
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 01 December 2020
Sec. Marine Biogeochemistry
Volume 7 - 2020 | https://doi.org/10.3389/fmars.2020.573268
 Amy E. Maas1*
Amy E. Maas1* Shuting Liu2
Shuting Liu2 Luis M. Bolaños3
Luis M. Bolaños3 Brittany Widner4
Brittany Widner4 Rachel Parsons1
Rachel Parsons1 Elizabeth B. Kujawinski4
Elizabeth B. Kujawinski4 Leocadio Blanco-Bercial1
Leocadio Blanco-Bercial1 Craig A. Carlson2
Craig A. Carlson2Particulate organic matter (POM) (fecal pellets) from zooplankton has been demonstrated to be an important nutrient source for the pelagic prokaryotic community. Significantly less is known about the chemical composition of the dissolved organic matter (DOM) produced by these eukaryotes and its influence on pelagic ecosystem structure. Zooplankton migrators, which daily transport surface-derived compounds to depth, may act as important vectors of limiting nutrients for mesopelagic microbial communities. In this role, zooplankton may increase the DOM remineralization rate by heterotrophic prokaryotes through the creation of nutrient rich “hot spots” that could potentially increase niche diversity. To explore these interactions, we collected the migratory copepod Pleuromamma xiphias from the northwestern Sargasso Sea and sampled its excreta after 12–16 h of incubation. We measured bulk dissolved organic carbon (DOC), dissolved free amino acids (DFAA) via high performance liquid chromatography and dissolved targeted metabolites via quantitative mass spectrometry (UPLC-ESI-MSMS) to quantify organic zooplankton excreta production and characterize its composition. We observed production of labile DOM, including amino acids, vitamins, and nucleosides. Additionally, we harvested a portion of the excreta and subsequently used it as the growth medium for mesopelagic (200 m) bacterioplankton dilution cultures. In zooplankton excreta treatments we observed a four-fold increase in bacterioplankton cell densities that reached stationary growth phase after five days of dark incubation. Analyses of 16S rRNA gene amplicons suggested a shift from oligotrophs typical of open ocean and mesopelagic prokaryotic communities to more copiotrophic bacterial lineages in the presence of zooplankton excreta. These results support the hypothesis that zooplankton and prokaryotes are engaged in complex and indirect ecological interactions, broadening our understanding of the microbial loop.
The daily migration of zooplankton from their night-time habitat in the euphotic zone to the mesopelagic during the day is a well-documented phenomenon referred to as diel vertical migration (DVM; Lampert, 1989). After consumption of phytoplankton during the night, these organisms release surface-derived carbon and nitrogen in the daytime at depth as respiratory carbon dioxide (CO2), dissolved organic and inorganic excreta, and fecal pellets (particulate organic matter; POM). These components comprise the “active flux” portion of the “biological pump,” a process by which organic matter from the ocean surface is exported to depth (Steinberg et al., 2000; Steinberg and Landry, 2017). Typical zooplankton metabolic experiments characterize respiratory activity (oxygen consumption or CO2 production), as well as the excretion of inorganic nitrogen and phosphorous compounds (Ikeda et al., 2001; Steinberg et al., 2002; Alcaraz et al., 2010; Ikeda, 2014), while direct measurements of the composition and quantity of total dissolved organic matter (DOM) excreta of individual zooplankton are rare. By simultaneously quantifying respiratory CO2 and dissolved organic carbon (DOC) production, Steinberg et al. (2000) reported that excreted DOC is on average ∼31% (range 5–72%) of the respiratory carbon of various zooplankton groups at the Bermuda Atlantic Time-series Study (BATS) site. Similar proportions have been estimated for zooplankton in the North Pacific by simultaneously measuring oxygen consumption and DOC excretion (Maas et al., in review), and pteropods in the Southern Ocean (Thibodeau et al., 2020). Applying this conversion factor to global estimates of zooplankton respiratory flux (Steinberg and Landry, 2017), migratory zooplankton DOM excretion is responsible for a flux of 0.26–10.08 mg C m–2 d–1, representing 0.5–18.5% of the particulate organic carbon (POC) flux from surface waters. This process appears to be particularly important to total carbon flux during periods of high thermal stratification in oligotrophic systems, where POC flux and DOC delivery via mixing are typically low or negligible (Steinberg et al., 2000).
Transport of carbon to depth is likely an important factor in controlling mesopelagic ecosystem structure and productivity. It has been previously demonstrated that the ocean twilight zone’s microbial food web is fueled primarily by delivery of dissolved or suspended organic matter. Pathways that have been well investigated include annual deep convective mixing of DOM, commonly referred to as the mixed layer pump (Carlson et al., 1994; Dall’Olmo et al., 2016), and the sinking and subsequent solubilization of POM (Ducklow et al., 2001). By the time ambient organic matter produced in the surface reaches the mesopelagic, the most labile fractions have been consumed, leaving behind compounds that have resisted or escaped rapid degradation by the epipelagic microbial community (the recalcitrant fraction; Carlson, 2002; Hansell, 2013). Thus, many mesopelagic prokaryotes are specialized to use this degraded organic matter (Giovannoni and Stingl, 2005; Morris et al., 2005; Landry et al., 2017; Saw et al., 2020).
We know, however, that zooplankton produce dissolved free amino acids (DFAA) and amino acid-like compounds such as taurine (Webb and Johannes, 1967; Clifford et al., 2017) that are highly labile, and as such are difficult to measure in hydrographic profiles due to their rapid uptake and use (Fuhrman, 1987). The excretion of these compounds in patchy dense pockets around zooplankton may serve to increase the niche diversity of the meso- and bathypelagic ocean, providing “hot spots” of more energetically rich and easy to metabolize carbon for prokaryotic communities (Azam, 1998; Stocker, 2012). In the Red Sea, a linkage between mesopelagic prokaryotic communities and the backscattering layer on a diel cycle suggests that the vertical migrating community is an energy and nutrient source to the mesopelagic microbial food web (Calleja et al., 2018; García et al., 2018). Additional studies demonstrate both bacterial growth and production based on the addition of compounds excreted by zooplankton (Arístegui et al., 2014; Valdés et al., 2017), although these studies focused on the response of epipelagic prokaryotic assemblages. The effects of zooplankton excreta on mesopelagic prokaryotes have not been deeply studied to date. It has been demonstrated that microbial communities grow better on zooplankton exudate than on inorganic nutrient additions (NH4+) alone (Arístegui et al., 2014); however, specific organic metabolite profiles produced by zooplankton have not been well characterized.
To explore the DOM composition of zooplankton excreta and interrogate the impact of excreta associated DOM on mesopelagic prokaryotic communities, we investigated the excreta of the copepod species Pleuromamma xiphias, a dominant member of the migratory zooplankton community in the Sargasso Sea (Steinberg et al., 2000). Readily identifiable and abundant in surface night tows (∼100–150 m), this species migrates down to depth (400–600 m) during the day (Buskey et al., 1989; Wiebe et al., 1992). Characterization of the excreta collected from P. xiphias and the response of prokaryotes to this zooplankton exudate thus provide insights into the influence of DOM active flux on the mesopelagic microbial community in the Sargasso Sea.
Copepod excreta and microbial remineralization experiments were conducted aboard the RV Atlantic Explorer on September 7–10th 2016 and July 9–10th 2017 in the vicinity of the BATS site (31° 40′N, 64° 10′W). Water for the copepod excreta production experiments was collected from the mesopelagic zone (200 m) using Niskin bottles and a CTD/rosette package (Table 1). A portion of this water was gravity-filtered through a 142-mm 0.2-μm Omnipore filter (Millipore) and the filtrate was stored in 8-L polycarbonate carboys (Nalgene) that were pre-cleaned with 10% HCl and rinsed with Milli-Q and 0.2 μm-filtered seawater. These carboys were stored at ambient mesopelagic temperature (∼19.5°C) in an onboard incubator until the excreta experiment.
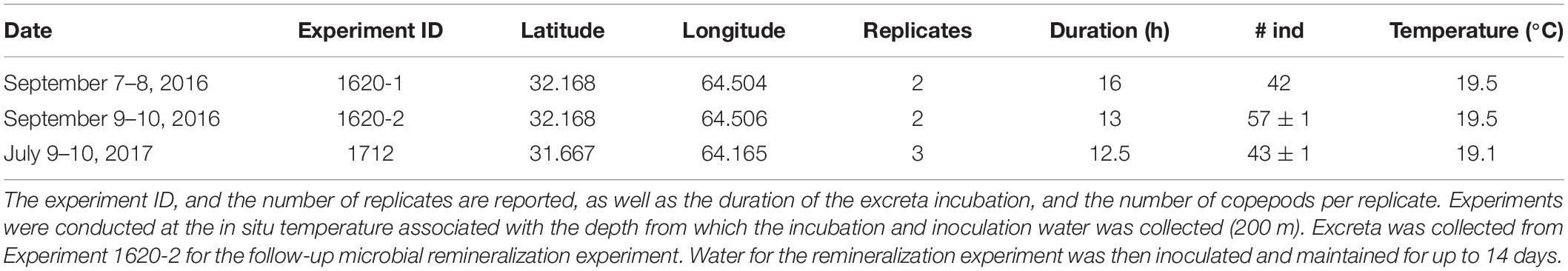
Table 1. Date and location of zooplankton sampling.
Copepods were captured with a custom 1 m Reeve net with 20-L cod end that was deployed after 22:00 (local time) through the deep chlorophyll max (visualized from prior CTD casts on station, ∼100–150 m). Two tows were conducted per experiment to ensure capture of a high number of individuals. After each net was retrieved, Pleuromamma copepods were picked from the sample using plastic pipettes, visually identified to species under a Leica stereomicroscope, and placed in the previously collected 0.2 μm-filtered seawater. After sorting, the copepods were each subjected to three consecutive rinses. During each rinse, ten individuals were placed into pre-cleaned glass dishes containing 60 mL of fresh, 0.2 μm-filtered seawater.
After cleaning, copepods were split among two (September 2016) or three (July 2017) replicates and placed into 2-L polycarbonate BiotainersTM (Nalgene), containing 0.2 μm-filtered water (Figure 1). For dissolved metabolite release experiments (Figure 1B) the initial volume in bottles of zooplankton excreta incubation (Figure 1A) was 1,200 mL, while for the microbial remineralization experiments (Figure 1C) the initial volume of zooplankton excreta incubation (Figure 1A) was 1,800 mL to support the necessarily larger water budget of this experiment. The number of copepods in the zooplankton treatments ranged between 42 and 58 individuals per replicate (Table 1). After the copepods were added, an initial time point in all treatments was sampled for DOC and DFAA concentrations (see below); excess water was then removed such that the final incubation volume was 1,000 mL for the dissolved metabolite release experiments and 1,500 mL for the remineralization experiments. This collection and picking process took >3 h, during which the organisms were held in 0.2 μm-filtered seawater. Control treatments comprised exclusively of 0.2 μm-filtered seawater were incubated and processed at the same time as treatments that included zooplankton. In the September 2016 experiment an additional control-type treatment was created, whereby the filtered seawater was inoculated with ∼5 mL of the water used to do the final rinse of the copepods. The objective of this treatment was to determine whether there was any “carryover” bacteria or DOM due to the unavoidable addition of a small volume of this rinsing seawater.
