are a considerable percentage of the 16S miTags were derived from candidate phyla such as GN04 and Hyd24_12, indicating a huge reservoir of rare prokaryotic species preserved in the carbonates. These rare species had been identified in global cold seep areas (Ruff et al., 2015). We extracted 4,144 18S miTags from MEX2 metagenome, while less than 100 in other metagenomes. A total of 132 18S OTUs were obtained using 97% similarity. After filtration of OTUs for animals, we selected 456 18S miTags for in a brief survey of eukaryotic communities in MEX2. Fungus is the most prevalent group, accounting for 37% of the miTags, followed by SAR superphylum (Figure 4). Most of the fungi were affiliated with Malassezia, Cladosporium, and Aspergillus. About 52% of the 18S miTags were unclassified, suggesting a large number of “dark matter” eukaryotes in deep-sea cold seep. Fungi are the dominant eukaryotic groups in carbonates in Lost City hydrothermal field (López-García et al., 2007). Fungi dominated deep-sea sediments globally and therefore they might be embedded during the carbonate formation (Wang et al., 2014; Rédou et al., 2015). Nevertheless, the presence of abundant fungi in the carbonates also implies the role of fungi in utilization of the fossil microbes as nutrient after the formation of the carbonate as has been argued previously (Bindschedler et al., 2016). Interestingly, the MEX2 samples also harbored a high percentage of minor prokaryotic groups, suggesting degradation of the embedded prokaryotic fossil microbes by fungi in the carbonates. However, this hypothesis warrants future efforts.
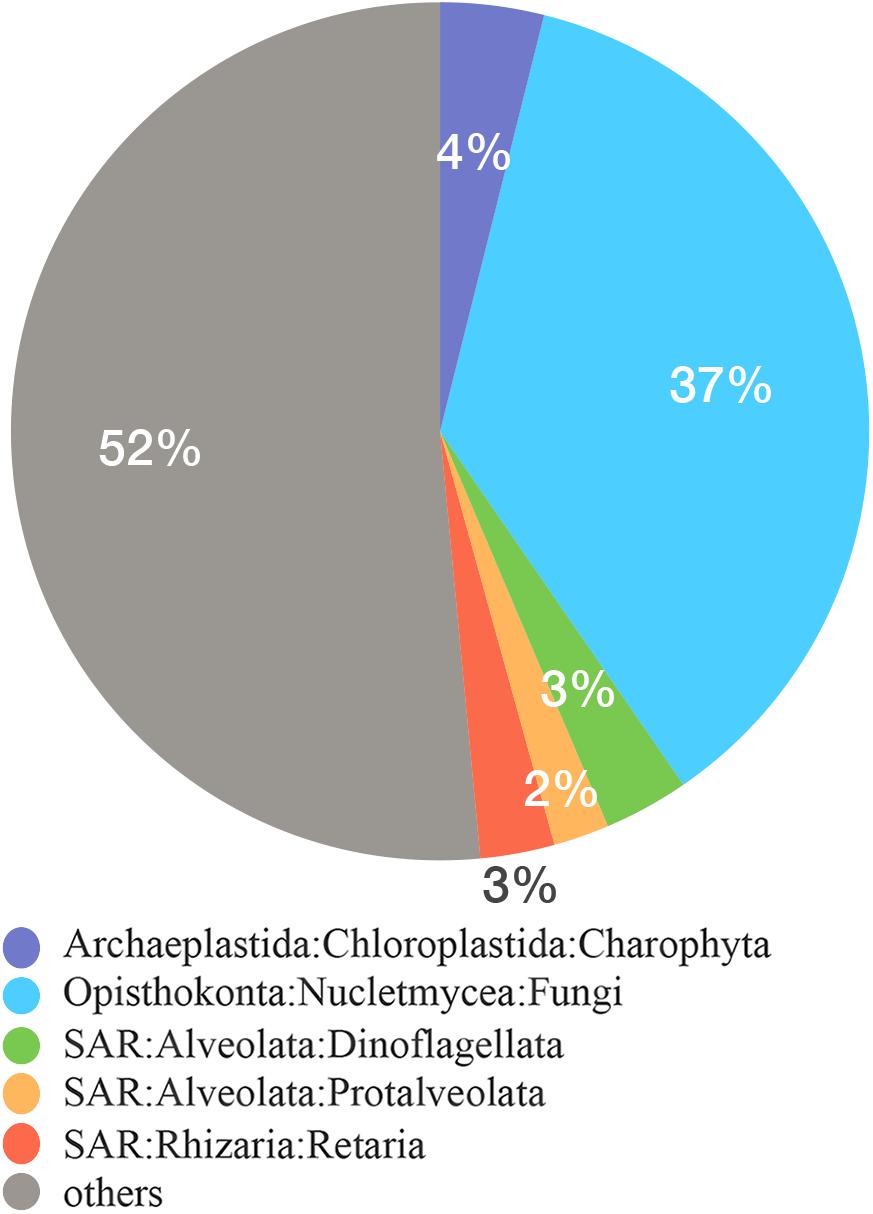
Figure 4. Eukaryotic community in MEX2 using 18S miTags. The 18S rRNA miTags were extracted from MEX2 metagenome. The classification was conducted using QIIME2 against SILVE 132 database for 18S rRNA genes.
