 MJosé Pérez-Alvarez1,2,3*
MJosé Pérez-Alvarez1,2,3* Sebastián Kraft2
Sebastián Kraft2 Nicolás I. Segovia2,4
Nicolás I. Segovia2,4 Carlos Olavarría3,5
Carlos Olavarría3,5 Sergio Nigenda-Morales6,7
Sergio Nigenda-Morales6,7 Jorge Urbán R.8
Jorge Urbán R.8 Lorena Viloria-Gómora8Frederick Archer9Rodrigo Moraga3
Lorena Viloria-Gómora8Frederick Archer9Rodrigo Moraga3 Maritza Sepúlveda3,10
Maritza Sepúlveda3,10 Macarena Santos-Carvallo3,10Guido Pavez3,10,11
Macarena Santos-Carvallo3,10Guido Pavez3,10,11 Elie Poulin2,11*
Elie Poulin2,11*- 1Facultad de Ciencias, Escuela de Medicina Veterinaria, Universidad Mayor, Santiago, Chile
- 2Laboratorio de Ecología Molecular, Facultad de Ciencias, Instituto de Ecología y Biodiversidad (IEB), Universidad de Chile, Santiago, Chile
- 3Centro de Investigación EUTROPIA, Santiago, Chile
- 4Departamento de Biología Marina, Facultad de Ciencias del Mar, Universidad Católica del Norte, Coquimbo, Chile
- 5Centro de Estudios Avanzados en Zonas Áridas (CEAZA), La Serena, Chile
- 6Laboratorio Nacional de Genómica para la Biodiversidad (LANGEBIO), Centro de Investigaciones y Estudios Avanzados (CINVESTAV), Unidad de Genómica Avanzada, Irapuato, Mexico
- 7Department of Ecology and Evolutionary Biology, University of California, Los Angeles, Los Angeles, CA, United States
- 8Departamento de Ciencias Marinas y Costeras, Universidad Autónoma de Baja California Sur, La Paz, Mexico
- 9Southwest Fisheries Science Center, La Jolla, CA, United States
- 10Centro de Investigación y Gestión de Recursos Naturales (CIGREN), Facultad de Ciencias, Universidad de Valparaíso, Valparaíso, Chile
- 11Departamento de Ciencias Ecológicas, Facultad de Ciencias, Universidad de Chile, Santiago, Chile
Four fin whale sub-species are currently considered valid: Balaenoptera physalus physalus in the North Atlantic, B. p. velifera in the North Pacific, B. p. quoyi and B. p. patachonica in the Southern Hemisphere. The last, not genetically validated, was described as a pygmy-type sub-species, found in low to mid latitudes of the Southern Hemisphere. Genetic analyses across hemispheres show strong phylogeographic structure, yet low geographic coverage in middle latitudes of the Southern Hemisphere impeded an assessment within the area, as well as evaluating the validity of B. p. patachonica. New mtDNA sequences from the Southeastern Pacific allowed an improved coverage of the species’ distribution. Our phylogenetic analyses showed three main lineages and contrasting phylogeographic patterns between Northern and Southern Hemispheres. Absence of recurrent female mediated gene flow between hemispheres was found; however, rare dispersal events revealing old migrations were noted. The absence of genetic structure suggests the existence of one single taxa within the Southern Hemisphere. Thus, until further evidence supporting this subspecies can be produced, such as genetic, ecological, behavioral, or morphological data, we propose that all fin whales from the Southern Hemisphere, including those from middle latitudes of the Southeastern Pacific belong to B. p. quoyi subspecies. This information is important for the current assessment of fin whales, contributing to the evaluation of the taxonomic classification and the conservation of the species.
Introduction
The fin whale (Balaenoptera physalus) is a widespread mysticete that occurs in all major oceans (Mizroch et al., 1984), particularly in middle and high latitudes (Mackintosh, 1966; Miyashita et al., 1995; Branch and Butterworth, 2001; Reilly et al., 2013) and mainly, but not exclusively, in offshore waters (Edwards et al., 2015). Four sub-species are currently considered valid by the Committee on Taxonomy (Taxonomy, 2020): (1) B. physalus physalus (Linnaeus, 1758) in the North Atlantic, the recently described (2) B. p. velifera in the North Pacific (Cope in Scammon, 1869), and (3) B. p. quoyi (Fischer, 1829) and (4) B. p. patachonica (Burmeister, 1865), which are found in the Southern Hemisphere (SH).
The taxonomic classification of the Northern (B. p. physalus) and Southern Hemisphere (B. p. quoyi) subspecies was proposed in the mid twentieth century (Tomilin, 1946), primarily based on size differences between fin whales from the Northern Hemisphere (NH) and Antarctic waters. This was later verified by comparisons of body measurements and organ weights of North Atlantic and Antarctic individuals (Lockyer and Waters, 1986). The fourth subspecies, B. p. patachonica, was defined as a pygmy-type (approximately 18–24 m total length) and dark in coloration, possibly with black baleen. Its distribution was described as being restricted to low to mid-latitudes in the SH, not extending further south than approximately 55°S, hence feeding at lower latitudes than most fin whales (Clarke, 2004). Aside from the original description, there has been no further evidence supporting this subspecies.
Strong genetic differentiation was detected in the Northern Hemisphere, between whales of the North Pacific (NP) and North Atlantic (NA) (Bérubé et al., 1998; Archer et al., 2013; Cabrera et al., 2019). Based on new genetic evidence, Archer et al. (2019) recently formally described fin whales in the North Pacific as a new subspecies, B. p. velifera, which was accepted by the Committee on Taxonomy (Taxonomy, 2020). Additionally, Jiménez López et al. (2019) presented evidence supporting previous findings that fin whales from the Gulf of California (GoC) are a small year-round resident population isolated from the rest of the NP (Bérubé et al., 2002; Urbán et al., 2005; Nigenda-Morales et al., 2008; Rivera-León et al., 2019). Genetic analyses between the Northern and Southern Hemisphere show a strong phylogeographic structure among NP, NA, and SH fin whale populations (Archer et al., 2013, 2019; Cabrera et al., 2019). However, the reduced sample size, restricted geographic coverage in the SH, and inability to positively associate morphological characteristics of putative pygmy fin whales with those sampled impeded the assessment of potential phylogeographic structure within this area and evaluation of the validity of the putative B. p. patachonica sub-species (Archer et al., 2013, 2019).
The occurrence of fin whales has recently become regular in mid-latitudes of Southeastern Pacific (SEP), in particular along the Chilean coast between 23°S and 29°S in spring and summer (Pérez et al., 2006; Pacheco et al., 2015; Toro et al., 2016; Sepúlveda et al., 2018). Foraging areas for the species have been identified within this zone (Pérez et al., 2006; Toro et al., 2016; Sepúlveda et al., 2018). If pygmy fin whales effectively inhabit the Southern Ocean from around 35°S (Burmeister, 1865) to ∼55°S, and from the Equator southwards along the Pacific coast of South America (Clarke, 2004), fin whales found in these lower SH latitudes may potentially correspond to the B. p. patachonica subspecies.
Here, we present a global phylogeographic study of fin whales, increasing the geographical coverage of previous studies by adding 37 new sequences from the Southeastern Pacific as well as longer sequences from the Gulf of California. We evaluate the genetic structure among fin whale populations within the Southern Hemisphere, the existence of two SH subspecies, the degree of genetic structure between North Pacific and Southeastern Pacific populations, and differentiation the GoC and the wider North Pacific. Finally, we discuss the existence of the putative pygmy fin whale sub-species B.p. patachonica.
Materials and Methods
Sample Collection and Laboratory Procedures
Skin samples of fin whales were obtained from free-swimming whales by biopsy darting (Harlin et al., 1999). Biopsy samples were collected under permits 1502/2013, 1803/2015, and 2982/2015 from the Chilean Undersecretariat of Fisheries and Aquaculture and with approval and permits issued by the Mexican Wildlife Agency (Dirección General de Vida Silvestre, Subsecretaría de Gestión para la Protección Ambiental, Secretaría del Medio Ambiente y Recursos Naturales). All methods were carried out in accordance with relevant guidelines and regulations. Experimental protocols were approved by the Bioethical Committee at the Institute of Ecology and Biodiversity, Chile (IEB). Potentially re-sampled individuals among the GoC samples were ruled out by microsatellite analysis comparisons (13 loci, Cervus v3.0, Kalinowski et al., 2007), while genetic sampling in the SEP was guided by simultaneous photo-identification of individuals.
