 Joshua G. Medina-Feliciano
Joshua G. Medina-Feliciano Stacy Pirro2
Stacy Pirro2 Jose E. García-Arrarás
Jose E. García-Arrarás Joseph F. Ryan
Joseph F. Ryan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Mar. Sci., 15 April 2021
Sec. Marine Molecular Biology and Ecology
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.603410
This article is part of the Research TopicMolecular Evolution and Genomics of Marine Megafauna: Methodological Solutions and Key AdvancesView all 8 articles
Regeneration is one of the most fascinating and yet least understood biological processes. Echinoderms, one of the closest related invertebrate groups to humans, can contribute to our understanding of the genetic basis of regenerative processes. Among echinoderms, sea cucumbers have the ability to grow back most of their body parts following injury, including the intestine and nervous tissue. The cellular and molecular events underlying these abilities in sea cucumbers have been most extensively studied in the species Holothuria glaberrima. However, research into the regenerative abilities of this species has been impeded due to the lack of adequate genomic resources. Here, we report the first draft genome assembly of H. glaberrima and demonstrate its value for future genetic studies. Using only short sequencing reads, we assembled the genome into 89,105 scaffolds totaling 1.1 gigabases with an N50 of 25 kilobases. Our BUSCO assessment of the genome resulted in 894 (91.4%) complete and partial genes from 978 genes queried. We incorporated transcriptomic data from several different life history stages to annotate 51,415 genes in our final assembly. To demonstrate the usefulness of the genome, we fully annotated the melanotransferrin (Mtf) gene family, which have a potential role in the regeneration of the sea cucumber intestine. Using these same data, we extracted the mitochondrial genome, which showed high conservation to that of other holothuroids. Thus, these data will be a critical resource for ongoing studies of regeneration and other studies in sea cucumbers.
Regeneration, the replacement of lost or damaged body parts, is one of the most fascinating biological processes. Regenerative abilities are widespread in the animal kingdom (Bely and Nyberg, 2010), but our understanding of the evolution of regenerative capacity is based primarily on a small number of animal species. As one of the closest invertebrate relatives to humans, echinoderms represent an important model for understanding the origin and evolution of vertebrates, as well as being a model for understanding human biology. Echinoderms, along with hemichordates make up the clade Ambulacraria, the sister lineage to chordates (Cannon et al., 2016). Unlike chordates, many echinoderms, especially sea cucumbers, have extensive regenerative abilities. Nevertheless, relatively few genomic resources exist for sea cucumber models (Long et al., 2016; Zhang et al., 2017).
Sea cucumbers (holothurians) have the ability to regenerate their intestine following evisceration (García-Arrarás et al., 1998) and their radial nerve cord following transection (San Miguel-Ruiz et al., 2009). Many cellular events that underlie these abilities have been best described in the species Holothuria glaberrima. For instance, details of cell migration, dedifferentiation, proliferation and apoptosis during intestinal regeneration have been published (García-Arrarás et al., 2018). While we know exactly where and when these events occur at the cellular level, the underlying molecular infrastructure has not yet been clearly elucidated (Mashanov and García-Arrarás, 2011). In recent years, we have reported gene expression profiles of various stages of regeneration, providing information on which genes play important roles in sea cucumber regenerative processes (Ortiz-Pineda et al., 2009; Mashanov et al., 2014). These RNA-Seq analyses have led to interesting findings, but the lack of a reference genome has limited the impact of these results.
Here we report the nuclear and mitochondrial genomes of H. glaberrima. We show the utility of our draft nuclear genome by using it to ascertain the genomic structure of melanotransferrins, proteins that might play important roles in the regeneration process (Rojas-Cartagena et al., 2007; Hernández-Pasos et al., 2017). Melanotransferrins are membrane-bound molecules involved in many vertebrate biological processes and diseases, including development, cancer, and Alzheimer’s disease (Rahmanto et al., 2012). Moreover, in the sea cucumber, melanotransferrin genes are highly expressed in the intestine after immune activation (Ramírez-Gómez et al., 2009). Previously it was reported, based on transcriptomic data, that H. glaberrima contains four different melanotransferrin (Mtf) genes (HgMTF1, HgMTF2, HgMTF3, and HgMTF4), while only one melanotransferrin is reported in all other echinoderms sampled to date (Hernández-Pasos et al., 2017). The continuous sequencing of new species has shown that certain Teleostei species appear to contain a single duplication of their Mtf gene, which is not surprising due to a whole genome duplication event that occurred in the lineage leading to the last common ancestor of extant teleosts (Jaillon et al., 2004). It has been hypothesized that the genomes from most species contain only one melanotransferrin because multiple copies may act as a dominant lethal (Lambert et al., 2005). The four melanotransferrin genes found in H. glaberrima are highly expressed at different stages of intestinal regeneration (Hernández-Pasos et al., 2017). For these reasons, analyzing the genomic characteristics and evolutionary history of these four melanotransferrin genes will shed light on this gene family and will facilitate further studies on their specific role and regulation.
Comparisons of mitochondrial genomes, the remnants of an ancient endosymbiotic event that led to the origin of eukaryotes (Martin et al., 2015), provide insight into the evolution of aerobic respiration across the tree of life. As part of our sequencing, assembly, and annotation of the nuclear genome, we have also done the same for the mitochondrial genome of H. glaberrima. In addition, we have compared the H. glaberrima mitochondrial genome with two other published holothuroid mitochondrial genomes.
The nuclear genome of H. glaberrima will be valuable resource for ongoing investigations into regenerative processes in this animal. In addition, both nuclear and mitochondrial genomic data will be useful for future studies in systematics, immunology, and evolution, among others.
