 Maximilian H. Ganser
Maximilian H. Ganser Dominik Forster
Dominik Forster Weiwei Liu
Weiwei Liu Xiaofeng Lin
Xiaofeng Lin Thorsten Stoeck
Thorsten Stoeck Sabine Agatha
Sabine Agatha
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 13 April 2021
Sec. Marine Evolutionary Biology, Biogeography and Species Diversity
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.643822
This article is part of the Research TopicCiliate Biodiversity and Evolution from Morphological, Genomic and Epigenomic ViewsView all 15 articles
Unraveling geographic distribution patterns of planktonic protists is a central goal in marine microbial ecology. Using a novel combination of recently developed phylogenetic and network analyses on a V4 18S rDNA metabarcoding dataset, we here analyzed the genetic diversity of marine planktonic ciliate communities in Chinese and European coastal waters. Thereby, our approach provided an unprecedented perspective on geographic patterns inferred from ciliate genetic diversity and accomplished a very fine resolution down to single nucleotides within operational taxonomic units (OTUs). While most OTUs (87%) exclusively contained sequences of either Chinese or European origin, those OTUs detected in both regions comprised the vast majority of reads (84%). Phylogenetic analyses of OTUs belonging to the same taxon revealed genetically distinct clades that were geographically restricted to either Chinese or European coastal waters. The detection of signature nucleotides emphasized this genetic distinction of Chinese and European clades. Second-level clustering of OTUs and reference sequences in two selected taxa (the oligotrichid Spirotontonia and the tintinnid Tintinnidium) revealed the presence of several potentially new species or ones lacking genetic reference data. Geographic patterns were also discovered by network analyses within 700 widespread and abundant OTUs; in 77 of these OTUs, European and Chinese sequences formed significantly assortative groups. These assortative groupings indicated a higher genetic similarity among sequences from the same region than between sequences from different regions. Our results demonstrate that detailed analyses of metabarcoding data down to single nucleotide differences expand our perception of geographical distribution patterns and provide insights into historic and ongoing effective dispersal in protists. The congruent discovery of geographic patterns at different levels of resolution (between and within OTUs) suggests that cosmopolitan distribution in marine planktonic ciliates is less common than previously postulated.
Marine planktonic ciliate communities contribute fundamentally to marine food webs by linking different trophic levels (Sherr and Sherr, 1988). This role is especially important in coastal environments that generally harbor a rich biodiversity and additionally host the breeding grounds of many multicellular organisms whose juvenile stages feed on ciliates (Stoecker and Capuzzo, 1990). Despite the importance of ciliates, our understanding of their distribution and speciation patterns is incomplete. Both kinds of patterns are strongly affected by past and ongoing effective dispersal through transport in active or passive state, which includes a subsequent successful establishment of individual organisms (Weisse, 2008). Likewise, dispersal limitations, species sorting caused by the absence of adequate ecological niches, and adaptations to local environmental conditions define a species’ geographic distribution.
Communities of marine planktonic ciliates are usually dominated by the oligotrichids and the choreotrichids comprising aloricate (naked) species and the house-forming tintinnids (Pierce and Turner, 1992; McManus and Santoferrara, 2013). Both are assigned to the Oligotrichea Bütschli, 1887 according to morphologic, molecular, and ontogenetic features (Adl et al., 2019). The prevalence of data on the diversity and distribution of the about 1,000 tintinnid morphospecies in the literature (e.g., Merkle, 1909; Laackmann, 1910; Hofker, 1931; Hada, 1938; Zeitzschel, 1966, 1969) is in sharp contrast to the comparatively few taxonomic and ecological papers on the about 140 oligotrichids and 60 aloricate choreotrichids. Moreover, geographic distribution patterns inferred from records of morphospecies are often influenced by investigation methods (e.g., sampling strategy, fixation blurring species-specific features) as well as taxonomic uncertainties caused by phenotypic plasticity and cryptic species (Agatha, 2011). In the latter cases, morphological divergences or convergences are incompletely linked to genetic relationships.
To circumvent the problems regarding species limitations in tintinnids, studies on their global distribution are largely restricted to genus level. Different geographic patterns emerged from the voluminous dataset: cosmopolitan, neritic, warm-temperate, boreal, austral, and tropical Pacific; yet, even in cosmopolitan genera (inhabiting the neritic and oceanic regions from the Arctic through the tropics to the Antarctic), none of its members covers the entire geographic range (Pierce and Turner, 1993). Furthermore, the composition of tintinnid communities also distinctly differs between neritic and oceanic regions (Pierce and Turner, 1993; Dolan and Pierce, 2013). In oligotrichids and aloricate choreotrichids, the records of morphospecies are widely restricted to coastal waters. Keeping the taxonomic impediments in mind, the data demonstrate a wide, possibly cosmopolitan distribution in most genera, while about one third of them seem to be geographically restricted and potentially contain few species with apparently limited distribution (Agatha, 2011).
The stepwise introduction of sequencing technologies into marine planktonic ciliate research complemented morphospecies analyses and allowed for investigating previously untouched aspects of genetic diversity. The first molecular study from coastal sites in the Northwest Atlantic on oligotrichids and choreotrichids used the V3 and V8 regions of the 18S rDNA and applied Sanger sequencing (Doherty et al., 2007). The molecular and morphological data were congruent regarding tintinnids, but revealed a much higher genetic diversity in oligotrichids, especially within the genus Strombidium Claparède & Lachmann, 1859 (Doherty et al., 2007, 2010). The comparison of molecular and morphological data on tintinnids by a subsequent study not only confirmed differences in the composition between coastal and adjacent open ocean communities, but also showed the advantages of sequencing studies for detecting less abundant species (Santoferrara et al., 2016b). Global records of 18S rDNA gene sequences also supported biogeographical patterns for tintinnid genera (Santoferrara et al., 2018).
Barcoding approaches, which combine morphological and molecular data, alleviated the limits of taxonomic resolution, and unveiled both phenotypic plasticity and (pseudo-) crypticity in oligotrichean ciliates (McManus et al., 2010; Santoferrara et al., 2015, 2017; Kim et al., 2020). The marker genes employed (e.g., 18S, ITS, and 28S rDNA gene sequences) generally enable to distinguish ciliates down to species rank (Warren et al., 2017). Ideally, a single cell is identified and documented, its marker genes are analyzed, and the sequences are deposited together with metadata in a reference database. Thereby, marker gene sequences are unambiguously linked with corresponding morphospecies (Santoferrara et al., 2016a). Reference databases that cover most of the diversity represent a valuable resource for “meta”-barcoding studies that depend on reliable taxonomic annotations of gene sequences obtained from environmental samples. Yet, species identification using reference sequences is hampered by overlapping interspecific and intraspecific sequence distances, different species divergence times, and arbitrary, a piori fixed divergence thresholds (Moritz and Cicero, 2004; Will and Rubinoff, 2004; DeSalle et al., 2005).
Identical marker gene sequences detected in independent samples revealed the occurrence of several marine planktonic ciliate species in different oceanic regions. For example, identical internal transcribed spacer sequences and 18S rDNA gene sequences of some oligotrichids were found to be distributed across the North Atlantic and adjacent sea regions or in the West Atlantic (Agatha et al., 2004; Katz et al., 2005; Doherty et al., 2010). Specimens of the tintinnid Antetintinnidium mucicola collected from the Northwest Atlantic are identical in their 18S rDNA sequences to one reference sequence from the Yellow Sea (Ganser and Agatha, 2019). However, even specimens with identical sequences in the rather conserved 18S rDNA might display genetic divergences in other, more variable marker genes (e.g., ITS, 28S, and COI), indicating distinct species (Xu et al., 2012; Santoferrara et al., 2015, 2017; Jung et al., 2018).
While the barcoding of ciliate species continues, metabarcoding studies, which employ high-throughput sequencing (HTS) technologies for rapidly investigating whole ciliate communities, are independent of taxonomic expertise. As outlined above, they make, however, use of the already existing taxonomic results from barcoding studies in publicly available databases. Despite their exclusive focus on molecular data, metabarcoding studies have considerably contributed to our current perception of geographic distribution patterns in marine planktonic ciliates, as well. The patterns were mainly analyzed via large-scale metabarcoding datasets comprising hyper-variable sequences of either the V4 or the V9 18S rDNA gene regions. Regarding ciliates, the genetic distances of the hyper-variable V4 regions are generally comparable to distances between near full-length 18S rDNA sequences, this applies, however, only to a lesser extent to the hyper-variable V9 region (Dunthorn et al., 2012). The findings of large-scale metabarcoding studies regarding planktonic ciliates in marine habitats suggest that European coastal waters exhibit a high regional genetic diversity and a strong habitat specificity based on V4 sequence data (Forster et al., 2015). By contrast, the global and regionally restricted genetic diversity of planktonic ciliate communities in the open oceans based on V9 sequence data was found to be low (Gimmler et al., 2016).
Moving from whole ciliate communities down to the level of individuals, metabarcoding also contributed to assess the intra- and interspecific genetic diversity (Forster et al., 2019). Whatever the targeted level of investigation, diversity assessments as well as the detection of geographic patterns and their underlying processes strongly depend on the molecular resolution (Hanson et al., 2012). This resolution, in turn, depends on the selected marker gene (Dunthorn et al., 2012) as well as on the approach with which metabarcoding sequences are clustered into operational taxonomic units (OTUs) (Forster et al., 2019). Traditionally, heuristic sequence clustering approaches that rely on global alignment scores (e.g., USEARCH), such as a given percentage of sequence similarity, were used to generate OTUs (Edgar, 2010). While these methods reduce the computational demand for processing a dataset (sequences are only compared with a centroid sequence), several studies have shown that OTUs generated by heuristic approaches lack accuracy and reproducibility (Sun et al., 2012; Bachy et al., 2013; Schmidt et al., 2015). Moreover, heuristically clustered OTUs are designed to assess community structure on the OTU level (e.g., α- and β-diversity), but provide little information about their internal structure and are therefore comparatively coarse in terms of genetic resolution. More recently developed hierarchical sequence clustering approaches that employ all-vs.-all pairwise sequence comparisons allow for a much more fine-grained resolution of diversity. In particular, the algorithm Swarm provides detailed information about the internal structure of its OTUs (Mahé et al., 2015). Swarm OTUs also grow from a centroid ‘seed’ sequence, but the sequences are linked to each other if their sequence alignment differs by not more than a given number of nucleotides (usually one nucleotide difference, which is likely to arise through biological processes). The internal structure of a Swarm OTU thus represents a network based on the information which sequences are linked to each other, which may be evaluated by means of graph theory (Forster et al., 2020). Evaluating the properties of the internal network structure distinctively increases the resolution of diversity down to single nucleotide differences and, therefore allows unprecedented insights into genetic diversity and geographical distribution patterns. Investigations of intraspecific genetic diversity are especially important within ciliates, which exhibit high copy numbers and sequence variation even within individual specimens (Gong et al., 2013; Wang et al., 2017; Zhao et al., 2019). An additional second-level clustering of Swarm OTUs enables an efficient differentiation of genetically divergent sequences into species-specific network sequence clusters. This strategy thus allows for a closer approximation between morphospecies and OTUs, i.e., a better estimation of the real species diversity from metabarcoding datasets (Forster et al., 2019). Owing to differences in evolutionary rates, however, a refinement of the similarity cut-off values for particular lineages is required (Wang et al., 2019; Zhao et al., 2019).
Current high-throughput sequencing technologies facilitate the rapid assessment of marine planktonic ciliate communities and thus the investigation of distribution patterns on various temporal and spatial scales. The two most common sampling strategies result in (i) a low spatial and high temporal resolution, i.e., the samples are collected several times at locally restricted sites, or (ii) a high spatial and low temporal resolution, i.e., the samples are collected usually only once at sites widely distributed over a coastal or oceanic region. The selection of a strategy depends on the aims of the study. Type 1 studies usually focused on the influence of environmental gradients on the community composition (Santoferrara et al., 2016b; Minicante et al., 2019), the distribution and composition of communities at varying depths (Grattepanche et al., 2015; Sun et al., 2019), or seasonal patterns (Piredda et al., 2017; Giner et al., 2019). However, type 2 studies investigated large-scale diversity and distribution patterns, for example, at a regional scale in European coastal waters (Logares et al., 2014) or at a global scale in the oceans (de Vargas et al., 2015; Logares et al., 2020). Usually, large-scale studies of the marine plankton either analyzed the entire community of eukaryotes or focused on certain size-fractions of protists.
Coastal waters are highly interesting environments for investigating geographic distribution patterns and their evolutionary origin in marine planktonic ciliates, particularly as data covering large spatial scales are lacking. Within these environments, assessments of ciliate diversity and distribution patterns were so far confined to whole communities, genera, morphospecies, or OTUs (Foissner et al., 2008; Doherty et al., 2010; Dolan and Pierce, 2013; Massana et al., 2015; Piredda et al., 2017). In the present study, we apply a novel combination of phylogenetic approaches (Hütter et al., 2020) and network analyses (Forster et al., 2019, 2020) to achieve an unparalleled increase of resolution down to within-OTU level for investigating geographic distribution patterns and their origin in marine planktonic ciliates. Hence, it overcomes the limitations of previous investigations by employing an increased genetic resolution and a unique sampling strategy that links a high spatiotemporal resolution (regional sampling at several sites, at different daytimes, and in different seasons) with a large geographic distance between the regions examined, namely, the Chinese and European coastal waters. The present analyses aim (i) to detect correlations between phylogenetic relationships and the geographic distribution based on metabarcoding data of the hyper-variable V4 region, (ii) to identify signature nucleotides for geographically restricted OTU clusters (Hütter et al., 2020), (iii) to determine the threshold values that reconcile secondary clusters of Swarm OTUs and morphospecies and allow estimating the diversity of taxa, and (iv) to discover geographic patterns of genetic diversity within Swarm OTUs by means of network analyses.
Following identical protocols, 80 samples were taken from Chinese and European coastal surface waters between October 2017 and November 2018, namely, one day and one night sample per season at each site (Supplementary Table 1); the high spatiotemporal sampling aimed to capture as much of the biodiversity of planktonic ciliates as possible. Our sampling sites belong to different Longhurst ecological provinces: the Northeast Atlantic Shelves, the Mediterranean Sea, and the China Seas Coastal provinces (Longhurst, 2007). The Chinese sampling (n = 32) was performed at four sites along the coast of southern China, i.e., in Aotou (coastal town), Donghai Island, Futian (mangrove wetland), and Hailing Island. The European sampling (n = 48) was conducted at the German North Sea coast, i.e., at two sites in Wilhelmshaven, and at two sites in the Baltic Sea, i.e., Warnemünde and Rostock, as well as at two sites in the Mediterranean Sea, i.e., Villefranche-sur-Mer (France) and Trieste (Italy). The spatial distances spanning the shortest sea routes along the coastlines amount to about 150–500 km between the Chinese sites, about 400–6,400 km between the European sites, and about 14,200–18,800 km between the Chinese and European sampling sites.
Surface water samples were collected with a bucket (5 L), and temperature and salinity were immediately measured. The collected water was pre-filtered through a 250 μm-mesh net to remove debris and metazoa and subsequently filtered through a 0.8 μm-pore size polyether sulfone membrane 47 mm across (Pall) in a filtration unit with a manual vacuum pump (Nalgene) until the membrane was clogged or a maximum of about 3.6 L was reached. The membrane filters were immediately placed in Cryo-Vials (Simport) and preserved with LifeGuard® buffer (Qiagen) at 0°C. In the laboratory, they were stored at −80°C until DNA extraction. Using a whole water sample, the entire ciliate community ranging from nano- to microplankton could be collected; plankton nets, which provide usually merely qualitative samples, were exclusively employed in sampling for microscopic inspection.
The total DNA from each membrane filter was extracted with the DNeasy® PowerSoil® Kit (Qiagen) with slight modifications of the manufacturer’s protocol (Supplementary Data 1). The extracted DNA of each sample was amplified and sequenced by Biomarker (Beijing, China). Amplification comprised a nested-PCR approach (Supplementary Data 1), using first ciliate-specific primers followed by eukaryotic primers targeting the hyper-variable V4 18S rDNA gene region (Lara et al., 2007; Stoeck et al., 2010). In a single run, paired-end sequencing (2 × 250 bp) of the library with the V4 sequences of all samples was conducted on an Illumina HiSeq 2500 platform. Sequence read data will be made accessible at the NCBI BioProject ID PRJNA714531 (https://www.ncbi.nlm.nih.gov/bioproject/).