DNA extraction of samples from north-central Chile (ca. 29°02′S, 71°36′W) (n = 36) followed the salt extraction method (Aljanabi and Martinez, 1997). The sex of each individual was identified by simultaneously amplifying a fragment of the ZFX/ZFY genes and a fragment of the SRY gene (Aasen and Medrano, 1990; Gilson et al., 1998). A 720 base pair (bp) fragment of both forward and reverse strands of the mitochondrial DNA control region (Dloop) was amplified, using the primers M13 Dlp1.5 5′-TGTAAAACGACAGCCAGTTCACCCAAAGCTGRARTTCTA-3′ and 8G 5′-GGAGTACTATGTCCTGTAACCA-3′ (Dalebout et al., 2005). Amplifications were performed in a total reaction volume of 25.6 μL, which included 12.7 μL water, 5 μL 10X Buffer (Invitrogen), 2 μL 50 mM MgCl2 (Invitrogen), 2 μL 10 pM dNTPs (Invitrogen), 1 μL 10 pM of each primer (2 μL total), 0.5 μL Taq polymerase (Invitrogen) and 100 ng of DNA. Sequences were edited and aligned by eye in ProSeq v3.0 (Filatov, 2009) and haplotypes were identified. Additionally, 107 sequences from the Gulf of California were obtained. For these samples, the DNA was extracted using the QIAmp DNA mini kit (Qiagen, Valencia, CA, United States) according to the manufacturer’s protocol. The primers used were the same as previously mentioned, but amplifications conditions and cycling were as follows: denaturation at 95°C for 3 min, followed by 34 cycles at 95°C for 30 s, 50°C for 45 s, 72°C for 45 s and a final extension at 72°C for 10 min. Reactions were performed in a total volume of 50 μl, with 2.5 mM MgCl2, 600 mM each deoxynucleotide triphosphate (dNTP), 4 pmol each primer, 1 unit of Taq DNA polymerase (Sigma-Aldrich, St Louis, MO, United States), 1 μl 10 mg/ml BSA and approximately 50–100 ng of template DNA. Amplified products were sequenced on an automated sequencer (ABI 3100, Applied Biosystems, Foster City, CA, United States). Sequence data were edited in SEQUENCHER v4.1 (Gene Codes Corporation, Ann Arbor, MI, United States).
Finally, 153 additional sequences from the NP (n = 92), GoC (n = 5), SEP (n = 1), Southern Ocean (n = 42), and NA (n = 13) were retrieved from the mitogenomes published by Archer et al. (2013), one by Arnason et al. (1991) (NA, n = 1), and another 8 sequences from Cabrera et al. (2019) (NP, n = 1; NA, n = 7). Our final global dataset of 305 sequences was then stratified into four regions according to the geographic origin of samples: NP (n = 93), SH = 79 (SEP, n = 37 + Southern Ocean and two samples from the Indian Ocean, n = 42), GoC (n = 112), and NA (n = 21) (Figure 1). We only included sequences from previous publications with a minimum of 624 bp.
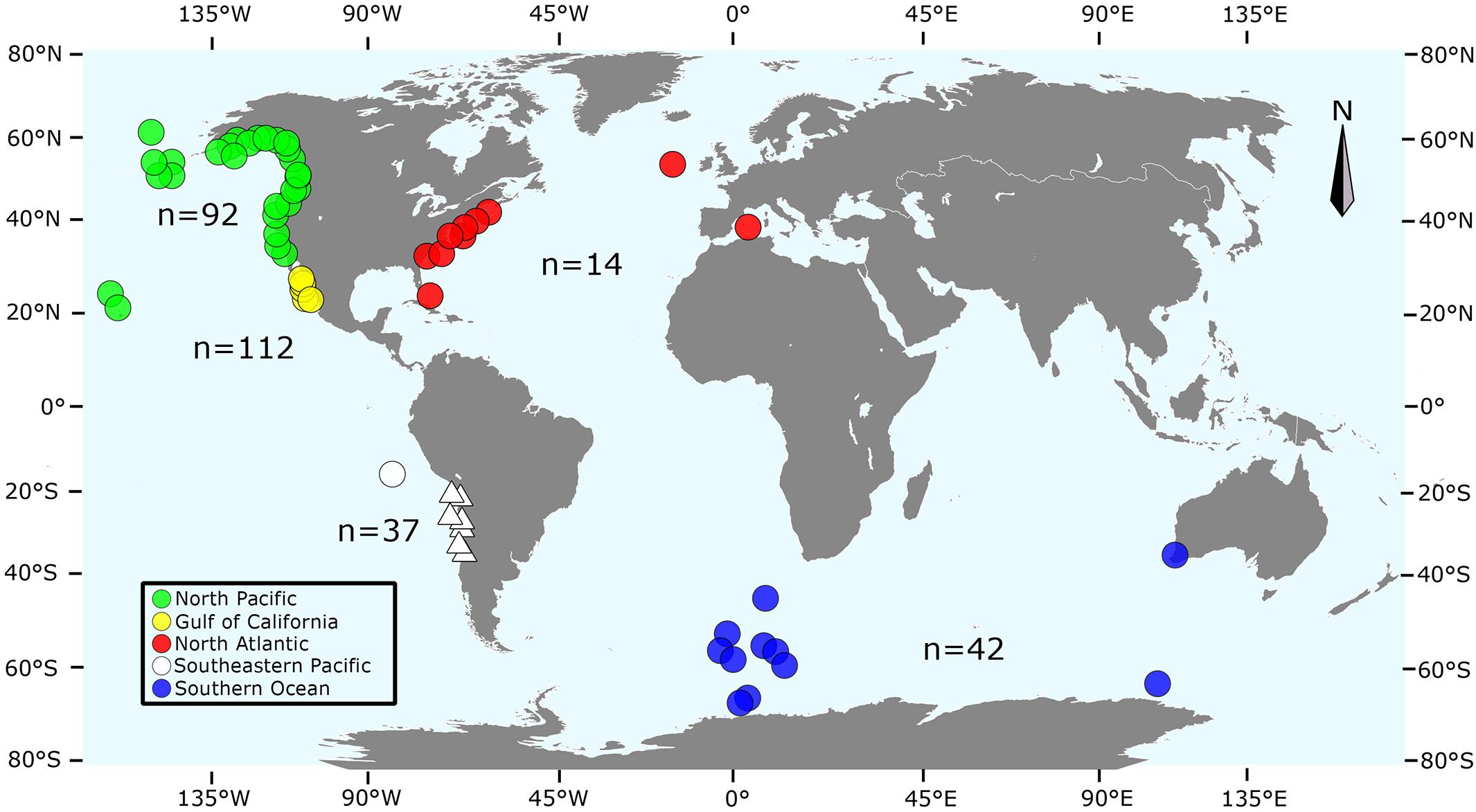
Figure 1. Geographic origin of the sequences included in this study: green (North Pacific, Archer et al., 2013), red (North Atlantic, Arnason et al., 1991; Archer et al., 2013), yellow (Gulf of California, this study), blue and white (Southern Hemisphere; circles are sequences from Archer et al. (2013), and triangles from this study). Numbers indicate the number of sequences within each locality. Map adapted from Archer et al. (2013), based on SVG SILH (https://svgsilh.com/image/306338.html), released as public domain under Creative Commons CC0 1.0. Figure generated by S.K. in Inkscape 0.92.4 (https://www.inkscape.org).
The eight mitogenome sequences from Cabrera et al. (2019) were trimmed to our D-loop segment length of 624 bp, and included in the construction of the haplotype network and the phylogenetic tree. However, these samples were not used for the genetic diversity and structure analyses. As explained by the authors, these sequences were deliberately selected for mitogenome amplification, which is inconsistent with a random sampling scheme.
Genetic Analyses
For all strata, we estimated the following genetic diversity indices: number of segregating sites (k), number of haplotypes (h), haplotype diversity (Hd), mean number of differences between two random sequences (Π) and nucleotide diversity (π). Analyses of phylogeographic structure (ΦST) that evaluate genetic structure considering both haplotype frequencies and genetic distance between haplotypes were conducted in Arlequin v3.5.2 (Excoffier et al., 2005) with 1,000 permutations and a significance level of 0.05. The haplotype network was constructed in Hapview1, a software package that uses phylogenetic trees to construct haplotype genealogies. For this, a neighbor-joining tree was constructed in MEGA v7 (Kumar et al., 2016) to serve as input file for the reconstruction of the haplotype network in Hapview.
To estimate the pairwise migration rates between geographic strata [NP, GoC, SH (SEP + SO) and NA], the software IMa2 was used (Hey, 2010). This was conducted only between groups that either shared haplotypes or showed discrepancy between genetic and geographic clusters (i.e., SH-NA, SH-NP, and GoC-NP). We carried out 10 preliminary runs in the M-mode of the software (Markov Chain Monte Carlo; MCMC mode), in order to select the set of priors that best improved mixing and convergence of the MCMC. These priors were then used to determine migration patterns based on calculations of effective population size (Θ1, Θ2, and ancestral Θa, Θ = 1,000) and splitting time (t = 100). We performed 10 × 108 MCMC steps using the HKY mutation model and sampled every 100 iterations, with an initial burn-in of 10%. Once MCMC convergence was achieved in the test runs (i.e., same results obtained with different runs through visual inspection of trace-plots and histograms of each parameter), simulated genealogies were used in the Load Tree mode (L-mode) to calculate the log of the maximum-likelihood and 95% HDP intervals for migration parameters. These were compared to a no migration scenario (null model), using a likelihood ratio-test.
For the phylogenetic reconstruction, we used multiple intra- and extra-specific calibration points, as proposed by Ho et al. (2008). This was done under the premise that if an estimate of evolutionary rate is calibrated using only an external calibration point, the intraspecific rate will be underestimated and the resulting divergence time will be overestimated. Thus, we performed a phylogenetic reconstruction of the fin whale haplotypes combining a phylogenetic mutation rate with a population substitution rate (Ho et al., 2008). For the former, we estimated divergence times among the three main clades using the phylogenetic rate, as it relates to the rate at which mutations are fixed among clades. For the latter, we used a population substitution rate to estimate the TMRCA of each clade, as it considers the rate at which new haplotypes appear in the clade.
