 Marta Turon
Marta Turon Maria J. Uriz
Maria J. Uriz- Centre d’Estudis Avançats de Blanes, CEAB-CSIC, Girona, Spain
Archaea is a poorly studied domain associated with sponges. Many questions that have been addressed for bacteria still remain largely unknown for archaea. In this study, we analyzed the archaeal communities of 17 tropical sponge species from Nha Trang Bay (Vietnam) using archaea specific primers. We recorded patterns of diversity and spatial stability of these microbial communities and compared the results obtained with the bacterial communities, already reported in our previous study. In our study species, Shannon diversity was always lower for archaeal than for bacterial communities. The differences in alpha diversity as well as the presence of indicator phyla reported for the bacterial domain in High Microbial Abundance (HMA) and Low Microbial Abundance (LMA) could not be confirmed for the archaeal communities. Host identity was the main factor structuring the archaeal assemblages. Sponge archaea core was formed by few but very abundant ZOTUs, which contribute with a high proportion to the relative archaea abundance. The inclusion of the obtained sequences into phylogenetic trees allowed finding out whether or not they belonged to the so-called sponge clusters (SC). Our results showed that most of the Thaumarchaeota and Euryarchaeota sequences were more closely related to environmental samples than to SC, suggesting that they might be acquired from the seawater, which need to be verified. However, representatives of Woesarchaeota, which were major members of the archaea microbiome of two sponge species, formed a monophyletic tree, distantly related to any known environmental sequence.
Introduction
Sponges, together with corals, are paradigmatic representatives of the holobiont concept (Dittami et al., 2019) in marine ecosystems. Sponges establish tight associations simultaneously with members of the three domains of life: Eukarya (Wulff, 2006; De Mares et al., 2017; Turon et al., 2019b), Bacteria (Taylor et al., 2007; Pita et al., 2018; Turon et al., 2018, 2019a) and Archaea (Margot et al., 2002; Simister et al., 2012; Zhang et al., 2014; Rodríguez-Marconi et al., 2015; De Mares et al., 2017). Sponge-associated bacteria are particularly abundant in the so-called bacteriosponges (Vacelet and Donadey, 1977) or High Microbial Abundant sponges. Other sponge species with lower bacteria richness, named Low Microbial Abundance -LMA- sponges (Hentschel et al., 2003), have also been deeply explored attempting to assign most of the extant sponge species to any of these two categorized groups (Alex and Antunes, 2015). The differences in bacteria communities between them have been attributed to the contrasting structural and ecological characteristics of the host sponges (Weisz et al., 2007; Gerçe et al., 2011; Ribes et al., 2012; Blanquer et al., 2013). Consistent generalizations on the composition, structure and functionality (genomes) of bacteria communities within sponges have arose from these studies (Webster and Thomas, 2016). Conversely, archaeal associations with sponges are largely unknown even for some basic aspects. Features that have been widely investigated for sponge bacteria such as presence of Sponge Clusters (SC), species specificity, microbe acquisition, environmental resilience, HMA-LMA dichotomy, or even functionality, remain poorly explored for the archaeal domain.
Sponge Clusters in the bacterial and archaeal domains have been reported in sponge hosts (Taylor et al., 2007; Simister et al., 2012). However, with the increase of sequencing depth, some SC bacterial sequences were also detected outside the sponges, albeit at low abundances. This made researchers to change the concept of SC to that of “Sponge Enriched Bacterial clusters” (Taylor et al., 2013). Whether the proposed SC for archaea (Simister et al., 2012) were also detected outside the hosts or not, needs to be confirmed after deep sequencing studies. For instance, Simister et al. (2012) identified five putative SC clusters within the Thaumarchaeota phylum, which were at that moment unknown from the seawater and from other (non-sponge) marine habitats. Revision of these archaea SC on the light of the new sequences deposited in the databases might confirm or discard the existence of these named archaea SC.
On the other hand, the available studies suggest that, as for bacteria, host identity is a main driver of archaeal community structure (Polónia et al., 2014; De Mares et al., 2017). These studies also suggest that archaea are stable across different environments [e.g., the archaeal microbiome of B. fortis does not change from marine lakes to open water habitats (Polónia and Cleary, 2019)], but additional species should be analyzed to confirm/discard the general character of the archaea resilience.
The research on how microsymbionts are acquired by the host sponges has run in parallel for bacteria and archaea. Archaea symbionts were first proposed to be vertically transmitted from progenitors to their progeny (Schmitt et al., 2008; Steger et al., 2008). However, later studies indicated that, as for bacteria microbiomes, host biogeography, and environment might also shape the sponge archaeal communities (Turque et al., 2010; Zhang et al., 2014).
Comparative studies on the two microbial domains in sponges often found archaeal cells at lower abundances than bacterial cells, although some exceptions have also been reported (Jackson et al., 2013). However, regardless the apparently lower abundance of archaea in sponge microbiomes with respect to bacteria, some archaea have been recognized to play chief roles in the holobiont functioning (Radax et al., 2012b; Tian et al., 2014; Moeller et al., 2019). Thaumarchaetoa is the most abundant phylum reported among the sponge-associated archaea (Margot et al., 2002; Hoffmann et al., 2009; Radax et al., 2012b; Moeller et al., 2019). Some members of this Phylum are key chemoautotrophic ammonia-oxidizers microorganisms in marine habitats (Pester et al., 2012) and have been suggested to play a role in ammonia detoxification within the host sponges (Steger et al., 2008; Hoffmann et al., 2009). This ability to metabolize nitrogenous waste products has been proposed to represent a common benefit for the mutualistic relationship between AOA and sponges (Steger et al., 2008; Tian et al., 2014). Indeed, adaptions to a symbiotic lifestyle have been found in the genome of the thaumarchaeal symbiont from Ianthella basta (Moeller et al., 2019). Ammonia oxidation in sponges has usually been linked to the presence of archaea through the amplification of the archaeal amoA gene (Hoffmann et al., 2009; Liu et al., 2011; Radax et al., 2012a) or metagenomic sequencing (Fan et al., 2012; Moeller et al., 2019), although its presence does not prove necessarily its activity (Mußmann et al., 2011).
In this study, we aimed to characterize the sponge archaeal communities of the most abundant sponges in Nha Trang Bay (Vietnam), to cast some light on some of the above-mentioned issues such as host specificity, archaeal core size, diversity and dominance of archaea taxa, and to confirm or reject the existence of SC archaeal clusters. To summarize, we attempt at discovering whether sponges are a reservoir of novel archaeal diversity and how the sponge archaeal communities are structured. Bacterial communities of these sponges have been already analyzed (Turon et al., 2018) thus, allowing for comparisons between the two prokaryote domains in terms of specificity, diversity, and stability.
Materials and Methods
Sponge Sampling and DNA Extraction
Sponge samples used in this study are the same used in Turon et al. (2018) to study the sponge bacterial communities. Sponge samples were collected in April 2015 by SCUBA diving along 13, 25 m-long transect lines, randomly placed between 3 and 9 m deep in three neighboring locations ∼2 km apart (i.e., Dambay, Hun Mun and Nock Island) within Nha Trang Bay (central Vietnam). Specimens that were crossed by the metric tape along the line transect were photographed and sampled. A piece of ca. 3 cm2 (whenever possible) was collected in a 50 mL Falcon tube with seawater, which was immediately replaced by 100% ethanol once on board. Back in the lab, the ethanol was replaced twice again with fresh absolute ethanol for a good sample preservation. As a result of this quantitative sampling methodology, the number of sponge replicates reflected their ecological distribution in the area, but did not allowed us to obtain the same number of replicates for all the sponge species collected. A total of 203 sponge samples were collected, although only 68 samples belonging to 19 sponge species were used for the study of sponge associated archaeal communities. DNA from the selected sponges was extracted following the DNeasy Blood & Tissue Kit (Qiagen) protocol.
Sponge Sampling and DNA Extraction
PCR and high-speed multiplexed SSU rRNA gene Illumina MiSeq sequencing (NGS) were carried out following the genomic core facilities and methods of the MrDNA Lab (Shallowater, TX, United States)1. From the 68 samples sent for amplification and sequencing, only 52 yielded proper results. The variable V4 region of the archaeal 16S rRNA gene was amplified using the primers 349F (5′-GYGCASCAGKCGMGAAW-3′) and 806R (5′-GGACTACVSGGGTATCTAAT-3′) (c.a. 380 nt). These archaea- specific primer pairs were chosen according to annealing temperatures, overall coverage of V4 region, and amplicon length (Klindworth et al., 2012). UPARSE pipeline (Edgar, 2017) was used to process the raw rRNA gene sequences. The fastq filter command with the arguments –fastq_trunclen 380 –fastq_maxee 0.25 was applied to our dataset for quality checking. After this filtering, we kept 67% of the raw original sequences. Sequences were then dereplicated with the – derep_fulllength command and sorted by size (-sortbysize command) in usearch 10.0.240 version. Denoising (error-correction) of amplicons was performed following the UNOISE pipeline (Edgar, 2016) using the – unosie3 command. This algorithm removed chimeras, reads with sequencing errors, PhiX, and low complexity sequences due to Illumina artifact, and generates ZOTUs (“zero-radius” OTUs) with 100% identity sequences. Finally, -usearch_global command with identity threshold set at 0.97 was applied to our dataset. Taxonomic assignment was done with SINA v1.2.11 (Pruesse et al., 2012) using SILVA 132 database. Sequences with low identity (<75%) and sequences identified as mitochondria or chloroplasts were removed from the analysis. In order to minimize biased effects for differences in sampling effort, the original bacterial ZOTU table was rarefied at a minimum reads threshold of 5000, using QIIME (Caporaso et al., 2010). Samples with less than 5000 reads were removed from the analysis. From the 68 sponge samples, 52 could be amplified and only 43 samples belonging to 17 sponge species met the quality requirements.
