 Marta Turon
Marta Turon Carlos Angulo-Preckler
Carlos Angulo-Preckler Adrià Antich
Adrià Antich Kim Præbel
Kim Præbel Owen S. Wangensteen
Owen S. Wangensteen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 09 December 2020
Sec. Marine Molecular Biology and Ecology
Volume 7 - 2020 | https://doi.org/10.3389/fmars.2020.605148
Sponges have recently been proposed as ideal candidates to act as natural samplers for environmental DNA due to their efficiency in filtering water. However, validation of the usefulness of DNA recovered from sponges to reveal vertebrate biodiversity patterns in Marine Protected Areas is still needed. Additionally, nothing is known about how different sponge species and morphologies influence the capture of environmental DNA and whether biodiversity patterns obtained from sponges are best described by quantitative or qualitative measures. In this study, we amplified and sequenced a vertebrate specific 12S barcode with a set of universal PCR primers (MiFish) for metabarcoding environmental DNA from fishes, to unveil fine-scale patterns of fish communities from natural-sampler DNA retrieved from 64 sponges (16 species) located in eutrophic and well-preserved coral reefs in Nha Trang Bay (central Vietnam). Ninety tropical fish species were identified from the sponges, corresponding to one third of the total local ichthyofauna reported from previous extensive conventional surveys. Significant differentiation in fish communities between eutrophic and well-preserved environments was observed, albeit eutrophication only explained a modest proportion of the variation between fish communities. Differences in efficiency of capturing fish environmental DNA among sponge species or morphologies were not observed. Overall, the majority of detected fish species corresponded to reef-associated small-sized species, as expected in coral reefs environments. Remarkably, pelagic, migratory, and deep-sea fish species were also recovered from sponge tissues, pointing out the ability of sponge natural sampled DNA to detect fishes that were not permanently associated to the biomes where the sponges were sampled. These results highlight the suitability of natural samplers as a cost-effective way to assess vertebrate diversity in hyper-diverse environments.
Environmental DNA (eDNA) refers to the DNA extracted directly form an environmental sample without the need of isolating the target organisms (Thomsen and Willerslev, 2015; Creer et al., 2016; Cristescu and Hebert, 2018). Environmental DNA metabarcoding is emerging as a fast and efficient method for biodiversity monitoring that provides increased sensitivity and reduced costs compared to conventional surveys (Kelly et al., 2014b; Thomsen and Willerslev, 2015; Creer et al., 2016; Boussarie et al., 2018; Sales et al., 2020). Using eDNA provides different opportunities, such as the detection of cryptic taxa or different life stages, the rapid quantification of species richness and the non-invasiveness of sampling procedures, among others (Deiner et al., 2017). However, challenges remain for its use in different fields of study such as biomonitoring, ecology, conservation and invasion biology (Deiner et al., 2017).
A persistent matter of debate in metabarcoding studies is the quantification of species abundances inferred from eDNA data (Kelly, 2016; Deiner et al., 2017). This quantification issue can be seen in a new light if the actual complexity of eDNA is considered. A natural eDNA sample is always a complex mix, comprised of two inseparable fractions with strikingly dissimilar concentrations. The trace-quantities of extra-organismal DNA released by macrofaunal individuals into their environment (which is what most researchers restrictively understand as “eDNA”) are embedded in an overwhelming background of genomic DNA fragments extracted from the community of living microorganisms (mostly bacteria, but also microeukaryotes) that were present in the environment when the sample was taken. Whether a method is targeting the trace-DNA of macrofauna or the community-DNA of microorganisms is determined by the specificity of the metabarcoding primers used. Methods relying on the detection of trace-DNA can be seriously hindered by non-specific amplification of hyper-abundant bacterial sequences if primers are not specific-enough (Collins et al., 2019). At the other end of the spectrum, primers with high specificity values applied on samples containing too low concentrations of the target sequences usually lead to stochastic amplification results and reproducibility issues (Kelly et al., 2019). The use of multiple PCR replicates has been proposed as a way to overcome this recurrent problem (Sigsgaard et al., 2019), but this solution leads to increased costs and needed sample volumes.
However, the real ability of the proposed methods based on natural sampler DNA (nsDNA) to provide robust ecological information, which could be translated into management decisions, is yet to be tested in a real-life scenario. Although promising, much work remains to be done to validate the use of sponges as natural samplers for marine biodiversity studies.
Environmental management and policy decision making for Marine Protected Areas (MPA) can greatly benefit from genetic monitoring (Kelly et al., 2014b; Shum et al., 2019), which can facilitate the assessment, among other biota, of fish community structure and distribution. Several traditional biodiversity studies have been carried in Nha Trang Bay MPA (Central Vietnam), although few have been published (Vo et al., 2004; Van Nguyen and Kim Phan, 2008). In the present study, we aim to (1) validate the usefulness of nsDNA recovered from sponges to reveal fine-scale fish biodiversity patterns in Nha Trang Bay, (2) assess the efficiency in capturing nsDNA between different sponge species and morphologies, and (3) evaluate whether quantitative or qualitative data represented better the fish communities. With the aim to work toward the validation of sponges as an alternative genetic tool for biodiversity assessments, we also compared our results with visual census data performed in the same area.
DNA extracts from sponge tissues were originally collected for research on microbiome structure (Turon et al., 2018, 2019) and were used in the present study as opportunistic samples for the analysis of nsDNA captured by the sponges. These DNA extracts had been previously stored in freezing facilities which also contained other samples from temperate species and which not followed specific routines to avoid cross-contamination by trace-DNA. However, no other tissue or DNA sample from Indo-Pacific origin were ever stored along with these samples. Sponges had been originally collected in April 2015 and intended to reflect the most common sponges found in coral reef areas of Nha Trang Bay (Central Vietnam) at depths between 3 and 9 m (Turon et al., 2019). DNA extractions from small fractions (0.5 cm3) of sponge samples were performed using DNeasy Blood & Tissue Kit (Qiagen). In total, 86 samples belonging to 24 different sponge species distributed across six different locations in Nha Trang Bay were processed (Figure 1). The number of replicates per sponge species ranged between 1 and 12 and sponge morphologies were classified as massive, branching, thick encrusting or encrusting (Table 1). Among the sampled locations, Dambay represented an eutrophic area, highly impacted by mariculture activities, and the five other locations represented relatively well-preserved areas within the Nha Trang Marine Protected area (Tkachenko et al., 2016).
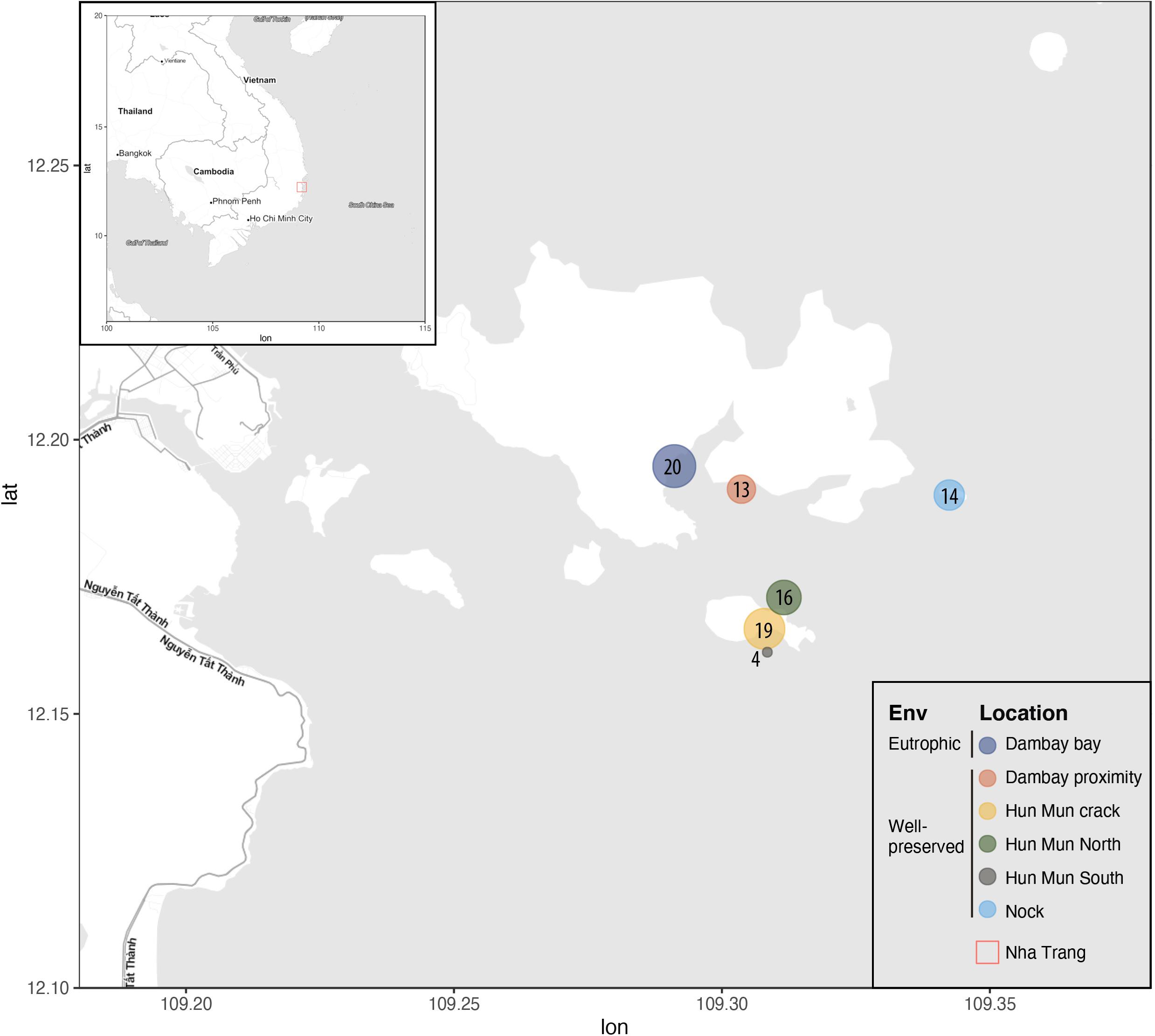
Figure 1. Map of the sampling locations of Nha Trang Bay. All the islands within the sampling region belonged to the Marine Protected area (MPA). Size and number of the circles correspond to the number of samples (sponges) taken from each location, represented with different colors.
