
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 21 April 2020
Sec. Marine Molecular Biology and Ecology
Volume 7 - 2020 | https://doi.org/10.3389/fmars.2020.00205
This article is part of the Research Topic Marine Environmental Epigenetics View all 14 articles
 Marie E. Strader*†
Marie E. Strader*† Logan C. Kozal
Logan C. Kozal Terence S. Leach
Terence S. Leach Juliet M. Wong
Juliet M. Wong Jannine D. Chamorro
Jannine D. Chamorro Madeline J. Housh
Madeline J. Housh Gretchen E. Hofmann
Gretchen E. Hofmann
Gene expression plasticity can confer physiological plasticity in response to the environment. However, whether epigenetic marks contribute to the dynamics of gene expression is still not well described in most marine invertebrates. Here, we explored the extent and molecular basis of intra- and intergenerational plasticity in the purple sea urchin, Strongylocentrotus purpuratus, by examining relationships between changes in DNA methylation, transcription, and embryo spicule length. Adult urchins were conditioned in the lab for 4 months to treatments that represent upwelling (∼1200 μatm pCO2, 13°C) and non-upwelling conditions (∼500 μatm pCO2, 17°C). Embryos spawned from conditioned adults were reared in either the same adult treatment or the reciprocal condition. Maternal conditioning resulted in significantly differentially methylated CpG sites and differential gene expression in embryos, despite no evidence of maternal effects on embryo spicule length. In contrast, conditions experienced during development resulted in significant differences in embryo spicule length. Intragenerational plasticity in spicule length was strongly correlated to transcriptomic plasticity, despite low levels of intragenerational plasticity in CpG methylation. We find plasticity in DNA methylation and gene expression in response to different maternal environments and these changes have similarities across broad functional groups of genes; yet exhibit little overlap on a gene-by-gene basis. Our results suggest that different forms of environmentally induced plasticity are observable across different time scales and that DNA methylation dynamics may be uncoupled from fast transcriptional responses to the environment and whole organism traits. Overall, this study illuminates the extent to which environmental differences can induce both intra- and intergenerational phenotypic plasticity in a common kelp forest herbivore.
Epigenetics, a suite of biochemical modifications to nucleic acids and proteins, has recently been highlighted as a potential mechanism by which environmental signals might modify transcription and thus drive phenotypic plasticity (Feil and Fraga, 2012). Detailed empirical work in plants indicates that epigenetic variation contributes to complex traits and can shape ecological and evolutionary processes (Richards et al., 2017). In addition, theoretical models suggest the timing and outcome of adaptive evolution can be impacted by epigenetic changes that are stable, assuming epigenetic mutations are heritable (Klironomos et al., 2013; Kronholm and Collins, 2016; Banta and Richards, 2018). In an ecological context, it has been proposed that environmentally responsive epigenetic marks could contribute to adaptive plasticity, a field now termed ‘environmental epigenetics’ (Johannes et al., 2009; Torda et al., 2017; Eirin-Lopez and Putnam, 2019). In order to explore this, it is critical to understand how environmentally induced trait-based, organismal changes may be causally linked to changes in DNA methylation.
Environmental epigenetics has been examined in few marine invertebrate species (Marsh and Pasqualone, 2014; Ardura et al., 2017; Gavery and Roberts, 2017; Gonzalez-Romero et al., 2017; Li et al., 2017; Dixon et al., 2018; Liew et al., 2018; Baums et al., 2019; Hawes et al., 2019). While epigenetics can encompass a variety of non-genetic mechanisms such as histone modifications and small non-coding RNAs, the majority of marine environmental epigenetics research has focused on DNA methylation, in which cytosines, typically in a CpG dinucleotide context, contain a methyl group (Eirin-Lopez and Putnam, 2019). In invertebrates, DNA methylation is confined to gene regions and associated with broad transcriptional patterns, potentially functioning in controlling spurious transcription or fine-tuning of the transcriptome as a whole (Riviere et al., 2013, 2017; Gavery and Roberts, 2014; Saint-Carlier and Riviere, 2015; Dixon et al., 2018; Liew et al., 2018). Epigenetic marks can arise by spontaneous epimutation during replication or repair, similar to mutation in the DNA sequence itself, and persist across generations within a population, or they can be induced to change in response to the environment (Richards, 2008; Shea et al., 2011). Both population-specific and environmentally inducible DNA methylation are evident in reef-building corals (Dixon et al., 2018; Liew et al., 2018). In response to environmental cues, changes in DNA methylation may link to effects on gene expression that could drive phenotypic plasticity in a manner that could be beneficial or deleterious, although key questions remain on mechanisms that may promote these changes across generations, which are illustrated in Eirin-Lopez and Putnam (2019). If shifts in environmentally induced gene expression can occur from one generation to the next, this could be ecologically significant as organisms “tune” to their respective, dynamic environments. In S. purpuratus, there is evidence of intergenerational plasticity in DNA methylation (Strader et al., 2019), indicating a potential link between DNA methylation and phenotypic plasticity in response to environmental changes.
Strongylocentrotus purpuratus are ecologically and environmentally important grazing herbivores in kelp forests (Pearse, 2006). In this ecosystem, the California Current System (CCS) is characterized by considerable variation in temperature, dissolved oxygen, and pH both latitudinally and through upwelling events (Feely et al., 2008; Chan et al., 2017). The dynamics of upwelling are likely to intensify with anthropogenic global change (Sydeman et al., 2014; Bakun et al., 2015), which will occur simultaneously with increased sea surface temperatures and the rapid progression of ocean acidification (Gruber et al., 2012). In the Santa Barbara Channel, long-term oceanographic sensor data have greatly informed the abiotic dynamics of these systems, giving insight into the water conditions within kelp forest ecosystems and how they have changed through time (Kapsenberg and Hofmann, 2016; Rivest et al., 2016; Hofmann and Washburn, 2019; Hoshijima and Hofmann, 2019). These data show seasonal and spatial variability in abiotic conditions, often as a result of upwelling events that bring cold, low pH waters from offshore. Upwelling can occur throughout the year, with events typically reaching their peak frequency, duration and intensity during the late winter and spring months (Kapsenberg and Hofmann, 2016; Hoshijima and Hofmann, 2019). These events can vary in length from days to weeks and are interspersed with relaxed, non-upwelling conditions characterized by ambient pH levels and temperatures. This knowledge of the natural abiotic environment is valuable for understanding the relationships between the environment and resident organisms, facilitating the development of ecologically relevant experiments to examine inter- and intragenerational plasticity.
The combination of the dynamic nature of the CCS and S. purpuratus phenology may lead to variable and complex organism–environment interactions. The peak upwelling season overlaps with the reproductive cycle of S. purpuratus populations near the Santa Barbara coast, which typically undergo gametogenesis during the fall and winter before spawning in the months between January and July (Strathmann, 1987). Therefore, adult S. purpuratus can experience upwelling conditions during gametogenesis, and depending on the timing in which spawning occurs, early development of S. purpuratus embryos and larvae is likely to occur during upwelling as well. Furthermore, because upwelling events vary in their duration, the sudden appearance or disappearance of an event will influence the environmental conditions adults and their offspring experience respectively. Simulating upwelling or relaxed, non-upwelling conditions that S. purpuratus experience in their kelp forest environment within a laboratory setting is therefore highly pertinent for understanding and predicting inter- and intragenerational plasticity in nature.
Previous studies demonstrate both intra- and intergenerational plasticity in S. purpuratus in response to pCO2 and temperature conditions associated with coastal upwelling. When S. purpuratus embryos and larvae developed under different pCO2 levels, there was substantial intragenerational plasticity in body size (Padilla-Gamiño et al., 2013; Kelly et al., 2016). Some forms of intergenerational plasticity are evident as well. When adult S. purpuratus were acclimated to different temperature and pCO2 regimes during gametogenesis in the lab and in the field, there were maternal effects on either egg protein and lipid content, larval body size, gene expression, or DNA methylation (Wong et al., 2018, 2019; Hoshijima and Hofmann, 2019; Strader et al., 2019). While these previous studies on purple urchins provided valuable insight into the potential for inter- and intragenerational plasticity, the knowledge of how gene expression and DNA methylation interact within this system has yet to be elucidated.
Given previous work on inter- and intragenerational plasticity in urchins, we hypothesized that variation in DNA methylation patterns would be associated with variation in the transcriptome, either within or across generations, in response to end-member upwelling index conditions naturally experienced in kelp forest ecosystems. We measured the larval methylome and transcriptome in response to two different pCO2 and temperature regimes experienced both during gametogenesis of the mothers and embryonic development of the offspring. In addition to assessing molecular-level changes that were induced by abiotic conditions, we measured embryonic spicule length as an organismal-level trait that might also vary with the experimental treatments. We found that variation in gene expression was largely driven by differences in developmental environments while variation in DNA methylation was largely driven by differences in maternal environments. Overall, this study identified plasticity of the transcriptome and the methylome as a result of environmental differences, but across different time scales, and maternal effects on DNA methylation and transcription likely target modulation of post-transcriptional and translational processes in the cell.
Adult purple sea urchins (Strongylocentrotus purpuratus, N = 83) were collected on September 20, 2017 off of Goleta Beach, CA, United States (34° 24.840′ N, 119° 49.742′ W), at a subtidal site near the University of California Santa Barbara (UCSB) under California Scientific Collection permit SC-1223. Urchins were transported to UCSB where they were held in flow-through seawater tables at ambient seawater conditions for 6 days. After this initial acclimation period, urchins were randomly assigned to one of two acclimation treatments, upwelling (U: 1390 ± 307 μatm pCO2 and 12.7 ± 0.5°C) or non-upwelling (N: 631 ± 106 μatm pCO2 and 16.8 ± 0.2°C) (Table 1 and Supplementary Figure 1). These conditions represent end points of those experienced in their natural environment (Kapsenberg and Hofmann, 2016; Rivest et al., 2016). Each treatment was replicated across three 20-gal flow-through aquarium tanks with each holding 12 urchins. Conditioning of adults was maintained for just over 4 months and animals were fed locally collected kelp (Macrocystis pyrifera) once a week.
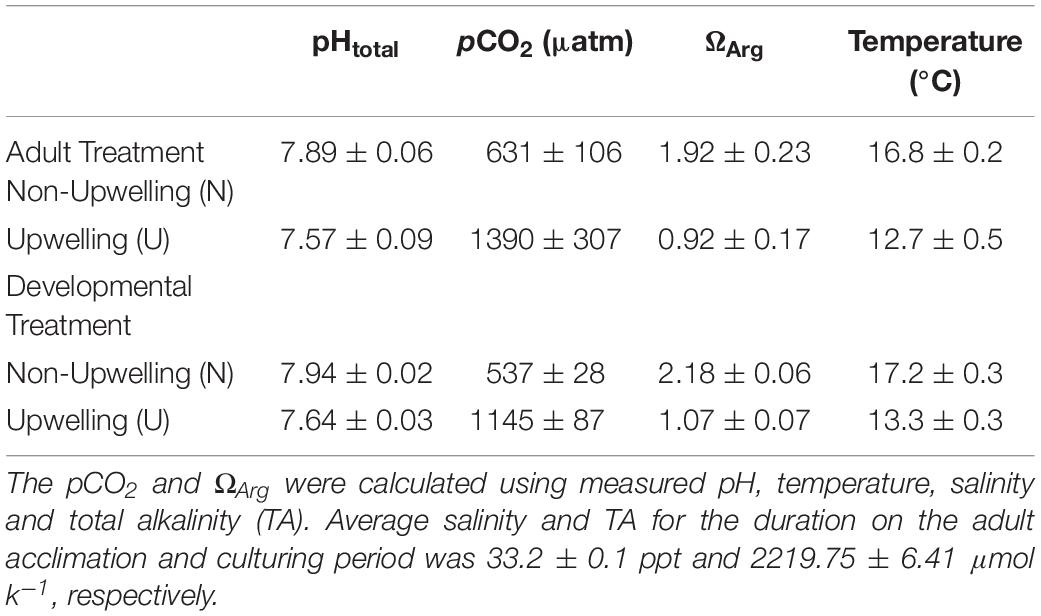
Table 1. Carbonate chemistry parameters for adult (U and N) and developmental (U and N) conditions.