A single adult male sea cucumber (Figure 1A) was collected from northeastern Puerto Rico and kept in aerated sea water until dissection. Gonads were collected into ethanol and sent to Iridian Genomes Inc. for DNA extraction, library preparation, and sequencing (Bethesda, MD). Library was prepared using the Illumina TruSeq kit with recommended 600 bp insert size following manufacturer’s standard procedure.
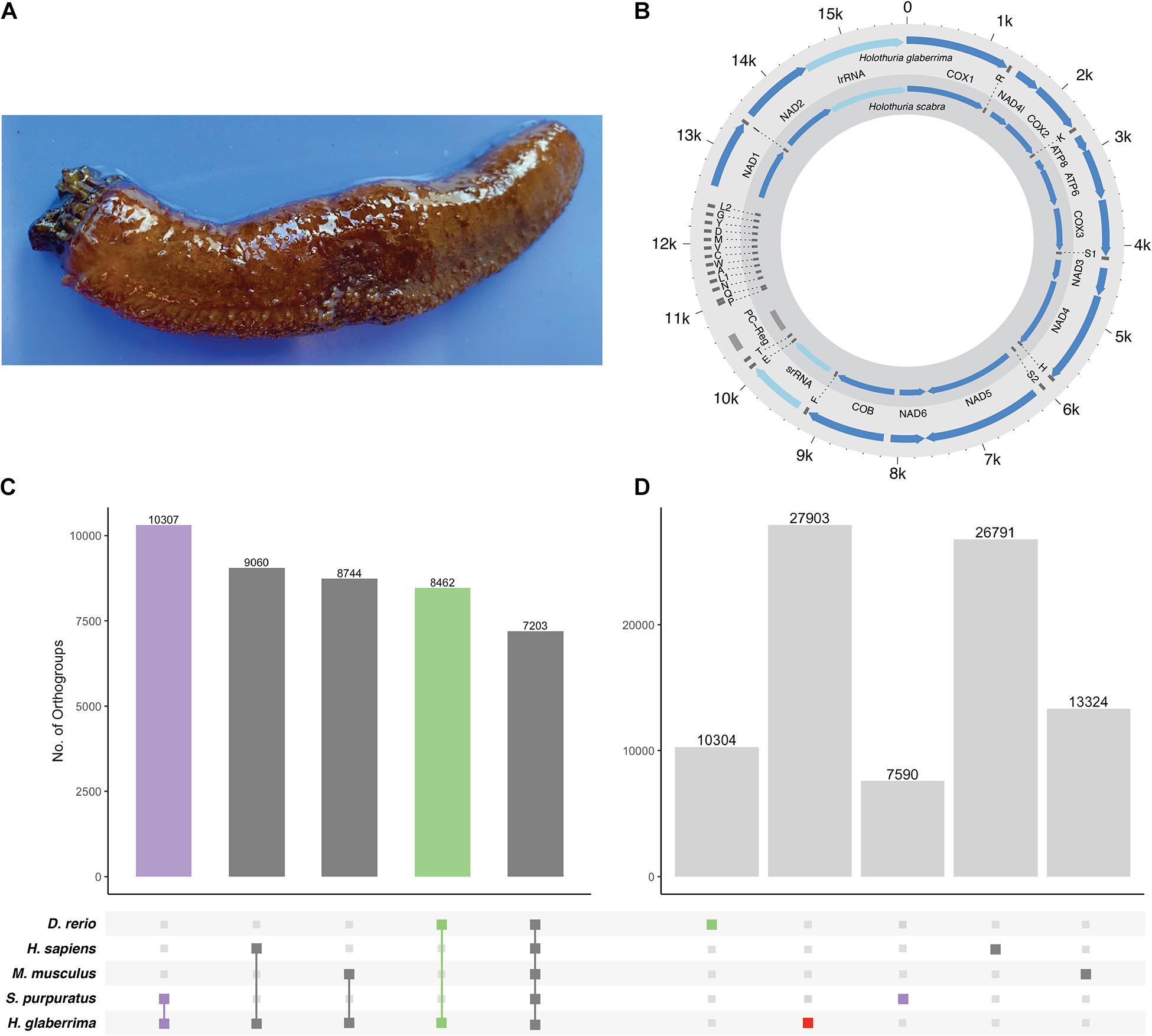
Figure 1. H. glaberrima mitochondrial genome and orthogroup analysis. (A) The sea cucumber H. glaberrima. (B) Comparison of mitochondrial features of H. glaberrima (outer) and H. scabra (inner). Genes = blue, rRNA = light blue, tRNA = dark gray, PC region = light gray. (C,D) Gene content distribution in echinoderm and vertebrate lineages. Filled circles in the bottom panel indicate (C) shared and (D) species-specific orthogroups in those lineages. Bar graphs indicate the number of orthogroups corresponding to each filled-circle pattern. (C) OrthoFinder analyses were performed with amino acid sequences of H. glaberrima protein models (based on predicted gene models), D. rerio, H. sapiens, M. musculus, and S. purpuratus. Total orthogroup overlap between H. glaberrima and each other species as well as total orthogroups shared between all species. (D) Species-specific orthogroups (OGs) for all species analyzed. These include singlets – sequences that didn’t form a group with any other sequence in the analysis and orthogroups consisting of only sequences from a single species.
Sequencing was performed using an Illumina HiSeq X Ten instrument, which generated a total of 421,770,588 paired-end reads (126.5 gigabases) of 150 nucleotides each. These reads were deposited in NCBI’s Short Read Archive (SRA) (Leinonen et al., 2011) under the accession SRR9695030 within 1 week of sequencing. Adapters from the raw sequencing reads were trimmed using Trimmomatic v0.39 (ILLUMINACLIP:2:30:10 LEADING:3 TRAILING:3 SLIDING-WINDOW:4:15 MINLEN:36) (Bolger et al., 2014). We used ALLPATHS-LG v.44837 ErrorCorrectReads.pl script (Gnerre et al., 2011) to apply error correction to reads that had been trimmed for adapters and genome size estimation.