The primer removal from each demultiplexed forward and reverse library was conducted in Cutadapt version 2.8 (Martin, 2011). Primers were clipped from both ends of each read, and only reads without mismatches in the primer regions were retained. Forward and reverse reads were then paired-end assembled by VSEARCH version 2.14.2 (Rognes et al., 2016) with high success rates (≥96.5% merged paired-end reads per sample; mean paired-end fragment lengths ≥344 nucleotides). Paired-end reads with ambiguous bases were discarded and identical reads were dereplicated to amplicons across all samples according to the recommendations for preparing sequence data for OTU clustering1. Our terminology follows Mahé et al. (2015) in that we refer to a single sequence obtained from raw HTS data as a ‘read,’ while an ‘amplicon’ comprises a set of identical reads obtained via dereplication. The term ‘sequence’ refers to the sequence of nucleotides of which a read or an amplicon consists.
Amplicons were used as input for OTU clustering by Swarm v. 3.0.0 (Mahé et al., 2015). Swarm is an efficient clustering algorithm for linking only those amplicons into one OTU which differ by a single nucleotide from at least one other amplicon in that OTU. Thereby, the OTUs grow iteratively from a central ‘seed’ amplicon. In each iteration, the algorithm searches for all amplicons that can be linked to the most recently added amplicon(s). If no more such amplicons can be found, the clustering process stops, the current OTU is closed, and a new OTU is initiated from another central amplicon. In our particular clustering approach, we enabled the options -d 1, -f (fastidious option), -i (output of a structure file), -j (output of all pairwise sequence connections into a file), and -s (output of a clustering statistics file). Since our goal was to achieve resolutions down to single nucleotide differences, we set the clustering threshold in Swarm accordingly (d = 1). Although not tested here, our methodological workflow allows for adjusting this threshold to the taxonomic group and marker gene under study. For ciliate metabarcoding datasets of the ribosomal V9 and V4 gene regions, a clustering threshold of one nucleotide difference has successfully been applied in previous studies (e.g., Forster et al., 2019). For other taxonomic groups and faster evolving marker genes, such as COI, though, higher clustering thresholds might be necessary for assessing the underlying genetic diversity within and between OTUs (see e.g., Bakker et al., 2019; Siegenthaler et al., 2019; Antich et al., 2020). Swarm uses all sequences in the input dataset and yields a very fine-scaled resolution of genetic diversity in a sample. The OTU networks generated by Swarm are used for downstream evaluations by means of graph theory.
Chimera detection was conducted after OTU clustering, using VSEARCH’s uchime_denovo option, an implementation of the UCHIME algorithm (Edgar et al., 2011). After removing chimeras and low-abundant OTUs (i.e., comprising less than three reads), all remaining OTUs were taxonomically annotated, using the PR2 reference database (Guillou et al., 2013) and the last common ancestor approach in VSEARCH, applying the sintax option with default parameters, which is an implementation of the SINTAX algorithm (Edgar, 2016). Those OTUs not assigned to the Ciliophora were removed from the dataset; the final ciliate-specific dataset contained 47,093 Swarm OTUs.
To detect geographic distribution patterns in marine planktonic ciliates, phylogenetic analyses were conducted. Those OTUs with an identical “taxon” annotation were grouped. “Taxon” labels derived from the PR2 reference database may represent environmental sequences, sequences of species identified to genus level, or reference sequences from known morphospecies (Guillou et al., 2013). All OTUs grouping to a “taxon” label might thus encompass several different but closely related ciliate taxa. Those groups assigned to the Oligotrichea with more than 15 OTUs in one of the two regions and a ratio of OTU numbers from China and Europe higher than 0.15 were chosen to assemble the final dataset. Next, each group was augmented by shared OTUs (i.e., OTUs consisting of amplicons/reads from both geographic regions) that matched the respective “taxon” labels. The final dataset (Supplementary Table 2, Supplementary Data 2) included 21 “taxon” groups containing in total 2,404 Chinese OTUs (51,369 reads), 2,679 European OTUs (71,085 reads), and 1,124 shared OTUs (1,076,655 reads).
The amplicon sequence of each Swarm OTU with the highest read number (representative Swarm OTU sequence) was chosen for the subsequent alignments, which were calculated for each “taxon” separately with MAFFT v. 7 (Katoh and Standley, 2013), using the globalpair command. The phylogenetic tree for each “taxon” was computed with FastTree v. 2.1.11 (Price et al., 2010) under the GTR + CAT model (Tavaré, 1986; Stamatakis, 2006). The trees were edited in FigTree v. 1.4.42. They have a polar tree layout, midpoint rooting, and increasing order of nodes. For displaying the different distribution patterns, the trees represent cladograms (Figure 4), whereas the detailed taxon trees show the original branch lengths (Supplementary Figure 1). The branches were colored according to the geographic affiliation of the respective OTU, i.e., those from China are marked red, those from Europe are blue, and the shared ones are purple.
Nucleotides characterizing certain tree branches geographically restricted to a large extent were identified with DeSignate (Hütter et al., 2020), using the web-interface3. In DeSignate, two sets of aligned sequences chosen by the user (=query and reference groups) are compared and each alignment position is ranked and categorized according to its diagnostic value. Alignment positions with a diagnostic value of 1 are categorized as binary or asymmetric if they are uniform within the query group. The nucleotides at these positions are termed “signature nucleotides” in the context of our study. At binary positions, a nucleotide base (e.g., A) is present in all sequences of the query group and another nucleotide base (e.g., G) is present in all sequences of the reference group. At asymmetric positions, the nucleotide bases in sequences of the reference group do not need to be uniform, but different from the nucleotide base in the query group (e.g., G, C, or T, but not A). These signature nucleotides thus indicate that part of the amplicon diversity of a ciliate taxon can be affiliated to a certain geographic region, namely, China or Europe. Suitable query and reference groups to test for the presence of signature nucleotides in aligned sequences were identified by eye, evaluating branching patterns and branch support values in each of the phylogenetic trees.
For two taxa in which reference sequences can be reliably linked to morphospecies, namely, the oligotrichid Spirotontonia and the tintinnid Tintinnidium, sequence similarity networks were calculated. The representative sequences of the phylogenetically analyzed Swarm OTUs affiliated to these taxa along with their closest related reference sequences of the PR2 database and sequences of known morphospecies (updated list of curated reference sequences from Santoferrara et al., 2017) were subjected to a second-level clustering by means of sequence similarity networks (SSNs). The pairwise sequence alignments by VSEARCH yielded similarity values for each “taxon” used for constructing the sequence similarity networks at different thresholds (Forster et al., 2019). The thresholds at which the morphologically distinct species separated into network sequence clusters (NSCs) were determined for elucidating the numbers of connected components that do not include reference sequences; they represent either new species or ones lacking genetic data.
The Swarm OTUs were subdivided into two categories: OTUs comprising sequence reads from only Europe or China and shared OTUs comprising sequence reads from both regions. A further subdivision of the European OTUs regarding their affiliation to the two different Longhurst ecological provinces and network analyses considering geographical distances were not performed. A case study on a test dataset of Scandinavian lakes detected no significant patterns on a regional scale (Forster et al., 2020). Therefore, we expanded the spatial scale toward two more distant geographical regions in the current study. For network analyses, only shared OTUs which comprised at least 0.0001% of all reads and included at least three amplicons from both geographic regions (China and Europe) were chosen. These thresholds were set to select those OTUs in which network analyses on the internal structure were feasible, as OTUs poor in reads and amplicons are prone to produce statistically insignificant network patterns. The resulting subset consisted of 700 OTUs.
Assortativity is a commonly used metric in graph theory (Newman, 2003). Within a particular OTU, it allows measuring preferential connections between amplicons originating from China or Europe, i.e., the assortativity is high (maximum 1) when amplicons from one region are genetically more similar among each other than to sequences from the other region. The -i and -j outputs of Swarm served as input for constructing a network which was then subjected to assortativity calculations.
For determining the statistical significance of the assortativity values each OTU network was randomized 1,000 times by shuffling the geographic attributes of the nodes (China or Europe), while the general network structure (i.e., same number of nodes and edges, same node degree) was retained. The assortativity values for the randomized networks were calculated. When the computed assortativity values of the original OTU network exceeded the 95% confidence interval of the 1,000 randomized values, amplicons from the same region were significantly more often connected to each other than expected by chance. The workflow and the bioinformatic commands used have previously been published (Forster et al., 2020) and were run in R Studio v. 3.5.1 (R Core Team, 2018), using the igraph package v. 1.2.2 (Csárdi and Nepusz, 2005).
The following results explicitly focus on the geographic patterns in OTUs from Chinese and European coastal waters representing two spatially distant regions. A detailed comparison of ciliate communities within regions and their variability through seasons and daytime will be the topic of a subsequent publication benefitting from the high spatiotemporal resolution of our sampling strategy. The ciliate-specific dataset shows that only 13% of OTUs consist of reads from both regions (shared OTUs), whereas 43% and 44% of OTUs exclusively consist of reads from China or Europe, respectively (Figure 1). On the contrary, most reads (84%) belong to shared OTUs, while the remaining reads are almost evenly divided between Chinese OTUs (7%) and European OTUs (9%). Out of the 6,227 shared OTUs, 663 OTUs (11%) consisted to at least 99% of Chinese reads and 744 OTUs (12%) consisted to at least 99% of European reads. Most OTUs (55%) were annotated to taxa assigned to the Oligotrichida, aloricate Choreotrichida, or Tintinnina, while the CONthreeP (e.g., peritrichs) were less often observed. Interestingly, 43% of all purely Chinese and European OTUs (8.4% of total reads) were annotated by the last common ancestor approach to the same taxon names, suggesting that there are considerable regional genetic deviations.
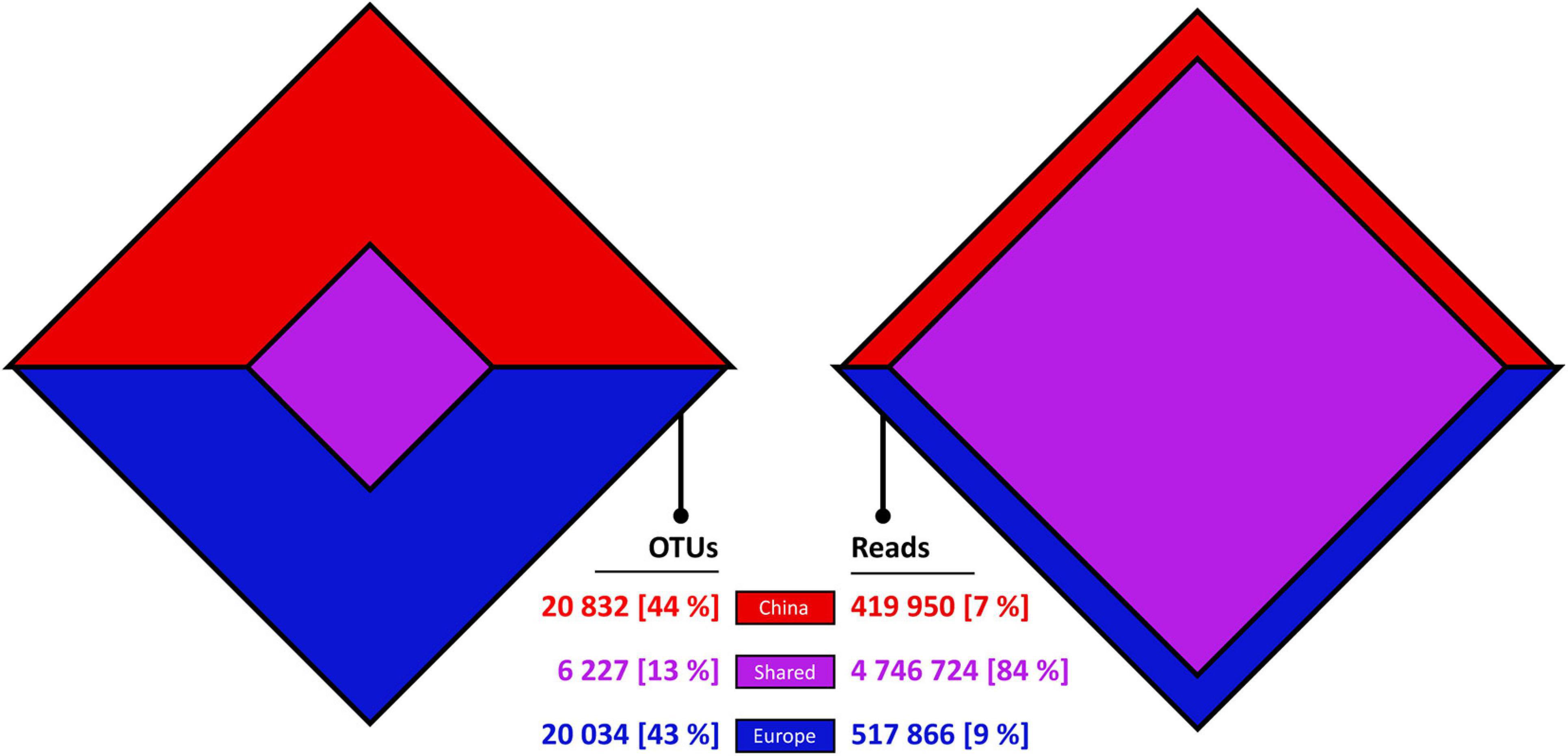
Figure 1. Venn-diamonds displaying the comparison between Chinese, European, and shared Swarm OTUs and their respective read numbers.
On closer inspection of the shared Swarm OTUs, three main patterns of geographic affiliation are distinguished for each enclosed amplicon (Figure 2): (i) the amplicon comprises reads from only one region, (ii) the vast majority of reads within the amplicon originate from one region, while only one or relatively few reads (<1%) originate from the respective other region and are interpreted as methodological artifacts (e.g., in the oligotrichid Spirotontonia sp.), and (iii) the amplicon contains a considerable number of reads from both regions, such that its occurrence in both regions is unlikely to be a methodological artifact (e.g., in the tintinnid Tintinnopsis baltica; Figure 3). The distinction of the different amplicon types was of utmost importance for the representative amplicons used in the phylogenetic analyses.
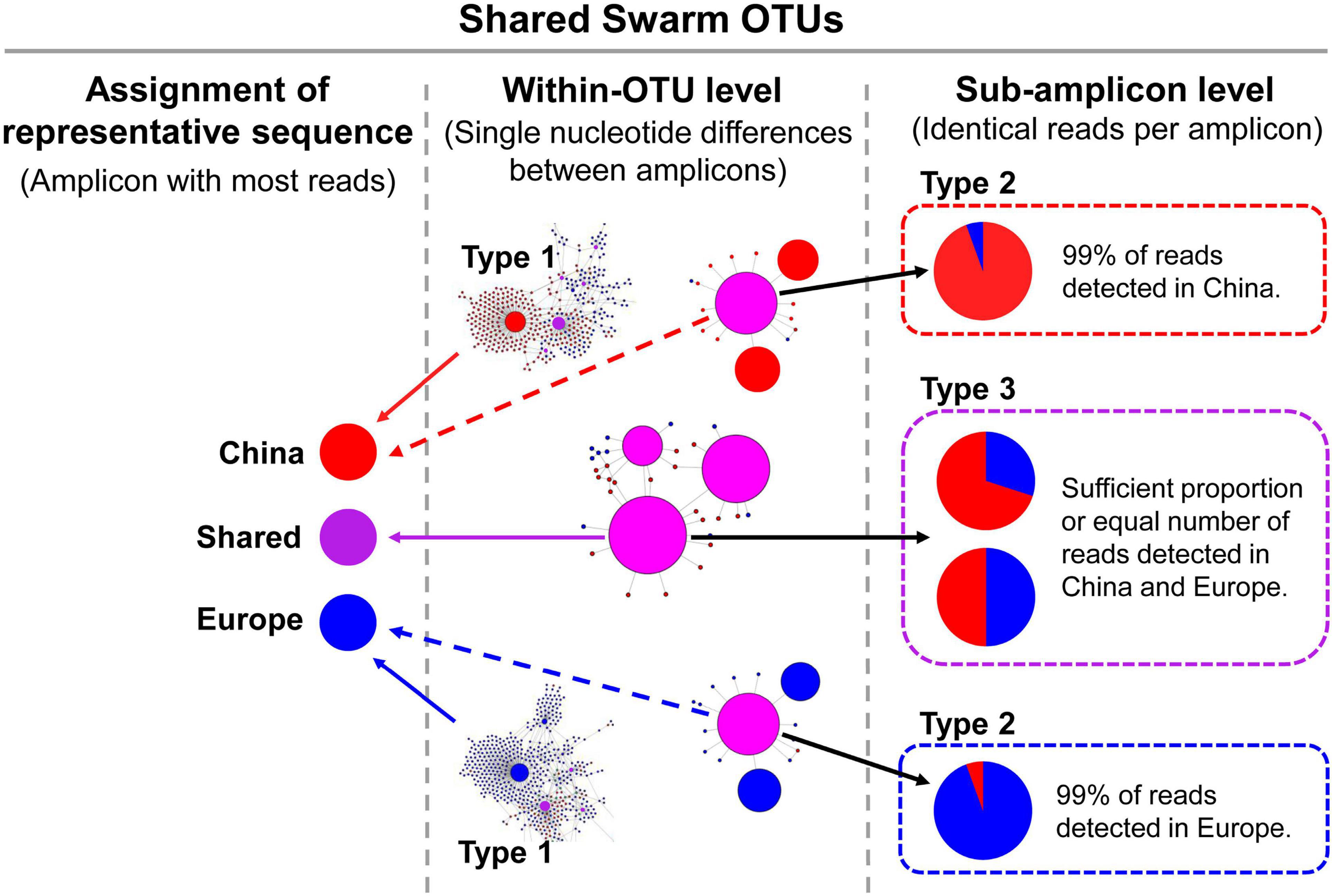
Figure 2. Types of shared Swarm OTUs. The circle sizes in the within-OTU networks correspond to the read numbers of the respective amplicons. The representative amplicons (with highest read number) of the shared OTUs might comprise reads from China and Europe in different proportions. In Type 1 of shared OTUs, the representative amplicon consists of reads only detected in China or in Europe. In Type 2 of shared OTUs, only a minute proportion of reads was detected in the respective other region and is attributed to methodological artifacts (sample bleeding; Mitra et al., 2015). Accordingly, Type 2 of shared OTUs is interpreted as purely consisting of reads from the dominating region. In Type 3 of shared OTUs, similar read numbers from China and Europe constitute the representative amplicon.