Adult acclimation conditions were controlled by a flow-through CO2-mixing system (modified from Fangue et al., 2010) and Delta Star heat pumps regulated by Nema 4x digital temperature controllers (AquaLogic San Diego, CA, United States). Each treatment tank was fed water from two 20-gal glass reservoir tanks supplied with 0.35 μm-filtered, UV sterilized seawater (FSW) with a flow rate of 10 L/h (20 L/h total into each tank). Target pCO2 levels were maintained in the reservoir tanks using mass-flow controllers (Sierra Instruments, United States) that mixed pure CO2 with dry air that was filtered and CO2 scrubbed.
Seawater chemistry measurements in each tank were taken every 1–2 days throughout the adult acclimation period (Table 1 and Supplementary Figure 2). Temperature and salinity were measured daily using an Omega HH81A thermocouple and YSI-3100 conductivity meter, respectively. pH was measured using a UV spectrophotometer (Shimadzu UV-1800) and m-cresol purple indicator dye (Sigma Aldrich) according to standard operating procedure (SOP) 6b (Dickson et al., 2007). Total Alkalinity (TA) was measured by titration following SOP 3b (Dickson et al., 2007). Water samples for total alkalinity were preserved prior to titration with 0.02% saturated mercuric chloride. CO2 calc (Robbins et al., 2010) was used to calculate the carbonate chemistry parameters pCO2, Ωara, and Ωcal with the equilibrium constants from Hansson (1973) refit by Dickson and Millero (1987; Table 1).
On January 30th, 2018 spawning of the acclimated adult urchins was induced by injection of 0.53 M KCl into the coelom. Sperm was collected by pipetting from the area around the gonopores of male urchins kept out of water and stored dry in 1.5 mL tubes on ice until activation. Eggs were collected from each female by inverting the female over a beaker filled with FSW so that the gonopores were submerged. Eggs were checked visually for quality under a microscope and counted. Females with eggs of abnormal shape or color or significant numbers of eggs containing visible germinal vesicles were excluded. Sperm from potential males was activated in FSW, checked for motility, and then used to fertilize a subset of eggs from each female to ensure male-female compatibility. Eggs from 9 females per treatment (N or U) were pooled in equal numbers and fertilized with the sperm from one high quality non-upwelling conditioned male (Supplementary Figure 1). As this study focused solely on maternal contributions to intergenerational plasticity, one male was used to sire all offspring to minimize the genetic diversity that might generate noise in the analysis. As such, all larvae in the experiment were a mix of full and half siblings. Dilute, activated sperm was slowly added to each pool of eggs until at least 95% fertilization was reached. The embryos from each treatment pool were divided into 3 non-upwelling culture vessels (N: 537 ± 28 μatm and 17.2 ± 0.3°C) and 3 upwelling culture vessels (U: 1145 ± 87 μatm and 13.3 ± 0.3°C) (Table 1 and Supplementary Figures 1, 2) in a flow-through seawater system for a final concentration of 10 embryos/mL (120,000 embryos/vessel). Therefore, embryos were raised either in the same treatment as the maternal acclimation or the reciprocal condition. This yielded four treatment groups, each with 3 replicate culture vessels: embryos from mothers conditioned to non-upwelling raised under non-upwelling (NN), embryos from mothers conditioned to non-upwelling raised under upwelling (NU), embryos from mothers conditioned to upwelling raised under non-upwelling (UN), embryos from mothers conditioned to upwelling raised under upwelling (UU).
Temperature and pCO2 conditions under which the embryos developed were maintained with the same CO2 mixing system and heat pumps used in the adult acclimation. Two reservoir tanks fed 6 culture vessels per treatment (non-upwelling or upwelling developmental conditions) at a rate of 6 L/hr using irrigation drippers (DIG Corporation). Each culture vessel consisted of two nested buckets with a 12L capacity in which the inner bucket had holes covered with 30 μm mesh, allowing water to flow from the inner to the outer bucket and overflow without losing any embryos. Flow-through and mixing was aided in each bucket by an acrylic paddle attached to a 12V motor. Following the same methods outlined for the adult acclimation treatments, temperature and pH were measured daily for each culture vessel to confirm uniform conditions across each treatment. Salinity and TA were measured daily as well (Table 1 and Supplementary Figure 2). Embryos were cultured in their respective treatments until the prism stage, an early echinopluteus larval stage, at which point they were sampled to assess morphometrics, DNA methylation, and transcription.
The prism stage was defined by the archenteron merging to one side of the body and becoming tripartite, the first development of skeletal rods, and a pyramid-like body shape. Prism larvae were preserved for morphometric analysis by adding 4% formalin buffered with 100mM NaBO3 (pH 8.7) to an equal volume of larvae and FSW for a final concentration of 2% formalin. Samples of 1000 larvae were stored at 4°C until measured. Larvae were photographed on a compound microscope (Olympus BX50) with an attached digital camera (Motic 10 MP) using the Motic Images Plus software. Prism larvae were photographed from a lateral view where both the tip of the body rod and postoral rod were in focus. Photographs were calibrated to a stage micrometer. Spicule length was measured as the length from the tip of the body rod to the branching point of the postoral rod (N = 35 larvae/culture vessel) (Supplementary Figure 4). The effect of each fixed factor (maternal condition or developmental condition) on spicule length was examined using a 2-way ANOVA.
Genomic DNA was extracted using an established CTAB protocol involving phenol chloroform purification. One sample per culture vessel was prepared for sequencing to generate 12 total samples (3 per treatment × 4 treatments). Each sample contained a pool of 6,000 larvae. Therefore, for each treatment, three replicates of 6,000 larvae were extracted and subsequently sequenced, totaling 18,000 larvae per treatment. In the case of the UU treatment, a missing sample from UU2 led us to process two unique pools (6,000 larvae each) of the UU3 culture. Integrity and quality of genomic DNA was assessed using gel electrophoresis and quantified using a Qubit fluorometer 3.0 (Life Technologies). Genomic DNA (∼2 μg per sample) was sent to the UC Davis Genome Center for RRBS library prep using the Diagenode Premium RRBS kit and sequencing on the Illumina 4000 with 50 bp single end reads across one lane of sequencing. Raw fastq sequences were trimmed and filtered using TrimGalore (V0.5.0)1, which utilizes cutadapt (V1.9.1) (Martin, 2011), specifying the -rrrbs option that trims one additional bp from the 3′ end of the read as is recommended for RRBS data. This trimming package removes low quality base pairs from the 3′ end of the read (Phred score = 20), Illumina adaptors and short reads. Read quality was visualized using FastQC. Bisulfite treated trimmed reads were mapped to a bisulfite converted genome [Strongylocentrotus purpuratus genome (V3.1)] using Bismark (V0.19.1) (Krueger and Andrews, 2011). Methylation output files were produced by running the bismark_methylation_extractor command and summary files were produced by the bismark2summary command.
Bismark coverage files were used for the subsequent analysis in the R environment (V3.4.2). Statistical analysis of CpG data was run using the package methylKit (Akalin et al., 2012). Samples were filtered by read coverage where base pairs with less than 10X coverage were removed as well as base pairs in the highest percentile (99.9). CpG sites were merged across samples into a methylBase object where base-pair locations were the same across all samples using the unite function. Two independent tests were run to identify differential methylation at each CpG site (DMCpG), one to determine DMCpGs by maternal condition (upwelling vs. non-upwelling) and a second by developmental condition (upwelling vs. non-upwelling). DMCpGs were identified using a logistic regression and significance was determined using p-values adjusted to q-values using a sliding linear model method (SLIM) (Akalin et al., 2012). A CpG was considered significantly differentially methylated (DMCpG) if the q-value < 0.01 and the difference in percent methylation was >25%. Multivariate analyses were performed using percent methylation values for each CpG site. Principal coordinates analysis (PCoA) using Manhattan distances was performed using the packages adegenet (Jombart, 2008) and vegan (Oksanen et al., 2013). To determine the proportion of variance explained by fixed factors a permutational multivariate ANOVA was run using the function adonis.
CpG annotation to genome features was performed using a custom R script. A CpG site was considered methylated if its median value across all samples was >1 (the site was methylated in over half of the samples). To examine percent methylation values per gene, genes with sequenced CpGs were filtered for those with at least 5 representative CpG sites within the gene region. The remaining genes had an average of 10.3 total CpGs represented within the gene region and percent methylation was calculated for each gene (methylated CpG sites/total CpG sites). Gene Ontology (GO) functional enrichment was performed using a Fisher’s Exact test on genes that contained at least one DMCpG for either experimental treatment (maternal or developmental) compared to all genes using the GO_MWU package (Wright et al., 2015).
Total RNA was extracted using TRIzol reagent (Invitrogen). One sample per culture vessel was prepared for sequencing to generate 12 total samples (3 per treatment X 4 treatments). Each sample contained a pool of 6,000 larvae. RNA quality and quantity were assessed using the Agilent TapeStation and Qubit fluorometer 3.0 (Life Technologies), respectively, and all samples had RIN values >8.0. Total RNA (∼2 μg per sample) was submitted to the UC Davis Genome Center for poly A enriched, strand specific RNA library preparation and sequencing on the Illumina 4000 with 50bp single end reads across one lane of sequencing. Raw sequences were filtered for quality and adaptor sequences were discarded using TimGalore (V0.5.0)1 which utilizes cutadapt (V1.9.1) (Martin, 2011). Trimmed reads were mapped to the Strongylocentrotus purpuratus genome (V3.1) using hisat2 (V2.0.0) (Kim et al., 2015). Gene counts were determined using featureCounts, a component of the subread package (V1.6.2) (Liao et al., 2014).
All statistical analyses were performed in R (V3.4.2). The package arrayQualityMetrics was used to identify potential sample outliers (Kauffmann et al., 2009). Gene normalization and models for differential gene expression analysis were performed using DESeq2 (Love et al., 2014). A Wald test [design = formula(∼ treat_maternal + treat_dev)] followed by pair-wise contrasts was run to identify differentially expressed genes (DEGs) associated with either maternal condition (upwelling vs. non-upwelling) or developmental condition (upwelling vs. non-upwelling). Multivariate analyses were performed using regularized log transformed gene count data. Principal coordinates analysis (PCoA) using Manhattan distances was performed using the packages adegenet (Jombart, 2008) and vegan (Oksanen et al., 2013). To determine the proportion of variance explained by fixed factors a permutational multivariate ANOVA was run using the function adonis.
Using only highly expressed genes (15,242 genes, mean counts >10), regularized log transformed counts data were used for a weighted gene co-expression network analysis (WGCNA) (Langfelder and Horvath, 2008). WGCNA analysis clusters genes in a manner that is un-biased toward the experimental design. A similarity matrix of gene expression was calculated using Pearson correlations for all gene pairs across samples and a “signed” network was specified, which allows the direction of the expression change to be retained. Using a power adjacency function and a soft thresholding power of 9, connectivities were transformed from the expression correlations. Genes were hierarchically clustered based on topological overlap which identifies groups of genes, or modules, whose expression covaried across samples. The minimum module size was set to 30 and similar modules were merged when their module eigengenes were highly correlated (R > 0.80). Experimental conditions and physiological larval trait data were then correlated to expression modules (Supplementary Figure 5). Gene Ontology (GO) enrichment analysis was performed for each module using a Fisher’s Exact Test, by presence or absence of each gene within a module using the GO_MWU package (Wright et al., 2015).
In order to examine how plasticity in the methylome and transcriptome correlate to a plastic, organismal trait, we measured the effect of different conditions on spicule length, a trait associated with larval fitness that is known to be sensitive to high pCO2 but not to temperature differences within the range manipulated in this experiment (Byrne et al., 2013; Padilla-Gamiño et al., 2013; Wong et al., 2018). For early life-history stages of echinoderms, the larval skeleton begins to form just following gastrulation (Okazaki, 1975) and is contingent on sufficient Ωara, and Ωcal to support biogenic calcification (Byrne et al., 2013). We found strong evidence that spicule length varied between development treatments (pANOVA < 0.001), but no evidence for intergenerational plasticity (Supplementary Figure 4). Specifically, developing under upwelling conditions (higher pCO2 and lower temperature) reduced average spicule length by 30.8% relative to those that developed under non-upwelling conditions. Thus, in the case of this organismal trait, we observed that abiotic conditions experienced during development were more influential than the environmental history of the mothers during gametogenesis.