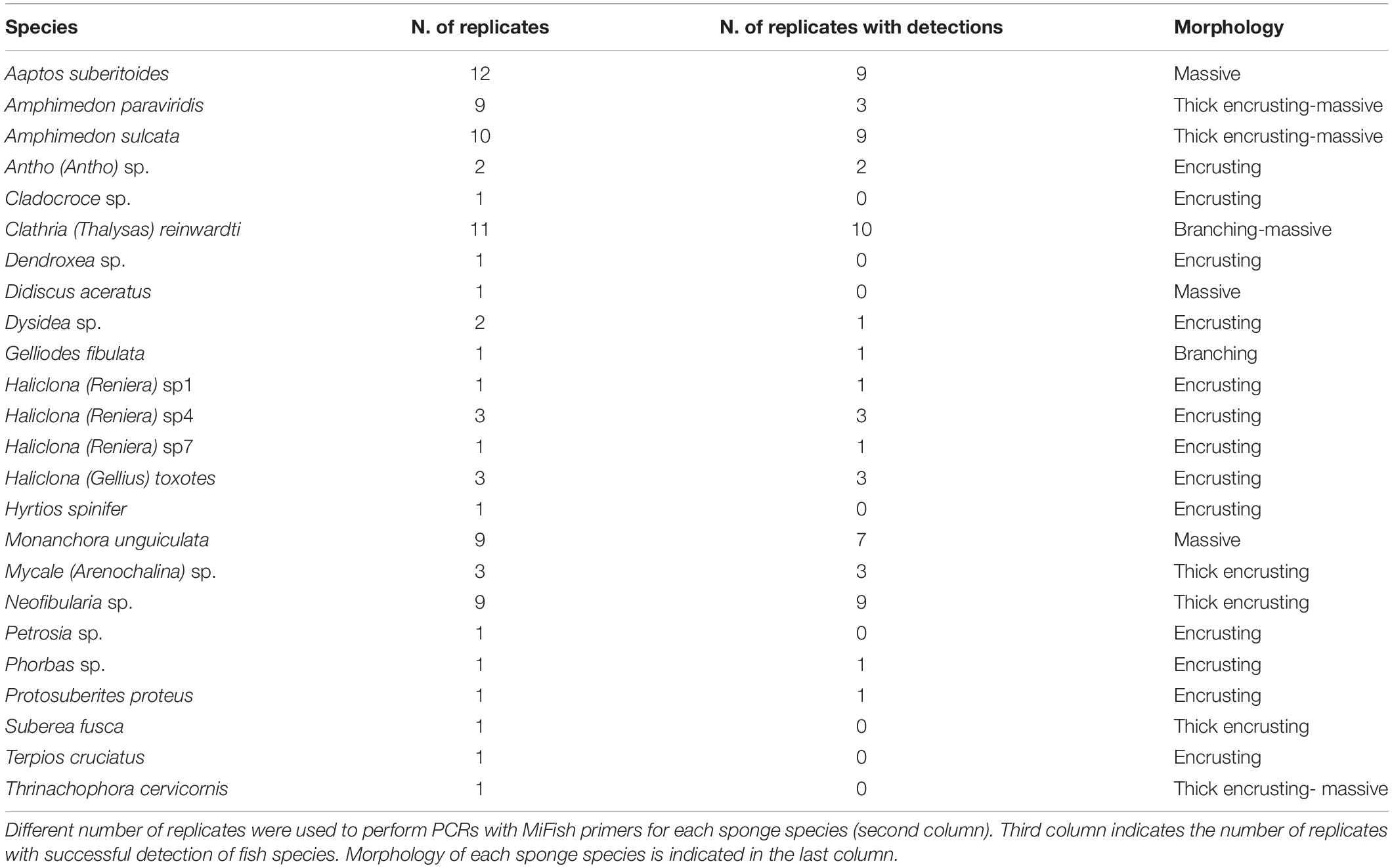
Table 1. Sponge species collected in Nha Trang Bay (Vietnam).
To assess the composition of fish assemblages recoverable from the sponge tissues, total DNA extracted from sponge samples was amplified using the high-specificity MiFish primer set targeting a hypervariable region in the mitochondrial 12S rRNA gene (163–185 bp), which specifically amplifies fish and other vertebrates (Miya et al., 2015). All amplification and library preparation works were performed following strict clean lab routines at the Norwegian College of Fishery Science. PCR amplifications were conducted in 15 μl reactions containing 2 μl of DNA template, 10 μl of AmpliTaq Gold Master mix, 0.16 μl of Bovine Serum Albumin (20 μg/μl), 1 μl of each forward (5′- GTCGGTAAAACTCGTGCCAGC-3′) and reverse (5′-CATAGTGGGGTATC TAATCCCAGTTTG−3′) primer (5 μM) and 5.84 μl of H2O. Both primers included 7-base tags in 5′ which uniquely identified amplicons belonging to the same sample, and a variable number (2–4) of leading Ns in order to increase sequence diversity within the sequencer flow-cell. The temperature profile was as follows: 94°C for 10 min; 40 cycles × (95°C/30 s, 60°C/30 s, 72°C/30 s); 72°C/5 min. Two PCRs were run per sample as technical replicates, and a negative control was run as above but without DNA template added. The success of PCR amplifications was checked by gel electrophoresis in 1% agarose and PCR products were then pooled together into two multiplex sample pools (one per PCR replicate). MinElute PCR purification columns (Qiagen) were used to concentrate the pooled DNA and to remove fragments below 70 bp. Library preparation was performed with the NEXTflex PCR-free library preparation kit (BIOO Scientific) and the exact library concentration was measured in a qPCR using the NEBNext Library Quant Kit (New England BioLabs). Finally, pools were sequenced along with 1% PhiX on an Illumina MiSeq platform using v2 chemistry (2 × 150 bp).
The OBITools v1.01.22 software suite (Boyer et al., 2016) was used for the initial steps of the bioinformatic analyses. Paired-end reads were aligned using illuminapairedend and only sequences with alignment quality score >40 were kept. Demultiplexing was done with ngsfilter, which also removed primer sequences. Aligned reads with length of 140–190 bp and without ambiguous positions were selected using obigrep and then dereplicated with obiuniq. Chimeric sequences were removed using the uchime-de novo algorithm implemented in vsearch v1.10.1 (Rognes et al., 2016). Clustering of sequences into molecular operational taxonomic units (MOTUs) was performed using SWARM 2.0 algorithm (Mahé et al., 2015) with a d value of 1. Taxonomic assignment of the most abundant (representative) sequence of each MOTU was done with the ecotag algorithm (Boyer et al., 2016) against the DUFA_MiFish v2020-05-01 reference database, compiled from sequences of the MiFish fragment available from Genbank and complemented with some in-house sequencing of selected taxa. This database is publicly available from github.com/uit-metabarcoding/DUFA. Further manual refining of the dataset consisted of removing some MOTUs identified as human or terrestrial vertebrates or as fish species clearly not distributed in the area, likely contaminations from other projects being conducted at the laboratories at Salford University (United Kingdom) and CEAB-CSIC (Spain), where samples had been previously stored.
Statistical analyses were performed in R version 3.1.31 with the vegan package (version 2.5-6; Oksanen et al., 2019) and graphic visualizations were done with ggplot2 package (Wickham, 2016). Species accumulation curve was drawn using the speacaccum function of the vegan package. Reads were first transformed to relative abundances to build a Bray-Curtis dissimilarity matrix (PCR replicates not pooled together), which was used to assess the variance in community composition using Permutational Multivariate Analyses of Variance (PERMANOVA). The samples were categorized as a function of Environment (eutrophic, normal), Location (6 levels), Species (16 levels), and Sponge morphology (4 levels) and the univariate effects of these factors on the community composition were tested using adonis function with 1,000 permutations. Additionally, PERMDISP analysis (betadisper function) was performed to determine if the significance of factors was due to different multivariate mean or to different heterogeneity of the groups. Non-metric multidimensional scaling (nMDS) representation with Bray-Curtis dissimilarities was performed with the metaMDS function with 500 iterations. Shannon diversity and MOTU richness per sample were calculated in vegan (Oksanen et al., 2019) and significant differences between sponge species and sponge morphologies were tested with Kruskal-Wallis test. The remaining analyses were made with presence/absence data (see Results). A heatmap was used to represent species with 0, 1, and 2 detections per sample. Only samples with positive counts were used for graphic representations.
All the fish MOTUs that were identified at species or genus level were grouped according to the following biological and ecological traits: schooling behavior (solitary, small, large), individual size (small (<15 cm), medium (15–50 cm), large (>50 cm), depth range (surface (0–10 m), shallow (10–60 m), deep (>60 m), habitat (reef-associated, pelagic) and migratory behavior (yes, no). We then calculated the percentage of total detections and total reads that belonged to the different categories of each trait. Moreover, we assessed the mean detections and the mean reads per MOTU according to the traits’ categories.
We compared our eDNA results with visual census data reported in Van Nguyen and Kim Phan (2008). In that study visual censuses were performed on coral reef fishes in 16 different locations in Nha Trang Bay during September-October 2008. Overall, these authors performed 32 transects. Each transect was 100 m long and 5 m wide, thus covering an area of 500 m2. Fish present within 5 m above the bottom were recorded. Venn diagrams were used to represent the fish families, genera and species recovered by both sampling methods. Additionally, we searched in Fishbase (Froese and Pauly, 2019)2 whether fish species or genera recovered in our eDNA survey were previously reported in Nha Trang or in other areas of Vietnam.