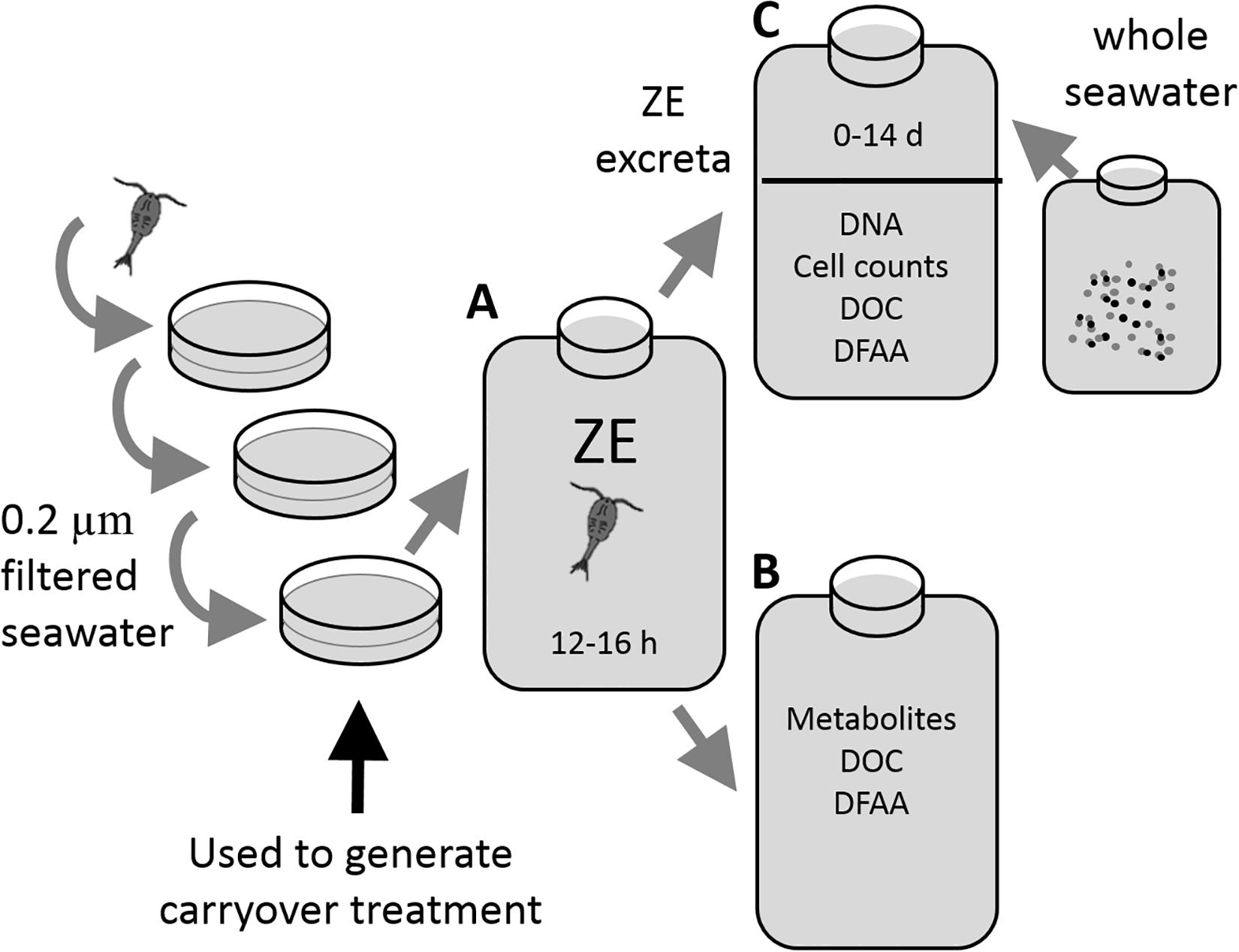
Figure 1. Experimental protocol for the zooplankton excreta production experiments (A), the water of which was used to either measure metabolites (B), or perform the microbial remineralization experiments (C). Copepods were serially rinsed and then incubated in filtered seawater for 12–16 h to generate excreta for analysis. For the microbial remineralization experiment 1,150 mL zooplankton excreta water was re-filtered through a 0.2 μm Omnipore filter and inoculated with 500 mL of whole seawater communities from 200 m and allowed to incubate for up to 14 days. At regular intervals the samples were collected for bacterioplankton cell abundance, 16S rRNA gene amplicon sequencing, DOC concentration, and dissolved free amino acids (DFAA) concentration.
In order to collect excreta, copepods were incubated for 12.5–16 h in the dark at in situ temperatures (Table 1). At the conclusion of the excreta incubation period samples were collected for various chemical and biological variables (see below). To minimize sample handling and reduce the possibility of DOC contamination, this was carried out using a custom positive-pressure system that enabled subsampling without removing caps from the Biotainers. An aquarium pump pumped air through a hydrocarbon trap, which pressurized the Biotainers and displaced sample water through submerged Teflon tubing as described in Liu et al. (2020). To separate copepods from the sample water, a piece of 80-μm Nytex screen was affixed to the intake end of the Teflon tube with a 1 cm piece of silicone tubing over the Nytex and Teflon tube as a collar.
Individual copepods were then recovered and counted under a stereomicroscope, and then frozen for return to shore. In the lab zooplankton were weighed wet on a Cahn microbalance then placed in a drying oven for >5 days at 65°C and reweighed for dry mass. Remaining water from the excreta experiments was then either used for metabolite analysis (1620-1 and 1712) or a remineralization experiment (1620-2; see below).
The change in the quantity of DOC in the incubation water was interrogated in all experiments by comparing the initial concentration (drawn from the BiotainersTM directly after the experiments had been set up) with final concentrations determined after the zooplankton excreted for 12.5–16 h. DOC samples (duplicate 30 mL aliquots) were filtered through double stacked GF-75 filters (Advantec, pore size 0.3 μm, pre-combusted at 450°C), packed in 25 mm Swinex filter holders attached directly to the sample line with a Luer-lock adaptor. DOC samples were acidified with 60 μL 4N HCl to a pH < 3 and stored at ∼14°C until analysis. DOC samples were analyzed using the high-temperature combustion method on a modified TOC-V or TOC-L analyzer (Shimadzu) at the University of California, Santa Barbara as described in Carlson et al. (2010). The precision for DOC analysis is ∼1 μmol L–1 or a CV of ∼2%. Daily reference waters were calibrated with DOC consensus reference material provided by D. Hansell (University of Miami1).
Concentrations of dissolved free amino acids and taurine (an amino-acid like compound) were measured for all samples (referred to as DFAA herein). DFAA samples were filtered through the same GF-75 filters used to collect DOC samples into 60 mL acid-washed high-density polyethylene bottles and stored at −20°C. These were analyzed by high performance liquid chromatography (HPLC, Dionex ICS 5000+) with a fluorescence detector (RF2000) after pre-column o-phthaldialdehyde (OPA) derivatization (Lindroth and Mopper, 1979; Lee et al., 2000; Clifford et al., 2017) following the modified protocol according to Liu et al. (2020). The DFAA that were resolved include aspartic acid, glutamic acid, histidine, serine, arginine, threonine, glycine, taurine, β-Alanine, tyrosine, alanine, γ-aminobutyric acid (GABA), methionine, valine, phenylalanine, isoleucine, leucine, and lysine. Statistical analysis of DFAA production rate was run as a paired t-test in SPSS.
The excreta product for a subset of the experiments was dedicated to dissolved metabolite analysis. Background concentrations were determined from water taken from the same cast and depth as the filtrate used for the excreta experiments. DOM sample collection was designed to minimize both contamination of the compounds measured as well as contact with plasticizers, which interfere with the analysis as described in Kido Soule et al. (2015). To that end, all chemicals were Fisher Scientific Optima grade, and sampling materials were glass or Teflon, when possible. At the termination of the zooplankton excreta experiments, an 800 mL aliquot from each bottle was filtered through a 0.2-μm Teflon filter (Omnipore, Millipore Sigma) placed in an acid-cleaned (10% HCl) Teflon filter holder. The filtrate was then acidified to pH ∼3 using HCl and loaded onto a methanol-rinsed solid phase extraction cartridge (1 g, 6 mL, Agilent Bond Elut PPL) using a vacuum pump as described in Longnecker (2015). The PPL cartridges were rinsed with acidified (pH ∼3, 0.01 M HCl) MilliQ water, and metabolites were eluted in 6 mL of methanol. Extracts were evaporated to near dryness using a Vacufuge (Eppendorf) and reconstituted in 250 μL of 95:5 (v/v) water/acetonitrile with deuterated biotin (0.05 mg mL–1).
Samples were analyzed using ultrahigh performance liquid chromatography (UPLC; Accela Open Autosampler and Accela 1250 Pump, Thermo Scientific) coupled to a heated electrospray ionization source (h-ESI) and a triple quadrupole mass spectrometer (TSQ Vantage, Thermo Scientific) operated under the selected reaction monitoring (SRM) mode (Kido Soule et al., 2015). Separation was performed at 40oC on a reverse phase column (Waters Aquity HSS T3, 2.1 × 100 mm, 1.8 μm) equipped with a Vanguard pre-column. Mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. The flow rate was maintained at 0.5 mL min–1. The gradient began at 1% B for 1 min, increased to 15% from 1 to 3 min, then increased to 50% B from 3 to 6 min, and then increased to 95% B from 6 to 9 min. The mobile phase was maintained at 95% B until 10 min and then decreased to 1% B from 10 to 10.2 min and held at 1% B for the remainder (12 min total run time). Samples were run in both positive and negative ion modes using a 5 μL injection for each.
Raw data files were converted to mzML format using msConvert (Chambers et al., 2012), and MAVEN (Melamud et al., 2010; Clasquin et al., 2012) was used to identify and integrate peaks for all samples and standards. Peaks were identified by their unique retention time, parent m/z, and product m/z. Two SRM transitions were monitored for quantification and confirmation. Metabolite concentrations were calculated using a standard curve of at least five points, and concentrations were corrected for the efficiency of the solid phase extraction step using data in Johnson et al. (2017).
Microbial remineralization experiments followed the basic design of inoculating a whole water aliquot into 0.2 μm filtrates of various treatments (i.e., control and zooplankton excreta) to reduce grazing pressure, followed by monitoring changes of biological and chemical variables throughout the dark incubation (see Carlson et al., 2002; Liu et al., 2020). Following the collection of DOC and DFAA samples at the termination time point during the 2016 (1620-2) zooplankton excreta production experiment, the remaining volume was filtered through a 0.2-μm Omnipore filter. This 1,150 mL of seawater, enriched with zooplankton dissolved excreta, was inoculated with 500 mL upper mesopelagic water (200 m) and then subsampled at multiple time points for DOC and DFAA concentrations as well as prokaryotic cell abundance for up to 14 days. The change in prokaryotic community composition was tested by comparing the 16S rRNA gene amplicon sequence of the community right after inoculation (T0) to that of the resulting communities at the beginning and middle of the stationary growth phase (days 5 and 14, respectively). Control blank (seawater only) and carryover control (seawater combined with “carryover” water from zooplankton washes) bottles were incubated and sampled using the same procedures. Each treatment included duplicate incubations.
The quantity of DOC consumed by the prokaryotic community was monitored throughout the 14-day incubation. The concentration, composition, and transformation of the DFAA pool was also monitored from lag phase through stationary phase to the final time point of the incubation. The DFAA composition (molar percent of each individual DFAA) was compared by constructing a Bray–Curtis similarity matrix in PRIMER version 7 (Clarke and Warwick, 2001; Clarke and Gorley, 2006). Principal coordinates analysis was run on this matrix to compare proportional composition of DFAA among samples, then SIMPROF cluster analysis was run to determine which factors contributed to the patterns in abundance. Pearson correlations were additionally run to determine which DFAA contributed to observed patterns in clustering.
Ten mL of sample were collected at each time point for prokaryotic cell abundance, fixed with 0.2 μm filtered formalin to a final concentration of 1%, and stored at −80°C. Samples were thawed and 10 mL were filtered onto Irgalan Black stained 25 mm, 0.2 μm polycarbonate filters (Nucleopore, Whatman) under a gentle vacuum (100 mm Hg) and stained with 0.5 mL of 4′, 6-diamidino-2-phenyl dihydrochloride (5 μg mL–1, DAPI, SIGMA-Aldrich, St. Louis, MO, United States) (Porter and Feig, 1980). The filters were mounted onto slides with Resolve immersion oil (high viscosity; Resolve, Richard-Allan Scientific, Kalamazoo, MI, United States) and stored at −20°C. We then enumerated slides using an AX70 epifluorescent microscope (Olympus, Tokyo, Japan) under ultraviolet excitation at 1,000× magnification. We counted at least 400 cells (10 fields) per sample for prokaryote abundance.
