 Bing-Jian Liu
Bing-Jian Liu Yu-Long Li1,3,4
Yu-Long Li1,3,4- 1CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2National Engineering Laboratory of Marine Germplasm Resources Exploration and Utilization, Zhejiang Ocean University, Zhoushan, China
- 3Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 4Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
The Japanese eel (Anguilla japonica) is a commercially important aquatic species in East Asia. The number of the Japanese eels has been dramatically declining over the last four decades, and it is now listed as an endangered species (International Union for Conservation of Nature [IUCN] 2014). To manage and conserve this endangered species, it is necessary to assess population genetic diversity, genetic structure, and identify regions of the genomes that are under selection. Here, we generated a catalog of novel single-nucleotide polymorphism (SNP) markers for the Japanese eel using restriction site-associated DNA (RAD) sequencing of 24 individuals from two geographic locations. The 73,557 identified SNPs were widely distributed across the draft genome of the Japanese eel. No genetic differentiation between the two populations was detected based on all loci or neutral loci. However, highly significant genetic differentiation was detected based on loci that appeared to be under selection. BLAST2GO annotations of the outlier SNPs yielded hits for 61 (72%) of 85 significant BLASTX matches. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified some of the putative targets of local selection, including genes in several important pathways such as calcium signaling pathway and intestinal immune network for IgA production. This SNP catalog will provide a valuable resource for future population genetic and genomic studies and allows for targeting specific genes and genomic regions under selection in the Japanese eel genome.
Introduction
Recent advances in next-generation sequencing (NGS) technologies have greatly improved the speed of genome sequencing, facilitating the application of genomic approaches in many research areas, including ecology, conservation, fisheries genetics, and so on (Hudson, 2008; Harismendy et al., 2009; Allendorf et al., 2010; Ekblom and Galindo, 2011; Dudgeon et al., 2012). The high-throughput sequencing methods in NGS can result in extremely large collections of data, which enables the identification of numerous sensitive markers for a high-resolution genetic analysis, such as single-nucleotide polymorphisms (SNPs) (Ekblom and Galindo, 2011). However, because of the large and complex genomes of research organisms, researchers have typically used complexity-reduction technology for SNP discovery (Slate et al., 2009). One promising approach is restriction site-associated DNA (RAD) sequencing, which reduces genome complexity by sequencing the same loci across the genomes of numerous individuals, enabling comparisons between individuals, and reducing sequencing cost. This approach consists of employing specific restriction enzymes to cleave double-stranded genomic DNA into random fragments and then amplifying and sequencing the regions flanking the restriction enzyme cut sites through specific adapters and NGS platforms, such as Illumina. The paired-end reads from each RAD tag can then be assembled into long contiguous sequences or aligned to a known reference to identify and score 1000s of genetic markers (Miller et al., 2007; Baird et al., 2008; Hohenlohe et al., 2010, 2011). To date, RAD sequencing has been successfully applied to SNP discovery in many species, such as three-spined stickleback (Gasterosteus aculeatus) (Baird et al., 2008; Hohenlohe et al., 2010), European eel (Anguilla anguilla) (Pujolar et al., 2013), sea mullet (Mugil cephalus) (Kruck et al., 2013), and rainbow trout (Oncorhynchus mykiss) (Palti et al., 2014). As sequencing cost continues to drop, these methods will have numerous applications in a genetic analysis of complex genomes.
The Japanese eel, Anguilla japonica, is an important economical marine fishery species distributed in East Asian countries, which is characterized as a temperate catadromous fish with a complex life cycle and an extensive migratory loop. The spawning area of this species is located in the western Mariana Islands, near 14°–16° N, 142°–143° E (Tsukamoto, 1992, 2006). After being spawned between April and November (Tsukamoto, 1992), leptocephalus larvae disperse via the North Equatorial Current (NEC) and the Kuroshio Current (KC). Four to six months later, they reach the coasts of East Asia and then develop into juvenile Japanese eels (which are typically referred to as “glass eels”). Glass eels then move into continental (freshwater, brackish, or coastal) growth habitats and become yellow eels. After more than four 4 years of feeding, they develop into mature silver eels and then migrate approximately 2,000 to 3,500 km back to the western Mariana Islands, spawn once, and die (Han et al., 2010b). The abundance of Japanese eels has sharply decreased by more than 90% since the 1970s, owing to factors such as commercial overfishing, habitat destruction, pollution, and other environmental changes (Han et al., 2010a). In March 2014, the Japanese eel was listed as an endangered species in the International Union for Conservation of Nature (IUCN) Red List of Threatened Species1. Hence, an assessment of population genetic structure, genetic diversity, and regions of the genome that are under selection for Japanese eel is urgently needed in order to better manage and conserve this endangered species.
Despite occupying a broad range of habitats spanning almost the entire coastal area of East Asia, the Japanese eel has been regarded as a panmictic species. The concept of panmictic populations for Japanese eel was originally established by genetic studies focusing on allozyme and mitochondrial DNA (mtDNA) markers (Sang et al., 1994; Ishikawa et al., 2001). In addition, Han et al. (2010a) used eight polymorphic microsatellite DNA loci to investigate the spatial population genetic structures of Japanese eels collected from nine locations in East Asia, resulting in no identified genetic differentiation between different geographical locations. Gong et al. (2014) used mtDNA and microsatellite DNA loci to investigate the population structure of Japanese glass eels collected from 10 locations in China, which provided further evidence for panmixia in Japanese eel. However, significant geographical clines have been detected in this species. For example, Chan et al. (1997) found that A. japonica in the western Pacific fringe exhibited clear geographic clines in two allozyme loci. Moreover, Tseng et al. (2006) conducted an analysis of eight microsatellite loci and found that Japanese eel populations could be divided into two major groups.