We next created filtered mitochondrial reads from the resulting trimmed/error-corrected reads. To do this we generated a preliminary assembly using Platanus Genome Assembler v1.2.4 (Kajitani et al., 2014) with k = 45 and then performed a BLASTN search against this assembly using the Holothuria scabra mitochondrial genome as a query. We identified a single contig that represented the entire mitochondrial genome. We then used FastqSifter v1.1.3 (Ryan, 2015a) to remove all trimmed/error-corrected reads that aligned to the H. glaberrima mitochondrial genome. FastqSifter removed 53,304 read pairs and 3,583 unpaired reads, leaving 386,920,975 read pairs and 33,176,161 unpaired reads, which were used to assemble the final nuclear genome.
We used Platanus Genome Assembler v1.2.4 using a range of kmer values (31, 45, 59, 73, and 87). From these assemblies we selected our optimal assembly (K31) based on the general statistics results (Supplementary Table 1). In order to incorporate contiguous regions brought together in suboptimal assemblies (i.e., K45, K59, K73, and K87) and absent from our K31 optimal assembly, we used Matemaker v1.0.0 (Ryan, 2015b) to generate artificial mate-pairs from our suboptimal assemblies using a range of insert sizes (Matemaker was previously used in Long et al., 2016; DeBiasse et al., 2020; Ketchum et al., 2020). In total, Matemaker produced ∼160 million artificial mate pairs from the suboptimal assemblies of which, insert sizes of 2 kb made up 42%, 5 kb made up 31%, 10 kb made up 19%, and 20 kb made up 8% (Supplementary Table 2). We then used SSPACE v3.0 (Boetzer et al., 2011) using default parameters to scaffold the optimal K31 contig assembly using the generated artificial mate-pairs. Finally, we applied Redundans v0.14a (Pryszcz and Gabaldón, 2016) with default parameters to eliminate redundancy and potentially generate additional scaffolding and gap closing.
We used Braker v2.1.5 (Stanke et al., 2006, 2008; Hoff et al., 2016, 2019; Brùna et al., 2020a) to predict gene models using GeneMark v4.62 (Lomsadze et al., 2005; Brùna et al., 2020b) and Augustus v3.3.3 (Stanke and Waack, 2003). As part of this process, we generated a sorted bam file from RNA sequencing data of non-regenerative and 1 and 3 days regenerative intestinal tissue (NCBI BioProject: PRJNA660762) by aligning unassembled reads to our genome assembly using STAR v.2.7.0e (Dobin et al., 2013). We also utilized protein sequences of Strongylocentrotus purpuratus (Spur_3.1) downloaded from ENSMBL to generate protein hints with ProtHint (Iwata and Gotoh, 2012; Gotoh et al., 2014; Buchfink et al., 2015; Brùna et al., 2020b). Both the RNA-Seq alignments and hints were utilized by Braker for its hybrid annotation pipeline. To test whether there was evidence for non-animal contamination in the resulting gene predictions, we screened predicted protein models using Alien_Index v2.1 (Ryan, 2014) using default parameters and the version 0.02 of the alien_index database. We identified 153 protein models with BLASTP hits to non-animal sequences with lower E-values than to hits to animal sequences. Of these, only 12 had alien index scores >45, a value deemed by Gladyshev et al. (2008) to be a good indicator of foreign origin. This result indicates that if there is non-animal contamination, it is minimal (it could be that these 12 are horizontally transferred genes). We assessed the assemblies and gene prediction models with BUSCO v3 (Simão et al., 2015) against the metazoan database of 978 genes using the web assessment tool gVolante (Nishimura et al., 2017).
While assembling the H. glaberrima nuclear genome, we identified the assembled mitochondrial genome. There was a 32-nucleotide overlap between the beginning and the end of the mitochondrial scaffold as would be expected from a circular genome. As such, we removed the overlap and reordered the scaffold so that the beginning of the scaffold corresponded with the beginning of the cox1 gene. We used Mitos (Bernt et al., 2013) to predict genes and tRNAscan-SE 2.0 (Chan and Lowe, 2019) to annotate tRNAs. Every annotated gene was additionally confirmed with BLAST against H. scabra mitochondrial genes. This approach resulted in more precise annotation as some of the stop codons were wrongly predicted by Mitos. Furthermore, tRNAscan-SE failed to predict some of the tRNAs in the H. glaberrima mitochondrial genome, in which case we used Mitos predictions.
We used OrthoFinder v2.2.7 (Emms and Kelly, 2019) with default parameters to generate orthogroups using protein models from the newly assembled H. glaberrima genome, as well as from Homo sapiens, Mus musculus, Danio rerio, and S. purpuratus. The datasets from the included species for this analysis were obtained from the Ensembl FTP database.
We further evaluated our genome assembly by closely examining H. glaberrima genes that had been previously described in studies based on expressed sequence tags (EST), RNA sequencing, and/or Sanger-based cDNA sequencing. This approach allowed us to manually assess the accuracy of our genome assembly and to determine gene structures. Specifically, we selected the melanotransferrin gene family which is significantly upregulated in H. glaberrima during intestine regeneration (Rojas-Cartagena et al., 2007). This group included HgMtf1 (NCBI: GQ243222), HgMtf2 (NCBI: KP861416), HgMtf3 (NCBID: KP861417), and HgMtf4 (NCBI: KP861418). We aligned four previously identified Mtf genes to our genome assembly using NCBI BLASTN (Altschul et al., 1990) (Figure 2A and Supplementary Figures 1, 2). Based on these alignments and 5′-GT and AG-3′ intron boundaries, we manually annotated the four Mtf genes across multiple scaffolds. The presence of these Mtf genes was further confirmed by BLAST searches against a previously unpublished transcriptome assembly1. Multiple sequence alignments were done using MAFFT v7.471 (Katoh and Standley, 2013).