Divergence times among the main lineages were first estimated in BEAST v2.6.0 (Bouckaert et al., 2014), using a relaxed molecular clock with a lognormal distribution and a phylogenetic substitution rate of 1.5% per million years (Ho et al., 2005). Additionally, given their close phylogenetic relationship (Sasaki et al., 2005; Archer et al., 2013), we used the humpback whale, Megaptera novaeangliae, as outgroup (GenBank accession mitochondrial DNA, complete genome Sequence ID: AP006467.1 Length: 16,398 bp), with an estimated divergence time from fin whales of 11 million years (Marx and Fordyce, 2015) as the primary calibration point. Bayesian Inference posterior probabilities were estimated using the Metropolis coupled MCMC mode, running four chains for 3 × 108 iterations, with trees sampled every 1,000 steps. The first 20% of the trees were discarded as burn-in and posterior probabilities were estimated as the fraction of trees in the clades of interest. Finally, posterior probability density was summarized as a maximum clade credibility tree using TREEANNOTATOR v.2.6.12 and visualized using FIGTREE v.1.43. The time to the Most Common Recent Ancestor (TMRCA) for each clade was estimated BEAST. For this, three independent Bayesian MCMC analyses with 100 × 106 iterations were carried out, considering a population substitution rate of 5.2% per million years (Alter and Palumbi, 2009) and a relaxed molecular clock analysis with an uncorrelated lognormal (ucln) distribution. Results of multiple runs were combined using LogCombiner v2.6.0 (Rambaut and Drummond, 2010), and convergence and TMRCA values were estimated with TRACER v1.6 (Rambaut et al., 2018). The final tree was constructed using the results of both approaches with the Chronos function in ape package (Paradis, 2013) in R v 4.0.5 (R Core Team, 2020), using a sequential secondary age calibration with the time of divergence of the main clades and TMRCA of each lineage.
Results
Genetic Diversity
All analyses were performed considering a final fragment of 624 bp of the mitochondrial DNA control region. We obtained 37 sequences from the SEP samples, which were composed of 26 different haplotypes. Of these haplotypes, 17 (65%) occurred only once, 7 (27%) were shared by two SEP individuals, and one haplotype (8%) was shared by three SEP individuals. Additionally, these samples presented high haplotype (Hd = 0.980) and nucleotide diversities (π% = 0.81). Compared to the rest of the SH, in a similar number of samples, the SEP had a lower number of haplotypes (SEP: n = 37, h = 26; rest of SH: n = 42, h = 40). SEP shared six haplotypes with the rest of SH and had similar haplotype diversities, only slightly lower in the SEP (Hd = 0.980 vs. 0.997) as well as nucleotide diversity (π% = 0.81 vs. 0.88). Overall, sequences from the SEP were mixed with those from other SH localities, with no separation visually evident in the haplotype network (Figure 2).
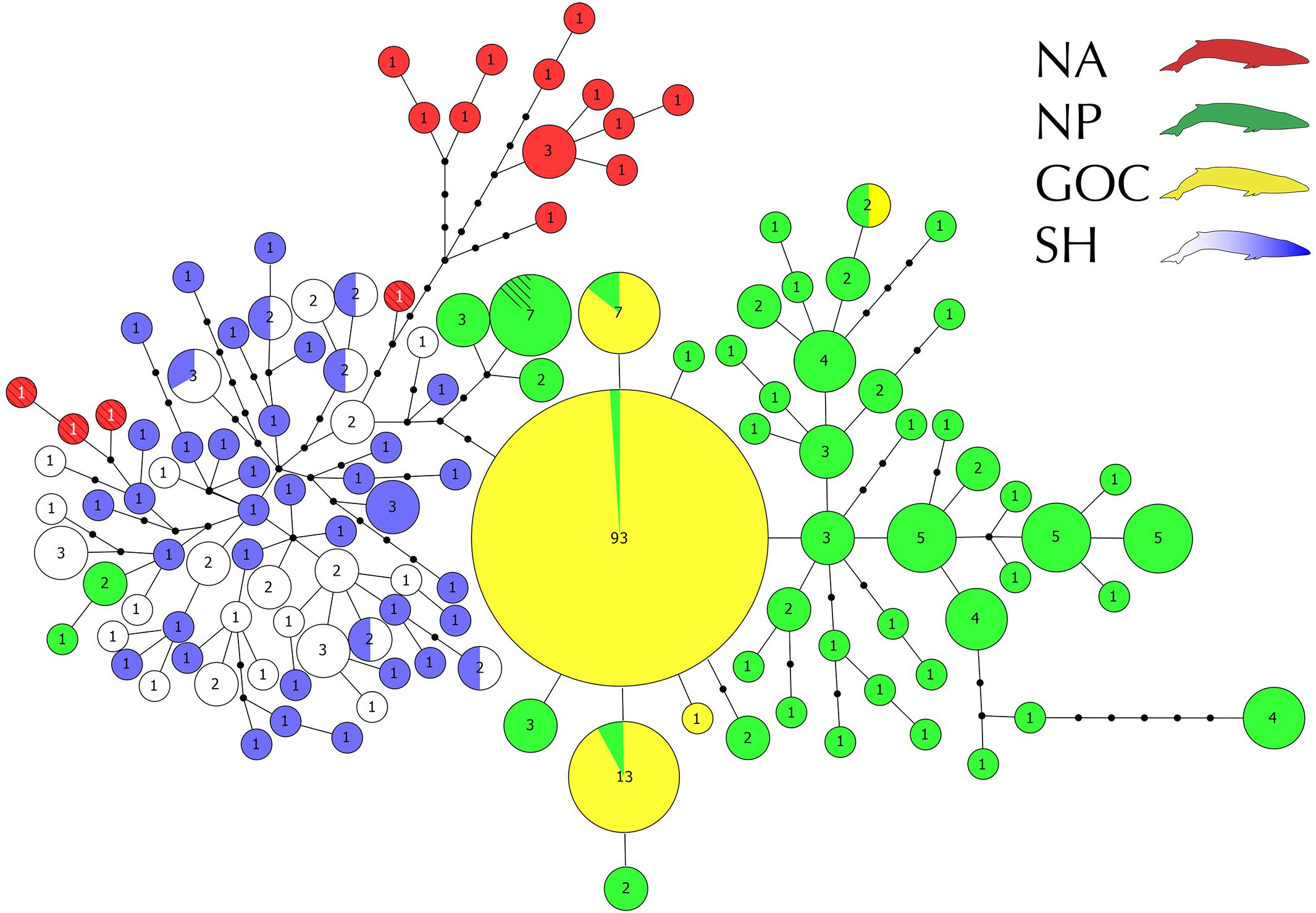
Figure 2. Global haplotype network of the 305 control region sequences constructed in Hapview. Localities are color coded as follows: North Pacific (green), Gulf of California (yellow), North Atlantic (red), Southern Ocean (blue) and southeastern Pacific (white). Circle size is proportional to the frequency, detailed within each haplotype. Black dots indicate one mutational step. The hatched haplotypes (four from NA red with white numbers and one from NP) were retrieved from Cabrera et al. (2019), their frequencies were not considered.
Comparing these results with the global control region diversity patterns of the species, similar high diversity was found in four of the five ocean basins (NP, NA, SEP, and SO, Hd range = 0.957–0.997) (Table 1). Haplotypic diversity in the GoC was approximately three times lower than in these regions (Hd = 0.314). This low diversity is the result of a single haplotype representing 93 of the 112 (83%) samples.
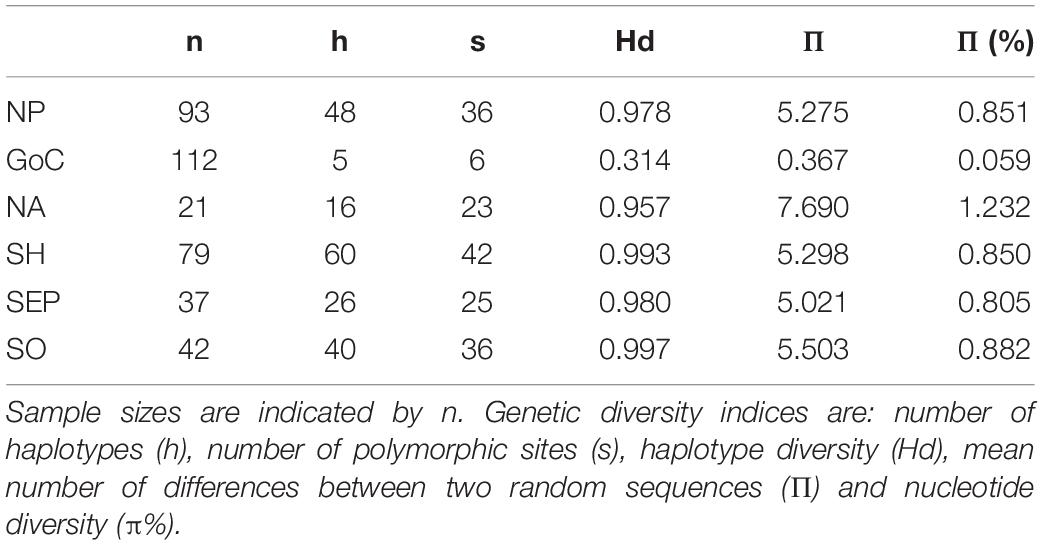
Table 1. Genetic diversity of fin whale populations by geographic origin: NP, North Pacific; GoC, Gulf of California; NA, North Atlantic; Southern Hemisphere [SH = southeastern Pacific (SEP) + Southern Ocean (SO)].