To identify the potential for ecologically relevant abiotic conditions to induce changes in DNA methylation, either in an inter- or intragenerational plasticity context, we characterized the larval methylome using RRBS sequencing. DNA methylation sequencing yielded an average of 24,800,247 reads per sample across 12 samples (Supplementary Table 1), with an average bisulfite conversion rate of 98.92%. After adaptor trimming, read mapping to the bisulfite converted genome resulted in an average of 38.13% uniquely mapped reads and 23.44% ambiguously aligned reads (Supplementary Table 1). Bismark revealed 164,062,595 ± 9,335,444 SE total cytosine calls across all sequenced samples. Of these total calls, three different types of cytosine methylation were quantified, CpG (32,803,060 ± 1,885,036 SE), CpH (26,641,240 ± 1,496,753 SE), and CHH (104,619,294 ± 5,954,888 SE). Of these methylation calls, an average of 22.12% of CpGs were methylated, with 1.72% and 1.41% of CpHs and CHHs methylated, respectively (Supplementary Table 1). After filtering low coverage and high coverage CpG sites, read coverage per CpG site was 30.26 ± 0.962 SE and percent methylation per base was 22.95% ± 0.3957 SE (Supplementary Table 1). After filtering and sample merging, 245,343 CpG sites were shared across all samples, which were used for subsequent analysis.
Using this set of shared CpG sites, we sought to identify whether there were significant changes in the methylome associated with maternal or developmental conditioning. Logistic regression models revealed multiple differentially methylated CpGs. In the context of maternal effects, models found 684 DMCpGs total, with 328 hypermethylated and 356 hypomethylated sites in the methylome when mothers were conditioned in upwelling conditions compared to non-upwelling (Supplementary Figure 3). With regard to developmental conditioning, models found 216 DMCpGs total, with 175 hypermethylated and 41 hypomethylated sites when larvae developed in upwelling conditions compared to non-upwelling conditions (Supplementary Figure 3). Only 13 of these DMCpGs were significant for both maternal and developmental conditioning of which the majority were hypermethylated with regards to developmental conditioning and hypomethylated with regards to maternal conditioning (Supplementary Figure 3).
Principal coordinates analysis revealed that variation in maternal conditioning separated the samples along PCo1 while developmental conditioning separated samples along PCo2 (Figure 1A). A permutational multivariate ANOVA revealed that 12.8% (p < 0.001) of the variance in percent CpG methylation was explained by maternal condition while 9.7% (not significant) of the variance was explained by developmental condition (Figure 1C). The interaction between these two factors explained 8.5% of the variance, but was non-significant (Figure 1C). However, the majority of variance was not explained by any of the factors in the model, an outcome that is similar to methylation work in corals (Dixon et al., 2018; Baums et al., 2019), suggesting this remaining epigenetic variance may be driven by genetics or spontaneous epimutations. In summary, we found that maternal conditioning, the abiotic conditions that female urchins experienced during gametogenesis, resulted in more pronounced changes in the methylome than when embryonic development occurred in different abiotic conditions.
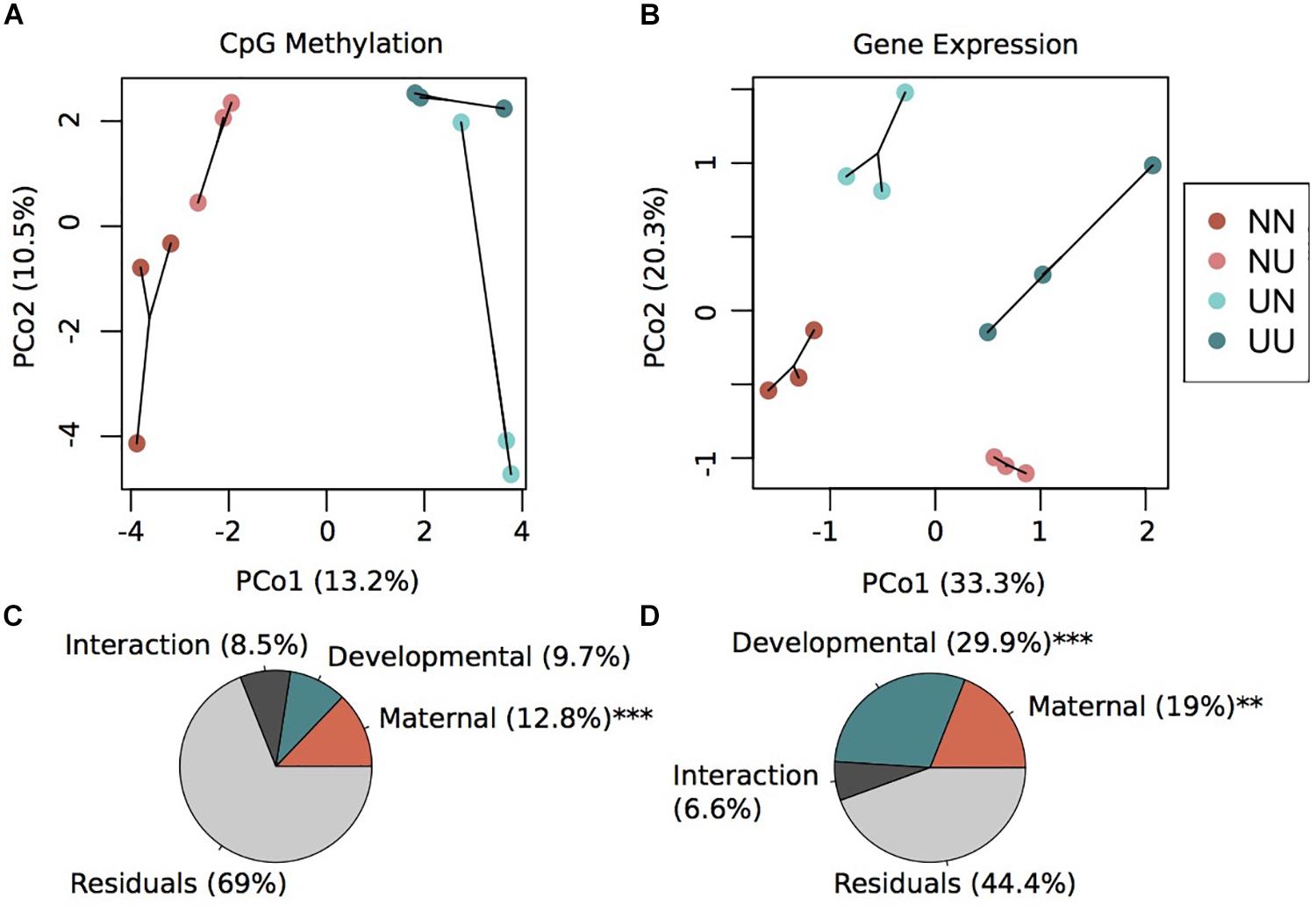
Figure 1. Environmental conditions induce changes in gene expression and DNA methylation. Principal coordinate analysis of CpG methylation (A) and gene expression (B). Variance explained by each principal coordinate is specified in the axes labels. Sample labels denote the maternal environment first and the developmental environment second. N, Non-Upwelling, U, Upwelling. Percentage of variation in CpG methylation (C) and gene expression (D) explained by fixed factors of maternal or developmental rearing environment using a permutational multivariate ANOVA. ***p < 0.001 and **p < 0.01.
In order to examine relationships between DMCpGs, transcription and environmental conditioning, we characterized gene bodies containing DMCpG sites. Research on DNA methylation in invertebrates has highlighted patterns between gene body methylation (GBM) and broad transcriptional patterns (Gavery and Roberts, 2013; Dixon et al., 2014, 2018). Therefore, we sought to identify genes with DMCpGs in comparisons of both maternal and developmental conditions. In addition, we performed functional enrichment tests, Gene Ontology (GO), to see if broad functional categories were over-represented in those genes. A total of 95,197 CpG sites were located within gene regions across 9,219 total genes and are further designated as gene specific CpGs (GS-CpGs). Across GS-CpG sites there was a strongly bimodal pattern in the percentage to which each site was methylated (Supplementary Figure 6A). For each GS-CpG site, we considered the site methylated if the site had methylated reads in over half of the samples (median > 1), a threshold that resulted in 41,940 methylated GS-CpG sites. Genes were filtered for only those that had at least five GS-CpG sites sequenced per gene, resulting in a total of 5,024 genes. Percentage methylation per gene also showed a bimodal pattern, with the majority of represented genes being 100% methylated (Supplementary Figure 6B).
We found a small fraction of total genes that contained DMCpGs. A total of 136 genes contained at least 1 DMCpG for maternal condition while 49 genes contained at least 1 DMCpG for developmental condition. Overall, a total of 258 DMCpGs were also GS-CpGs across 177 unique genes (Supplementary Table 2). Notably, the majority of these 177 genes include those that function in protein modification including hydrolase activity (Sp-Vps35, Sp-Abhd3, Sp-Abhd3, Sp-Asah2L3), protein phosphorylation (Sp-Dusp6/7/9, Sp-Mekk4a, Sp-Melkl, Sp-Frap_1, Sp-Ck1g), and ubiquitination (Sp-Ube3cL, Sp-Uba1, Sp-Traf3). In addition to genes with protein modification processes, there were also genes involved in binding (Sp-Myef2, Sp-Z371, Sp-Z163, Sp-Ciz, Sp-Traf3, Sp-Pcbp2) and modification of nucleic acids (Sp-Endrvt33, Sp-Tsen34L_1). Cytoskeletal components such as myosin and kinesin are also represented across all genes containing DMCpGs (Sp-Non/MmyIIhcph, Sp-Unk_109, Sp-Krp85_1, Sp-Kif13A2, Sp-Kif1a, Sp-Kif13BL) as well as DNA binding proteins such as transcription factors (Sp-FoxK, Sp-Mlxip, and Sp-Myc) (Supplementary Table 2). GO enrichment tests were run on genes with DMCpGs for maternal condition, developmental condition, and all genes containing a DMCpG. Only tests for maternal DMCpG genes and all genes yielded significant GO enrichments, with the majority of the terms being the same (Supplementary Table 3). Among all genes with DMCpGs, there was significant GO enrichment of terms that reflect functions mentioned above including endonuclease activity (GO:0004519), endopeptidase activity (GO:0004175), DNA metabolic processes (GO:0006259), and kinesin complex (GO:0005871), among others (Supplementary Table 3). It should be noted that due to the low representation of genic CpGs in our RRBS data, we are missing what could be key information about CpG methylation in gene regions. Therefore, future studies should further examine CpG methylation across gene regions using a whole genome bisulfite sequencing approach. However, this dataset reveals what could be important genes and functional categories associated with DNA methylation in sea urchins. In summary, we identified multiple genic CpGs that varied by environmental context, particularly maternal, with functions indicative of DNA integration, modification and turnover of proteins and nucleic acids as well as cytoskeletal components.
To further examine potential connections between environmentally induced changes in DNA methylation and plastic phenotype, such as spicule length, we also sequenced transcriptomes of prism stage embryos. RNA sequencing yielded an average of 29,722,500 reads per sample across 12 samples (Supplementary Table 1). After size and quality trimming, an average of 29,713,377 reads per sample remained. Trimmed reads were mapped to the Strongylocentrotus purpuratus genome (V3.1) with an average mapping efficiency of 62.1%. Mapped reads were converted to gene counts across 30,284 gene meta-features with an average of 7,897,000 counts per sample (Supplementary Table 1). The outlier detection package, arrayQualityMetrics, did not detect any sample outliers so all were retained. Genes with low mean counts (less than 3) were discarded from further analysis, leaving 18,015 genes total.