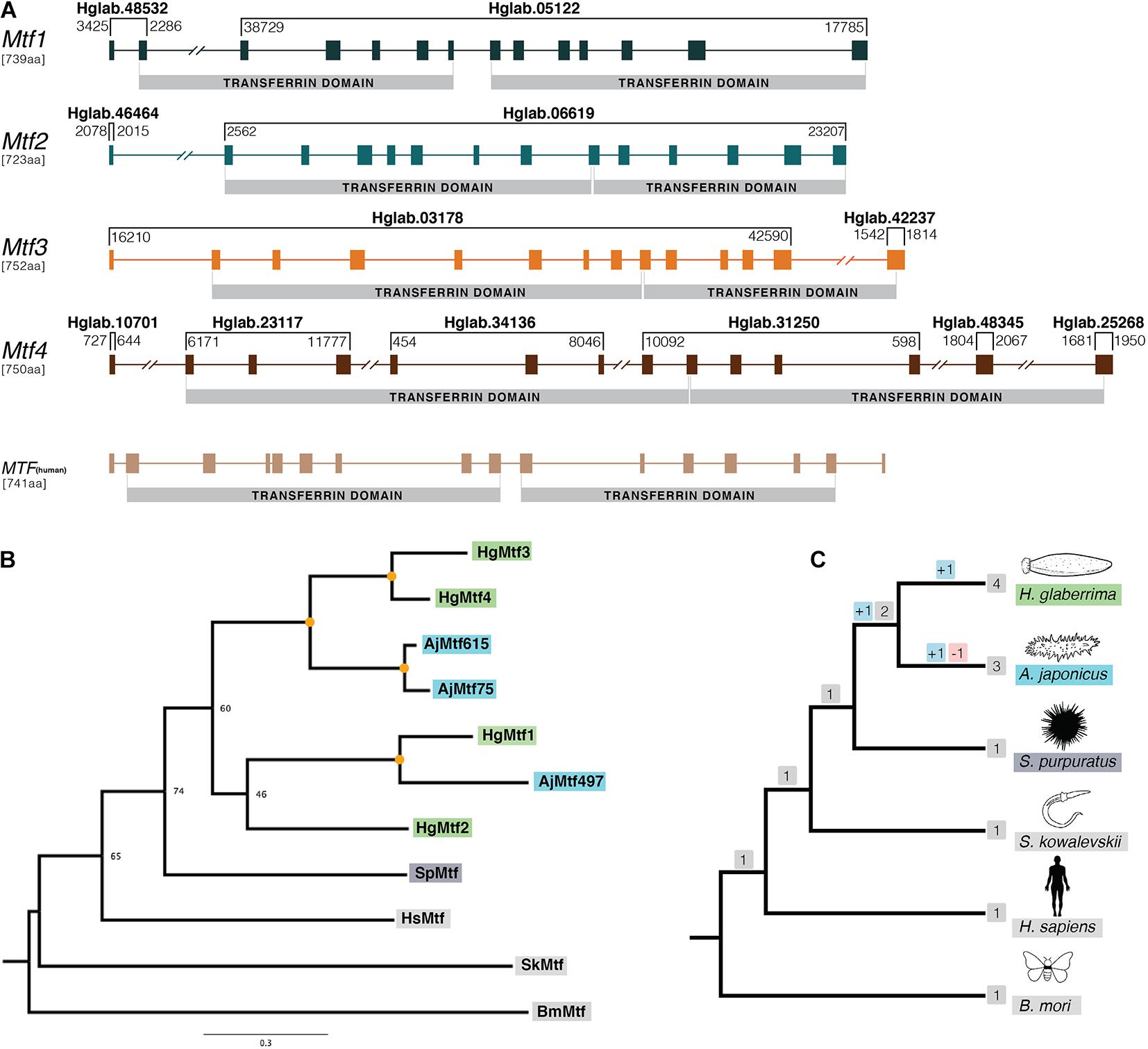
Figure 2. Gene structure and phylogenetic analysis of H. glaberrima melanotransferrin (Mtf) genes. (A) Genomic structure was determined using H. glaberrima Mtf gene and protein sequences present in NCBI from previous studies from Hernández-Pasos et al. (2017). Scaffolds containing specific exons of the genes are shown at the top of each bracket (e.g., Hglab.48532). Coordinates of the sequence in each scaffold are shown among brackets based on genome nucleotide sequence. Human Mtf gene structure was acquired from NCBI genome browser (MELTF NCBI: NM_005929.6). Transferrin domains are shown at the bottom of each gene. (B) Gene tree of all Mtf genes from H. glaberrima (Hg), A. japonicus (Aj), S. purpuratus (Sp), H. sapiens (Hs), S. kowalevskii (Sk), and B. mori (Bm). Mtf gene sequences of the last three species were acquired from NCBI. A. japonicus Mtf gene names represent the scaffold number where they are found in its genome assembly. Orange circles in nodes indicate bootstrap values of 100. (C) Species tree of organisms used in the phylogenetic analysis of the Mtf genes. Gene duplication and loss are shown in red and blue squares, respectively.