At three time points (Day 0, 5, and 14), 250 ml samples were drawn from each incubation vessel via positive pressure displacement through an in line 0.2-μm Sterivex filter and stored frozen until DNA extraction. DNA was extracted using the phenol, isoamylalcohol and chloroform method described by Giovannoni et al. (1990). Amplicon sequencing was conducted targeting the V1–V2 region of the 16S rRNA gene. The forward primer consisted of the “general” Illumina overhang adapter and the 27F forward primer (5′-AGAGTTTGATCNTGGCTCAG-3) (Lane, 1991). The reverse primer consisted of the “general” Illumina overhang adapter and the reverse primer 338 RPL (5′- GCWGCCWCCCGTAGGWGT-3′) (Daims et al., 1999; Vergin et al., 2013a). 25 μL polymerase chain reactions (PCR) using 4 μL (∼2.5 ng/μL) of genomic DNA as template were performed with the following concentrations: amplicon PCR forward and reverse primers 0.2 nM each, 0.2 mM dNTPs, 5 U of Taq DNA Polymerase (Invitrogen, Carlsbad, CA, United States), 1X Taq polymerase buffer and 1.5 mM MgCl2. Thermocycling conditions used for amplification reactions consisted of: denaturation of 94°C for 230 s; 30 cycles of 95°C for 60 s, 55°C for 45 s, 72°C for 60 s; and a final elongation step of 72°C for 480 s. Correct V1–V2 size was confirmed in a Bioanalyzer DNA 1000 chip. PCR purification was performed using AMPure XP beads following manufacturer’s protocol. Libraries for each amplicon were constructed by attaching dual indices and Illumina sequencing adaptors with the Nextera XT Index Kit and a second PCR amplification. 50 μL indexed PCR were performed with the following volumes: DNA 5 μL, Nextera XT Index Primer 1 (N7xx) 5 μL, Nextera XT Index Primer 2 (S5xx) 5 μL, 2x KAPA HiFi HotStart ReadyMix 25 μL, PCR Grade water 10 μL. Thermocycling conditions used for amplification reactions consisted of: denaturation of 95°C for 180 s; 8 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; and a final elongation step of 72°C for 300 s. Library purification and size validation were done following the 16S rRNA gene amplicon library preparation protocol of the illumina MiSeq Sequencer. Purified libraries were pooled in equimolar concentrations. The pool of libraries was sequenced using one 2 × 250 Paired-End lane with a MiSeq Reagent Kit v2 at the Center for Genome Research and Biocomputing (Oregon State University), Corvallis, Oregon. Files containing the raw reads of the sequenced samples are available under BioSample accession numbers: SAMN14605589, SAMN14605590, and SAMN14605591.
Primer sequences from de-multiplexed raw paired-end fastq files were cropped using the CutAdapt software (Martin, 2011). A fixed number of bases was removed, 20 bases from forward files and 18 from reverse files matching the length of the 27F and 338 RPL primers, respectively. Trimmed fastq files were quality-filtered using the fastqPairedFilter command within the DADA2 R package, version 1.2 (Callahan et al., 2016) with following parameters: truncLen = c(220,190), maxN = 0, maxEE = c(2,2), truncQ = 2, rm.phix = TRUE, compress = TRUE, and verbose = TRUE. Quality-filtered reads were dereplicated using derepFastq command. Paired dereplicated fastq files were joined using the mergePairs function with the default parameters. An amplicon sequence variant (ASV) table was constructed with the makeSequenceTable command. Potential chimeras were removed de novo using removeBimeraDenovo. Taxonomic assignment was done by aligning the sequences to the silva_nr_v123 dataset of ribosomal genes with the assignTaxonomy command in DADA2 package. The phylogenetic taxonomic placement program Phyloassigner (Vergin et al., 2013a) was used to place SAR11 and SAR202 (Landry et al., 2017) sequences in the nodes of highly resolved reference phylogenetic trees. The abundances from the final version were standardized by total number of reads per sample and a Bray–Curtis similarity matrix was constructed in PRIMER version 7 (Clarke and Warwick, 2001; Clarke and Gorley, 2006). Time points were compared using Principal coordinates analysis. SIMPROF cluster analysis and Pearson correlations were run to determine which ASVs and factors contributed to observed patterns in abundance.
Net DOC production (ΔDOC) in the three zooplankton excreta-enriched experiments was calculated as the change in DOC concentration in each zooplankton enriched treatment relative to that of the average of the control treatments at the final time point (Figure 2A and Supplementary Material 1). When normalized to the number of copepods in the incubation, a net DOC production rate per individual (ΔDOCind) ranging between 10 and 27 nmol C h–1 ind–1 was observed (Table 2). The ΔDOC in the zooplankton excreta treatments did not correlate with duration of the experiment, number of individuals, nor the overall dry mass of the individuals. When net DOC production was converted to DOC produced per unit mass (normalized by dry mass per individual) an average of 0.55 ± 0.06 SE μg C h–1 mg–1 was obtained.
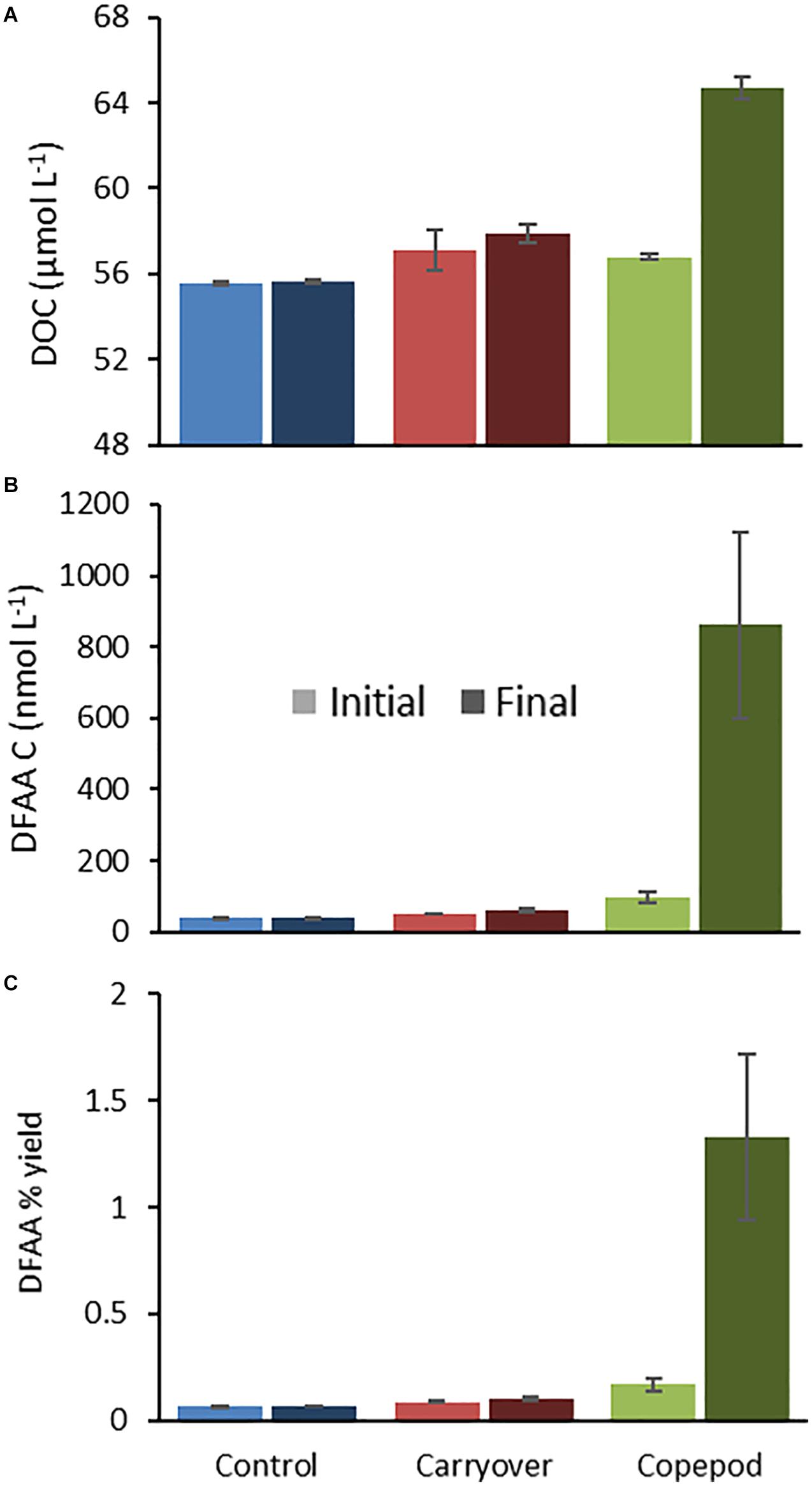
Figure 2. Representative plots of DOC (A) and DFAA (B) concentration, as well as changes in diagenetic state (as indicated by DFAA yield) over the period of zooplankton excreta production (from 2016; experiment 1620-2) in duplicate bottles demonstrating a less altered diagenetic state of the copepod excreta (C). Light bars represent T0 and dark bars represent TF.
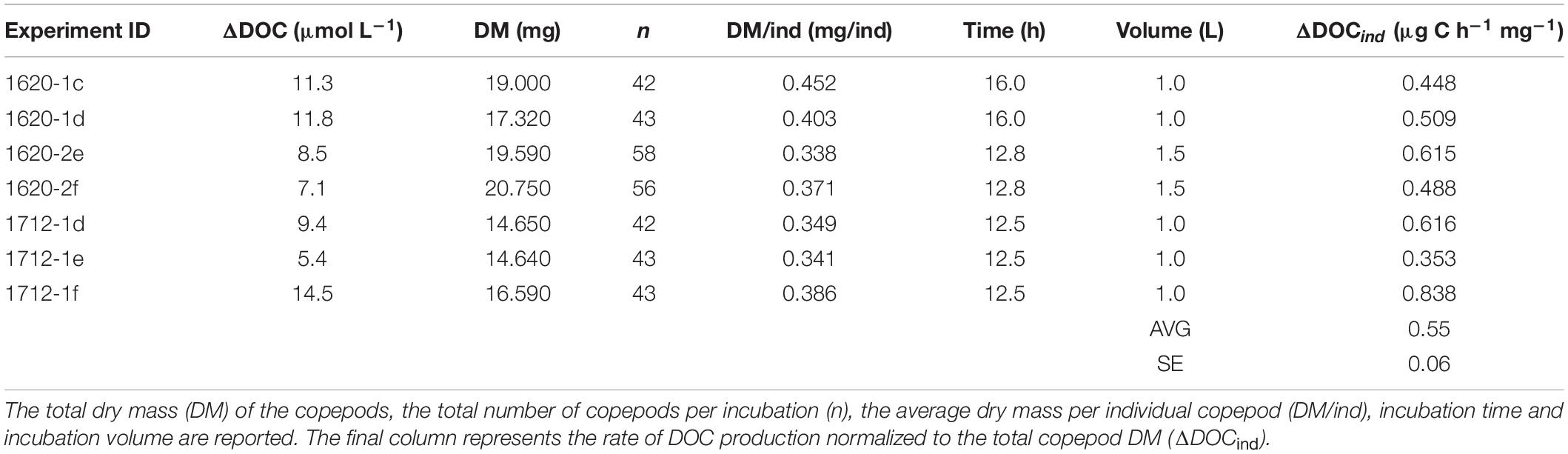
Table 2. Zooplankton excreta production experiment: production of dissolved organic carbon (ΔDOC) by copepods during the incubations.
