
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 30 March 2021
Sec. Aquatic Physiology
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.658091
 Wenxiao Cui1,2Qin Yang1,2Yin Zhang1,2Ardavan Farhadi1,2Huan Fang1,2
Wenxiao Cui1,2Qin Yang1,2Yin Zhang1,2Ardavan Farhadi1,2Huan Fang1,2 Huaiping Zheng1,2
Huaiping Zheng1,2 Shengkang Li1,2
Shengkang Li1,2 Yueling Zhang1,2
Yueling Zhang1,2 Mhd Ikhwanuddin2,3
Mhd Ikhwanuddin2,3 Hongyu Ma1,2*
Hongyu Ma1,2*The mud crab Scylla paramamosain is a species with significant sexual dimorphism in growth rate and body size, of which the females are of higher economic and nutritional values than the males. Accordingly, there is an urgent need to explore the molecular mechanism underlying sex determination and gonadal differentiation. The single-molecule long-read technology combining with RNA sequencing was employed to construct a full-length transcriptome for gonads of S. paramamosain. In total, 1,562,819 FLNC reads were obtained from 1,813,758 reads of inserts (ROIs). Among them, the 10,739 fusion isoforms corresponded to 23,634 reads and were involved in 5,369 genes in the reference annotation. According to the criteria for new transcripts, a total of 213,809 isoforms were recognized as novel transcripts and then matched against Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), NR, Swissprot, and KOG databases. We also identified 22,313 SSRs, 169,559 lncRNAs, and 25,451 SNPs. Additionally, 349,854 alternative splicing (AS) events from 8,430 gene models were detected, and 5,129 polyadenylation sites were profiled from 3,090 genes. GO and KEGG annotation indicated that AS and APA probably play important roles in the gonadal development and maturation. Besides, the DEGs associated with gonadal development and maturation were identified and analyzed based on the RNA-Seq data.
The mud crab (Scylla paramamosain), which is widely distributed along the southeast Asia coasts, is one of the most prevalent aquaculture crustaceans in China due to its huge economic value and superior growth performance (Shi et al., 2019). In China, the artificial culture of mud crab can be traced back to more than 100 years ago (Shen and Lai, 1994), and the aquaculture production has reached 150,000 tons in the recent years (Wu et al., 2019b). As a species provided with significant sexual dimorphism in growth rate and body size, females are of higher economic and nutritional values than males (Ma et al., 2013). Accordingly, there is an urgent need to improve the artificial breeding and farming techniques of female crabs.
The molecular mechanism underlying sex determination and gonadal differentiation of the mud crab has received considerable attention due to the remarkable biological and economic differences between sexes (Yang et al., 2018). Consequently, multiple omics and sequencing technologies have recently focused on the understanding of sexual differentiation, gonadal maturation, and gametogenesis in mud crab. Restriction site-associated DNA sequencing (RAD-seq) was employed to isolate sex-specific single-nucleotide polymorphism (SNP) markers for S. paramamosain, which provided powerful genetic evidence for a WZ/ZZ sex determination system in mud crabs S. paramamosain, Scylla tranquebarica, and Scylla serrata (Shi et al., 2018). Comparative profiling of ovarian and testicular piRNAs in the S. paramamosain, which provided baseline information of mud crab piRNAs and their differential expression, suggested that piRNAs play an essential role in regulating gametogenesis and gonadal development (Waiho et al., 2019). The de novo gonad transcriptome sequencing was performed to identify differentially expressed genes (DEGs) and long non-coding RNAs (lncRNAs) between male and female S. paramamosain, in which homologous genes and lncRNAs involved in sex determination and gonadal development pathways (Sxl-Tra/Tra-2-Dsx/Fru, Wnt4, and thyroid hormone synthesis pathway) were identified (Yang et al., 2018). Although some achievements have been implemented, the understanding of gonadal development and sexual differentiation which facilitates monosexual crab breeding and culture, remains lacking.
The performance of single-molecule real-time (SMRT) sequencing provides a third-generation sequencing platform widely used in genome sequencing because of its long reads (Li et al., 2018). Isoform sequencing (Iso-Seq) for single-molecule long reads provides directly a considerable advantage in the transcriptome-wide comprehensive analysis of full-length splice isoforms and other forms of post-transcriptional regulatory events such as alternative splicing (AS) and alternative polyadenylation (APA) (Thomas et al., 2014; Tilgner et al., 2015). Using Iso-Seq, novel alternative post-transcriptional events were identified in primates at the whole-transcript level, and the combination of these events, which has contributed to complex isoform evolution, is largely independent along with the transcript (Zhang et al., 2017). Other eukaryotes from plants to animals, such as bamboo (Wang T. et al., 2017), sorghum (Abdel-Ghany et al., 2016), mouse (Gupta et al., 2018), and pig (Tang et al., 2018), showed high diversity during the transcriptome-wide identification of AS or APA events. However, little information about alternative mRNA processing events is available in crustaceans, even though they might push forward an immense influence on studying genes associated with sexual differentiation and gonadal development.
In this study, PacBio single-molecule long-read technology was conducted to construct and sequence full-length transcriptome for gonads of S. paramamosain. Meanwhile, Illumina RNA-Seq was performed to correct PacBio long reads and calculate the gene expression levels. We aim to establish a comprehensive profile of gonad transcriptome in S. paramamosain, and provide insights into post-transcriptional processing events in the regulatory mechanism of sexual differentiation and gonadal maturation.
All animal experiments were performed according to recommendations in the Guide for the Care and Use of Laboratory Animals of Shantou University and with the approval of the Institutional Animal Care and Use Ethics Committee of Shantou University. All crabs were anesthetized on ice before dissection.
Healthy sexually mature and juvenile crabs were purchased from a local market in Shantou City, China. The mature testis at stage 2 (Te 2) and ovary at stage 4 (Ov 4) were obtained from 12 individuals (six individuals of each sex), and immature testis (Te 0) and ovary (Ov 0) from 12 juvenile crabs (six individuals of each sex) were excised for SMRT sequencing and RNA-Seq. At stage 2, testes are swollen, opaque, and white; vas deferens are swollen and pink, contains spermatophore, which is ready for mating (Mhd et al., 2018). As for the stage 4 ovary, which is in the late-maturing or secondary vitellogenic (stage 4) stage, the oocyte grows rapidly, and the nucleus reached the maximum at this stage or the end of primary vitellogenesis (Islam et al., 2010).
Total RNA from 24 individuals including four different groups (Te2, Ov4, Te0, and Ov0) was extracted using Qiagen Kit (QIAGEN GmbH, Germany). In total, 12 samples were obtained by mixing two individuals in each sample for sufficient specimen on the transcriptome sequence, given that the immature testis or ovary is small. The quality of the RNA was examined by quantifying concentration and integrity analysis. Agilent 2100 Bioanalyzer (Agilent Technologies, United States) was used to analyze RNA integrity, and total RNA with a 28S/18S value ≥ 1.5 and RNA Integrity Number (RIN) ≥8 were used for library construction.
Each three RNA samples for a single group were mixed to prepare a hybrid cDNA library for the PacBio Sequel platform using SMARTer PCR cDNA Synthesis Kit (Takara, Japan). PCR amplification with an optimal number of PCR cycles, testing by the PCR cycle optimization, was performed on the first-strand cDNA. The amplified cDNA products were carried out for size selection. Then, a large-scale cDNA was generated by PCR amplification for the PacBio Sequel library. Using the SMRTbell template prep kit, four single-group constructed libraries were prepared and then sequenced on the PacBio Sequel platform by Nextomics Biosciences Co., Ltd. (Wuhan, China).
In total, 12 RNA-Seq libraries (three libraries for each group) for gonads of S. paramamosain were prepared. PCR amplification of cDNA fragments was performed, and the purified PCR products were sequenced by Nextomics Biosciences Co., Ltd. (Wuhan, China). More than 10 G raw reads were yielded from each library during sequencing.
To get full-length consensus reads for further analysis, raw data generated by single molecular sequencing were processed using SMRT Analysis Software v2.3 Suite1. First, removing the invalid raw reads with adapters, chimeric, and low-quality bases, post-filter polymerase reads from each movie can be obtained. Next, quality control was performed on the reads of insert (ROI). With a minimum full pass of 1 and a minimum of 80% read accuracy, high-quality ROIs were obtained in polymerase reads. ROIs were then categorized into full-length non-chimeric (FLNC) reads, non-full-length reads, full-length chimeric reads, and filtered short reads based on primer and poly (A) tail detection. FLNC reads were clustered by the ICE algorithm module to get unpolished consensus sequence for the alignment of non-full-length reads, then the polished consensus sequences were yielded by arrow correcting. To avoid and correct high error rates of the long-read sequencing, all the polished consensus sequences were compared with RNA-Seq Illumina platform paired-end reads using LoRDEC software with parameter: -k 19 -s 3. Finally, the corrected consensus reads, which can be recognized as unique isoforms, were obtained.
The mud crab whole-genome sequences and any gene model annotations (data unpublished) were used as a reference genome. The corrected isoforms were mapped to a reference genome using the Genome Mapping and Alignment Program (GMAP) with option: -f samse -n 1 (Wu and Watanabe, 2005). Following error correction and alignment, a software fusion_finder.py in cDNA Cupcake2 was used to extract candidate fusion transcript through four criteria as follows: (a) read map to two or more isolated loci; (b) transcript coverage for each locus should be at least 5%; (c) comparative region coverage ≥99%; (d) the mapped locus must be at least 10 kb apart. Apart from the fusion transcripts, isoform mapping to the genome with a low identity and coverage were discarded. By using another program, collapse_isoforms_by_sam.py, collapsed isoforms were obtained and matched to the gene model using MatchAnnot3.
The gene structure annotation results were compared with those of reference annotation to determine the new gene following these criteria: (1) the exon length between the isoform and the reference gene is differential; (2) the part or little of exons were one-to-one overlaps; (3) exon showed no overlap; (4) and the isoform was annotated on the reference genome but no gene found. Then, open reading frame (ORFs) prediction and function annotation were performed on novel transcripts. The coding potential was estimated using the ANGEL (Shimizu et al., 2006). To run this program, genome sequences with any annotations are needed to set up a training dataset for novel transcripts. The ORF prediction was then processed after model training. Databases Non-redundant Protein Sequence Database (NR), Swissprot, clusters of orthologous groups (COG/KOG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) were used for functional annotation of all novel transcripts using the BLASTX program with E-Value < 10–5. From the reads representing a novel transcript with overall annotation, we characterized those overlap no annotated gene loci as read cluster for novel gene identification.
The most widely used methods Predictor of LncRNAs and Messenger RNAs based on an improved K-mer scheme (PLEK) (Li et al., 2014), Coding Potential Calculator (CPC) (Kong et al., 2007), Coding-Non-coding Index (CNCI)4, and Coding Potential Assessment Tool (CPAT) (Deng et al., 2006) were conducted to predict coding capacity on the unannotated transcripts and those classified to likely-NA and suspicious-NA, which show low reliability of coding sequences (CDs) when running ANGEL (Shimizu et al., 2006). Meanwhile, Swissprot protein database annotations also supported the reliability of non-coding RNA. As a result of non-coding from prediction and database annotations, candidate non-coding RNA was screened out with a length longer than 200 bp.
The identification and location of simple sequence repeat (SSR) maker in all unique isoform sequences were implemented using MISA software with the default parameters (Thiel et al., 2003). Microsatellites (mononucleotide repeats ≥16, dinucleotide repeats ≥6, and trinucleotide to hexanucleotide repeats ≥5) and compound microsatellites consisting of two individual SSR interrupted by a certain number of bases (≤100) were allowed to identify. SNPs and short indel sequence variation identification were conducted with samtools/bcftools5.