Owing to the inconclusive and often conflicting results of earlier research, a better understanding of crucial aspects of the population genetic structure in the Japanese eel is still needed. Here, we developed a genome-wide catalog of SNPs for the Japanese eel by employing Illumina HiSeq 2500 paired-end sequencing of RAD tags (Baird et al., 2008). Previous studies in this area either sequenced single-end reads (Hohenlohe et al., 2011; Palti et al., 2014; Guo et al., 2015) or sequenced paired-end reads but only used the single reads (Baird et al., 2008; Wang et al., 2013; Pujolar et al., 2014), which resulted in less accurate alignments. In the present study, we compared the number of SNPs generated from single-end and paired-end data, and we found that paired-end sequencing resulted in more high-quality SNPs. The RAD tags in this study were generated from a total of 24 glass eels from two separate sampling locations, enabling the discovery of novel candidate SNP markers for this widespread, highly fecund marine fish species. The SNPs were then used in a population genetic analysis of two Japanese eel populations, which improved the current understanding of population structure and identified genomic regions that are subject to selection. This improved our understanding of Japanese eel populations, which have significant implications for the sustainable management and utilization of these important resources.
Materials and Methods
Ethics Statement
All methods were performed in strict accordance with the relevant guidelines and regulations in China.
Sample Preparation and Restriction Site-Associated DNA Tag Sequencing
Samples of glass eels were collected from two geographically distant locations: one is in Dandong, Liaoning Province (39°58′ N; 124°12′ E), and the other is in Xiapu, Fujian Province (26°55′ N; 120°13′ E). Tissues were kept in 95% ethanol and stored at −80°C. Genomic DNA was extracted from a total of 24 individuals (12 from each location) using standard phenol–chloroform extraction. The quality of the DNA samples was checked on 1% agarose gels and measured using a Nanodrop 2000 spectrophotometer and Qubit fluorometric quantitation, in order to ensure that the concentration was more than 25 ng/μl and the total DNA weight was no less than 1 μg. RAD libraries were prepared as described by Zhang et al. (2016).
Restriction Site-Associated DNA Data Analysis and Single-Nucleotide Polymorphism Identification
Sequencing reads from the Illumina runs were filtered to generate clean reads and then were aligned to the Japanese eel genome draft2 using BWA version 0.7.12 (Li and Durbin, 2010). The BWA-MEM algorithm was used for mapping with default parameters except for adjusting the seeding length to 32. SNP calling was performed by the SAMTOOLS 0.1.19 software package (Li et al., 2009) with a maximum read depth of 1,000. This package consists of two key programs, SAMTOOLS and BCFTOOLS, which incorporate genotype likelihood and call variants using Bayesian inference. In order to remove false positives, putative SNPs were required to fit the following criteria: (i) only bi-allelic SNPs were retained; (ii) SNPs with an average phred-scaled quality score > 30 (Q30) were retained; (iii) a minimum read depth of 10 for each individual was required; (iv) the minimum individual coverage for each population was set to 80%; v) a global minor allele frequency (MAF) of 0.05 was applied; vi) SNPs with FIS values < -0.3 or > 0.3 and observed heterozygosity (HO) values > 0.5 were discarded to exclude false SNPs, which might have resulted from the paralog clusters (Hohenlohe et al., 2011). All subsequent datasets used were reformatted with PGDSPIDER version 2.0.5.2 (Lischer and Excoffier, 2012).
Outlier Detection
FST-based outlier tests implemented in ARLEQUIN v3.5.2.2 (Excoffier and Lischer, 2010) were applied to test for selection between the two samples. This approach obtains the distribution of FST across loci as a function of heterozygosity between populations by performing coalescent simulations under a symmetrical two island model assuming near-random mating in order to reduce the number of false-positive loci (Beaumont and Nichols, 1996). For each outlier locus, FST values located above the 99.5th quantile of the simulated distribution were selected.
Population Genetic Analyses
The package VCFTOOLS (Danecek et al., 2011) was used to estimate genome-wide nucleotide FST values, as well as observed (HO) and expected (HE) heterozygosity at each SNP for all individuals. Population pairwise FST values were calculated using ARLEQUIN v3.5.2.2 (Excoffier and Lischer, 2010) and significance was determined using 10,000 permutations. Furthermore, ADMIXTURE v1.23 (Alexander et al., 2009) was used for maximum likelihood estimation of individual ancestries from multi-locus SNP genotype datasets. The analysis was performed with values of K ranging from 1 to 3, and the K with the lowest cross-validation error was selected. NETVIEW P 0.7.1 (Steinig et al., 2016), which is an implementation of NETVIEW (Neuditschko et al., 2012) in Python, was used to infer population structure including individual- and family-level relationships of populations on the basis of putative neutral SNPs and outlier datasets. A K-nearest neighbor (KNN) value of 10 was selected for visualization in CYTOSCAPE 3.2.1 (Shannon et al., 2003) as suggested by the author. The analyses of population pairwise FST, ADMIXTURE, and NETVIEW were conducted based on all SNPs, neutral SNPs, and outlier datasets.
BLAST Analyses and Gene Ontology Annotation
Contigs containing outlier SNPs were used as queries in nucleotide searches using local BLASTX against the Genome Reference Consortium Zebrafish Build 10 (GRCz10) proteins with an E-value threshold of 1E-5. Next, functional annotations of these genes were obtained using the BLAST2GO suite (Götz et al., 2008), with parameters set according to previous recommendations (Pujolar et al., 2013). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis implemented in the Database for Annotation, Visualization and Integrated Discovery (DAVID) web-server v6.73 was used for a more systematic functional interpretation of the set of candidate genes. A gene functional analysis in DAVID was conducted with default settings.
Comparison of the Number of Single-Nucleotide Polymorphisms Generated From Paired and Unpaired Reads
In order to compare the number of SNPs generated from paired-end reads with that of SNPs generated from single-end reads, SNP calling was performed on each single-end read group separately in addition to SNP calling on reads mapped as pairs. Single-end BAM files were extracted from paired-end BAM files by using SAMTOOLS. SNPs were called by using SAMTOOLS and BCFTOOLS, followed by additional filtering based on the following criteria: (i) retained only bi-allelic SNPs; (ii) retained SNPs with a minimum quality score of 30; and (iii) retained SNPs with a minimum read depth of 10. In order to increase read depth, individuals were pooled together before filtering.