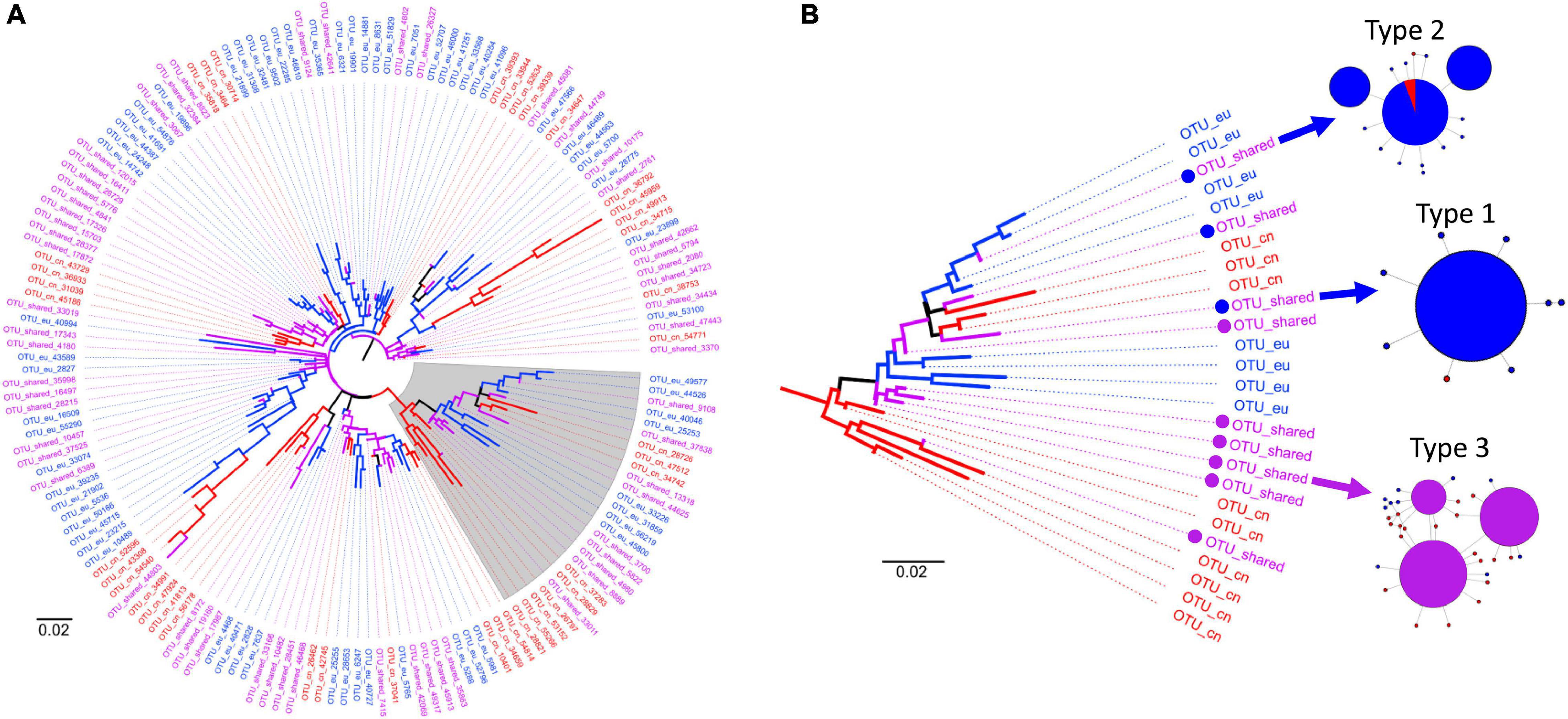
Figure 3. Phylogenetic tree of the tintinnid Tintinnopsis baltica (A) and highlighted sub-clade (B) displaying various branching patterns with different types of shared Swarm OTUs. In Type 1, the representative amplicon of the shared OTUs was exclusively detected in one region but is associated to one or more amplicons from the respective other region. In Type 2, the representative amplicon consists of many reads from one region and only one or very few reads from the respective other region, which are interpreted as methodological artifacts (cp. Figure 2). In Type 3, the representative amplicon comprises a considerable number of reads from both regions.
The phylogenetic trees of the 21 oligotrichean taxa display a spectrum of topologies ranging from distinct geographic to heterogeneous distribution patterns generated by the representative amplicon sequences of the Swarm OTUs (Figure 4). Distinct patterns thereby suggest that the amount of geographically restricted genetic diversity is the result of past effective dispersal events in specific taxa as indicated by long and highly supported branches (Supplementary Figure 1). On the other hand, multiple effective dispersal events are repeatedly ongoing in several other taxa as indicated by heterogeneous distribution patterns and short branch lengths.
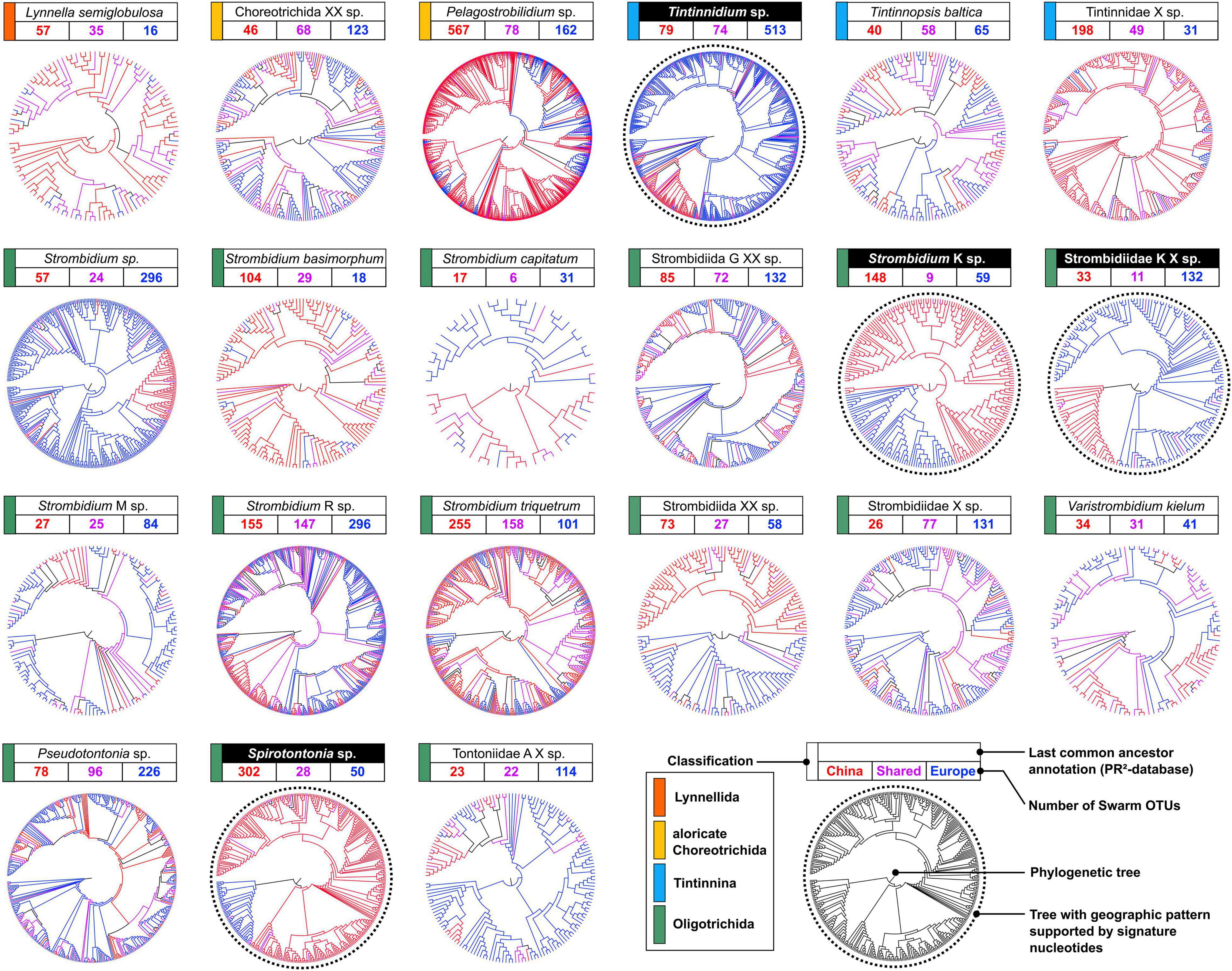
Figure 4. Phylogenetic trees based on the representative sequences of the Swarm OTUs. Only trees that display a distinct geographic pattern supported by signature nucleotides are marked by “taxon” names highlighted in black and dotted circles.
Most of the phylogenetic trees show various degrees of heterogeneous branching patterns with no clear split between Chinese and European OTUs. Several different branching patterns are exemplified in the tree of the tintinnid T. baltica (Figure 3A). Please, note that the last common ancestor approach assigned the V4 sequences only to the reference sequence of T. baltica although the reference sequences of Tintinnopsis acuminata and Tintinnopsis nana are identical in their V4 regions. Hence, the tree shown might actually comprise several species. The various subclades in the tree mainly consist of closely related Chinese, European, and shared OTUs, which display an alternating and irregular branching (Figure 3B). The representative sequences of the shared OTUs were assigned to one of the three types described above (Figure 2), depending on the percentages of reads from the particular regions in its representative amplicon (Figure 3B).
Distinct geographic distribution patterns are apparent in trees of four taxa (Figure 4), in which Chinese and European OTUs are entirely (the oligotrichids Spirotontonia sp. and Strombidium K sp.) or largely separated (the oligotrichid Strombidiidae K X sp. and the tintinnid Tintinnidium sp.). The representative sequences of shared OTUs are evenly distributed in the phylogenies of the four taxa and either fall into clades of European or Chinese OTUs. They belong to Type 2 of shared OTUs, and their representative amplicons can be assigned to the geographic region of their respective clade (Figure 2).
The bifurcation forming the conspicuous split between Chinese and European OTUs in the trees of the four abovementioned taxa (Supplementary Figure 1) exhibit high support values (Shimodaira–Hasegawa test ≥ 0.9; Shimodaira, 2002) and are further corroborated by signature nucleotides (see section “Materials and Methods”). In general, the number of signature nucleotides greatly varies (Supplementary Figure 3). Further geographic patterns emerged, e.g., in Strombidium sp.; yet, they are not supported by signature nucleotides. In the individual alignments, all signature nucleotides are restricted to an otherwise conserved section of the V4 (positions 59–152; Supplementary Figure 2), which comprises the highest read quality scores for each nucleotide position. In the same V4 section, signature nucleotides also occurred in other taxa (e.g., the oligotrichids Strombidium sp., Strombidiida XX sp., and the aloricate choreotrichid Pelagostrobilidium sp.; data not shown). However, they are not signature nucleotides characteristic for either geographic region as the separation of the OTUs is ambiguous; they represent phylogenetic signatures. These findings demonstrate the significance of this particular section of the V4 for ascribing amplicons to a certain geographic region.
In the two trees with nearly complete geographic separation (the oligotrichids Spirotontonia sp. and Strombidium K sp.), the majority of OTUs originate from China. The genealogy of Spirotontonia sp. including reference sequences (Supplementary Figures 1A,B) shows that a single shared OTU in the European clade (OTU 1377; 472 reads) is identical to one unidentified environmental sequence collected from the East Pacific Rise (KF129779) and nearly identical to another unidentified environmental sequence collected in the South China Sea (KJ760553). On the other hand, the reference sequences of Spirotontonia turbinata (FJ422994) and Spirotontonia taiwanica (FJ715634) described from coastal waters of China and north-eastern Taiwan, respectively, group closely together with a few basally branching Chinese OTUs, while the remaining Chinese OTUs are more distantly related to any reference sequence and display an intense radiation. Each reference sequence shares the signature nucleotides of the respective European or Chinese OTU clades they are assigned to. Interestingly, the reference sequence of Spirotontonia grandis from the South China Sea (KU525755) branching between both clades is not closely related to any particular OTU. Further, it shares 13 binary signature nucleotides with the European OTUs and two binary signature nucleotides with the Chinese OTUs.
The PR2 reference sequences of the oligotrichid “taxa” Strombidium K sp. and Strombidiidae K X sp. are unidentified environmental sequences, but are related with sequences of two species sampled from Chinese coastal waters and deposited as Strombidium triquetrum (KJ609052; Gao et al., 2016) and Strombidium capitatum (KP260510; Song et al., 2015) in GenBank. The sequences of the four “taxa” are aggregated into the “Strombidiidae K cluster” in the PR2 database. In the phylogenetic tree of the Strombidium K sp. OTUs and reference sequences (Supplementary Figures 1C,D), the S. capitatum sequence is related with the clade of European OTUs, while the sequence of S. triquetrum groups together with two PR2 sequences in the cluster of Chinese OTUs. Both clades feature a huge genetic diversity as indicated by the numerous branches and partially considerable branch lengths. Signature nucleotides for the Chinese or European clades are shared by the respective reference sequences, except for the sequence of S. capitatum, which shares one binary signature with each of the two clades and two asymmetric signatures with the European clade.
In the phylogenetic tree of Strombidiidae K X sp. (Supplementary Figures 1E,F), two reference sequences from the eastern North Pacific (AF372790 and AF372789) and one sequence from the Mediterranean Sea (HQ394045) fall within the clade of Chinese OTUs, while one reference sequence from the eastern North Pacific (KJ762996) as well as one sequence from the North Atlantic (KC488382) are highly similar to a shared OTU (OTU 409) in the clade of mainly European OTUs. The latter clade comprises most sequences (including a single Chinese OTU) and is clearly separated from the clade of mostly Chinese OTUs by a long bifurcation. Additionally, both clades are characterized by 14 binary signatures.