Archaeal Community Analysis
A distance-based multivariate analysis of the rarefied ZOTU table was carried out using the vegan package (Oksanen et al., 2018) in R. The Bray-Curtis dissimilarity distance matrix of sponge archaeal communities was used to build a hierarchical cluster dendogram (Ward method). PERMANOVA (Anderson, 2001) based on 999 permutations as implemented in adonis function was used to test the effect of host identity (species) on structuring the sponge archaeal communities. Archaeal composition at the lowest taxonomic level and the relative abundance of the most abundant ZOTU were represented for each sponge sample. Moreover, Shannon diversity (vegan package) and the abundance-based species richness estimator, Chao 1, as implemented in the iNEXT package (Hsieh et al., 2019), with the dissimilarity level set to 0.05, were calculated for sponge archaeal communities. Shannon diversity indices of archaeal communities were compared with those obtained for bacterial communities [already reported in Turon et al. (2018)].
We defined the archaeal sponge core as those ZOTUs present across all samples and the archaeal species core as those ZOTUs present across all replicates of each species. For sponge species with replicates, we calculated the relative abundance of the core ZOTUs to the overall species archaeal community.
Phylogenetic Analysis
The most abundant ZOTU sequences (>0.1% relative abundance) were added to the original tree containing the previously identified archaeal clusters (Simister et al., 2012) by using the Parsimony Interactive tool in the ARB program (Ludwig et al., 2004). The SILVA database and alignment of the archaeal sequences, used for the identification of SC clusters, was kindly provided by the authors of the study (Simister et al., 2012). Only ZOTUs belonging to Thaumarchaetoa and Euryarchaeota were included in those trees. Woesarchaetoa representative sequences (>0.1% rel. abundance) were imported into the SILVA SSU Ref NR 132 database (Quast et al., 2013) and inserted in the original tree keeping the overall tree topology by using the Parsimony Interactive tool implemented in ARB (Ludwig et al., 2004). Although nowadays the SC are better referred to as “Sponge-Enriched Clusters” (Taylor et al., 2013) we will keep the term SC to facilitate comparisons with Simister’ results.
Functional Predictions of Archaeal Sponge Community
Tax4Fun2 (Aßhauer et al., 2015; Wemheuer et al., 2018) was used to predict the metabolic pathways of the sponge archaeal communities. This software predicts the functional profiles of 16S rRNA gene data by aligning 16S sequences with reference genomes and subsequently calculating the functional predictions based on KEGG pathways. Our data was first aligned with the reference dataset provided (275 archaeal genomes available through NCBI RefSeq database) with the runRefBlast function in the tax4fun R package, and the functional predictions were then calculated with the makefunctionalprediction function (Wemheuer et al., 2018). Only the samples for which at least 75% of the reads were used in the prediction (close genome available) were considered for further analysis and interpretation.
Results
Sponge Archaeal Communities
Nine sponge samples had less than 5000 archaea reads and were excluded from the analysis. Thereby, we present the results based on 43 sponge samples belonging to 17 sponge species with replicates varying from 1 to 8 (see Table 1).
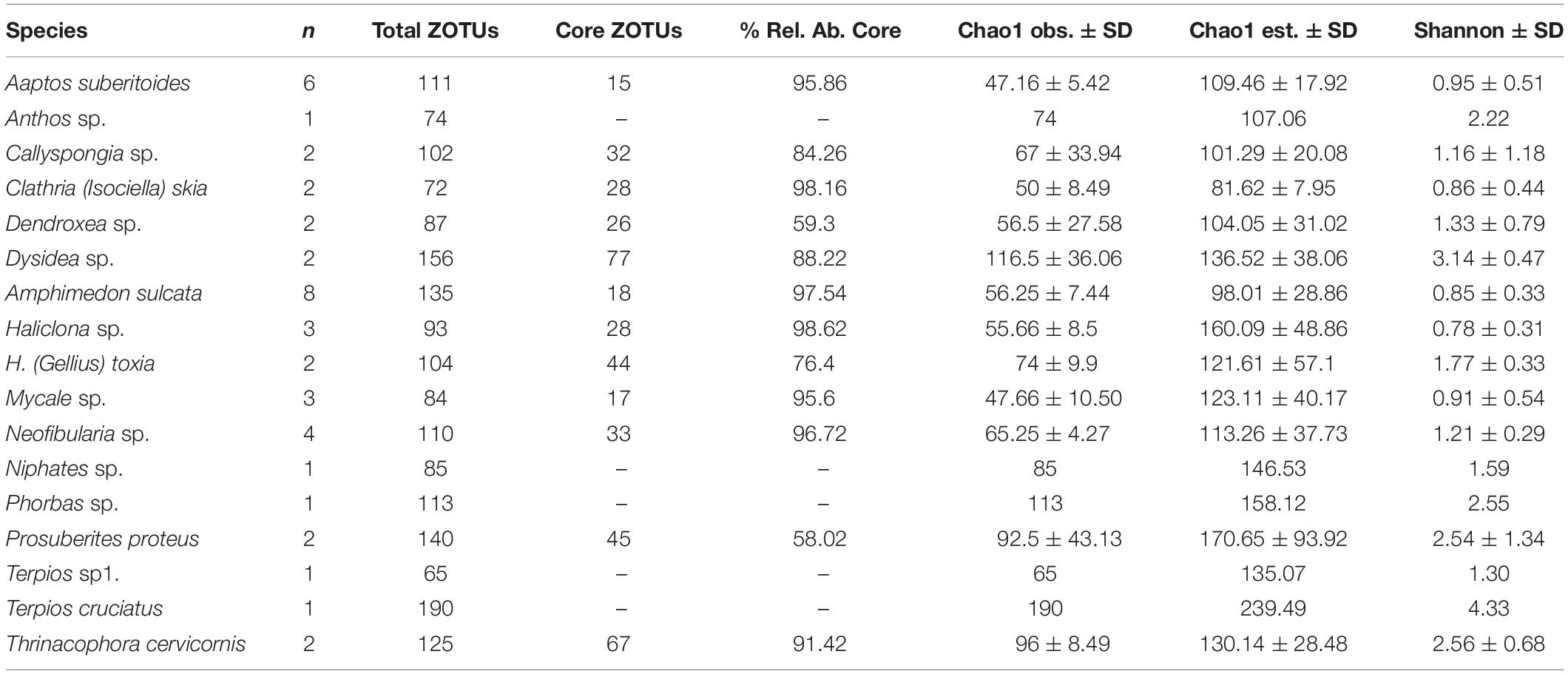
Table 1. Number of replicates, total ZOTUs, number and relative abundance of core ZOTUs, and diversity indices of the study species.
We obtained a total of 316 archaeal high-quality ZOTUs belonging to four different phyla Thaumarchaeota (∼85%), Nanoarchaeota (∼10%), Euryarchaeota (∼4%), and Diapherotrites (<0.5%). The most dominant genus were Candidatus Nitrosopumilus (∼60%), Nitrosopumilaceae unclassified (∼16%), Woesearchaeia unclassified (∼10%), Cenarchaeum (∼8%), Thermoplasmata Marine group II (∼4%) and Candidatus Nitrosopelagicus (∼1.5%). Of the 316 ZOTUs, 119 ZOTUs were found at relative abundances higher than 0.01% in at least one sample but only 39 ZOTUs were kept when the relative abundance threshold was set to 0.1%.
Clustering of sponge host species as a function of similarity among their archaeal communities showed that, in most cases, species replicates were more similar to each other than to other sponge species, with the few exceptions of Protosuberites proteus, Dendroxea sp. and Haliclona (Gellius) toxia (Figure 1). Overall, host identity was the main factor structuring the sponge archaeal communities (Adonis, Pseudo-F: 4.67, R2 = 0.74, p-value < 0.001), indicating a species-specific component of sponge archaeal composition.