Twenty-five MAGs were obtained from the four metagenomes, and most of them (85%) were binned from SCS1 and MEX2. Five of them were contaminants and had been removed. The 20 clean MAGs with completeness higher than 50% were classified with GTDB-tk (Chaumeil et al., 2019; Table 1). The taxonomic novelty could be indicated by RED value that may serve as an estimator of genome novelty in GTDB (Parks et al., 2018). The average of the RED values is 0.80 ± 0.11, which means that most of the MAGs fall into the range of RED values for new families (Parks et al., 2018). The lowest RED value of 0.48 in our MAGs indicates identification of novel class in Gammaproteobacteria.
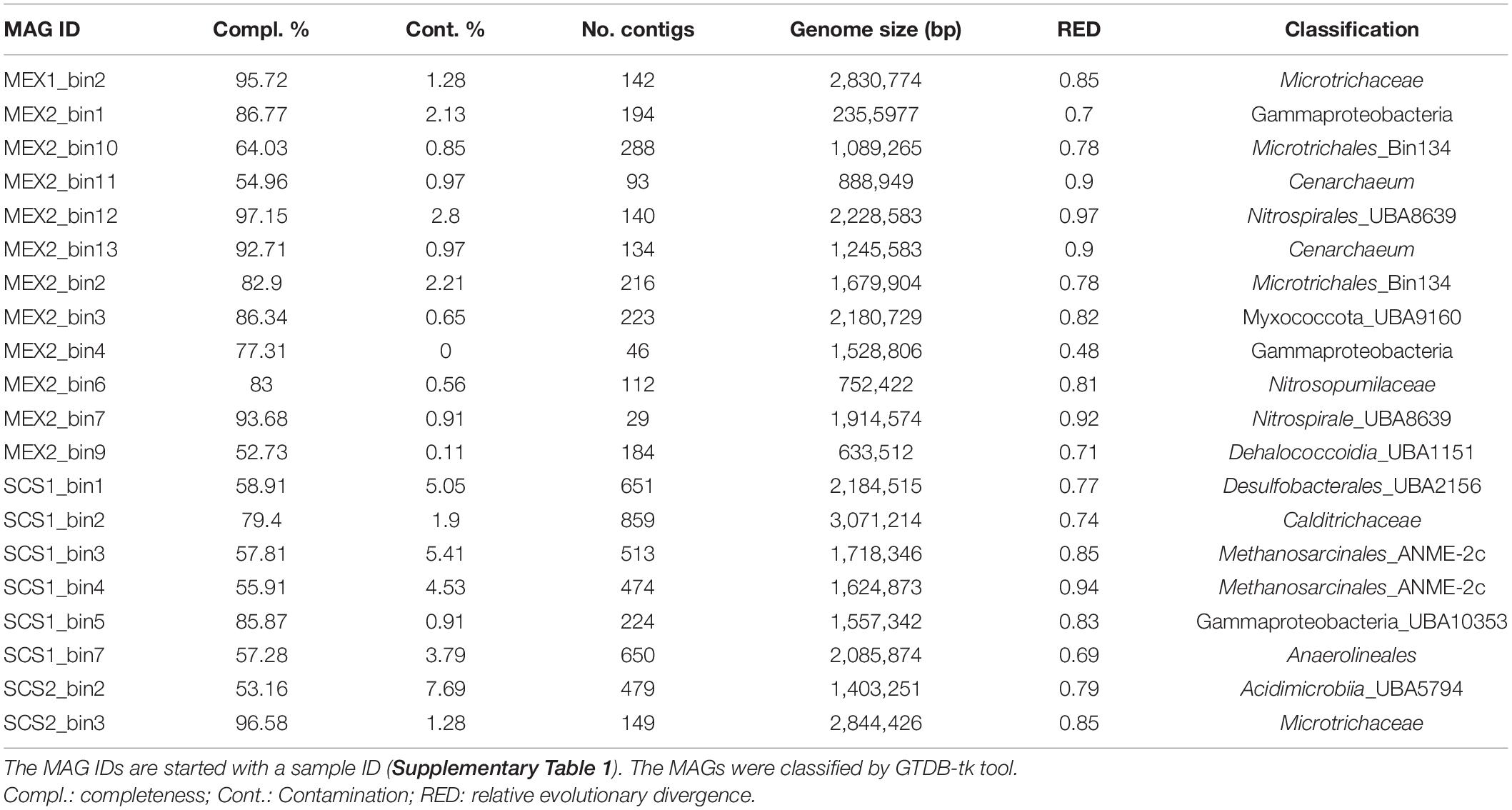
Table 1. Information of MAGs.