Our final dataset consisted of 90 fish MOTUs recovered from 172 PCR samples (86 sponge samples with 2 PCR replicates each). Of these, only 16 sponge species (64 samples), from the original 24 species selected, had positive PCR counts in at least one of the PCR sample replicates after removal of contaminants (Table 1). Indeed, the species accumulation curve did not reach an asymptote, suggesting that more sampling effort is needed to increase the accumulation rate of new MOTUs and accurately capture the whole fish diversity in the area (Figure 2). MOTU richness per sample was rather low, with the majority of samples having only one or two MOTUs. The maximum MOTU richness was 12 in a single sample (Supplementary Figure S1). No significant differences were observed in the number of MOTUs recovered from different sponge species or sponge morphologies (Supplementary Figure S2 and Supplementary Table S1). Within species, no coherent pattern was detected either, as different sponge replicates of a given species presented varying values of MOTU richness (Supplementary Figure S2).
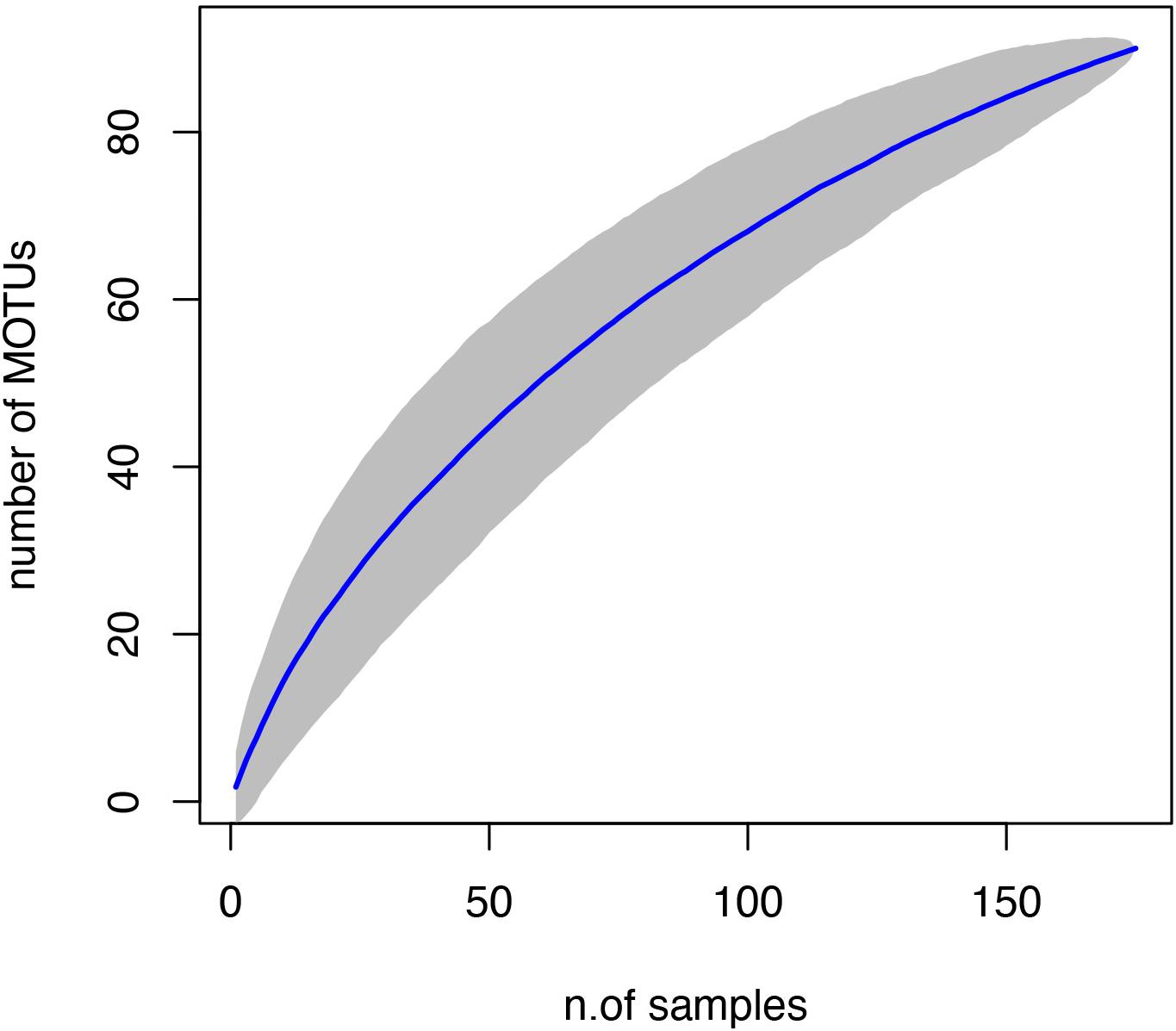
Figure 2. Species accumulation curve with 95% confidence intervals (gray area) for the 172 samples (considering 2 PCR replicates per sample).
The sequencing produced a highly differential number of reads per sample. In general, the number of reads from samples that only had one positive PCR replicate was lower than the number of reads from samples that had two positive PCR replicates (Supplementary Figure S3). However, the difference in number of reads between the two PCR replicates was high, suggesting an important stochastic component of the PCR amplification of the 12S MiFish primer set (Supplementary Figure S3).
The majority of the MOTUs were identified at the species (76 MOTUs) or genus (11 MOTUs) level, with only three MOTUs that could not be classified at lower taxonomic level than class (Supplementary Table S2). In our dataset, each MOTU belonged to a different fish species, so both terms (MOTU and species) will be used interchangeably hereafter. We identified 40 different families, among which Pomacentridae (11 MOTUs), Labridae (7 MOTUs), Gobiidae (5 MOTUs), and Apogonidae (5 MOTUs) were the ones with most representatives.
The non-metric multidimensional scaling (nMDS) representation based on relative read abundance showed slightly different fish communities between eutrophic and well-preserved environments, although with a large overlap (Figure 3). PERMANOVA analyses of the environment (adonis: R2 = 0.02, p < 0.001) and sponge morphology (adonis: R2 = 0.04, p < 0.001) factors were significant but only explained a small proportion of the total variation in the nsDNA fish communities (Supplementary Table S3). Similarly, sampling location (adonis: R2 = 0.08, p < 0.001) was a significant factor in structuring fish communities, but only explained a low amount of the total variation (Supplementary Table S3 and Supplementary Figure S4). However, PERMDISP test showed that location effect could be explained by differences in dispersion levels across locations. In a similar manner, differences in the nsDNA from fish communities between sponge species (adonis: R2 = 0.21, p < 0.001) were significant, but this effect could be due to significant heterogeneity in dispersion across species (Supplementary Table S3).
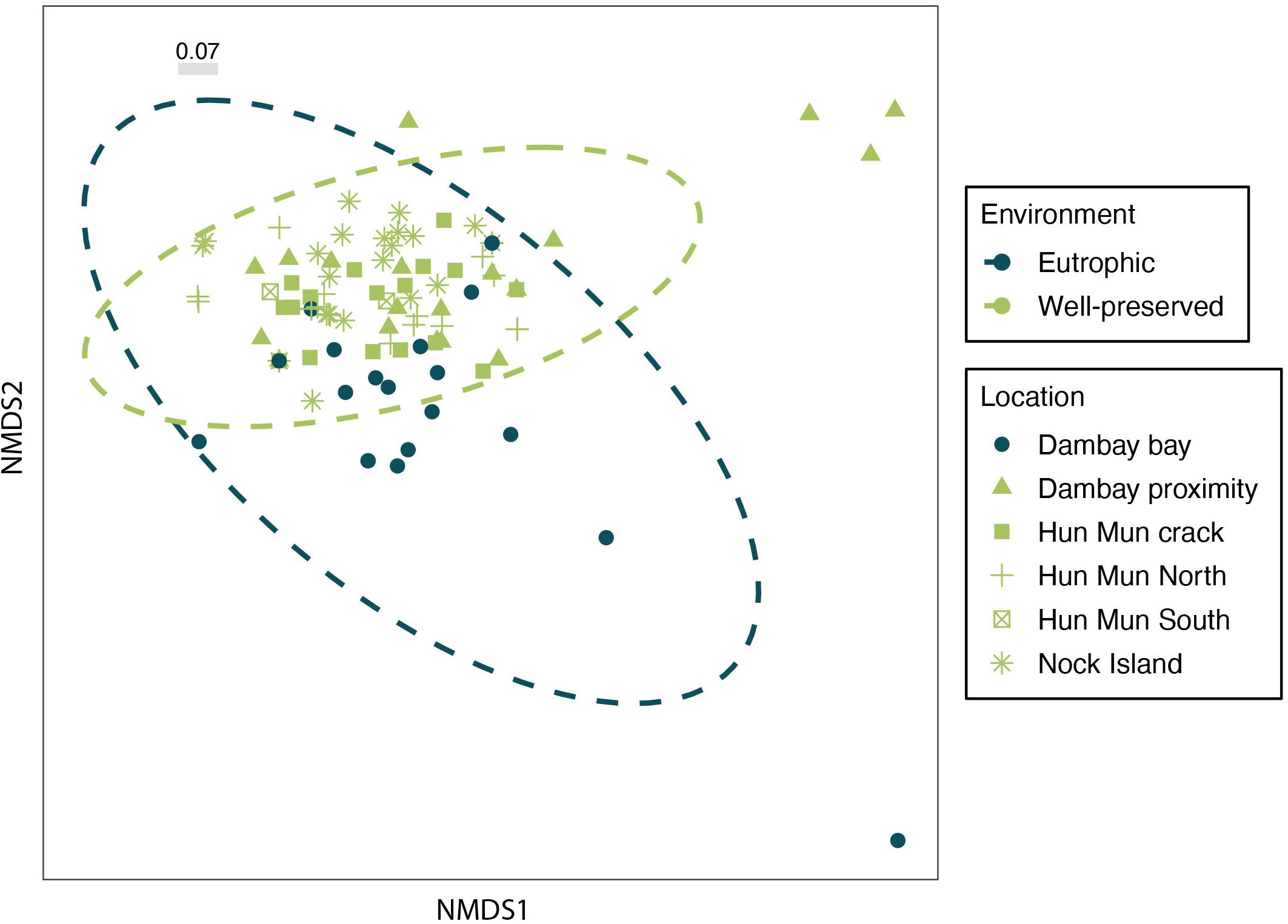
Figure 3. Non-metric multidimensional scaling (nMDS) ordination of fish communities obtained from sponge nsDNA based on Bray-Curtis distances. Colors show differences between Eutrophic (blue) and Well-Preserved (green) environments. Each location is represented by a different shape. Gray bar represents the percentage of stress.