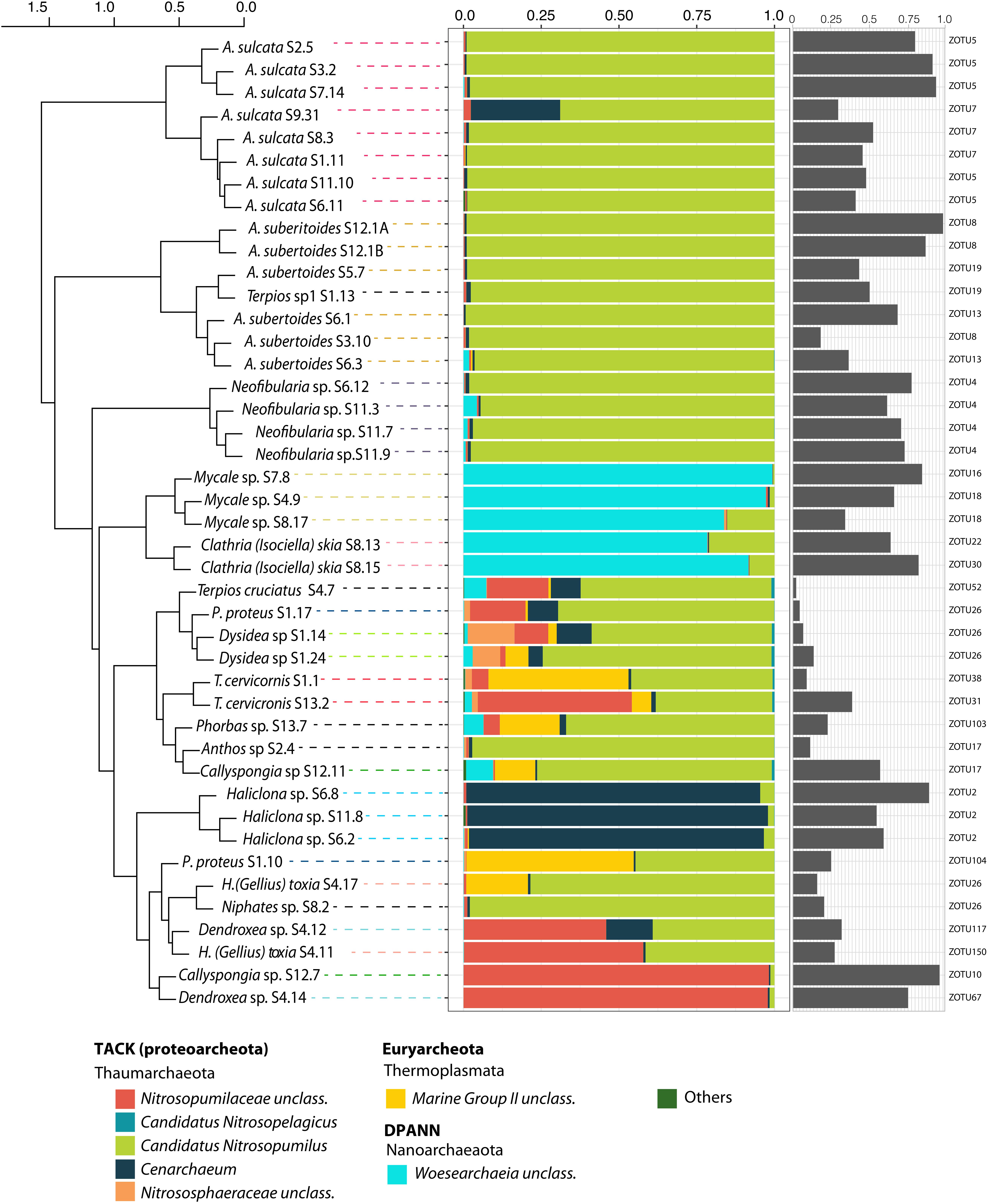
Figure 1. Relative abundance of archaeal community at the lowest taxonomic level for each sponge sample. Gray bars represent the relative abundance of the most dominant ZOTU for each sample. The dominant ZOTU number is indicated. Sponge samples are organized according to a hierarchical clustering (Ward method) based on Bray-Curtis dissimilarity matrix (at the level of ZOTU).
Most of the species analyzed were dominated by different genera of the Thaumarchaeota phylum. Among them, Candidatus Nitrosopumilus was the dominant genus in the majority of the species analyzed (e.g., Amphimedon sulcata, Aaptos suberitoides, Neofibularia sp., Terpios sp.1 and Anthos sp.) (Figure 1). Cenarchaeum was dominant only in the three replicates of Haliclona sp. with more than 50% of its relative abundance due to a single ZOTU (ZOTU2). Unclassified members of the Nitrosopumilaceae family were the major components of some replicates of Dendroxea sp., Callyspongia sp. and Haliclona (Gellius) toxia. Among the Nanoarchaetoa phylum, unclassified members of Woesarchaeia were the major components of Mycale sp. and Clathria (Isociella) skia.
The Dominant ZOTU
The relative abundance of the most abundant archaeal ZOTU per sponge species varied from 2% (Terpios cruciatus) to 98% (Aaptos suberitoides). Some microbiomes were dominated by a single ZOTU, while in some others; the most abundant archaeal ZOTU did not reach 15% of relative abundance. Intraspecies variation in the dominant ZOTU occurred in Amphimedon sulcata, A. suberitoides, Mycale sp. and Clathria (Isociella) skia, but consistency among replicates was found for some species, such as Neofibularia sp. (ZOTU 4), Dysidea sp. (ZOTU26) and Haliclona sp. (ZOTU2) (Figure 1). Most of the dominant ZOTUs belonged to Thaumarchaeota, except for those of Mycale sp. and C. (Isociella) skia, which belonged to Woesarchaeia and ZOTU104 (Euryarchaeota), which dominated in a replicate of Prosuberites proteus.
The Core Community
We detected five ZOTUs present across all sponge individuals (ZOTU 10, 4, 26, 17, 2), which constituted the archaeal core community. All of them belonged to the phylum Thaumarchaeota, family Nitrosopumilaceae. ZOTU 4, 26, and 17 belonged to Candidatus Nitrosopumilus, ZOTU 10 was an unclassified member of the Nitrosopumilaceae, and ZOTU 2 belonged to Cenarchaeum.
The number of ZOTUs that constituted the archaeal core community of each sponge species varied from 15 (Aaptos suberitoides) to 77 (Dysidea sp.) (Table 1). However, the relative abundance contribution of those core ZOTUs to the overall community was relatively high in most cases, accounting for more than 60% in the majority of the species. Thus, although the number of replicates analyzed influenced the number of ZOTUs that constitute the species core community, the relative abundance of the core ZOTUs remained stable regardless of the number of replicates. In this sense, the sponges with a higher number of replicates (A. suberitoides and Amphimedon sulcata), had a lower number (15 and 17, respectively) of core ZOTUs, but these ZOTUs accounted for more than 95% of the archaea relative abundance in the species. Thus, the small variable fraction was formed by low abundance ZOTUs, which indicated a high contribution of the few core ZOTUs to the overall archaeal community (Table 1).
Diversity Metrics
The Shannon diversity of sponge archaeal communities was in general low compared to the values obtained for bacterial communities (Figure 2), except for the species Terpios cruciatus that showed similar diversity for both biological domains. Most archaeal Shannon diversity ranged between 1 and 2, whereas values of Shannon diversity of the sponge bacterial communities ranged from 3 to 4. The Chao estimates showed that most species had values between 100 and 150 archaeal ZOTUs, except T. cruciatus with more than 200 ZOTUs (Table 1).