DFAA-C production (nM C) was significantly higher [Paired t-test, t(6) = 4.080, p = 0.007] in the zooplankton excreta treatment compared to the minimal change in control or carryover treatments and made up on average 18% (range 11–25%) of the ΔDOC by copepods (Figure 2B). The Mol% of individual amino acids to total DFAA in zooplankton excreta treatments revealed enrichment with glycine (∼40%), arginine, lysine, and taurine (∼10% each). The concentrations of specific amino acids and total DFAA at the end of the zooplankton incubation were typically highly positively correlated (R2 > 0.8), indicating a similar proportional excreta makeup for DFAA among the three zooplankton excreta experiments (Supplementary Material 1). DFAA yield (sum of all individual DFAA concentrations in carbon units divided by DOC concentration) is used as a metric to describe the diagenetic state of organic matter, with a relative increasing DFAA yield being indicative of less diagenetically altered organic matter (Cowie and Hedges, 1994; Davis et al., 2009; Kaiser and Benner, 2009). Here we showed that after the incubation the DFAA yield in the zooplankton excreta was significantly higher than that observed in the unamended control or carryover treatments, suggesting the production of less diagenetically altered and potentially more labile compounds by zooplankton (Figure 2C).
Dissolved metabolite analysis identified a total of fourteen quantifiable compounds in the zooplankton excreta (Supplementary Material 2). In addition to three of the amino acids that were quantified with the DFAA measurement, metabolites were characterized as highly labile compounds including vitamins (B10, B9, and B5) and nucleosides. In the 2016 experiment the copepod excreta contained five known metabolites, two of which were significantly enriched relative to the control [folic acid (vitamin B9) and thymidine; Table 3]. In 2017, thirteen targeted metabolites were detected, of which five were statistically enriched in copepod excreta relative to the control (4-aminobenzoic acid, pantothenic acid, guanosine, inosine, and thymidine). The mass spectrometry data and its associated metadata are available at MetaboLights under accession number MTBLS18222.
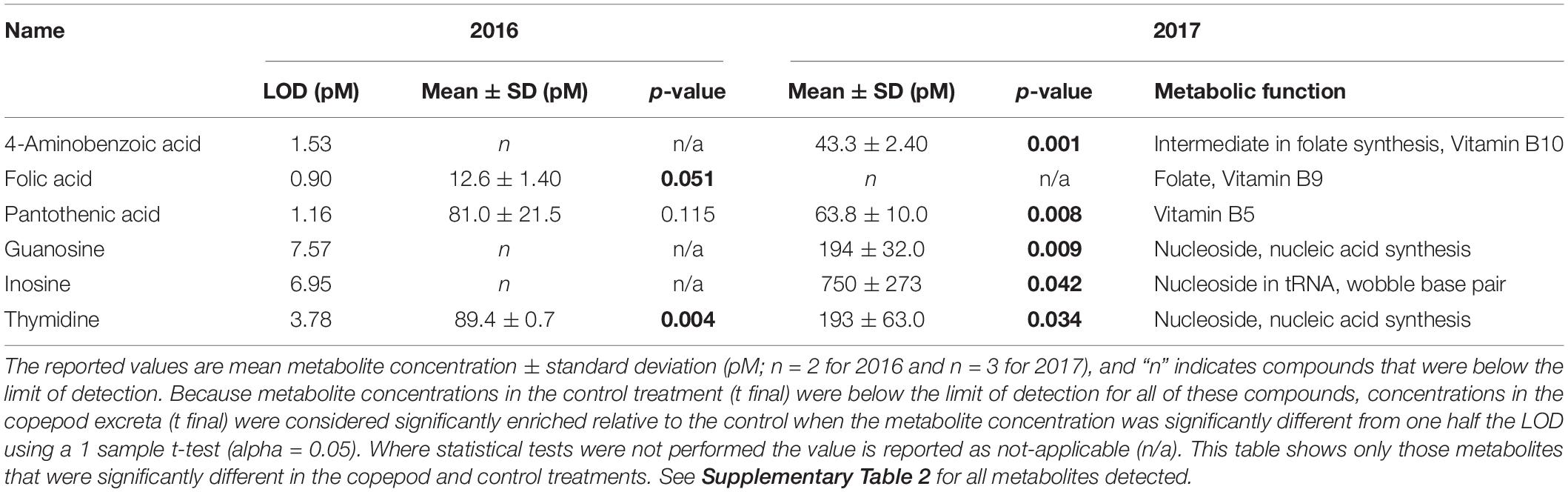
Table 3. Dissolved metabolites that were enriched in copepod excreta relative to the control treatment in at least one year.
Excreta generated from the 2016 zooplankton production experiment (1620-2) that had been filtered through a 0.2-μm filter and inoculated with a mesopelagic prokaryotic assemblage showed a clear prokaryotic growth response over the 14-day incubation. The initial incubation conditions of the zooplankton excreta treatment demonstrated a DOC enrichment of 7 μmol C L–1 and 4 μmol C L–1 greater than the unamended control and the “carryover” treatments, respectively. Prokaryotic growth was enhanced in this zooplankton excreta enriched treatment, with cell densities reaching a carrying capacity that was fourfold and twofold greater than that of the unamended control treatment and “carryover” treatments, respectively, by the fifth day of incubation (Figure 3A). By day 14 the cell abundance in the copepod treatment returned back to baseline. This pronounced death phase observed in the zooplankton treatment is typical during late stage in seawater culture experiments due to mortality by grazers and viruses introduced from the whole water inoculum. Over the course of the 14-day incubation net DOC consumption (at the μmol l–1 resolution) was not detected in the unamended control or “carryover” treatments. However, by day 5 a measured 43% of the ΔDOC in the excreta treatments relative to the control treatments had been removed while 100% of the amended DOC in the excreta treatments relative to the “carryover” treatments had been removed (Figure 3B). The concentrations of DFAA + taurine in the zooplankton excreta enriched treatment were also drawn down to background levels by day 5 of the remineralization incubation (Figure 3C). In addition to the removal of total DFAA, there was a change in the Mol% composition of the DFAA pool in the zooplankton excreta enriched incubation. Principal coordinate analysis of the DFAA composition indicated that the initial composition of the DFAA in the zooplankton excreta-enriched treatment was distinctly different from that of the unamended control and carryover treatments. Incubation time correlated with axis 1, which explained 43% of the variation among samples, while the source of the water (i.e., experimental versus control) correlated with axis 2, which explained 23% of the variance in proportional DFAA composition. By day 5 the proportional composition was similar to that of the carryover (Figure 4). The initial zooplankton excreta treatment was enriched in arginine and taurine, while the initial unamended control and carryover treatments were enriched with serine. Although the concentrations of all DFAA decreased rapidly, glycine, valine, arginine, and taurine were preferentially consumed over the course of the remineralization experiment, while phenylalanine comprised a higher proportion of the remaining DFAA composition (Figure 5 and Supplementary Material 3).
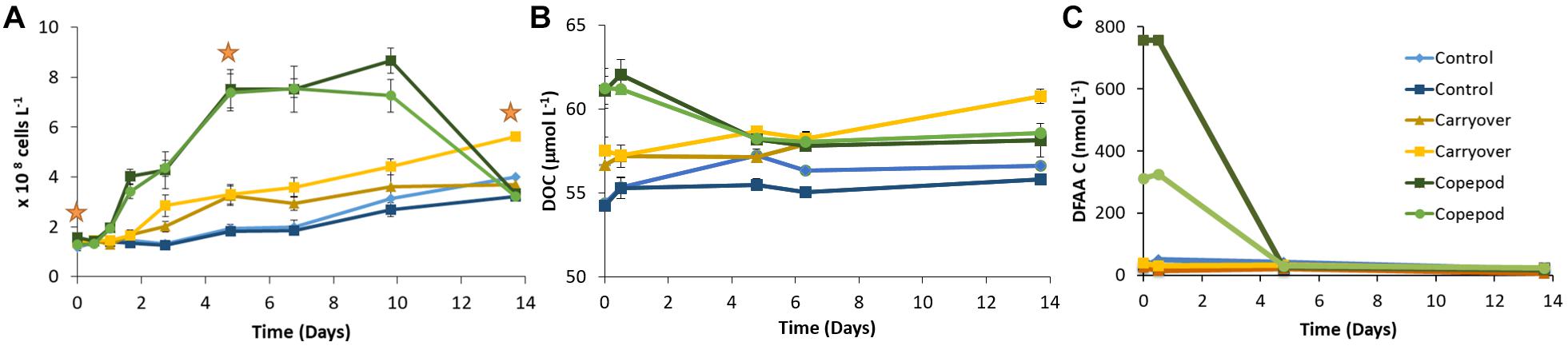
Figure 3. Change in DAPI cell counts ± SD (A), DOC ± SD (B), and DFAA + Tau C (C) over the time course of the remineralization experiment (1620-2). DNA for metabarcoding of the prokaryotic community was collected at three time points [orange stars; (A)].
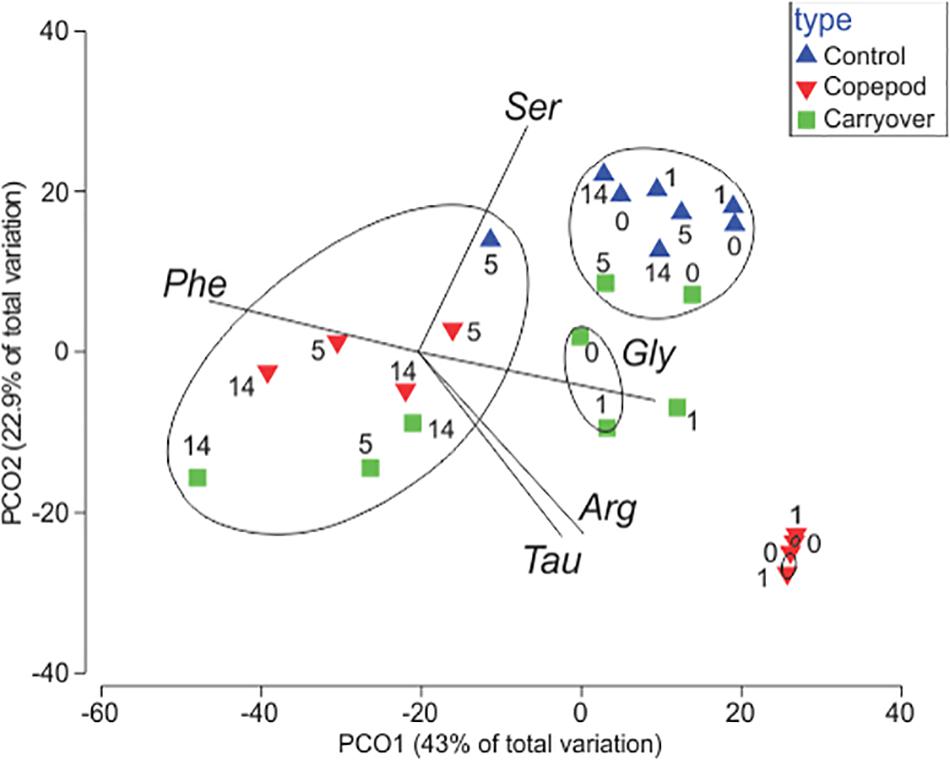
Figure 4. Principal coordinates analysis assessing the change in Mol% of DFAA + Tau in the remineralization experiments over the duration of experiment (numbers represent days of incubation). SIMPROF analysis suggests five clusters (p < 0.05; black circles). A Pearson correlation indicates which DFAAs [phenylalanine (Phe), serine (Ser), glycine (Gly), arginine (Arg), and taurine (Tau)] contributed to observed patterns in clustering (denoted as vectors; R > 0.95).