The MAGs were used to build a phylogenomics tree with the reference genomes of the closest relatives to examine the classification result and to show genetic distance. These MAGs were affiliated with eight phyla (Figure 5). Four of them were assigned to ANME-2c and AOA, which is consistent with our 16S rRNA gene amplicon result. For the ANME MAGs from the site F, they highly resemble those obtained from the cold seep sediment of the same sampling site, regardless of a short genetic distance. In the ANME-2 MAGs, most of the functional genes involved in AOM were identified, including those coding for methyl coenzyme M reductase system (Mcr), anaerobic carbon-monoxide dehydrogenase, CODH/ACS complex (Cdh), 5,10-methylenetetrahydromethanopterin reductase (Mer), tetrahydromethanopterin S-methyltransferase (Mtr) and heterodisulfide reductase (Hdr) (Supplementary Table 3). The products of the AOM process are probably formate, CO2 and acetate as indicated by the presence of the related genes in the MAGs. The AOA MAGs of MEX1 sample were grouped with Nitrosopumilus maritimus (Figure 5), which belongs to alpha type AOA that was isolated from marine sediment (Wang et al., 2019).
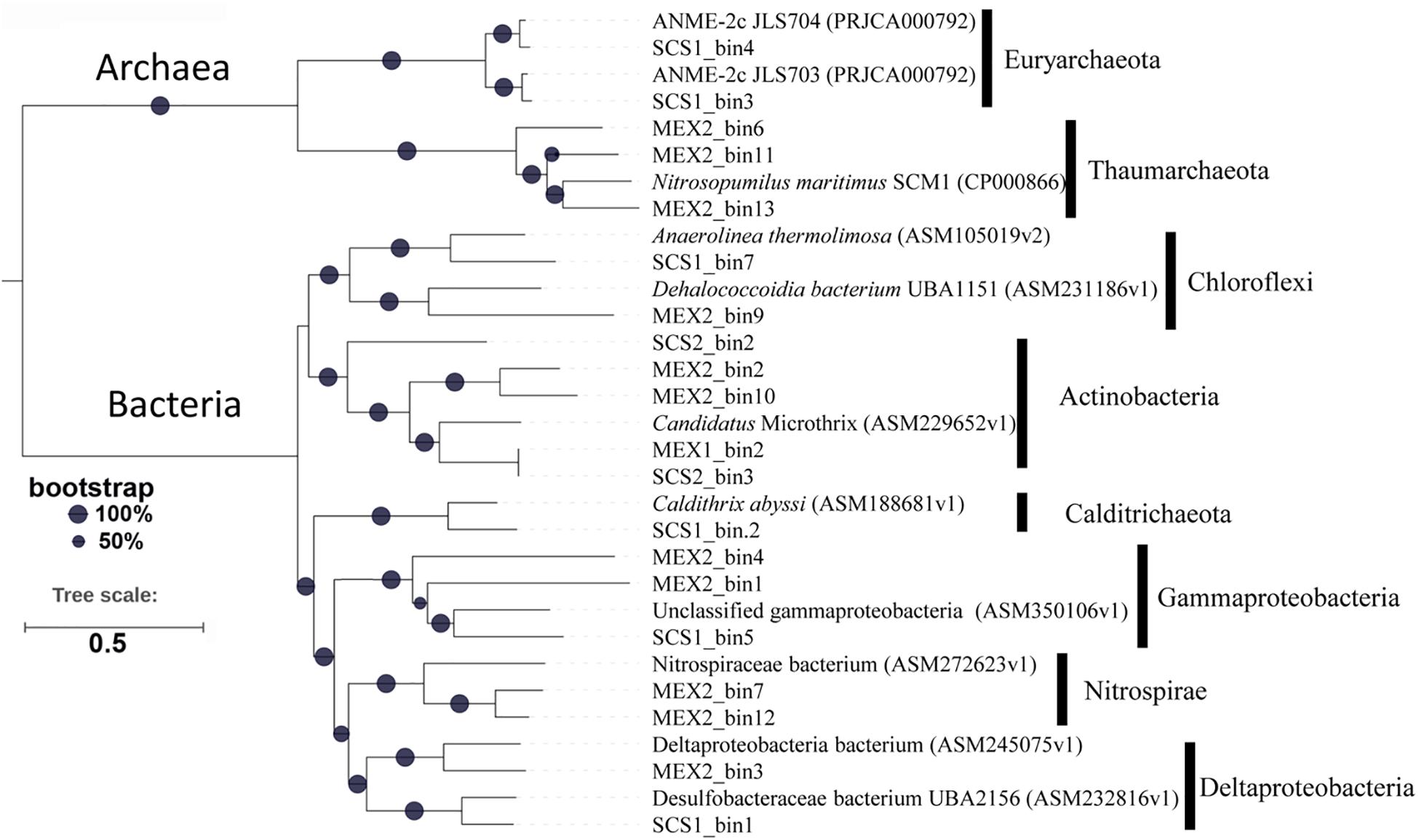
Figure 5. Phylogenomics tree of MAGs. 43 conserved proteins of the MAGs and the reference genomes in NCBI were used to construct the maximum-likelihood phylogenetic tree. The bootstrap values were based on 1,000 replicates. The MAGs from this study are referred to Table 1.
The biochemical catalysis between CO2 and HCO3– is crucial for the formation of carbonates. ANME-1 and ANME-2 archaeal genomes harbor diversified AOM processes that convert methane into CO2, acetate and probably propanoate (Timmers et al., 2017; Li et al., 2020). Carbonate dehydratase is a bidirectional enzyme that acts on a balance between CO2 and HCO3–, which is critical for local pH condition and carbonate