Results
Restriction Site-Associated DNA Tag Sequencing and Data Filtering
Sequencing of the RAD libraries generated a total of 42.10 Gb of raw data with an average of 1.75 Gb per individual, ranging from 1.02 to 7.84 Gb (Table 1). A total of 40.00 Gb of clean data was retained after quality filtering, with an average effective rate (clean reads compared with raw reads) of 95.01%. The clean data had a guanine–cytosine (GC) content that ranged from 40.02 to 41.78%, and the proportion of Q20 (the bases of one individual with Phred Q > 20) ranged from 89.97 to 95.27%, whereas that of Q30 ranged from 81.96 to 91.09%.
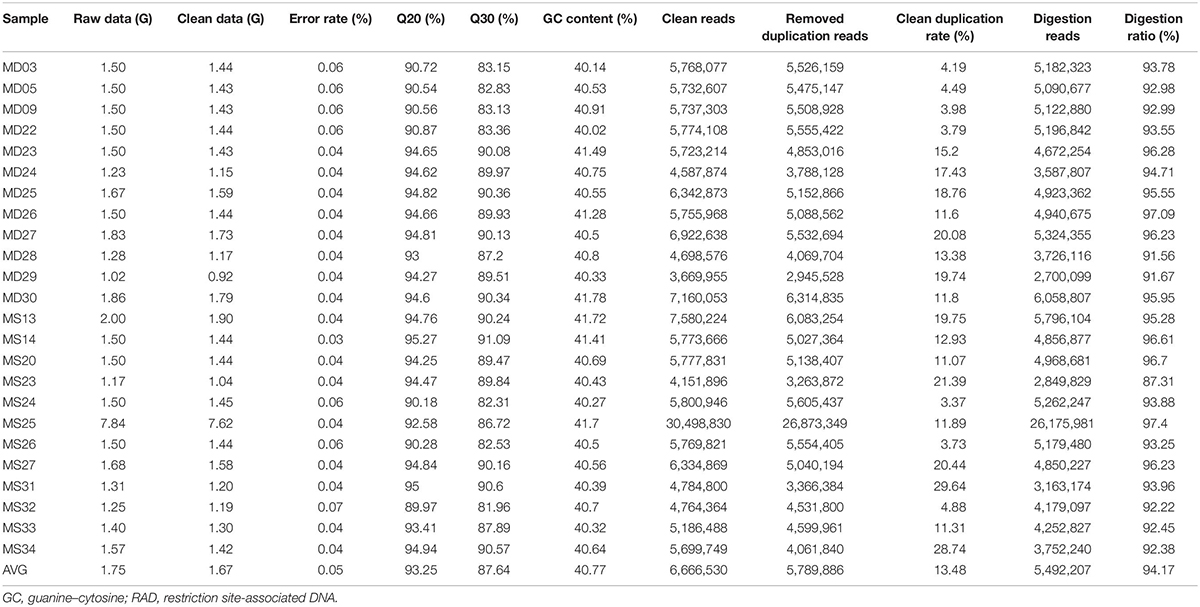
Table 1. Statistics describing the distribution of different properties of RAD sequences after each step of filtering.
Single-Nucleotide Polymorphism Discovery and Population Genetic Structure
Of the retained sequences, an average of 10.51 million (97.35%) reads per individual were aligned to the Japanese eel draft genome contigs, and then 9,905,533 SNPs were called from 24 individuals using SAMTOOLS. After quality filtering, a total of 73,557 SNPs were retained. The final retained SNPs were widely distributed across the genome and were found in a total of 28,407 contigs.
The expected and observed heterozygosity (HE and HO) per individual ranged from 0.194 to 0.198 and from 0.157 to 0.312, respectively (Table 2). The FST values between the two populations ranged from −0.0667 to 0.4354 for each SNP. The final dataset had a transition:transversion ratio of 1.83:1 (Figure 1).
Table 2. Statistics of the expected and observed heterozygosity (HE and HO) per individual.
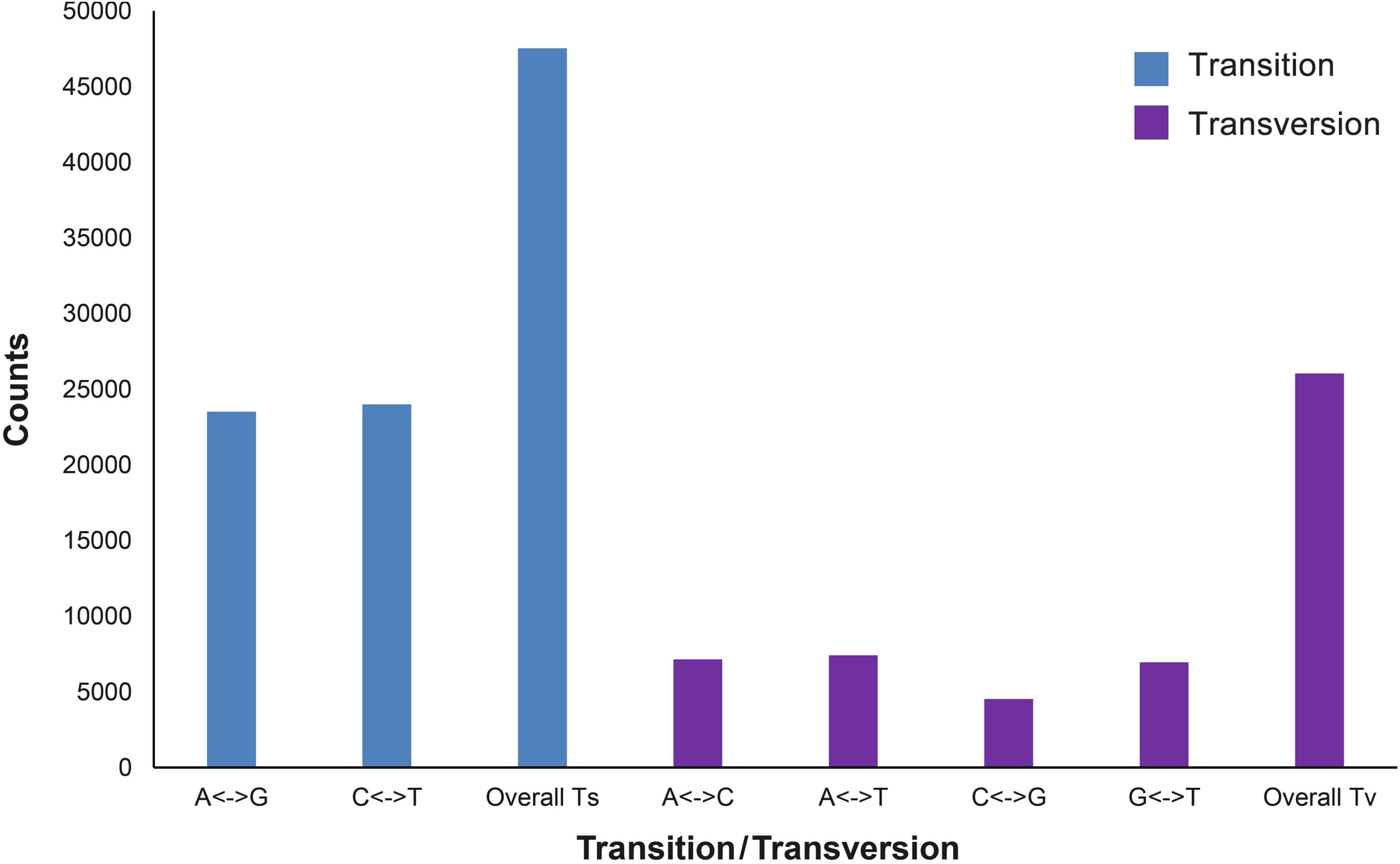
Figure 1. Transitions and transversions occurring within a set of 73,557 single-nucleotide polymorphisms (SNPs).