The phylogenetic tree of Tintinnidium sp. (Supplementary Figures 1G,H) was complemented by reference sequences of known morphospecies belonging to the family Tintinnidiidae plus relevant sequences from the PR2 database and an outgroup sequence belonging to the choreotrichid family Strobilidiidae (Rimostrombidium lacustre). The majority of OTUs form a predominantly Chinese and an exclusively European clade. Shared OTUs that group within these two clades contain 99% of reads from the respective geographic clade affiliation (Type 2 cp. Figure 2). Several OTUs branch between the outgroup sequence and the first reference sequences. The representative sequences of these OTUs are highly different from sequences of the Chinese and European clades (e.g., partly containing gap regions), indicating that they do not belong to the Tintinnidiidae at all and were thus excluded from the subsequent analysis of signature nucleotides. The clade of Chinese OTUs also contains eight European OTU sequences. Both the Chinese and European clades are supported by seven binary signatures. Interestingly, all reference sequences of known morphospecies (Antetintinnidium mucicola, Tintinnidium fluviatile, Tintinnidium pusillum, and Tintinnidium balechi) exclusively fall into the Chinese clade. The latter therefore comprises most of the currently known diversity of the Tintinnidiidae. Four unidentified environmental sequences, namely, from the Baltic Sea (FN690031), from two freshwater lakes in France and Germany (EU162620 and HM135052), and from the Columbia River estuary at the north-eastern Pacific coast (KJ925310), fall into a rather basally branching group of the European clade, while the remaining European and related shared OTUs display a broad diversification into numerous subclades.
Those phylogenetically analyzed representative amplicons of Swarm OTUs that displayed clear geographical distribution patterns were subjected to a second-level clustering via sequence similarity networks (SSNs) along with reference sequences of the PR2 database and closely related sequences of known morphospecies (Figures 5, 6). The cut-off divergence at which the morphospecies separated into different network sequence clusters (NSCs) was determined for elucidating the number of NSCs without reference sequences; these may represent new species or ones lacking genetic data and await further validation, for example, by targeted recovery approaches (Gimmler and Stoeck, 2015). In both the oligotrichid Spirotontonia and the tintinnid Tintinnidium, a threshold of 98.5% sequence similarity was determined for separating the reference sequences of known morphospecies in the SSNs. The Spirotontonia network (Figure 5) contained five NSCs, which comprised 369 Swarm OTUs and 39,113 reads, indicating potential species. The largest of these NSCs was associated with S. turbinata; the smallest was associated with S. grandis. Both of these NSCs as well as two NSCs without any reference sequence predominantly included sequence data from Chinese samples. Only one NSC of a potential species included sequences predominantly from European samples as well as two unidentified environmental sequences.
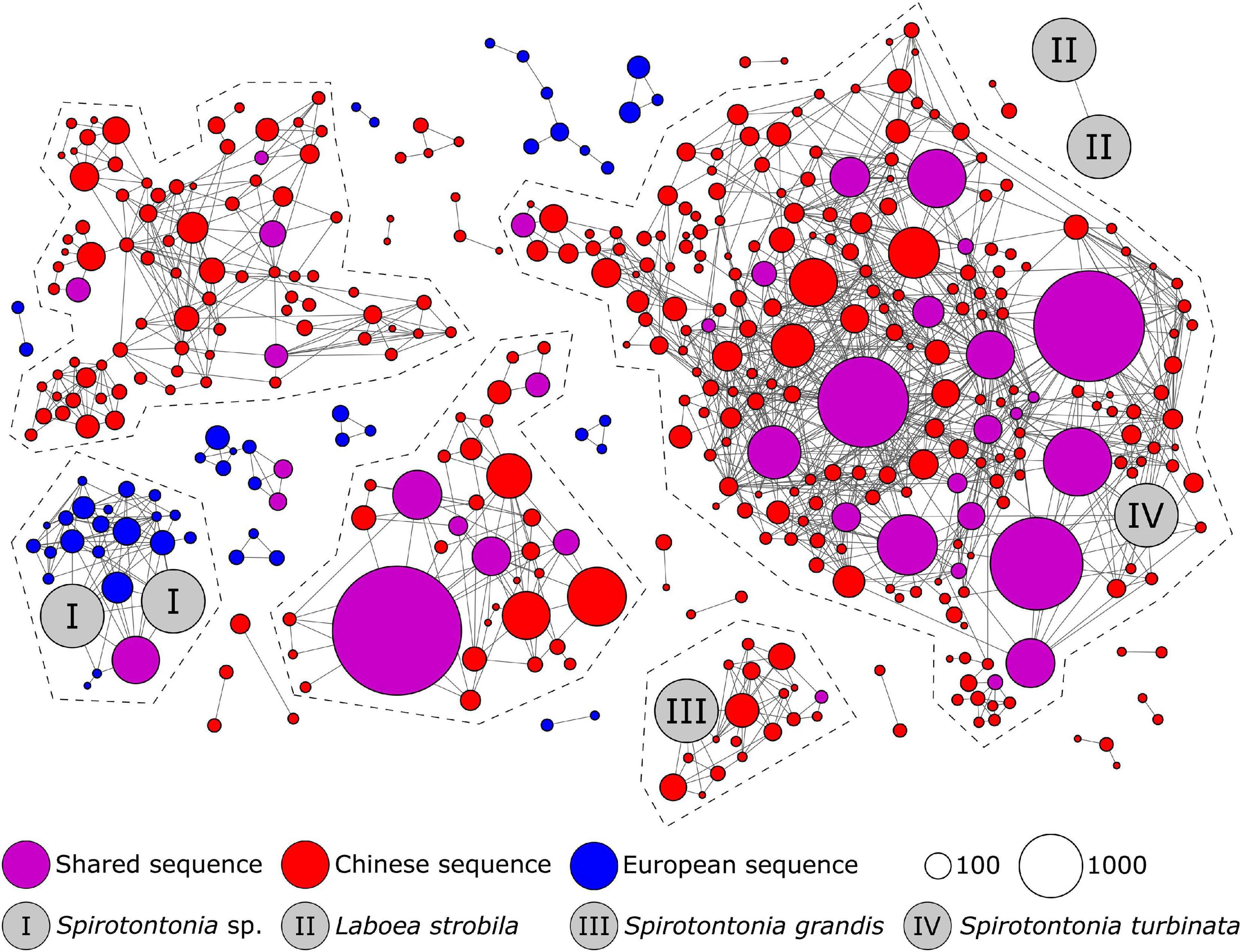
Figure 5. Second-level clustering of the representative amplicons of Swarm OTUs detected in the present study and reference sequences from the PR2 database assigned to Spirotontonia oligotrichids. At a threshold of 98.5%, the OTUs clustered into five network sequence clusters (NSC), two of which did not contain a reference sequence. Laboea strobila, a supposedly secondarily tail-less tontoniid with a somatic ciliary pattern identical to that in Spirotontonia species, remains separate. The circle sizes correspond to the read numbers of the representative amplicon sequences of the Swarm OTUs. NSCs which indicate potential species are framed by a dashed line.
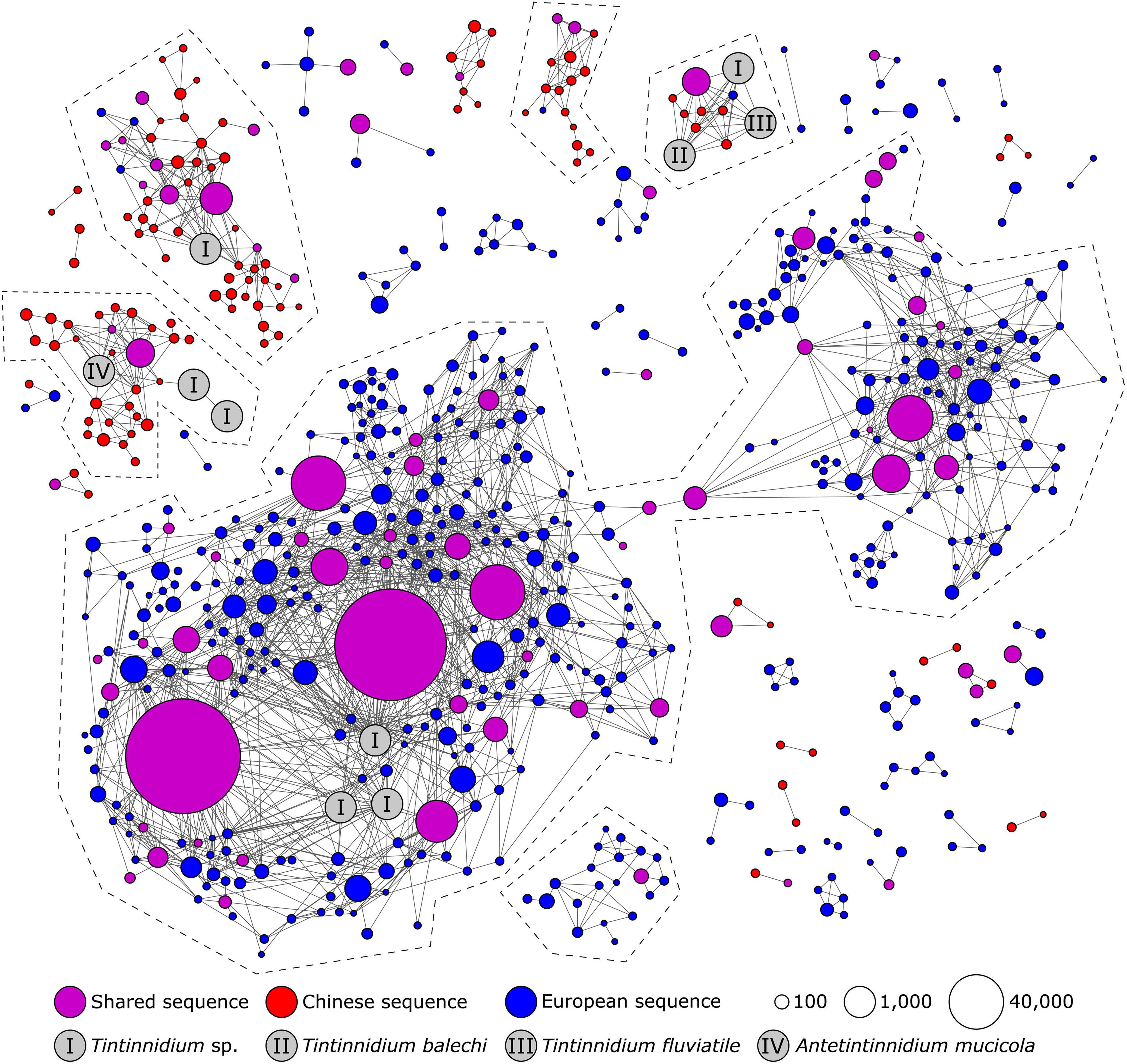
Figure 6. Second-level clustering of the representative amplicons of Swarm OTUs detected in the present study and reference sequences from the PR2 database assigned to the Tintinnidiidae tintinnids. At a threshold of 98.5%, the OTUs grouped into six larger network sequence clusters (NSCs), two of which did not contain any reference sequences and another two NSCs which contained unidentified congeneric reference sequences (Tintinnidium sp.). The circle sizes correspond to the read numbers of the representative amplicon sequences of the Swarm OTUs. NSCs which indicate potential species are framed by a dashed line.
The Tintinnidium network (Figure 6) contained at least six NSCs that indicated potential species. Four of these NSCs included unidentified congeneric environmental sequences, while two NSCs were not affiliated with any reference sequence. While the largest NSC and one without reference sequences predominantly included sequences from Europe, three smaller NSCs with reference sequences and the other NSC without affiliated reference sequences contained mostly sequences from China. One of the latter NSCs persistently contained reference sequences of the marine T. balechi and the freshwater T. fluviatile above thresholds of 98% because these two reference sequences share 99.5% V4 18S rDNA sequence similarity to another. The largest NSC of the Tintinnidium network consisted of two nearly separated groups of nodes (separation at 98.7% sequence similarity): one group with affiliations to reference sequences (Tintinnidium sp.), the other without affiliations to reference sequences. This pattern might indicate further potential species in the Tintinnidium network in addition to the six potential ones mentioned above.
For 700 widespread (occurring in Chinese and European coastal waters) and abundant (comprising ≥ 0.0001% of the reads in the dataset) Swarm OTUs, assortativity analyses on the internal network structures were conducted. In 149 of these OTUs, assortativity values that were significantly higher than expected by chance for amplicons detected in Chinese samples were observed. Furthermore, 166 OTUs with assortativity values that were significantly higher than expected by chance for amplicons detected in European samples were found.
In another 211 of the 700 OTUs analyzed, the assortativity values were significantly higher than expected by chance for each geographic region simultaneously. Only patterns within these 211 OTUs are considered in the following and their results are displayed in Figure 7. Besides these significantly higher assortativity values, a disassortative grouping (assortativity ≤ 0) of amplicons from either region was discovered in 12 out of the 211 OTUs, most of which were taxonomically affiliated to the CONthreeP clade. In these OTUs, the slight tendency of amplicons to be connected to amplicons from the respective other region indicates that the effective dispersal between China and Europe does not seem to be strongly limited (Figure 7; lower right quadrant).
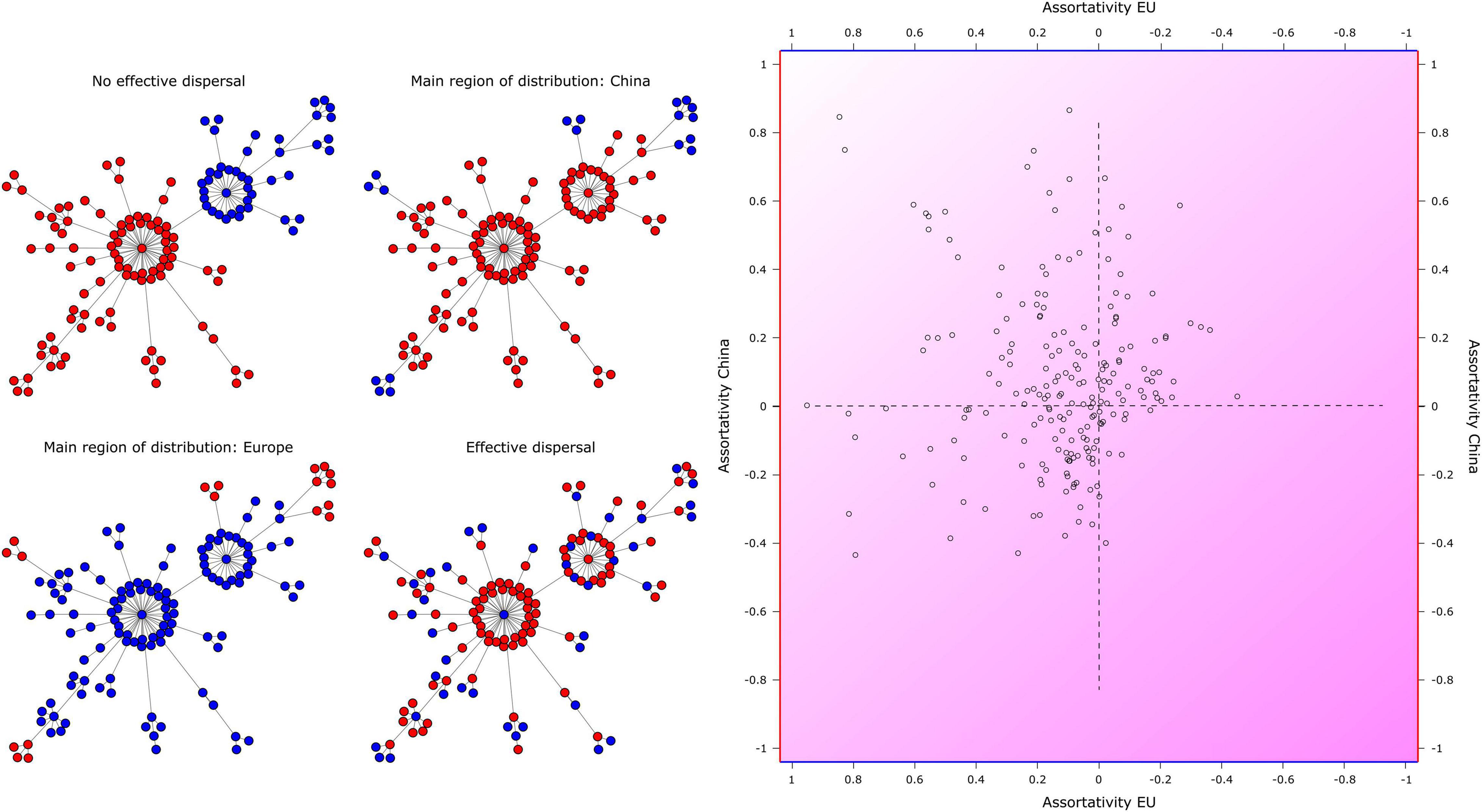
Figure 7. Significant assortativity patterns of amplicons within 211 shared Swarm OTUs detected in Chinese and European (EU) coastal waters. (Left panel) Mock shared Swarm OTUs representing the four combinations of assortative and disassortative patterns (in these mock Swarms, node sizes were not scaled to read abundances). (Right panel) Each data point refers to a shared OTU and the respective assortativity values of its amplicons determine its placement in one of the four quadrants. (Upper left) Assortative grouping of amplicons from each region (indicating limited effective dispersal; 77 OTUs); (upper right) assortative grouping of Chinese amplicons, disassortative grouping of European amplicons (China main distribution area; 49 OTUs); (lower left) assortative grouping of European amplicons, disassortative grouping of Chinese amplicons (Europe main distribution area; 73 OTUs); (lower right) disassortative grouping of amplicons from each region (unlimited effective dispersal; 12 OTUs). Data points in the latter quadrant are scarce, since our analyses focused on OTUs which exhibited significantly higher assortativity values than expected by chance for sequences of both regions.