stabilization. A total of 83 carbonate dehydratase genes were identified in the assembled contigs of the metagenomes. Five of them were associated with >1× coverage (Supplementary Table 4). With respect to the highest similarity to known homologs, we predicted that these carbonate dehydratase genes were probably derived from ANME-2 (84% identity), Pseudoalteromonas (100% identity), Novosphingobium (99.5% identity), and candidate division FCPU426 (69.9% identity). The finding of the species that encode the carbonate dehydratases likely revealed the major microbial groups that played an important role in formation and/or dissolution of carbonates.
In this study, we detected microbial communities and their representative genomes from carbonate samples of two cold seep areas. Although the two sites were far from each other, they all contained similar microbial consortia of cold seep regardless of different percentages of dominant species. The difference in the community structure was probably resulted from the discrepancy in carbonate age, cold seep type, and microenvironment. Aside from ANME, there were perhaps unnoticed microbial inhabitants mediating carbonate formation in oxic layer above methane-sulfate transition zone. Dissolution and reprecipitation of carbonate under effect of both biological and chemical processes on an existing carbonate might also have changed the embedded microbial communities. More samples with clear geochemical background may help explain the variation of fossil communities in carbonates and highlight their potential significance to global climate change.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA723199).
YW conceived and designed the study and wrote the manuscript. WL performed the experiment and analysis. QL, YZ, and ZG provided the analysis pipelines. DF provided the samples and associated metadata. All authors agree with the final version of the manuscript.