Alternative splicing analysis was conducted using the Python script alternative_splice.py6 by aligning full-length consensus reads to gene model in the existing genome (Wang M. et al., 2017). The unique isoforms mapped to the same gene model were considered undergoing AS events. The AS events including alternative donor site (AD), alternative acceptor site (AA), alternative position (AP), exon skipping (ES), and intron retention (IR) were identified by the TAPIS pipelines (Abdel-Ghany et al., 2016). KEGG and GO enrichments were conducted on the genes undergoing AS events to analyze the potential functions.
The full-length consensus reads generated by SMRT sequencing were used to identify multiple poly (A) positions. An analysis of APA was carried out using TAPIS (Arefeen et al., 2018). All reads associated with the gene model were processed to distinguish variant poly (A) sites in the reference genome sequence. By aligning reads to annotated gene, reliable alternative poly (A) sites supported by at least two trusted reads were then characterized by candidates with a minimum distance of 20 nucleotides (nt) between two poly (A) sites. The nucleotide composition profile of poly (A) sites was analyzed, and MEME was conducted on 50 nucleotides upstream of multiple poly (A) sites to search for polyadenylation signal motif.
RNA-Seq with the Illumina HiSeq X ten platform was used to generate paired-end (PE) short reads to correct PacBio long reads. Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina. The resulting libraries were finally sequenced on a HiSeq X ten sequencing system as 150-nt paired-end reads. All the clean reads were then aligned to the reference genome (unpublished) using HISAT2 (Pertea et al., 2016). DEGs were identified using DESeq2 package with an adjusted p-value (Padj) ≤ 0.05 and fold change value (FC) > 2. Furthermore, GO classification and KEGG pathway analyses were performed on DEGs.
Total RNA was extracted using the TRIzol reagent (Invitrogen), and its quality and integrity were checked using NanoDrop spectrophotometers and 1% agarose gel, respectively. First-strand cDNA was synthesized using TranScript First-Stand cDNA Synthesis SuperMix kit (Beijing TransGen Biotech, Beijing, China) for PCR and quantitative real-time PCR (qPCR). Primers (Supplementary Table 1) were designed using Primer Premier 5.0 software. PCR amplification was performed to validated AS events using EasyTaq DNA Polymerase (TransGen) and monitored on one agarose gel, which was followed by Sanger sequencing. The 3′-RACE (Rapid Amplification of cDNA ends) was performed to research APA using 3′-RACE Kit (Sangon, Shanghai, China). cDNA was prepared using 3′-RACE adaptor primer, and nest-PCR was carried out using 3′-RACE (R) primer and gene-specific forward (F) primer. We performed qPCR on 10 randomly selected DEGs using Talent qPCR Premix (SYBR Green) kit (TIANGEN Biotech, Beijing, China) following the manufacturer’s instructions to investigate the expression profiles on LightCycler® 480 System (Roche Applied Science, Germany). Each experiment was performed in three biological replicates. Normalization of the expression level of transcripts to that of 18S rRNA was conducted using the optimized comparative Ct (2–ΔΔCt) value method, respectively. Results are expressed as mean values with the standard error mean (SEM) of three replicates. All qPCR data were analyzed by Student’s t-test (two-tailed) for the determination of significant statistical changes in pair with p < 0.05.
To establish a full-length transcriptome of gonads in male and female S. paramamosain, four hybrid libraries of 0.5–6 k were constructed for single-molecule long-read sequencing. In total, 417,594, 492,228, 481,038, and 422,898 ROIs were yielded from four SMRT cells (Supplementary Table 2). By identifying 5′ primer and 3′ primer as well as poly (A) tail, the Iso-Seq analysis pipeline classified ROIs in terms of FLNC reads, non-full-length reads leaving out full-length chimeric reads, and filtered short reads (Table 1 and Figure 1). After classification and clustering, a total of 777,438 consensus sequences were then received by arrow polishing. Among them, 126,761 high-quality quivered reads with the length of 291,728,132 bp (2,301 bp per reads) were characterized by a minimum 99% accuracy and at least two consensus reads supporting (Supplementary Table 3). All consensus sequences were corrected using paired-end reads generated by the RNA-Seq Illumina X ten platform to avoid high error rates of single-molecule long-read sequencing. Finally, 777,438 corrected consensus reads with an average length of 2,358 bp were obtained.
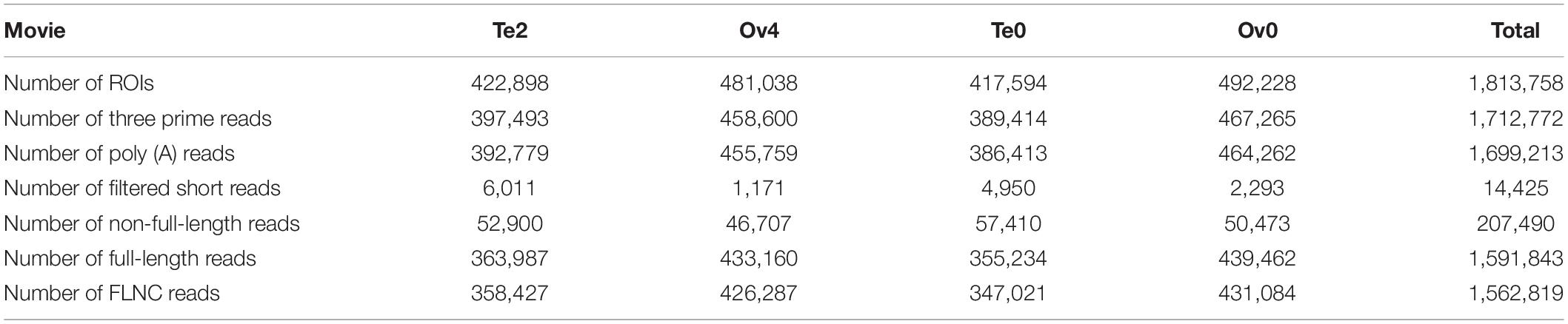
Table 1. Classification of reads of insert (ROIs).
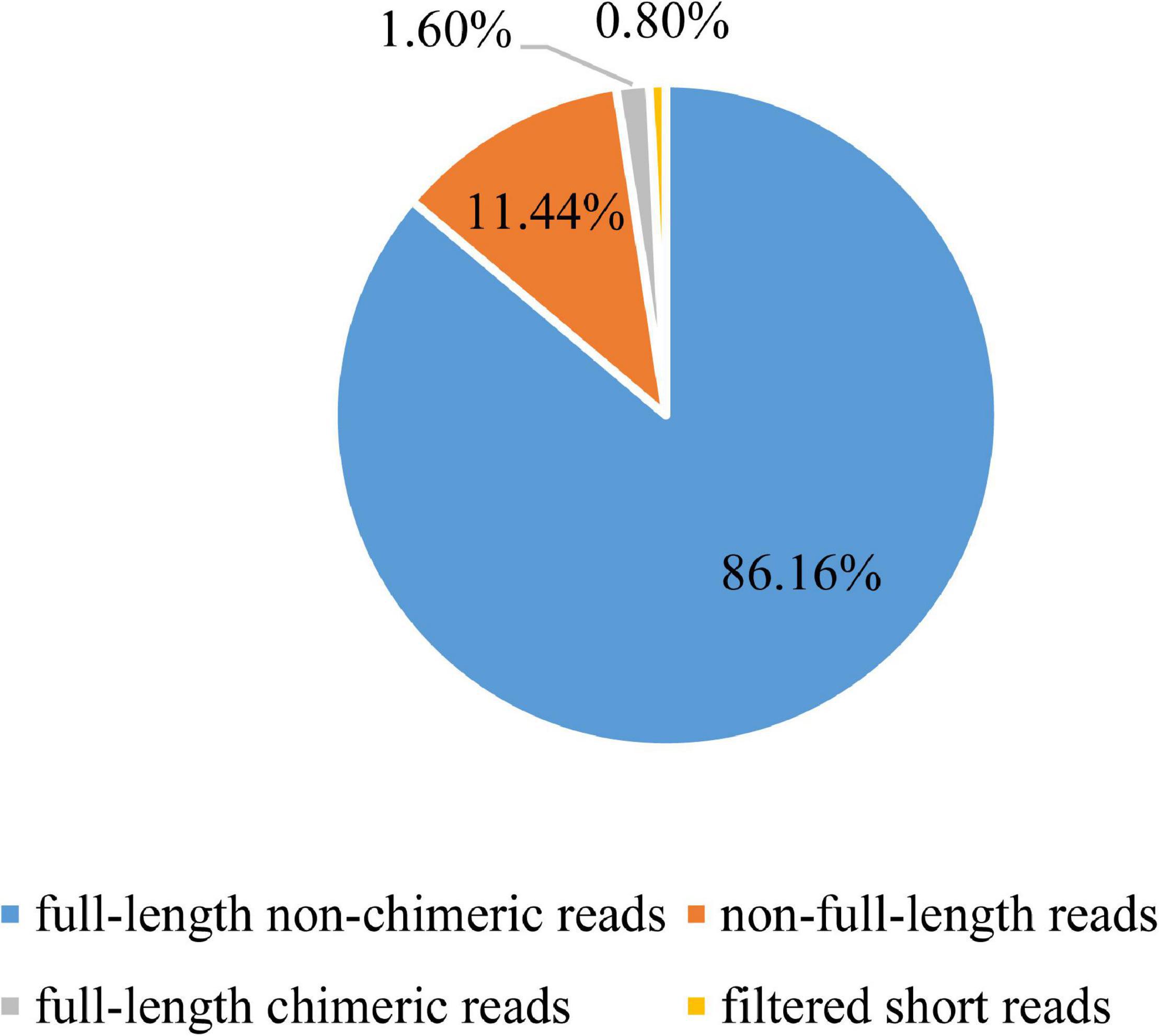
Figure 1. Classification of reads of insert (ROIs).
Blasted with the gene model in the reference genome with the condition mentioned above in the “Materials and Methods” section, we identified 10,739 isoforms, and they could simultaneously cover two or more annotated genes. The 10,739 fusion isoforms corresponded to 23,634 reads and were involved in 5,369 genes in the reference annotation. Most of the fusion transcripts were a combination of genes with other genes or no-genes (Supplementary Figure 1). After filtering out the putative fusion transcripts and the collapse isoforms (539,442 isoforms), the remaining 214,361 isoforms were then matched with gene annotation of the reference genome. After aligning with the reference genome, 114,062 isoforms mapped to 9,513 known gene covering more than half of the total annotated gene, while 100,299 isoforms were not found in the gene region (Supplementary Table 4).
According to the criteria for new transcripts mentioned in the “Method and Materials” section, a total of 213,809 isoforms were recognized as unannotated production of full-length transcriptome sequencing. The ANGEL software was used to predict CDS of novel transcripts, which were categorized into nine types (Supplementary Table 5). The majority of novel transcripts with initiation or termination codons showed high confidence in coding capacity. Nevertheless, 17,460 (likely-NA) and 69,236 (suspicious-NA) CD sequences had lower reliability, and a total of 1,146 sequences with little coding potential were left out when running ANGEL. The novel transcripts were then matched against GO, KEGG, NR, Swissprot, and KOG databases. A total of 152,151 isoforms (71.16%) were overall annotated in five databases (Figure 2). It showed that 106,312 (49.72%), 83,210 (38.92%), 148,677 (69.54%), 118,823 (55.57%), and 73,374 (34.32%) of 213,809 new genes could be found in KOG, KEGG, NR, SwissProt, and GO databases, respectively. The annotations on GO and KOG showed that all of these isoforms were classified into 42 and 26 functional categories, respectively. KEGG analysis indicated that the cluster of “signal transduction” represented the major group, followed by “Folding, sorting and degradation.” According to the results of NR database annotation, novel transcripts matched to known non-redundant sequences from up to 427 species. Most of them belong to the invertebrates including Penaeus vannamei, S. paramamosain, Eriocheir sinensis, Procambarus clarkia, and Zootermopsis nevadensis.