Phylogeographic Structure
Three main haplogroups can be distinguished in the resulting haplotype network (Figure 2). Each group is associated with a main geographic area (i.e., SH, NA, and NP + GoC). Samples from the SEP (the new sequences generated here plus one from Archer et al., 2013) and from the Southern Ocean (Archer et al., 2013) formed a single Southern Hemisphere haplogroup, sharing six haplotypes and showing a homogeneous distribution within the SH network topology. Fourteen NA haplotypes (13 sequences from Archer et al., 2013, one from Arnason et al., 1991) formed a separate haplogroup, while the 4 NA haplotypes from Cabrera et al. (2019) joined the SH haplogroup. Finally, the NP formed a third haplogroup and, as with the NA, a few NP haplotypes were associated with the SH haplogroup. Similarly, in the Northern Hemisphere, haplotypes sampled in GoC were associated with the NP haplogroup. However, most of the GoC haplotypes were found at higher frequencies than in other areas, contrasting with the high diversity patterns observed in the NP, NA and SH. Forty-eight haplotypes were identified among the 93 NP individuals, while only five haplotypes with few mutational steps among them (from 1 to 4) were found among the 112 GoC samples (Figure 2).
High and significant phylogeographic structure was detected among all geographic areas considered here (Figure 3), except within the Southern Hemisphere (ΦST = 0.007, p = 0.157). The highest ΦST values were obtained comparing the GoC population to NA and SH (ΦST = 0.800 and 0.656, respectively). Phylogeographic structure indices among NP, NA and SH were lower, ranging from 0.305 to 0.441, while ΦST between NP and GoC showed the lowest value (0.278). This value was highly significant, probably due to the higher frequency of three GoC haplotypes (Figure 3).
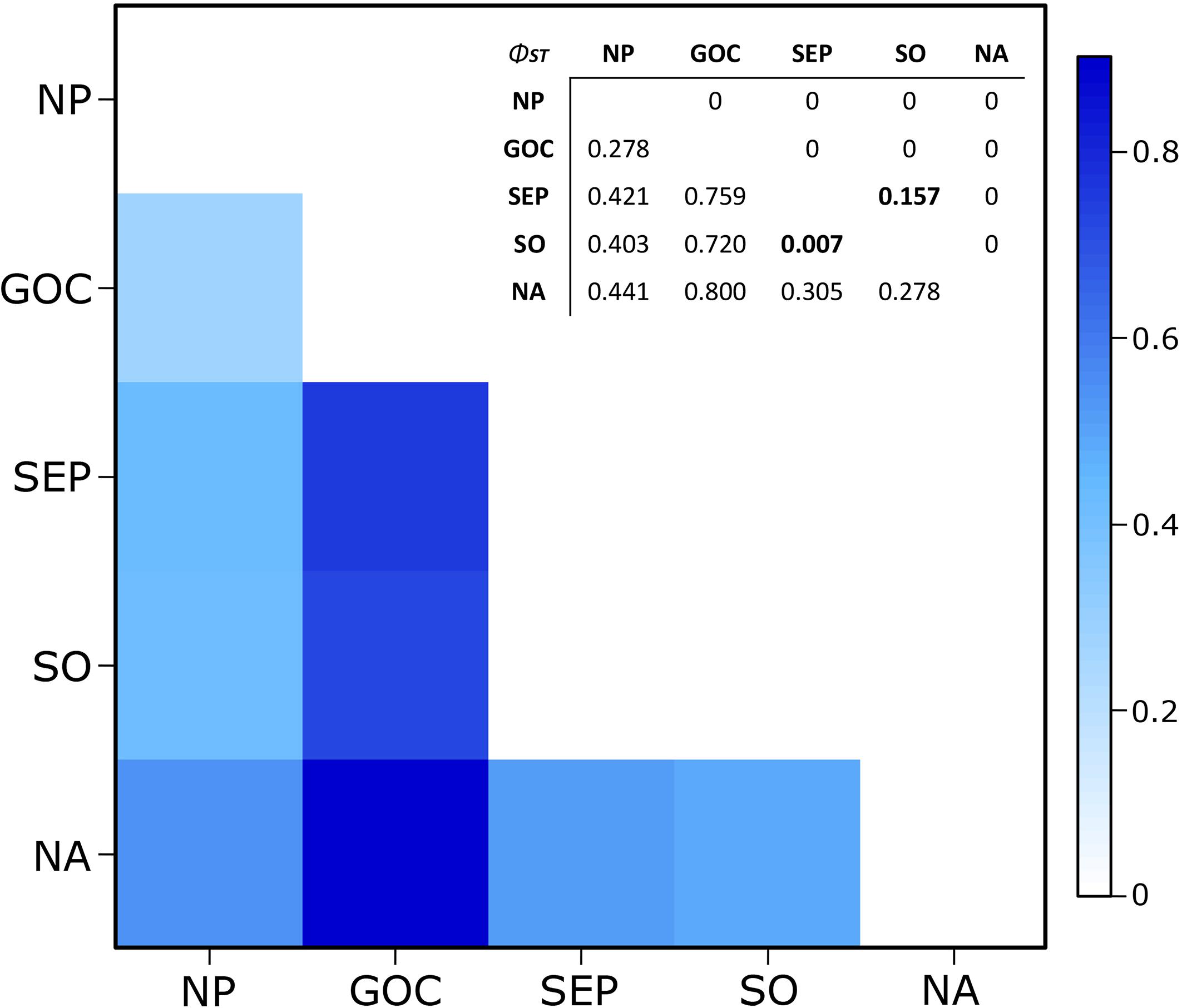
Figure 3. Heatmap for the pairwise phylogeographic structure (ΦST) comparisons between the five geographic areas NP, GoC, SEP, SO, and NA. In the embedded table, values are given below the diagonal and p-values above it. Non-significant values are in bold. Note: 0 represent p < 0.00001.
Regarding migration patterns, GoC-NP was the only pair that showed a statistically significant and asymmetrical migration rate. A statistically significant migration rate was obtained from the GoC to the NP, yet even though the migration rate in the opposite direction (from the NP to the GoC) showed non-zero values, it was not significantly different from a non-migration model (Figure 4). For the SH-NP and SH-NA pairs, near zero and non- statistically significant migration rate values were obtained.
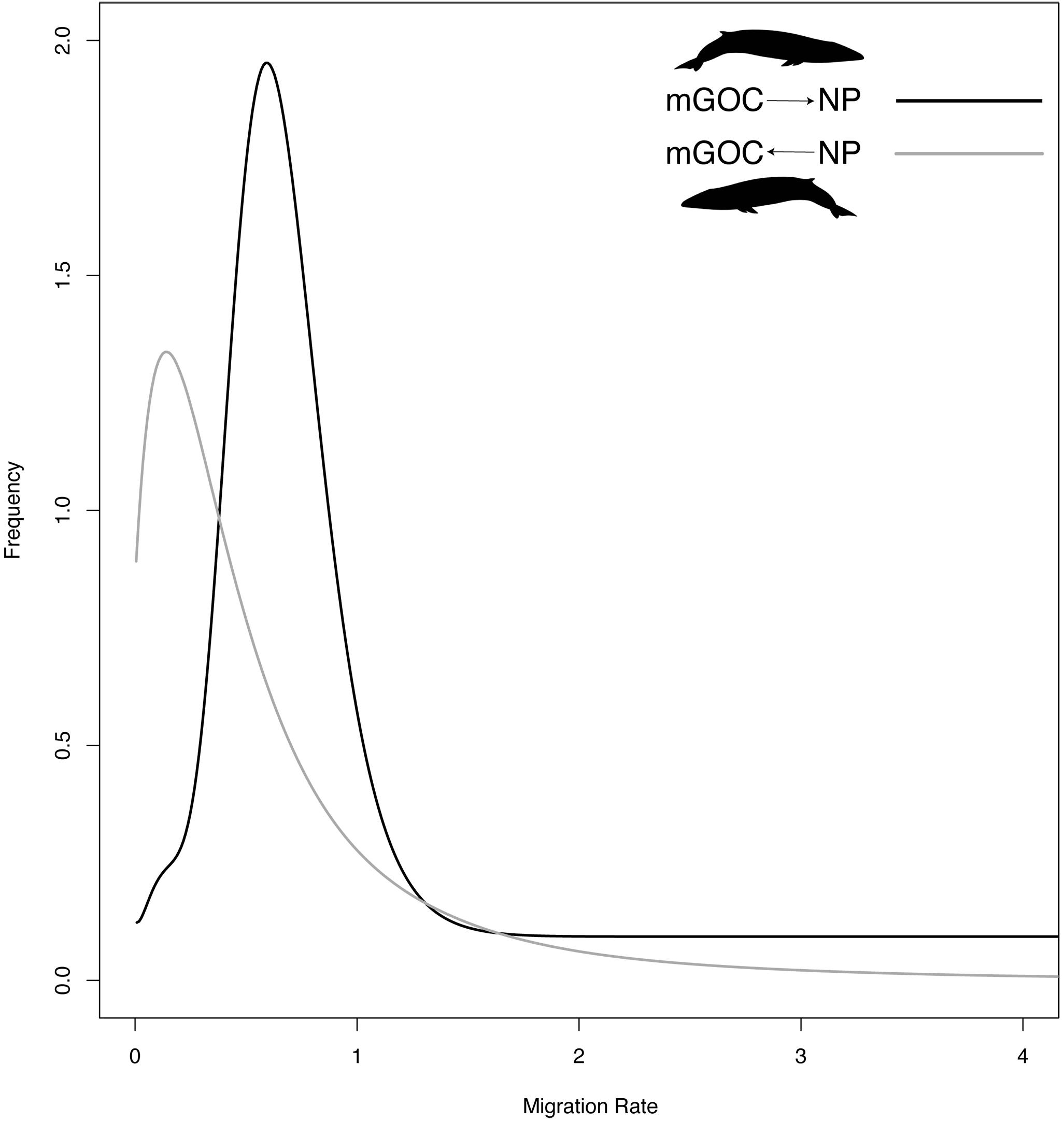
Figure 4. Marginal posterior probability distribution (frequency) of asymmetric migration rate estimates of GoC and NP, using an Isolation with Migration model implemented in IMa2.