Most of the PCR replicates from the same sample clustered together in Bray-Curtis distance of relative abundance data (Supplementary Figure S5), although there was a high variation in the number of reads recovered from each PCR replicate (Supplementary Figure S3). That is why we thus chose to work with presence/absence data with replicates pooled together, considering “detection” whenever a MOTU was detected in at least one of the PCR replicates from the same sample (Figure 4). Ordination of sponge samples based on Jaccard distances (Supplementary Figure S6) did not show any specific pattern related to sponge species, location or environment (Figure 4). Spratelloides gracilis and Pterocaesio digramma were the fish species most frequently occurring across samples (with more than 15 detections), but the majority of fish species (MOTUs) were detected just once in a single sample (Figure 4). Nonetheless, the majority of sponge samples had between one and five fish detections when pooling the two PCR replicates per sample.
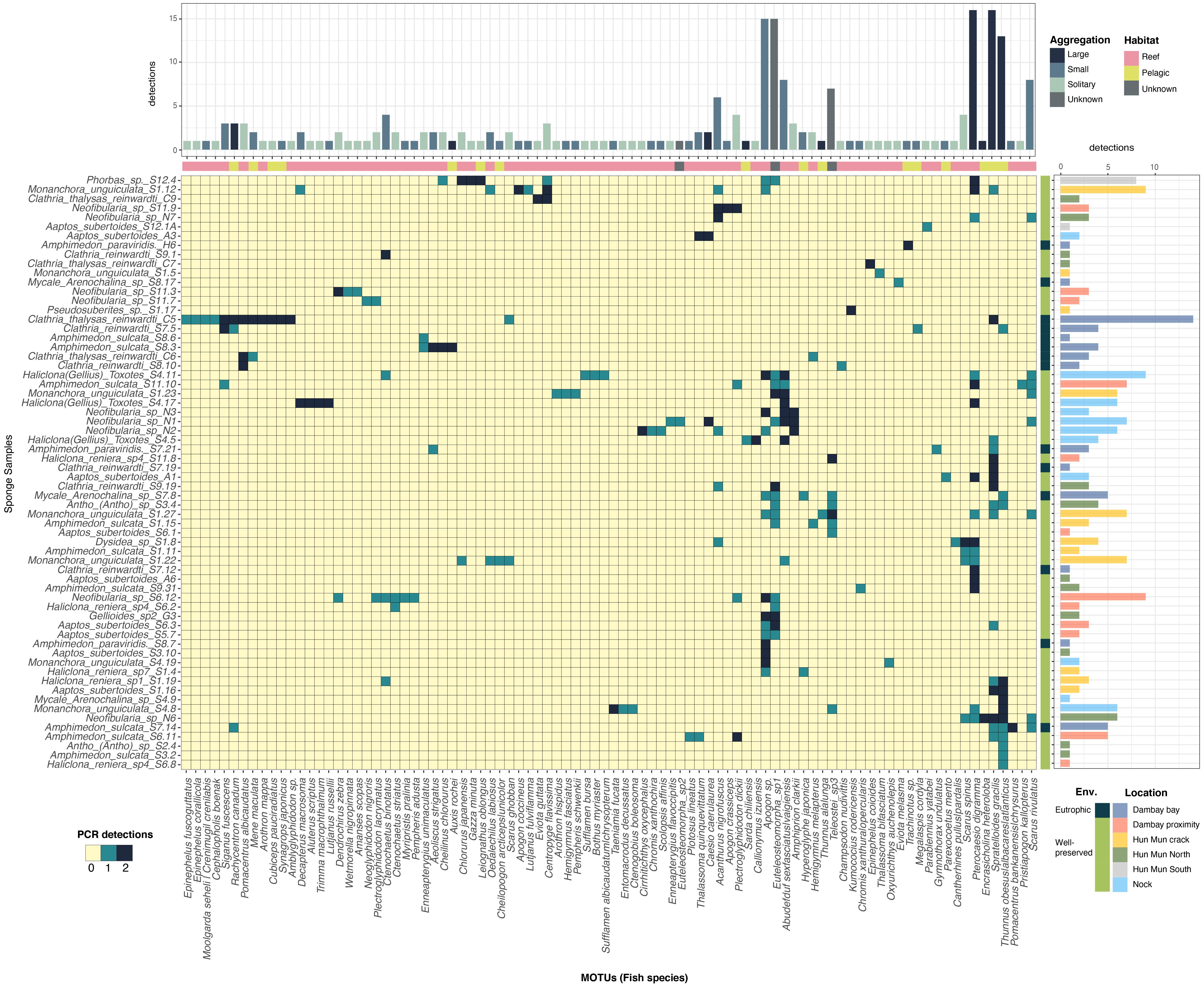
Figure 4. Heatmap based on presence/absence data. The heatmap scale color indicates whether a MOTU is detected in 0, 1, or 2 PCR replicates of a sample. Sponges samples (y-axis) and fish species (x-axis) are ordered according to cluster dendrogram based on Jaccard distances (shown in Supplementary Figure S6). Bars indicate total detections (considering one detection independently of the number of replicates) of (Upper panel) Fish species (MOTUs) in all samples and (Right panel) Fish species in each sample. Bar color for y-axis indicates the sampling location and environment. Bar color for x-axis indicates the aggregation behavior and the habitat of fish species.
We were able to classify the 87 MOTUs identified at genus or species level according to their schooling behavior, individual size, depth range, habitat, and migratory behavior. We found a differential distribution of the total detections and reads that belonged to different trait categories, considering the whole sampling area (Figure 5). For the schooling behavior, we detected a similar frequency fishes of the three schooling categories: large groups, small groups or solitary individuals (Figure 5A), although a higher percentage of reads was found for fishes that are usually found in large schools, such as Spratelloides gracilis (∼24%) and Thunnus obesus (∼15%). Similarly, individuals of the three size classes were present in our dataset, but small- and medium-sized fishes were more frequently detected, with the highest percentage of the total reads from small fishes (Figure 5B). This was mostly due to the high percentage of reads retrieved from Spratelloides gracilis (∼24%) and Plectroglyphidodon dickii (∼17%), two species represented by individuals < 15 cm. Although the collected sponges had a surface distribution (3–9 m), the majority of the fish species retrieved from the sponge samples have a shallow depth preference (10–60 m). Moreover, there was a non-negligible proportion (∼15% detections and reads) of fish species that are associated with a deeper depth range (>60 m) (Figure 5C). Finally, the majority of the MOTUs in our sponge nsDNA survey belonged to non-migratory reef-associated fish species (Figures 5D,E). Among the most common reef species, Apogon sp. and Pterocaesio digramma had the highest proportion of detections, and Acanthurus nigrofuscus and Plectroglyphidodon dickii had the highest proportion of reads. Overall, the majority of fish detected in the samples were reef-associated small individuals, with shallow depth range and non-migratory behavior.
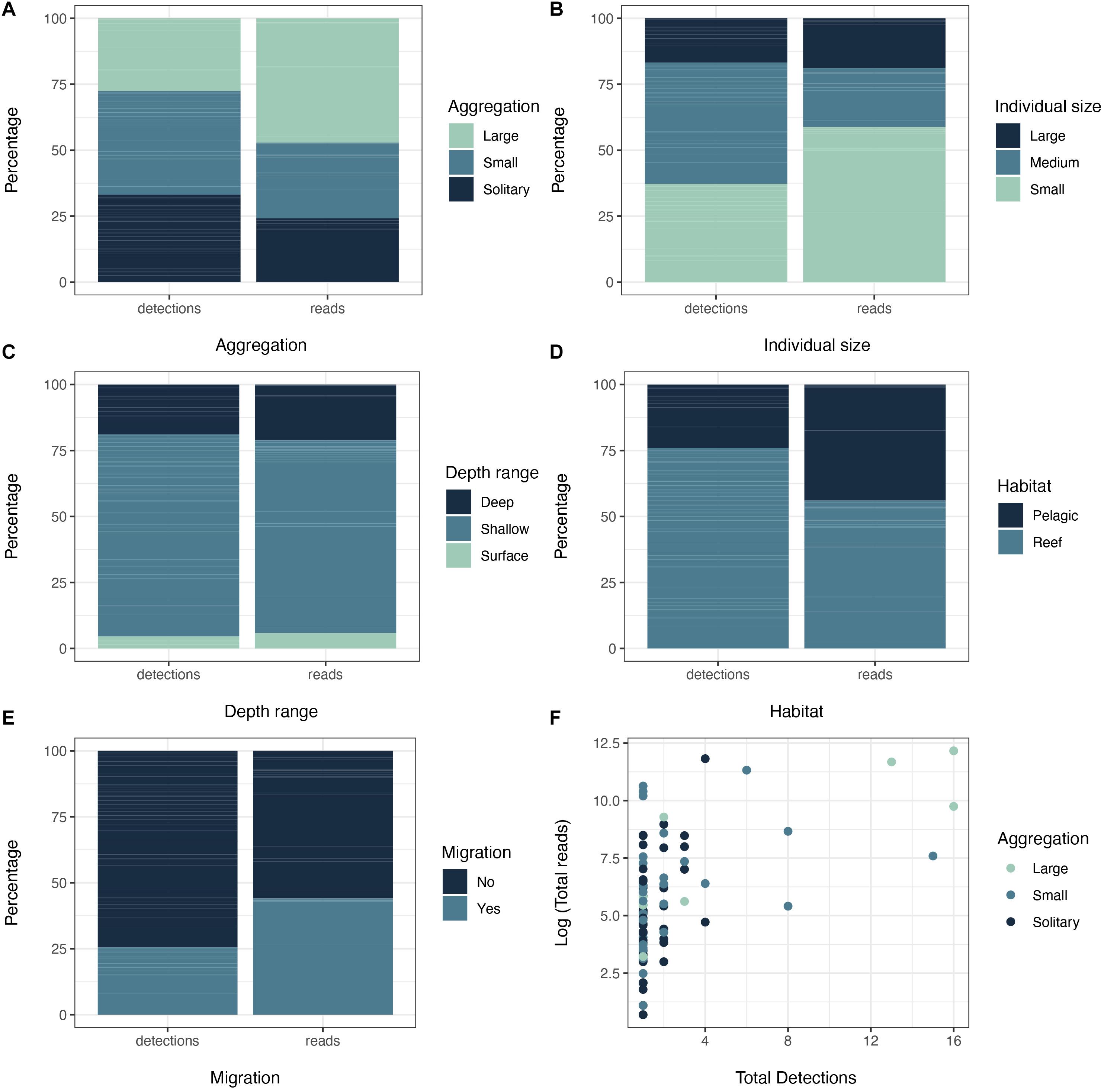
Figure 5. Percentage of total detections and total reads associated to specific categories within the studied fish features: (A) Aggregation behavior, (B) Individual size, (C) Depth range, (D) Habitat, and (E) Migration. (F) Relationship between total reads (log transformed) and total detections colored by the aggregation behavior of the represented MOTUs (fish species).