There were 49 out of the 211 OTUs in which amplicons detected in China displayed a significant assortative grouping (assortativity > 0), but for which amplicons detected in Europe displayed a disassortative grouping (assortativity ≤ 0). For these OTUs (for an example see Supplementary Figure 4A), the assortativity of the amplicons in the internal OTU structure indicates a main area of distribution regarding genetic diversity in Chinese waters (Figure 7; upper right quadrant). Of those 49 OTUs, 23 were taxonomically affiliated to the Oligotrichida, eight to each the aloricate Choreotrichida and members of the CONthreeP, and four to the Tintinnina; taxonomic groups with less than three affiliated OTUs are not mentioned.
By contrast, there were 73 out of the 211 OTUs in which amplicons detected in Europe displayed a significant assortative grouping, but for which amplicons detected in China displayed a disassortative grouping. For these OTUs (for an example see Supplementary Figure 4B), the assortativity results indicate a main area of distribution regarding genetic diversity in European waters (Figure 7; lower left quadrant). The Oligotrichida were again the most abundant group among them, contributing 26 OTUs. Other abundant groups were the CONthreeP (23 OTUs), aloricate Choreotrichida (six OTUs), and Tintinnina (four OTUs).
Significant assortative groupings of amplicons from both Chinese and European waters occurred in 77 out of the 211 OTUs. Since the amplicons were mostly connected to amplicons detected in the same region, we suppose a limited effective dispersal between the regions for all these OTUs (Figure 7; upper left quadrant). Once more, the Oligotrichida were by far the most abundant group (42 OTUs), followed by the CONthreeP (14 OTUs), by OTUs that could not be assigned further than to the class Spirotrichea (nine OTUs), and by the aloricate Choreotrichida and the Tintinnina (four OTUs each).
Coasts are the ideal region for combining knowledge on ciliate plankton obtained by various methodological approaches. Apart from their easy accessibility, coastal habitats are characterized by high temporal dynamics and spatial heterogeneity, both of which are assumed to foster speciation (Norris, 2000). Climate variation and sea level changes severely affect the highly productive shallow coastal areas with their brackish water habitats. Sea level changes cause dramatic shifts in the distribution, the dimensions, and the connectivity of these regions (Palumbi, 1994). A drop in sea level would thus restrict the neritic region to a narrow strip along the upper continental slope and thus reduce the habitat for specialized communities of coastal planktonic ciliates. Therefore, such regions are predestined for a bridge building approach like ours that aims to reconcile morphospecies, barcoded reference sequences, and OTUs in investigating geographic distribution patterns and their evolution, focusing on the community of oligotrichean ciliates. A comprehensive body of literature exists on neritic oligotrichean morphospecies (Cariou et al., 1999; Agatha, 2011; Liu et al., 2017; Hu et al., 2019). Furthermore, the majority of barcodes are from neritic taxa (Doherty et al., 2007; Bachy et al., 2012; Zhang et al., 2017; Santoferrara et al., 2018), and metabarcoding data are available (Santoferrara et al., 2014; Massana et al., 2015; Tucker et al., 2017).
Previous metabarcoding studies analyzed the genetic material obtained from entire microzooplankton or ciliate plankton communities and investigated community changes on higher taxonomic ranks (e.g., Massana et al., 2015). In our interdisciplinary multi-species approach, we combined the expertises of molecular ecologists and ciliate taxonomists to focus on the geographic patterns of particular ciliophoran Swarm OTUs and the interpretation of these patterns under evolutionary aspects. Thereby, our sampling strategy stands out from commonly applied approaches. In contrast to previous studies, our dataset is based on a material collection that complements a high temporal resolution (seasonal and daytime shifts) and large geographic distances between samples. Since the sea and the neritic regions are dynamic (Abboud-Abi Saab and Owaygen, 1998; Marteinsson et al., 2016; Huang et al., 2021) and highly structured horizontally and vertically (McManus and Woodson, 2012), environmental patchiness is a question of scale. Additionally, these regions are very heterogeneous for protists effecting together with community assembly processes a patchy distribution highly likely prone to undersampling. Previous data from our sampling regions indicate a pronounced seasonality in the ciliate community compositions (Mozetič et al., 1998; Witek, 1998; Gómez and Gorsky, 2003; Jiang et al., 2013; Yang et al., 2014). Despite the comparatively intensive sampling, however, only a small part of the pronounced seasonality with short abundance peaks and a rapid turnover of the dominating taxa could be caught. Likewise, the limitation to Chinese and European sites prevents an investigation of the entire taxon ranges.
Metabarcoding is a powerful complement to taxonomic surveys, especially when comprehensive and taxonomically validated reference databases enable unequivocal species identification. In this study, we contribute a novel approach combining phylogenetics, signature nucleotide analyses, and network analyses to enable an unprecedented investigation of genetic diversity patterns on multiple scales. Swarm is an ideal de novo clustering method to generate OTUs from metabarcoding data based on single nucleotide differences while retaining the underlying diversity in the amplicon networks. Different timescales can be investigated on between- and within-OTU levels: phylogenetic trees of the representative amplicon sequences (between-OTU level) visualize comparatively older processes (e.g., past effective dispersal and speciation events), whereas the internal network structure of Swarm OTUs (within-OTU level) may display more recent divergences on sub-species level. Albeit we are pretty sure to have ascertained genetic differentiations that evolved on different time scales, the distinction between intra- and interspecific sequence deviations remains blurry (Santoferrara et al., 2020).
In Swarm OTUs, those sequences resulting from natural genetic variation as well as some artificially created ones (see discussion below) group around the amplicon with the highest read number (the ‘representative,’ ‘seed,’ or ‘dominant’ amplicon). We interpret the numerous amplicons with much fewer reads (the ‘minor’ amplicons) than the ‘dominant’ one as intraspecific V4 variants generated by imperfect concerted evolution of the ribosomal DNA tandem repeats (Ganley and Kobayashi, 2007). In marine planktonic diatoms, Gaonkar et al. (2020) found a general correlation between the number of haplotypes comprising a taxon and the number of reads in its ‘dominant’ haplotype. Interestingly, the ‘minor’ amplicons of our dataset frequently displayed some peculiarities, namely, some were exclusively detected either in China or in Europe and thus exhibited a clear affiliation to one geographic region. It is unclear whether they originated from an ongoing splitting process or a recent merging of conspecific populations; yet, we favor a diverging scenario in cases where the dominant amplicon is truly shared. Estimating the extent of this intraspecific variation is crucial for metabarcoding studies, in which the ‘minor’ amplicons might indicate a considerable rare diversity. In the present study, the number of supposed ‘minor’ amplicon variants surpasses by far the findings obtained from sequencing up to three conspecific specimens and subsequent cloning steps in most major ciliate groups (Gong et al., 2013; Wang et al., 2017, 2019). We agree with these and previous studies that most genetic diversity originates from the high number of low-divergent sequence variants present in individuals of single ciliate species (Forster et al., 2019; Zhao et al., 2019). OTU clustering by Swarm in combination with second-level clustering turned out to represent an elegant solution to this problem, since the approach allows for inferring species-specific thresholds (Forster et al., 2019; Wang et al., 2019; Zhao et al., 2019).
Nevertheless, not all low-divergent (‘minor’) amplicons necessarily originate from real biological variation, but may instead reflect artificial sequences caused by methodological errors. A perfect distinction between these two types of sequences is currently impossible, but, of course, important for accurate diversity estimations (Brown et al., 2015). Potential error sources that generate non-biological sequences occur in the PCR amplification process, for instance, by a decrease in polymerase fidelity, thermal damage (Pienaar et al., 2006), or by the formation of chimeric sequences from two or multiple parental sequences (Lahr and Katz, 2009). Artificial sequences may also be generated during high-throughput sequencing (HTS) by erroneous base calls (Bokulich et al., 2013). Another often ignored error source in HTS is sample bleeding, which is caused by incorrect assignment of small numbers of reads to multiplexed samples processed on the same sequencing lane (Mitra et al., 2015). Because of the fine-scaled resolution in our approach, we could observe events of sample bleeding and attribute Swarm OTUs with more than 99% of reads from one region and less than 1% of reads from the other region to this phenomenon.
To exclude artificially generated sequences from potential HTS error sources, we took several precautions and followed advices for best practices in sequence quality filtering4. Thus, except for sample bleeding (which may assign real biological sequences to the wrong sample), we conclude that only a negligible part of the genetic variation in our dataset is of non-biological origin. As a consequence, we further presume that the effect of artificial sequences on assortativity analyses will be of minor relevance. Since artificially generated sequences are highly unlikely to be generated from scratch, but rather from a dominant parental amplicon, both the artificial and dominant sequence will carry the same geographic information. This scenario is therefore not altering results of Swarm OTUs that consist completely or nearly completely of amplicons from the same region. In Swarm OTUs that include amplicons from different regions, artificial sequences might increase the number of edges between amplicons from the same region and therefore lead to more assortative patterns. Regarding the latter, though, we refer again to our quality filtering strategy, which removes low-quality sequences before clustering in Swarm is conducted. In addition, the patterns observed by within-OTU analyses were further corroborated by between-OTU analyses, which imply that they are not the result of artificially created sequences.
Denoising algorithms are an alternative to our sequence quality filtering strategy and have gained in popularity for analyzing metabarcoding datasets (e.g., Callahan et al., 2017). While denoising metabarcoding data prior to OTU clustering in Swarm is possible, too many of the low-abundant ‘minor’ sequence variants are aggregated into central consensus sequences or discarded by the denoising algorithms (Antich et al., 2021). The denoised dataset thus contains only few amplicons that can be further clustered by Swarm. Instead, the majority of these denoised amplicons are placed in standalone Swarm OTUs (i.e., singleton OTUs of high read abundance) without any other amplicon. Denoising a dataset before clustering in Swarm is therefore counter-effective for evaluating underlying network topologies of Swarm OTUs and exploring distribution patterns of genetic diversity with our newly introduced strategy.
The vast majority of Swarm OTUs in the present study were detected either in China or in Europe, whereas only a small proportion represents shared OTUs, admittedly, though, with high read numbers. Several phylogenetic relationships between the OTUs confirmed the occurrence of geographic patterns and indicate continuing limitations of effective dispersal. Some of the highly diversified clusters found only in one region branch basally and suggest historic cladogeneses with a high degree of genetic divergence possibly due to large population sizes. The geographic patterns are further corroborated by region-specific signature nucleotides detected by means of DeSignate (Hütter et al., 2020). The analyses focused on the most abundant ciliates in coastal waters, namely, the Oligotrichea. Four of them displayed geographic patterns, namely, the oligotrichid Spirotontonia sp., two further oligotrichids of the genus Strombidium (Strombidium K sp. and Strombidiidae K X sp.), and the tintinnid Tintinnidium sp.
The genus Spirotontonia Agatha, 2004 currently comprises three species (S. grandis, S. taiwanica, and S. turbinata). Like all Tontoniidae Agatha, 2004, it is characterized by a highly contractile tail. The single somatic kinety forms a sinistral spiral around the cell. The genus Laboea Lohmann, 1908 shares this somatic ciliary pattern and is considered a secondarily tail-less tontoniid based on ribosomal DNA phylogenies (Gao et al., 2009). Based on literature data about morphospecies, the genus is distributed in the Arctic Sea, the Mediterranean Sea, the North Atlantic, the North Pacific, and the Indian Ocean (SA, unpubl. data). Although no Chinese OTU grouped within the clade of European OTUs, the close relation between reference sequences from China and few European OTUs with identical signature nucleotides indicates that similar sequences occur in China, but were not detected in the present study. Yet, Chinese OTUs could not be detected in Europe, considering that the single or few reads from the respective other region in the shared OTUs are methodological artifacts caused by sample bleeding. The Chinese OTUs probably represent more than the currently known species (S. turbinata and S. taiwanica) given the numerous short branches, which suggest a radiation with a high diversification rate. Spirotontonia grandis might be of hybrid origin as its reference sequence shares signature nucleotides with amplicon sequences from both the Chinese and European clades. In the related euplotid ciliates, hybridization for instance caused by an overlap in the pheromone structures and inter-specific mating further increases the complexity of the interpretations (Luporini et al., 2016). The phylogenetic tree features a high diversity beyond reference sequences of known morphospecies, which is corroborated by the second-level clustering proposing at least two new Spirotontonia species in Chinese coastal waters.
The Strombidium K cluster contains numerous reference sequences labeled as Strombidium K sp. and Strombidiidae K X sp., respectively, and two reference sequences of morphologically identified species. Considering the original descriptions, however, the identification of these morphospecies is doubtful, and their sequences probably belong to different species. Strombidium capitatum (Leegaard, 1915) Kahl, 1932 was discovered in the Northeast Atlantic (reported as Laboea capitata). Seravinella pectinata Alekperov and Mamaeva, 1992 is based on a completely deformed and destroyed S. capitatum cell as revealed by the reinvestigation of the type slide. Further reports of the morphospecies come from the Arctic Sea, the North Atlantic, the Mediterranean Sea, the South Atlantic, the North Pacific, the Indian Ocean, and the Antarctic Sea (SA, unpubl. data). The GenBank sequence KP260510 was linked to a specimen identified as Strombidium capitatum sampled from Chinese coastal waters (Song et al., 2015). The gene sequence is accompanied by a short description, line drawings, and micrographs. The line drawings and one micrograph come from a previous study on S. capitatum; the remaining micrographs do not provide many details and primarily depict a conspicuous apical protrusion. Conspecificity of the sequenced specimens with S. capitatum is questioned by a much larger cell size (90–135 μm × 70–100 μm vs. 45–70 μm × 40–60 μm) and much longer extrusomes (22–24 μm vs. about 15 μm; Song et al., 2015). Hence, a cryptic species different from S. capitatum might have been sequenced. Strombidium triquetrum Agatha and Riedel-Lorjé, 1997 was described from the North Sea; further reports of this morphospecies do not exist. Gao et al. (2016) sequenced specimens identified as Strombidium cf. capitatum; the sequences were deposited in GenBank and included in the PR2 database under the name S. triquetrum. Since the associated stamp-sized micrograph merely displays the anterior cell portion of a protargol-stained specimen, conspecificity is uncertain. The high amount of genetic diversity present in the Strombidium K cluster, which was partly restricted to one region, suggests several potentially new species. Nevertheless, attention should be paid to the reliable identification of species, using original descriptions or authoritative redescriptions to avoid an incorrect linkage of reference sequences and morphospecies.
The family Tintinnidiidae currently comprises the genera Antetintinnidium Ganser and Agatha, 2019, Membranicola Foissner et al., 1999, and Tintinnidium Kent, 1881 with in total 16 species (SA, own data). Considering the genetic divergence between known morphospecies (Antetintinnidium mucicola, T. fluviatile, T. pusillum, and T. balechi), the enormous genetic diversity recognizable in the European clade of the Tintinnidium sp. phylogeny suggests the presence of several as yet unknown species, which very likely differ distinctly in their cell morphologies. Likewise, the second-level clustering of Tintinnidium sp. yielded two network sequence clusters (NSC) with known morphospecies and additionally three clusters of probably novel Tintinnidiidae taxa. In addition, the NSC which contained the most amplicons and reads could only be linked to Tintinnidium sp. reference sequences, suggesting another unknown morphospecies. The relationships in the phylogenetic trees propose several independent introductions of Tintinnidiidae from brackish waters into freshwater (Ganser and Agatha, 2019).