This study was supported by the Key Science and Technology Program of Hainan Province (ZDKJ2019011) and National Natural Science Foundation of China (42076136).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to J. Li, J. Chen, D. S. Cai, S. X. Wang, and Y. Z. Xin for their assistance of the cold seep sampling. We thank H. H. Roberts for providing the Gulf of Mexico samples. We also thank crews of Xiangyanghong-09 as well as the Jiaolong team for collecting the South China Sea samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.698945/full#supplementary-material
Alneberg, J., Bjarnason, B. S., De Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/s0022-2836(05)80360-2
Bian, Y., Feng, D., Roberts, H. H., and Chen, D. (2013). Tracing the evolution of seep fluids from authigenic carbonates: Green Canyon, northern Gulf of Mexico. Mar. Petrol. Geol. 44, 71–81. doi: 10.1016/j.marpetgeo.2013.03.010
Bindschedler, S., Cailleau, G., and Verrecchia, E. (2016). Role of fungi in the biomineralization of calcite. Minerals 6:41. doi: 10.3390/min6020041
Birgel, D., Feng, D., Roberts, H. H., and Peckmann, J. (2011). Changing redox conditions at cold seeps as revealed by authigenic carbonates from Alaminos Canyon, northern Gulf of Mexico. Chem. Geol. 285, 82–96. doi: 10.1016/j.chemgeo.2011.03.004
Boetius, A., and Wenzhofer, F. (2013). Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6, 725–734. doi: 10.1038/ngeo1926
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Caporaso, J. G., Bittinger, K., Bushman, F. D., Desantis, T. Z., Andersen, G. L., and Knight, R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Cavagna, S., Clari, P., and Martire, L. (1999). The role of bacteria in the formation of cold seep carbonates: geological evidence from Monferrato (Tertiary, NW Italy). Sediment. Geol. 126, 253–270. doi: 10.1016/S0037-0738(99)00044-5
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461.
Feng, D., and Chen, D. (2015). Authigenic carbonates from an active cold seep of the northern South China Sea: new insights into fluid sources and past seepage activity. Deep Sea Res. II 122, 74–83. doi: 10.1016/j.dsr2.2015.02.003
Feng, D., Qiu, J.-W., Hu, Y., Peckmann, J., Guan, H., Tong, H., et al. (2018). Cold seep systems in the South China Sea: an overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Ge, L., Jiang, S. Y., Blumenberg, M., and Reitner, J. (2015). Lipid biomarkers and their specific carbon isotopic compositions of cold seep carbonates from the South China Sea. Mar. Petrol. Geol. 66, 501–510. doi: 10.1016/j.marpetgeo.2015.02.005
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504.
Heijs, S. K., Aloisi, G., Bouloubassi, I., Pancost, R. D., Pierre, C., Damste, J. S. S., et al. (2006). Microbial community structure in three deep-sea carbonate crusts. Microb. Ecol. 52, 451–462. doi: 10.1007/s00248-006-9099-8
Huang, Y., Gilna, P., and Li, W. (2009). Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340. doi: 10.1093/bioinformatics/btp161
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119.
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kuczynski, J., Stombaugh, J., Walters, W. A., Gonzalez, A., Caporaso, J. G., and Knight, R. (2011). Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinformatics 10:10.17. doi: 10.1002/0471250953.bi1007s36
Leprich, D. J., Flood, B. E., Schroedl, P. R., Ricci, E., Marlow, J. J., Girguis, P. R., et al. (2021). Sulfur bacteria promote dissolution of authigenic carbonates at marine methane seeps. ISME J. [Epub ahead of print]. doi: 10.1038/s41396-021-00903-3
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li, J. W., Peng, X. T., Bai, S. J., Chen, Z. Y., and Van Nostrand, J. D. (2018). Biogeochemical processes controlling authigenic carbonate formation within the sediment column from the Okinawa trough. Geochim. Cosm. Acta 222, 363–382. doi: 10.1016/j.gca.2017.10.029
Li, W.-L., Wu, Y.-Z., Zhou, G.-W., Huang, H., and Wang, Y. (2020). Metabolic diversification of anaerobic methanotrophic archaea in a deep-sea cold seep. Mar. Life Sci. Technol. 2, 431–441. doi: 10.1007/s42995-020-00057-9
López-García, P., Vereshchaka, A., and Moreira, D. (2007). Eukaryotic diversity associated with carbonates and fluid–seawater interface in Lost City hydrothermal field. Environ. Microbiol. 9, 546–554. doi: 10.1111/j.1462-2920.2006.01158.x