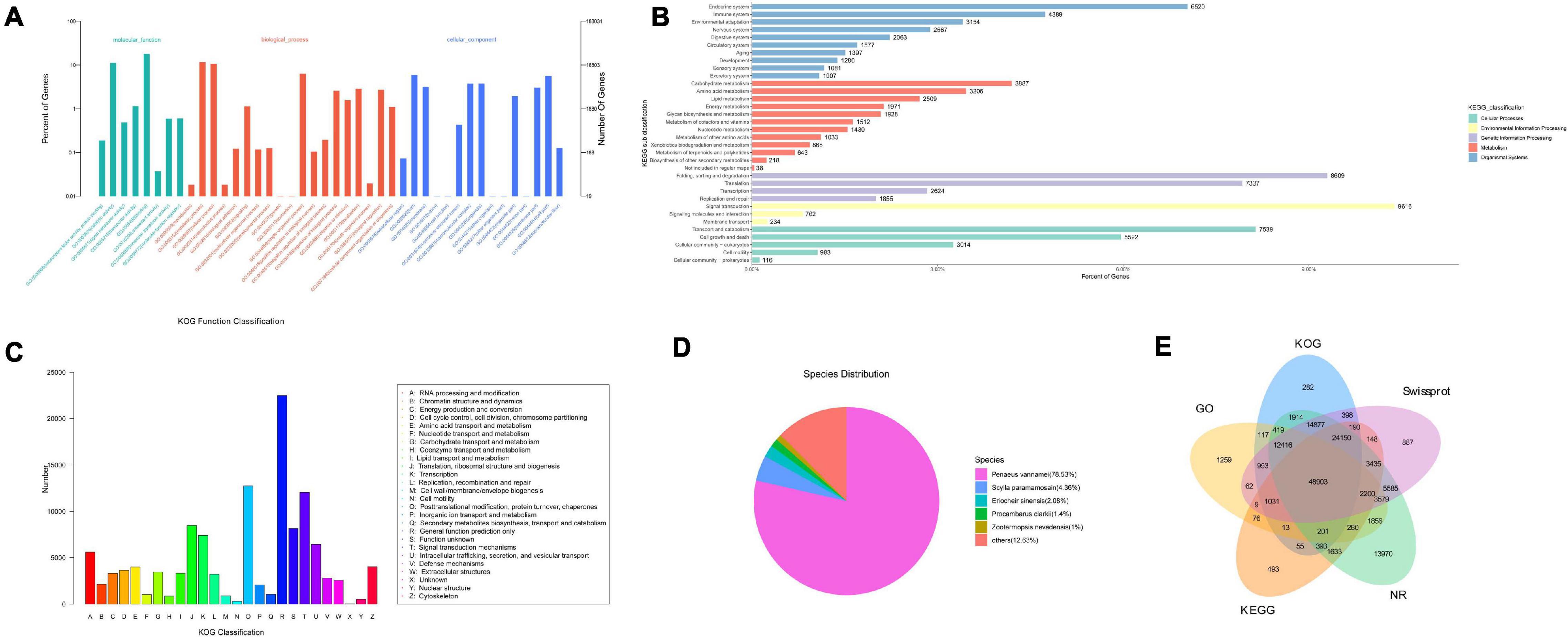
Figure 2. Functional classification of novel transcripts of Scylla paramamosain. (A) Gene ontology (GO) annotation; (B) KEGG pathway classification; (C) KOG classification; (D) Species distribution of NR annotation; and (E) overall results of annotation on GO, KEGG, NR, Swissprot, and KOG databases. The number represents novel genes identified in NR, KOG, KO, Swissprot, and GO databases.
Rarefaction analysis was performed to detect the connection between the PacBio reads and the gene number and the gene-discovery rate was further observed. As the saturation curves show (Supplementary Figure 2), the number of genes has a trend of slow growth with the rising number of sequenced reads, which indicated that sequencing depth had not reached saturation of gene and isoform discovery within those size ranges.
All the unpredictable and lower-reliable sequences were used to identified non-coding RNA. The results showed that 36,584 candidates were identified with PLEK, 34,176 with CPC, and 36,388 with CNCI. Based on the Swissprot database, a total of 45,137 candidate sequences with the annotated result of non-coding were identified. According to the common results of four predicting methods, a total of 22,313 candidate non-coding RNAs with the shortest length of 268 bp were identified (Figure 3). Therefore, all 22,313 candidates could be characterized as lncRNA.
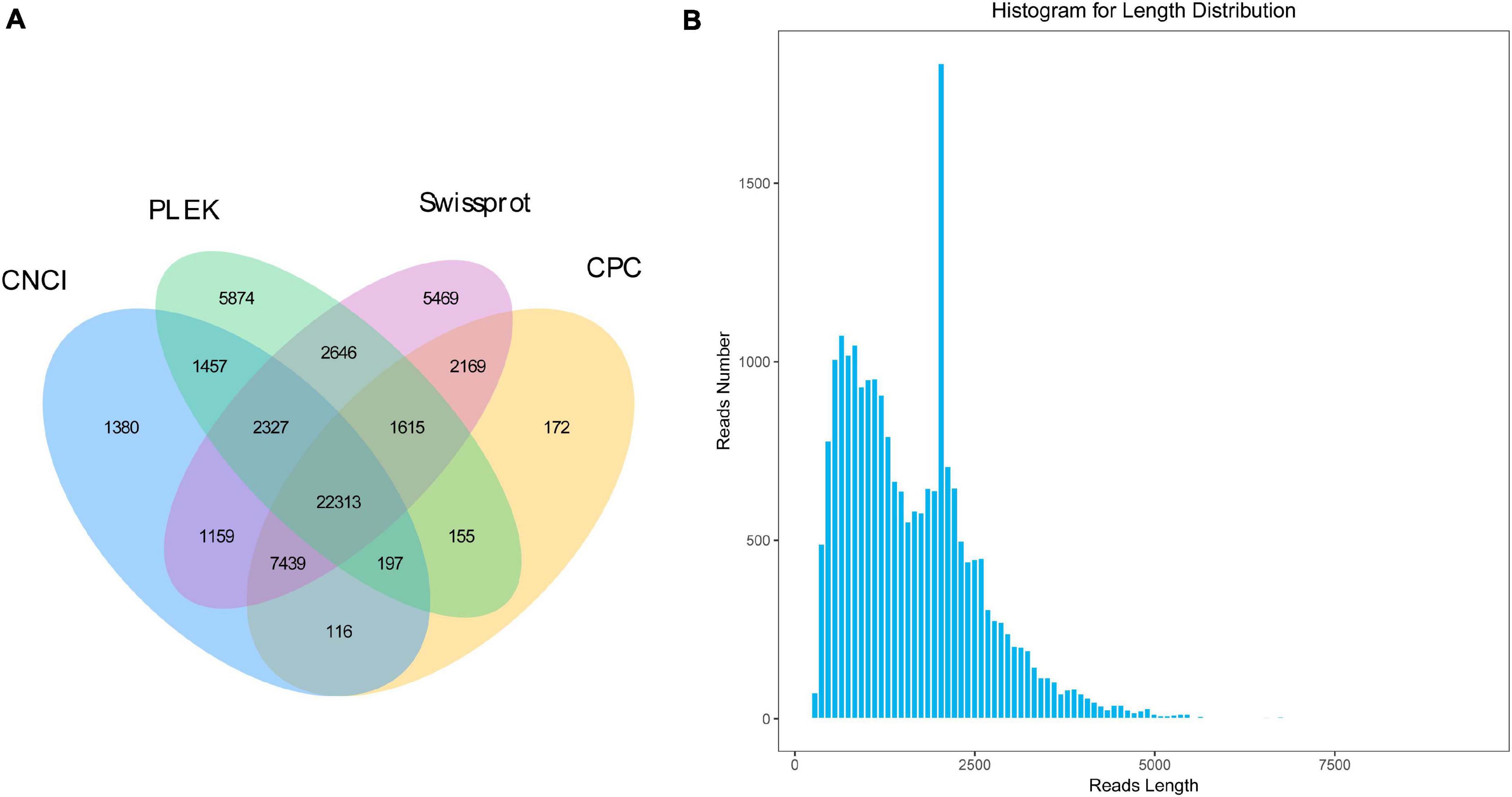
Figure 3. The identification and length distribution of long non-coding RNA (LncRNA). (A) Overlapping results of LncRNA from the different method, (B) Length distribution of LncRNA.
A total of 169,559 SSRs were generated from our transcriptome dataset, which contained 44,884 (42,815 and 2,069), 42,125, 39,427, 39,574, 2,932, 478, and 139 for complex repeats type (c and c∗), mononucleotide repeats (p1), dinucleotide repeats (p2), trinucleotide repeats (p3), tetra nucleotides repeats (p4), penta-nucleotides repeats (p5), and hexa-nucleotides repeats (p6), respectively (Figure 4). SNP loci were predicted based on the variation bases at one position in the reference from a unique isoform sequence. In total, 25,451 SNPs and 45 indels were identified.
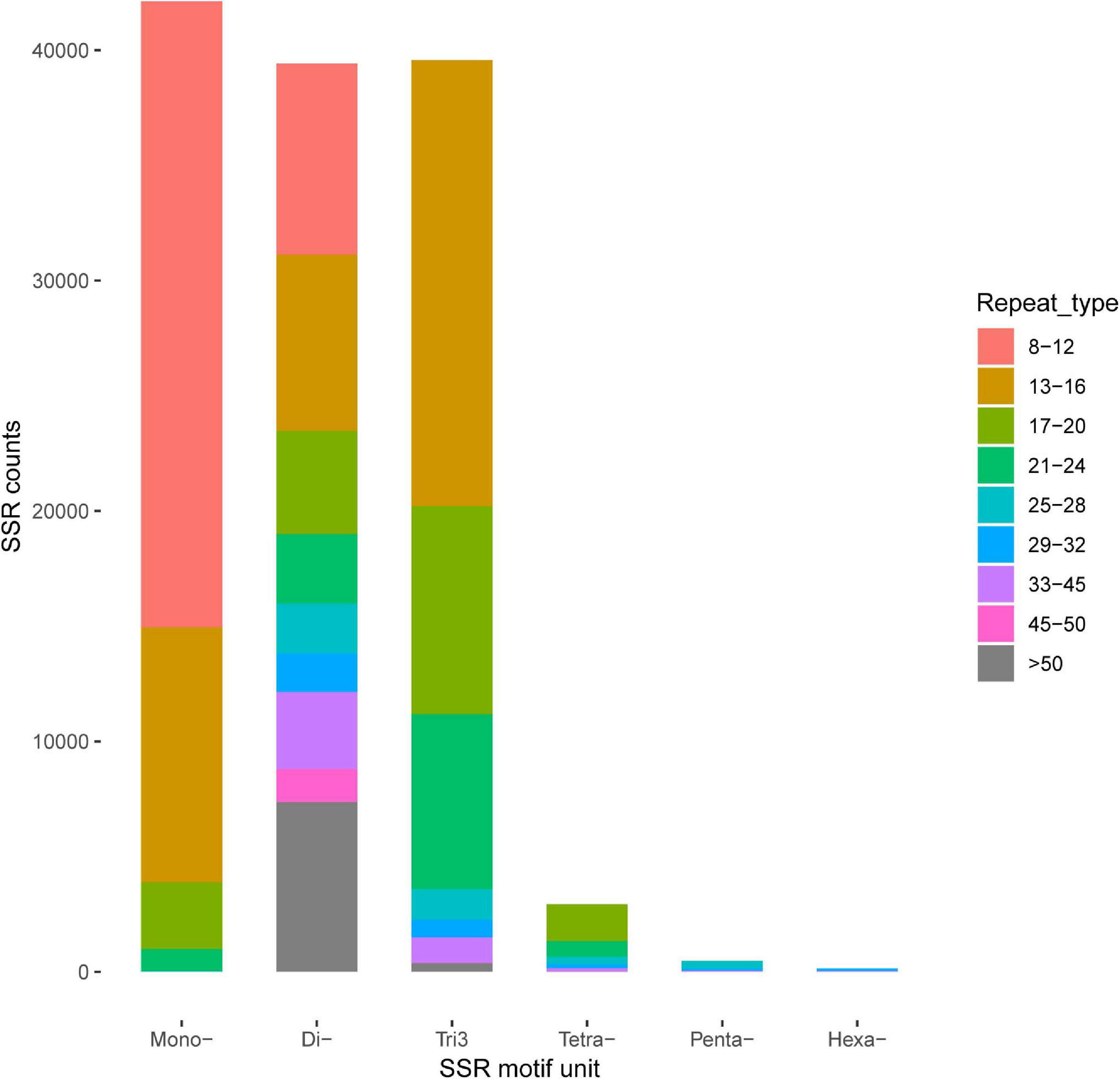
Figure 4. Identification of simple sequence repeats (SSR).