To deduce the relationship between the four paralogous H. glaberrima Mtf genes with those of other animals, we conducted a phylogenetic analysis (Figures 2B,C). We used Mtf genes from H. glaberrima, Apostichopus japonicus, S. purpuratus, Saccoglossus kowalevskii, H. sapiens, and Bombyx mori. We also performed a phylogenetic analysis including Parus major (bird), Ciona intestinalis (tunicate), Etheostoma cragini (teleostei), and Exaiptasia diaphana (cnidaria) (Supplementary Figure 3). For both phylogenetic trees performed, we used a comparative approach using IQ-TREE v2.03 and RAxML v8.2.12 (Stamatakis, 2014; Nguyen et al., 2015). The phylotocol (DeBiasse and Ryan, 2019) laying out the planned phylogenetic analyses a priori can be found in the article’s GitHub repository (josephryan/Holothuria_glaberrima_draft_genome).
After removing adapter sequence with Trimmomatic, 400,075,279 (95%) read pairs were retained and 4,200,427,854 (3.5%) bases from these retained pairs were trimmed. After error correction of reads with the ErrorCorrectReads.pl script from ALLPATHS-LG we removed a total of 1,104,390 read pairs (0.1%), orphaned (removed 1 from a pair) an additional 12,545,131 read pairs and made 117,247,709 corrections to 95,509,388 (11.6%) reads (an average of 1.2 corrections per corrected read). The genome size estimation generated by ErrorCorrectReads.pl was 1.2 gigabases (1,171,092,004 bases) with a coverage estimate of 84×. This number is close to the estimate obtained with Feulgen densitometry (∼1.4 gigabases) (Supplementary Figure 4).
We generated five assemblies using Platanus with a range of kmer parameters (31, 45, 59, 73, and 87). The assembly with the kmer parameter K = 31 resulted in the highest N50 (11,332) (Supplementary Table 1). After scaffolding the optimal K31 assembly with artificial mate-pairs from the sub-optimal assemblies (K45, K59, K73, and K87), we produced an assembly consisting of 2,960,762 scaffolds comprising 1,468,331,553 bases with an N50 of 15,069 bases. We next ran the program Redundans to reduce the overall redundancy due to heterozygosity in the data. After Redundans, our assembly consisted of 89,105 scaffolds comprising 1,074,346,208 bases (close to the size estimations from both kmers and Feulgen densitometry) and an N50 of 25,282 bases. The longest scaffold was 244,429 nucleotides. After applying Redundans, there was an improvement in the percentage of scaffolds greater than 50 kb from 11.9 to 18.7%. BUSCO assessment of our final assembly resulted in recovering 750 (76.7%) complete and 144 (14.7%) partial genes for a total of 894 (91.4%) found genes from the 978 queried genes in the metazoa dataset (Supplementary Table 3). Comparative statistics and BUSCO assessments for assemblies before and after applying Redundans can be found in Supplementary Table 3.
We predicted 53,080 gene models with a mean exon length of 185 bp and mean intron length of 1916 bp as calculated using GenomeFeatures v3.10 (Lawrence et al., 2013). Of these 53,080 gene models, 1,665 represented additional isoforms, and the total number of uniquely predicted genes was 51,415. We estimate that more than 40% of the H. glaberrima genes are represented by more than one gene model and infer that this is likely due to the fractured nature of the genome. We therefore suspect the number of genes is likely substantially lower than the number of gene models (Supplementary Figure 5). Our BUSCO assessment of translated gene models resulted in the recovery of 798 (81.6%) complete and 164 (16.8%) partial genes for a total of 962 (98.4%) from the 978 queried genes in the Metazoa dataset (Supplementary Table 4). We also performed gene predictions on our initial assembly, after manually eliminating scaffolds shorter than 200 bp, to make comparisons to that of our predictions using our final assembly after Redundans. To validate our removal of redundant scaffolds, we annotated the assembly before and after applying Redundans and assessed these annotations with BUSCO. The BUSCO assessment of the 58,944 protein models produced from the assembly prior to running Redundans resulted in 778 (79.6%) complete and 181 (18.5%) partial genes, which is substantially inferior to our final annotation (Supplementary Table 4).
The H. glaberrima mitochondrial genome is 15,617 bp, similar in size to that of previously reported holothuroids (H. scabra = 15,779 bp, H. leucospilata = 15,904 bp) (Perseke et al., 2010). Likewise, mitochondrial genome features of H. glaberrima showed the same gene order reported for H. scabra and similar characteristics to those of H. scabra and H. leucospilata (Figure 1B and Supplementary Table 5) (Xia et al., 2016; Yang et al., 2019). Differences included the length of the putative control region (PC region) which was smaller in H. glaberrima with 272 bp compared to 456 bp and 551 bp for H. scabra and H. leucospilata, respectively. Moreover, the total number of intergenic nucleotides in H. glaberrima (113 bp) was similar to H. scabra (109 bp). Total intergenic nucleotides were distributed among 15 intergenic spacers, ranging from 1 to 20 bp in size, comparable to H. scabra and H. leucospilata which contain 15 and 16 intergenic spacers, respectively. There were 5 regions with overlapping genes within the genome, the most significant being those between atp8 and atp6, and nad4 and tRNA-His, with overlaps of 7 bp, and tRNA-P and tRNA-Q with overlap of 4 bp. From the 5 overlapping regions, H. glaberrima shares all the mentioned overlapping areas with H. scabra and H. leucospilata suggesting that these are ancient overlaps. In addition, as reported for both H. scabra and H. forskali, H. glaberrima has an incomplete stop codon T of cox2, supporting the likeliness of it being a distinctive feature of Holothuria (Perseke et al., 2010; Xia et al., 2016).