ARLEQUIN 3.5 obtained an FST value of 0.00271 (P > 0.05) between the two populations using all the retained SNPs. The population pairwise FST was 0.00112 (P > 0.05) based on the neutral SNPs and 0.28473 (P < 0.001) based on the outlier SNPs. Results of ADMIXTURE and NETVIEW showed that all individuals from Dandong and Xiapu were grouped into a single cluster when using the neutral or all SNPs. However, when the analysis was based on the outlier SNPs, the two populations were clearly separated (Figure 2).
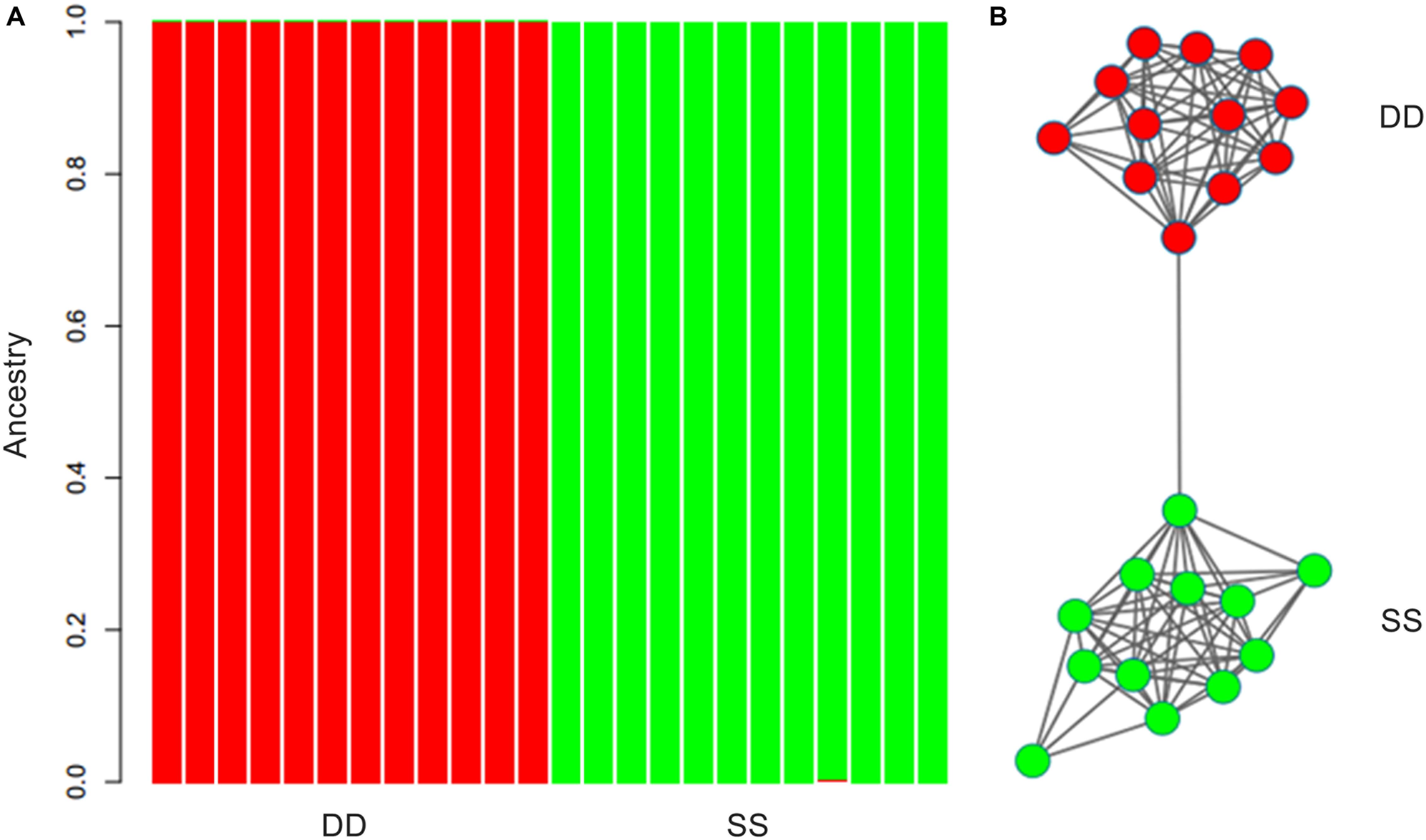
Figure 2. Results of ADMIXTURE (A) and NETVIEW (B) based on outlier dataset.