DESeq2 models revealed significant differentially expressed genes for both maternal and developmental conditions. Differences in maternal environment resulted in 1,811 DEGs while developmental environment resulted in 3,765 DEGs (FDR < 0.05), with 645 DEGs overlapping between the two factors (Supplementary Figure 3). PCoAs of gene counts revealed strong differences in gene expression between each of the four treatments where developmental condition separated variation along PCo1 and maternal condition separated variation along PCo2 (Figure 1B). A permutational multivariate ANOVA revealed that 29.9% (p = 0.001) of the variation in gene expression was explained by developmental condition while 19% (p = 0.002) was explained by maternal condition (Figure 1D). The interaction between the two factors explained 6.6% of the variation, although this was not significant. Overall, a large proportion of the prism-stage transcriptome was responsive to different environmental conditions; this is not likely due to developmental delay as staging was performed using developmental cues not body size.
To characterize environmentally responsive transcriptional changes, we performed a weighted gene co-expression network analysis (WGCNA). WGCNA revealed 12 gene expression modules with all showing significance for at least one experimental treatment or physiological trait (Supplementary Figure 5). Each module contained between 2,676 (turquoise) and 84 genes (royalblue) and the majority of modules showed significant enrichment of GO categories. Significant modules were binned as either strongly representative of changes due to developmental condition (turquoise, pink, red, black, and yellow) (Figure 2) or maternal condition (lightyellow, cyan, royalblue, lightgreen, blue, and red) (Figure 3). There was a trend for maternal modules to have a higher percentage of genes with DMCpGs in them, although this trend was not significant (Supplementary Figure 5). The red module represents genes that show significant correlations for both experimental treatments and therefore represents interaction genes, however, there was no significant enrichment of GO terms within this module (Supplementary Table 4).
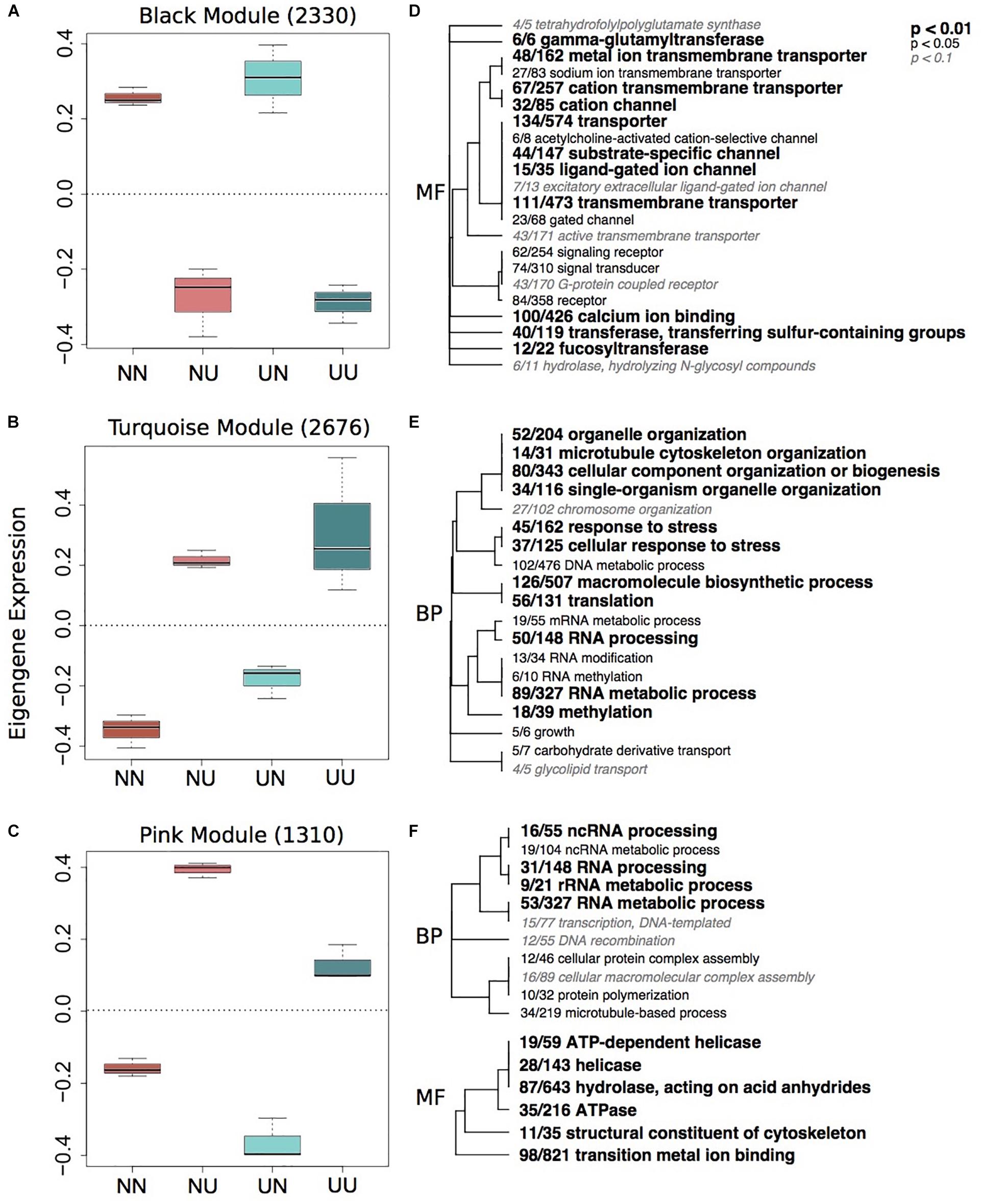
Figure 2. Gene expression modules strongly correlated to developmental environment. Boxplots of eigenegene expression for the black (A), turquoise (B), and pink (C) modules. The number of genes represented in each module is in parentheses. Hierarchical clustering of Gene Ontology (GO) terms showing significant enrichment in the black (D), turquoise (E) and pink (F) modules. Level of significance indicated with bold text. BP, biological processes; MF, molecular function.
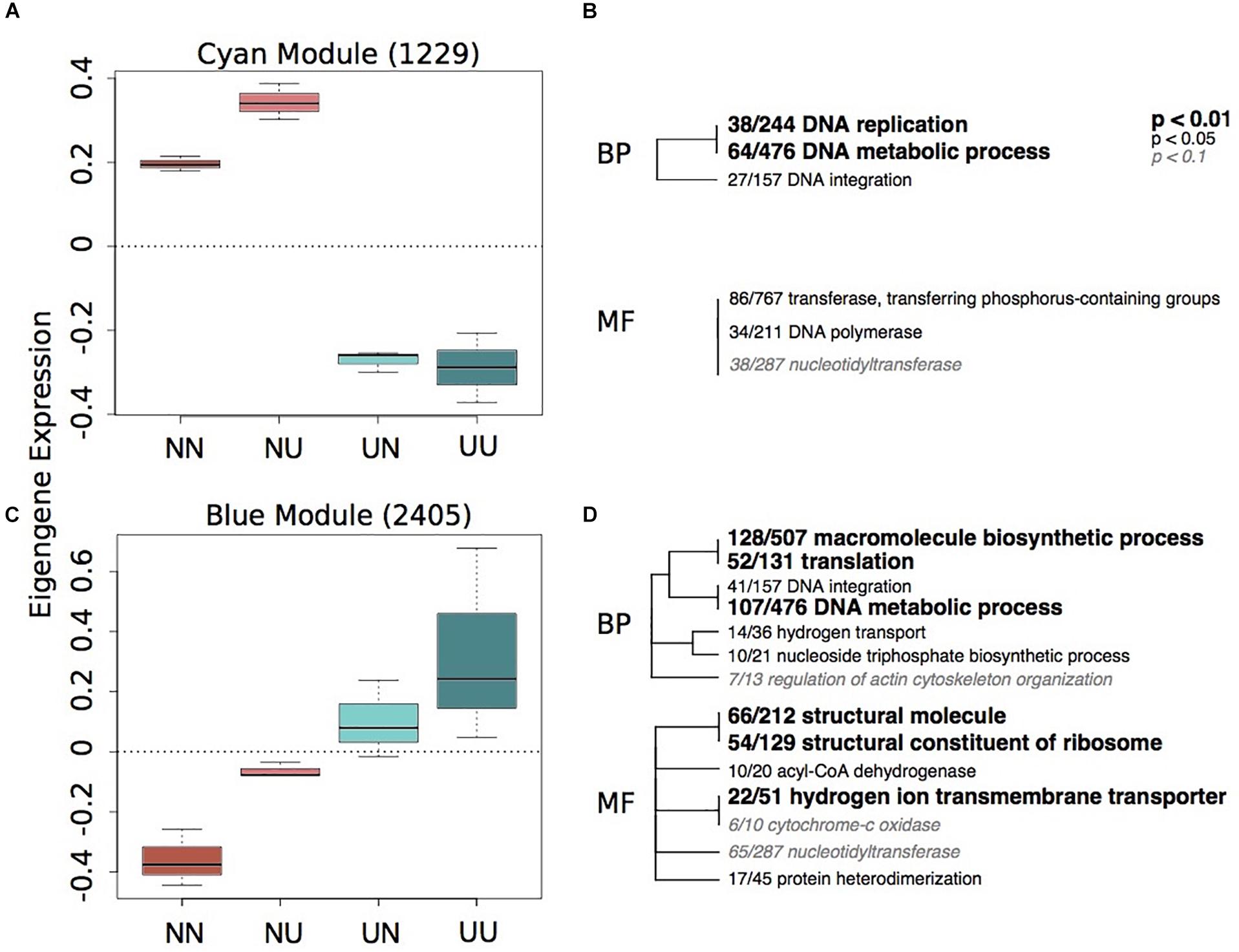
Figure 3. Gene expression modules strongly correlated to maternal environment. Boxplots of eigenegene expression for the cyan (A) and blue (C) modules. The number of genes represented in each module is in parentheses. Hierarchical clustering of Gene Ontology (GO) terms showing significant enrichment in the cyan (B) and blue (D) modules. Level of significance indicated with bold text. BP, biological processes; MF, molecular function.
Developmental modules include those with genes that were up-regulated when embryos were reared in upwelling as compared to non-upwelling conditions (turquoise and pink, Figure 2) and those with genes that were down-regulated when reared in upwelling as compared to non-upwelling (black, Figure 2). The turquoise and pink modules show enrichment of GO categories associated with RNA processing, methylation and metabolic processes, as well as cellular response to stress and growth. In contrast, genes in the black module show larvae reared under upwelling conditions down-regulated multiple neurological and signaling processes, suggesting these physiological processes are suppressed under upwelling stress. Calcium ion binding genes were also reduced when larvae were reared in upwelling conditions, which correlate strongly with the noticeable reduction in spicule length of these embryos (Supplementary Figure 4). The yellow module showed no significant enrichment of GO terms (Supplementary Table 4).
Of the modules associated with differences in maternal environment, only two of them showed significant enrichment of GO terms, the cyan and blue modules (Figure 3). The cyan module represents genes down-regulated in larvae when mothers were reared in upwelling conditions and includes changes in DNA replication and metabolic processes (Figure 3B). The blue module represents genes upregulated in larvae when mothers were reared in upwelling conditions and also includes GO categories associated with DNA metabolism, translation and hydrogen ion transport.
Due to environmental effects on DNA methylation, transcription, and spicule length, we further examined patterns that might indicate causality. Percent methylation values per gene were associated with overall gene expression. There was a strong significant correlation between mean gene expression counts and percent methylation (Adjusted R2 = 0.1836, plm < 2.2e–16) (Supplementary Figure 7). A total of 3,359 genes had >5 sequenced CpG sites and gene expression counts >3. However, there was little association between differences in GS-CpG methylation and differential expression of genes (Figures 4, 5). Differential gene expression was evident in both genes with low methylation as well as high methylation (Figure 4); however, DEGs showed higher log2FC differences amongst lowly methylated genes, particularly when considering differences in maternal treatment (Figure 4B). Genes with DMCpGs were almost exclusively those with high methylation, regardless of experimental treatment (Figure 4). There was a significant negative relationship between developmental DEGs and percent methylation; lowly methylated genes were more likely to be upregulated when larvae were reared under upwelling conditions (R2 = 0.11, plm < 0.001, Figure 4A).