Our OrthoFinder analysis resulted in a total of 23,126 orthogroups. We identified 7,203 orthogroups shared between all species, 10,307 that contained only sequences from H. glaberrima and S. purpuratus, and close to 9,000 for each of the groups that included only H. glaberrima and one of the vertebrates (Figure 1C). The high number of orthogroups shared between S. purpuratus and H. glaberrima highlights the quality of the predicted gene models. We identified 27,903 H. glaberrima-specific orthogroups; this included 107 that consisted of multiple H. glaberrima protein models per orthogroup and 27,796 consisting of only a single H. glaberrima protein model (“singlets”). There were far more species-specific orthogroups in H. glaberrima than there were in any of the other species—H. sapiens was next with 26,791, which we attribute to the high amount of peptide sequences in the dataset (102,915) (Figure 1D).
The human Mtf gene (MELTF) is large, coding for 749 amino acids and its 16 exons span more than 28 kb. The H. glaberrima Mtf genes are similarly large (HgMtf1 = 739 aa, HgMtf2 = 723 aa, HgMtf3 = 752 aa, HgMtf4 = 750 aa), with all four genomic loci having 14 exons and spanning many kilobases. Due to the large size of these genes and the fragmented nature of our assembly, all four of the Mtf genes occur on multiple scaffolds (Figure 2A). Nevertheless, we were able to account for all exons and capture each gene structure. Besides differing from the human Mtf gene in the number of exons, variations occur in the position of several introns suggesting that there have been substantial structural changes in these genes since the last common ancestor of vertebrates and echinoderms.
Our assembly captures considerable variability between the HgMtf1 and the three other H. glaberrima Mtf genes. For example, the sequence that codes for the N-terminal transferrin domains of HgMtf1 is interrupted by 5 introns, whereas the corresponding coding regions in HgMtf2–4 include 7 introns. Likewise, the sequence that codes for the C-terminal transferrin domain of HgMtf1 includes 6 introns, whereas these same coding regions in HgMtf2–4 include 5 introns (Figure 2A). These results show that there have been considerable evolutionary changes in the genomic architecture HgMtf1, suggesting that HgMtf1 did not arise recently. A sequence alignment of all the annotated Mtf proteins is shown in Supplementary Figure 2.
The 3′ sequence of HgMtf1 in the genome differs from the previously reported cDNA sequence in GenBank (ACS74869) (Supplementary Figure 1). There appears to be a single nucleotide present in the genome that is absent from the previously reported sequence. This frameshift leads to 65 fewer amino acids in the translation of the genomic HgMtf1. We surmise that the genome is correct and that the previously published sequence includes an erroneous single-nucleotide deletion based on the following evidence: a BLASTP of the 65 amino-acid portion against the GenBank non-redundant database produces only a single match to itself. The region in question is exactly the same in all of our optimal and suboptimal assemblies. Moreover, TBLASTN of the 65 amino acids against our transcriptome produced no hits. Alignment of HgMtf1 against the previous GenBank sequence and the HgMtf1 recovered from our transcriptome also confirms this erroneous frameshift (Supplementary Figure 1).
The other sea cucumber species, A. japonicus, in our orthogroup analysis, included more than one Mtf gene in its genome (Zhang et al., 2017). Initial pre-genomic studies reported only one melanotransferrin in A. japonicus (Qiu et al., 2014), but the latest genome data showed that there were multiple copies in its genome. We searched the predicted gene models of A. japonicus and found three Mtf genes, which we included in our phylogenetic analysis (our criteria for inclusion were the presence of two full transferrin domains). The gene tree shown in Figure 2B, demonstrate the relationship between the A. japonicus and H. glaberrima Mtf genes. As such, our phylogenetic analysis suggests that some Mtf gene-family expansion occurred in the stem of the holothuroid lineage but that additional duplications occurred independently in the lineages leading to A. japonicus and H. glaberrima (Figures 2B,C). Recent evidence of data deposited in NCBI suggest that also certain teleost species might contain a duplicated Mtf gene (i.e., E. cragini). However, it has been predicted that one of these E. cragini Mtf genes is no longer functional (Glasauer and Neuhauss, 2014). Our phylogenetic results suggest that the four A. japonicus and four H. glaberrima Mtf genes are not one-to-one orthologs, which would suggest that they each inherited these from the last common ancestor of these two lineages. Instead, under parsimony, our results suggest the following: this last common ancestor had at least 3 Mtf genes (i.e., MTF1, MTF2, and MTF3/4), MTF2 was lost in A. japonicus, MTF1 and MTF3/4 duplicated in A. japonicus, and MFTF3/4 duplicated in H. glaberrima.
We have sequenced, assembled, and annotated the first draft genome of H. glaberrima, one of the best studied regenerative model invertebrates. We demonstrated the potential of the assembled genome by analyzing the Mtf gene family genomic loci. We also assembled and annotated the H. glaberrima mitochondrial genome showing conservation with other mitochondrial genomes from the genus Holothuria. Our work provides a key resource for future studies in animal regeneration and beyond.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) are as follows: the raw genomic reads are available at NCBI’s SRA (Accession: SRR9695030). The genome assembly is available at http://ryanlab.whitney.ufl.edu/genomes/Holothuria_glaberrima/ and at NCBI (PRJNA497079). The RNA sequencing reads used for the annotation are available in NCBI under the BioProject PRJNA660762. Custom scripts, command lines, and data used in these analyses and alignment and tree files are available at https://github.com/josephryan/Holothuria_glaberrima_draft_genome.
JR, JG-A, JM-F, and VM conceived the project. JG-A collected the animal and tissue samples and contributed to the manuscript. JM-F and JR performed all assemblies, annotations, and analyses. JM-F wrote the first draft of the manuscript with input from JR. SP performed the DNA extraction and sample processing. VM performed the experimental genome size estimation and contributed to the manuscript. All authors approved the final version of the manuscript.