There was a significant correlation between the number of detections and the number of reads (log transformed) (Pearson, Rp = 0.58, p < 0.001, Figure 5F). In general trends, the most frequently detected MOTUs also had the highest number of reads and belonged to fish species with schooling behavior. However, it is important to note that there were many MOTUs with just a single detection that presented a wide range of variability in their number of reads (Figure 5F). The number of total detections and the number of total reads (log transformed) were classified into three abundance categories for each MOTU (Figure 5F): high (> 10), medium (5–10), low (2–5). Thunnus obsesus and Spratelloides gracilis were the two species found at high abundances considering both, detections and reads (Figure 6). Other highly abundant species were Apogon sp. and Pterocaesio digramma in terms of detections (Figure 7F), and Acanthurus nigrofuscus and Plectroglyphidodon dickii in terms of number of reads (Figure 6).
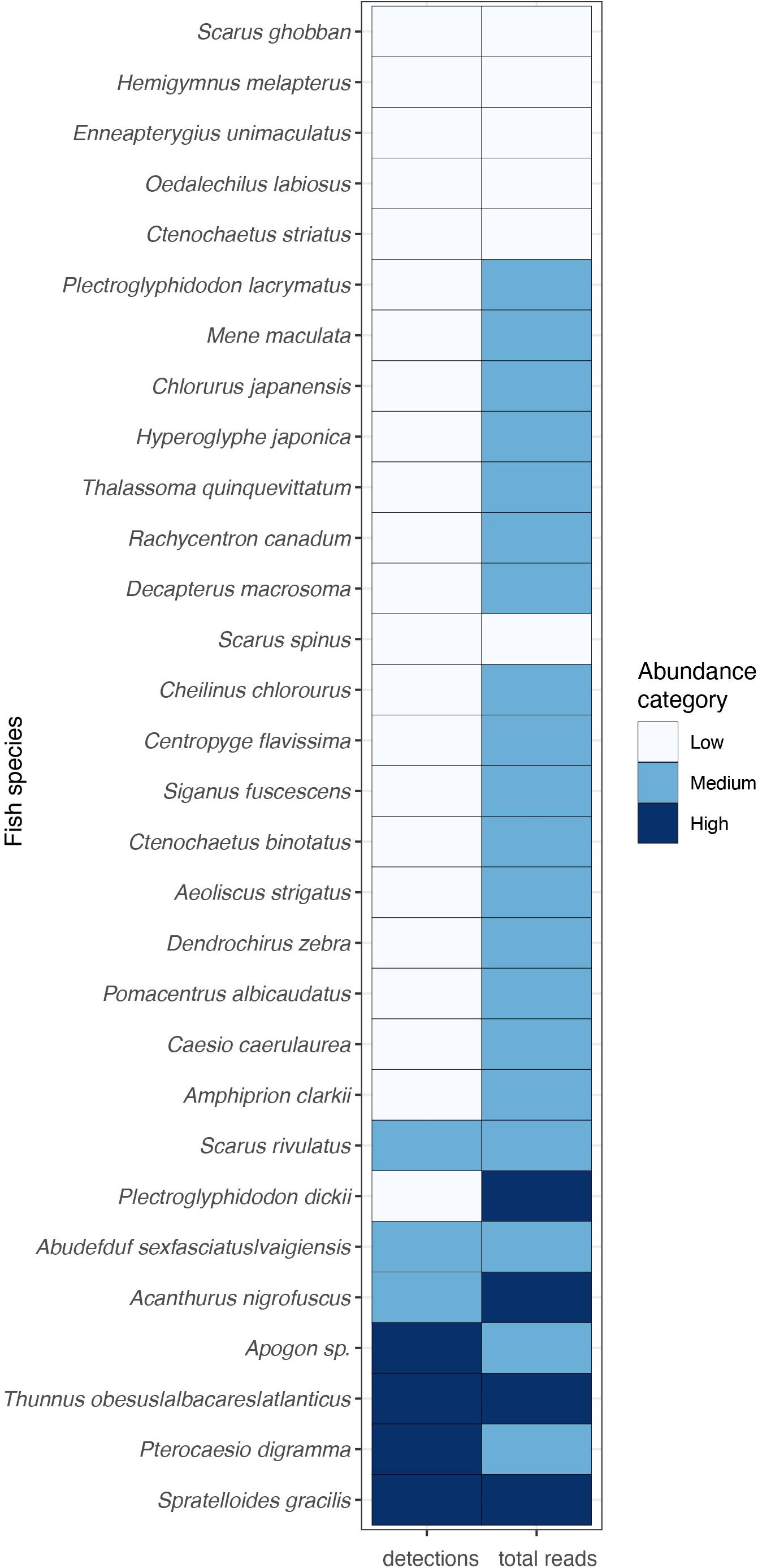
Figure 6. The most abundant fish species recovered from sponge nsDNA in Nha Trang Bay. The abundance categories correspond to number of total detections and number of total reads (log transformed) classified as: high (>10), medium (5–10), and low (2–5). Only MOTUs detected at least twice are represented.
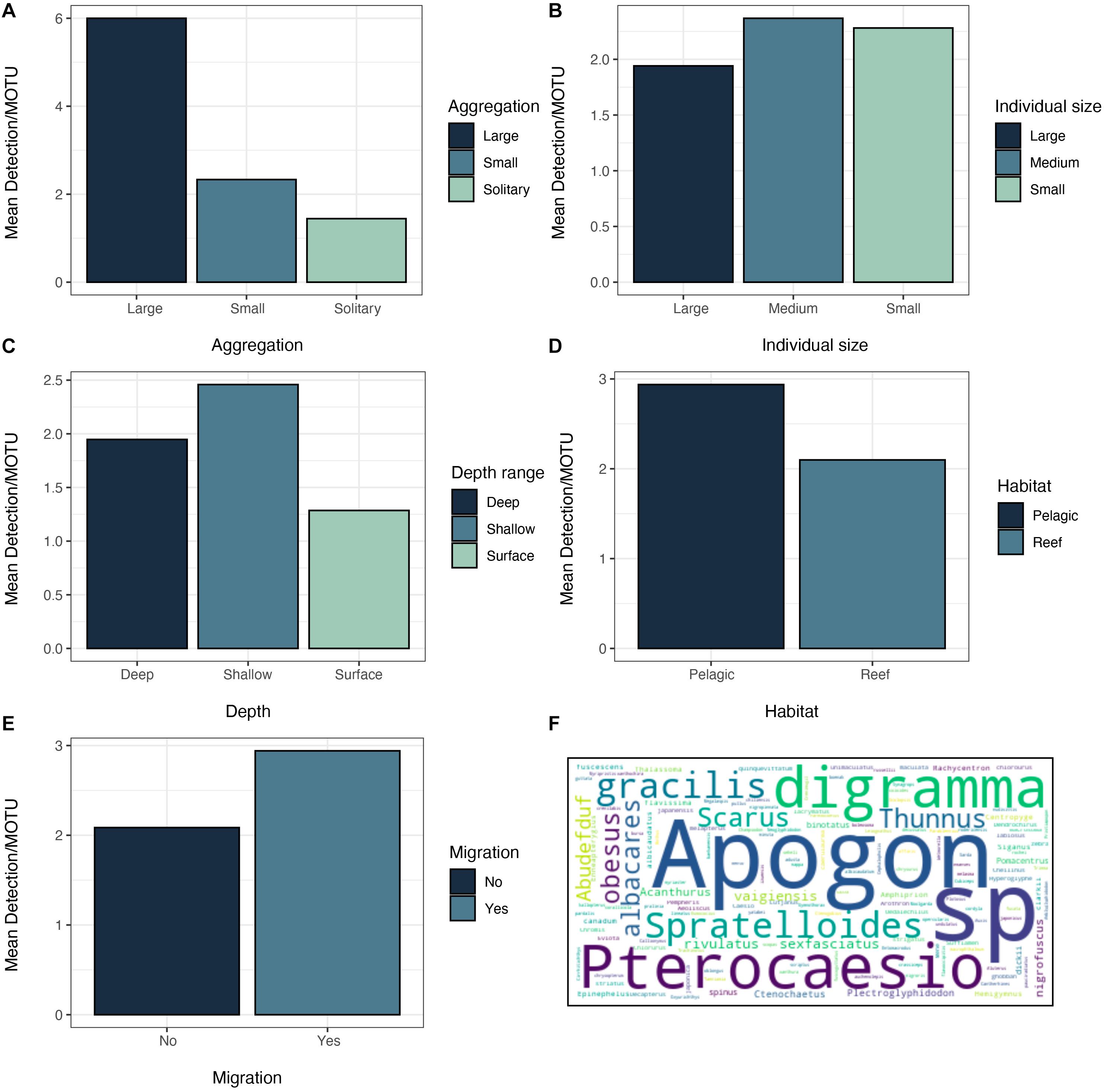
Figure 7. Mean detection per MOTU of the studied fish features: (A) Aggregation behavior, (B) Individual size, (C) Depth range, (D) Habitat, and (E) Migration. (F) Names of the fish species recovered from sponge nsDNA sized according to their number of detections.