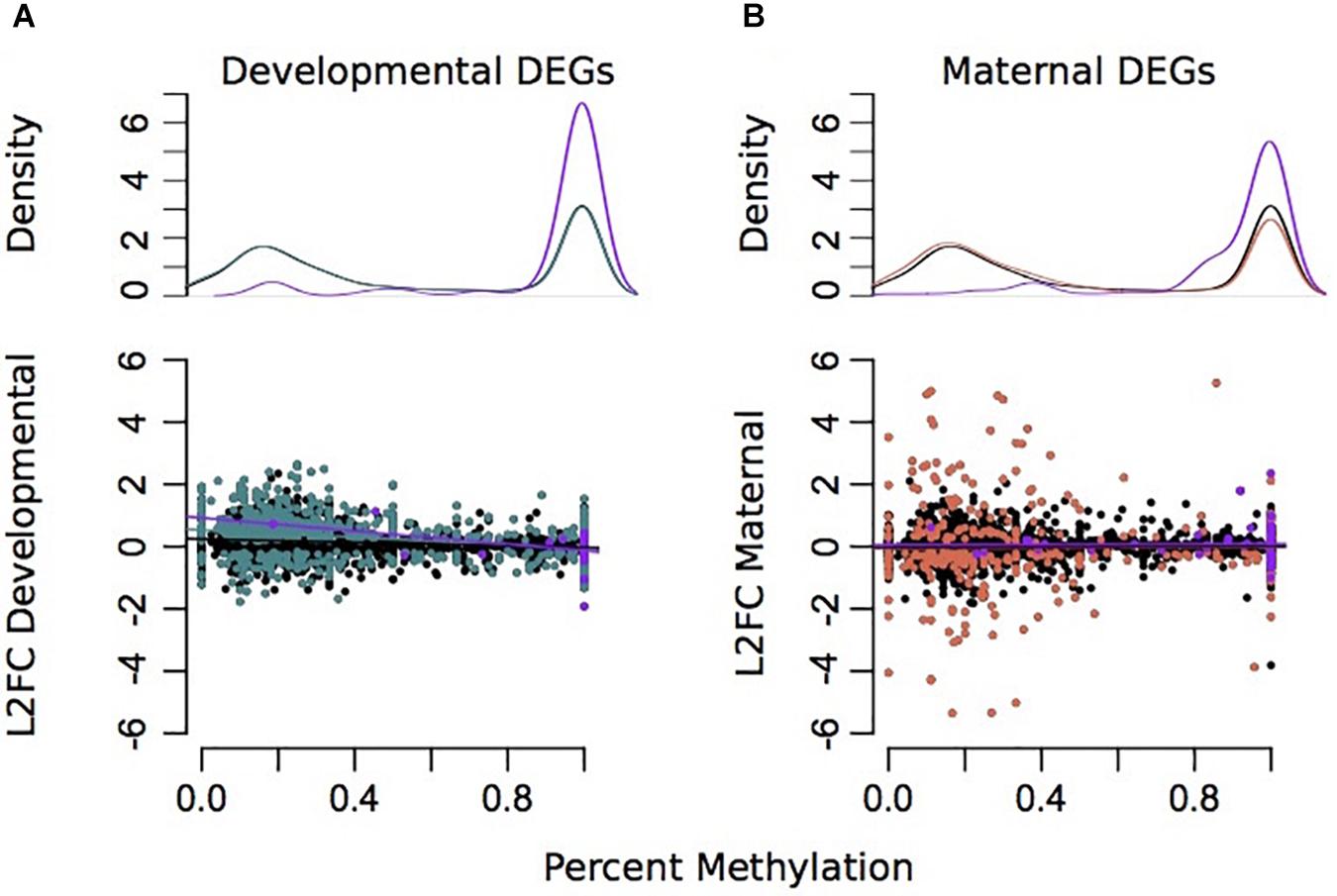
Figure 4. Associations between differential gene expression and percent methylation for comparisons between developmental (A) and maternal (B) environments. The Y-axis represents the log2 fold change for each comparison. The X-axis represents the percent methylation for each gene. Gray dots represent each gene. Turquoise genes represent DEGs by developmental environment (FDR < 0.05) and coral genes represent DEGs by maternal environment (FDR < 0.05). Purple genes denote DEGs with differentially methylated CpGs.
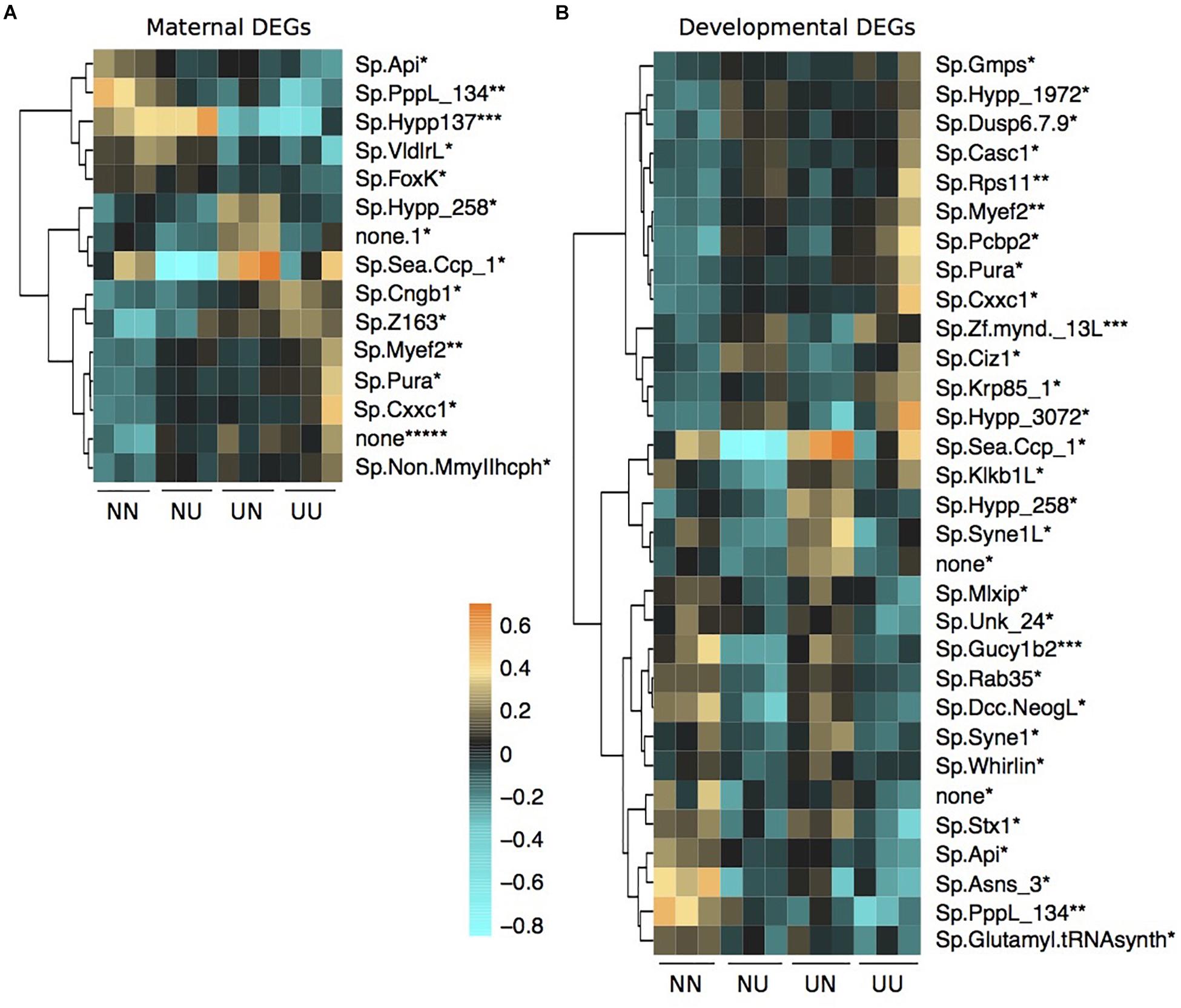
Figure 5. Differentially expressed genes by maternal environment (A) and developmental environment (B) containing differentially methylated CpGs. Asterisks by each gene name denote the number of DMCpGs within that gene region. Scale is log2fold change relative to the gene mean.
There was some overlap between genes with DMCpGs and DEGs from the DESeq2 models in response to the treatments (Figure 5). Of the 136 genes with maternal DMCpGs, 12 were differentially expressed with respect to maternal environment and 22 were differentially expressed with respect to developmental environment (Figure 5 and Supplementary Table 5). Of the 49 genes with developmental DMCpGs, 5 were differentially expressed with respect to maternal environment and 12 were differentially expressed with respect to developmental environment (Figure 5 and Supplementary Table 5). Overall, very few individual genes exhibited both differential expression and DMCpGs, suggesting it is unlikely that changes in DNA methylation at the level of gene bodies impart regulatory control on transcription, at least across the time frames examined in this study.
Intragenerational and intergenerational plasticity of physiological and molecular traits in response to global change are well characterized in many marine systems (Marshall, 2008; Munday, 2014; Donelson et al., 2017), including in S. purpuratus (Wong et al., 2018, 2019; Hoshijima and Hofmann, 2019; Strader et al., 2019). However, molecular pathways that underlie phenotypic plasticity in response to global change are not well understood. We hypothesized that environmentally induced changes in DNA methylation variation may play a role in transcriptional change in response to the environment, and that this would correspond to changes in a known phenotypically plastic trait, spicule length (Byrne, 2011; Byrne et al., 2013). Overall, this study found that environmental differences impacted plasticity of the transcriptome and the methylome, but across different time scales, and that DNA methylation variation within gene bodies is not likely to modulate transcription directly, despite similarities in broad functional categories between DEGs and genes with DMCpGs. To explore the significance of these patterns further, below we discuss: (1) differences in timing of changes and the relationship between DNA methylation and the transcriptome, (2) maternal effects on gene expression and DNA methylation, (3) how conditions during development affect phenotype and gene expression, and (4) potential consequences of epigenetic mechanisms on intergenerational plasticity.
The transcriptome is highly sensitive to environmental differences, ultimately changing organismal traits from the molecular level up to higher-level processes such as behavior. Environmental perturbations can induce substantial changes in the transcriptome within short time frames (on the scale of minutes). These fast reaction times are necessary to alter cellular machinery in response to change. DNA methylation, on the other hand, is relatively stable and while able to change in response to the environment, does so at a lesser magnitude than gene expression and across longer time scales. Therefore, in the context of our experimental design, we see the biggest differences in larval CpG methylation when mothers were conditioned in different environments for 4 months, while the developmental treatment was only ∼72 h. Because of the stability and proposed functions of DNA methylation in gene regions, modulating CpG methylation during periods of short-term environmental differences is not likely to be an evolutionarily favored response, since short-term stress might not be indicative of persistent environmental change. Longer-term chronic environmental differences, such as maternal conditioning in the current experiment, may be more predictive of the future environment, so modulation of DNA methylation in response to this may be an adaptive response that occurs across longer time scales and generations.
In this experiment, we chose to use conditions representative of upwelling, which is variable and periodic in the Santa Barbara Channel. This design employed was intentionally simplified from the in situ environmental conditions in an attempt to identify the potential for environmentally induced methylation and gene-expression changes and query any mechanistic relationship between methylation changes driven by the maternal and developmental environment and any corresponding change in transcription. Through field experiments, we know that even variable in situ conditions can drive maternal effects (Hoshijima and Hofmann, 2019). Therefore, going forward it will be important to investigate whether offspring DNA methylation patterns are modulated in response to non-static maternal conditioning or if the induction of methylation changes is dependent upon the predictability of the environmental changes (Burgess and Marshall, 2014).
Mechanisms connecting DNA methylation with transcription are less well understood in invertebrates with sparsely methylated genomes as opposed to vertebrate systems where promoter methylation is able to directly alter transcription and downstream phenotypes (reviewed in Li and Zhang, 2014). Generally, gene body methylation is positively correlated with gene expression levels across invertebrate taxa (Zemach et al., 2010) including, bivalves (Gavery and Roberts, 2013; Wang et al., 2014), ascidians (Suzuki et al., 2007), and social insects (Bonasio et al., 2012; Sarda et al., 2012; Wang et al., 2013; Patalano et al., 2015). In corals specifically, it has been suggested that DNA methylation fine tunes gene expression in response to environmental change via reduction in spurious transcription (Li et al., 2017; Liew et al., 2018), genome-wide balance of gene expression (Dixon et al., 2018) and through shaping codon usage over evolutionary time scales (Dixon et al., 2016). Similar studies have been shown in oysters as well (Gavery and Roberts, 2010; Roberts and Gavery, 2012; Wang et al., 2014; Jiang et al., 2015; Saint-Carlier and Riviere, 2015). While these functions are not mutually exclusive, consensus on how actively DNA methylation regulates transcription in response to environmental change is not well agreed upon across many invertebrate systems. That being said, our data is suggestive that DNA methylation changes in genes rarely correspond to gene expression on a gene by gene level, despite broad genome-wide patterns: similarly enriched GO terms are found between DEGs and genes with DMCpGs associated with different maternal environments, highly methylated genes are more likely to be highly transcribed, and genes with DMCpGs are always highly methylated.