The project was partially funded by the National Institutes of Health (NIH) (Grant Nos. R15NS01686 and R21AG057974) and the University of Puerto Rico. Also, funding support was provided by the National Science Foundation (NSF) (1542597 to JR and 1826558 to JM-F). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF grant number ACI-1548562. Specifically, it used the Bridges System, which is supported by the NSF award number ACI-1445606, at the Pittsburgh Supercomputing Center (PSC).
SP was employed by Iridian Genomes, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the high-performance computing facility of the University of Puerto Rico making their resources available to us, sponsored by the University of Puerto Rico and the Institutional Development Award (IDeA) INBRE grant P20 GM103475 from the National Institute for General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH) and the Bioinformatics Research Core of INBRE.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.603410/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Bely, A. E., and Nyberg, K. G. (2010). Evolution of animal regeneration: re-emergence of a field. Trends Ecol. Evol. 25, 161–170. doi: 10.1016/j.tree.2009.08.005
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brùna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2020a). BRAKER2: automatic eukaryotic genome annotation with genemark-EP+ and augustus supported by a protein database. bioRxiv [preprint] doi: 10.1101/2020.08.10.245134
Brùna, T., Lomsadze, A., and Borodovsky, M. (2020b). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics Bioinform. 2:lqaa026. doi: 10.1093/nargab/lqaa026
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cannon, J. T., Vellutini, B. C., Smith, J., Ronquist, F., Jondelius, U., and Hejnol, A. (2016). Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93. doi: 10.1038/nature16520
Chan, P. P., and Lowe, T. M. (2019). “tRNAscan-SE: searching for tRNA genes in genomic sequences,” in Gene Prediction Methods in Molecular Biology, ed. M. Kollmar (New York, NY: Springer New York), 1–14. doi: 10.1007/978-1-4939-9173-0_1
DeBiasse, M. B., Colgan, W. N., Harris, L., Davidson, B., and Ryan, J. F. (2020). Inferring tunicate relationships and the evolution of the tunicate hox cluster with the genome of corella inflata. Genome Biol. Evol. 12, 948–964. doi: 10.1093/gbe/evaa060
DeBiasse, M. B., and Ryan, J. F. (2019). Phylotocol: promoting transparency and overcoming bias in phylogenetics. Syst. Biol. 68, 672–678. doi: 10.1093/sysbio/syy090
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
García-Arrarás, J. E., Lázaro-Peña, M. I., and Díaz-Balzac, C. A. (2018). “Holothurians as a model system to study regeneration,” in Marine Organisms as Model Systems in Biology and Medicine, eds M. Kloc and J. Z. Kubiak (Cham: Springer International Publishing), 255–283. doi: 10.1007/978-3-319-92486-1_13
García-Arrarás, J. E., Estrada-Rodgers, L., Santiago, R., Torres, I. I., Díaz-Miranda, L., and Torres-Avillán, I. (1998). Cellular mechanisms of intestine regeneration in the sea cucumber, Holothuria glaberrima Selenka (Holothuroidea:echinodermata). J. Exp. Zool. 281, 288–304. doi: 10.1002/(SICI)1097-010X(19980701)281:4<288::AID-JEZ5<3.0.CO;2-K
Gladyshev, E. A., Meselson, M., and Arkhipova, I. R. (2008). Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213. doi: 10.1126/science.1156407
Glasauer, S. M. K., and Neuhauss, S. C. F. (2014). Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060. doi: 10.1007/s00438-014-0889-2
Gnerre, S., MacCallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Gotoh, O., Morita, M., and Nelson, D. R. (2014). Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC Bioinform. 15:189. doi: 10.1186/1471-2105-15-189
Hernández-Pasos, J., Valentín-Tirado, G., and García-Arrarás, J. E. (2017). Melanotransferrin: new homolog genes and their differential expression during intestinal regeneration in the sea cucumber holothuria glaberrima: melanotransferrin homologs in holothurians. J. Exp. Zool. (Mol. Dev. Evol.) 328, 259–274. doi: 10.1002/jez.b.22731
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M., and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS: Table 1. Bioinformatics 32, 767–769. doi: 10.1093/bioinformatics/btv661
Hoff, K. J., Lomsadze, A., Borodovsky, M., and Stanke, M. (2019). “Whole-genome annotation with braker,” in Gene Prediction Methods in Molecular Biology, ed. M. Kollmar (New York, NY: Springer New York), 65–95. doi: 10.1007/978-1-4939-9173-0_5
Iwata, H., and Gotoh, O. (2012). Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 40, e161–e161. doi: 10.1093/nar/gks708
Jaillon, O., Aury, J.-M., Brunet, F., Petit, J.-L., Stange-Thomann, N., Mauceli, E., et al. (2004). Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957. doi: 10.1038/nature03025
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Ketchum, R. N., Smith, E. G., DeBiasse, M. B., Vaughan, G. O., McParland, D., Leach, W. B., et al. (2020). Population genomic analyses of the sea urchin echinometra sp. EZ across an extreme environmental gradient. Genome Biol. Evol. 12, 1819–1829. doi: 10.1093/gbe/evaa150
Lambert, L. A., Perri, H., and Meehan, T. J. (2005). Evolution of duplications in the transferrin family of proteins. Compar. Biochem. Physiol. Part B: Biochem. Mol. Biol. 140, 11–25. doi: 10.1016/j.cbpc.2004.09.012