BLAST Analyses of the Divergent Loci
A total of 250 outlier SNPs representing 238 unique contigs in the draft genome were detected, with all showing high FST values (0.209–0.435). BLASTX similarity searches demonstrated that 85 contigs (36%) significantly matched to zebrafish proteins, with 61 of these contigs corresponding to zebrafish proteins Gene Ontology (GO) functional annotations (Figure 3). These annotations are represented by GO terms including signaling, response to stimulus, metabolic process, biological regulation, catalytic activity, development process, and others (Figure 4). Classification of biological and molecular functionalities for these hits is listed in Supplementary Table S1. A total of 228 zebrafish genes, which were homologous to the 85 contigs with significant BLASTX hits, were loaded into the DAVID database and were found to correspond to 224 DAVID IDs. Enriched KEGG pathways including 52 genes classified into nine pathways were summarized in Table 3. The pathway with the highest number of genes was endocytosis (10 genes), including several genes such as those encoding chemokine (C-X-C motif) receptor 4, fibroblast growth factor receptor, and par-6 partitioning defective 6 homolog. Other pathways of particular interest were neuroactive ligand–receptor interaction (nine genes), calcium signaling pathway (eight genes), intestinal immune network for IgA production (three genes), and mitogen-activated protein kinase (MAPK) signaling pathways (eight genes).

Figure 3. The numbers of the annotated contigs.
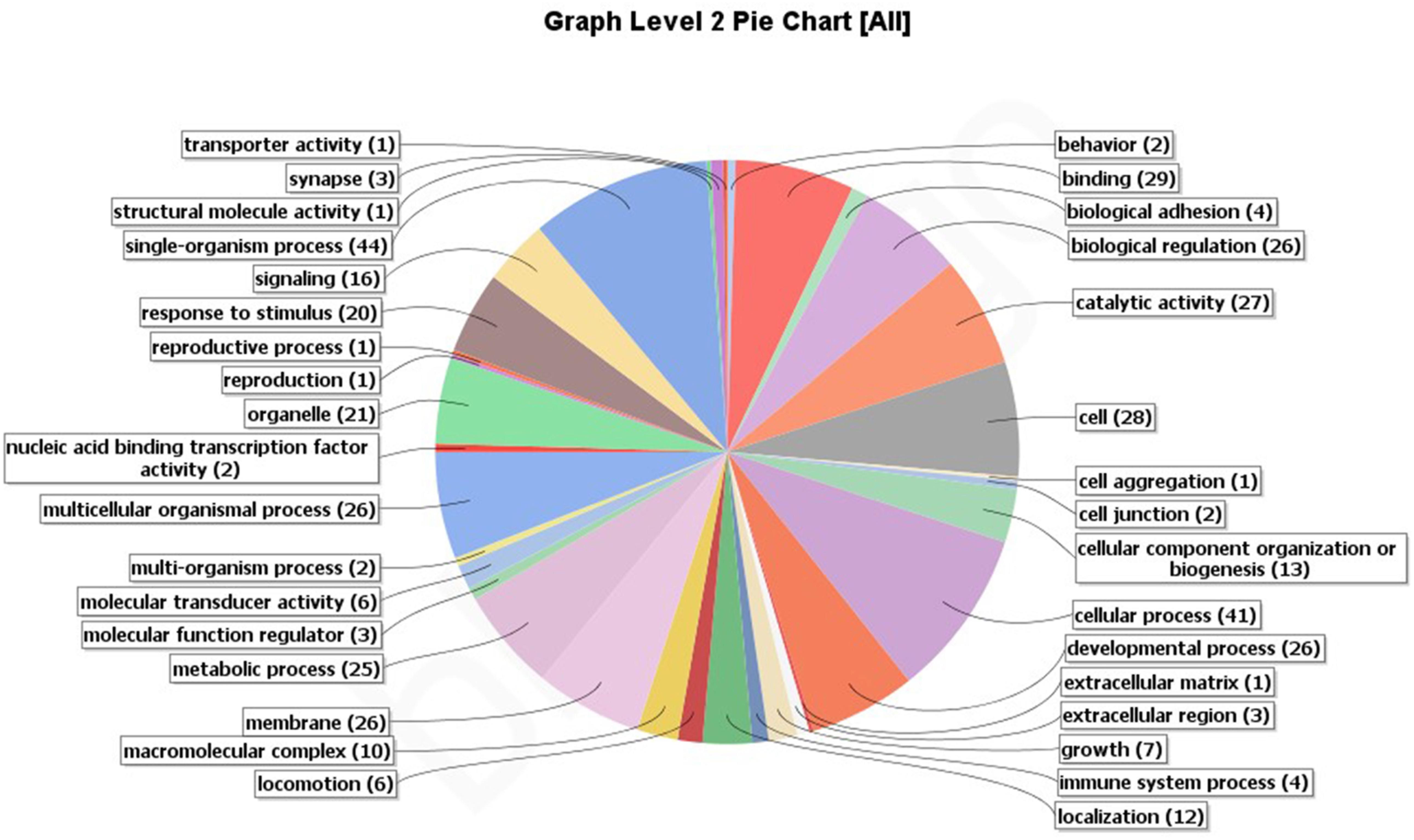
Figure 4. Putative functional categorization and distribution of 61 significant BLAST hits (E-value ≤ 1E-5) of Anguilla japonica according to BLAST2GO annotation.

Table 3. KEGG pathways and genes identified using the FST outlier approach implemented in ARLEQUIN.
The Number of Single-Nucleotide Polymorphisms Generated Based on Single Reads or Paired Reads
A total of 4,227,450 SNPs were called using the first reads before filtering, whereas 7,741,237 SNPs were called when using the second reads. A total of 2,449,359 for the first reads, 3,724,441 for the second reads, and 5,515,225 for both of the paired reads were retained after applying filtration. However, much fewer SNPs were retained for the second reads while applying the filter on each individual, suggesting low depth of the second reads. This is a common phenomenon in RAD, as the randomly sheared fragments are expected to be sequenced at a much lower depth than the other ends. However, the longer fragment coverage of the second reads increases the chance for targeting loci under local adaptation and is also suitable for annotations.