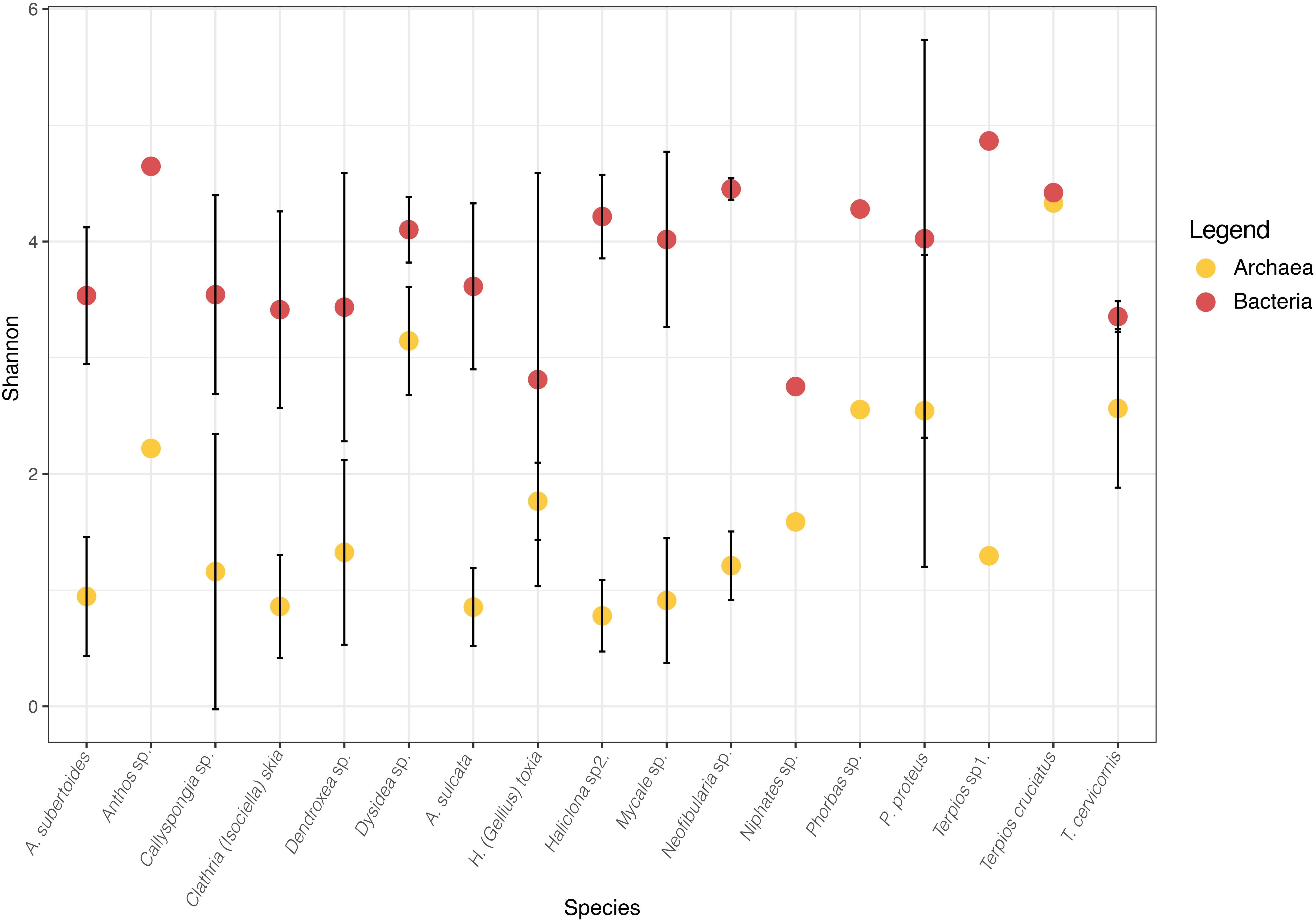
Figure 2. Mean Shannon diversity for Bacteria (red) and Archaea (yellow) of each sponge species. Bars represent Standard deviation. Bacteria data was obtained from Turon et al. (2018).
Phylogenetic Analysis
Only 3 (ZOTU 19, 58, and 150) out of the 39 ZOTUs added to the phylogenetic trees fell into monophyletic sponge enriched clusters within the Thaumarchaeota phylum (Figure 3). ZOTU19, the dominant ZOTU in the species Aaptos suberitoides and Terpios sp1 fell, together with ZOTU58, within the SC174 while ZOTU150, dominant in a replicate of H. (Gellius) toxia, fell within the SC175 (Figure 3). None of the five ZOTUs of the sponge core belonged to a SC cluster. However, ZOTU2 (Cenarchaeum), was related (96.5% identity) to the Cenarchaeum symbiosum cluster, formed by clones derived from Axinella mexicana. Blast search of the other core ZOTUs (ZOTUs 4, 26, 17, and 10) revealed that they were more similar (>96% identity) to archaeal clones retrieved from environmental samples (data not shown) than to other sponge derived clones (Figure 3). Whilst some of the Thaumarchaeota abundant ZOTUs were also similar to other archaeal sequences retrieved from sponges, they did not fell within SC clusters (Figure 3). None SC existed within Euryarchaeota phylum (Simister et al., 2012) and the four abundant Euryarchaeota ZOTUs from our dataset were distantly related to other sponge derived sequences (Supplementary Figure S1).
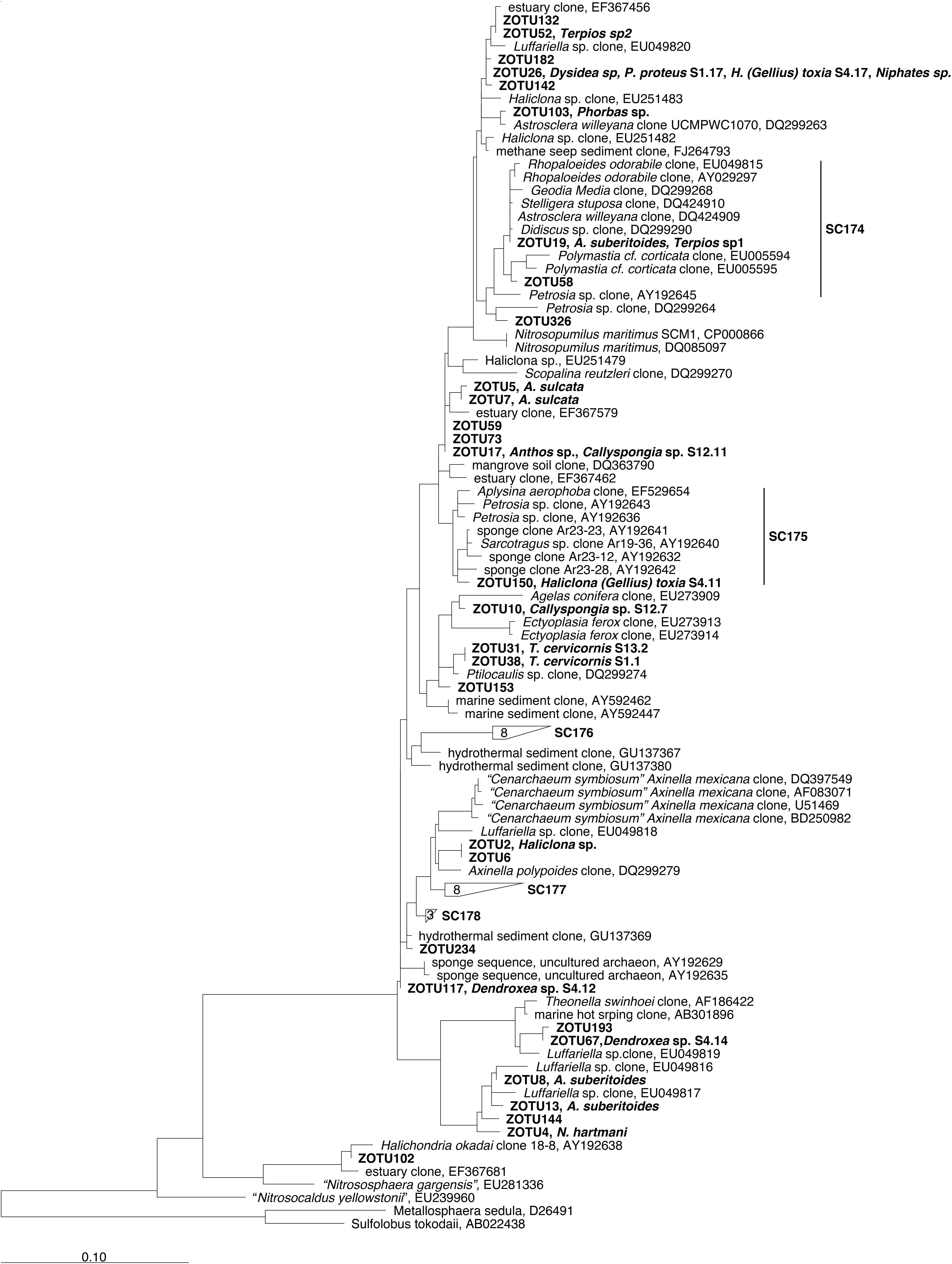
Figure 3. 16S rRNA-based phylogeny of sponge associated Thaumarchaeota reported in Simister et al. (2012). The displayed phylogeny is a maximum likelihood tree based on long sequences only (>1200 bp). Sponge-specific monophyletic clusters are indicated (SC num). ZOTUs obtained in our study were added to this tree using the parsimony interactive tool in ARB and are indicated in Bold. Sponge species replicate is indicated next to the ZOTU if that ZOTU is the dominant in this sponge. Bar indicates sequence divergence.
Noteworthy, all the Woesarchaeota ZOTUs, highly abundant in the species Mycale sp. and Clathria (Isociella) skia, fell all together and were far related to any existing Woesarchaeota sequence available in SILVA132 database (Figure 4). Additionally, blast search of these ZOTUs against NCBI database did not retrieve any sequence with higher identity than 85% (data not shown).