There is contention as to whether gene expression or DNA methylation imparts control on the other. While the overall magnitude of gene expression responses to environmental conditions is larger, there was a stronger influence of maternal conditioning on DNA methylation than of the abiotic conditions under which the embryos developed. This might suggest that maternally mediated changes in DNA methylation may translate to differential expression in offspring. However, we found minimal overlap between maternally driven differences in DNA methylation and transcription of those genes in their offspring; the majority of genes with DMCpGs by maternal condition were not differentially expressed. However, genes with DMCpGs showed functional enrichment of DNA metabolic processes and DNA integration (Supplementary Table 3), similar to terms enriched in modules showing strong correlation with maternal conditions (Figure 3 and Supplementary Table 4). This is evidence that while there are environmentally inducible changes in both CpG methylation and gene expression, they appear to be only broadly linked. This suggests that environmentally induced changes in DNA methylation may play a different functional role on a cellular level than regulating transcription directly as is observed in vertebrate systems, potentially through controlling spurious transcription. However, it should be noted that the reduced representation approach limits our ability to examine methylation patterns in all genes and a full genomic picture connecting transcription and DNA methylation is warranted.
While the focus of our study was to examine the role of DNA methylation in response to environmental conditions, it is worth noting that DNA methylation is also believed to be necessary to ensure successful development in invertebrates (Regev and Lamb, 1998; Riviere et al., 2013). Indirect evidence of this in sea urchins includes observations that DNA methylation of broad functional groups and developmentally regulated genes vary by life history stage in S. purpuratus (Fronk et al., 1992; Strader et al., 2019). Additionally, DNA methyltransferase activity has been shown to change dramatically during the early development of the sea urchin Sphaerechinus granularis (Tosi et al., 1995). Treatment with 5-azacytidine, a nuclear methylation inhibitor, at any stage prior to blastula inhibits development beyond the blastula stage in sea urchin embryos, Paracentrotus lividus and Sphaerechinus granularis, causing embryos to arrest at the blastula stage though some continue to develop spicules with no other morphological change (Crkvenjakov et al., 1970; Maharajan et al., 1986). Although details regarding the specific role that DNA methylation plays in sea urchin development are generally lacking, there is evidence that it is critical to the development of other invertebrates such as the oyster, Crassostrea gigas (Riviere et al., 2013), the wasp, Nasonia vitripennis (Zwier et al., 2012), and the ascidian, Phallusia manzmilata (Maharajan et al., 1986). Thus, in order for development to proceed successfully, we might expect certain limitations on how much DNA methylation can change in developing embryos and larvae. Our previous work showed minimal differential methylation in S. purpuratus as a function of developmental conditions (Strader et al., 2019), although the experiment included developmental environments that only varied by pCO2 level. In this study, the combined temperature and pCO2 levels of our non-upwelling versus upwelling developmental treatments resulted in differential methylation, but to a lesser extent than the effects of maternal environment on differential methylation. Therefore, some alterations in DNA methylation can occur in response to external factors in spite of potential constraints on methylation during early development, although this likely varies by the multitude and intensity of the environmental stressor(s).
The blue module shows an upregulation of genes associated with hydrogen ion transmembrane transporters and hydrogen transport when larvae had mothers that were reared in upwelling conditions. This suggests that there is a strong maternal effect of these genes, potentially priming these larvae to express more H+ transporters in the high pCO2 environment associated with the upwelling treatment. While extracellular tissue in larval urchins maintains the pH of the environment, there is internal control of pH within intracellular compartments, including those surrounding the principal calcifying cells. This internal control is mediated by transporters that regulate the concentrations of H+ and HCO ions on either side of the membrane (Evans et al., 2013). Several studies on gene expression responses to high pCO2 in larval urchins and other marine invertebrates find either no differential expression or downregulation of hydrogen ion transporters (Todgham and Hofmann, 2009; Moya et al., 2012; Evans and Watson-Wynn, 2014). These studies, however, were all performed by exposing early life-history stages to different pCO2 environments, similar to the developmental treatment in the present study, which found no differential expression of H+ transporters in response to upwelling developmental conditions (Figure 2 and Supplementary Table 5). This suggests that modulation of H+ transporters is not likely a fast response to environmental change, as in, within the course of embryonic and larval development. However, longer-term exposure of adult urchins to high pCO2 and low temperatures (upwelling) can impart an upregulation of these genes that are important in maintaining the balance between H+ and HCO ions in offspring. The upregulation of these genes, however, does not seem to generate a benefit to the larvae when exposed to upwelling conditions themselves with regards to larval spicule length (Supplementary Figure 4) and expression of cellular stress response genes (Figure 2).
Black and cyan modules showed enrichment of genes associated with DNA metabolic processes, macromolecule biosynthetic processes and DNA integration, suggesting overall differential regulation of these processes when mothers are exposed to different conditions. Functional enrichment of genes with DMCpGs, the majority differentially methylated with respect to maternal environment, also reveals enrichment of DNA integration, macromolecule biosynthetic processes, and DNA metabolic processes, among others (Supplementary Table 3). Despite similarities in GO terms, maternal modules did not contain more genes with DMCpGs than developmental modules (Supplementary Figure 5) and there were very few genes that exhibited both DMCpGs and DEGs by maternal condition (12 genes, Supplementary Table 5). The result that maternal responses to upwelling imparted differential methylation and expression in genes with similar functionality suggests that modulating the activity of these genes may be a critical long-term response to upwelling conditions. While there are caveats in extrapolating GO categories from only a subset of genes with sequenced CpGs, in Aiptasia sea anemones, similar GO categories are enriched in DM genes, including “DNA-dependent DNA replication” and “DNA integration” (Li et al., 2017), the same top GO category enriched for genes with DMCpGs in this dataset (Supplementary Table 3). DNA integration is the process by which one segment of DNA is inserted into another, one example being transposable elements. Epigenetic and transposable elements strongly interact with each other and both can lead to changes in phenotypes and genotypes in response to stress (Rey et al., 2016). For example, epigenetic components are key in repressing transposable element activity. Therefore, it is possible that interactions between DNA methylation and transposable elements can drive rapid changes in phenotypes in response to global change across short time scales in taxa other than vertebrates and plants (Rey et al., 2016). Our results, as well as others reporting differential methylation of genes associated with DNA integration, posit that more research into potential effects of transposable elements on rapid phenotypic change is warranted in marine systems exposed to a global change multi-stressor scenario. In addition, DNA methylation can impact phenotypes by changing the mutability of gene sequences, which has been suggested to impact codon bias evolution (Dixon et al., 2016). These mechanisms might be better representative of down-stream effects of environmentally induced changes in DNA methylation than directly changing transcription in invertebrates with sparsely methylated genomes.
The developmental environment had a more marked impact on spicule length and the transcriptome than did the adult environment during gametogenesis (Figure 1 and Supplementary Figure 4). This is in contrast to a previous study in S. purpuratus in which the maternal environmental conditions appeared to have a greater impact on the offspring phenotype and transcriptome than the conditions during development (Wong et al., 2018). This dissimilarity could be in part due to differences in developmental stage, as organismal responses to the environment have been shown to vary greatly by developmental stage (Kurihara, 2008; Ross et al., 2011; Byrne, 2012). Wong et al. (2018) examined the body size and transcriptomic patterns of the gastrula stage, during which major developmental landmarks distinguish this from later larval stages. Another noteworthy distinction is that in Wong et al. (2018), the embryos were raised under two different conditions that only varied by a single abiotic factor: pCO2 level. In the study presented here, combinations of both temperature and pCO2, intended to reflect ecologically relevant upwelling or non-upwelling conditions, were manipulated during early development so as to generate similar or reciprocal treatments as the adults experienced. Thus, the dominance of phenotypic and transcriptomic plasticity as a result of developmental conditions may be a result of the inclusion of temperature as a factor during development, or due to the multi-stressor nature of including combinations of different temperature and pCO2 levels.
Offspring that were reared in upwelling conditions exhibited phenotypic and transcriptomic patterns associated with a response to stress (Figure 2 and Supplementary Table 4). In concert with this stress response, there was an up-regulation of numerous RNA modification processes as well as processes involved in translation and potentially post-translational modifications (protein polymerization). For example, one gene in the turquoise module, also with a significantly differentially methylated CpG (Figure 5B), Sp.Dusp6.7.9, is known to be involved with protein dephosphorylation. Finally, there was also upregulation of genes involved in carbohydrate and lipid transport. These results strongly suggest larvae reared in upwelling conditions exhibit a stress response and a potential reallocation of energy resources, mostly likely to maintain calcification and growth. Larvae reared in upwelling conditions also exhibit a significant reduction in spicule length compared to those reared in non-upwelling conditions (Supplementary Figure 4). Differences in RNA metabolic and processing pathways suggest an attempt to modulate cellular functions in response to upwelling stress. In addition, the majority of DMCpGs were hypermethylated in the upwelling condition compared to the non-upwelling condition, which is a similar response to other organisms exposed to pH stress (Liew et al., 2018). Overall, these results show clear compensatory mechanisms in response to upwelling stress.
Stress as a result of developing under the simulated upwelling treatment could be due to low pH, low temperature, or the combination of both. In regards to low pH environments, sea urchin species have generally been shown to display reduced skeletal growth and calcification (Kurihara and Shirayama, 2004; Clark et al., 2009; Dupont et al., 2010; Byrne and Przeslawski, 2013), similar to what we found in this study. The increased expression of genes related to calcium homeostasis has been suggested as a means by which developing sea urchin larvae are able to cope and maintain skeletogenesis in low pH environments (Evans et al., 2013; Evans and Watson-Wynn, 2014). Thus, failure to upregulate calcium-related genes could result in decreased skeletogenesis. Indeed, larvae in this study exhibited a relative downregulation of calcium ion binding genes, a similar pattern to that observed by Todgham and Hofmann (2009) in S. purpuratus larvae raised under elevated pCO2 levels. In regards to temperature effects, sea urchin larvae across a variety of species and latitudes generally exhibit greater growth and calcification when raised under warmer temperatures, which occasionally provides a compensatory effect that helps offset the negative effect of low pH (Sheppard Brennand et al., 2010; Byrne, 2012; Byrne and Przeslawski, 2013; Wangensteen et al., 2013). Throughout upwelling events, however, no such compensatory effect is expected because, in addition to higher pCO2, cold temperatures are associated with upwelling seawater. Therefore, the down-regulation of calcium ion binding genes and the significant reduction of spicule length under upwelling conditions align with our expectations for larvae developing under low pH and/or low temperature conditions.
Epigenetic inheritance, in which epigenetic marks are intergenerationally transmitted through the germline, is a major study question within the field of environmental epigenetics (Verhoeven et al., 2016; Eirin-Lopez and Putnam, 2019), as it may contribute to rapid acclimation and resilience to changing environments, thereby influencing the fitness landscape and resulting evolutionary processes (Bonduriansky and Day, 2009; Bonduriansky et al., 2012). Although this study does not provide direct evidence of this, the pattern in which maternal conditions had a greater influence on differential methylation between larvae than developmental conditions supports the potential for epigenetic inheritance. In this case, epigenetic marks in the form of DMCpGs may have been incorporated by the mothers into their eggs, which then persisted in their offspring during early development despite differences in the environmental conditions under which the embryos and larvae were raised. While further study is required to determine if these epigenetic changes are truly integrated into the germline and are capable of remaining across generations, this study provides support for the potential of epigenetic inheritance in this system.