Discussion
Compared with microsatellites, SNPs are the most widespread type of sequence variation in genomes. Despite microsatellites presenting higher diversity per locus (with a mutation rate of 10–4 per generation), the large number of unlinked SNPs may overcome their relatively low mutation rates (10–8–10–9 per generation), and panels of several 1000 SNPs are likely to be more informative than the 10–20 microsatellite loci used in standard population genetic studies (Helyar et al., 2011; Seeb et al., 2011). Here, we reported the discovery of a large number of genome-wide SNPs in the Japanese eel using RAD sequencing. After quality filtering, 73,557 high-quality SNPs were retained, which further improved the amount of genomic resources available for this endangered species. Most previous population genetic studies of A. japonica were conducted using less than 20 microsatellite loci, mitochondrial sequence, or isozyme markers, resulting in inconsistent findings (Sang et al., 1994; Chan et al., 1997; Tseng et al., 2003, 2006, 2009; Han et al., 2010a). Compared with other studies of eels on the basis of RAD (Pujolar et al., 2013, 2014), our study made full use of the paired-end reads, which resulted in significantly more markers and enabled the identification of genomic regions under selection.
The transition:transversion ratio of the SNPs in our dataset was 1.83:1, which is in keeping with similar ratios that have been reported in the study of European eel (Anguilla anguilla) (1.6:1) (Pujolar et al., 2013) and great tit (Parus major) (1.7:1) (Bers et al., 2010). The overall non-significant pairwise FST (0.00271, P > 0.05) indicated that most of the genome is homogenized by gene flow, supporting the theory of panmixia in the Japanese eel. However, the FST based on the outlier SNPs was 0.285 and significant (P < 0.001). In addition, both ADMIXTURE and NETVIEW analyses based on the outlier SNPs demonstrated strong genetic differentiation between the two populations. Taken together, these results indicated that some genomic regions of the Japanese eel have undergone divergent natural selection.
Furthermore, RAD sequencing-based population genomic studies can also detect signatures of selection and local adaptation. Candidate genes and genomic regions were identified using an FST outlier approach by detecting loci showing increased or decreased differentiation across populations compared with neutral expectations, which are suggestive of directional or purifying natural selection. For a panmictic species like Japanese eel, SNP-based genome scans can be used for test selection within a single generation, which results from geographically varying environmental conditions encountered by glass eels across different regions. Such outlier detection methods have been illustrated by several recent population genomic studies in marine fishes displaying extensive gene flow (Russello et al., 2012; Milano et al., 2014). For instance, Hess et al. (2013) detected fine-scale population structure in Pacific lamprey (Entosphenus tridentatus) by using 162 putatively adaptive SNPs identified by RAD sequencing. Larson et al. (2014) found three potential regions of adaptive divergence in Chinook salmon (Oncorhynchus tshawytscha) using RAD sequencing, showing that RAD sequencing is a promising approach for marine species with large population sizes and shallow structures.
The two populations of A. japonica analyzed were collected from two environmentally heterogeneous locations, and different environmental factors may lead to local adaptation. BLASTX and BLAST2GO revealed that 61 out of 238 highly divergent contigs were located in functional genes or genomic regions, which might indicate divergent natural selection (Foll and Gaggiotti, 2008). The low frequency of BLASTX-annotated outliers suggested that substantial outlier polymorphisms detected might be in the intergenic or intronic regions of the genome. The KEGG pathway analysis showed that some of the putative targets under local selection were associated with genes involved in several important pathways, such as calcium signaling pathway, intestinal immune network for IgA production, cytokine–cytokine receptor interaction, and MAPK signaling pathways. Differences in these pathways might indicate local adaptation of A. japonica to different geographical environments, which vary in salinity and temperature. For instance, the calcium signaling pathway plays an important role in fertilization, development, learning and memory, ion exchange, and osmoregulation (Berridge et al., 2000; Daverat et al., 2006). The intestine can generate large amounts of non-inflammatory immunoglobulin A (IgA) antibodies that serve as the first line of defense against microorganisms (Reinking et al., 1999) and is also crucial for adaptive inflammatory host defenses, cell growth, differentiation, cell death, and angiogenesis (Lackie, 2010). Similar results were also reported in other studies. For instance, Guo et al. (2015) investigated genome-wide patterns of genetic variability of Atlantic herring using the RAD sequencing approach, and outlier analyses uncovered 100s of directionally selected loci that were associated with salinity and temperature. Pujolar et al. (2014) found a small set of SNPs showing high genetic differentiation, which is consistent with single-generation signatures of spatially varying selection acting on European eel (glass eels) using RAD sequencing. After 50,354 SNPs were screened, a total of 754 potentially locally selected SNPs were identified. Candidate genes for local selection include those involved in calcium signaling, neuroactive ligand–receptor interaction, circadian rhythm, and others.
It remains an open question whether the adaptive loci inferred in this study were directly related to the local adaptation of A. japonica populations to specific environments or were linked to genes underlying specific traits. Further analyses are necessary to confirm divergent selection on outliers through a correlation analysis between allele frequencies and environmental factors among populations (Seeb et al., 2011; Limborg et al., 2012; DeFaveri et al., 2013; Teacher et al., 2013).
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Supplementary Information Files.
Ethics Statement
The animal study was reviewed and approved by the Institutional Ethics Committee of Zhejiang Ocean University.
Author Contributions
J-XL and B-JL conceived and designed the research. B-JL, Y-LL, and B-DZ conducted the experiments, analyzed the data, and wrote the manuscript. All authors critically reviewed and approved the manuscript.