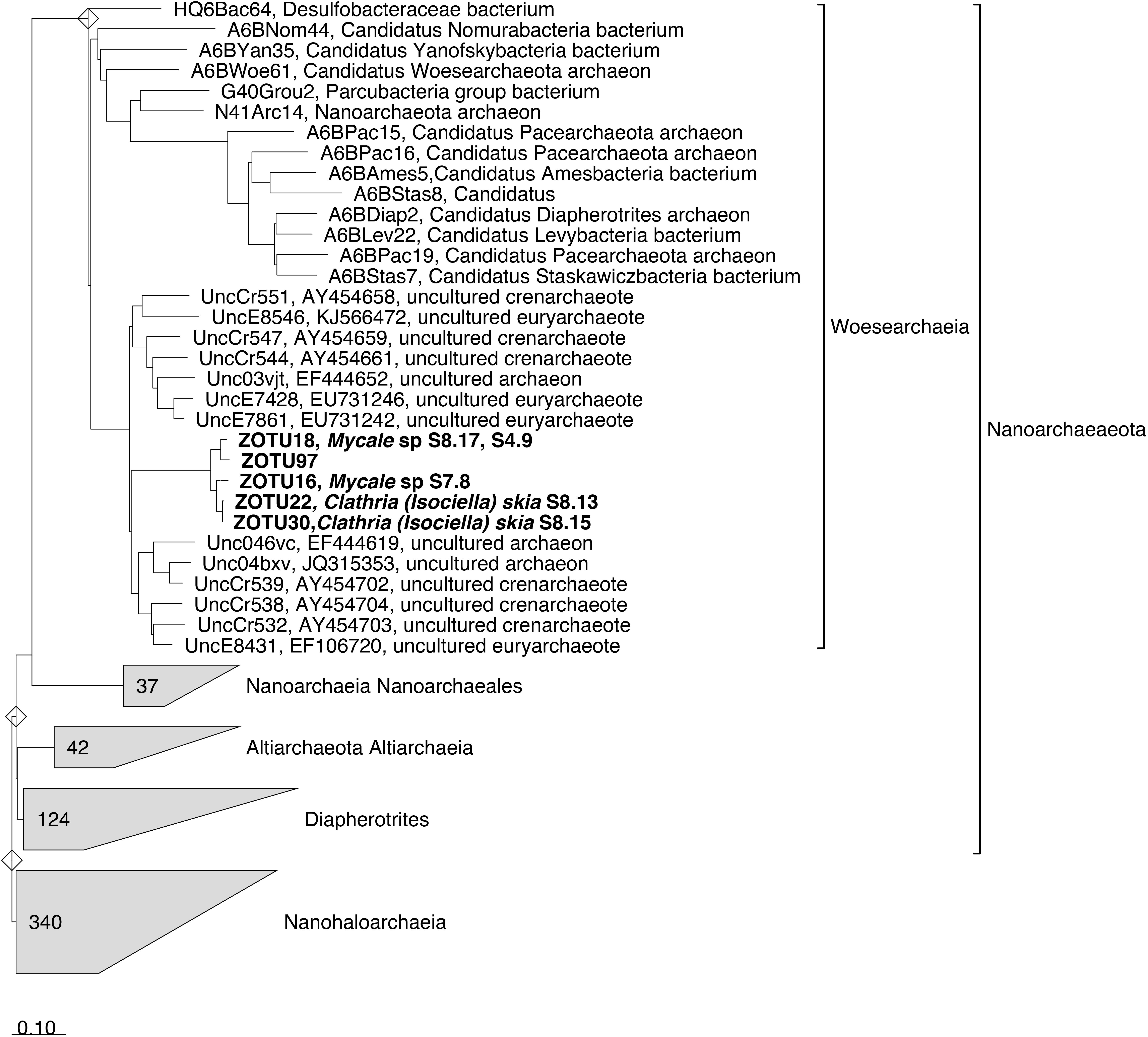
Figure 4. 16S rRNA Woesarchaeia tree imported from ARB (SILVA SSU Ref NR 132 database). ZOTUs obtained in this study were added using the parsimony interactive tool in ARB and are indicated in Bold. Sponge species replicate is indicated next to the ZOTU if that ZOTU is the dominant in this sponge. Bar indicates sequence divergence.
Functional Prediction
The amount of sequences used in the prediction of functionalities was fairly high in the majority of our species (Supplementary Figure S2). Most of the species had a total fraction of at least 75% of reads that matched a “known” genome in the reference database. However, Haliclona sp., Mycale sp. and Clathria sp. had to be removed from the analysis as more than 80% of their reads could not be predicted (Supplementary Figure S2). This poor prediction rate was likely due to the dominant archaea in these sponges, Woesarchaeota and Crenarchaetota, for which no genomes were available in the reference database used. Several members of Thaumarchaetoa, for which genomes were available, dominated in all other sponge species and most of their reads could be used in the functional predictions.
All the study sponge species showed a similar pattern of archaea functionalities. The predicted metagenomes of archaeal communities revealed that pathways related to the metabolism (carbohydrate, cofactors and vitamins, amino acid and energy), environmental processing (membrane transport), translation and cellular processes (cellular community-prokaryotes) were the most abundant (Figure 5). However, functions related with defense and competition mechanisms such as metabolisms related to biosynthesis of secondary metabolites (terpenoids and polyketides) and drug resistance (antimicrobial) functions were also present.
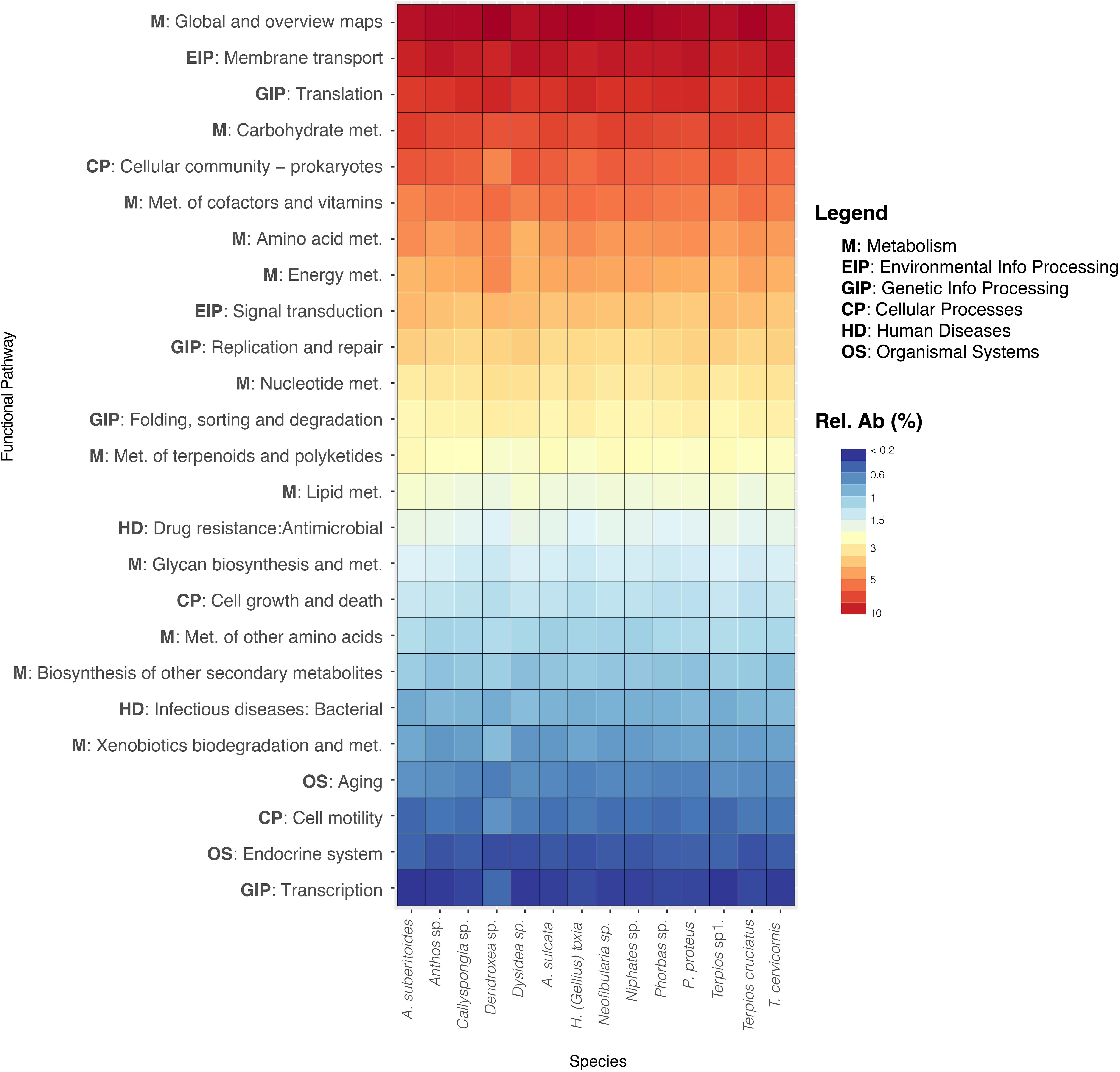
Figure 5. Heatmap of the most abundant functional pathways obtained from Tax4fun for each sponge species. Abundance is represented in the color temperature bar.