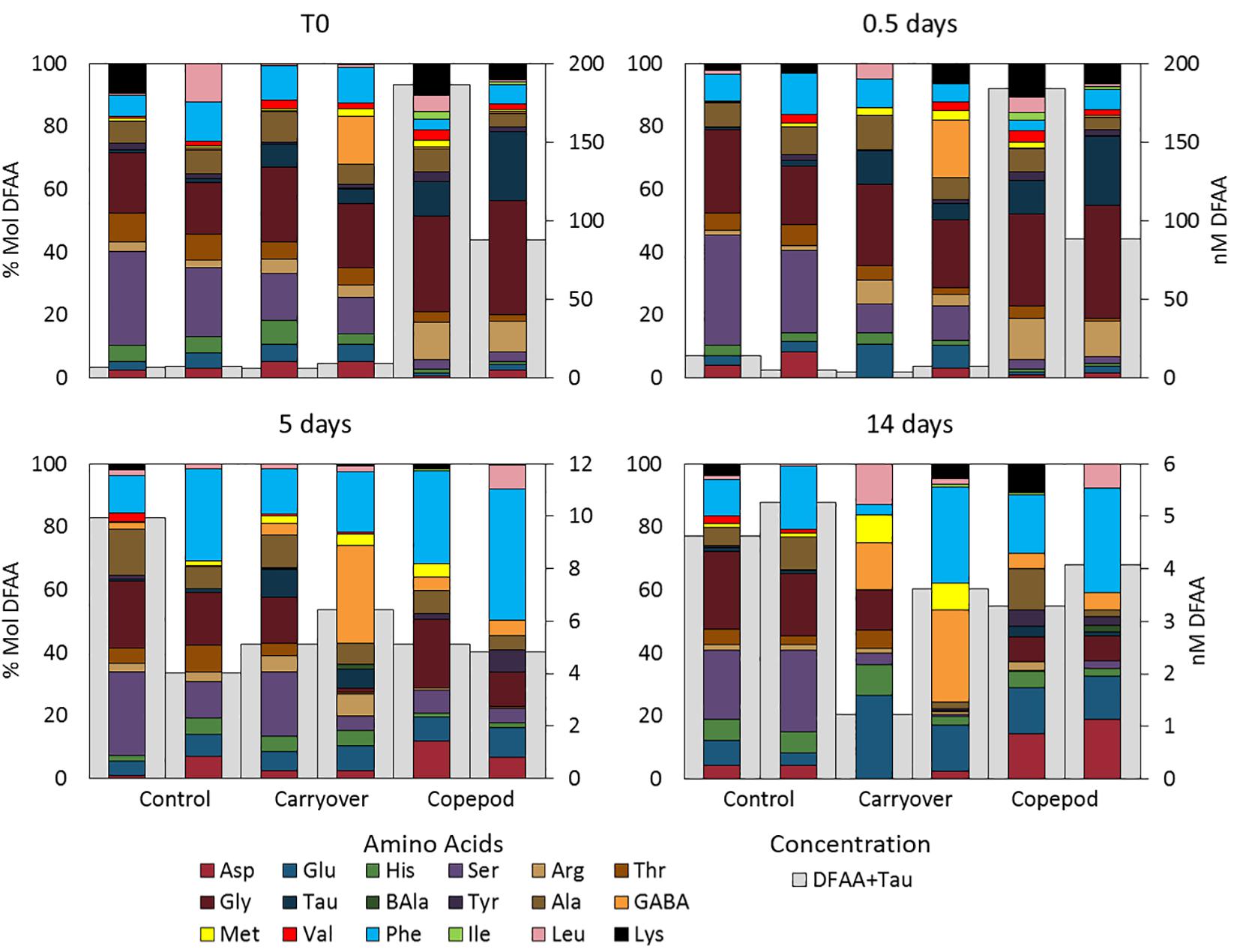
Figure 5. Total DFAA + Taurine concentration (nM; right axis) and %Mol contribution (left axis) of each of the amino acids over the course of the remineralization experiment. Note the different scale bars for total concentration.
Bacterioplankton community composition changed substantially over the course of the remineralization experiment, responding to both experimental handling and the presence of zooplankton excreta (Figure 6). The initial inoculation community was clearly dominated by known representatives of mesopelagic bacterioplankton lineages with ASVs representing members of the SAR202, SAR11deep, SAR324, and SAR406 clades and making up >64% of the amplicons (Supplementary Material 4). By day 5, the bacterioplankton community structure in control and carryover treatments shifted from communities dominated by oligotrophs, described above, to communities where the relative contribution appears to be dominated by putative copiotrophs i.e., Alteromonas and Thiotrichales (Figure 6). The response by these representative Gammaproteobacteria is a bottle effect that has been observed previously in various incubation studies when trace amounts of labile organic matter is available (Eilers et al., 2000; McCarren et al., 2010; Stewart et al., 2012; James et al., 2019). While we did observe an increase in Alteromonas in all treatments, the main point is that the responding bacterioplankton community in the zooplankton excreta enriched treatment was significantly different from the responding community observed in the control and carryover treatments. ASVs associated with the Flavobacteriales and Rhodobacterales clades became enriched in the zooplankton excreta treatment compared to the other treatments. We also note that, in addition to change in bacterial community structure, the total cell abundance change in the control and carryover is four and two times less than that in the copepod treatment respectively.
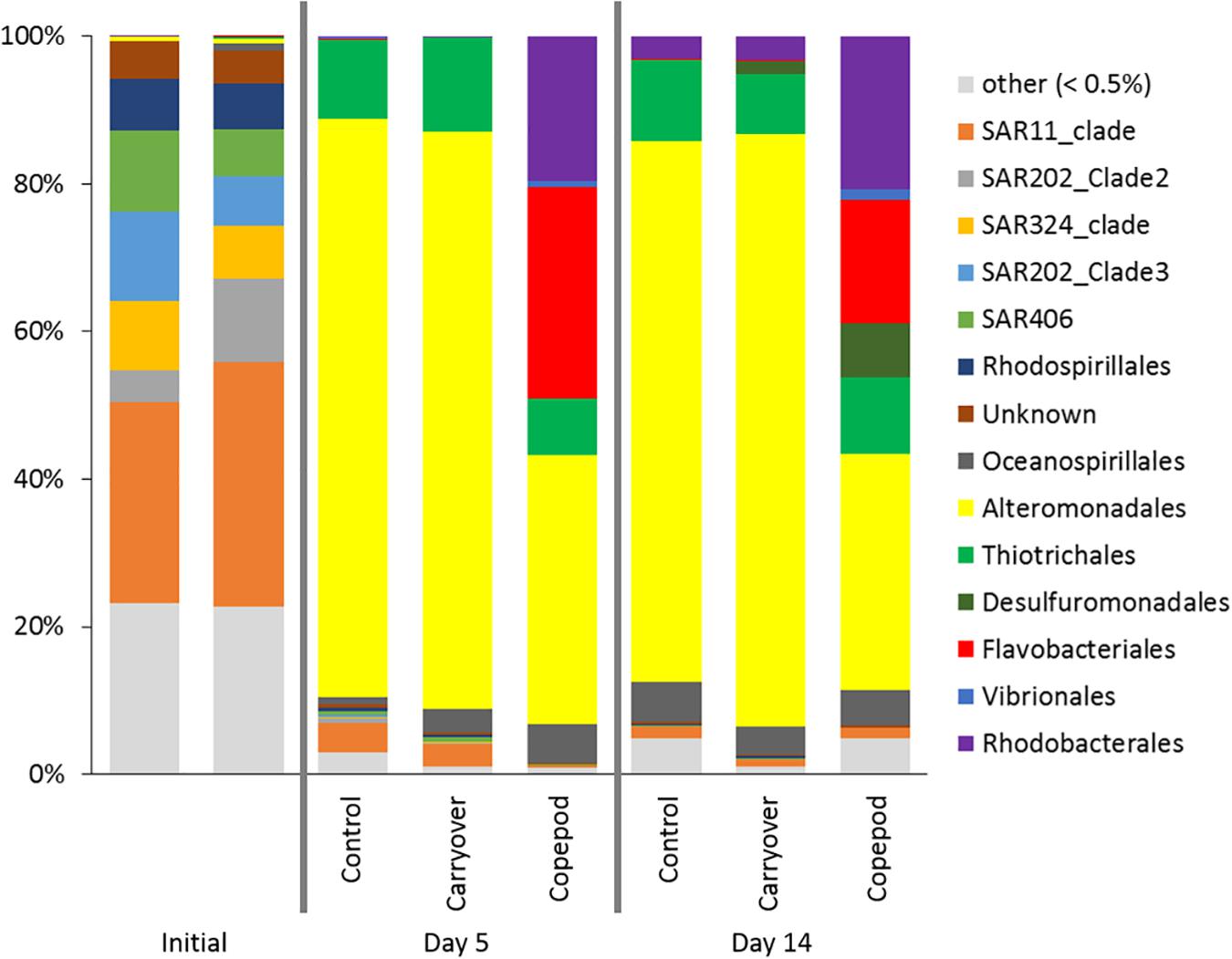
Figure 6. 16S rRNA gene amplicons were used to determine community composition at the Order level at the initiation of the experiment (T0), and after Day 5 and Day 14 of the incubation for all treatments in the microbial remineralization experiment. The proportion of reads in a sample is grouped by ASV order, with “other” being all groups contributing to less than <0.5% of the total reads.
SIMPROF clustering grouped communities significantly (p < 0.05) based on treatment type, with another cluster containing the initial time points, irrespective of treatment. Principal coordinates analysis indicated that the difference between initial time points and later experimental time points accounted for 48% of the variation in the communities (PCO#1; Figure 7). The Pearson correlations suggested that the difference among these groups was driven by significant (p < 0.05) enrichment of SAR202, SAR11, SAR324, Acidimicrobiales, and Salinisphaerales ASVs at the initial time point, while later time points were characterized by an increasing relative abundance of Alteromonadales (statistics in Supplementary Material 4). Treatment type (source of the inoculation water) was associated with PCO#2, which accounted for 26% of the variance. Pearson correlations indicated that the ASV types that were statistically significant (p < 0.05) in driving the observed differences among the treatments were an enrichment in Flavobacteriales, Rhodobacterales, and Pseudoalteromonadaceae in the zooplankton excreta enriched treatment (Figure 7 and statistics in Supplementary Material 4).