Lawrence, M., Huber, W., Pagès, H., Aboyoun, P., Carlson, M., Gentleman, R., et al. (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9:e1003118. doi: 10.1371/journal.pcbi.1003118
Leinonen, R., Sugawara, H., Shumway, M., and International Nucleotide Sequence Database Collaboration (2011). The sequence read archive. Nucleic Acids Res. 39, D19–D21. doi: 10.1093/nar/gkq1019
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Long, K. A., Nossa, C. W., Sewell, M. A., Putnam, N. H., and Ryan, J. F. (2016). Low coverage sequencing of three echinoderm genomes: the brittle star Ophionereis fasciata, the sea star Patiriella regularis, and the sea cucumber Australostichopus mollis. GigaSci 5:20. doi: 10.1186/s13742-016-0125-6
Martin, W. F., Garg, S., and Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Phil. Trans. R. Soc. B 370:20140330. doi: 10.1098/rstb.2014.0330
Mashanov, V. S., and García-Arrarás, J. E. (2011). Gut regeneration in holothurians: a snapshot of recent developments. Biol. Bull. 221, 93–109. doi: 10.1086/BBLv221n1p93
Mashanov, V. S., Zueva, O. R., and García-Arrarás, J. E. (2014). Transcriptomic changes during regeneration of the central nervous system in an echinoderm. BMC Genomics 15:357. doi: 10.1186/1471-2164-15-357
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nishimura, O., Hara, Y., and Kuraku, S. (2017). gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 33, 3635–3637. doi: 10.1093/bioinformatics/btx445
Ortiz-Pineda, P. A., Ramírez-Gómez, F., Pérez-Ortiz, J., González-Díaz, S., Santiago-De Jesús, F., Hernández-Pasos, J., et al. (2009). Gene expression profiling of intestinal regeneration in the sea cucumber. BMC Genomics 10:262. doi: 10.1186/1471-2164-10-262
Perseke, M., Bernhard, D., Fritzsch, G., Brümmer, F., Stadler, P. F., and Schlegel, M. (2010). Mitochondrial genome evolution in Ophiuroidea, Echinoidea, and Holothuroidea: insights in phylogenetic relationships of Echinodermata. Mol. Phylogenet. Evol. 56, 201–211. doi: 10.1016/j.ympev.2010.01.035
Pryszcz, L. P., and Gabaldón, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113–e113. doi: 10.1093/nar/gkw294
Qiu, X., Li, D., Cui, J., Liu, Y., and Wang, X. (2014). Molecular cloning, characterization and expression analysis of Melanotransferrin from the sea cucumber Apostichopus japonicus. Mol. Biol. Rep. 41, 3781–3791. doi: 10.1007/s11033-014-3243-1
Rahmanto, Y. S., Bal, S., Loh, K. H., Yu, Y., and Richardson, D. R. (2012). Melanotransferrin: search for a function. Biochim. Biophys. Acta (BBA)-General Subjects 1820, 237–243. doi: 10.1016/j.bbagen.2011.09.003
Ramírez-Gómez, F., Ortiz-Pineda, P. A., Rivera-Cardona, G., and García-Arrarás, J. E. (2009). LPS-Induced genes in intestinal tissue of the sea cucumber Holothuria glaberrima. PLoS One 4:e6178. doi: 10.1371/journal.pone.0006178
Rojas-Cartagena, C., Ortíz-Pineda, P., Ramírez-Gómez, F., Suárez-Castillo, E. C., Matos-Cruz, V., Rodríguez, C., et al. (2007). Distinct profiles of expressed sequence tags during intestinal regeneration in the sea cucumber Holothuria glaberrima. Physiol. Genomics 31, 203–215. doi: 10.1152/physiolgenomics.00228.2006
Ryan, J. (2015a). FastqSifter. Available online at: https://github.com/josephryan/FastqSifter (accessed February 26, 2020).
Ryan, J. (2015b). Matemaker. Available online at: https://github.com/josephryan/matemaker (accessed February 26, 2020).
Ryan, J. F. (2014). Alien Index: Identify Potential Non-Animal Transcripts or Horizontally Transferred Genes in Animal Transcriptomes. Available online at: http://dx.doi.org/10.5281/zenodo.21029 (accessed March 3, 2021).
San Miguel-Ruiz, J. E., Maldonado-Soto, A. R., and García-Arrarás, J. E. (2009). Regeneration of the radial nerve cord in the sea cucumber Holothuria glaberrima. BMC Dev. Biol. 9:3. doi: 10.1186/1471-213X-9-3
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Stanke, M., Schöffmann, O., Morgenstern, B., and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 7:62. doi: 10.1186/1471-2105-7-62
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225. doi: 10.1093/bioinformatics/btg1080
Xia, J., Ren, C., Yu, Z., Wu, X., Qian, J., and Hu, C. (2016). Complete mitochondrial genome of the sandfish Holothuria scabra (Holothuroidea, Holothuriidae). Mitochondrial DNA Part A 27, 4174–4175. doi: 10.3109/19401736.2014.1003899
Yang, Q., Lin, Q., Wu, J., Tran, N. T., Huang, R., Sun, Z., et al. (2019). Complete mitochondrial genome of Holothuria leucospilata (Holothuroidea, Holothuriidae) and phylogenetic analysis. Mitochondrial DNA Part B 4, 2751–2752. doi: 10.1080/23802359.2019.1644226
Keywords: holothuroid, echinoderm, regeneration, melanotransferrin, mitochondrial genome
Citation: Medina-Feliciano JG, Pirro S, García-Arrarás JE, Mashanov V and Ryan JF (2021) Draft Genome of the Sea Cucumber Holothuria glaberrima, a Model for the Study of Regeneration. Front. Mar. Sci. 8:603410. doi: 10.3389/fmars.2021.603410
Received: 07 September 2020; Accepted: 12 March 2021;
Published: 15 April 2021.
Edited by:
Oleg Simakov, University of Vienna, AustriaReviewed by:
Gustavo Sanchez, Hiroshima University, JapanCopyright © 2021 Medina-Feliciano, Pirro, García-Arrarás, Mashanov and Ryan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph F. Ryan, am9zZXBoLnJ5YW5Ad2hpdG5leS51ZmwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.