Niemann, H., Losekann, T., De Beer, D., Elvert, M., Nadalig, T., Knittel, K., et al. (2006). Novel microbial communities of the Haakon Mosby mud volcano and their role as a methane sink. Nature 443, 854–858. doi: 10.1038/nature05227
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Olm, M. R., Brown, C. T., Brooks, B., and Banfield, J. F. (2017). dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868. doi: 10.1038/ismej.2017.126
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patel, R. K., and Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rédou, V., Navarri, M., Meslet-Cladière, L., Barbier, G., and Burgaud, G. (2015). Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl. Environ. Microbiol. 81, 3571–3583. doi: 10.1128/AEM.04064-14
Reitner, J., Peckmann, J., Reimer, A., Schumann, G., and Thiel, V. (2005). Methane-derived carbonate build-ups and associated microbial communities at cold seeps on the lower Crimean shelf (Black Sea). Facies 51, 66–79. doi: 10.1007/s10347-005-0059-4
Roberts, H. H., Feng, D., and Joye, S. B. (2010). Cold-seep carbonates of the middle and lower continental slope, northern Gulf of Mexico. Deep Sea Res. II 57, 2040–2054. doi: 10.1016/j.dsr2.2010.09.003
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Ruff, S. E., Biddle, J. F., Teske, A. P., Knittel, K., Boetius, A., and Ramette, A. (2015). Global dispersion and local diversification of the methane seep microbiome. Proc. Natl. Acad. Sci. U.S.A. 112, 4015–4020. doi: 10.1073/pnas.1421865112
Salter, S., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Skarke, A., Ruppel, C., Kodis, M., Brothers, D., and Lobecker, E. (2014). Widespread methane leakage from the sea floor on the northern US Atlantic margin. Nat. Geosci. 7, 657–661. doi: 10.1038/ngeo2232
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Subrahmanyam, G., Vaghela, R., Bhatt, N. P., and Archana, G. (2012). Carbonate-dissolving bacteria from ‘miliolite’, a bioclastic limestone, from Gopnath, Gujarat, Western India. Microbes Environ. 27, 334–337. doi: 10.1264/jsme2.me11347
Timmers, P. H. A., Welte, C. U., Koehorst, J. J., Plugge, C. M., Jetten, M. S. M., and Stams, A. J. M. (2017). Reverse methanogenesis and respiration in methanotrophic archaea. Archaea 2017:1654237. doi: 10.1155/2017/1654237
Uritskiy, G. V., Diruggiero, J., and Taylor, J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Wang, Y., Huang, J.-M., Cui, G.-J., Nunoura, T., Takaki, Y., Li, W.-L., et al. (2019). Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ. Microbiol. 21, 716–729. doi: 10.1111/1462-2920.14518
Wang, Y., and Qian, P.-Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401. doi: 10.1371/journal.pone.0007401
Wang, Y., Zhang, W. P., Cao, H. L., Shek, C. S., Tian, R. M., Wong, Y. H., et al. (2014). Diversity and distribution of eukaryotic microbes in and around a brine pool adjacent to the Thuwal cold seeps in the Red Sea. Front. Microbiol. 5:37. doi: 10.3389/fmicb.2014.00037
Wu, Y., Qiu, J. W., Qian, P. Y., and Wang, Y. (2018). The vertical distribution of prokaryotes in the surface sediment of Jiaolong cold seep at the northern South China Sea. Extremophiles 22, 499–510. doi: 10.1007/s00792-018-1012-0
Wu, Y. W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Xu, H., Luo, X., Qian, J., Pang, X., Song, J., Qian, G., et al. (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7:e52249. doi: 10.1371/journal.pone.0052249
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina paired-end read merge R. Bioinformatics 30, 614–620.
Keywords: methane oxidation, microbial community, ammonia-oxidizing archaea, metagenome-assembled genomes, carbonate weathering
Citation: Wang Y, Li W, Li Q, Zhou Y, Gao Z and Feng D (2021) Deep-Sea Carbonates Are a Reservoir of Fossil Microbes Previously Inhabiting Cold Seeps. Front. Mar. Sci. 8:698945. doi: 10.3389/fmars.2021.698945
Received: 22 April 2021; Accepted: 30 June 2021;
Published: 21 July 2021.
Edited by:
Hao Chen, Institute of Oceanology, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Yu Zhang, Shanghai Jiao Tong University, ChinaCopyright © 2021 Wang, Li, Li, Zhou, Gao and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Wang, d2FuZ3lAaWRzc2UuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.