Discussion
Archaea Amplification Constrains
A critical step for accurate rDNA amplicon analysis is still the choice of appropriate primers (Klindworth et al., 2012), which has been proven particularly difficult for Archaea (Dupont et al., 2013). The use of inappropriate primer pairs, can lead to under-representation or selection against single species or even whole groups. Thus, between studies comparisons should be taken with care, as they could lead to equivoque conclusions, in particular when archaea composition and abundance are inferred from massive amplicon sequencing using primers primarily designed for bacteria (Klindworth et al., 2012) and vice-versa. The use of two couple of primers, one for bacteria amplification exclusively and the other for bacteria plus archaea identification has been recommended (Dupont et al., 2013) to obtain a more complete microbiome landscape. This approach was followed in our study, but amplification can be problematic even with archaea directed primers, as it occurred with our study sponges. Only 52 out from 68 samples belonging to 17 sponge species provided good-quality archaea sequences and from these, only 43 sponge samples retrieved over 5000 reads. However, despite inevitable primer constrains, the Archaea sequences obtained may provide useful information on the Kingdom characteristics where associated to sponges.
Sponge Archaeal Communities
The archaeal communities of our study sponges showed a relatively high species-specific component, as previously reported for other sponge species from other locations (Kennedy et al., 2014; Polónia et al., 2014; Zhang et al., 2014; Rodríguez-Marconi et al., 2015; De Mares et al., 2017) and for sponge-associated bacteria (Lee et al., 2011; Schmitt et al., 2012; Turon et al., 2018). However, a higher intra-species variability for both domains was found in the sponges Dendroxea sp. and Haliclona (Gellius) toxia, with some replicates that did not cluster together [also see Figure 3 in Turon et al. (2019a, b), for comparison with bacteria]. Conversely, Clathria (Isociella) skia and Mycale sp., showed a much higher species-specific component for the archaea than for bacteria communities. The opposite was reported for sponges from the Red Sea (Lee et al., 2011) in which high species specificity was recorded for bacterial communities but not for archaeal communities (Lee et al., 2011).
Taxonomic affiliation of the archaeal community showed that Candidatus Nitrosopumilus was the most abundant taxon in the majority of the study species, especially, in Amphimedon sulcata, Aaptos suberitoides and Neofibularia sp., where a few ZOTUs of Candidatus Nitrosopumilus accounted for most of the archaeal relative abundance. Dominance of this archaea species has also been recorded in a calcareous sponge (Lopes et al., 2019), in several Antarctic sponges (Rodríguez-Marconi et al., 2015), and in three Ircinia species and Mycale laxissima from the Mediterranean and Caribbean seas (Zhang et al., 2014). C. Nitrosopumilus was also highly abundant in other 18 sponges from the same region in Vietnam amplified with bacteria primers (Dat et al., 2018). These authors found many aerobic nitrifiers among both symbiotic bacteria and archaea [C. Nitrosopumilus (AOA), Nitrosococcus, Nitrosomonadaceae (AOB), and Nitrospira (NOB)] and pointed out the putative relevance of aerobic nitrification in those sponges. In addition to C. Nitrosopumilus, we also retrieved members of Cenarchaeaum, which had not been previously detected in the Vietnamese species (Dat et al., 2018). Particulary, ZOTU2, affiliated to Cenarchaeaum, was consistently found in all the sponge samples studied, although it was solely abundant in Haliclona sp., where it accounted for more than 50% of the archaea relative abundance. Members of Cenarchaeaum have been widely reported to be associated with sponges. Moreover, Thaumarchaeota phylum is the single widely reported archaea taxon in the Sponge Microbiome Project that includes sponges from all over the world (Moitinho-Silva et al., 2017).
Usually, few archaeal OTUs dominated the archaeal community of sponges from deep-sea environments (Jackson et al., 2013; Kennedy et al., 2014), polluted estuarine waters (Turque et al., 2010), arctic deep-waters (Pape et al., 2006), and Indonesian reefs (Polónia et al., 2014, this study). Indeed, the description of Cenarchaeum symbiosum associated to the marine sponge Axinella mexicana represented the first described symbiosis between sponges and archaea (Preston et al., 1996). Since then, many other sequences retrieved from sponge samples had been found to be identical or similar to that of C. symbiosum. The closest sequence retrieved by blast search of our ZOTU2 (Cenarchaeum) had 96.5% identity to the C. symbiosum clone (Ar20-3) found in an unidentified marine sponge from Korea (AY192631.1). Thus, although different phylotypes of this archaeon might exist (Margot et al., 2002), its presence in all sponge samples gives support to the cosmopolitan distribution of Cenarchaeum in sponges and points to a relevant role in the holobiont. However, the exclusive presence of C. symbiosum reported in some sponges, such as Petromica citrina (Turque et al., 2010) and A. mexicana (Preston et al., 1996), is rather an exceptional pattern in our samples.
Few ZOTUs belonging to unclassified members of order Woesearchaeia dominated the microbiomes of Mycale sp. and Clathria (Isociella) skia. To our knowledge, this linage has been only reported previously in two sponge species from the Mediterranean and two more species from Caribbean Sea (De Mares et al., 2017). These authors detected up to eight archaeal phyla, with Crenarchaetoa, Euryarchaeota, Thaumarchaeota, and Woesarchaeota being present in all samples. However, Cenarchaeaceae family was by far, the most dominant in their study species (De Mares et al., 2017). Blast search of our Woesearchaeia abundant ZOTUs did not retrieve any sequence with similarity higher than 85%, emphasizing the novelty of this recently described group (Castelle et al., 2015). Moreover, the dominant Woesarchaeia ZOTUs found in our sponges clustered together within the same monophyletic group. This group was far from any environmental archaeal clone sequence, suggesting the existence of a Sponge Enriched Cluster in the Nanoarchaeota phylum. Members of Woesarchaeia have only been detected in sponges with the advent of new sequencing technologies (Illumina), and thus only short amplicon reads are available. More studies are required to obtain long sequences of this group allowing for phylogenetic reconstructions to ascertain whether or not a Sponge Enriched Cluster in the Woesarchaeia exists.
Finally, Euryarchaeota have been typically reported in seawater (Lee et al., 2011; Jackson et al., 2013; Alex and Antunes, 2015) and are usually absent (Turque et al., 2010; Jackson et al., 2013) or detected at low abundances in sponges (Webster et al., 2001; Holmes and Blanch, 2007; Dupont et al., 2013; Kennedy et al., 2014; Polónia and Cleary, 2019). Only 10 Euryarchaeota sequences were retrieved from sponges in the revision from 2010 data in Simister et al. (2012), and no SC clusters were detected for this group (Taylor et al., 2007; Simister et al., 2012). However, the first pyrosequencing study of sponge archaeal communities (Lee et al., 2011) contributed to increase the number of Euryarchaeota sequences from sponges, and since then, they have been more frequently reported (De Mares et al., 2017). In our study sponges, Euryarchaeota of the Marine Group II were detected in several species being particularly abundant in Thrinacophora cervicornis, Phorbas sp., Haliclona (Gellius) toxia and Protosuberites proteus.
Archaeal Diversity
Many of the studied species presented an archaeal community mostly dominated by just few ZOTUs, as already reported for other sponge species (Jackson et al., 2013; Kennedy et al., 2014; Polónia et al., 2014; Polónia and Cleary, 2019). However, some other species such as Terpios cruciatus, Protosuberites proteus, Dysidea sp.1 and Thricanophora cervicornis, presented highest Shannon diversity indices, with equal contribution of many ZOTUs to the overall archaeal community. The number of observed ZOTUs per sample varied from 47 to 190, which agrees with the number of archaea OTUs reported for the Red Sea sponges Hyrtios erectus, Stylissa carteri, and Xestospongia muta (Lee et al., 2011), but it was much lower than the number of archaea of two Dysidea and Aplysina species, the latter (A. cauliformis) with up to 370 archaeal OTUs (De Mares et al., 2017).
In our study species, Shannon diversity was always higher for bacteria than for archaeal communities. The dichotomy between HMA and LMA, which was clear for the bacterial domain in terms of alpha diversity and indicator phyla (Turon et al., 2018), could not be confirmed for the archaeal communities. In contrast to sponge-associated bacteria, which show contrasting composition and specific bacterial phyla in HMA and LMA species, neither specific archaeal phyla nor Shannon diversity differentiated the archaea assemblages in HMA and LMA sponges in our study. However, only two HMA species (Aaptos suberitoides and Neofibularia sp.) could be analyzed in the present study, and thus, these conclusions should be taken with care. Bayer et al. (2014) found up to four orders of magnitude higher archaeal gene numbers in HMA than in LMA sponges, although they also found an exception in S. carteri, a LMA sponge with a number of archaeal OTUs within the range of HMA sponges. Moreover, the correspondence between low abundance and low phylum-level diversity of microorganisms found for the bacterial communities of two LMA Dysidea species was not confirmed for the archaeal and eukaryote domains (De Mares et al., 2017).