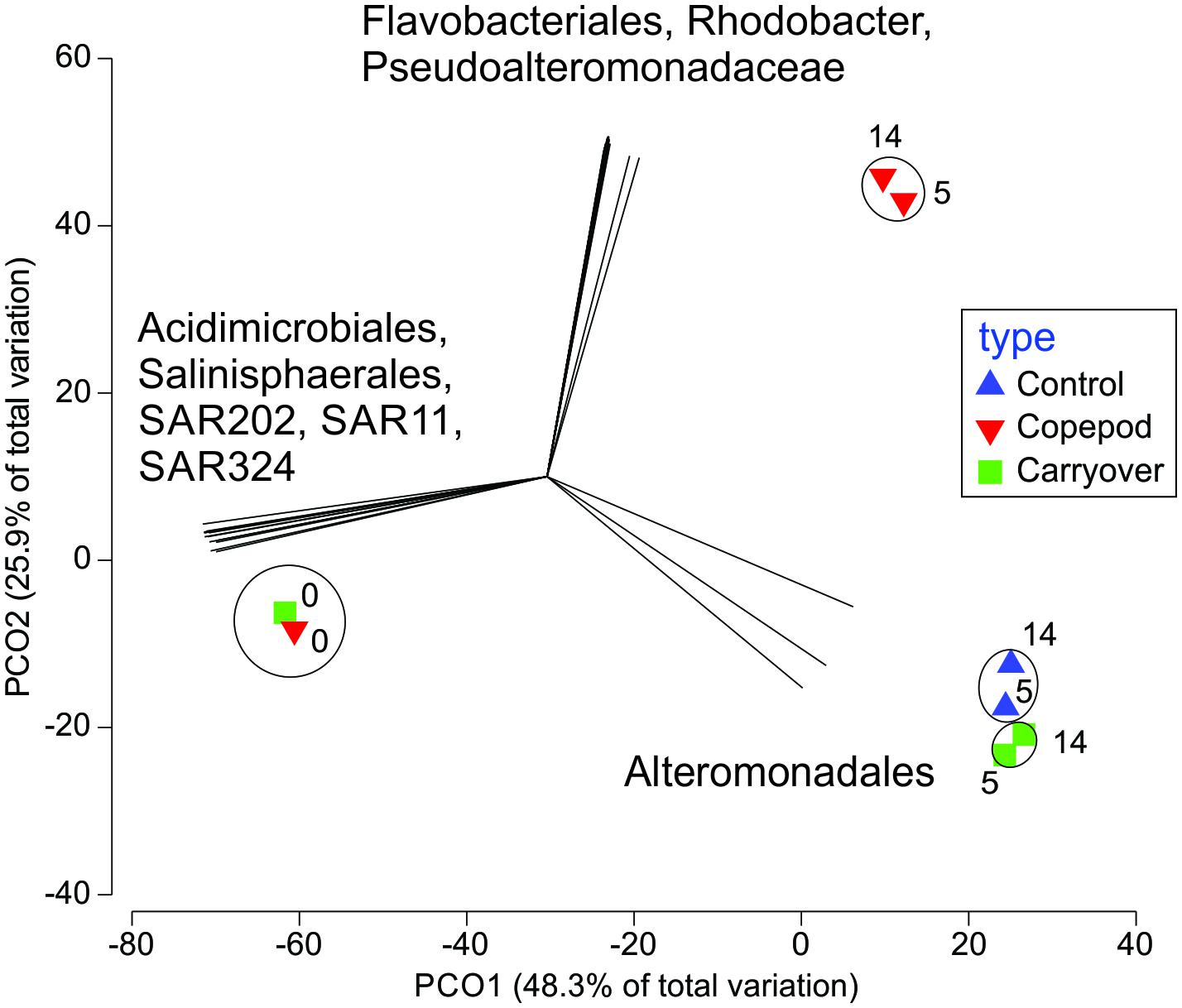
Figure 7. Principal coordinates analysis assessing the change in bacterioplankton community over the 14-day incubation (numbers represent days of the incubation) in the microbial remineralization experiment. The comparison is constructed using a Bray–Curtis similarity matrix of the ASVs relative contribution per sample. A SIMPROF cluster analysis show statistically similar communities (p < 0.05; black circles), while a Pearson correlation indicates which ASVs contributed to observed patterns in abundance (denoted as vectors; R > 0.95).
Our data indicate that the DOM produced by migrating zooplankton is composed of labile compounds, including DFAA, nucleosides, and vitamins, and is sufficient to result in a significant increase in the abundance of prokaryotic plankton. Previous studies of the DOC production of P. xiphias in the Sargasso Sea estimated 0.75 μg C h–1 mg dry mass(DM) –1 in experiments conducted at 26°C (Steinberg et al., 2000), which when converted to a comparable temperature as our experiments (using a Q10 of 2; Hochachka and Somero, 2002), is similar to our measurement of 0.55 ± 0.06 μg C h–1 mg DM–1 at 20°C. DFAA composition in the zooplankton excreta was relatively consistent between experiments. It was dominated by glycine, arginine, lysine, and taurine and was rapidly taken up by the prokaryotic community. This is similar to observations in prior work (Webb and Johannes, 1967; Fuhrman, 1987; Suttle et al., 1991; Clifford et al., 2017).
The dissolved metabolites measured here (nucleosides and vitamins) are not commonly resolved in open ocean mesopelagic samples. This is partially due to tight coupling between production and consumption processes whereby these labile compounds are readily and rapidly scavenged by the free-living prokaryotic community. This makes assessment of their contributions to carbon cycling and mesopelagic bacterial production difficult to trace without manipulation experiments. These results draw attention to the difficulty of balancing carbon budgets between the surface and the mesopelagic waters (Burd et al., 2010), because zooplankton excreta is presumed to be released and immediately “lost” both to dilution and to a rapacious mesopelagic prokaryotic heterotrophic community. Field evidence for this tight coupling is limited largely due to methodological challenges. Prior work in coastal communities, however, demonstrated diel and seasonal changes in the Mol% composition of DFAA in the water column (Lu et al., 2014). They observed evidence of zooplankton contributions (high glycine and taurine production) in the mesopelagic in the middle of the day and in the surface in the early morning, right before zooplankton would make their diel migration to depth.
Excreta-derived DOM appeared to meet a portion of the energy and nutritional demands of the prokaryotic community in this study (Figure 3A), indicating that the components of this DOM have an important ecological role. Vitamins have been proposed to be a currency for ecological interactions in prokaryotic marine communities to dictate biogeographic ranges (Giovannoni, 2012; Sañudo-Wilhelmy et al., 2014; Morris, 2015). Folic acid (folate; vitamin B9), which was detected in our exudate as well as its precursor molecule, 4-aminobenzoic acid (vitamin B10), is needed to make, repair, and methylate DNA. Christine et al. (1998) measured folate at nM concentrations along a transect in the Northeast Atlantic, with a peak in concentration corresponding to the depth of the chlorophyll maximum layer, and a second concentration peak in the mesopelagic zone. That study made no measurements of zooplankton, but the mesopelagic folate peak, located between 500 and 800 m, is consistent with depths typical for the deep scattering layer of vertical migrators. These migrating zooplankton may contribute to mesopelagic imbalances between production and uptake of this compound. Christine et al. (1998) estimated a folic acid removal rate of 10% per hour by subsurface (50–60 m) prokaryotic communities, resulting in the removal of 92% of the folic acid within a sample after 10 h (temperature not reported). Similar data concerning pantothenic acid (vitamin B5) is lacking, but it has been demonstrated that members of the SAR11 and SAR86 clades lack biosynthesis pathways for this compound (Giovannoni et al., 2005), suggesting that these organisms meet their nutritional demand for pantothenic acid from external sources such as vertical migrating zooplankton. Generally not much is known about the distribution and fate of nucleosides in the environment; however, some nucleosides and nucleotides have been studied in seawater and are known to be used by marine bacteria (Berg and Jørgensen, 2006; Li et al., 2020). These results suggest that as our ability to characterize the dissolved compounds in the water column improves and routine sampling of mesopelagic biological signatures becomes more common, it will be important to conduct both day- and night-time sampling to observe contributions of migratory metazoans to the available substrates for mesopelagic prokaryotic communities.
Although there were some consistent patterns in the production of various compounds (particularly the relative contribution by specific amino acids), the proportion of DOC made up of DFAA and the quantified metabolites (measured by LC MS/MS) differed among the zooplankton excreta production experiments. In particular, some of the metabolites measured in the September 2016 experiments were not present in the July 2017 experiments and vice versa. While we cannot rule out experimental or handling artifacts, it is quite possible that the observed variability is due to seasonal changes in the composition of the prey field or in the metabolism of the copepods whose life history may have changed between the two seasons. Furthermore, recent work suggests that zooplankton metabolism changes over the diel cycle due to intrinsic circadian rhythms (Teschke et al., 2011; De Pitta et al., 2013; Maas et al., 2018). Our experiments were conducted over a 12–16 h period, generally starting in the middle of the night (0200–0300) and ending in the afternoon or early evening (1300–1900). Thus, the compounds we measured reflect daytime excretion that occurs when these organisms occupy the mesopelagic zone, while having a substantial contribution of compounds that would have been excreted exclusively in the photic zone (during the 0300–0800 period). Isolating and quantifying the compounds that are excreted during the day when the organisms are in the mesopelagic will be important in determining the relevance of zooplankton excreta to deeper prokaryotic communities.
Our remineralization experiments demonstrate that prokaryotic cell densities increased by approximately four-fold over 5 days when a natural prokaryotic community from 200 m was mixed with filtrate enriched with zooplankton excreta (Figure 3A). Over this same period the bacterioplankton community structure in all treatments shifted significantly (SIMPROF and Pearson Correlation; p < 0.05) from mesopelagic lineages such as SAR11deep and SAR202 clades, members of which specialize in the metabolism of more recalcitrant DOM (Carlson et al., 2009; Liu et al., 2020; Saw et al., 2020), to larger copiotrophic groups including Alteromonadales, Pseudoalteromonadaceae, Rhodobacterales, and Flavobacteriales. The increase in the relative contribution of Alteromonadales lineages (specifically Marinobacter and Alteromonas) to total 16S rRNA amplicons was ubiquitous across the control, carryover, and copepod treatments. This shift has been observed in incubation studies before (e.g., Eilers et al., 2000; McCarren et al., 2010; Stewart et al., 2012; James et al., 2019) and appears to be a common bottle effect. The disconnect between only a slight increase in total cell counts and a substantial increase in relative contribution of Alteromonadales to total amplicons additionally likely reflect bias associated with multiple copies of the 16S rRNA gene (Klappenbach et al., 2000; Math et al., 2012; Větrovský and Baldrian, 2013). This has been observed previously in similar manipulation experiments (Valdés et al., 2017; Lund Paulsen et al., 2018). Specifically, Gammaproteobacteria (which includes Alteromonas and Thiotrichales) typically have a 16S rRNA/genome ratio of ∼6 ± 3, while Alphaproteobacteria (e.g., SAR11 and Rhodospirillales) are ∼2 ± 1 (Větrovský and Baldrian, 2013). Thus, a modest increase in the number of Gammaproteobacterial cells could result in a substantial over-representation of their ASV in the community composition analysis. Bottle incubations cannot completely simulate in situ conditions and microbes may thus be affected by confinement to some extent. Despite potential containment artefacts, bottle incubation experiments that simulate environmental conditions are still historically an effective approach to investigate roles of specific groups in the ocean and link potential factors contributing to the change of what we observe in the real ambient environment.
Unlike Alteromonadales, members of the Pseudoalteromonadaceae, Rhodobacterales, and Flavobacteriales clades increased in relative abundance exclusively and significantly (SIMPROF and Pearson Correlation; p < 0.05) in the zooplankton excreta enriched treatment. Members of these clades have been shown to increase in other studies that inoculated microbial communities with zooplankton (Valdés et al., 2017). Additionally, Calleja et al. (2018), who incubated water from three depths in the Red Sea, found substantial increases in the proportion of Rhodobacter from all depths of their incubations, and increases in Gammaproteobacteria (which include Alteromonadales and Pseudoalteromonadaceae) from their mesopelagic treatments at depths where migrators were known to congregate (based on acoustic backscattering). Therefore, although these groups are often considered opportunistic lineages that are typically of low relative abundance in subtropical field samples, point sources of DOM such as those provided by migratory zooplankton excreta may be important to sustain their presence in mesopelagic ocean communities. Thus, although the ability of prokaryotes to sense and take advantage of transient gradients in nutrients has been hypothesized and described mechanistically (Stocker et al., 2008; Lauro et al., 2009; Stocker, 2012), our study experimentally demonstrates that DOM by vertical migratory zooplankton can support growth of seed populations of specific copiotrophic responders that are found in the mesopelagic. The overwhelming response of bacterioplankton production in the copepod treatment relative to the carryover and control treatments highlights the unique role of copepod excretory release of DOM (and associated inorganic nutrients) in controlling bacterioplankton growth and shaping the prokaryotic communities.