By aligning the full-length isoforms with the reference genome, optimization on the gene models can be conducted. We extended the 5′ or 3′ ends of 8,411 annotated genes based on 84,346 reads, which would make a great improvement on the genome annotation. Furthermore, long-read data can be used to predict the mRNA processing events, which lead to transcriptome diversity.
As a result of AS, mRNA with different structures were generated from a single gene. In the present study, a total of 349,854 AS events from 8,430 gene models were detected (Table 2). According to the description for AS analysis programs, AS events were divided into five major types including ES, IR, AD, AA, and AP (Figure 5). As a result, AP, which refers to introns overlapping but with both different 5′ end and 3′ end, was the most common AS pattern in gonad transcriptome of mud crabs. We also found that some splice isoforms are differentially expressed between the testis and ovary or their different stages of development in mud crab (Figure 6). For instance, one of the transcripts from the gene.Scaffold17_g66 has presented only in control gonadal differentiation while another only in testis development. Apart from that, the overwhelming majority of the alternative spliceosomes had not been annotated, which also function in the sex determination and gonadal differentiation (Figure 6).

Table 2. Classification of alternative splicing (AS) events.
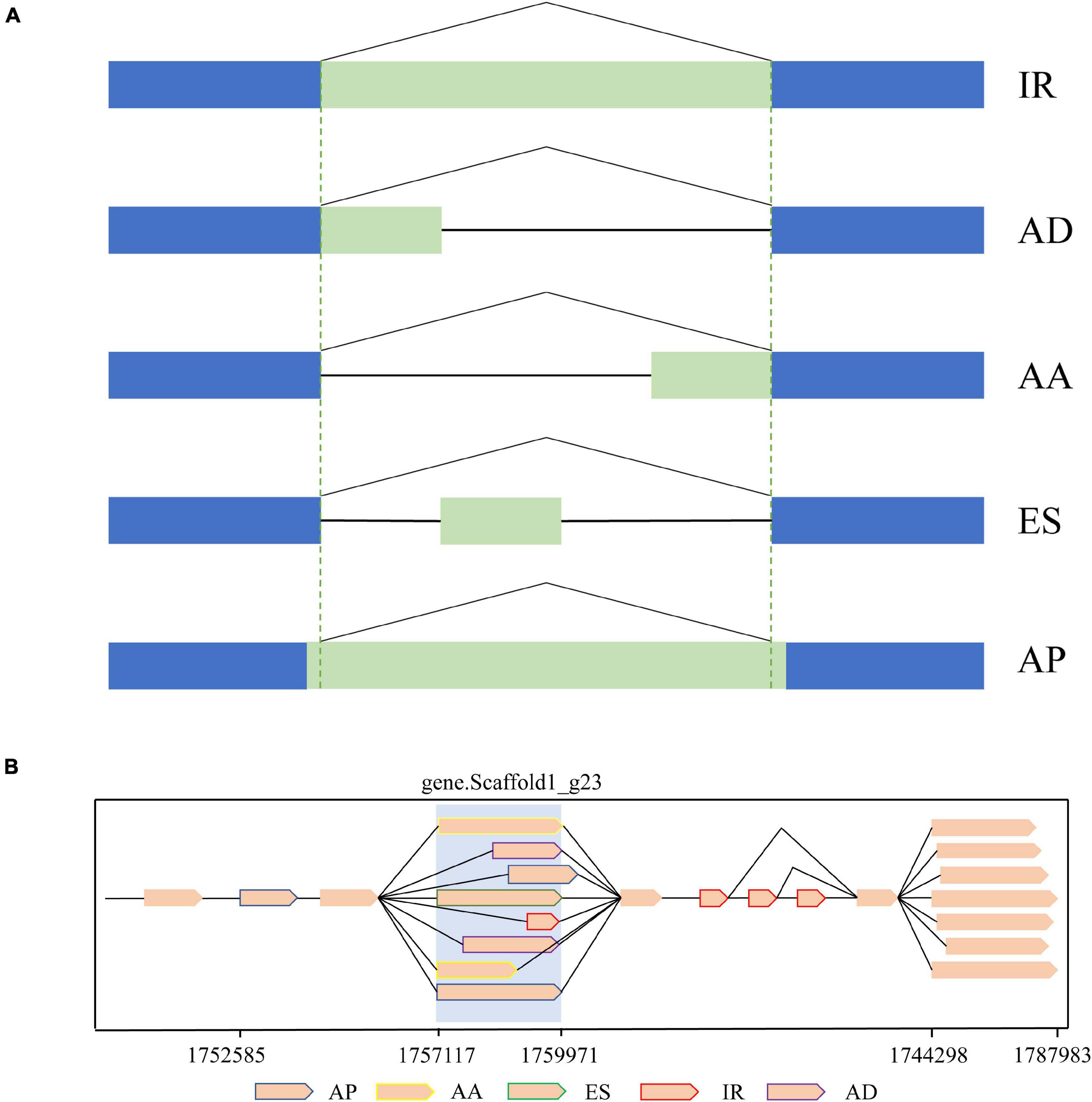
Figure 5. Schematic diagram of Alternative Splicing (AS). (A) Structure of AS types classified using alternative splice. (B) Schematic illustration of alternative isoforms within the PacBio transcriptome.
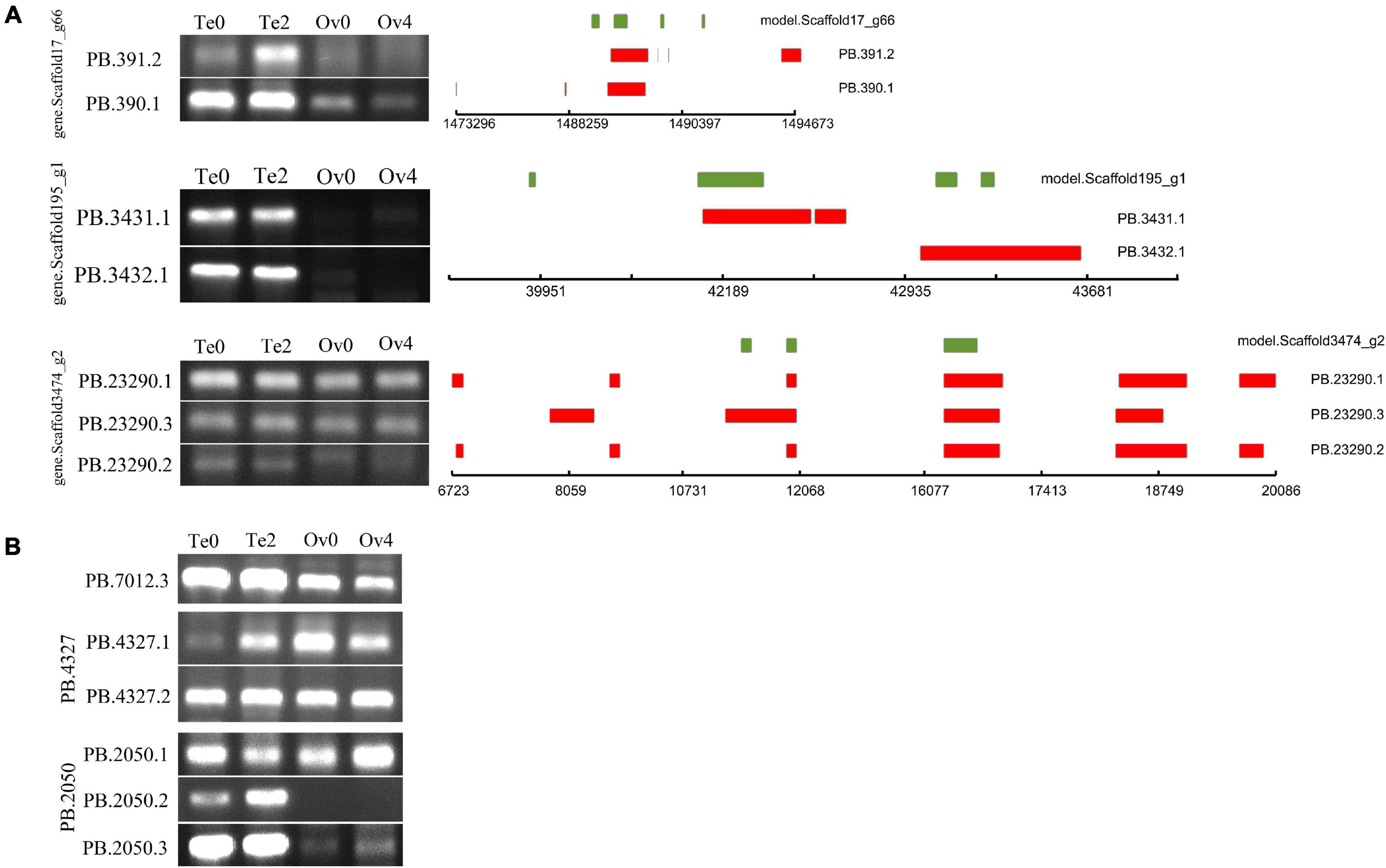
Figure 6. PCR validation of AS events identified by Iso-Seq. The green box represents the gene model; the exons in transcripts are represented by red boxes. Gene ID is shown at the left for each panel. (A) AS events of annotated genes, (B) AS events of unannotated genes.
To understand the roles of genes with AS, GO function enrichment and KEGG pathway classification were conducted. GO annotation analysis indicated that these genes act mainly on the variant physiological processes related to binding function. Additionally, the GO term “protein folding” and “unfold protein binding” possess the high enrichment factor. KEGG enrichment showed that these genes were significantly enriched in “cell cycle–yeast” and “RNA transport” followed by “meiosis–yeast” (Figure 7).