In order to investigate which fish species were more likely to be detected depending on their behavior or physiological characteristics, we calculated the mean detections and reads per MOTU and grouped them according the above explained categories. The most relevant differences were observed for the schooling behavior, for which, species that are usually found in large schools had a much higher detections and reads per individual than solitary individuals (Figure 7A and Supplementary Figure S7A). No differences were observed regarding the individual size for detections (Figure 7B), although small individuals presented a higher number of reads than larger individuals. This latter case is mostly due to the high number of reads belonging to the herring (S. gracilis), which is usually found in large schools. Moreover, shallow, pelagic and migratory fishes had a higher number of reads and detection rates than reef-associated non-migratory species (Figures 7C–E and Supplementary Figures S7D,E).
With the aim of assessing to what extend the nsDNA recovered from sponges captured the fish diversity reported in the same area, we compared our results with the visual census data reported in Van Nguyen and Kim Phan (2008). Although the study area was similar for both studies (Nha Trang Bay), the sampling effort was not comparable, being much higher in the visual survey, where 16 different locations were surveyed and thus covering a much larger area of the bay than our study. The total fish richness recovered was higher for the visual survey than for the nsDNA data with 266 and 90 species, respectively. Among them, 24 species were detected by both methods (Figure 8) and all of them belonged to reef-associated fishes. Thirty different genera were detected by both methods, with 79 genus exclusively detected in visual surveys and 38 genus exclusively detected with nsDNA. At the family level, up to 40 different families were captured by both methods and more than 50% of them were shared. Of the 90 species detected by nsDNA, 62 were reported for Vietnam in the FishBase, and 52 of them had specific reports in Nha Trang Bay. Up to 16 species had other representatives of the same genus reported in Vietnam and only 12 species had no genus representative described in Vietnam according to the FishBase.
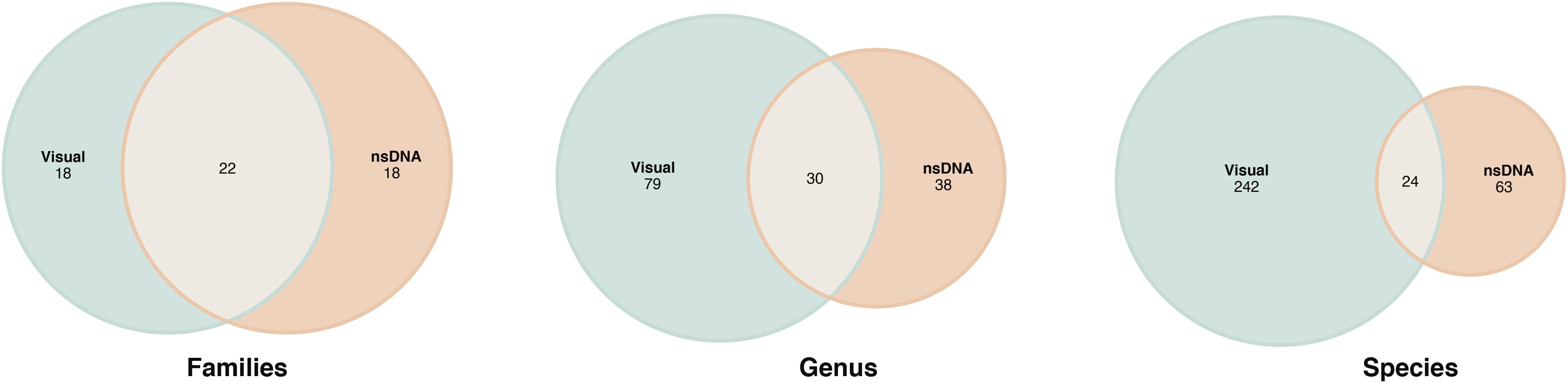
Figure 8. Venn diagrams of the common families, genus and species recorded by visual census data reported in Van Nguyen and Kim Phan (2008) and nsDNA recovered from the study sponges in Nha Trang Bay.
Sponges are cosmopolitan and the most efficient water filterers in the world (Van Soest et al., 2012). Because of that, they have been recently proposed as ideal candidates to act as natural environmental samplers (Mariani et al., 2019). In this study, we used specific vertebrate primers (Miya et al., 2015) to unveil fine-scale fish patterns from the nsDNA retrieved from the sponge tissues. We were able to identify up to 90 fish species from the sponges collected, although species accumulation curves showed that more replicates are needed to accurately capture the fish diversity of the area. Indeed, a significant proportion of our samples (34%) did not retrieve any positive PCR counts. This result suggests that fish nsDNA is not always present in sponges, in accordance with previous results from Mariani et al. (2019), in which two out of nine samples contained too few reads/MOTUs to be retained for further analysis. Keep in mind that our sampling was originally intended for microbiome studies, so the samples comprised ca. 0.5 cm3 of sponge material, which may be too small for other purposes. Specific studies should be carried out by repeatedly sampling the same sponge to determine the right sample volume necessary to reliably detect nsDNA and accurately capture the fish diversity in a given locality. From our results, it is clear that fish nsDNA was not abundant or ubiquitous in the sponges analyzed, leading to a relatively high stochasticity in detection levels and in number of reads obtained from the samples. Whether this observed stochasticity is lower or higher than those that would be obtained from comparable filtered seawater, or if it is an artifact due to the high amount of contamination reads detected, is yet to be studied. Moreover, we did not have a control for extraction kit contamination, although this is unlikely to affect our results. Regardless of these constraints, significant differentiation in the fish communities was observed between the eutrophic and the well-preserved environments and between sampling locations, albeit these factors only explained a modest proportion of the observed variation. Sponge species proved also to be a significant factor for detecting fish, albeit a high dispersion between species prevented drawing definite conclusions. The morphological characteristics of the sponge species (encrusting, massive, branching), did not seem to affect the capturing of nsDNA, as no sponge species performed significantly better or worse than others in terms of MOTU richness. Although sponges can have interspecific differences in filtration efficiencies, it has been shown that similar high efficiency is obtained after a few hours of incubation by species with contrasting biological characteristics (Turon et al., 1998).
Reassuringly, consistency was found between PCR replicates of the same sample, indicating that there was no technical artifact related to amplification. Notwithstanding, given that our extracts in general have little DNA of the target group, PCR bias due to the non-linear nature of the amplification reaction can stochastically lead to markedly different number of reads between PCR replicates. In the case of our dataset, with the aim of not adding more sources of variation than the ones already intrinsic to our sampling (environment, location, sponge species), we chose to focus on presence/absence data rather than focusing on differences in the relative abundance of reads. Indeed, presence/absence data has been proven to better differentiate fish communities between sites than read abundances (Port et al., 2016; DiBattista et al., 2017). The majority of sponges only contained between one and five fish MOTUs considering the two PCR replicates per sponge, with the exception of C. (Thalysas) reinwardti, for which up to 12 MOTUs were retrieved. However, other replicates of this species had low MOTU richness, comparable to other sponge species, preventing the consideration of this species as a “better” natural sampler than others. Considering our data, MOTU richness per sponge species is a rather stochastic factor, although a more detailed study focusing in less species and more replicates should better address this issue. No specific pattern for fish communities could be ascribed to sampling location, which were closely located (max ∼2 km apart). Water movements can highly affect the distribution of eDNA in the environment (Goldberg et al., 2016), possibly explaining this lack of specific patterns between fine-scale sampling locations. However, Jeunen et al. (2019) demonstrated that eDNA was able to discriminate diverse marine habitats within a small spatial scale subject to tidal and water currents. Indeed, some studies have already shown that eDNA surveys are able to detect fine-scale community composition patterns in heterogeneous aquatic habitats (Port et al., 2016; O’Donnell et al., 2017; Pont et al., 2018; Bakker et al., 2019; Stat et al., 2019; Nguyen et al., 2020; Oka et al., 2020). In our study, the residency time and degradation rate of nsDNA in the sponge tissues is an important factor that can hamper the detection of community patterns. To date, only one other study has assessed vertebrate communities using nsDNA from Mediterranean and Antarctic sponges (Mariani et al., 2019). Thus, further studies are needed to prove the viability of sponges as natural samplers to detect community patterns at small geographical scales.
With the aim to describe the main features of the fish sequences retrieved from the sponges of Nha Trang Bay we considered several ecological and biological traits in our dataset. A high proportion of the fishes observed were reef-associated small individuals with shallow depth range and non-migratory behavior, reflecting the general characteristics of the fishes one may expect to find in a coral reef environment. Pterocaesio digramma, Acanthurus nigrofuscus, Plectroglyphidodon dickii, and Apogon sp. were the most common reef fish species detected in the sponge tissues. It is relevant to point out that we were able to detect eDNA from fishes with features not typical of reef-associated species and thus, not permanently associated to the environment where the sponges were sampled, such as pelagic, migratory or deep-sea species. According to our molecular survey, pelagic fishes might have a more important presence in coral reefs than previously assessed by visual surveys, such as the large schools of tuna and herring. Detection of certain species can be influenced by the efficiency of detecting rare species, and tiny individuals or larvae (Yamamoto et al., 2017) but also, by fish movement patterns and residence times (Stat et al., 2019). In that sense, if residence time of nsDNA in sponges is long enough, using these biological samplers can provide a picture of the ichthyofauna integrated over time, rather than a snapshot that can be acquired by a punctual visual sampling, or by collecting eDNA directly from water. Clearly, the dynamics of nsDNA in sponges (and other filter feeders as well) is an interesting field for further research.
In this analysis we have considered both detections and total number of reads to assign a certain abundance level to the observed MOTUs. Although there is a relationship between the number of reads and the number of detections, it is important to note the high variability found in the number of reads for single detected fish species, probably due to PCR bias discussed above. Overall, among the most abundant fishes for both parameters we could discern two main categories; solitary reef-associated fishes and schooling behavior fishes.