The Archaeal Core Community
The archaeal core community of the sponge species that had replicates was similar to the bacterial core community in terms of relative abundance; however, the former was mostly dominated by a few abundant symbionts. We are aware that having a variable number of replicates per species might influence the number of core-species ZOTUs, however, the core size (in terms of relative abundance) remained stable regardless of the number of replicates (i.e., sponge species with higher number of replicates still had a core size over 90%). Only five ZOTUs were found in the archaeal core community of the 17 study sponge, although none of them belonged to the known SC archaeal clusters. The ubiquity of C. symbiosum in sponges has been long recognized (Preston et al., 1996; Turque et al., 2010; Jackson et al., 2013; Kennedy et al., 2014) and ammonia oxidation within the sponge, which could play an important role in ammonia detoxification, has been related to this Cenarchaeal symbiont (Steger et al., 2008).
The presence of AOA in sponge larvae suggested the vertical inherence of C. symbiosum (Steger et al., 2008). However, we found four ZOTUs belonging to Nitrosopumilales in the sponge core, whose closest sequences were clones derived from environmental samples, which advises to investigate more in deep a purported horizontal acquisition of these archaea from the surrounding seawater. Overall, a combination of both, vertical and horizontal transmission of archaeal symbionts might exist in sponges, as already reported for the bacterial domain (Sipkema et al., 2015; Björk et al., 2019) although comparisons with seawater archaea from the sponge neighboring could contribute to support this issue.
Predictive Metagenomic Analysis
Functional prediction of sponges dominated by Woesarchaeia and Cenarchaeum could not be made, as sequences of those groups did not match any close genome in the database used. This fact puts into relevance the lack of knowledge of the archaeal domain, specifically for the newly described Woesarchaeia group, for which no genome is available yet.
Most of our Archaea ZOTUs showed similar predicted functional patterns, which is likely due to a similar archaeal composition in the species analyzed, as archaea functional capabilities have been predicted from closely related metagenomes. Similar approaches have been used to predict functionalities of sponge archaeal communities in the sponges Biemna fortis (Polónia and Cleary, 2019) and P. magna (Lopes et al., 2019). Members of the Thaumarchaeota phylum also dominated those species and the most abundant pathways were related to metabolisms of nitrogenated compounds, energy metabolism, and cellular maintenance (Lopes et al., 2019). Our results also predict a high abundance of heterotrophic metabolisms, followed by environmental and genetic information processing, and secondary metabolisms, likely involved in allopatric microbe-microbe interactions.
Conclusion
The analysis of the archaeal communities of 17 tropical sponge species allowed us to record patterns of diversity and stability, which, as for bacterial communities, depended on the sponge species. Other traits such as high relative abundance of core communities run in parallel for archaea and bacteria in sponge microbiomes. In contrast, the number of ZOTUs that formed the core species community was lower for archaea than for bacteria and the dichotomy between HMA and LMA, which was clear for the bacterial domain could not be confirmed for the archaeal communities. The lack of seawater samples from the sponge surroundings prevented us to analyze the possible acquisition of archaea from the seawater by the sponge. However, many Thaumarchaeota and Euryarchaeota sequences obtained from the study sponges were close to sequences from environmental samples and, thus, their acquisition from the seawater cannot be discarded. However, a few ZOTUs belonging to unclassified members of order Woesearchaeia, which dominated the microbiomes of two poecilosclerid species analyzed, formed a monophyletic group far from any environmental archaeal clone sequence. This group might correspond to a Sponge Enriched Cluster within the Woesearchaeia, although long sequences of these ZOTUs are required to be used in phylogenetic reconstructions to confirm its monophyly.
Data Availability Statement
Raw 16S rRNA gene amplicon sequences are available in the SRA archive under the project number PRJNA453898 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA453898). Accession numbers for sponge eukaryotic sequences are reported in the author’s previous publication (Turon et al., 2018).
Author Contributions
MT and MU conceived the study and wrote the manuscript. MT performed the analyses.
Funding
This research was partially funded by projects MarSymbiOmics (CTM2013-43287-P) and PopCOmics (CTM2017-88080, MCIU/AEI/FEDER/UE) from the Spanish MINECO.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank D. Martin (CEAB, Blanes), T. A. Britayev (RAS, Russia), and E. S. Mekhova (RAS, Russia) for the help provided in the sponge sampling. Special thanks to R. L. Simister and C. Astudillo for kindly providing the ARB SILVA database containing the SC archaeal clusters.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00789/full#supplementary-material
Footnotes
References
Alex, A., and Antunes, A. (2015). Pyrosequencing characterization of the microbiota from Atlantic intertidal marine sponges reveals high microbial diversity and the lack of co-occurrence patterns. PLoS One 10:e0127455. doi: 10.1371/journal.pone.0127455
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Bayer, K., Moitinho-Silva, L., Brümmer, F., Cannistraci, C. V., Ravasi, T., and Hentschel, U. (2014). GeoChip-based insights into the microbial functional gene repertoire of marine sponges (high microbial abundance, low microbial abundance) and seawater. FEMS Microbiol. Ecol. 90, 832–843. doi: 10.1111/1574-6941.12441
Björk, J. R., Díez-Vives, C., Astudillo-García, C., Archie, E. A., and Montoya, J. M. (2019). Vertical transmission of sponge microbiota is inconsistent and unfaithful. Nat. Ecol. Evol. 3, 1172–1183. doi: 10.1038/s41559-019-0935-x
Blanquer, A., Uriz, M. J., and Galand, P. E. (2013). Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ. Microbiol. 15, 3008–3019. doi: 10.1111/1462-2920.12261
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 7, 335–336.
Castelle, C. J., Wrighton, K. C., Thomas, B. C., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Dat, T. T. H., Steinert, G., Thi, Kim Cuc N, Smidt, H., Sipkema, D., and Sipkema, D. (2018). Archaeal and bacterial diversity and community composition from 18 phylogenetically divergent sponge species in Vietnam. Peer J 6:e4970. doi: 10.7717/peerj.4970
De Mares, M. C., Sipkema, D., Huang, S., Bunk, B., Overmann, J., and van Elsas, J. D. (2017). Host specificity for bacterial, archaeal and fungal communities determined for high- and low-microbial abundance sponge species in two genera. Front. Microbiol. 8:2560. doi: 10.3389/fmicb.2017.02560
Dittami, S. M., Arboleda, E., Auguet, J.-C., Bigalke, A., Briand, E., Cardenas, P., et al. (2019). A community perspective on the concept of marine holobionts: current status, challenges, and future directions. PeerJ Prepr. 7:e27519v3. doi: 10.7287/peerj.preprints.27519v3
Dupont, S., Corre, E., Li, Y., Vacelet, J., and Bourguet-Kondracki, M. L. (2013). First insights into the microbiome of a carnivorous sponge. FEMS Microbiol. Ecol. 86, 520–531. doi: 10.1111/1574-6941.12178
Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing.. bioRxiv. [preprint]. doi: 10.1101/081257
Edgar, R. C. (2017). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. bioRxiv. [preprint]. doi: 10.1101/192211
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Gerçe, B., Schwartz, T., Syldatk, C., and Hausmann, R. (2011). Differences between bacterial communities associated with the surface or tissue of mediterranean sponge species. Microb. Ecol. 61, 769–782. doi: 10.1007/s00248-011-9802-2
Hentschel, U., Fieseler, L., Wehrl, M., Gernert, C., Steinert, M., Hacker, J., et al. (2003). Microbial diversity of marine sponges. Prog. Mol. Subcell. Biol. 37, 59–88. doi: 10.1007/978-3-642-55519-0_3
Hoffmann, F., Radax, R., Woebken, D., Holtappels, M., Lavik, G., Rapp, H. T., et al. (2009). Complex nitrogen cycling in the sponge Geodia barretti. Environ. Microbiol. 11, 2228–2243. doi: 10.1111/j.1462-2920.2009.01944.x
Holmes, B., and Blanch, H. (2007). Genus-specific associations of marine sponges with group I crenarchaeotes. Mar. Biol. 150, 759–772. doi: 10.1007/s00227-006-0361-x
Hsieh, T. C., Ma, K. H., and Chao, A. (2019). iNEXT: iNterpolation and EXTrapolation for species diversity. R Package Version 2.0.19. Available at: http://chao.stat.nthu.edu.tw/wordpress/software_download/inext-online/
Jackson, S. A., Flemer, B., McCann, A., Kennedy, J., Morrissey, J. P., O’Gara, F., et al. (2013). Archaea appear to dominate the microbiome of Inflatella pellicula Deep sea sponges. PLoS One 8:e84438. doi: 10.1371/journal.pone.0084438
Kennedy, J., Flemer, B., Jackson, S. A., Morrissey, J. P., O’Gara, F., and Dobson, A. D. W. (2014). Evidence of a putative deep sea specific microbiome in marine sponges. PLoS One 9:e91092. doi: 10.1371/journal.pone.0091092
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, 1–11. doi: 10.1093/nar/gks808
Lee, O. O., Wang, Y., Yang, J., Lafi, F. F., Al-Suwailem, A., and Qian, P. Y. (2011). Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 5, 650–664. doi: 10.1038/ismej.2010.165
Liu, M. Y., Kjelleberg, S., and Thomas, T. (2011). Functional genomic analysis of an uncultured δ-proteobacterium in the sponge Cymbastela concentrica. ISME J. 5, 427–435. doi: 10.1038/ismej.2010.139
Lopes, M. F., Klautau, M., Esteves, E. L., Albano, R. M., Janeiro, R., Janeiro, R., et al. (2019). Microbiota of the alien species Paraleucilla magna (Porifera, Calcarea) from the Southwestern Atlantic, and a comparison with that of other calcareous sponges. bioRxiv. [preprint]. doi: 10.1101/626192
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Margot, H., Acebal, C., Toril, E., Amils, R., and Fernandez Puentes, J. L. (2002). Consistent association of crenarchaeal Archaea with sponges of the genus Axinella. Mar. Biol. 140, 739–745. doi: 10.1007/s00227-001-0740-2
Moeller, F. U., Webster, N. S., Herbold, C. W., Behnam, F., Domman, D., Albertsen, M., et al. (2019). Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella basta. Environ. Microbiol. 21, 3831–3854. doi: 10.1111/1462-2920.14732
Moitinho-Silva, L., Nielsen, S., Amir, A., Gonzalez, A., Ackermann, G. L., Cerrano, C., et al. (2017). The sponge microbiome project. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix077
Mußmann, M., Brito, I., Pitcher, A., Sinninghe Damsté, J. S., Hatzenpichler, R., Richter, A., et al. (2011). Thaumarchaeotes abundant in refinery nitrifying sludges express amoA but are not obligate autotrophic ammonia oxidizers. Proc. Natl. Acad. Sci. U.S.A. 108, 16771–16776. doi: 10.1073/pnas.1106427108
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., Mcglinn, D., et al. (2018). Vegan: Community Ecology Package. R package version 2.5–1.