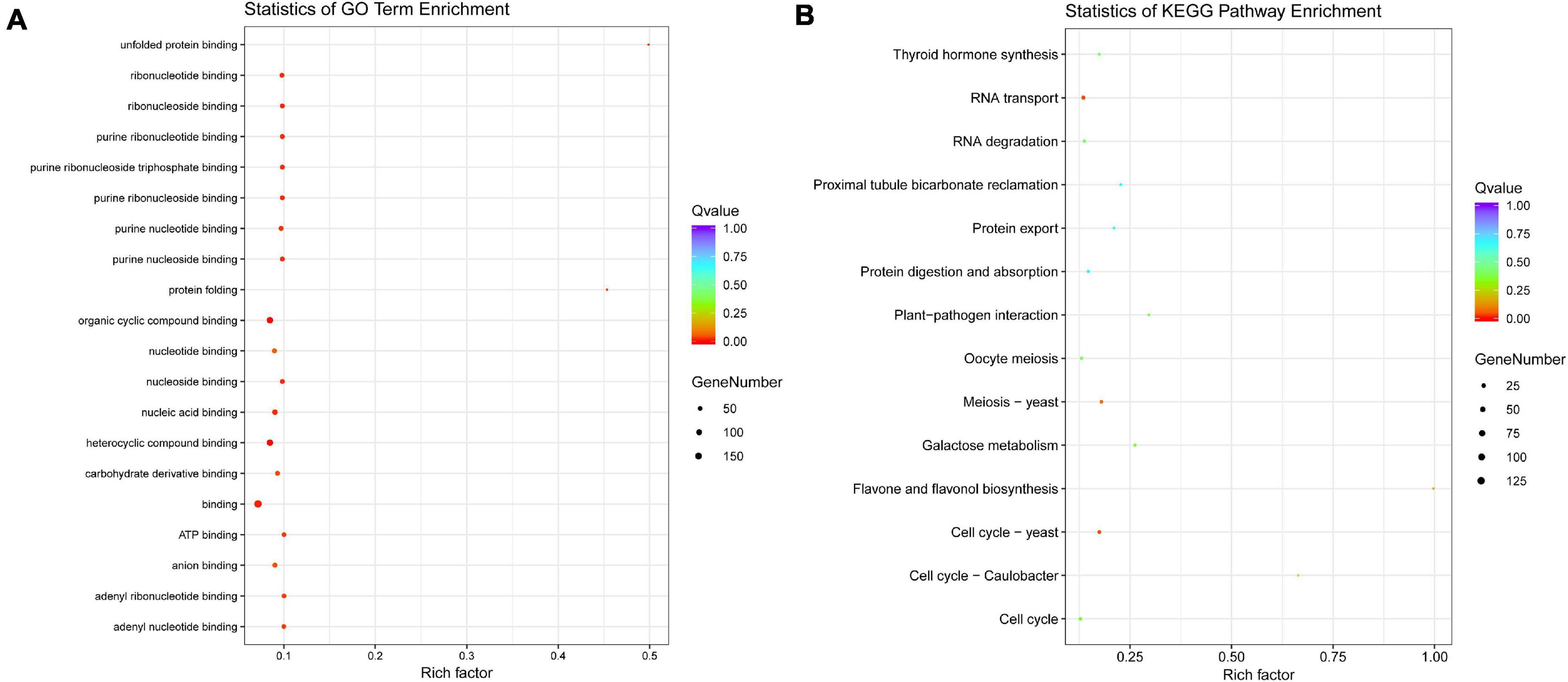
Figure 7. Functional annotation of the genes with AS events. (A) GO term enrichment, (B) KEGG pathway enrichment.
As for APA, a total of 5,129 polyadenylation sites were profiled from 3,090 genes. By polyadenylation analysis, we detected 1,265 genes containing two or more polyadenylation sites (Figure 8). The nucleotide composition around poly (A) sites (+50 nts and −50 nts) was analyzed for nucleotide bias, in which the clear bias of nucleotides upstream and downstream of the cleavage site in the 3′ UTRs was represented by the low content of GC confirming that our identified poly (A) sites are reliable (Figure 8). We searched for the polyadenylation signal motif on the sequences 50 nucleotides upstream and downstream of all 5,129 poly (A) sites using MEME. The 2,061 sites were contributed to the construction of an AAUAAA motif, which is known as the canonical poly (A) signal in eukaryotes (Figure 8; Tian et al., 2005). Additionally, another conserved motif UGUA was found, and the following two nucleotides (C and A) appeared with a high possibility in our study (Figure 8). Based on the 3′ RACE, we have observed differential APA because of sex difference (Figure 8).
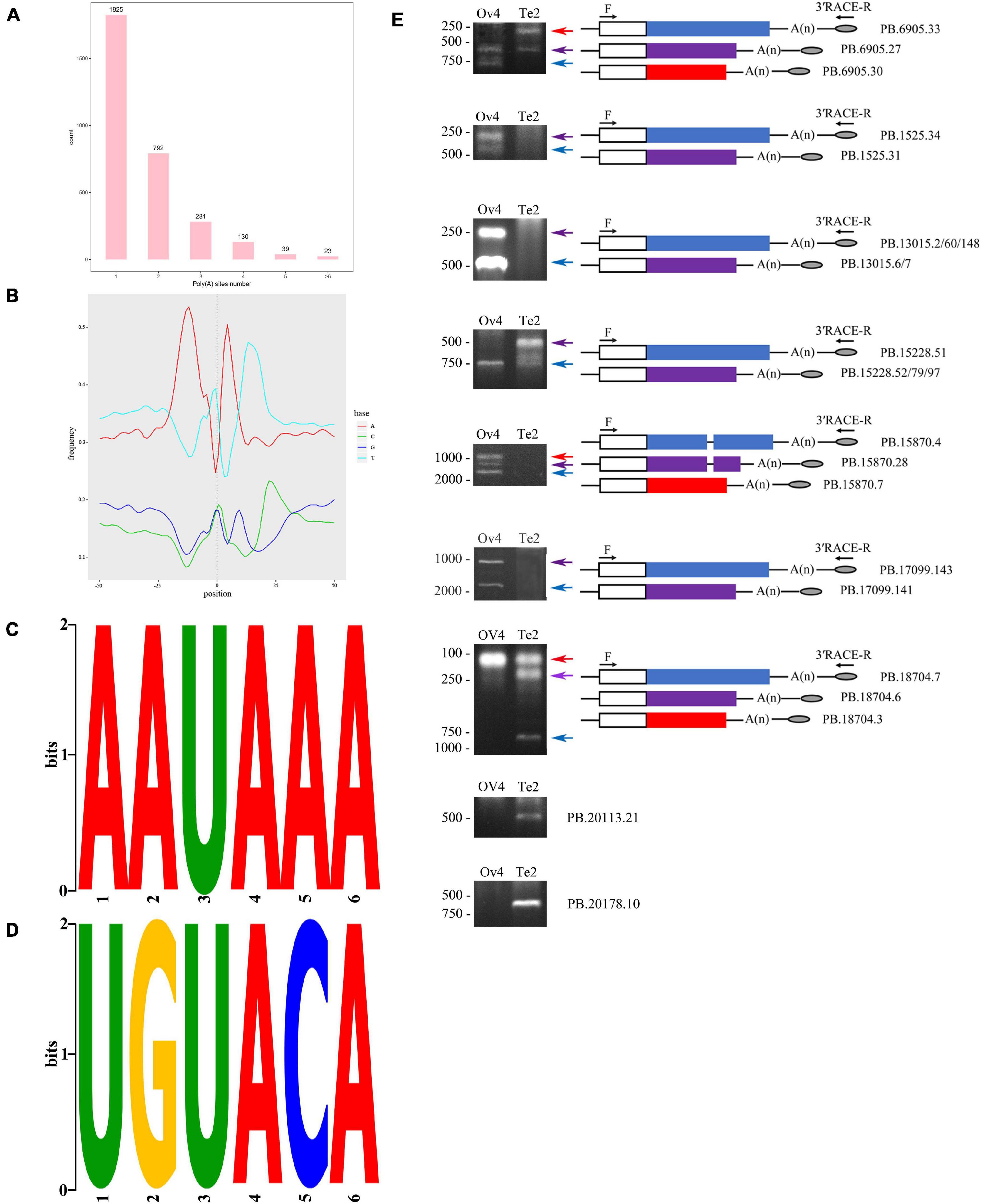
Figure 8. Alternative polyadenylation analysis. (A) Result of poly (A) sites analysis using TAPIS. (B) Nucleotide bias analysis on 50 nt up- and down-stream of poly (A) sites. (C) MEME analysis identified a conserved poly (A) signal. (D) Another overrepresented motif (UGUA) is found. (E) Validation of polyadenylation sites by nest-PCR. R, 3′ RACE reverse primer; F, gene-specific primer; consensus and specific sequence of isoforms were represented by open boxes and colored boxes, respectively.
The expression level of genes in each group was analyzed to screen the DEGs using fragments per kilobase of exons per million fragments mapped (FPKM). The principal component analysis (PCA) was performed to determine that there was no separation or outliers in the sample set (Supplementary Figure 3). The comparative analyses of DEGs among groups were performed. The results showed that the 302 and 5,606 DEGs were identified from the development process of testis and ovary, respectively (Figure 9). Furthermore, 14,619 and 18,175 DEGs were screened in the developmental initiation and maturation stages of the ovary compared with the testis. Among these genes, the 64 common DEGs acted on the maturation both of the testis and ovary, and the 29 genes showed different expression profiles during all stages of ovarian and testicular development and maturation (Figure 9).
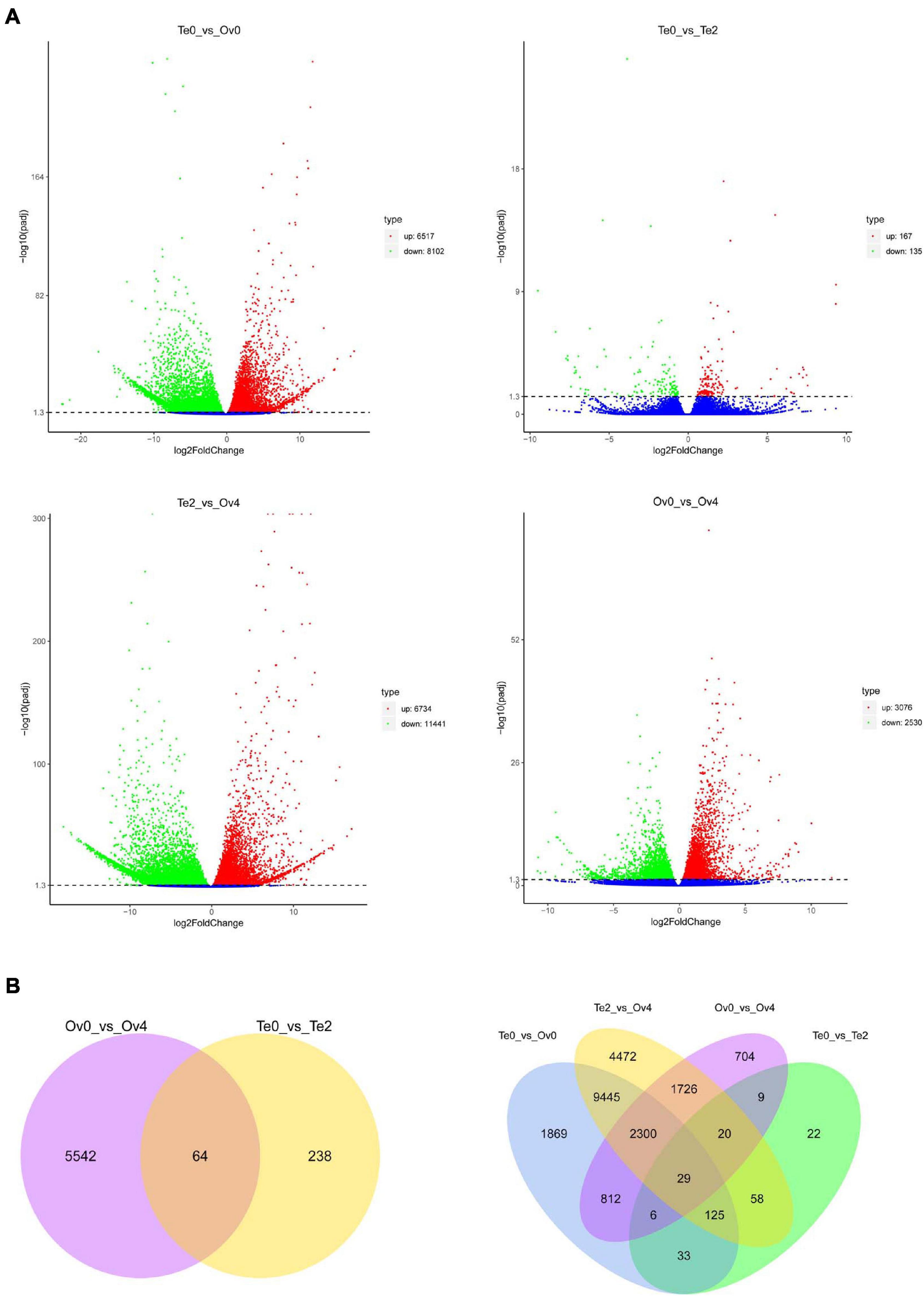
Figure 9. Comparative analyses of differentially expressed genes (DEGs) among groups. Volcano plot (A) and Venn diagram (B) of DEGs.