In order to better predict the abundances of fish from eDNA concentrations, it has been proposed that sampling should be standardized for a given species by considering seasonal variation in species behavior such as aggregation, migrations, spawning or vertical movements, among others (Lacoursière-Roussel et al., 2016). Moreover, quantitative estimates would also benefit from the understanding of the variability in the DNA shedding rates across species (Port et al., 2016). In the present study, we standardized the number of detections and the number of reads according to some physiological and behavior fish features, to gain insights into possible variability in their shedding rates. The main differences in the number of detections and reads were found when considering the aggregation behavior of fish species. Clearly, fishes found in large or small schools had higher mean detections and reads per individual than solitary individuals, suggesting that schools of fish would shed more genetic material into the environment than single individuals, as already suggested (Kelly et al., 2014a). On the contrary, no differences in the number of detections according to the fish body size were found in our dataset. This is probably due to the increased detection of small fishes that are found in schools. Overall, a combination of both, size and number of individuals should be considered for quantitative estimates of molecular data. In line with our findings, the highest number of fish reads from a tropical area was also attributed to a schooling species, the anchovy (Nguyen et al., 2020), being somehow analogous to the herring (Spratelloides gracilis) in our sampling area. Moreover, the mean detections and reads per individual was higher for pelagic and migratory species than for reef-associated non-migratory species. In this sense, although in our sampling area reef-associated species represent a higher proportion of the detected fishes, once a pelagic fish appears in the reef, it is more likely to be detected due to its higher DNA shedding rate. This is in line with the increased eDNA shedding rate found for larger fish biomass (Jo et al., 2019), although many other factors have to be taken into account when assessing DNA shedding rates (Maruyama et al., 2014; Klymus et al., 2015; Dunn et al., 2017).
A key point when assessing the use of eDNA data for species surveys is the comparison with traditional visual surveys. Although we did not have visual data corresponding to our sampling period (April 2015), we compared the results obtained with visual censuses performed in the area in September–October 2008 (Van Nguyen and Kim Phan, 2008). In that study, up to 266 species were detected, and fish communities could be differentiated depending on the substrate were the survey took place. It is noteworthy that, with a spatially and temporally restricted sampling of 86 sponges, we could detect one third of the species found in extensive conventional surveys. We were able to detect the same number of fish families as in the visual survey and half of them were coincident. Indeed, the higher density of Pomacentriidae and Labridae species found in Hun Mun area by visual censuses (Van Nguyen and Kim Phan, 2008) is coherent with the higher number of representatives detected for these families in our nsDNA survey. The majority of species detected by nsDNA from sponges had been described in Vietnam, and the majority of them had specific detections in Nha Trang according to FishBase (Froese and Pauly, 2019). The few species for which no representatives were found in the database can be indicative of the incompleteness of the molecular reference database or might represent new reports of species never detected or misidentified before in the area.
It has to be considered that results obtained by both sampling methods should be standardized by their cost-efficiency (time and expertise needed). Performing 32 transects of 500 m2 each is much more time consuming than collecting sponges in a single (or a few) dives. Moreover, traditional censuses (visual or captures) are usually invasive, selective, and can be limited by environmental conditions and lack of taxonomic expertise (Thomsen et al., 2012; DiBattista et al., 2017). Some recent studies have pointed out the differences between eDNA metabarcoding methods and visual or capture methods (Tkachenko et al., 2016; Yamamoto et al., 2017; Fujii et al., 2019; Stat et al., 2019). The main advantages of metabarcoding rely on its time efficiency (Yamamoto et al., 2017; Nguyen et al., 2020), its detection capability of eDNA signatures of fishes that cannot be observed during visual surveys (Nguyen et al., 2020) and the dispensable need of taxonomic expertise (DiBattista et al., 2017). In this sense, censuses made by different observers may not be comparable, while results from eDNA are more objective and, particularly, better suited for archiving and future reanalyses (the sequences remain in databases, visual observations are hardly archivable). However, the main drawbacks of the eDNA approach lie on the accuracy of the results obtained, which are biased by the coverage of the reference database (Nguyen et al., 2020), the number of replicates (Yamamoto et al., 2017), the possible PCR inhibition (Fujii et al., 2019), and the differences in the species DNA shedding rates (Port et al., 2016).
With the aim of avoiding the problems associated with water filtration and DNA isolation in eDNA research, the use of natural samplers, such as shrimps (Siegenthaler et al., 2019) or sponges (Mariani et al., 2019) has recently been proposed for surveying marine ecosystems. However, their usefulness remains to be tested in different environmental settings and community types. This study contributed to this goal by using nsDNA obtained from tropical sponges under different eutrophication levels. We were able to capture fish diversity from the area with reasonable resolution considering the limited effort required to sample sponge biopsies compared to traditional visual survey techniques. Although we were not able to discriminate fine-scale geographic patterns, for which a more extensive replication would be needed, we observed significant differences in fish communities between an eutrophic area and the five relatively well-preserved areas within the Nha Trang Marine Protected area. Additionally, fish communities were also observed to be structured significantly different among sample locations, suggesting that sponges are effective in monitoring the stability of fish communities and their ecosystems. Our results also show that sponge species and morphology does not impact the efficiency of capturing nsDNA, as also suggested by Mariani et al. (2019).
Overall, our results show that sponges are promising natural samplers: a small number of sponges provided one third the species found in visual censuses of more than 32 transects performed over 2 months. Most species detected correspond to local ichthyofauna and reef fish were the best represented. However, further studies based on properly designed sampling schemes (as compared to our opportunistic study) are required to determine the right sample volumes and replication levels to obtain reliable quantitative information. Investigation on the trace-DNA dynamics and residence time in the sponges is also necessary. For the time being, results are better interpreted in a qualitative way. Comparative studies between sponges and other filter-feeders (such as ascidians or bivalves) can also shed light on the utility of natural samplers for community characterization.
The raw sequence data and the final datasets generated for this study can be found in the Mendeley Data repository with the DOI number: 10.17632/3btg5jz9t8.1.
MT performed the sampling and wrote the first manuscript draft. MT, KP, and OW designed the study. KP acquired the funding. MT, CA-P, and OW performed the lab work. OW performed the bioinformatics pipeline. MT, CA-P, and AA performed the analyses. All authors contributed significantly to later versions of the manuscript and approved submission.
This research was partially funded by the project MArSymbiOmics (CTM2013-43287-P) from the Spanish MINECO and by a small research grant from UiT the Arctic University of Norway to KP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank to Dr. Nguyen Van Long for kindly providing the list of species visually recorded in the Nha Trang Bay in 2008. We thank T. A. Britayev (RAS, Russia), and E. S. Mekhova (RAS, Russia) for the help provided in the sponge sampling. We also thank Maria Jesús Uriz (CEAB-CSIC, Blanes) for providing the funding for the sampling and for her invaluable help in the sponge identification.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.605148/full#supplementary-material
Supplementary Figure 1 | Bar plot showing the richness per sample. Y-axis indicates the number of samples (replicates pooled) with the same richness (Number of MOTUs).
Supplementary Figure 2 | Box plot of the richness per sponge species colored by the sponge morphology. Each dot represents a sample replicate.
Supplementary Figure 3 | Number of reads (log transformed) of samples with one positive PCR replicate (red) and two positive PCR replicates (green). Blue violin plot shows the difference in number of reads between two PCR replicates of the same sample.
Supplementary Figure 4 | Non-metric multidimensional scaling (nMDS) ordination of fish communities obtained from sponge nsDNA based on Bray-Curtis distances. Colors show differences between locations. Each environment is represented by a different shape.
Supplementary Figure 5 | Heatmap based on relative abundance data of fish communities (x-axis) recovered from sponge nsDNA. Replicates of the same sample (y-axis) are indicated by “a” or “b.” Cluster dendrograms of MOTUs and samples are based on Bray-Curtis distances. Scale bar represents the relative abundance of the fish MOTUs in each sample.
Supplementary Figure 6 | Cluster dendrogram based on Jaccard distances of fish communities recovered from sponge nsDNA. Sample replicates pooled together.
Supplementary Figure 7 | Mean reads per MOTU of the studied fish features: (A) Aggregation behavior, (B) Individual size, (C) Depth range, (D) Habitat, and (E) Migration. (F) Names of the fish species recovered from sponge nsDNA sized according to their number of reads.
Bakker, J., Wangensteen, O. S., Baillie, C., Buddo, D., Chapman, D. D., Gallagher, A. J., et al. (2019). Biodiversity assessment of tropical shelf eukaryotic communities via pelagic eDNA metabarcoding. Ecol. Evol. 9, 14341–14355. doi: 10.1002/ece3.5871
Boussarie, G., Bakker, J., Wangensteen, O. S., Mariani, S., Bonnin, L., Juhel, J.-B., et al. (2018). Environmental DNA illuminates the dark diversity of sharks. Sci. Adv. 4:eaa9661. doi: 10.1126/sciadv.aap9661
Boyer, F., Mercier, C., Bonin, A., Le Bras, Y., Taberlet, P., and Coissac, E. (2016). obitools: a unix -inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176–182. doi: 10.1111/1755-0998.12428
Collins, R. A., Bakker, J., Wangensteen, O. S., Soto, A. Z., Corrigan, L., Sims, D. W., et al. (2019). Non-specific amplification compromises environmental DNA metabarcoding with COI. Methods Ecol. Evol. 10, 1985–2001. doi: 10.1111/2041-210X.13276
Creer, S., Deiner, K., Frey, S., Porazinska, D., Taberlet, P., Thomas, W. K., et al. (2016). The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 7, 1008–1018. doi: 10.1111/2041-210X.12574
Cristescu, M. E., and Hebert, P. D. N. (2018). Uses and misuses of environmental DNA in biodiversity science and conservation. Annu. Rev. Ecol. Evol. Syst. 49, 209–230. doi: 10.1146/annurev-ecolsys-110617-062306
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., et al. (2017). Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895. doi: 10.1111/mec.14350
DiBattista, J. D., Coker, D. J., Sinclair-Taylor, T. H., Stat, M., Berumen, M. L., and Bunce, M. (2017). Assessing the utility of eDNA as a tool to survey reef-fish communities in the Red Sea. Coral Reefs 36, 1245–1252. doi: 10.1007/s00338-017-1618-1
Dunn, N., Priestley, V., Herraiz, A., Arnold, R., and Savolainen, V. (2017). Behavior and season affect crayfish detection and density inference using environmental DNA. Ecol. Evol. 7, 7777–7785. doi: 10.1002/ece3.3316
Froese, R., and Pauly, D. (2019). FishBase. World Wide Web Electron. Publ. Available online at: www.fishbase.org (accessed July, 2020).