The phylogenetic tree (Figure 5) shows a clear assortment of haplotypes according to their geographic origin (NA, NP, SH), and as previously observed in the haplotype network (Figure 2), five haplotypes sampled in the NP and three in the NA were placed in the predominantly SH clade. Using sequential calibration points, the divergence times estimated among the main clades of fin whales suggest the NP linage as the first clade-group to diverge, around 1.8 ± 0.4 million years ago (Mya), followed by the divergence between NA and SH linages around 1.2 ± 0.3 Mya. TMRCA estimates for the main clades were NA: 0.14 Mya (95% HDP = 0.06–0.24 Mya); NP: 0.13 Mya (95% HDP = 0.06–0.25 Mya) and SH: 0.17 Mya (95% HDP = 0.09–0.29 Mya, Figure 5).
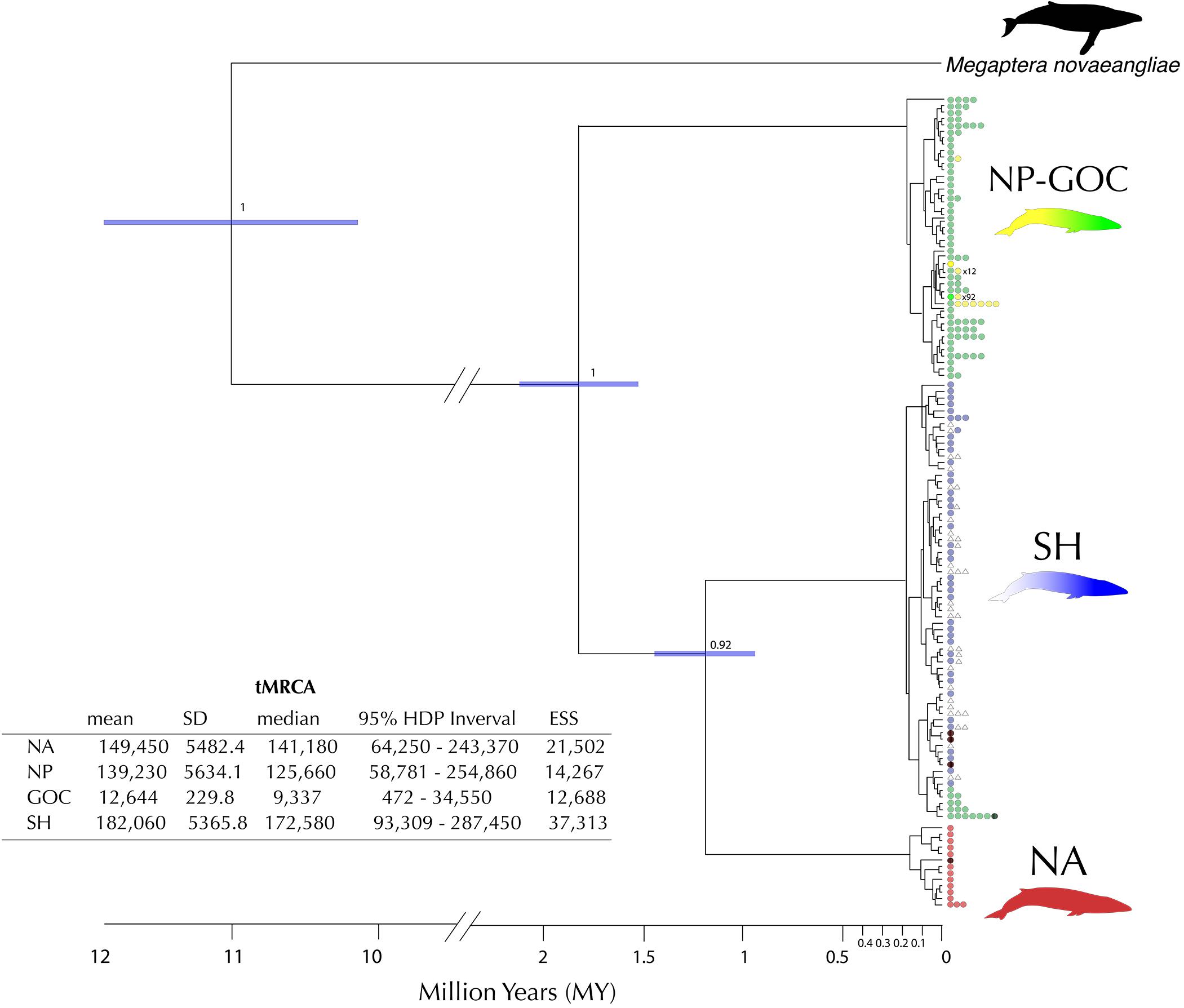
Figure 5. Phylogenetic tree using extraspecific and intraspecific calibration points (as proposed by Ho et al., 2008). Node ages are the median values from both Bayesian molecular clock analyses and TMRCA of each clade estimated with a Bayesian Skyline plot. In each clade of interest (North Pacific NP, Southern Hemisphere SH and North Atlantic NA), nodes indicate the posterior probability support for the Bayesian Inference. The embedded table shows the time to Most Recent Common Ancestor (TMRCA) estimates for the different linages. Mean, standard deviation (SD), median, 95% Highest Posterior Density (HPD) and Estimated Sample Size (ESS) for Bayesian Inference are reported. Number of colored dots represents the frequency of that particular haplotype. The hatched haplotypes were retrieved from Cabrera et al. (2019).
Discussion
The addition of new control region sequences from the waters off of Chile to the global data set of fin whale (Balaenoptera physlaus) sequences has allowed us to contribute new insights to our growing understanding of phylogeographic structure within this species. These sequences provide the first estimates of genetic diversity for a previously unsurveyed area in the Southern Hemisphere. Our analysis showed that SEP fin whales have high levels of genetic diversity similar to, albeit slightly lower than, that in the rest of the Southern Hemisphere. The haplotypes, of which six were shared, appeared homogeneously distributed in the network with no clear structure within the SH. This was further supported by the statistically non-significant phylogeographic structure observed between samples from the SEP and the rest of the Southern Hemisphere. Diversity within the SEP was similar to that in the neighboring North Pacific. Nevertheless, except for five haplotypes nested within the Southern Hemisphere, the NP haplotypes were more clearly separated in the network.
Our expanded dataset also confirms previously described patterns of global fin whale differentiation (Archer et al., 2013, 2019; Cabrera et al., 2019). The three previously described fin whale maternal linages, each corresponding to an ocean basin (NA, NP + GoC, and SH) were also recovered in our phylogenetic analyses. This is notable because in previous analyses, the SH was primarily represented by a set of 40 samples collected from the same area in the Southern Ocean over a period of 2 years (Archer et al., 2013). The addition of a comparable number of SEP samples, from the opposite side of the Southern Hemisphere does not appreciably change the structure of previously published phylogenetic trees (Archer et al., 2013; Cabrera et al., 2019). Estimated divergence times suggest that the diversification of the three maternal linages (1,8 ± 0,4 Mya) occurred during the Lower (Early) Pleistocene (dated from 2.58 to 0.77 Mya; Gibbard, 2015). This estimate is concordant with Archer et al. (2013), who estimate a divergence time of approximately 2 Mya. This overall pattern of divergence may be part of a complete oceanographic re-organization and climate variation that occurred at that time, including the gradual closure of the Panama Seaway, between 13 and 2.5 Mya (Lunt et al., 2008). The complete process promoted species diversification not only in cetaceans (Rosenbaum et al., 2000; Pastene et al., 2007), but also in many pelagic marine organisms (Jones and Hasson, 1985; Lima et al., 2020). The non-continuous distribution of the species across Southern and Northern Hemispheres may relate to the equatorial warm-water belt that operates as a variable but efficient barrier to dispersal, leading to antitropical distribution patterns (Davies, 1963).
Strong genetic differentiation was observed between the NA and NP, with each ocean basin being characterized by different haplogroups as previously described (Archer et al., 2013, 2019). Given that continents separate ocean basins and prevent inter-oceanic dispersal in the NH, this pattern of structure has been reported for many taxa, including humpback whales (Jackson et al., 2014), sei whales (Balaenoptera borealis; Baker et al., 2004; Huijser et al., 2018), as well as odontocetes, including bottlenose dolphins (Tursiops truncatus; Dowling and Brown, 1993) and orcas (Orcinus orca; Morin et al., 2015). Additionally, absence of recurrent gene flow was observed between the two Northern Hemisphere fin whale subspecies (NP and NA) and the SH populations. However, few haplotypes sampled in the North Pacific nested genetically in the Southern Hemisphere clade, suggesting the existence of discrete dispersal events from SP to NP as previously proposed by Archer et al. (2013). Similarly, the finding of three haplotypes in NA that nested in the SH clade may indicate the existence of dispersal events from SH to NA, as suggested by Cabrera et al. (2019). However, asymmetrical migration was not supported by our IMa2 analyses and may indicate that migrations from SH to NA and NP are the result of rare or even exceptional dispersal events from the Southern to Northern Hemisphere. This is also supported by close genetic relationship between some of these geographically misplaced haplotypes, corresponding to small post-migration lineages. Although we note that incomplete lineage sorting cannot be discarded as a possible explanation of this pattern. Nonetheless, other occasional gene flow between baleen whale populations across hemispheres has been reported at least for southern minke whales (Balaenoptera bonaerensis; Glover et al., 2010) and humpback whales. These species perform synchronous seasonal migrations to feeding areas, which differ in timing between hemispheres and thus precluding them from mixing (Jackson et al., 2014).