The GO and KEGG annotation showed that the DEGs identified from Ov0 vs. Ov4 were significantly enriched in “ribosome” and “ion binding” (Figure 10). The GO and KEGG annotation analysis for the DEGs identified from Te0 vs. Te2 indicated that the physiological activities related with ferrous ion involving “heme metabolic process,” “heme biosynthetic process,” and “ferrochelatase activity” were inhibited, while the “cytokine-cytokine receptor interaction” and “cytokine receptors” were enhanced during the testis development and maturation (Figure 10). In addition, for Ov0 vs. Ov4, a total of 65 DEGs were enriched in the KEGG pathway “Spliceosome.”
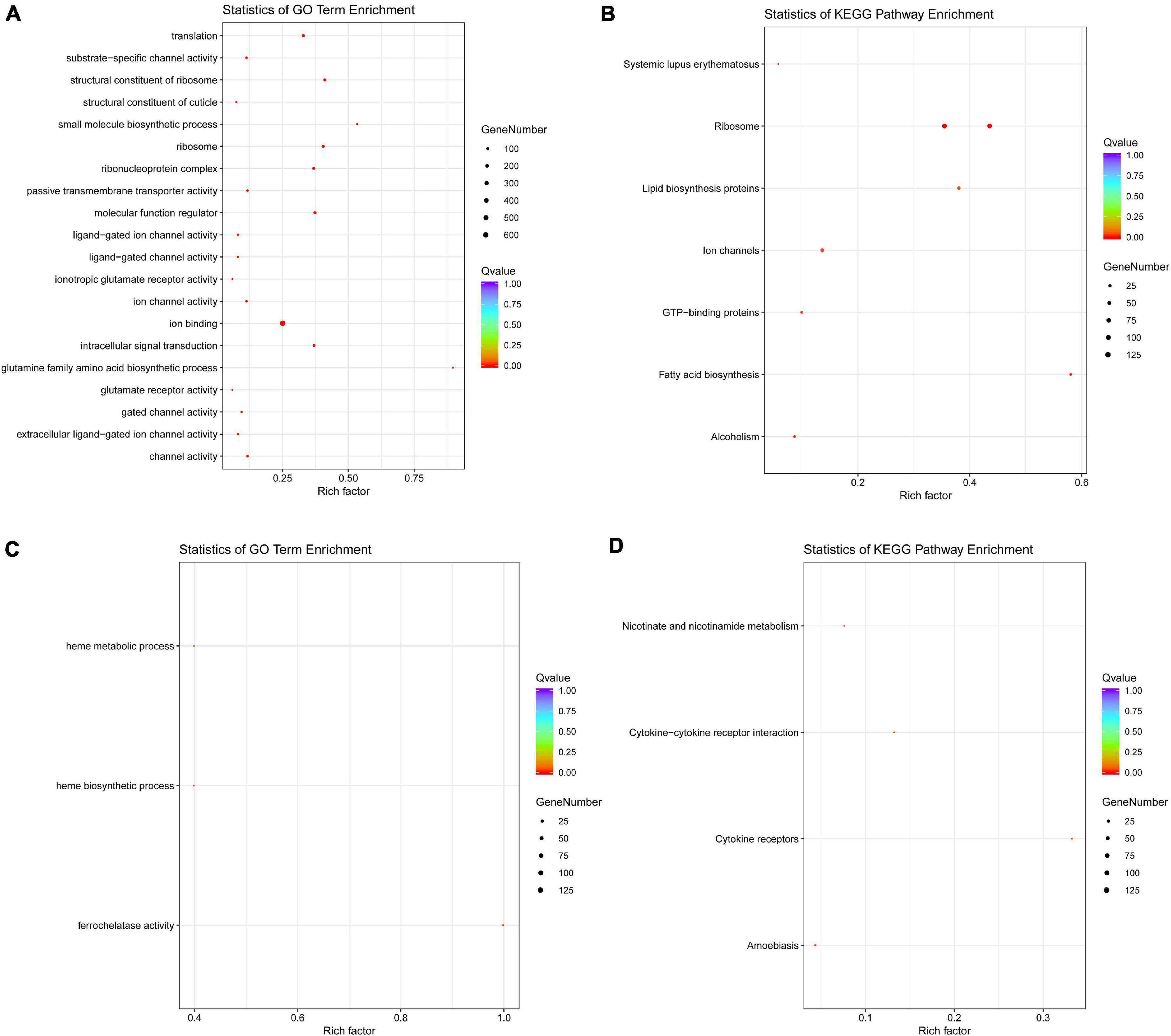
Figure 10. The GO and KEGG annotation of DEGs. The GO (A) and KEGG (B) annotation of DEGs from Ov0 vs. Ov4, and the GO (C) and KEGG (D) annotation of DEGs from Te0 vs. Te2.
Combined with the published literature and the gene annotation in the KEGG term “oocyte meiosis,” the several genes contributing to sex determination and gonad development were identified (Figure 11). In the present study, a large number of unique isoform sequences that annotated to SXL were found, and the numbers of splice isoforms generated by AS were predicted from them, which showed the complexity of post-transcriptional processing events for SXL (Supplementary Table 6). In our data, variant isoforms (dmrt1, DMRT3, and dmrta2) with the annotated result of dmrt can be found and identified, in which the dmrta2 with AS events were expressed only in the testis. Additionally, qPCR indicated that the dmrt1 and dmrt3 genes were higher expressed in testis development with sharp fluctuation (Figure 11). Furthermore, several genes contributed to oocyte meiosis such as cell division cycle 20 (CDC20), cell division cycle (CDC23), serine/threonine-protein kinase MOS-like (mox), structural maintenance of chromosomes protein 1A (Smc1a), structural maintenance of chromosomes protein 1A (Smc3), separin (Espl1), and F-box only protein 43 (FBXO43) can be found in the top 5% of the genes with the highest frequency of AS events. Also, genes annotated to cell division cycle-associated protein 3-like (cdca3), cell division control protein 45 homolog (CDC45), and cyclin-dependent kinase 14-like (CDC14) could be identified during polyadenylation site analysis with variant transcripts. From DEG cluster, as defined in our study, mitotic interactor and substrate of PLK1-like (PLK1) were found.
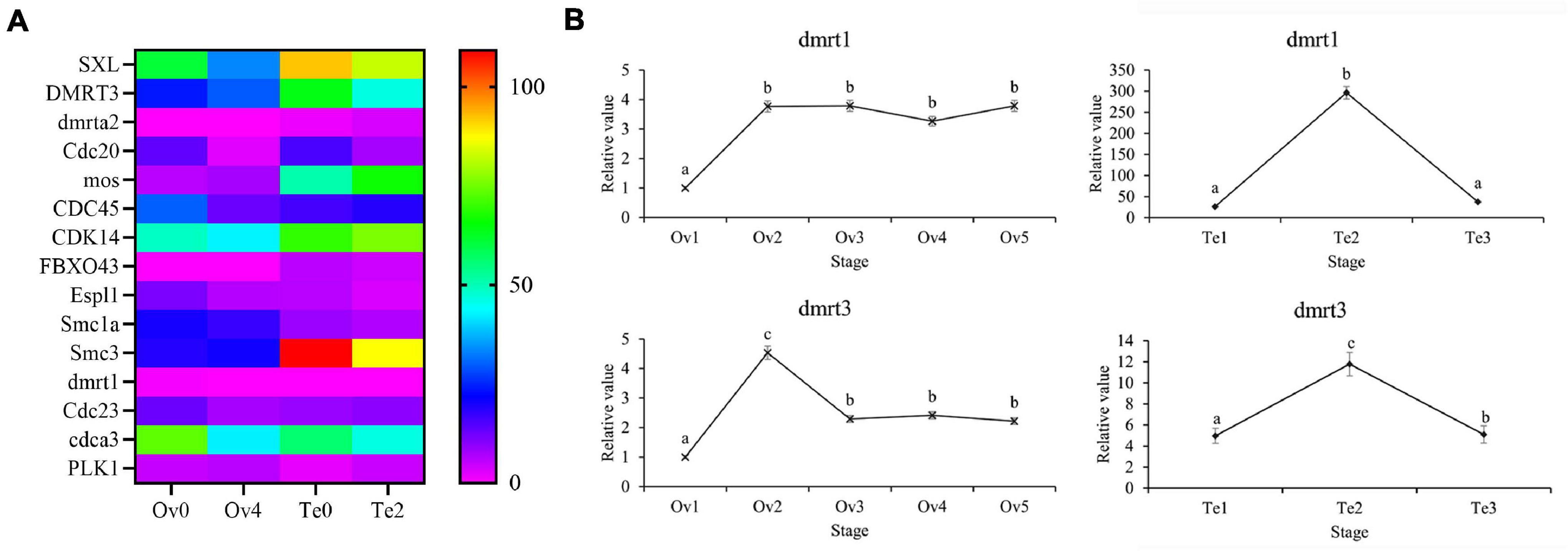
Figure 11. The DEGs associated with sex determination and gonad development in the transcriptome. SXL, sex-lethal homolog; DMRT3, doublesex and mab-3-related transcription factor 3; dmrta2, doublesex- and mab-3-related transcription factor A2; Cdc20, cell division cycle protein 20 homolog; mos, serine/threonine-protein kinase mos; CDC45, cell division control protein 45 homolog; CDK14, cyclin-dependent kinase 14; FBXO43, F-box only protein 43; Espl1, separin; Smc1a, structural maintenance of chromosomes protein 1A; Smc3, structural maintenance of chromosomes protein 3; dmrt1, doublesex- and mab-3-related transcription factor 1; Cdc23, cell division cycle protein 23; cdca3, cell division cycle-associated protein 3-like; PLK1, mitotic interactor and substrate of PLK1-like isoform X1-like. (A) Heatmap of DEGs, (B) The expression of DEGs in the different stages of the gonad. Different letters represent significant differences (p < 0.05).
For validation, qPCR was performed on 10 randomly selected sequences. Their gene relative expression, depicted in Figure 12, shows the similarity of the expression profiles by qPCR to those by bioinformatic analysis.