Funding
The study was supported by the National Natural Science Foundation of China (NSFC) (Nos. 41676137 and 41806156); Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology (2017-2B05); Natural Science Foundation of Zhejiang Province (LY17C200010 and LY15D060004); Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes (No. 2019J00048); Starting Research Fund from the Zhejiang Ocean University; The Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes; and Open Foundation from Marine Sciences in the First-Class Subjects of Zhejiang Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JZ declared a shared affiliation, with no collaboration, with one of the authors, B-DZ, to the handling Editor at the time of review.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00782/full#supplementary-material
Footnotes
References
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Allendorf, F. W., Hohenlohe, P. A., and Luikart, G. (2010). Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709. doi: 10.1038/nrg2844
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD Markers. PLoS One 3:e3376. doi: 10.1371/journal.pone.0003376
Beaumont, M. A., and Nichols, R. A. (1996). Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B Biol. Sci. 263, 1619–1626. doi: 10.1098/rspb.1996.0237
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bers, N. E., Oers, K. V., Kerstens, H. H., Dibbits, B. W., Crooijmans, R. P., Visser, M. E., et al. (2010). Genome-wide SNP detection in the great tit Parus major using high throughput sequencing. Mol. Ecol. 19, 89–99. doi: 10.1111/j.1365-294X.2009.04486.x
Chan, I. K. K., Chan, D. K. O., Lee, S. C., and Tsukamoto, K. (1997). Genetic variability of the Japanese eel Anguilla japonica (Temminck & Schlegel) related to latitude. Ecol Freshw Fish. 6, 45–49. doi: 10.1111/j.1600-0633.1997.tb00141.x
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Daverat, F., Limburg, K. E., Thibault, I., Shiao, J. C., Dodson, J. J., Caron, F. O., et al. (2006). Phenotypic plasticity of habitat use by three temperate eel species, Anguilla anguilla, A-japonica and A-rostrata. Mar. Ecol. Prog. Ser. 308, 231–241. doi: 10.3354/meps308231
DeFaveri, J., Shikano, T., Shimada, Y., and Merila, J. (2013). High degree of genetic differentiation in marine three-spined sticklebacks (Gasterosteus aculeatus). Mol. Ecol. 22, 4811–4828. doi: 10.1111/mec.12430
Dudgeon, C. L., Blower, D. C., Broderick, D., Giles, J. L., Holmes, B. J., Kashiwagi, T., et al. (2012). A review of the application of molecular genetics for fisheries management and conservation of sharks and rays. J. Fish Biol. 80, 1789–1843. doi: 10.1111/j.1095-8649.2012.03265.x
Ekblom, R., and Galindo, J. (2011). Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107, 1–15. doi: 10.1038/hdy.2010.152
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Foll, M., and Gaggiotti, O. E. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Gong, X. L., Ren, S. J., Cui, Z. K., and Yue, L. J. (2014). Genetic evidence for panmixia of Japanese eel (Anguilla japonica) populations in China. Genet. Mol. Res. 13, 768–781. doi: 10.4238/2014.January.31.3
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Guo, B., DeFaveri, J., Sotelo, G., Nair, A., and Merilä, J. (2015). Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks. BMC Biol. 13:19. doi: 10.1186/s12915-015-0130-8
Han, Y. S., Hung, C. L., Liao, Y. F., and Tzeng, W. N. (2010a). Population genetic structure of the Japanese eel Anguilla japonica: panmixia at spatial and temporal scales. Mar. Ecol. Prog. Ser. 401, 221–232. doi: 10.3354/meps08422
Han, Y.-S., Iizuka, Y., and Tzeng, W.-N. (2010b). Does Variable Habitat Usage by the Japanese Eel Lead to Population Genetic Differentiation? Zool. Stud. 49, 392–397.
Harismendy, O., Ng, P. C., Strausberg, R. L., Wang, X., Stockwell, T. B., Beeson, K. Y., et al. (2009). Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 10:R32. doi: 10.1186/gb-2009-10-3-r32
Helyar, S. J., Hemmer-Hansen, J., Bekkevold, D., Taylor, M. I., Ogden, R., Limborg, M. T., et al. (2011). Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol. Ecol. Resour. 11:36. doi: 10.1111/j.1755-0998.2010.02943.x
Hess, J. E., Campbell, N. R., Close, D. A., Docker, M. F., and Narum, S. R. (2013). Population genomics of Pacific lamprey: adaptive variation in a highly dispersive species. Mol. Ecol. 22, 2898–2916. doi: 10.1111/mec.12150
Hohenlohe, P. A., Amish, S. J., Catchen, J. M., Allendorf, F. W., and Luikart, G. (2011). Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol. Ecol. Resour. 11, 117–122. doi: 10.1111/j.1755-0998.2010.02967.x
Hohenlohe, P. A., Bassham, S., Etter, P. D., Stiffler, N., Johnson, E. A., and Cresko, W. A. (2010). Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Gene. 6:e1000862. doi: 10.1371/journal.pgen.1000862
Hudson, M. E. (2008). Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol. Ecol. Resour. 8, 3–17. doi: 10.1111/j.1471-8286.2007.02019.x
Ishikawa, S., Aoyama, J., Tsukamoto, K., and Nishida, M. (2001). Population structure of the Japanese eel Anguilla japonica as examined by mitochondrial DNA sequencing. Fish Sci. 67, 246–253. doi: 10.1046/j.1444-2906.2001.00227.x
Kruck, N. C., Innes, D. I., and Ovenden, J. R. (2013). New SNPs for population genetic analysis reveal possible cryptic speciation of eastern Australian sea mullet (Mugil cephalus). Mol. Ecol. Resour. 13, 715–725. doi: 10.1111/1755-0998.12112
Lackie, J. (2010). “Cytokine,” in John Lackie. A Dictionary of Biomedicine, ed. C. A. O’Callaghan, (Oxford: Oxford University Press).