Within the North Pacific, analyses of additional Gulf of California (GoC) fin whale sequences in this study were consistent with the known significant genetic differentiation and isolated nature of this population (Bérubé et al., 2002; Rivera-León et al., 2019). Haplotype diversity within the GoC was the lowest of all regions examined (Hd = 0.314). However, this value was twice as high as that previously reported by Rivera-León et al. (2019) (Hd = 0.15). Notwithstanding the higher number of samples used in this previous study (n = 253 vs. n = 112), this difference can be explained by the longer sequences used in our study (624 vs. 280 base pairs), which also generated a new GoC haplotype. We estimate the founding of the GoC from the North Pacific around 9,000 years ago (TMRCA estimates 95% HPD: 472–34,550 years; ESS 12,688). These results refine the TMRCA estimates and confidence interval between NP and GoC obtained by Rivera-León et al. (2019) of approximately 1,300 years ago (95% HPD: 0–2,700 kya; ESS: 559,148). This new estimate is now also more consistent with a sea-level rise associated with the deglaciation process, about 15,000 years ago (Keigwin and Jones, 1990). Paleohabitat modeling has shown strong variations in estuarine habitats since the Last Glacial Maximum (LGM), approximately 20 kya, when the sea level was approximately 130 m lower. The colonization of the Gulf of California by marine species may be related to this sea level rise, as mentioned by Dolby et al. (2016), who identified and assessed a sea-level driven recolonization process after the LGM along the coast of California (United States) and Baja California (Mexico). The colonization of the GoC by fin whales would have been followed by a switch from a migratory to resident behavior, sustained by the permanently highly productive ecosystem of the GoC (Mercado-Santana et al., 2017). The year-round foraging activity recorded for fin whales in the GoC (Jiménez López et al., 2019) is evidence of the low movement of whales from this region. Similarly, a suspension of the southward migration in humpback whales in Cape Columbine, South Africa, was observed in response to locally abundant prey (Best et al., 1995). Also, an exception to the seasonal migrations of humpback whales by year-round sightings as well as records of feeding and breeding behavior in a limited area of the Arabian sea has been reported (Pomilla et al., 2010). However, although such a residence pattern may explain the genetic structure detected between NP and GoC, the geographic distribution of the shared, closely related haplotypes, suggests the existence of low and asymmetric gene flow from the GoC to the NP. This migration pattern and the distribution of haplotypes between the two regions fits a continent-island migration model, where migrants coming from a small population have no noticeable effect on the gene frequencies of the large one (Nagylaki, 1992; Bürger and Akerman, 2011). Regardless, our migration rate estimates should be taken with caution, since they are derived from a single mtDNA marker. Further studies are needed in order to confirm patterns of asymmetrical migration in NP. Nevertheless, even if no gene flow was detected between NP and GoC, one haplotype detected in the GoC clearly has a genetic affinity with NP haplotypes, consistent with rare or even exceptional migration events from the NP to the GoC, as previously proposed by Rivera-León et al. (2019).
One of the most important contributions of this study is that our findings challenge the validity of the pygmy fin whale sub-species, B. p. patachonica (Burmeister, 1865), which has previously been called into question (Archer et al., 2019). Pygmy fin whales are described as being relatively short (total length approximately < 20 m), dark in color with black baleen, and occurring from approximately 55°S to the Equator along the South American Pacific coast (Clarke, 2004). We could not positively assign any of the SH samples to either B. p. quoyi or B. p. patachonica, as we did not have total body length measurements, or observed significantly darker individuals, nor a whale with black baleen among the over 50 fin whales sighted around the area. Additionally, aside from the type specimen residing in the Museo Argentino de Ciencias Naturales Bernardino Rivadavia (MACN) in Buenos Aires, Argentina, there are no known specimens that have previously been positively identified as pygmy fin whales for morphological comparison. However, given the time scale and geographic scope of our SH samples, we would have expected to have sampled at least some pygmy fin whales off Chile. Our samples were obtained during the austral summer season, which is when fin whales of the subspecies B. p. quoyi are expected to be southbound, migrating to sub-Antarctic and Antarctic waters (Mackintosh, 1942). Moreover, five of the six individuals that were satellite tagged by Sepúlveda et al. (2018), which were also included in our study, remained in lower latitudes during the summer months (sampled period), within the latitudinal distribution range that is proposed for pygmy fin whales (Clarke, 2004). Additionally, Cooke (2018) reviewed information on body size of fin whales collected in modern whaling activity off the northern (∼20°S), central (30–38°S), and southern (44°S) coasts of Chile. The author mentions that these whales were smaller than those of higher latitudes, potentially meeting the small size criterion pygmy fin whales of Clarke (Clarke, 2004).
Although significant control region differentiation was obtained in all other comparisons, none was detected between SEP fin whales and those from the rest of the Southern Hemisphere. Thus, it is evident that the differentiation that might exist between regions within the SH is not at the same level as that between subspecies within the Northern Hemisphere, between Hemispheres, or even between the Gulf of California and the greater North Pacific suggesting the existence of a single evolutionary unit in SH. Similarly, low genetic differentiation and high gene flow among regions within the SH has been reported for humpback whales (Jackson et al., 2014). By contrast, Bryde’s whales (Balaenoptera edeni) show strong genetic differentiation between the South Pacific and South Atlantic oceans, however this species is not distributed further south than approximately 40°S (Pastene et al., 2015) and would be effectively separated by the South American continent.
Based on the absence of any detectable structure within the Southern Hemisphere and the insufficient support of the initial subspecies description by Clarke (2004) (see review in Archer et al., 2019), it appears that the pygmy fin whale, B. p. patachonica, may not be a valid subspecies. Alternatively, it is possible that pygmy fin whales are not present in the study area or we have not yet sampled it. Thus, until evidence that positively supports the validity of B. p. patachonica can be produced, such as genetic, ecological, behavioral, or morphological data (Crandall et al., 2000), we propose that all Southern Hemisphere fin whales, including those present in middle latitudes of southeastern Pacific belong to the earlier described subspecies B. p. quoyi (Tomilin, 1946). The smaller size of fin whales along the central Chilean coast may be because these individuals are juveniles of B. p. quoyi, and the different migration behavior they present could be related to energy saving, as proposed by Cooke (2018). The existence of a single taxa within the Southern Hemisphere is important for the assessment of fin whales, primarily relevant to the long-term management issues defining conservation priorities (Moritz, 1994) being crucial in conservation legislation (Robertson et al., 2014).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank (accession: MW266395–MW266537).
Ethics Statement
The animal study was reviewed and approved by the Permits 1502/2013, 1803/2015, and 2982/2015 from Chilean Undersecretariat of Fisheries and Aquaculture and with approval and permits issued by the Mexican Wildlife Agency (Dirección General de Vida Silvestre, Subsecretaría de Gestión para la Protección Ambiental, Secretaría del Medio Ambiente y Recursos Naturales).
Author Contributions
MJP-A, RM, SN-M, JU, MS, MS-C, CO, and GP collected the samples. MJP-A, EP, SK, MS, NS, MS-C, GP, FA, and JU contributed to the reagents, materials, and analysis tools. SK, MJP-A, SN-M, and LV-G did the laboratory work. MJP-A, SK, NS, SN-M, LV-G, and FA acquired the data. MJP-A, EP, SK, and NS conceived, designed the analysis, and analyzed the data. MJP-A, EP, SK, NS, FA, and CO wrote the manuscript. All authors read, edited, commented on, and approved the final manuscript.
Funding
This project was supported by the Agencia Nacional de Investigación y Desarrollo (ANID) under Grant Program FONDECYT Iniciación 11170182, and Postdoctoral FONDECYT program 3190482, CONICYT-PCHA/Doctorado Nacional/2016-21161109 Ph.D. scholarship; CONICYT III Fortalecimiento Puente R16A10003 and Projects PIA CONICYT APOYO CCTE AFB170008 and PIA CONICYT ACT172065, Project Innova-Corfo 14BPCR-33451, and UCMEXUS-CONACYT collaborative grant 2006.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the following people for help with fieldwork Paula López, Aurelio Aguirre, Patricio Ortiz, and Marinella Maldonado. We also thank Robert Wayne and Sergio Flores-Ramírez for the initial support in the Gulf of California project design and Lafayette Eaton for the English correction.