Fujii, K., Doi, H., Matsuoka, S., Nagano, M., Sato, H., and Yamanaka, H. (2019). Environmental DNA metabarcoding for fish community analysis in backwater lakes: a comparison of capture methods. PLoS One 14:e0210357. doi: 10.1371/journal.pone.0210357
Goldberg, C. S., Turner, C. R., Deiner, K., Klymus, K. E., Thomsen, P. F., Murphy, M. A., et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307. doi: 10.1111/2041-210X.12595
Jeunen, G., Knapp, M., Spencer, H. G., Lamare, M. D., Taylor, H. R., Stat, M., et al. (2019). Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol. Ecol. Resour. 19, 426–438. doi: 10.1111/1755-0998.12982
Jo, T., Murakami, H., Yamamoto, S., Masuda, R., and Minamoto, T. (2019). Effect of water temperature and fish biomass on environmental DNA shedding, degradation, and size distribution. Ecol. Evol. 9, 1135–1146. doi: 10.1002/ece3.4802
Kelly, R. P. (2016). Making environmental DNA count. Mol. Ecol. Resour. 16, 10–12. doi: 10.1111/1755-0998.12455
Kelly, R. P., Port, J. A., Yamahara, K. M., and Crowder, L. B. (2014a). Using Environmental DNA to Census Marine Fishes in a Large Mesocosm. PLoS One 9:e86175. doi: 10.1371/journal.pone.0086175
Kelly, R. P., Port, J. A., Yamahara, K. M., Martone, R. G., Lowell, N., Thomsen, P. F., et al. (2014b). Harnessing DNA to improve environmental management. Science 344, 1455–1456. doi: 10.1126/science.1251156
Kelly, R. P., Shelton, A. O., and Gallego, R. (2019). Understanding PCR processes to draw meaningful conclusions from environmental DNA studies. Sci. Rep. 9:12133. doi: 10.1038/s41598-019-48546-x
Klymus, K. E., Richter, C. A., Chapman, D. C., and Paukert, C. (2015). Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix. Biol. Conserv. 183, 77–84. doi: 10.1016/j.biocon.2014.11.020
Lacoursière-Roussel, A., Rosabal, M., and Bernatchez, L. (2016). Estimating fish abundance and biomass from eDNA concentrations: variability among capture methods and environmental conditions. Mol. Ecol. Resour. 16, 1401–1414. doi: 10.1111/1755-0998.12522
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2015). Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ 3:e1420. doi: 10.7717/peerj.1420
Mariani, S., Baillie, C., Colosimo, G., and Riesgo, A. (2019). Sponges as natural environmental DNA samplers. Curr. Biol. 29, R401–R402. doi: 10.1016/j.cub.2019.04.031
Maruyama, A., Nakamura, K., Yamanaka, H., Kondoh, M., and Minamoto, T. (2014). The release rate of environmental DNA from juvenile and adult fish. PLoS One 9:e114639. doi: 10.1371/journal.pone.0114639
Miya, M., Sato, Y., Fukunaga, T., Sado, T., Poulsen, J. Y., Sato, K., et al. (2015). MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2:150088. doi: 10.1098/rsos.150088
Nguyen, B. N., Shen, E. W., Seemann, J., Correa, A. M. S., O’Donnell, J. L., Altieri, A. H., et al. (2020). Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci. Rep. 10:6729. doi: 10.1038/s41598-020-63565-9
O’Donnell, J. L., Kelly, R. P., Shelton, A. O., Samhouri, J. F., Lowell, N. C., and Williams, G. D. (2017). Spatial distribution of environmental DNA in a nearshore marine habitat. PeerJ 5:e3044. doi: 10.7717/peerj.3044
Oka, S., Doi, H., Miyamoto, K., Hanahara, N., Sado, T., and Miya, M. (2020). Environmental DNA metabarcoding for biodiversity monitoring of a highly diverse tropical fish community in a coral reef lagoon: estimation of species richness and detection of habitat segregation. Environmental DNA 1–15. doi: 10.1002/edn3.132
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., Mcglinn, D., et al. (2019). Vegan: Community Ecology Package. R-Package Version 25-6.
Pont, D., Rocle, M., Valentini, A., Civade, R., Jean, P., Maire, A., et al. (2018). Environmental DNA reveals quantitative patterns of fish biodiversity in large rivers despite its downstream transportation. Sci. Rep. 8:10361. doi: 10.1038/s41598-018-28424-8
Port, J. A., O’Donnell, J. L., Romero-Maraccini, O. C., Leary, P. R., Litvin, S. Y., Nickols, K. J., et al. (2016). Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541. doi: 10.1111/mec.13481
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Sales, N. G., Wangensteen, O. S., Carvalho, D. C., Deiner, K., Præbel, K., Coscia, I., et al. (2020). Space-time dynamics in monitoring neotropical fish communities using eDNA metabarcoding. Sci. Total Environ. 754:142096. doi: 10.1016/j.scitotenv.2020.142096
Shum, P., Barney, B. T., O’Leary, J. K., and Palumbi, S. R. (2019). Cobble community DNA as a tool to monitor patterns of biodiversity within kelp forest ecosystems. Mol. Ecol. Resour. 19, 1470–1485. doi: 10.1111/1755-0998.13067
Siegenthaler, A., Wangensteen, O. S., Soto, A. Z., Benvenuto, C., Corrigan, L., and Mariani, S. (2019). Metabarcoding of shrimp stomach content: harnessing a natural sampler for fish biodiversity monitoring. Mol. Ecol. Resour. 19, 206–220. doi: 10.1111/1755-0998.12956
Sigsgaard, E. E., Torquato, F., Frøslev, T. G., Moore, A. B. M., Sørensen, J. M., Range, P., et al. (2019). Using vertebrate environmental DNA from seawater in biomonitoring of marine habitats. Conserv. Biol. 34, 697–710. doi: 10.1111/cobi.13437
Stat, M., John, J., DiBattista, J. D., Newman, S. J., Bunce, M., and Harvey, E. S. (2019). Combined use of eDNA metabarcoding and video surveillance for the assessment of fish biodiversity. Conserv. Biol. 33, 196–205. doi: 10.1111/cobi.13183
Thomsen, P. F., Kielgast, J., Iversen, L. L., Wiuf, C., Rasmussen, M., Gilbert, M. T. P., et al. (2012). Monitoring endangered freshwater biodiversity using environmental DNA: species monitoring by environmental DNA. Mol. Ecol. 21, 2565–2573. doi: 10.1111/j.1365-294X.2011.05418.x
Thomsen, P. F., and Willerslev, E. (2015). Environmental DNA – an emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18. doi: 10.1016/j.biocon.2014.11.019
Tkachenko, K. S., Britayev, T. A., Huan, N. H., Pereladov, M. V., and Latypov, Y. Y. (2016). Influence of anthropogenic pressure and seasonal upwelling on coral reefs in Nha Trang Bay (Central Vietnam). Mar. Ecol. 37, 1131–1146. doi: 10.1111/maec.12382
Turon, M., Cáliz, J., Garate, L., Casamayor, E. O., and Uriz, M. J. (2018). Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 8:15201. doi: 10.1038/s41598-018-33545-1
Turon, M., Cáliz, J., Triadó-Margarit, X., Casamayor, E. O., and Uriz, M. J. (2019). Sponges and their microbiomes show similar community metrics across impacted and well-preserved reefs. Front. Microbiol. 10:1961. doi: 10.3389/fmicb.2019.01961
Turon, X., Galera, J., and Uriz, M. J. (1998). Clearance rates and aquiferous systems in two sponges with contrasting life-history strategies. J. Exp. Zool. 278, 22–36. doi: 10.1002/(sici)1097-010x(19970501)278:1<22::aid-jez3>3.0.co;2-8
Van Nguyen, L., and Kim Phan, H. (2008). Distribution and factors influencing on structure of reef fish communities in Nha Trang bay marine protected area, South-Central Vietnam. Environ. Biol. Fishes 82, 309–324. doi: 10.1007/s10641-007-9293-7
Van Soest, R. W. M., Boury-Esnault, N., Vacelet, J., Dohrmann, M., Erpenbeck, D., De Voogd, N. J., et al. (2012). Global Diversity of Sponges (Porifera). PLoS One 7:e35105. doi: 10.1371/journal.pone.0035105
Vo, S. T., Nguyen, V. L., Hua, T. T., and Nguyen, X. H. (2004). “Coral reefs of the hon mun marine protected area, nha trang bay, Vietnam, 2002. Species composition, community structure, status and management recommendations,” in Proceeding of the National Conference “Bien Dong–2002,”, Vietnam, 649–690.
Keywords: natural samplers, environmental DNA, sponges, fish diversity, coral reefs
Citation: Turon M, Angulo-Preckler C, Antich A, Præbel K and Wangensteen OS (2020) More Than Expected From Old Sponge Samples: A Natural Sampler DNA Metabarcoding Assessment of Marine Fish Diversity in Nha Trang Bay (Vietnam). Front. Mar. Sci. 7:605148. doi: 10.3389/fmars.2020.605148
Received: 11 September 2020; Accepted: 10 November 2020;
Published: 09 December 2020.
Edited by:
Sandie M. Degnan, The University of Queensland, AustraliaReviewed by:
Chunsheng Wang, State Oceanic Administration, ChinaCopyright © 2020 Turon, Angulo-Preckler, Antich, Præbel and Wangensteen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Turon, bWFydGEudHVyb25AdWl0Lm5v; bXR1cm9ucm9kcmlnb0BnbWFpbC5jb20=; Owen S. Wangensteen, b3dlbi53YW5nZW5zdGVlbkB1aXQubm8=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.