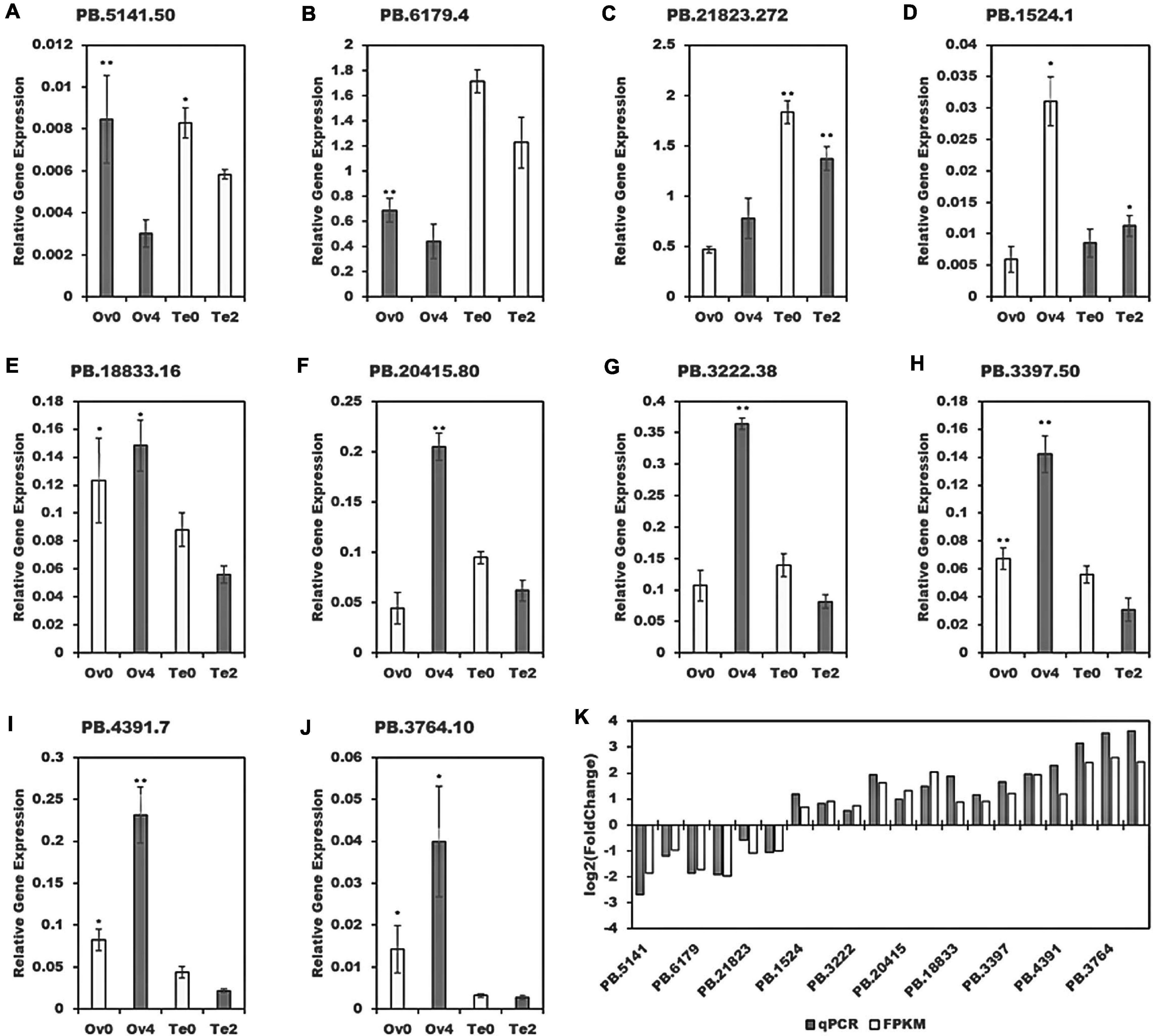
Figure 12. Validation of 10 randomly selected sequences. *p < 0.05, **p < 0.01. (K) The qPCR results were achieved by log2 (Fold Change) and bioinformatic results by that based on FPKM. (A–J) The relative expression of 10 randomly selected sequences.
Although SMRT sequencing has been widely used to generate full-length transcriptome in crustaceans (Zeng et al., 2018; Lin et al., 2019; Wan et al., 2019), little full-length transcriptome resources are available for gonads of mud crab. In this study, unique isoform sequences were obtained from the gonad transcriptome by using single molecular long-read sequencing with the PacBio Sequel platform. The high annotation rate of transcripts indicated several advantages of SMRT sequencing over RNA-Seq (Zeng et al., 2018; Wan et al., 2019). Additionally, we identified fusion transcripts based on alignment to gene model annotation; prediction of coding potential is needed to be conducted, and the function of fusion transcripts should be analyzed in a further study. In our findings, a total of 22,313 lncRNAs were characterized. Interestingly, the number of lncRNAs was similar to the recent report of full-length transcriptome sequencing for S. paramamosain (Wan et al., 2019), but less remarkably than previous research on lncRNAs from the gonad transcriptome using RNA-Seq (Yang et al., 2018).
Alternative splicing, contributing to the high level of mRNA complexity, plays important roles in the development, differentiation, or other physiological processes in a variety of organisms (Kalsotra and Cooper, 2011). Full-length sequences with alternative mRNA event identification set the stage for detailed research on the post-transcriptional mechanism involved in gonadal development for S. paramamosain. As the saturation curves show, the number of isoforms is still increasing with the rising number of sequenced reads, which indicates that alternative post-transcriptional events generate various isoforms leading to higher transcriptome diversity of gonads (Wang et al., 2016). Behind such a huge number of AS events, S. paramamosain transcriptome also shows extreme diversity during AS event identification. The GO annotation analysis demonstrated that AS regulates probably the developmental initiation and maturation of the gonad by mediating the post-translational protein modification or folding. The result was consistent with the previous study: AS promotes dynamic remodeling of the transcriptome to promote physiological changes by binding to proteins or a motif (Kalsotra and Cooper, 2011). The KEGG annotation suggested that most alternative spliced isoforms in gonadal function affect the regulation of gametogenesis, especially in female crabs. These results were confirmed by the founding in which AS in the testes may be involved in the control of the mitotic and meiotic cell cycles in spermatocytes (Venables, 2002). Consistent with the previous study (Chen et al., 2017), the results of mRNA-processing events indicated that genes undergoing AS events were remarkably more than those with APA. Nevertheless, recent discoveries have revealed that APA is a widespread phenomenon, which contributes to the complexity of the transcriptome by generating isoforms that differ either in their CD, thereby potentially regulating the spermatogenesis (Liu et al., 2007; Giammartino et al., 2011). In the present study, the UGUACA sequence, which is frequent in testis-specific mRNAs, was found in addition to the canonical AAUAAA sequence (McMahon et al., 2006). This finding verified the importance of APA events in the regulation of gametogenesis as well.
In the present study, SMRT-seq data also yielded many novel findings including the uncovering of molecular markers (169,559 SSRs and 25,451 SNPs) and identification of over 213,800 novel genes that were not previously annotated. Among these novel genes, many are putative long non-coding transcripts. These data are useful for the development of polymorphic genetic markers in S. paramamosain and other crab species.
The regulatory mechanisms involved in sex determination, gametogenesis, and gonadal differentiation and maturation of the crustacean were modulated by multiple genes and pathways (Zhang et al., 2018). Combination with Illumina RNA-seq data, the DEGs associated with sex differential and gonad maturation among different tissues and developmental stages were identified and analyzed in the present study. DEGs between adults and juveniles of the same sex were less than those between males and females, while the number of DEGs (Ov0 vs. Ov4) functioning in the maturation of the ovary was more than that of the testis. This phenomenon can be explained by the reality that ovary development goes through more developmental stages, which is more complicated than the testis (Mhd et al., 2018; Wu et al., 2019a). The functional classifications in GO and KEGG of DEGs between testis (Te0 vs. Te2) and ovary (Ov0 vs. Ov4) maturation demonstrated the differences in the gonadal developmental regulatory pathways. In the female mud crab, the DEGs were enriched in the KEGG pathway “ribosome,” “ion binding,” and “spliceosome,” which are in conformity with the previous study (Wahl et al., 2009), indicating that genes that function in catalyzing pre-mRNA splicing were found in the present dataset. This result supports the role of AS in the regulatory network of gonadal development in female crabs (Deloffre et al., 2009). In the male, the upregulated DEGs were enriched significantly in “cytokine-cytokine receptor interaction” and “cytokine receptors,” which suggested that the cytokines and their pathways were implicated in the gonad axis, especially in the testis development (Stratone et al., 2001). The inhibition of DEGs enriched in “heme metabolic process,” “heme biosynthetic process,” and “ferrochelatase activity” may be due to the increased requirement of male crabs of non-heme iron protein to synthesize steroid hormones in the process of testis development (Ford, 1969).
Identification of DEGs between sexes and during developmental stages helps to uncover the molecular mechanism underlying sexual differentiation and gonadal development. Contrary to the research of the well-studied model species Drosophila melanogaster (Cline et al., 2010), Sxl (sex-lethal) controlling sex determination in early embryogenesis and resulting in feminization was higher expressed in the testis, which provided insights into a WZ/ZZ sex determination system in mud crabs S. paramamosain (Shi et al., 2018). In the female drosophila, Sxl protein regulates the AS events of the downstream genes tra and dsx, which are key to maintain sex differential pathway (Salz and Erickson, 2010). The same as previous transcriptome sequencing for crabs S. paramamosain (Yang et al., 2018) and E. sinensis (Liu et al., 2015), tra-2, which interacts with tra to regulate the downstream gene dmrt (same family of dsx) was identified. In both male and female mud crab S. paramamosain, there may be some specific spliceosome or RNA-binding proteins for dmrt, resulting in sex-specific isoforms, which are similar to the published articles (Zheng et al., 2019). The genes contributing to oocyte meiosis with a high frequency of AS and APA events were screened in the present study as well, such as mos, Cdc23, FBXO43, Espl1, Smc1a, and Smc3. It implies that the regulatory mechanism of germ cell maturation relies on AS events, which are extensively used to regulate gene expression (Venables, 2002; Wolgemuth et al., 2002).
As previously discussed, germ cell development and maturation are controlled by cell cycle regulators in a wide variety of animals (Wolgemuth et al., 2002). Consistent with this, the DEGs (Cdc20, CDC45, and CDK14) related to the cell cycle were identified in the present study, which meets the need for faster-proliferating cells to produce more proteins (Giammartino et al., 2011). Some studies suggested that Polo-like kinase 1 (PLK1) may play an essential role in protein phosphorylation of CDC25C, which is required for activating of the Cyclin B–CDC2 complex (Qian et al., 2001; Toyoshima et al., 2002). The variant transcripts of PLK1-like generated by AS were more abundant in adult female mud crabs, which demonstrated that target genes of PLK1 may have a sexually dimorphic expression pattern as well. Alternative post-transcriptional events were identified on these genes to show their potential functions on the gonadal development and maturation in crustaceans. It has been reported that isoforms undergoing AS and APA might contribute to cell cycle and transition to meiosis in both vertebrates and invertebrates (Liu et al., 2015, 2017). However, little information is available in crustaceans, and further studies are required to confirm whether it is common in crustaceans or not.
In summary, the present study demonstrated the long-read sequencing corrected by short-read sequencing for cataloging and quantifying gonadal transcripts in mud crabs. We reported that the large-scale comprehensive profiling of full-length splice isoforms and novel genes, lncRNA, molecular marker (SSR and SNP), alternative mRNA-processing events (AS and APA), and DEGs act on gonadal development and maturation by full-length transcriptome sequencing. These findings not only improve the understanding of the molecular mechanism underlying sex determination and gonadal differentiation of the mud crab but also revealed important rules and generated novel resource and information with positive implications for the monosexual crab-breeding techniques.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA667583.
This animal study was reviewed and approved by the Institutional Animal Care and Use Ethics Committee of Shantou University.
WC participated in the experimental design, investigations, data analyses, interpretation, and drafted the manuscript. QY participated in the experimental design and data analysis. YiZ and HF participated in the investigations. AF participated in language editing. HZ, SL, YuZ, and MI participated in the manuscript modification. HM participated in the experimental design, and provided all materials and reagents. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.658091/full#supplementary-material
Supplementary Figure 1 | Identification of fusion transcripts.
Supplementary Figure 2 | Saturation curve of full-length sequencing for gonads of S. paramamosain. The cyan line represents the number of isoforms and the red line represents the number of genes.
Supplementary Figure 3 | Principal component analysis of DGEs.
Supplementary Table 1 | Primers for quantitative real-time PCR (qPCR), RT-PCR, and nest-PCR.
Supplementary Table 2 | Summary of ROIs.
Supplementary Table 3 | Summary of the polished consensus sequence.
Supplementary Table 4 | Summary of match annotation result.
Supplementary Table 5 | CDS types classification of prediction using ANGEL.
Supplementary Table 6 | The number of gene transcripts.