Footnotes
- ^ http://www.cibiv.at/∼greg/haploviewer
- ^ http://beast.bio.ed.ac.uk/TreeAnnotator
- ^ http://tree.bio.ed.ac.uk/software/figtree
References
Aasen, E., and Medrano, J. F. (1990). Amplification of the Zfy and Zfx Genes for Sex identification in humans, cattle, sheep and goats. Nat. Biotechnol. 8, 1279–1281. doi: 10.1038/nbt1290-1279
Aljanabi, S. M., and Martinez, I. (1997). Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693. doi: 10.1093/nar/25.22.4692
Alter, S. E., and Palumbi, S. R. (2009). Comparing evolutionary patterns and variability in the mitochondrial control region and cytochrome b in three species of baleen whales. J. Mol. Evol. 68, 97–111. doi: 10.1007/s00239-008-9193-2
Archer, F. I., Brownell, R. L. Jr., Hancock-Hanser, B. L., Morin, P. A., Robertson, K. M., Sherman, K. K., et al. (2019). Revision of fin whale Balaenoptera physalus (Linnaeus, 1758) subspecies using genetics. J. Mammal. 100, 1653–1670. doi: 10.1093/jmammal/gyz121
Archer, F. I., Morin, P. A., Hancock-Hanser, B. L., Robertson, K. M., Leslie, M. S., Bérubé, M., et al. (2013). Mitogenomic phylogenetics of fin whales (Balaenoptera physalus spp.): genetic evidence for revision of subspecies. PLoS One 8:e63396. doi: 10.1371/journal.pone.0063396
Arnason, U., Gullberg, A., and Widegren, B. (1991). The complete nucleotide sequence of the mitochondrial DNA of the fin whale. Balaenoptera physalus. J. Mol. Evol. 33, 556–568. doi: 10.1007/BF02102808
Baker, C. S., Dalebout, M. L., Funahashi, N., Ma, Y. U., Steel, D., and Lavery, S. (2004). “Market surveys of whales, dolphins and porpoises in Japan and Korea, 2003–2004, with reference to stock identity of sei whales,” in IWC Scientific Committee meeting document SC/56/BC3, London, 8.
Bérubé, M., Aguilar, A., Dendanto, D., Larsen, F., Notarbartolo Di Sciara, G., Sears, R., et al. (1998). Population genetic structure of North Atlantic, Mediterranean Sea and Sea of Cortez fin whales, Balaenoptera physalus (Linnaeus, 1758): analysis of mitochondrial and nuclear loci. Mol. Ecol. 7, 585–599. doi: 10.1046/j.1365-294x.1998.00359.x
Bérubé, M., Urbán, J., Dizon, A. E., Brownell, R. L., and Palsbøll, P. J. (2002). Genetic identification of a small and highly isolated population of fin whales (Balaenoptera physalus) in the Sea of Cortez, Mexico. Conserv. Genet. 3, 183–190. doi: 10.1023/A:1015224730394
Best, P. B., Sekiguchi, K., and Findlay, K. P. (1995). A suspended migration of humpback whales Megaptera novaeangliae on the west coast of South Africa. Mar. Ecol. Prog. Ser. 118, 1–12. doi: 10.3354/meps118001
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comp. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Branch, T. A., and Butterworth, D. S. (2001). Estimates of abundance south of 60°S for cetacean species sighted frequently on the 1978/79 to 1997/98 IWC/IDCR-SOWER sighting surveys. J. Cetacean Res. Manage. 3, 251–270.
Bürger, R., and Akerman, A. (2011). The effects of linkage and gene flow on local adaptation: a two-locus continent–island model. Theor. Popul. Biol. 80, 272–288. doi: 10.1016/j.tpb.2011.07.002
Burmeister, H. (1865). Letter on a new species of whale Balaenoptera patachonica from Argentina. Proc. Zool. Soc. Lond. 13, 190–195.
Cabrera, A. A., Hoekendijk, J. P. A., Aguilar, A., Barco, S. G., Berrow, S., Bloch, D., et al. (2019). Fin whale (Balaenoptera physalus) mitogenomics: a cautionary tale of defining sub-species from mitochondrial sequence monophyly. Mol. Phylogen. Evol. 135, 86–97. doi: 10.1016/j.ympev.2019.02.003
Clarke, R. (2004). Pygmy fin whales. Mar. Mamm. Sci. 20, 329–334. doi: 10.1111/j.1748-7692.2004.tb01161.x
Cooke, J. G. (2018). “A note on the size distribution of fin whales (Balaenoptera physalus) in the southeast Pacific and implications for the comprehensive assessment of SH fin whales,” in IWC Scientific Committee WP5-SH. Bled, Slovenia.
Crandall, K. A., Bininda-Emonds, O. R. P., Mace, G. M., and Wayne, R. K. (2000). Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 15, 290–295. doi: 10.1016/S0169-5347(00)01876-0
Dalebout, M. L., Robertson, K. M., Frantzis, A., Engelhaupt, D., Mignucci-Giannoni, A. A., Rosario-Delestre, R. J., et al. (2005). Worldwide structure of mtDNA diversity among Cuvier’s beaked whales (Ziphius cavirostris): implications for threatened populations. Mol. Ecol. 14, 3353–3371. doi: 10.1111/j.1365-294X.2005.02676.x
Davies, J. L. (1963). The antitropical factor in cetacean speciation. Evolution 17, 107–116. doi: 10.2307/2406339
Dolby, G. A., Hechinger, R., Ellingson, R. A., Findley, L. T., Lorda, J., and Jacobs, D. K. (2016). Sea-level driven glacial-age refugia and post-glacial mixing on subtropical coasts, a palaeohabitat and genetic study. Proc. R. Soc. Ser. B Biol. Sci. 283:20161571. doi: 10.1098/rspb.2016.1571
Dowling, T. E., and Brown, W. M. (1993). Population structure of the bottlenose dolphin (Tursiops truncatus) as determined by restriction endonuclease analysis of mitochondrial DNA. Mar. Mamm. Sci. 9, 138–155.
Edwards, E. F., Hall, C., Moore, T. J., Sheredy, C., and Redfern, J. V. (2015). Global distribution of fin whales Balaenoptera physalus in the post−whaling era (1980–2012). Mamm. Rev. 45, 197–214. doi: 10.1111/mam.12048
Excoffier, L., Laval, G., and Schneider, S. (2005). ARLEQUIN ver. 3.0: an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50. doi: 10.1177/117693430500100003
Filatov, D. A. (2009). Processing and population genetic analysis of multigenic datasets with ProSeq3 software. Bioinformatics 25, 3189–3190. doi: 10.1093/bioinformatics/btp572
Gibbard, P. (2015). The quaternary System/Period and its major subdivisions. Russian Geol. Geophys. 56, 686–688. doi: 10.1016/j.rgg.2015.03.015
Gilson, A., Syvanen, M., Levine, K., and Banks, J. (1998). Deer gender determination by polymerase chain reaction: validation study and application to tissues, bloodstains, and hair forensic samples from California. Calif. Fish Game 84, 159–169.
Glover, K. A., Kanda, N., Haug, T., Pastene, L. A., Øien, N., Goto, M., et al. (2010). Migration of Antarctic minke whales to the Arctic. PLoS One 5:e15197. doi: 10.1371/journal.pone.0015197
Harlin, A. D., Würsig, B., Baker, C. S., and Markowitz, T. M. (1999). Skin swabbing for genetic analysis: application to dusky dolphins (Lagenorhynchus obscurus). Mar. Mamm. Sci. 15, 409–425. doi: 10.1111/j.1748-7692.1999.tb00810.x
Hey, J. (2010). Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920. doi: 10.1093/molbev/msp296
Ho, S. Y., Phillips, M. J., Cooper, A., and Drummond, A. J. (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568. doi: 10.1093/molbev/msi145
Ho, S. Y. W., Saarma, U., Barnett, R., Haile, J., and Shapiro, B. (2008). The effect of inappropriate calibration: three case studies in molecular ecology. PLoS One 3:e1615. doi: 10.1371/journal.pone.0001615
Huijser, L. A. E., Bérubé, M., Cabrera, A. A., Prieto, R., Silva, M. A., Robbins, J., et al. (2018). Population structure of North Atlantic and North Pacific sei whales (Balaenoptera borealis) inferred from mitochondrial control region DNA sequences and microsatellite genotypes. Conserv. Genet. 19, 1007–1024. doi: 10.1007/s10592-018-1076-5
Jackson, J. A., Steel, D. J., Beerli, P., Congdon, B. C., Olavarría, C., Leslie, M. S., et al. (2014). Global diversity and oceanic divergence of humpback whales (Megaptera novaeangliae). Proc. R. Soc. Ser. B Biol. Sci. 281:20133222. doi: 10.1098/rspb.2013.3222
Jiménez López, M. E., Palacios, D. M., Jaramillo Legorreta, A., Urbán, J., and Mate, B. R. (2019). Fin whale movements in the Gulf of California. Mexico, from satellite telemetry. PLoS One 14:e0209324. doi: 10.1371/journal.pone.0209324
Jones, D. S., and Hasson, P. F. (1985). “History and development of the marine invertebrate faunas separated by the Central American Isthmus,” in The Great American Biotic Interchange, eds F. G. Stehli and S. D. Webb (Boston, MA: Springer), 325–355. doi: 10.1007/978-1-4684-9181-4_13
Kalinowski, S. T., Taper, M. L., and Marshall, T. C. (2007). Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106. doi: 10.1111/j.1365-294X.2007.03089.x
Keigwin, L. D., and Jones, G. A. (1990). Deglacial climatic oscillations in the Gulf of California. Paleoceanography 5, 1009–1023. doi: 10.1029/PA005i006p01009
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lima, F. D., Strugnell, J. M., Leite, T. S., and Lima, S. M. (2020). A biogeographic framework of octopod species diversification: the role of the Isthmus of Panama. PeerJ 8:e8691. doi: 10.7717/peerj.8691
Linnaeus, C. (1758). Systema Naturae Per Regna Tria Naturae: Secundum Classes, Ordines, Genera, Species, Cum Characteribus, Differentiis, Synonymis, Locis, 10th Edn. Stockholm: Laurentius Salvius.