Different environmental conditions revealed plasticity of the transcriptome and the methylome, but across different time scales within the life history of these marine invertebrates. Namely, DNA methylation variation changes over longer time scales, such as between generations, and is not likely to modulate transcription directly in response to abiotic, physical cues. We found significant plasticity in DNA methylation and gene expression in response to different maternal environmental conditions. While this pattern affected similar broad functional groups, there was little overlap between differential methylation and differential expression on a gene-by-gene basis. We did observe potential maternal priming of larvae with enhanced physiological capacity to function in low pH seawater via increased ion transporter activity, although priming was not evident in larval phenotypes: development under upwelling conditions inhibited skeletal growth and activated genes associated with stress response. The over-arching results of the study point to further examination of the connection between changes in the methylome and changes in phenotypes modulated by global change.
Datasets and analysis scripts can be accessed through a Github repository (https://github.com/mariestrader/S.purp_RRBS_RNAseq_2019) and fastq sequences are available through the NCBI Short Read Archive under the accession PRJNA548926.
Adult urchins were collected in the Santa Barbara Channel under the California Scientific Collecting Permit to GEH (SC-1223).
GH designed the study. MS, LK, TL, JW, JC, MH, and GH executed the experiment and generated the samples. MS analyzed the data with contributions from LK, TL, JW, JC, MH, and GH. MS wrote the manuscript with contributions from LK, TL, JW, JC, MH, and GH.
This research was funded by a United States National Science Foundation award (IOS-1656262) to GH. In addition, diving and boating resources were provided by Santa Barbara Coastal Long-Term Ecological Research program (NSF award OCE-1232779; Director: Dr. Daniel Reed).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Clint Nelson of the Santa Barbara Coastal Long-Term Ecological Research (SBC LTER) program for assistance with boating and sea urchin collection. We also thank Christoph Pierre, Director of Marine Operations at the UC Santa Barbara, for kelp collections during the course of the experiment. We also thank Drs. Umi Hoshijima and Groves Dixon for advice on data analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00205/full#supplementary-material
Akalin, A., Kormaksson, M., Li, S., Garrett-Bakelman, F. E., Figueroa, M. E., Melnick, A., et al. (2012). MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13:R87. doi: 10.1186/gb-2012-13-10-R87
Ardura, A., Zaiko, A., Morán, P., Planes, S., and Garcia-Vazquez, E. (2017). Epigenetic signatures of invasive status in populations of marine invertebrates. Sci. Rep. 7:42193. doi: 10.1038/srep42193
Bakun, A., Black, B. A., Bograd, S. J., García-Reyes, M., Miller, A. J., Rykaczewski, R. R., et al. (2015). Anticipated effects of climate change on coastal upwelling ecosystems. Curr. Clim. Chang. Rep. 1, 85–93. doi: 10.1007/s40641-015-0008-4
Banta, J. A., and Richards, C. L. (2018). Quantitative epigenetics and evolution. Heredity 121, 210–224. doi: 10.1038/s41437-018-0114-x
Baums, I. B., Devlin-Durante, M. K., Williams, D. W., and Kemp, D. W. (2019). What drives phenotypic divergence among coral clonemates? Mol. Ecol. 28, 3208–3224. doi: 10.1101/514430
Bonasio, R., Li, Q., Lian, J., Mutti, N. S., Jin, L., Zhao, H., et al. (2012). Genome-wide and caste-specific DNA methylomes of the ants camponotus floridanus and harpegnathos saltator. Curr. Biol. 22, 1755–1764. doi: 10.1016/j.cub.2012.07.042
Bonduriansky, R., Crean, A. J., and Day, T. (2012). The implications of nongenetic inheritance for evolution in changing environments. Evol. Appl. 5, 192–201. doi: 10.1111/j.1752-4571.2011.00213.x
Bonduriansky, R., and Day, T. (2009). Nongenetic inheritance and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 40, 103–125. doi: 10.1146/annurev.ecolsys.39.110707.173441
Burgess, S. C., and Marshall, D. J. (2014). Adaptive parental effects: the importance of estimating environmental predictability and offspring fitness appropriately. Oikos 123, 769–776. doi: 10.1111/oik.01235
Byrne, M. (2011). Impact of ocean warming and ocean acidification on marine invertebrate life history stages: vulnerabilities and potential for persistence in a changing ocean. Oceanogr. Mar. Biol. An Annu. Rev. 49, 1–42. doi: 10.1016/j.marenvres.2011.10.00
Byrne, M. (2012). Global change ecotoxicology: identification of early life history bottlenecks in marine invertebrates, variable species responses and variable experimental approaches. Mar. Environ. Res. 76, 3–15. doi: 10.1016/j.marenvres.2011.10.004
Byrne, M., Lamare, M., Winter, D., Dworjanyn, S. A., and Uthicke, S. (2013). The stunting effect of a high CO2 ocean on calcification and development in sea urchin larvae, a synthesis from the tropics to the poles. Philos. Trans. R. Soc. B Biol. Sci. 368:20120439. doi: 10.1098/rstb.2012.0439
Byrne, M., and Przeslawski, R. (2013). Multistressor impacts of warming and acidification of the ocean on marine invertebrates’ life histories. Integr. Comp. Biol. 53, 582–596. doi: 10.1093/icb/ict049
Chan, F., Barth, J. A., Blanchette, C. A., Byrne, R. H., Chavez, F., Cheriton, O., et al. (2017). Persistent spatial structuring of coastal ocean acidification in the California current system. Sci. Rep. 7, 1–7. doi: 10.1038/s41598-017-02777-y
Clark, D., Lamare, M., and Barker, M. (2009). Response of sea urchin pluteus larvae (Echinodermata: Echinoidea) to reduced seawater pH: a comparison among a tropical, temperate, and a polar species. Mar. Biol. 156, 1125–1137. doi: 10.1007/s00227-009-1155-8
Crkvenjakov, R., Bajkovic, N., and Glisin, V. (1970). The effect of 5-azacytidine on development, nucleic acid and protein metabolism in sea urchin embryos. Biochem. Biophys. Res. Commun. 39, 655–660.
Dickson, A. G., and Millero, F. J. (1987). A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep Sea Res. 34, 1733–1743.
Dickson, A. G., Sabine, C. L., and Christian, J. R. (2007). Guide to Best Practices for Ocean CO 2 Measurements. Sidney: North Pacific Marine Science Organization.
Dixon, G., Bay, L. K., Matz, M. V., Biology, M., Science, M., Centre, A. R. C., et al. (2016). Evolutionary consequences of DNA methylation in a basal metazoan. Mol. Biol. Evol. 33:043026. doi: 10.1101/043026
Dixon, G., Liao, Y., Bay, L. K., and Matz, M. V. (2018). Role of gene body methylation in acclimatization and adaptation in a basal metazoan. PNAS 115, 13342–13346. doi: 10.1073/pnas.1813749115
Dixon, G. B., Bay, L. K., and Matz, M. V. (2014). Bimodal signatures of germline methylation are linked with gene expression plasticity in the coral Acropora millepora. BMC Genomics 15:1109. doi: 10.1186/1471-2164-15-1109
Donelson, J. M., Salinas, S., Munday, P. L., and Shama, L. N. S. (2017). Transgenerational plasticity and climate change experiments: where do we go from here? Glob. Chang. Biol. 24, 13–34. doi: 10.1111/gcb.13903
Dupont, S., Ortega-Martínez, O., and Thorndyke, M. (2010). Impact of near-future ocean acidification on echinoderms. Ecotoxicology 19, 449–462. doi: 10.1007/s10646-010-0463-6
Eirin-Lopez, J. M., and Putnam, H. M. (2019). Marine environmental epigenetics. Ann. Rev. Mar. Sci. 11, 335–368. doi: 10.1146/annurev-marine-010318-095114
Evans, T. G., Chan, F., Menge, B. A., and Hofmann, G. E. (2013). Transcriptomic responses to ocean acidification in larval sea urchins from a naturally variable pH environment. Mol. Ecol. 22, 1609–1625. doi: 10.1111/mec.12188
Evans, T. G., and Watson-Wynn, P. (2014). Effects of seawater acidification on gene expression: resolving broader-scale trends in sea urchins. Biol. Bull. 226, 237–254. doi: 10.1086/BBLv226n3p237
Fangue, N. A., O’Donnell, M. J., Sewell, M. A., Matson, P. G., MacPherson, A. C., and Hofmann, G. E. (2010). A laboratory-based, experimental system for the study of ocean acidification effects on marine invertebrate larvae. Limnol. Oceanogr. Methods 8, 441–452. doi: 10.4319/lom.2010.8.441
Feely, R. A., Sabine, C. L., Hernandez-Ayon, J. M., Ianson, D., and Hales, B. (2008). Evidence for upwelling of corrosive “acidified” water onto the continental shelf. Science 320, 1490–1492. doi: 10.1126/science.1155676
Feil, R., and Fraga, M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109. doi: 10.1038/nrg3142
Fronk, J., Tank, G. A., and Langmore, J. P. (1992). DNA methylation pattern changes during development of a sea urchin. Biochem. J. 283, 751–753. doi: 10.1042/bj2830751
Gavery, M. R., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 11:483. doi: 10.1186/1471-2164-11-483
Gavery, M. R., and Roberts, S. B. (2013). Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215. doi: 10.7717/peerj.215
Gavery, M. R., and Roberts, S. B. (2014). A context dependent role for DNA methylation in bivalves. Brief. Funct. Genomics 13, 217–222. doi: 10.1093/bfgp/elt054
Gavery, M. R., and Roberts, S. B. (2017). Epigenetic considerations in aquaculture. PeerJ 5:e4147. doi: 10.7717/peerj.4147
Gonzalez-Romero, R., Suarez-Ulloa, V., Rodriguez-Casariego, J., Garcia-Souto, D., Diaz, G., Smith, A., et al. (2017). Effects of Florida Red Tides on histone variant expression and DNA methylation in the Eastern oyster Crassostrea virginica. Aquat. Toxicol. 186, 196–204. doi: 10.1016/j.aquatox.2017.03.006
Gruber, N., Hauri, C., Lachkar, Z., Loher, D., Frolicher, T. L., and Plattner, G.-K. (2012). Rapid progression of ocean acidification in the California current system. Science 337, 220–223. doi: 10.1126/science.1216773
Hansson, I. (1973). A new set of acidity constants for carbonic acid and boric acid in sea water. Deep Sea Res. Oceanogr. Abstr. 20, 461–478.
Hawes, N. A., Amadorou, A., Tremblay, L. A., Pochon, X., Dunphy, B., Fidler, A. E., et al. (2019). Epigenetic patterns associated with an ascidian invasion: a comparison of closely related clades in their native and introduced ranges. Sci. Rep. 9:14275. doi: 10.1038/s41598-019-49813-7
Hofmann, G. E., and Washburn, L. (2019). SBC LTER: Ocean: Time-series: Mid-Water SeaFET pH and CO2 System Chemistry with Surface and Bottom Dissolved Oxygen at Mohawk Reef(MKO), Ongoing Since 2012-01-11. Available online at: https://pasta.lternet.edu/package/eml/knb-lter-sbc/6003/3 (accessed August 2017).
Hoshijima, U., and Hofmann, G. E. (2019). Variability of seawater chemistry in a kelp forest environment is linked to in situ transgenerational effects in the purple sea urchin, Strongylocentrotus purpuratus. Front. Mar. Sci. 6:62. doi: 10.3389/fmars.2019.00062
Jiang, L., Lei, X.-M., Liu, S., and Huang, H. (2015). Fused embryos and pre-metamorphic conjoined larvae in a broadcast spawning reef coral. F1000Research 4:44. doi: 10.12688/f1000research.6136.1
Johannes, F., Porcher, E., Teixeira, F. K., Saliba-colombani, V., Albuisson, J., Heredia, F., et al. (2009). Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 5:e1000530. doi: 10.1371/journal.pgen.1000530
Jombart, T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Kapsenberg, L., and Hofmann, G. E. (2016). Ocean pH time-series and drivers of variability along the northern Channel Islands, California, USA. Limnol. Oceanogr. 61, 953–968. doi: 10.1002/lno.10264
Kauffmann, A., Gentleman, R., and Huber, W. (2009). arrayQualityMetrics–a bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416. doi: 10.1093/bioinformatics/btn647
Kelly, M. W., Padilla-Gamiño, J. L., and Hofmann, G. E. (2016). High pCO2 affects body size, but not gene expression in larvae of the California mussel (Mytilus californianus). ICES J. Mar. Sci. 73, 962–969. doi: 10.1093/icesjms/fsv184
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Klironomos, F. D., Berg, J., and Collins, S. (2013). How epigenetic mutations can affect genetic evolution: model and mechanism. BioEssays 35, 571–578. doi: 10.1002/bies.201200169
Kronholm, I., and Collins, A. D. (2016). Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 25, 1856–1868. doi: 10.1111/mec.13296
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Kurihara, H. (2008). Effects of CO2 driven ocean acidification on the early developmental stages of invertebrates. Mar. Ecol. Prog. Ser. 373, 275–284. doi: 10.3354/meps07802
Kurihara, H., and Shirayama, Y. (2004). Effects of increased atmospheric CO2 on sea urchin early development. Mar. Ecol. Prog. Ser. 274, 1–9.