In contrast to the abovementioned taxa, the majority did not display distinct geographic patterns. Exemplary for these taxa, OTU sequences assigned to the tintinnid Tintinnopsis baltica Brandt, 1896 were analyzed. The species had been established based on material from the Baltic Sea. Further reports of the morphospecies are from the Antarctic/Sub-Antarctic Sea, the North Atlantic, the Mediterranean Sea, the Black Sea, the Sea of Azov, the Southwest Atlantic, the North Pacific, the Southeast Pacific, and the Indian Ocean (SA, unpubl. data). Its tree topology might be interpreted as alternating multiple invasions, suggesting that this species belongs to a group of organisms with effective long-distance dispersal. Thus, allopatric speciation is probably a slow process in this tintinnid which might be indicated by the identical V4 regions in T. baltica, T. acuminata, and T. nana.
All phylogenetic analyses corroborate each other and shed light on previously undetected aspects of diversity. On the one hand, novel diversity, i.e., potentially new species or ones for which genetic data are not available, emerged in both the phylogenetic trees and the second-level clustering. On the other hand, geographical distribution patterns of OTUs in Chinese and European coastal waters were revealed by phylogenetic trees and second-level clustering and, in several cases, were further underpinned by signature nucleotides. In the future, ‘dominant’ amplicon sequences that are not closely related to sequences of a known morphospecies may enable the identification and proper description of new species, following a ‘reverse taxonomy’ or ‘targeted recovery’ approach (Markmann and Tautz, 2005; Gimmler and Stoeck, 2015). The sampling site and season at which the amplicon sequence was recorded together with the signature nucleotides detected by DeSignate (see above) further facilitate this aim.
Magnifying the resolution of sequence analyses from between Swarm OTU-level to within Swarm OTU-level further emphasized the existence of distribution patterns. Compared to results from phylogenetic analyses, which focus on sequences between which a sufficiently high sequence divergence has led to the establishment of genetically distinct units (OTUs), the assortativity analyses explore relatively recent or even ongoing processes (e.g., effective dispersal and gene flow), in which the genetic divergence is momentarily insufficient for establishing distinct OTUs.
Since our network analyses aimed at revealing dispersal limitations, the interpretations focused on those 211 widespread and abundant Swarm OTUs within which amplicons from each region had a significantly higher assortativity value than expected by chance. Using our novel approach, we distinguish three different scenarios by interpreting the assortative and disassortative patterns of amplicons within these shared OTUs. (i) No effective dispersal takes place. The assortative grouping of both the Chinese and the European amplicons indicates genetically different amplicon clusters that presumably experience limited gene flow. With increasing genetic divergence, it may eventuate in an allopatric speciation. One or very few edges (connections) between distinct clusters of amplicons from different regions suggest founder events followed by bottlenecks and genetic drift. This scenario was observed in 77 OTUs. (ii) The main area of distribution is apparently restricted to one particular region. This distribution pattern might be attributed to species sorting, i.e., the presence or absence of an appropriate niche, a rather recent dispersal limitation, or an extinction event without considerable resettlement. This scenario dominated in the present dataset on planktonic ciliates from European and Chinese coastal waters. It was observed in 122 OTUs: 49 OTUs with main area of distribution in China and 73 OTUs with main area of distribution in Europe. (iii) Effective dispersal takes place. The disassortative grouping of amplicons in the OTUs does not follow a clear geographic pattern owing to the lack of a strong genetic separation between Chinese and European amplicons. Possibly because the ciliates are capable of effective dispersal or because their evolution rates are so low that they did not genetically diverge after spatial separation. This scenario was rarely found owing to our selection of OTUs (see section “Materials and Methods”). Although the patterns perceived are highly interesting, they necessitate further interpretations by intertwining them with future data, for instance, on the species’ autecology (see section “Outlook”). By demonstrating geographic patterns on within-OTU level, we contradict Caron and Hu (2019) in that the knowledge far beyond species-level distinctions has no practical value.
The “everything is everywhere, but the environment selects” tenet states that protist taxa are dispersed globally, but form populations only where the environmental conditions (abiotic and biotic) are suitable for them; otherwise, they might be present merely as resting cysts (see below). Since the autecology (including dispersal rates) and thus the niche parameters are unknown for the majority of Oligotrichea (Maselli et al., 2020), it is impossible to tell apart ecological and historical effects that cause distribution patterns (Bass and Boenigk, 2011).
Studies on the effective dispersal capabilities of marine planktonic ciliates are currently missing. The sparse evidence are records of identical marker gene sequences at the east and west coasts of the North Atlantic plus adjacent sea regions and in the Northwest Atlantic and Northeast Pacific (see section “Introduction”). For pelagic cyanobacteria, copepods, and foraminifera (Darling et al., 1999, 2000; de Vargas et al., 1999), genetic data indicate a strong effective dispersal across ocean basins as well as between them (Norris, 2000). Interestingly, almost identical gene sequences (three base pairs difference in the 18S) of planktonic foraminifera were detected in the Northeast Pacific and Mediterranean Sea, and a transport from east to west is supposed owing to the prevailing surface currents (Darling et al., 1999). These data imply that many foraminiferan genotypes have either evolved only slightly after their spatial separation or they are still capable of an effective dispersal. Molecular data on bipolar planktonic foraminifera and benthic ciliates as well as morphologic data on a planktonic tintinnid (Acanthostomella norvegica) suggest a trans-tropical effective dispersal (Darling et al., 2000; Di Giuseppe et al., 2013; Dolan and Pierce, 2013).
The metacommunity structure of small organisms with relatively high dispersal rates is probably dominated by species sorting and mass effects rather than by dispersal limitation and patch dynamics (De Meester, 2011). Likewise, Norris (2000) concluded that ranges of many pelagic groups are much more limited by their ability to maintain viable populations than by any inability to disperse across tectonic and hydrographic barriers. Oceanographic and climatic changes may create new habitats with new niches instead of preventing dispersal. Such shifting selective regimes have been found to generate large genetic differences very quickly, even between large populations that are not completely isolated (Palumbi, 1994). Three scenarios of species-niche interactions were discussed (Weiner et al., 2014). (i) Dispersal is limited. Bottlenecks, genetic drift, and abiotic boundaries may cause in concert the differentiation of allopatric sister lineages. (ii) Dispersal is rather unlimited and species interaction are negligible; the distribution of species thus probably reflects the spatial realization of appropriate niches. (iii) Species interactions are considerable; competitive exclusion hence influences the pattern of niche occupation. The present heterogeneous data on marine planktonic ciliates from Chinese and European coastal waters propose that all three scenarios are realized.
Dispersal limitation can, however, not be concluded from the absence of an active ciliate species at a particular site, i.e., a more intensive sampling might prove at least some of the Oligotrichea to be more widely dispersed and distributed, resulting in an increased number of shared amplicons in the Swarm OTUs. In non-random processes, planktonic Oligotrichea might be dispersed as active states by water currents over large areas or possibly even globally; but, the timespans are long compared to their generation times, questioning whether it is possible for a species to be cosmopolitan. In response to external triggers, many protists are, however, capable to form more or less resistant resting cysts (Foissner, 2011). Actually, some oligotricheans are known to generate at least occasionally resting cysts, and several similar shaped, but unidentified forms had been described (Meunier, 1910; Kim et al., 2002; Rubino et al., 2002; Agatha et al., 2005). Yet, the viability of the cysts and hence the ability to excyst decreases with time (Kim and Taniguchi, 1997). The factors controlling encystment, excystment, and viability of cysts may differ among closely related protist species and probably even between clones of the same species (Weisse, 2008). Since cysts also seem to sediment rather rapidly (Kim et al., 2008), they are very rarely found in plankton samples collected in the upper 2 m of the water column (SA, own observ.). Therefore, long-distance dispersal of suspended cysts and a subsequent population growth are probably rare events for oceanic taxa (Agatha, 2011), while cysts might easily be resuspended in shallow coastal waters during storm events. In the present study, the probability is low that considerable quantities of vital resting cysts were among the sequenced material, which was confirmed by microscopic inspection of some samples.
In the phylogenies based on our data, we detected comparatively deep and more recent splits; in addition, the assortativity values of some shared OTUs indicated ongoing divergence, possibly speciation. The origin of the Choreotrichida and Oligotrichida took place ca. 462 million years ago (mya) after an extreme boost on speciation rates in the stem branch linking these taxa to the hypotrichs (Fernandes and Schrago, 2019). Based on the ribosomal data set comprising the 18S, 5.8S, and 28S rDNA, the divergence time of the oligotrichids was calculated to 199–443 mya and that of the choreotrichids to 256–447 mya. A significant diversification rate shift commenced at the stem branch leading to the Perilemmaphora (= hypotrichs + oligotrichids + choreotrichids) at the Ediacaran period (about 550–650 mya) with the development of numerous new lineages. While the mean rate of nucleotide substitutions per 100 million years is rather low (μ = 0.0185) for the ciliate ribosomal dataset, the substitution and speciation rates are markedly higher in the Perilemmaphora with on average 0.779 (0.635–0.956) species per million years (my). Their extinction rates were distinctly (5×) lower compared to the speciation rates and have remained nearly constant throughout time (0.011–0.396 species/my). At that time, the climate and the water currents were totally different from those prevailing today. Generally, the combination of high speciation rates with cladogenesis and low extinction rates generates an accumulation of lineages with long branches. However, the ribosomal time-calibrated tree of Fernandes and Schrago (2019) demonstrated an exceptional pattern in the cluster of hypotrichs, oligotrichids, and choreotrichids with several radiation events concentrated at the tips of the tree. The cladogenetic success of the Perilemmaphora is thus probably most likely a consilience of accelerated speciation rates and lineage accumulation. The combination of sexual reproduction and cell divisions might strongly promote the generation of local adaptations according to De Meester et al. (2002). The first reliable tintinnid fossils are from the Jurassic. Since they seem to be fully developed, the tintinnids apparently have an even longer history (Lipps et al., 2013), matching the abovementioned calculations by Fernandes and Schrago (2019).
In contrast to the conception that long-distance dispersal mitigates speciation by swamping the gene pool of the resident population, Norris (2000) suggests that long-distance dispersal may actually promote evolution by regularly carrying variants of a species across major oceanic fronts and thus exposing them to very different selection pressures than those occurring in their home range. Accordingly, large populations may accumulate mutations beneficial in particular environments in which natural selection and further adaptation take place, resulting in a differentiation from the parental population and finally a speciation (Shapiro et al., 2016). The experimental evolution of dispersal and movement in the freshwater ciliate Tetrahymena illustrated an increase in individual movement velocity and overall dispersal rates of populations at the margins of their distribution range over time (Fronhofer and Altermatt, 2015). Soft hydrographic barriers might cause populations to experience selection sufficiently strong on either side that speciation may take place, although they do not inhibit dispersal completely (Martín et al., 2020). Actually, individual populations might be more isolated than currently accepted as patterns of dispersal and connectivity could be highly structured (McManus and Woodson, 2012). Even sympatric speciation is possibly more widespread than previously assumed and is facilitated by environmental gradients of intermediate slope (Dieckmann and Doebeli, 1999; Doebeli and Dieckmann, 2003). An isolation by ecology, namely, regarding the depth or season of reproduction, had been described in planktonic foraminifera. In euplotid and hypotrich ciliates, any mutations in the coding sequence of a pheromone results in a new pheromone and a new pheromone receptor and thus in a new mating type (Luporini et al., 2016). Likewise, shifts in the seasonal occurrence or isolation in confined coastal areas may have been involved in the observed rapid speciation of the Oligotrichea.
Studies on the marine biogeography date back to more than 150 years and described a close linkage between faunistic and hydrogeographic features of the major oceanic water masses, resulting in a circumglobal warm water region with subtropical and tropical provinces and Northern and Southern cold water regions with polar and subpolar provinces (Zeitzschel, 1990). Van der Spoel and Heyman (1983) defined planktonic faunal centers with taxa sharing the same ecological preferences or ancestral forms, which simultaneously represent the main spots of speciation and dispersal. Many studies based on morphospecies discovered rather similar patterns in the distribution of planktonic organisms (Zeitzschel, 1990). In contrast, the Longhurst (2007) ecological provinces are primarily based on the primary production and frontal systems. Likewise, the preliminary metabarcoding studies revealed geographic patterns on community level. Yet, the rationales differ: some publications ascribe the observed patterns to abundance-dependent dispersal, while others regard species sorting, community interactions, or dispersal limitations as crucial. The present findings covering data from three Longhurst provinces suggest that marine planktonic ciliates, specifically, the Oligotrichea, might display similar geographic zonations as other planktonic organisms.
With the present study, a new perspective to this discussion is added by analyzing distribution patterns on within-OTU level down to single nucleotide differences. The impressive number of Swarm OTUs with records restricted to China or Europe and the geographical distribution patterns of sequences within OTUs revealed by assortativity analyses suggest an inverted scenario of the moderate endemicity model concerning the distribution of planktonic ciliates in coastal waters. This implies that the majority of marine planktonic ciliates has a restricted distribution, while only few have a wide, potentially cosmopolitan distribution.
While we discovered a considerable variation in geographic distribution patterns of planktonic ciliates in Chinese and European coastal waters, the causes remain unknown. It is crucial to bring life-history traits into play, which are highly likely linked to (effective) dispersal. Much more experiments have to be conducted for comprehending the dispersal of active and resting stages, the formation of resting cysts and their viability, as well as the autecology regarding abiotic factors, food items and preferences, functional, numerical and thermal traits. For addressing these topics, we have to go beyond the simple assumption that geographical patterns are size-dependent (Jenkins et al., 2011). Nevertheless, our novel approach can uncover variational patterns at a resolution up to single nucleotide differences and is applicable to investigate any group of interest.
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MG, DF, and SA conceived the study and wrote the manuscript. MG, SA, and XL selected the sampling sites. WL performed the DNA extraction. MG and DF analyzed the data. WL, XL, and TS helped to improve the manuscript. All authors approved the final version of the manuscript.
This study was financially supported by the FWF project I3268 given to SA and the NSFC project 31761133001 given to XL. DF was supported by a postdoctoral research grant of the Carl Zeiss Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks go to Michael Gruber, Margit Kagerer, and Yong Zhang for taking samples. Further thanks go to John Dolan at the Observatoire Océanologique de Villefranche (France), Mona Hoppenrath at the German Centre for Marine Biodiversity Research in Wilhelmshaven (Germany), and Klaus Jürgens at the Leibniz Institute for Baltic Sea Research in Warnemünde (Germany) for providing lab space during the sampling campaign.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.643822/full#supplementary-material
Supplementary Figure 1 | Phylogenetic analyses of the oligotrichids Spirotontonia sp. (A,B), Strombidium K sp. (C,D), Strombidiidae K X sp. (E,F), and the tintinnid Tintinnidium sp. (G,H). (A,C,E,G) Phylogenetic trees based on the representative Swarm OTU amplicons obtained in the present study. The green dots mark the branches selected as query (Q) and reference groups (R) in the DeSignate analyses for identifying signature nucleotides. (B,D,F,H) Phylogenetic trees based on the representative Swarm OTU amplicons obtained by the present study and reference sequences from GenBank and the PR2 database.
Supplementary Figure 2 | Distribution of signature nucleotides detected by DeSignate (Hütter et al., 2020) in the Swarm OTU sequence alignments of the four taxa displaying distinct geographic patterns.
Supplementary Figure 3 | Signature nucleotides identified by DeSignate (Hütter et al., 2020). The upper line displays the V4 sequence with the position of the signature nucleotides. The middle and lower left portions demonstrate the entropy plot (blue line) of the alignment shown below. The upper portion of the alignment comprises the query group sequences, the lower portion the reference group sequences. In the lower right portion, the alignment positions are ranked according to their relevance. Green, binary signatures; orange, asymmetric signatures; gray, noisy characters.
Supplementary Figure 4 | Examples of Swarm OTU networks. The networks display the internal structure of Swarm OTUs, in which nodes represent amplicons and edges represent one nucleotide differences between the amplicons. The node size was scaled to the read abundance comprised within an amplicon. (A) Swarm OTU with main area of distribution in Chinese coastal waters. (B) Swarm OTU with main area of distribution in European coastal waters.
Supplementary Table 1 | Sampling data.
Supplementary Table 2 | List of the 21 oligotrichean taxa included in the phylogenetic analyses (1); signature nucleotides detected in the four taxa with distinct geographic patterns (2); list of reference sequences obtained from GenBank and the PR2 database (3).
Supplementary Data 1 | Modifications to the DNA extraction protocol and details on the nested-PCR.
Supplementary Data 2 | Alignments and tree files of the 21 oligotrichean taxa included in the phylogenetic analyses.
Abboud-Abi Saab, M., and Owaygen, M. (1998). Day-to-day fluctuations of microplankton during an autumnal period at a fixed station off the Lebanese coast. Leb. Sci. Bull. 11, 45–65.
Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., et al. (2019). Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 66, 4–119.
Agatha, S. (2011). Global diversity of aloricate Oligotrichea (Protista, Ciliophora, Spirotricha) in marine and brackish sea water. PLoS One 6:e22466. doi: 10.1371/journal.pone.0022466
Agatha, S., Strüder-Kypke, M. C., and Beran, A. (2004). Morphologic and genetic variability in the marine planktonic ciliate Laboea strobila Lohmann, 1908 (Ciliophora, Oligotrichia), with notes on its ontogenesis. J. Eukaryot. Microbiol. 51, 267–281. doi: 10.1111/j.1550-7408.2004.tb00567.x
Agatha, S., Strüder-Kypke, M. C., Beran, A., and Lynn, D. H. (2005). Pelagostrobilidium neptuni (Montagnes and Taylor, 1994) and Strombidium biarmatum nov. spec. (Ciliophora, Oligotrichea): phylogenetic position inferred from morphology, ontogenesis, and gene sequence data. Eur. J. Protistol. 41, 65–83. doi: 10.1016/j.ejop.2004.09.005
Antich, A., Palacín, C., Cebrian, E., Golo, R., Wangensteen, O. S., and Turon, X. (2020). Marine biomonitoring with eDNA: can metabarcoding of water samples cut it as a tool for surveying benthic communities? Mol. Ecol. (early view). doi: 10.1111/mec.15641
Antich, A., Palacin, C., Wangensteen, O. S., and Turon, X. (2021). To denoise or to cluster? That is not the question. optimizing pipelines for COI metabarcoding and metaphylogeography. bioRxiv [preprint] doi: 10.1101/2021.01.08.425760
Bachy, C., Dolan, J. R., López-García, P., Deschamps, P., and Moreira, D. (2013). Accuracy of protist diversity assessments: morphology compared with cloning and direct pyrosequencing of 18S rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. ISME J. 7, 244–255. doi: 10.1038/ismej.2012.106
Bachy, C., Gómez, F., López-García, P., Dolan, J. R., and Moreira, D. (2012). Molecular phylogeny of tintinnid ciliates (Tintinnida, Ciliophora). Protist 163, 873–887. doi: 10.1016/j.protis.2012.01.001
Bakker, J., Wangensteen, O. S., Baillie, C., Buddo, D., Chapman, D. D., Gallagher et al. (2019). Biodiversity assessment of tropical shelf eukaryotic communities via pelagic eDNA metabarcoding. Ecol. Evol. 9, 14341–14355. doi: 10.1002/ece3.5871
Bass, D., and Boenigk, J. (2011). “Everything is everywhere: a twenty-first century de-/reconstruction with respect to protists,” in Biogeography of Microscopic Organisms: is Everything Small Everywhere?, ed. D. Fontaneto (Cambridge: Cambridge University Press), 88–110. doi: 10.1017/cbo9780511974878.007
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Brown, S. P., Veach, A. M., Rigdon-Huss, A. R., Grond, K., Lickteig, S. K., Lothamer, K., et al. (2015). Scraping the bottom of the barrel: are rare high throughput sequences artifacts? Fungal Ecol. 13, 221–225. doi: 10.1016/j.funeco.2014.08.006
Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643. doi: 10.1038/ismej.2017.119
Cariou, J.-B., Dolan, J. R., and Dallot, S. (1999). A preliminary study of tintinnid diversity in the NW Mediterranean Sea. J. Plankton Res. 21, 1065–1075. doi: 10.1093/plankt/21.6.1065
Caron, D. A., and Hu, S. K. (2019). Are we overestimating protistan diversity in nature? Trends Microbiol. 27, 197–205. doi: 10.1016/j.tim.2018.10.009
Csárdi, G., and Nepusz, T. (2005). The igraph software package for complex network research. InterJ. Complex Syst. 1695, 1–9.
Darling, K. F., Wade, C. M., Kroon, D., Brown, A. J. L., and Bijma, J. (1999). The diversity and distribution of modern planktic foraminiferal small subunit ribosomal RNA genotypes and their potential as tracers of present and past ocean circulations. Paleoceanography 14, 3–12. doi: 10.1029/1998pa900002
Darling, K. F., Wade, C. M., Stewart, I. A., Kroon, D., Dingle, R., and Brown, A. J. L. (2000). Molecular evidence for genetic mixing of Arctic and Antarctic subpolar populations of planktonic foraminifers. Nature 405, 43–47. doi: 10.1038/35011002
De Meester, L. (2011). “A metacommunity perspective on the phylo- and biogeography of small organisms,” in Biogeography of Microscopic Organisms: is Everything Small Everywhere?, ed. D. Fontaneto (Cambridge: Cambridge University Press), 324–334. doi: 10.1017/cbo9780511974878.017
De Meester, L., Gómez, A., Okamura, B., and Schwenk, K. (2002). The monopolization hypothesis and the dispersal-gene flow paradox in aquatic organisms. Acta Oecologica 23, 121–135. doi: 10.1016/s1146-609x(02)01145-1
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1–11. doi: 10.1126/science.1261605
de Vargas, C., Norris, R., Zaninetti, L., Gibb, S. W., and Pawlowski, J. (1999). Molecular evidence of cryptic speciation in planktonic foraminifers and their relation to oceanic provinces. Proc. Natl. Acad. Sci. USA 96, 2864–2868. doi: 10.1073/pnas.96.6.2864
DeSalle, R., Egan, M. G., and Siddall, M. (2005). The unholy trinity: taxonomy, species delimitation and DNA barcoding. Phil. Trans. R. Soc. B 360, 1905–1916. doi: 10.1098/rstb.2005.1722
Di Giuseppe, G., Barbieri, M., Vallesi, A., Luporini, P., and Dini, F. (2013). Phylogeographical pattern of Euplotes nobilii, a protist ciliate with a bipolar biogeographical distribution. Mol. Ecol. 22, 4029–4037. doi: 10.1111/mec.12363
Dieckmann, U., and Doebeli, M. (1999). On the origin of species by sympatric speciation. Nature 400, 354–357. doi: 10.1038/22521
Doebeli, M., and Dieckmann, U. (2003). Speciation along environmental gradients. Nature 421, 259–264. doi: 10.1038/nature01274
Doherty, M., Costas, B. A., McManus, G. B., and Katz, L. A. (2007). Culture-independent assessment of planktonic ciliate diversity in coastal northwest Atlantic waters. Aquat. Microb. Ecol. 48, 141–154. doi: 10.3354/ame048141
Doherty, M., Tamura, M., Costas, B. A., Ritchie, M. E., McManus, G. B., and Katz, L. A. (2010). Ciliate diversity and distribution across an environmental and depth gradient in Long Island Sound. USA. Environ. Microbiol. 12, 886–898. doi: 10.1111/j.1462-2920.2009.02133.x
Dolan, J. R., and Pierce, R. W. (2013). “Diversity and distributions of tintinnids,” in The Biology and Ecology of Tintinnid Ciliates: Models for Marine Plankton, eds J. R. Dolan, D. J. S. Montagnes, S. Agatha, D. W. Coats, and D. K. Stoecker (Oxford: John Wiley & Sons, Ltd), 214–243. doi: 10.1002/9781118358092.ch10
Dunthorn, M., Klier, J., Bunge, J., and Stoeck, T. (2012). Comparing the hyper-variable V4 and V9 regions of the small subunit rDNA for assessment of ciliate environmental diversity. J. Eukaryot. Microbiol. 59, 185–187. doi: 10.1111/j.1550-7408.2011.00602.x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2016). SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv [Preprint] doi: 10.1101/074161
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fernandes, N. M., and Schrago, C. G. (2019). A multigene timescale and diversification dynamics of Ciliophora evolution. Mol. Phylogenet. Evol. 139:106521. doi: 10.1016/j.ympev.2019.106521
Foissner, W. (2011). “Dispersal of protists: the role of cysts and human interactions,” in Biogeography of Microscopic Organisms: is Everything Small Everywhere?, ed. D. Fontaneto (Cambridge: Cambridge University Press), 61–87. doi: 10.1017/cbo9780511974878.006
Foissner, W., Chao, A., and Katz, L. A. (2008). Diversity and geographic distribution of ciliates (Protista: Ciliophora). Biodivers. Conserv. 17, 345–363. doi: 10.1007/s10531-007-9254-7
Forster, D., Bittner, L., Karkar, S., Dunthorn, M., Romac, S., Audic, S., et al. (2015). Testing ecological theories with sequence similarity networks: marine ciliates exhibit similar geographic dispersal patterns as multicellular organisms. BMC Biol. 13:16. doi: 10.1186/s12915-015-0125-5
Forster, D., Lentendu, G., Filker, S., Dubois, E., Wilding, T. A., and Stoeck, T. (2019). Improving eDNA-based protist diversity assessments using networks of amplicon sequence variants. Environ. Microbiol. 21, 4109–4124. doi: 10.1111/1462-2920.14764
Forster, D., Lentendu, G., Wilson, M., Mahé, F., Leese, F., Andersen, T., et al. (2020). Evaluating geographic variation within molecular operational taxonomic units (OTUs) using network analyses in Scandinavian lakes. bioRxiv [preprint] doi: 10.1101/2020.08.06.240267
Fronhofer, E. A., and Altermatt, F. (2015). Eco-evolutionary feedbacks during experimental range expansions. Nat. Commun. 6:6844. doi: 10.1038/ncomms7844
Ganley, A. R. D., and Kobayashi, T. (2007). Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 17, 184–191. doi: 10.1101/gr.5457707
Ganser, M. H., and Agatha, S. (2019). Redescription of Antetintinnidium mucicola (Claparède and Lachmann, 1858) nov. gen., nov. comb. (Alveolata, Ciliophora, Tintinnina). J. Eukaryot. Microbiol. 66, 802–820. doi: 10.1111/jeu.12728
Gao, F., Li, J., Song, W., Xu, D., Warren, A., Yi, Z., et al. (2016). Multi-gene-based phylogenetic analysis of oligotrich ciliates with emphasis on two dominant groups: cyrtostrombidiids and strombidiids (Protozoa, Ciliophora). Mol. Phylogenet. Evol. 105, 241–250. doi: 10.1016/j.ympev.2016.08.019
Gao, S., Gong, J., Lynn, D., Lin, X., and Song, W. (2009). An updated phylogeny of oligotrich and choreotrich ciliates (Protozoa, Ciliophora, Spirotrichea) with representative taxa collected from Chinese coastal waters. Syst. Biodivers. 7, 235–242. doi: 10.1017/s1477200009002989
Gaonkar, C. C., Piredda, R., Sarno, D., Zingone, A., Montresor, M., and Kooistra, W. H. C. F. (2020). Species detection and delineation in the marine planktonic diatoms Chaetoceros and Bacteriastrum through metabarcoding: making biological sense of haplotype diversity. Environ. Microbiol. 22, 1917–1929. doi: 10.1111/1462-2920.14984
Gimmler, A., and Stoeck, T. (2015). Mining environmental high-throughput sequence data sets to identify divergent amplicon clusters for phylogenetic reconstruction and morphotype visualization. Environ. Microbiol. Rep. 7, 679–686. doi: 10.1111/1758-2229.12307
Gimmler, A., Korn, R., de Vargas, C., Audic, S., and Stoeck, T. (2016). The Tara Oceans voyage reveals global diversity and distribution patterns of marine planktonic ciliates. Sci. Rep. 6:33555. doi: 10.1038/srep33555
Giner, C. R., Balagué, V., Krabberød, A. K., Ferrera, I., Reñé, A., Garcés et al. (2019). Quantifying long-term recurrence in planktonic microbial eukaryotes. Mol. Ecol. 28, 923–935. doi: 10.1111/mec.14929
Gómez, F., and Gorsky, G. (2003). Annual microplankton cycles in Villefranche Bay, Ligurian Sea, NW Mediterranean. J. Plankton Res. 25, 323–339. doi: 10.1093/plankt/25.4.323
Gong, J., Dong, J., Liu, X., and Massana, R. (2013). Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 164, 369–379. doi: 10.1016/j.protis.2012.11.006
Grattepanche, J.-D., Santoferrara, L. F., McManus, G. B., and Katz, L. A. (2015). Distinct assemblage of planktonic ciliates dominates both photic and deep waters on the New England shelf. Mar. Ecol. Prog. Ser. 526, 1–9. doi: 10.3354/meps11256
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Hada, Y. (1938). Studies on the Tintinnoinea from the western tropical Pacific. J. Fac. Sci. Hokkaido Imp. Univ. Ser. VI Zool. 6, 87–190.
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Hu, X., Lin, X., and Song, W. (2019). Ciliate Atlas: Species Found in the South China Sea. Singapore: Science Press & Springer Nature.
Huang, H., Yang, J., Huang, S., Gu, B., Wang, Y., Wang, L., et al. (2021). Spatial distribution of planktonic ciliates in the western Pacific Ocean: along the transect from Shenzhen (China) to Pohnpei (Micronesia). Mar. Life Sci. Technol. 3, 103–115. doi: 10.1007/s42995-020-00075-7
Hütter, T., Ganser, M. H., Kocher, M., Halkic, M., Agatha, S., and Augsten, N. (2020). DeSignate: detecting signature characters in gene sequence alignments for taxon diagnoses. BMC Bioinformatics 21:151. doi: 10.1186/s12859-020-3498-6
Jenkins, D. G., Medley, K. A., and Franklin, R. B. (2011). “Microbes as a test of biogeographic principles,” in Biogeography of Microscopic Organisms: is Everything Small Everywhere?, ed. D. Fontaneto (Cambridge: Cambridge University Press), 309–323. doi: 10.1017/cbo9780511974878.016
Jiang, Y., Xu, H., Zhu, M., and Al-Rasheid, K. A. S. (2013). Temporal distributions of microplankton populations and relationships to environmental conditions in Jiaozhou Bay, northern China. J. Mar. Biol. Ass. U.K. 93, 13–26. doi: 10.1017/s0025315412001324
Jung, J.-H., Moon, J. H., Park, K.-M., Kim, S., Dolan, J. R., and Yang, E. J. (2018). Novel insights into the genetic diversity of Parafavella based on mitochondrial CO1 sequences. Zool. Scr. 47, 743–755. doi: 10.1111/zsc.12312
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katz, L. A., McManus, G. B., Snoeyenbos-West, O. L. O., Griffin, A., Pirog, K., Costas, B., et al. (2005). Reframing the ‘everything is everywhere’ debate: evidence for high gene flow and diversity in ciliate morphospecies. Aquat. Microb. Ecol. 41, 55–65. doi: 10.3354/ame041055
Kim, S. Y., Xu, D., Jung, J.-H., and Choi, J. K. (2020). Phylogeny and genetic/morphological variation of Strombidinopsis minima-like species (Ciliophora: Choreotrichia). J. Eukaryot. Microbiol. 67, 115–124. doi: 10.1111/jeu.12761
Kim, Y.-O., and Taniguchi, A. (1997). Seasonal variation of excystment pattern of the planktonic oligotrich ciliate Strombidium conicum. Mar. Biol. 128, 207–212. doi: 10.1007/s002270050084
Kim, Y.-O., Ha, S., and Taniguchi, A. (2008). Morphology and in situ sedimentation of the cysts of a planktonic oligotrich ciliate, Strombidium capitatum. Aquat. Microb. Ecol. 53, 173–179. doi: 10.3354/ame01241
Kim, Y.-O., Suzuki, T., and Taniguchi, A. (2002). A new species in the genus Cyrtostrombidium (Ciliophora, Oligotrichia, Oligotrichida): its morphology, seasonal cycle and resting stage. J. Eukaryot. Microbiol. 49, 338–343. doi: 10.1111/j.1550-7408.2002.tb00380.x
Laackmann, H. (1910). Die Tintinnodeen der Deutschen Südpolar-Expedition 1901-1903. Deutsche Südpolar-Expedition 1901-1903 im Auftrage des Reichsamtes des Innern. XI. Zoologie 3, 342–496.