Moving forward from this work, it is important to recognize that, although we have no measurements of the frequency, duration, or physical size of excretion pulses of P. xiphias, it is likely that in nature the excreta in a micro-“hot spot” (Azam, 1998; Stocker, 2012) is much more concentrated than in our zooplankton excreta and remineralization experiments. In reality, zooplankton-generated “hot spots” of organic and inorganic nutrients would likely be smaller, farther apart, and more concentrated. This likely creates even greater heterogeneity on the submicron scale and more pronounced succession patterns of bacterioplankton communities than was measured in our experiments. A primary consideration of the experimental design was to minimize the incubation period but obtain a large enough DOC, TDAA, and metabolite signal that could be resolved given the current analytical methods. We cannot rule out the possibility that the high densities of organisms during the excreta production period may have influence the amount of excreta produced by copepods in our study. However, our protocol is consistent and comparable with prior studies (Steinberg et al., 2000; Thibodeau et al., 2020; Maas et al., in review) used to both generate sufficient exudate for the remineralization and dissolved metabolite analysis. Until there are substantial increases in the sensitivity of DOC and metabolite analyses this provides our best estimate of production rates.
An additional consideration is that the excreta measured here from diel vertical migrators is also likely produced by resident mesopelagic zooplankton that ingest particles delivered from passive flux or consume each other and then release waste in ambient seawater. Although the focus is typically on the activity and biomass of migratory species, as in this study, the resident mesopelagic mesozooplankton and fish community are likely to make a similarly substantial contribution to point sources of inorganic and organic nutrients and merit further investigation.
This study, along with a suite of others (Clifford et al., 2017; Valdés et al., 2017; Calleja et al., 2018; De Corte et al., 2018), suggests that there are free living midwater prokaryotic communities that are specialized in the use of labile compounds produced by metazoans. Although the exact mechanisms of prokaryotic use of this excreta are not yet well understood, our results provide a novel dataset on the particular DOM compounds, such as amino acids and metabolites produced by zooplankton. By showing which bacterial groups respond to those excreta, we have an insight into what lineages are influenced, providing targets for more directed investigations. Characterization of these organisms and their biogeochemical significance is complicated by the differences in sampling strategies typically used to assess the prokaryotic and zooplankton communities. As we begin to map ocean nutrients and metabolic cycling via molecular methods, including proteomics, metatranscriptomics, and metagenomics (Venter et al., 2004; Sowell et al., 2009; Vergin et al., 2013b; Guidi et al., 2016; Saito et al., 2019; Santoro, 2019), it will be important to recognize that the complex network of ecological interactions includes chemical transactions between prokaryotes and metazoans. Our results demonstrate that process studies and targeted searches for suites of metabolomics biomarkers associated with zooplankton products are promising tools by which we can begin to quantify these pathways.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject – PRJNA625717, MetaboLights – MTBLS1822.
AM, LB-B, and CC conceived the experiments. AM, LB-B, CC, and SL conducted the experiments. AM, SL, LB, BW, and RP analyzed the samples. AM wrote the manuscript with contributions and review by all authors. All authors contributed to the article and approved the submitted version.
Funding for this research was provided by Simons Foundation International as part of the BIOS-SCOPE project to AM, LB-B, CC, and EK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the entire BIOS-SCOPE team for their enthusiasm and comradery. In particular we must thank Joseph Jones, Joe Cope, Samm Newton, and Keri Opalk for copepod collection, Nick Baetge for helping set up experiments, and Keri Opalk for DOC analyses. We would also like to thank Krista Longnecker, Gretchen Swarr, and Melissa Kido Soule for assistance with metabolomics sampling and analysis. We are indebted to the captains, crew, and marine technicians of the R/V Atlantic Explorer for making our collections and experiments run smoothly and safely. We thank Mark Dasenko and members of the Center for Genome Research and Biocomputing at Oregon State University for DNA sequencing services. This work benefited from discussion among members within the JETZON community.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.573268/full#supplementary-material
Alcaraz, M., Almeda, R., Calbet, A., Saiz, E., Duarte, C. M., Lasternas, S., et al. (2010). The role of arctic zooplankton in biogeochemical cycles: respiration and excretion of ammonia and phosphate during summer. Polar Biol. 33, 1719–1731. doi: 10.1007/s00300-010-0789-9
Arístegui, J., Duarte, C. M., Reche, I., and Gómez-Pinchetti, J. L. (2014). Krill excretion boosts microbial activity in the Southern Ocean. PLoS One 9:e89391. doi: 10.1371/journal.pone.0089391
Azam, F. (1998). Microbial control of oceanic carbon flux: the plot thickens. Science 280, 694–696. doi: 10.1126/science.280.5364.694
Berg, G. M., and Jørgensen, N. O. (2006). Purine and pyrimidine metabolism by estuarine bacteria. Aquat. Microb. Ecol. 42, 215–226. doi: 10.3354/ame042215
Burd, A. B., Hansell, D. A., Steinberg, D. K., Anderson, T. R., Arístegui, J., Baltar, F., et al. (2010). Assessing the apparent imbalance between geochemical and biochemical indicators of meso-and bathypelagic biological activity: what the@ $#! is wrong with present calculations of carbon budgets? Deep Sea Res. Part II Top. Stud. Oceanogr. 57, 1557–1571. doi: 10.1016/j.dsr2.2010.02.022
Buskey, E. J., Baker, K. S., Smith, R. C., and Swift, E. (1989). Photosensitivity of the oceanic copepods Pleuromamma gracilis and Pleuromamma xiphias and its relationship to light penetration and daytime depth distribution. Mar. Ecol. Prog. Ser. 55, 207–216. doi: 10.3354/meps055207
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13:581. doi: 10.1038/nmeth.3869
Calleja, M. L., Ansari, M. I., Røstad, A., da Silva, L. R., Kaartvedt, S., Irigoien, X., et al. (2018). The mesopelagic scattering layer: a hotspot for heterotrophic prokaryotes in the Red Sea twilight zone. Front. Mar. Sci. 5:259. doi: 10.3389/fmars.2018.00259
Carlson, C. (2002). “Production and removal processes,” in Biogeochemistry of Marine Dissolved Organic Matter, eds D. Hansell and C. Carlson, (San Diego: Academic Press), 91–151. doi: 10.1016/b978-012323841-2/50006-3
Carlson, C. A., Ducklow, H. W., and Michaels, A. F. (1994). Annual flux of dissolved organic carbon from the euphotic zone in the northwestern Sargasso Sea. Nature 371, 405–408. doi: 10.1038/371405a0
Carlson, C. A., Giovannoni, S. J., Hansell, D. A., Goldberg, S. J., Parsons, R., Otero, M. P., et al. (2002). The effect of nutrient amendments on bacterioplankton growth, DOC utilization, and community structure in the northwestern Sargasso Sea. Aquat. Microb. Ecol. 30, 19–36. doi: 10.3354/ame030019
Carlson, C. A., Hansell, D. A., Nelson, N. B., Siegel, D. A., Smethie, W. M., Khatiwala, S., et al. (2010). Dissolved organic carbon export and subsequent remineralization in the mesopelagic and bathypelagic realms of the North Atlantic basin. Deep Sea Res. Part II 57, 1433–1445. doi: 10.1016/j.dsr2.2010.02.013
Carlson, C. A., Morris, R., Parsons, R., Giovannoni, S. J., and Vergin, K. (2009). Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 3, 283–295. doi: 10.1038/ismej.2008.117
Chambers, M. C., Maclean, B., Burke, R., Amodei, D., Ruderman, D. L., Neumann, S., et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920.
Christine, A., Gall, L., and Van Den Berg, C. M. (1998). Folic acid and glutathione in the water column of the North East Atlantic. Deep Sea Res. Part I Oceanogr. Research Papers 45, 1903–1918. doi: 10.1016/s0967-0637(98)00042-9
Clarke, K. R., and Warwick, R. M. (2001). Changes in Marine Communities: An Approach to Statistical Analysis and Interpretation, 2nd Edn. Plymouth: PRIMER-E.
Clasquin, M. F., Melamud, E., and Rabinowitz, J. D. (2012). LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 37, 1–23.
Clifford, E. L., Hansell, D. A., Varela, M. M., Nieto-Cid, M., Herndl, G. J., and Sintes, E. (2017). Crustacean zooplankton release copious amounts of dissolved organic matter as taurine in the ocean. Limnol. Oceanogr. 62, 2745–2758. doi: 10.1002/lno.10603
Cowie, G. L., and Hedges, J. I. (1994). Biochemical indicators of diagenetic alteration in natural organic matter mixtures. Nature 369:304. doi: 10.1038/369304a0
Daims, H., Brühl, A., Amann, R., Schleifer, K.-H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/s0723-2020(99)80053-8
Dall’Olmo, G., Dingle, J., Polimene, L., Brewin, R. J., and Claustre, H. (2016). Substantial energy input to the mesopelagic ecosystem from the seasonal mixed-layer pump. Nat. Geosci. 9:820. doi: 10.1038/ngeo2818
Davis, J., Kaiser, K., and Benner, R. (2009). Amino acid and amino sugar yields and compositions as indicators of dissolved organic matter diagenesis. Org. Geochem. 40, 343–352. doi: 10.1016/j.orggeochem.2008.12.003
De Corte, D., Srivastava, A., Koski, M., Garcia, J. A. L., Takaki, Y., Yokokawa, T., et al. (2018). Metagenomic insights into zooplankton-associated bacterial communities. Environ. Microbiol. 20, 492–505. doi: 10.1111/1462-2920.13944
De Pitta, C., Biscontin, A., Albiero, A., Sales, G., Millino, C., Mazzotta, G. M., et al. (2013). The Antarctic krill Euphausia superba shows diurnal cycles of transcription under natural conditions. PLoS One 8:e68652. doi: 10.1371/journal.pone.0068652
Ducklow, H. W., Steinberg, D. K., and Buesseler, K. O. (2001). Upper ocean carbon export and the biological pump. Oceanogr. Washington Oceanogr. Soc. 14, 50–58. doi: 10.5670/oceanog.2001.06
Eilers, H., Pernthaler, J., and Amann, R. (2000). Succession of pelagic marine bacteria during enrichment: a close look at cultivation-induced shifts. Appl. Environ. Microbiol. 66, 4634–4640. doi: 10.1128/aem.66.11.4634-4640.2000
Fuhrman, J. (1987). Close coupling between release and uptake of dissolved free amino acids in seawater studied by an isotope dilution approach. Mar. Ecol. Prog. Ser. 37, 45–52. doi: 10.3354/meps037045
García, F. C., Calleja, M. L., Al-Otaibi, N., Røstad, A., and Morán, X. A. G. (2018). Diel dynamics and coupling of heterotrophic prokaryotes and dissolved organic matter in epipelagic and mesopelagic waters of the central Red Sea. Environ. Microbiol. 20, 2990–3000. doi: 10.1111/1462-2920.14336
Giovannoni, S. J., Britschgi, T. B., Moyer, C. L., and Field, K. G. (1990). Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60. doi: 10.1038/345060a0
Giovannoni, S. J., and Stingl, U. (2005). Molecular diversity and ecology of microbial plankton. Nature 437:343. doi: 10.1038/nature04158
Giovannoni, S. J., Tripp, H. J., Givan, S., Podar, M., Vergin, K. L., Baptista, D., et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245. doi: 10.1126/science.1114057
Guidi, L., Chaffron, S., Bittner, L., Eveillard, D., Larhlimi, A., Roux, S., et al. (2016). Plankton networks driving carbon export in the oligotrophic ocean. Nature 532:465. doi: 10.1038/nature16942
Hansell, D. A. (2013). Recalcitrant dissolved organic carbon fractions. Annu. Rev. Mar. Sci. 5, 421–445. doi: 10.1146/annurev-marine-120710-100757
Hochachka, P. W., and Somero, G. N. (2002). Biochemical Adaptation: Mechanism and Process in Physiological Evolution. New York, NY: Oxford University Press.