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Li, E., and Zhang, Y. (2014). DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 6:a019133. doi: 10.1101/cshperspect.a019133
Li, Y., Liew, Y. J., Cui, G., Cziesielski, M. J., Zahran, N., Michell, C. T., et al. (2017). DNA methylation regulates transcriptional homeostasis of algal endosymbiosis in the coral model Aiptasia. Sci. Adv. 4:eaat2142. doi: 10.1101/213066
Liao, Y., Smyth, G. K., and Shi, W. (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Liew, Y. J., Zoccola, D., Li, Y., Tambutté, E., Venn, A. A., Craig, T., et al. (2018). Epigenome-associated phenotypic acclimatization to ocean acidification in a reef-building coral. Sci. Adv. 216:188227. doi: 10.1101/188227
Love, M. I., Anders, S., and Huber, W. (2014). Differential Analysis of Count Data - The DESeq2 Package. Heidelberg: EMBL.
Maharajan, P., Maharajan, V., Branno, M., and Scarano, E. (1986). Effects of 5 azacytidine on DNA methylation and early development of sea urchins and ascidia. Differentiation 32, 200–207.
Marsh, A. G., and Pasqualone, A. A. (2014). DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 5:173. doi: 10.3389/fphys.2014.00173
Marshall, D. J. (2008). Transgenerational plasticity in the sea: context- dependent maternal effects across the life history. Ecology 89, 418–427. doi: 10.1890/07-0449.1
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Moya, A., Huisman, L., Ball, E. E., Hayward, D. C., Grasso, L. C., Chua, C. M., et al. (2012). Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO2-driven acidification during the initiation of calcification. Mol. Ecol. 21, 2440–2454. doi: 10.1111/j.1365-294X.2012.05554.x
Munday, P. L. (2014). Transgenerational acclimation of fishes to climate change and ocean acidification. F1000Prime Rep. 6, 1–7. doi: 10.12703/P6-99
Okazaki, K. (1975). Spicule formation by isolated micromeres of the sea urchin embryo. Integr. Comp. Biol. 15, 567–581. doi: 10.1093/icb/15.3.567
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., et al. (2013). Package ‘vegan.’ Community Ecol. Packag. Version 2.
Padilla-Gamiño, J. L., Kelly, M. W., Evans, T. G., Hofmann, G. E., Padilla-Gamiño, J. L., Kelly, M. W., et al. (2013). Temperature and CO 2 additively regulate physiology, morphology and genomic responses of larval sea urchins, Strongylocentrotus purpuratus. Proc. R. Soc. B 280:20130155. doi: 10.1098/rspb.2013.0155
Patalano, S., Vlasova, A., Wyatt, C., Ewels, P., Camara, F., Ferreira, P. G., et al. (2015). Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies. Proc. Natl. Acad. Sci. U.S.A. 112, 13970–13975. doi: 10.1073/pnas.1515937112
Pearse, J. S. (2006). Ecological role of purple sea urchins. Science 314, 940–941. doi: 10.1126/science.1131888
Regev, A., and Lamb, M. J. (1998). The role of DNA methylation in invertebrates: developmental regulation or genome defense? Mol. Biol. Evol. 15, 880–891.
Rey, O., Danchin, E., Mirouze, M., Loot, C., and Blanchet, S. (2016). Adaptation to global change: a transposable element-epigenetics perspective. Trends Ecol. Evol. 31, 514–526. doi: 10.1016/j.tree.2016.03.013
Richards, C. L., Alonso, C., Becker, C., Bossdorf, O., Bucher, E., Colomé-Tatché, M., et al. (2017). Ecological plant epigenetics: evidence from model and non-model species, and the way forward. Ecol. Lett. 20, 1576–1590. doi: 10.1111/ele.12858
Richards, E. J. (2008). Population epigenetics. Curr. Opin. Genet. Dev. 18, 221–226. doi: 10.1016/j.gde.2008.01.014
Rivest, E. B., Brien, M. O., Kapsenberg, L., Gotschalk, C. C., Blanchette, C. A., Hoshijima, U., et al. (2016). Ecological Informatics Beyond the benchtop and the benthos?: dataset management planning and design for time series of ocean carbonate chemistry associated with Durafet ®-based pH sensors. Ecol. Inform. 36, 209–220. doi: 10.1016/j.ecoinf.2016.08.005
Riviere, G., He, Y., Tecchio, S., Crowell, E., Sourdaine, P., Guo, X., et al. (2017). Dynamics of DNA methylomes underlie oyster development. PLoS Genet. 13:e1006807. doi: 10.1371/journal.pgen.1006807
Riviere, G., Wu, G. C., Fellous, A., Goux, D., Sourdaine, P., and Favrel, P. (2013). DNA methylation Is crucial for the early development in the oyster C. gigas. Mar. Biotechnol. 15, 739–753. doi: 10.1007/s10126-013-9523-2
Robbins, L. L., Hansen, M. E., Kleypas, J. A., and Meylan, S. C. (2010). CO2calc: A User-Friendly Seawater Carbon Calculator for Windows, Mac OS X, and iOS (iPhone). Reston, VA: USGS.
Roberts, S. B., and Gavery, M. R. (2012). Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiol. 2:116. doi: 10.3389/fphys.2011.00116
Ross, P. M., Parker, L., O’Connor, W. A., and Bailey, E. A. (2011). The impact of ocean acidification on reproduction, early development and settlement of marine organisms. Water 3, 1005–1030. doi: 10.3390/w3041005
Saint-Carlier, E., and Riviere, G. (2015). Regulation of Hox orthologues in the oyster Crassostrea gigas evidences a functional role for promoter DNA methylation in an invertebrate. FEBS Lett. 589, 1459–1466. doi: 10.1016/j.febslet.2015.04.043
Sarda, S., Zeng, J., Hunt, B. G., and Yi, S. V. (2012). The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 29, 1907–1916. doi: 10.1093/molbev/mss062
Shea, N., Pen, I., and Uller, T. (2011). Three epigenetic information channels and their different roles in evolution. J. Evol. Biol. 24, 1178–1187. doi: 10.1111/j.1420-9101.2011.02235.x
Sheppard Brennand, H., Soars, N., Dworjanyn, S. A., Davis, A. R., and Byrne, M. (2010). Impact of ocean warming and ocean acidification on larval development and calcification in the sea urchin Tripneustes gratilla. PLoS One 5:e0011372. doi: 10.1371/journal.pone.0011372
Strader, M. E., Wong, J. M., Kozal, L. C., Leach, T. S., and Hofmann, G. E. (2019). Parental environments alter DNA methylation in offspring of the purple sea urchin, Strongylocentrotus purpuratus. J. Exp. Mar. Biol. Ecol. 517, 54–64. doi: 10.1016/j.jembe.2019.03.002
Strathmann, M. F. (1987). Reproduction and development of marine invertebrates of the northern Pacific coast:?: Data and Methods for the Study of Eggs, Embryos, and Larvae. Seattle: University of Washington Press.
Suzuki, M. M., Kerr, A. R. W., De Sousa, D., and Bird, A. (2007). CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 17, 625–631. doi: 10.1101/gr.6163007
Sydeman, W. J., García-Reyes, M., Schoeman, D. S., Rykaczewski, R. R., Thompson, S. A., Black, B. A., et al. (2014). Climate change. Climate change and wind intensification in coastal upwelling ecosystems. Science 345, 77–80. doi: 10.1126/science.1251635
Todgham, A. E., and Hofmann, G. E. (2009). Transcriptomic response of sea urchin larvae Strongylocentrotus purpuratus to CO2-driven seawater acidification. J. Exp. Biol. 212, 2579–2594. doi: 10.1242/jeb.032540
Torda, G., Donelson, J. M., Aranda, M., Barshis, D. J., Bay, L., Berumen, M. L., et al. (2017). Rapid adaptive responses to climate change in corals. Nat. Clim. Chang. 7, 627–636. doi: 10.1038/nclimate3374
Tosi, L., Aniello, F., Geraci, G., and Branno, M. (1995). DNA methyltransferase activity in the early stages of a sea urchin embryo Evidence of differential control. FEBS Lett. 361, 115–117. doi: 10.1016/0014-5793(95)00160-B
Verhoeven, K. J. F., VonHoldt, B. M., and Sork, V. L. (2016). Epigenetics in ecology and evolution: what we know and what we need to know. Mol. Ecol. 25, 1631–1638. doi: 10.1111/mec.13617
Wang, X., Li, Q., Lian, J., Li, L., Jin, L., Cai, H., et al. (2014). Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genomics 15:1119. doi: 10.1186/1471-2164-15-1119
Wang, X., Wheeler, D., Avery, A., Rago, A., Choi, J. H., Colbourne, J. K., et al. (2013). Function and Evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 9:e1003872. doi: 10.1371/journal.pgen.1003872
Wangensteen, O. S., Dupont, S., Casties, I., Turon, X., and Palacín, C. (2013). Some like it hot: temperature and pH modulate larval development and settlement of the sea urchin Arbacia lixula. J. Exp. Mar. Bio. Ecol. 449, 304–311. doi: 10.1016/j.jembe.2013.10.007
Wong, J. M., Johnson, K. M., Kelly, M. W., and Hofmann, G. E. (2018). Transcriptomics reveal transgenerational effects in purple sea urchin embryos: adult acclimation to upwelling conditions alters the response of their progeny to differential pCO2levels. Mol. Ecol. 27, 1120–1137. doi: 10.1111/mec.14503
Wong, J. M., Kozal, L. C., Leach, T. S., Hoshijima, U., and Hofmann, G. E. (2019). Transgenerational effects in an ecological context: conditioning of adult sea urchins to upwelling conditions alters maternal provisioning and progeny phenotype. J. Exp. Mar. Biol. Ecol. 57, 65–77. doi: 10.1016/j.jembe.2019.04.006
Wright, R. M., Aglyamova, G. V., Meyer, E., and Matz, M. V. (2015). Gene expression associated with white syndromes in a reef building coral, Acropora hyacinthus. BMC Genomics 16:2. doi: 10.1186/s12864-015-1540-2
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Keywords: gene expression, DNA methylation, intragenerational plasticity, intergenerational plasticity, Strongylocentrotus purpuratus
Citation: Strader ME, Kozal LC, Leach TS, Wong JM, Chamorro JD, Housh MJ and Hofmann GE (2020) Examining the Role of DNA Methylation in Transcriptomic Plasticity of Early Stage Sea Urchins: Developmental and Maternal Effects in a Kelp Forest Herbivore. Front. Mar. Sci. 7:205. doi: 10.3389/fmars.2020.00205
Received: 31 August 2019; Accepted: 16 March 2020;
Published: 21 April 2020.
Edited by:
Hollie Putnam, University of Rhode Island, United StatesReviewed by:
Alexandre Fellous, Alfred-Wegener-Institute Helmholtz Center for Polar and Marine Research (AWI), GermanyCopyright © 2020 Strader, Kozal, Leach, Wong, Chamorro, Housh and Hofmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie E. Strader, c3RyYWRlcm1hcmllQGdtYWlsLmNvbQ==
†present address: Marie E. Strader, Department of Biological Sciences, Auburn University, Auburn, AL, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.