Lahr, D. J. G., and Katz, L. A. (2009). Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. BioTechniques 47, 857–866. doi: 10.2144/000113219
Lara, E., Berney, C., Harms, H., and Chatzinotas, A. (2007). Cultivation-independent analysis reveals a shift in ciliate 18S rRNA gene diversity in a polycyclic aromatic hydrocarbon-polluted soil. FEMS Microbiol. Ecol. 62, 365–373. doi: 10.1111/j.1574-6941.2007.00387.x
Lipps, J. H., Stoeck, T., and Dunthorn, M. (2013). “Fossil tintinnids,” in The Biology and Ecology of Tintinnid Ciliates: Models for Marine Plankton, eds J. R. Dolan, D. J. S. Montagnes, S. Agatha, D. W. Coats, and D. K. Stoecker (Oxford: John Wiley & Sons, Ltd), 186–197. doi: 10.1002/9781118358092.ch8
Liu, W., Jiang, J., Xu, Y., Pan, X., Qu, Z., Luo, X., et al. (2017). Diversity of free-living marine ciliates (Alveolata, Ciliophora): faunal studies in coastal waters of China during the years 2011-2016. Eur. J. Protistol. 61, 424–438. doi: 10.1016/j.ejop.2017.04.007
Logares, R., Audic, S., Bass, D., Bittner, L., Boutte, C., Christen, R., et al. (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 24, 813–821. doi: 10.1016/j.cub.2014.02.050
Logares, R., Deutschmann, I. M., Junger, P. C., Giner, C. R., Krabberød, A. K., Schmidt et al. (2020). Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8:55. doi: 10.1186/s40168-020-00827-8
Luporini, P., Alimenti, C., Pedrini, B., and Vallesi, A. (2016). “Ciliate communication via water-borne pheromones,” in Biocommunication of Ciliates, eds G. Witzany and M. Nowacki (Cham: Springer), 159–174. doi: 10.1007/978-3-319-32211-7_10
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2015). Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ 3:e1420. doi: 10.7717/peerj.1420
Markmann, M., and Tautz, D. (2005). Reverse taxonomy: an approach towards determining the diversity of meiobenthic organisms based on ribosomal RNA signature sequences. Philos. Trans. R. Soc. B 360, 1917–1924. doi: 10.1098/rstb.2005.1723
Marteinsson, V. Þ., Groben, R., Reynisson, E., and Vannier, P. (2016). “Biogeography of marine microorganisms,” in The Marine Microbiome, eds L. J. Stal and M. S. Cretoiu (Cham: Springer), 187–207. doi: 10.1007/978-3-319-33000-6_6
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martín, P. V., Bucek, A., Bourguignon, T., and Pigolotti, S. (2020). Ocean currents promote rare species diversity in protists. bioRxiv [preprint] doi: 10.1101/2020.01.10.901165
Maselli, M., Altenburger, A., Stoecker, D. K., and Hansen, P. J. (2020). Ecophysiological traits of mixotrophic Strombidium spp. J. Plankton Res. 42, 485–496. doi: 10.1093/plankt/fbaa041
Massana, R., Gobet, A., Audic, S., Bass, D., Bittner, L., Boutte, C., et al. (2015). Marine protist diversity in European coastal waters and sediments as revealed by high-throughput sequencing. Environ. Microbiol. 17, 4035–4049. doi: 10.1111/1462-2920.12955
McManus, G. B., and Santoferrara, L. F. (2013). “Tintinnids in microzooplankton communities,” in The Biology and Ecology of Tintinnid Ciliates: Models for Marine Plankton, eds J. R. Dolan, D. J. S. Montagnes, S. Agatha, D. W. Coats, and D. K. Stoecker (Oxford: John Wiley & Sons, Ltd), 198–213. doi: 10.1002/9781118358092.ch9
McManus, G. B., Xu, D., Costas, B. A., and Katz, L. A. (2010). Genetic identities of cryptic species in the Strombidium stylifer/apolatum/oculatum cluster, including a description of Strombidium rassoulzadegani n. sp. J. Eukaryot. Microbiol. 57, 369–378. doi: 10.1111/j.1550-7408.2010.00485.x
McManus, M. A., and Woodson, C. B. (2012). Plankton distribution and ocean dispersal. J. Exp. Biol. 215, 1008–1016. doi: 10.1242/jeb.059014
Merkle, H. (1909). Untersuchungen an Tintinnodeen der Ost- und Nordsee. Wiss. Meeresunters., Abt. Kiel 11, 139–186 + Plates II, III.
Meunier, A. (1910). Microplankton des Mers de Barents et de Kara. Campagne Arctique de 1907. Bruxelles: C. Bulens.
Minicante, S. A., Piredda, R., Quero, G. M., Finotto, S., Aubry, F. B., Bastianini, M., et al. (2019). Habitat heterogeneity and connectivity: effects on the planktonic protist community structure at two adjacent coastal sites (the Lagoon and the Gulf of Venice, Northern Adriatic Sea, Italy) revealed by metabarcoding. Front. Microbiol. 10:2736.
Mitra, A., Skrzypczak, M., Ginalski, K., and Rowicka, M. (2015). Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using Illumina platform. PLoS One 10:e0120520. doi: 10.1371/journal.pone.0120520
Moritz, C., and Cicero, C. (2004). DNA barcoding: promise and pitfalls. PLoS Biol. 2:e354. doi: 10.1371/journal.pbio.0020354
Mozetič, P., Fonda Umani, S., Cataletto, B., and Malej, A. (1998). Seasonal and inter-annual plankton variability in the Gulf of Trieste (northern Adriatic). ICES J. Mar. Sci. 55, 711–722. doi: 10.1006/jmsc.1998.0396
Newman, M. E. J. (2003). Mixing patterns in networks. Phys. Rev. E 67:026126. doi: 10.1103/PhysRevE.67.026126
Norris, R. D. (2000). Pelagic Species Diversity, Biogeography, and Evolution. Cambridge: Cambridge University Press
Palumbi, S. R. (1994). Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 25, 547–572. doi: 10.1146/annurev.es.25.110194.002555
Pienaar, E., Theron, M., Nelson, M., and Viljoen, H. J. (2006). A quantitative model of error accumulation during PCR amplification. Comput. Biol. Chem. 30, 102–111. doi: 10.1016/j.compbiolchem.2005.11.002
Pierce, R. W., and Turner, J. T. (1992). Ecology of planktonic ciliates in marine food webs. Rev. Aquat. Sci. 6, 139–181.
Pierce, R. W., and Turner, J. T. (1993). Global biogeography of marine tintinnids. Mar. Ecol. Prog. Ser. 94, 11–26. doi: 10.3354/meps094011
Piredda, R., Tomasino, M. P., D’Erchia, A. M., Manzari, C., Pesole, G., Montresor, R., et al. (2017). Diversity and temporal patterns of planktonic protist assemblages at a Mediterranean Long Term Ecological Research site. FEMS Microbiol. Ecol. 93:fiw200. doi: 10.1093/femsec/fiw200
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
R Core Team (2018). R: a Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rubino, F., Moscatello, S., Saracino, O. D., Fanelli, G., Belmonte, G., and Boero, F. (2002). Plankton-derived resting stages in marine coastal sediments along the Salento Peninsula (Apulia, south-eastern Italy). Mar. Ecol. 23, (Suppl. 1), 329–339. doi: 10.1111/j.1439-0485.2002.tb00031.x
Santoferrara, L. F., Alder, V. V., and McManus, G. B. (2017). Phylogeny, classification and diversity of Choreotrichia and Oligotrichia (Ciliophora, Spirotrichea). Mol. Phylogenet. Evol. 112, 12–22. doi: 10.1016/j.ympev.2017.03.010
Santoferrara, L. F., Bachy, C., Alder, V. A., Gong, J., Kim, Y.-O., Saccà, A., et al. (2016a). Updating biodiversity studies in loricate protists: the case of the tintinnids (Alveolata, Ciliophora, Spirotrichea). J. Eukaryot. Microbiol. 63, 651–656. doi: 10.1111/jeu.12303
Santoferrara, L. F., Grattepanche, J.-D., Katz, L. A., and McManus, G. B. (2014). Pyrosequencing for assessing diversity of eukaryotic microbes: analysis of data on marine planktonic ciliates and comparison with traditional methods. Environ. Microbiol. 16, 2752–2763. doi: 10.1111/1462-2920.12380
Santoferrara, L. F., Grattepanche, J.-D., Katz, L. A., and McManus, G. B. (2016b). Patterns and processes in microbial biogeography: do molecules and morphologies give the same answers? ISME J. 10, 1779–1790. doi: 10.1038/ismej.2015.224
Santoferrara, L. F., Rubin, E., and McManus, G. B. (2018). Global and local DNA (meta)barcoding reveal new biogeography patterns in tintinnid ciliates. J. Plankton Res. 40, 209–221. doi: 10.1093/plankt/fby011
Santoferrara, L. F., Tian, M., Alder, V. A., and McManus, G. B. (2015). Discrimination of closely related species in tintinnid ciliates: new insights on crypticity and polymorphism in the genus Helicostomella. Protist 166, 78–92. doi: 10.1016/j.protis.2014.11.005
Santoferrara, L., Burki, F., Filker, S., Logares, R., Dunthorn, M., and McManus, G. B. (2020). Perspectives from ten years of protist studies by high-throughput metabarcoding. J. Eukaryot. Microbiol. 67, 612–622. doi: 10.1111/jeu.12813
Schmidt, T. S. B., Matias Rodrigues, J. F., and von Mering, C. (2015). Limits to robustness and reproducibility in the demarcation of operational taxonomic units. Environ. Microbiol. 17, 1689–1706. doi: 10.1111/1462-2920.12610
Shapiro, B. J., Leducq, J.-B., and Mallet, J. (2016). What is speciation? PLoS Genet. 12:e1005860. doi: 10.1371/journal.pgen.1005860
Sherr, E., and Sherr, B. (1988). Role of microbes in pelagic food webs: a revised concept. Limnol. Oceanogr. 33, 1225–1227. doi: 10.4319/lo.1988.33.5.1225
Shimodaira, H. (2002). An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508. doi: 10.1080/10635150290069913
Siegenthaler, A., Wangensteen, O. S., Soto, A. Z., Benvenuto, C., Corrigan, L., and Mariani, S. (2019). Metabarcoding of shrimp stomach content: harnessing a natural sampler for fish biodiversity monitoring. Mol. Ecol. Resour. 19, 206–220. doi: 10.1111/1755-0998.12956
Song, W., Zhao, X., Liu, W., Hu, X., Al-Farraj, S. A., Al-Rasheid, K. A. S., et al. (2015). Biodiversity of oligotrich ciliates in the South China Sea: description of three new Strombidium species (Protozoa, Ciliophora, Oligotrichia) with phylogenetic analyses. Syst. Biodivers. 13, 608–623. doi: 10.1080/14772000.2015.1081992
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H.-W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, (Suppl. 1), 21–31. doi: 10.1111/j.1365-294x.2009.04480.x
Stoecker, D. K., and Capuzzo, J. M. (1990). Predation on protozoa: its importance to zooplankton. J. Plankton Res. 12, 891–908. doi: 10.1093/plankt/12.5.891
Sun, P., Huang, L., Xu, D., Warren, A., Huang, B., Wang, Y., et al. (2019). Integrated space-time dataset reveals high diversity and distinct community structure of ciliates in mesopelagic waters of the northern South China Sea. Front. Microbiol. 10:2178. doi: 10.3389/fmicb.2019.02178
Sun, Y., Cai, Y., Huse, S. M., Knight, R., Farmerie, W. G., Wang, X., et al. (2012). A large-scale benchmark study of existing algorithms for taxonomy-independent microbial community analysis. Brief. Bioinform. 13, 107–121. doi: 10.1093/bib/bbr009
Tavaré, S. (1986). Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math. Life Sci. 17, 57–86.
Tucker, S. J., McManus, G. B., Katz, L. A., and Grattepanche, J.-D. (2017). Distribution of abundant and active planktonic ciliates in coastal and slope waters off New England. Front. Microbiol. 8:2178.
Van der Spoel, S., and Heyman, R. P. (1983). A Comparative Atlas of Zooplankton - Biological Patterns in the Oceans. Berlin: Springer-Verlag.
Wang, C., Zhang, T., Wang, Y., Katz, L. A., Gao, F., and Song, W. (2017). Disentangling sources of variation in SSU rDNA sequences from single cell analyses of ciliates: impact of copy number variation and experimental error. Proc. R. Soc. London Ser. B 284:20170425. doi: 10.1098/rspb.2017.0425
Wang, Y., Wang, C., Jiang, Y., Katz, L. A., Gao, F., and Yan, Y. (2019). Further analyses of variation of ribosome DNA copy number and polymorphism in ciliates provide insights relevant to studies of both molecular ecology and phylogeny. Sci. China Life Sci. 62, 203–214. doi: 10.1007/s11427-018-9422-5
Warren, A., Patterson, D. J., Dunthorn, M., Clamp, J. C., Achilles-Day, U. E. M., Aescht, E., et al. (2017). Beyond the “Code”: a guide to the description and documentation of biodiversity in ciliated protists (Alveolata, Ciliophora). J. Eukaryot. Microbiol. 64, 539–554. doi: 10.1111/jeu.12391
Weiner, A. K. M., Weinkauf, M. F. G., Kurasawa, A., Darling, K. F., Kucera, M., and Grimm, G. W. (2014). Phylogeography of the tropical planktonic foraminifera lineage Globigerinella reveals isolation inconsistent with passive dispersal by ocean currents. PLoS One 9:e92148. doi: 10.1371/journal.pone.0092148
Weisse, T. (2008). Distribution and diversity of aquatic protists: an evolutionary and ecological perspective. Biodivers. Conserv. 17, 243–259. doi: 10.1007/s10531-007-9249-4
Will, K. W., and Rubinoff, D. (2004). Myth of the molecule: DNA barcodes for species cannot replace morphology for identification and classification. Cladistics 20, 47–55. doi: 10.1111/j.1096-0031.2003.00008.x
Witek, M. (1998). Annual changes of abundance and biomass of planktonic ciliates in the Gdańsk basin, southern Baltic. Int. Rev. Hydrobiol. 83, 163–182. doi: 10.1002/iroh.19980830207
Xu, D., Sun, P., Shin, M. K., and Kim, Y. O. (2012). Species boundaries in tintinnid ciliates: a case study - morphometric variability, molecular characterization, and temporal distribution of Helicostomella species (Ciliophora, Tintinnina). J. Eukaryot. Microbiol. 59, 351–358. doi: 10.1111/j.1550-7408.2012.00625.x
Yang, J., Löder, M. G. J., and Wiltshire, K. H. (2014). A survey of ciliates at the long-term sampling station “Helgoland Roads”, North Sea. Helgol. Mar. Res. 68, 313–327. doi: 10.1007/s10152-014-0392-5
Zeitzschel, B. (1966). Die Verbreitung der Tintinnen im Nordatlantik. Veröff. Inst. Meeresforsch. Bremerh. Sonderbd. 2, 293–300.
Zeitzschel, B. (1969). Tintinnen des westlichen Arabischen Meeres, ihre Bedeutung als Indikatoren für Wasserkörper und Glied der Nahrungskette. ”Meteor”. Forschungsergeb 4, 47–101.
Zeitzschel, B. (1990). “Zoogeography of marine protozoa: an overview emphasizing distribution of planktonic forms,” in Ecology of Marine Protozoa, ed. G. M. Capriulo (Oxford: Oxford University Press), 139–185.
Zhang, Q., Agatha, S., Zhang, W., Dong, J., Yu, Y., Jiao, N., et al. (2017). Three rDNA loci-based phylogenies of tintinnid ciliates (Ciliophora, Spirotrichea, Choreotrichida). J. Eukaryot. Microbiol. 64, 226–241. doi: 10.1111/jeu.12354
Zhao, F., Filker, S., Xu, K., Li, J., Zhou, T., and Huang, P. (2019). Effects of intragenomic polymorphism in the SSU rRNA gene on estimating marine microeukaryotic diversity: a test for ciliates using single-cell high-throughput DNA sequencing. Limnol. Oceanogr. Meth. 17, 533–543. doi: 10.1002/lom3.10330
Keywords: distribution, metabarcoding, network analyses, phylogenetic analyses, protists, second-level clustering, Swarm OTU
Citation: Ganser MH, Forster D, Liu W, Lin X, Stoeck T and Agatha S (2021) Genetic Diversity in Marine Planktonic Ciliates (Alveolata, Ciliophora) Suggests Distinct Geographical Patterns – Data From Chinese and European Coastal Waters. Front. Mar. Sci. 8:643822. doi: 10.3389/fmars.2021.643822
Received: 18 December 2020; Accepted: 24 March 2021;
Published: 13 April 2021.
Edited by:
Thomas Wilke, University of Giessen, GermanyReviewed by:
Xavier Turon, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright © 2021 Ganser, Forster, Liu, Lin, Stoeck and Agatha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maximilian H. Ganser, bWF4aW1pbGlhbi5nYW5zZXJAc2JnLmFjLmF0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.