Ikeda, T. (2014). Respiration and ammonia excretion by marine metazooplankton taxa: synthesis toward a global-bathymetric model. Mar. Biol. 161, 2753–2766. doi: 10.1007/s00227-014-2540-5
Ikeda, T., Kanno, Y., Ozaki, K., and Shinada, A. (2001). Metabolic rates of epipelagic marine copepods as a function of body mass and temperature. Mar. Biol. 139, 587–596. doi: 10.1007/s002270100608
James, A. K., Kelly, L. W., Nelson, C. E., Wilbanks, E. G., and Carlson, C. A. (2019). Elevated pCO2 alters marine heterotrophic bacterial community composition and metabolic potential in response to a pulse of phytoplankton organic matter. Environ. Microbiol. 21, 541–556. doi: 10.1111/1462-2920.14484
Johnson, W. M., Kido Soule, M. C., and Kujawinski, E. B. (2017). Extraction efficiency and quantification of dissolved metabolites in targeted marine metabolomics. Limnol. Oceanogr. Methods 15, 417–428. doi: 10.1002/lom3.10181
Kaiser, K., and Benner, R. (2009). Biochemical composition and size distribution of organic matter at the Pacific and Atlantic time-series stations. Mar. Chem. 113, 63–77. doi: 10.1016/j.marchem.2008.12.004
Kido Soule, M. C., Longnecker, K., Johnson, W. M., and Kujawinski, E. B. (2015). Environmental metabolomics: analytical strategies. Mar. Chem. 177, 374–387. doi: 10.1016/j.marchem.2015.06.029
Klappenbach, J. A., Dunbar, J. M., and Schmidt, T. M. (2000). rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66, 1328–1333. doi: 10.1128/aem.66.4.1328-1333.2000
Lampert, W. (1989). The adaptive significance of diel vertical migration of zooplankton. Funct. Ecol. 3, 21–27. doi: 10.2307/2389671
Landry, Z., Swan, B. K., Herndl, G. J., Stepanauskas, R., and Giovannoni, S. J. (2017). SAR202 genomes from the dark ocean predict pathways for the oxidation of recalcitrant dissolved organic matter. mBio 8, e00413–e00417.
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt, and M. Goodfellow (Chichester, UK: Wiley), 115–148.
Lauro, F. M., McDougald, D., Thomas, T., Williams, T. J., Egan, S., Rice, S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 106, 15527–15533. doi: 10.1073/pnas.0903507106
Lee, C., Wakeham, S. G., and Hedges, J. I. (2000). Composition and flux of particulate amino acids and chloropigments in equatorial Pacific seawater and sediments. Deep Sea Res. Part I Oceanogr. Res. Pap. 47, 1535–1568. doi: 10.1016/s0967-0637(99)00116-8
Li, J., Zhang, X., Fan, W.-Y., Yao, M.-C., and Sheng, G.-P. (2020). Dissolved organic matter dominating the photodegradation of free DNA bases in aquatic environments. Water Res. 179:115885. doi: 10.1016/j.watres.2020.115885
Lindroth, P., and Mopper, K. (1979). High performance liquid chromatographic determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization with o-phthaldialdehyde. Anal. Chem. 51, 1667–1674. doi: 10.1021/ac50047a019
Liu, S., Parsons, R., Opalk, K., Baetge, N., Giovannoni, S., Bolaños, L. M., et al. (2020). Different carboxyl-rich alicyclic molecules proxy compounds select distinct bacterioplankton for oxidation of dissolved organic matter in the mesopelagic Sargasso Sea. Limnol. Oceanogr. 65, 1532–1553. doi: 10.1002/lno.11405
Longnecker, K. (2015). Dissolved organic matter in newly formed sea ice and surface seawater. Geochim. Cosmochim. Acta 171, 39–49. doi: 10.1016/j.gca.2015.08.014
Lu, X., Zou, L., Clevinger, C., Liu, Q., Hollibaugh, J. T., and Mou, X. (2014). Temporal dynamics and depth variations of dissolved free amino acids and polyamines in coastal seawater determined by high-performance liquid chromatography. Mar. Chem. 163, 36–44. doi: 10.1016/j.marchem.2014.04.004
Lund Paulsen, M., Müller, O., Larsen, A., Møller, E. F., Middelboe, M., Sejr, M. K., et al. (2018). Biological transformation of Arctic dissolved organic matter in a NE Greenland fjord. Limnol. Oceanogr. 64, 1014-1033.
Maas, A. E., Blanco-Bercial, L., Lo, A., Tarrant, A. M., and Timmins-Schiffman, E. (2018). Variations in copepod proteome and respiration rate in association with diel vertical migration and circadian cycle. Biol. Bull. 235, 30–42. doi: 10.1086/699219
Maas, A. E., Miccoli, A., Stamieszkin, K. S., Carlson, C. A., and Steinberg, D. K. (in review). Allometry and the calculation of zooplankton metabolism in the subarctic Northeast Pacific Ocean. J. Plankton Res.
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12.
Math, R. K., Jin, H. M., Kim, J. M., Hahn, Y., Park, W., Madsen, E. L., et al. (2012). Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS One 7:e35784. doi: 10.1371/journal.pone.0035784
McCarren, J., Becker, J. W., Repeta, D. J., Shi, Y., Young, C. R., Malmstrom, R. R., et al. (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc. Natl. Acad. Sci. U.S.A. 107, 16420–16427. doi: 10.1073/pnas.1010732107
Melamud, E., Vastag, L., and Rabinowitz, J. D. (2010). Metabolomic Analysis and Visualization Engine for LC-MS Data. Anal. Chem. 82, 9818–9826. doi: 10.1021/ac1021166
Morris, J. J. (2015). Black Queen evolution: the role of leakiness in structuring microbial communities. Trends Genet. 31, 475–482. doi: 10.1016/j.tig.2015.05.004
Morris, R. M., Vergin, K. L., Cho, J.-C., Rappé, M. S., Carlson, C. A., and Giovannoni, S. J. (2005). Temporal and spatial response of bacterioplankton lineages to annual convective overturn at the Bermuda Atlantic Time-Series Study Site. Limnol. Oceanogr. 50, 1687–1696. doi: 10.4319/lo.2005.50.5.1687
Porter, K. G., and Feig, Y. S. (1980). The use of DAPI for identifying and counting aquatic microflora. Limnol. Oceanogr. 25, 943–948. doi: 10.4319/lo.1980.25.5.0943
Saito, M. A., Bertrand, E. M., Duffy, M. E., Gaylord, D. A., Held, N. A., Hervey, I. V. W. J., et al. (2019). Progress and challenges in ocean metaproteomics and proposed best practices for data sharing. J. Prot. Res. 18, 1461–1476. doi: 10.1021/acs.jproteome.8b00761
Santoro, A. E. (2019). Crystal ball: the microbial map of the ocean. Environ. Microbiol. Rep. 11, 35–37. doi: 10.1111/1758-2229.12721
Sañudo-Wilhelmy, S. A., Gómez-Consarnau, L., Suffridge, C., and Webb, E. A. (2014). The role of B vitamins in marine biogeochemistry. Annu. Rev. Mar. Sci. 6, 339–367. doi: 10.1146/annurev-marine-120710-100912
Saw, J. H., Nunoura, T., Hirai, M., Takaki, Y., Parsons, R., Michelsen, M., et al. (2020). Pangenomics reveal diversification of enzyme families and niche specialization in globally abundant SAR202 bacteria. mBio 11, e02975–02919.
Sowell, S. M., Wilhelm, L. J., Norbeck, A. D., Lipton, M. S., Nicora, C. D., Barofsky, D. F., et al. (2009). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 3, 93–105. doi: 10.1038/ismej.2008.83
Steinberg, D. K., Carlson, C. A., Bates, N. R., Goldthwait, S. A., Madin, L. P., and Michaels, A. F. (2000). Zooplankton vertical migration and the active transport of dissolved organic and inorganic carbon in the Sargasso Sea. Deep Sea Res. Part I 47, 137–158. doi: 10.1016/s0967-0637(99)00052-7
Steinberg, D. K., Goldthwait, S. A., and Hansell, D. A. (2002). Zooplankton vertical migration and the active transport of dissolved organic and inorganic nitrogen in the Sargasso Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 49, 1445–1461. doi: 10.1016/s0967-0637(02)00037-7
Steinberg, D. K., and Landry, M. R. (2017). Zooplankton and the Ocean Carbon Cycle. Ann. Rev. Mar. Sci. 9, 413–444. doi: 10.1146/annurev-marine-010814-015924
Stewart, F. J., Dalsgaard, T., Young, C. R., Thamdrup, B., Revsbech, N. P., Ulloa, O., et al. (2012). Experimental incubations elicit profound changes in community transcription in OMZ bacterioplankton. PLoS One 7:e37118. doi: 10.1371/journal.pone.0037118
Stocker, R. (2012). Marine microbes see a sea of gradients. Science 338, 628–633. doi: 10.1126/science.1208929
Stocker, R., Seymour, J. R., Samadani, A., Hunt, D. E., and Polz, M. F. (2008). Rapid chemotactic response enables marine bacteria to exploit ephemeral microscale nutrient patches. Proc. Natl. Acad. Sci. U.S.A. 105, 4209–4214. doi: 10.1073/pnas.0709765105
Suttle, C. A., Chan, A. M., and Fuhrman, J. A. (1991). Dissolved free amino acids in the Sargasso Sea: uptake and respiration rates, turnover times, and concentrations. Mar. Ecol. Prog. Ser. 70, 189–199. doi: 10.3354/meps070189
Teschke, M., Wendt, S., Kawaguchi, S., Kramer, A., and Meyer, B. (2011). A circadian clock in antarctic krill: an endogenous timing system governs metabolic output rhythms in the euphausid species Euphausia superba. PLoS One 6:e26090. doi: 10.1371/journal.pone.0026090
Thibodeau, P. S., Steinberg, D., and Maas, A. E. (2020). Effects of temperature and food concentration on pteropod metabolism along the Western Antarctic Peninsula. J. Exp. Mar. Biol. Ecol. 53:151412. doi: 10.1016/j.jembe.2020.151412
Valdés, V. P., Fernandez, C., Molina, V., Escribano, R., and Joux, F. (2017). Dissolved compounds excreted by copepods reshape the active marine bacterioplankton community composition. Front. Mar. Sci. 4:343. doi: 10.3389/fmars.2017.00343
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74. doi: 10.1126/science.1093857
Vergin, K. L., Beszteri, B., Monier, A., Thrash, J. C., Temperton, B., Treusch, A. H., et al. (2013a). High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic Time-series Study site by phylogenetic placement of pyrosequences. ISME J. 7, 1322–1332. doi: 10.1038/ismej.2013.32
Vergin, K. L., Done, B., Carlson, C. A., and Giovannoni, S. J. (2013b). Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat. Microb. Ecol. 71, 1–13. doi: 10.3354/ame01661
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Webb, K. L., and Johannes, R. (1967). Studies of the release of dissolved free amino acids by marine zooplankton. Limnol. Oceanogr. 12, 376–382. doi: 10.4319/lo.1967.12.3.0376
Keywords: DOC, dissolved metabolites, diel vertical migration, biogeochemistry, copepod
Citation: Maas AE, Liu S, Bolaños LM, Widner B, Parsons R, Kujawinski EB, Blanco-Bercial L and Carlson CA (2020) Migratory Zooplankton Excreta and Its Influence on Prokaryotic Communities. Front. Mar. Sci. 7:573268. doi: 10.3389/fmars.2020.573268
Received: 16 June 2020; Accepted: 02 November 2020;
Published: 01 December 2020.
Edited by:
Hongbin Liu, Hong Kong University of Science and Technology, Hong KongReviewed by:
Taichi Yokokawa, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), JapanCopyright © 2020 Maas, Liu, Bolaños, Widner, Parsons, Kujawinski, Blanco-Bercial and Carlson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy E. Maas, YW15Lm1hYXNAYmlvcy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.