Pape, T., Hoffmann, F., Quéric, N. V., Von Juterzenka, K., Reitner, J., and Michaelis, W. (2006). Dense populations of Archaea associated with the demosponge Tentorium semisuberites schmidt, 1870 from Arctic deep-waters. Polar Biol. 29, 662–667. doi: 10.1007/s00300-005-0103-4
Pester, M., Rattei, T., Flechl, S., Gröngröft, A., Richter, A., Overmann, J., et al. (2012). AmoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions. Environ. Microbiol. 14, 525–539. doi: 10.1111/j.1462-2920.2011.02666.x
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Polónia, A. R. M., and Cleary, D. F. R. (2019). Archaeal communities in sponge, sediment and water from marine lakes and open water habitats. Mar. Biol. Res. 15, 259–274. doi: 10.1080/17451000.2019.1633469
Polónia, A. R. M., Cleary, D. F. R., Duarte, L. N., de Voogd, N. J., and Gomes, N. C. M. (2014). Composition of Archaea in seawater, sediment, and sponges in the Kepulauan Seribu reef system. Indonesia. Microb. Ecol. 67, 553–567. doi: 10.1007/s00248-013-0365-2
Preston, C. M., Wu, K. Y., Molinski, T. F., and Delong, E. F. (1996). A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc. Natl. Acad. Sci. U.S.A. 93, 6241–6246. doi: 10.1073/pnas.93.13.6241
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Radax, R., Hoffmann, F., Rapp, H. T., Leininger, S., and Schleper, C. (2012a). Ammonia-oxidizing archaea as main drivers of nitrification in cold-water sponges. Environ. Microbiol. 14, 909–923. doi: 10.1111/j.1462-2920.2011.02661.x
Radax, R., Rattei, T., Lanzen, A., Bayer, C., Rapp, H. T., Urich, T., et al. (2012b). Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ. Microbiol. 14, 1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x
Ribes, M., Jiménez, E., Yahel, G., López-Sendino, P., Diez, B., Massana, R., et al. (2012). Functional convergence of microbes associated with temperate marine sponges. Environ. Microbiol. 14, 1224–1239. doi: 10.1111/j.1462-2920.2012.02701.x
Rodríguez-Marconi, S., De La Iglesia, R., Díez, B., Fonseca, C. A., Hajdu, E., Trefault, N., et al. (2015). Characterization of bacterial, archaeal and eukaryote symbionts from antarctic sponges reveals a high diversity at a three-domain level and a particular signature for this ecosystem. PLoS One 10:e0138837. doi: 10.1371/journal.pone.0138837
Schmitt, S., Angermeier, H., Schiller, R., Lindquist, N., and Hentschel, U. (2008). Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 74, 7694–7708. doi: 10.1128/AEM.00878-08
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576. doi: 10.1038/ismej.2011.116
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524. doi: 10.1111/j.1462-2920.2011.02664.x
Sipkema, D., de Caralt, S., Morillo, J. A., Al-Soud, W. A. B., Sørensen, S. J., Smidt, H., et al. (2015). Similar sponge-associated bacteria can be acquired via both vertical and horizontal transmission. Environ. Microbiol. 17, 3807–3821. doi: 10.1111/1462-2920.12827
Steger, D., Ettinger-Epstein, P., Whalan, S., Hentschel, U., De Nys, R., Wagner, M., et al. (2008). Diversity and mode of transmission of ammonia-oxidizing archaea in marine sponges. Environ. Microbiol. 10, 1087–1094. doi: 10.1111/j.1462-2920.2007.01515.x
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-Associated Microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Taylor, M. W., Tsai, P., Simister, R. L., Deines, P., Botte, E., Ericson, G., et al. (2013). Sponge-specific bacteria are widespread (but rare) in diverse marine environments. ISME J. 7, 438–443. doi: 10.1038/ismej.2012.111
Tian, R. M., Wang, Y., Bougouffa, S., Gao, Z. M., Cai, L., Bajic, V., et al. (2014). Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 16, 3548–3561. doi: 10.1111/1462-2920.12586
Turon, M., Cáliz, J., Garate, L., Casamayor, E. O., and Uriz, M. J. (2018). Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 8:15201. doi: 10.1038/s41598-018-33545-1
Turon, M., Cáliz, J., Triadó-Margarit, X., Casamayor, E. O., and Uriz, M. J. (2019a). Sponges and their microbiomes show similar community metrics across impacted and well-preserved reefs. Front. Microbiol. 10:1961. doi: 10.3389/fmicb.2019.01961
Turon, M., Uriz, M. J., and Martin, D. (2019b). multipartner symbiosis across biological domains: looking at the eukaryotic associations from a microbial perspective. mSystems 4, 1–14. doi: 10.1128/mSystems.00148-19
Turque, A. S., Batista, D., Silveira, C. B., Cardoso, A. M., Vieira, R. P., Moraes, F. C., et al. (2010). Environmental shaping of sponge associated archaeal communities. PLoS One 5:e15774. doi: 10.1371/journal.pone.0015774
Vacelet, J., and Donadey, C. (1977). Electron microscope study of the association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 30, 301–331. doi: 10.1016/0022-0981(77)90038-7
Webster, N. S., Watts, J. E. M., and Hill, R. T. (2001). Detection and phylogenetic analysis of novel crenarchaeote and euryarchaeote 16s ribosomal RNA gene sequences from a Great Barrier Reef sponge. Mar. Biotechnol. 3, 600–608.
Weisz, J. B., Hentschel, U., Lindquist, N., and Martens, C. S. (2007). Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar. Biol. 152, 475–483. doi: 10.1007/s00227-007-0708-y
Wemheuer, F., Taylor Jessica, A., Daniel, R., Johnston, E., Meinicke, P., Thomas, T., et al. (2018). Tax4fun2: a R-based tool for the rapid prediction of habitat-specific funtional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. bioRxiv. [preprint]. doi: 10.1101/490037
Wulff, J. L. (2006). Ecological interactions of marine sponges. Can. J. Zool. 84, 146–166. doi: 10.1139/z06-019
Keywords: archaea, symbiosis, sponges, diversity, central pacific
Citation: Turon M and Uriz MJ (2020) New Insights Into the Archaeal Consortium of Tropical Sponges. Front. Mar. Sci. 6:789. doi: 10.3389/fmars.2019.00789
Received: 17 October 2019; Accepted: 06 December 2019;
Published: 08 January 2020.
Edited by:
Russell T. Hill, University of Maryland, United StatesReviewed by:
Zhiyong Li, Shanghai Jiao Tong University, ChinaLucia Pita, GEOMAR Helmholtz Centre for Ocean Research Kiel, Germany
Copyright © 2020 Turon and Uriz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria J. Uriz, aW9zdW5lQGNlYWIuY3NpYy5lcw==