Lockyer, C., and Waters, T. (1986). Weights and anatomical measurements of northeastern Atlantic fin (Balaenoptera physalus, Linnaeus) and sei (B. borealis, Lesson) whales. Mar. Mamm. Sci. 2, 169–185.
Lunt, D. J., Valdes, P. J., Haywood, A., and Rutt, I. C. (2008). Closure of the Panama Seaway during the Pliocene: implications for climate and Northern Hemisphere glaciation. Clim. Dyn. 30, 1–18. doi: 10.1007/s00382-007-0265-6
Mackintosh, N. A. (1966). “The distribution of southern blue and fin whales,” in Whales, Dolphins, and Porpoises, ed. K. S. Norris (Berkley: University of California Press), 125–144. doi: 10.1525/9780520321373-010
Marx, F. G., and Fordyce, R. E. (2015). Baleen boom and bust: a synthesis of mysticete phylogeny, diversity and disparity. R. Soc. Open Sci. 2:140434. doi: 10.1098/rsos.140434
Mercado-Santana, J. A., Santamaría-Del-Ángel, E., González-Silvera, A., Sánchez-Velasco, L., Gracia-Escobar, M. F., Millán-Núñez, R., et al. (2017). Productivity in the Gulf of California large marine ecosystem. J. Environ. Dev. 22, 18–29. doi: 10.1016/j.envdev.2017.01.003
Miyashita, T., Kato, H., and Kasuya, T. (1995). Worldwide Map of Cetacean Distribution Based on Japanse Sighting Data. Shimizu: National Research Institute of Far Sea Fisheries.
Mizroch, S. A., Rice, D. W., and Breiwick, J. M. (1984). The fin whale, Balaenoptera physalus. Mar. Fish. Rev. 46, 20–24.
Morin, P. A., Parsons, K. M., Archer, F. I., Ávila-Arcos, M. C., Barrett-Lennard, L. G., Dalla Rosa, L., et al. (2015). Geographic and temporal dynamics of a global radiation and diversification in the killer whale. Mol. Ecol. 24, 3964–3979. doi: 10.1111/mec.13284
Moritz, C. (1994). Defining ‘evolutionarily significant units’ for conservation. Trends Ecol. Evol. 9, 373–375. doi: 10.1016/0169-5347(94)90057-4
Nagylaki, T. (1992). Introduction to Theoretical Population Genetics. Berlin: Springer-Verlag Berlin Heidelberg.
Nigenda-Morales, S., Flores-Ramírez, S., Urbán, J., and Vazquez-Juarez, R. (2008). MHC DQB-1 polymorphism in the Gulf of California fin whale (Balaenoptera physalus) population. J. Hered. 99, 14–21. doi: 10.1093/jhered/esm087
Pacheco, A. S., Villegas, V. K., Riascos, J. M., and Van Waerebeek, K. (2015). Presence of fin whales (Balaenoptera physalus) in Mejillones Bay, a major seaport area in northern Chile. Rev. Biol. Mar. Ocean. 50, 383–389. doi: 10.4067/S0718-19572015000300017
Paradis, E. (2013). Molecular dating of phylogenies by likelihood methods: a comparison of models and a new information criterion. Mol. Phylogen. Evol. 67, 436–444. doi: 10.1016/j.ympev.2013.02.008
Pastene, L. A., Acevedo, J., Siciliano, S., Sholl, T. G., De Moura, J. F., Ott, P. H., et al. (2015). Population genetic structure of the South American Bryde’s whale. Rev. Biol. Mar. Oceanogr. 50, 453–464. doi: 10.4067/S0718-19572015000400005
Pastene, L. A., Goto, M., Kanda, N., Zerbini, A. N., Kerem, D., Watanabe, K., et al. (2007). Radiation and speciation of pelagic organisms during periods of global warming: the case of the common minke whale. Balaenoptera acutorostrata. Mol. Ecol. 16, 1481–1495. doi: 10.1111/j.1365-294X.2007.03244.x
Pérez, M., Thomas, F., Uribe, F., Sepúlveda, M., Flores, M., and Moraga, R. (2006). Fin Whales (Balaenoptera physalus) Feeding on euphausia mucronata in nearshore waters off North-Central Chile. Aquat. Mamm. 32, 109–113. doi: 10.1578/AM.32.1.2006.109
Pomilla, C., Collins, T., Minton, G., Findlay, K., Leslie, M. S., Ponnampalam, L., et al. (2010). “Genetic distinctiveness and decline of a small population of humpback whales (Megaptera novaeangliae) in the Arabian Sea (Region X),” in IWC Scientific Committee SC/62/SH6. Agadir, Morocco.
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 65, 901–904. doi: 10.1093/sysbio/syy032
Reilly, S. B., Bannister, J. L., Best, P. B., Brown, M., Brownell, R. L. Jr., Butterworth, D. S., et al. (2013). Balaenoptera physalus. IUCN Red List Threat. Spec. 2013:e.T2478A44210520.
Rivera-León, V. E., Urbán, J., Mizroch, S., Brownell, R. L., Oosting, T., Hao, W., et al. (2019). Long-term isolation at a low effective population size greatly reduced genetic diversity in Gulf of California fin whales. Sci. Rep. 9:12391. doi: 10.1038/s41598-019-48700-5
Robertson, J. M., Langin, K. M., Sillett, T. S., Morrison, S. A., Ghalambor, C. K., and Funk, W. C. (2014). Identifying evolutionarily significant units and prioritizing populations for management on islands. Monogr. West. N. Am. Nat. 7, 397–411. doi: 10.3398/042.007.0130
Rosenbaum, H. C., Brownell, R. L. Jr., Brown, M. W., Schaeff, C., Portway, V., White, B. N., et al. (2000). World-wide genetic differentiation of Eubalaena: questioning the number of right whale species. Mol. Ecol. 9, 1793–1802. doi: 10.1046/j.1365-294x.2000.01066.x
Sasaki, T., Nikaido, M., Hamilton, H., Goto, M., Kato, H., Kanda, N., et al. (2005). Mitochondrial phylogenetics and evolution of mysticete whales. Syst. Biol. 54, 77–90. doi: 10.1080/10635150590905939
Scammon, C. M. (1869). On the cetaceans of the western coast of North America. Proc. Acad. Nat. Sci. Philadelphia 21, 13–63.
Sepúlveda, M., Pérez-Álvarez, M. J., Santos-Carvallo, M., Pavez, G., Olavarría, C., Moraga, R., et al. (2018). From whaling to whale watching: identifying fin whale critical foraging habitats off the Chilean coast. Aquat. Conserv.: Mar. Freshwat. Ecosyst. 28, 821–829. doi: 10.1002/aqc.2899
Taxonomy, C. O. (2020). List of Marine Mammal Species and Subspecies. Lawrence, KS: Society for Marine Mammalogy.
Tomilin, A. G. (1946). Thermoregulation and the geographical races of cetaceans. Doklady Akademii Nauk S.S.S.R. 54, 465–472.
Toro, F., Vilina, Y. A., Capella, J. J., and Gibbons, J. (2016). Novel Coastal Feeding Area for Eastern South Pacific Fin Whales (Balaenoptera physalus) in Mid-Latitude Humboldt Current Waters off Chile. Aquat. Mamm. 42, 47–55. doi: 10.1578/AM.42.1.2016.47
Urbán, R. J., Rojas-Bracho, L., Guerrero-Ruíz, M., Jaramillo-Legorreta, A., and Findley, L. T. (2005). “Cetacean diversity and conservation in the Gulf of California,” in Biodiversity, Ecosystems, and Conservation in Northern Mexico, eds J. E. Cartron, G. Ceballos, and R. S. Felger (New York, NY: Oxford University Press), 276–297.
Keywords: fin whale, genetics, Chile, Balaenoptera physalus, pygmy fin whale
Citation: Pérez-Alvarez MJ, Kraft S, Segovia NI, Olavarría C, Nigenda-Morales S, Urbán RJ, Viloria-Gómora L, Archer F, Moraga R, Sepúlveda M, Santos-Carvallo M, Pavez G and Poulin E (2021) Contrasting Phylogeographic Patterns Among Northern and Southern Hemisphere Fin Whale Populations With New Data From the Southern Pacific. Front. Mar. Sci. 8:630233. doi: 10.3389/fmars.2021.630233
Received: 17 November 2020; Accepted: 31 May 2021;
Published: 05 July 2021.
Edited by:
Phil J. Bouchet, University of St Andrews, United KingdomReviewed by:
Andrea A. Cabrera, University of Copenhagen, DenmarkMorten Olsen, University of Copenhagen, Denmark
Copyright © 2021 Pérez-Alvarez, Kraft, Segovia, Olavarría, Nigenda-Morales, Urbán, Viloria-Gómora, Archer, Moraga, Sepúlveda, Santos-Carvallo, Pavez and Poulin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: MJosé Pérez-Alvarez, bWFyaWEucGVyZXpAdW1heW9yLmNs; Elie Poulin, ZXBvdWxpbkB1Y2hpbGUuY2w=