Larson, W. A., Seeb, L. W., Everett, M. V., Waples, R. K., Templin, W. D., and Seeb, J. E. (2014). Genotyping by sequencing resolves shallow population structure to inform conservation of Chinook salmon (Oncorhynchus tshawytscha). Evol. App. 7:69. doi: 10.1111/eva.12128
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Limborg, M. T., Helyar, S. J., Bruyn, M. D., Taylor, M. I., Nielsen, E. E., Ogden, R., et al. (2012). Environmental selection on transcriptome-derived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus). Mol. Ecol. 21, 2355–2356. doi: 10.1111/j.1365-294X.2012.05639.x
Lischer, H., and Excoffier, L. (2012). PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299. doi: 10.1093/bioinformatics/btr642
Milano, I., Babbucci, M., Cariani, A., Atanassova, M., Bekkevold, D., Carvalho, G. R., et al. (2014). Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 23, 118–135. doi: 10.1111/mec.12568
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A., and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248. doi: 10.1101/gr.5681207
Neuditschko, M., Khatkar, M. S., and Raadsma, H. W. (2012). NETVIEW: a high-definition network-visualization approach to detect fine-scale population structures from genome-wide patterns of variation. PLoS One 7:e48375. doi: 10.1371/journal.pone.0048375
Palti, Y., Gao, G. T., Miller, M. R., Vallejo, R. L., Wheeler, P. A., Quillet, E., et al. (2014). A resource of single-nucleotide polymorphisms for rainbow trout generated by restriction-site associated DNA sequencing of doubled haploids. Mol. Ecol. Resour. 14, 588–596. doi: 10.1111/1755-0998.12204
Pujolar, J. M., Jacobsen, M. W., Als, T. D., Frydenberg, J., Munch, K., Jonsson, B., et al. (2014). Genome-wide single-generation signatures of local selection in the panmictic European eel. Mol. Ecol. 23, 2514–2528. doi: 10.1111/mec.12753
Pujolar, J. M., Jacobsen, M. W., Frydenberg, J., Als, T. D., Larsen, P. F., Maes, G. E., et al. (2013). A resource of genome-wide single-nucleotide polymorphisms generated by RAD tag sequencing in the critically endangered European eel. Mol. Ecol. Resour. 13, 706–714. doi: 10.1111/1755-0998.12117
Reinking, R. R., Raedler, A., and Möckel, G. (1999). The intestinal immune system. Medizinische Monatsschrift Für Pharmazeuten. 22, 198–204.
Russello, M. A., Kirk, S. L., Frazer, K. K., and Askey, P. J. (2012). Detection of outlier loci and their utility for fisheries management. Evol. App. 5, 39–52. doi: 10.1111/j.1752-4571.2011.00206.x
Sang, T. K., Chang, H. Y., Chen, C. T., and Hui, C. F. (1994). Population structure of the japanese eel. Anguilla japonica. Mol. Biol. Evol. 11, 250–260.
Seeb, J. E., Carvalho, G., Hauser, L., Naish, K., Roberts, S., and Seeb, L. W. (2011). Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Mol. Ecol. Resour. 11(Suppl. 1), 1–8. doi: 10.1111/j.1755-0998.2010.02979.x
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Slate, J., Gratten, J., Beraldi, D., Stapley, J., Hale, M., and Pemberton, J. M. (2009). Gene mapping in the wild with SNPs: guidelines and future directions. Genetica 136, 97–107. doi: 10.1007/s10709-008-9317-z
Steinig, E. J., Neuditschko, M., Khatkar, M. S., Raadsma, H. W., and Zenger, K. R. (2016). netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol. Ecol. Resour. 16, 216–227. doi: 10.1111/1755-0998.12442
Teacher, A. G. F., Andre, C., Jonsson, P. R., and Merila, J. (2013). Oceanographic connectivity and environmental correlates of genetic structuring in Atlantic herring in the Baltic Sea. Evol. App. 6, 549–567. doi: 10.1111/eva.12042
Tseng, M. C., Tzeng, W. N., and Lee, S. C. (2003). Historical decline in the Japanese eel Anguilla japonica in northern Taiwan inferred from temporal genetic variations. Zool. Stud. 42, 556–563.
Tseng, M. C., Tzeng, W. N., and Lee, S. C. (2006). Population genetic structure of the Japanese eel Anguilla japonica in the northwest Pacific Ocean: evidence of non-panmictic populations. Mar. Ecol. Prog. Ser. 308, 221–230. doi: 10.3354/meps308221
Tseng, M. C., Tzeng, W. N., and Lee, S. C. (2009). Genetic differentiation of the Japanese eel. Am. Fish. Soc. Sym.. 58, 59–69.
Tsukamoto, K. (1992). Discovery of the spawning area for japanese eel. Nature 356, 789–791. doi: 10.1038/356789a0
Tsukamoto, K. (2006). Oceanic biology: spawning of eels near a seamount. Nature 439:929. doi: 10.1038/439929a
Wang, X. Q., Zhao, L., Eaton, D. A. R., Li, D. Z., and Guo, Z. H. (2013). Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol. Ecol. Resour. 13, 938–945. doi: 10.1111/1755-0998.12136
Zhang, B., Xue, D., Wang, J., Li, Y. L., Liu, B. J., Liu, J. X., et al. (2016). Development and preliminary evaluation of a genomewide single nucleotide polymorphisms resource generated by RAD -seq for the small yellow croaker (Larimichthys polyactis). Mol. Ecol. Resour. 16, 755–768. doi: 10.1111/1755-0998.12476
Keywords: population genomics, genetic diversity, restriction site-associated DNA sequencing, single-nucleotide polymorphism discovery, local adaptation
Citation: Liu B-J, Li Y-L, Zhang B-D and Liu J-X (2020) Genome-Wide Discovery of Single-Nucleotide Polymorphisms and Their Application in Population Genetic Studies in the Endangered Japanese Eel (Anguilla japonica). Front. Mar. Sci. 6:782. doi: 10.3389/fmars.2019.00782
Received: 09 September 2019; Accepted: 04 December 2019;
Published: 14 January 2020.
Edited by:
Dongdong Xu, Marine Fisheries Research Institute of Zhejiang, ChinaReviewed by:
Xiubao Li, Hainan University, ChinaJunlong Zhang, Institute of Oceanology (CAS), China
Copyright © 2020 Liu, Li, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing-Jian Liu, YmpldGJqQDE2My5jb20=; Jin-Xian Liu, amlueGlhbmxpdUBnbWFpbC5jb20=