Abdel-Ghany, S. E., Hamilton, M., Jacobi, J. L., Ngam, P., Devitt, N., Schilkey, F., et al. (2016). A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 7:11706. doi: 10.1038/ncomms11706
Arefeen, A., Liu, J., Xiao, X., and Jiang, T. (2018). TAPAS: tool for alternative polyadenylation site analysis. Bioinformatics 34, 2521–2529. doi: 10.1093/bioinformatics/bty110
Chen, S. Y., Deng, F., Jia, X., Li, C., and Lai, S. J. (2017). A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep. 7:7648. doi: 10.1038/s41598-017-08138-z
Cline, T. W., Dorsett, M., Sun, S., Harrison, M. M., Dines, J., Sefton, L., et al. (2010). Evolution of the Drosophila feminizing switch gene Sex-lethal. Genetics 186, 1321–1336. doi: 10.1534/genetics.110.121202
Deloffre, L. A. M., Martins, R. S. T., Mylonas, C. C., and Canario, A. V. M. (2009). Alternative transcripts of DMRT1 in the european sea bass: expression during gonadal differentiation. Aquaculture 293, 89–99. doi: 10.1016/j.aquaculture.2009.03.048
Deng, Y., Li, J. Q., Wu, S. F., Zhu, Y. P., Chen, Y. W., and He, F. C. (2006). Integrated nr database in protein annotation system and its localization. Compu. Eng. 32, 71–74.
Ford, H. C. (1969). Biosynthesis of Steroid Hormones in Human Endocrine Tissue and in the Rat Testis. Vancouver: University of British Columbia, doi: 10.14288/1.0102216
Giammartino, D. C. D., Nishida, K., and Manley, J. L. (2011). Mechanisms and consequences of alternative polyadenylation. Mol. Cell 43, 853–866. doi: 10.1016/j.molcel.2011.08.017
Gupta, I., Collier, P. G., Haase, B., Mahfouz, A., Joglekar, A., Floyd, T., et al. (2018). Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 36, 1197–1202. doi: 10.1038/nbt.4259
Islam, M. S., Kodama, K., and Kurokora, H. (2010). Ovarian development of the mud crab Scylla paramamosain in a tropical mangrove swamps, Thailand. J. Sci. Res. 2, 380–389. doi: 10.3329/jsr.v2i2.3543
Kalsotra, A., and Cooper, T. A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729. doi: 10.1038/nrg3052
Kong, L., Zhang, Y., Ye, Z. Q., Liu, X. Q., Zhao, S. Q., Wei, L. P., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Li, A. M., Zhang, J. Y., and Zhou, Z. Y. (2014). PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 15:311. doi: 10.1186/1471-2105-15-311
Li, Y., Fang, C., Fu, Y., Hu, A., Zou, C., Li, X., et al. (2018). A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 25, 421–437. doi: 10.1093/dnares/dsy014
Lin, J. L., Shi, X., Fang, S. B., Zhang, Y., You, C. H., Ma, H. Y., et al. (2019). Comparative transcriptome analysis combining SMRT and NGS sequencing provides novel insights into sex differentiation and development in mud crab (Scylla paramamosain). Aquaculture 513:734447. doi: 10.1016/j.aquaculture.2019.734447
Liu, D., Brockman, J. M., Dass, B., Hutchins, L. N., Singh, P., McCarrey, J. R., et al. (2007). Systematic variation in mRNA 3’-processing signals during mouse spermatogenesis. Nucleic Acids Res. 35, 234–246. doi: 10.1093/nar/gkl919
Liu, W., Wang, F., Xu, Q., Shi, J., Zhang, X., Lu, X., et al. (2017). BCAS2 is involved in alternative mRNA splicing in spermatogonia and the transition to meiosis. Nat. Commun. 8:14182. doi: 10.1038/ncomms14182
Liu, Y., Hui, M., Cui, Z., Luo, D., Song, C., Li, Y., et al. (2015). Comparative transcriptome analysis reveals sex-biased gene expression in juvenile Chinese mitten crab Eriocheir sinensis. PLoS One 10:e0133068. doi: 10.1371/journal.pone.0133068
Ma, H. Y., Ma, C. Y., Ma, L. B., Xu, Z., Feng, N. N., and Qiao, Z. G. (2013). Correlation of growth-related traits and their effects on body weight of the mud crab (Scylla paramamosain). Genet. Mol. Res. 12, 4127–4136. doi: 10.4238/2013.October.1.3
McMahon, K. W., Hirsch, B. A., and MacDonald, C. C. (2006). Differences in polyadenylation site choice between somatic and male germ cells. BMC Mol. Biol. 7:35. doi: 10.1186/1471-2199-7-35
Mhd, I., Ghazali, A., Nahar, S. F., Wee, W., and Abol-Munafi, A. B. (2018). Testis maturation stages of mud crab (Scylla olivacea) broodstock on different diets. Sains Malays 47, 427–432. doi: 10.17576/jsm-2018-4703-01
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Qian, Y. W., Erikson, E., Taieb, F. E., and Maller, J. L. (2001). The polo-like kinase Plx1 is required for activation of the phosphatase Cdc25C and cyclin B-Cdc2 in Xenopus oocytes. Mol. Biol. cell 12, 1791–1799. doi: 10.1091/mbc.12.6.1791
Salz, H. K., and Erickson, J. W. (2010). Sex determination in Drosophila: the view from the top. Fly 4, 60–70. doi: 10.4161/fly.4.1.11277
Shen, Y., and Lai, Q. (1994). Present status of mangrove crab (Scylla serrata (Forskal)) culture in China. Naga ICLARM Q. 17, 28–29.
Shi, X., Lu, J., Wu, Q., Waiho, K., Aweya, J. J., Fazhan, H., et al. (2019). Comparative analysis of growth performance between female and male mud crab Scylla paramamosain crablets: evidences from a four-month successive growth experiment. Aquaculture 505, 351–362.
Shi, X., Waiho, K., Li, X., Ikhwanuddin, M., Miao, G., Lin, F., et al. (2018). Female-specific SNP markers provide insights into a WZ/ZZ sex determination system for mud crabs Scylla paramamosain, S. tranquebarica and S. serrata with a rapid method for genetic sex identification. BMC Genomics 19:981. doi: 10.1186/s12864-018-5380-8
Shimizu, K., Adachi, J., and Muraoka, Y. (2006). ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinf. Comput. Biol. 04, 649–664. doi: 10.1142/S0219720006002260
Stratone, A., Stratone, C., Chiruţă, R., and Topoliceanu, F. (2001). Normal and pathologic implication of cytokines. Rev. Med. Chir. Soc. Med. Nat. Iasi. 105, 657–661.
Tang, T. T., Ran, X. Q., Mao, N., Zhang, F. P., Xi, N., Ruan, Y. Q., et al. (2018). Analysis of alternative splicing events by RNA sequencing in the ovaries of Xiang pig at estrous and diestrous. Theriogenology 119, 60–68. doi: 10.1016/j.theriogenology.2018.06.022
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Thomas, S., Underwood, J. G., Tseng, E., Holloway, A. K., and on behalf of the Bench To Basinet CvDC Informatics Subcommittee (2014). Long-read sequencing of chicken transcripts and identification of new transcript isoforms. PLoS One 9:e94650. doi: 10.1371/journal.pone.0094650
Tian, B., Hu, J., Zhang, H., and Lutz, C. S. (2005). A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201–212. doi: 10.1093/nar/gki158
Tilgner, H., Jahanbani, F., Blauwkamp, T., Moshref, A., Jaeger, E., Chen, F., et al. (2015). Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat. Biotechnol. 33, 736–742. doi: 10.1038/nbt.3242
Toyoshima, M. F., Taniguchi, E., and Nishida, E. (2002). Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 3, 341–348. doi: 10.1093/embo-reports/kvf069
Venables, J. P. (2002). Alternative splicing in the testes. Curr. Opin. Genet. Dev. 12, 615–619. doi: 10.1016/S0959-437X(02)00347-7
Wahl, M. C., Will, C. L., and Lührmann, R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. doi: 10.1016/j.cell.2009.02.009
Waiho, K., Fazhan, H., Zhang, Y., Li, S., Zhang, Y., Zheng, H., et al. (2019). Comparative profiling of ovarian and testicular piRNAs in the mud crab Scylla paramamosain. Genomics 112, 323–331. doi: 10.1016/j.ygeno.2019.02.012
Wan, H., Jia, X., Zou, P., Zhang, Z., and Wang, Y. (2019). The Single-molecule long-read sequencing of Scylla paramamosain. Sci. Rep. 9:12401. doi: 10.1038/s41598-019-48824-8
Wang, B., Tseng, E., Regulski, M., Clark, T. A., Hon, T., Jiao, Y., et al. (2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7:11708. doi: 10.1038/ncomms11708
Wang, T., Wang, H., Cai, D., Gao, Y., Zhang, H., Wang, Y., et al. (2017). Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 91, 684–699. doi: 10.1111/tpj.13597
Wang, M., Wang, P., Liang, F., Ye, Z., Li, J., Shen, C., et al. (2017). A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 217, 163–178. doi: 10.1111/nph.14762
Wolgemuth, D. J., Laurion, E., and Lele, K. M. (2002). Regulation of the mitotic and meiotic cell cycles in the male germ line. Recent Prog. Horm. Res. 57, 75–101. doi: 10.1210/rp.57.1.75
Wu, Q., Khor, W., Huang, Z., Li, S., Zheng, H., Zhang, Y., et al. (2019a). Growth performance and biochemical composition dynamics of ovary, hepatopancreas and muscle tissues at different ovarian maturation stages of female mud crab. Scylla paramamosain. Aquaculture 515:734560. doi: 10.1016/j.aquaculture.2019.734560
Wu, Q., Shi, X., Fang, S., Xie, Z., Guan, M., Li, S., et al. (2019b). Different biochemical composition and nutritional value attribute to salinity and rearing period in male and female mud crab Scylla paramamosain. Aquaculture 513:734417. doi: 10.1016/j.aquaculture.2019.734417
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Yang, X., Ikhwanuddin, M., Li, X., Lin, F., Wu, Q., Zhang, Y., et al. (2018). Comparative transcriptome analysis provides insights into differentially expressed genes and long non-coding RNAs between ovary and testis of the mud crab (Scylla paramamosain). Mar. Biotechnol. 20, 20–34. doi: 10.1007/s10126-017-9784-2
Zeng, D., Chen, X., Peng, J., Yang, C., Peng, M., Xie, D., et al. (2018). Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 8:16920. doi: 10.1038/s41598-018-35066-3
Zhang, S., Wang, C., Yan, S., Fu, A., Luan, X., Li, Y., et al. (2017). isoform evolution in primates through independent combination of alternative RNA processing events. Mol. Biol. Evol. 34, 2453–2468. doi: 10.1093/molbev/msx212
Zhang, Y., Miao, G., Fazhan, H., Waiho, K., Zheng, H., Li, S., et al. (2018). Transcriptome-seq provides insights into sex-preference pattern of gene expression between testis and ovary of the crucifix crab (Charybdis feriatus). Physiol. Genomics 50, 393–405. doi: 10.1152/physiolgenomics.00016.2018
Keywords: Scylla paramamosain, full-length transcriptome, gonad, alternative splicing, alternative polyadenylation, differentially expressed genes
Citation: Cui W, Yang Q, Zhang Y, Farhadi A, Fang H, Zheng H, Li S, Zhang Y, Ikhwanuddin M and Ma H (2021) Integrative Transcriptome Sequencing Reveals the Molecular Difference of Maturation Process of Ovary and Testis in Mud Crab Scylla paramamosain. Front. Mar. Sci. 8:658091. doi: 10.3389/fmars.2021.658091
Received: 25 January 2021; Accepted: 05 March 2021;
Published: 30 March 2021.
Edited by:
Haihui Ye, Jimei University, ChinaCopyright © 2021 Cui, Yang, Zhang, Farhadi, Fang, Zheng, Li, Zhang, Ikhwanuddin and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyu Ma, bWFoeUBzdHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.