 Yi Lan1
Yi Lan1 Jin Sun1
Jin Sun1 Weipeng Zhang1
Weipeng Zhang1 Ting Xu2
Ting Xu2 Yu Zhang3
Yu Zhang3 Chong Chen4
Chong Chen4 Dong Feng5
Dong Feng5 Hongbin Wang6Jun Tao6
Hongbin Wang6Jun Tao6 Jian-Wen Qiu2
Jian-Wen Qiu2 Pei-Yuan Qian1*
Pei-Yuan Qian1*- 1Department of Ocean Science, Division of Life Science and Hong Kong Branch of the Southern Marine Science and Engineering Guangdong Laboratory, The Hong Kong University of Science and Technology, Hong Kong, China
- 2Department of Biology, Hong Kong Baptist University, Kowloon Tong, Hong Kong
- 3College of Life Sciences and Oceanography, Shenzhen University, Shenzhen, China
- 4X-STAR, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), Yokosuka, Japan
- 5CAS Key Laboratory of Ocean and Marginal Sea Geology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 6MLR Key Laboratory of Marine Mineral Resources, Guangzhou Marine Geological Survey, Guangzhou, China
In deep-sea hydrothermal vents and hydrocarbon seeps, chemoautotrophic bacteria use chemical substances as energy resources for primary production, ultimately supporting dense communities of megafauna, including charismatic giant vesicomyid clams. These clams inherit their endosymbionts from their parents and house them intracellularly in their gills. How these organisms maintain their unique symbiotic relationship at the cellular level, however, remains largely unclear. In the present study, transcriptomes of different organs in Phreagena okutanii collected from a hydrothermal vent and in Archivesica marissinica collected from a methane seep were sequenced in order to decipher their host–symbiont relationships. Expressional analyses of the transcriptomes showed that the tricarboxylic acid (TCA) cycle-related genes, the Rab gene family, and the lysozyme genes were highly expressed in the gills. Furthermore, genes related to vesicle trafficking, lysosomes, and mitochondrial and energy metabolism were positively selected. The endosymbiont genes involved in sulfur oxidation, oxidative phosphorylation, and adenosine triphosphate (ATP) synthesis were highly expressed. The results suggest that the vesicomyid clams provide intermediates to fulfill the metabolic needs of their endosymbionts, and in return the endosymbionts actively generate nutrients for the hosts through being digested by the lysozymes of the host. Furthermore, the positive selection of genes related to vesicle trafficking, lysosomes, and mitochondrial and energy metabolism indicates molecular adaptations of the host in order to benefit from symbiosis. Overall, the present study provides the first set of transcriptomes for deep-sea chemosymbiotic vesicomyid clams, facilitating a better understanding of the host–symbiont relationship that has allowed them to become dominant animals in deep-sea hydrothermal vents and cold seeps.
Introduction
Deep-sea hydrothermal vents and methane seeps are environments characterized by high hydrostatic pressure, high and variable temperatures compared with those of the surrounding seafloor, and high concentrations of hydrogen sulfide, methane, and heavy metals (Van Dover, 2000; Levin, 2005). These habitats often support chemosynthesis-based ecosystems dominated by megafauna that host symbionts and rely on chemosynthetic bacteria to help them utilize chemical energy derived from various reduced substances such as hydrogen sulfide and methane (Sibuet and Olu, 1998). Illuminating host-symbiotic relationships in holobiont animals living in these ecosystems can provide fundamental knowledge of biological adaptation to these “extreme” deep-sea habitats.
Vesicomyid clams in the subfamily Pliocardiinae host symbionts and are often dominant in both vents and seeps across the global oceans (Sibuet and Olu, 1998). Like other chemosynthetic holobionts, endosymbiotic bacteria provide the hosts with energy and nutrients (Newton et al., 2008). Some holobiont taxa inhabiting vents and seeps, such as mussels and tubeworms, take in free-living bacteria from their ambient environment each generation in a process called horizontal transmission that is independent of the reproduction of the host (Won et al., 2003; Nussbaumer et al., 2006; Funkhouser and Bordenstein, 2013). In contrast, vesicomyid clams inherit the symbionts from their parents, a process referred to as maternal or vertical transmission (Bright and Bulgheresi, 2010; Funkhouser and Bordenstein, 2013; Ozawa et al., 2017). Although host switching or horizontal transmission may occasionally occur in some vesicomyid clams (Stewart et al., 2008; Decker et al., 2013; Ozawa et al., 2017), maternal transmission is the predominant way for symbiont acquisition. Previous studies have revealed approximately 400 sulfur-oxidizing bacterial cells extracellularly attached to the outer surface of each egg spawned by Phreagena okutanii (originally described as Calyptogena okutanii by Kojima and Ohta, 1997), indicating the transmission of symbiotic bacteria via eggs from the parent to their offspring (Ikuta et al., 2016). It is essential for these megafauna to adopt a mutualistic relationship with their symbionts acquired through either horizontal or vertical transmission mode in order to maintain fundamental metabolism. For example, Bathymodiolus mussels are likely to utilize the lysosomal enzymes to digest the symbiont to obtain nutrients (Ponnudurai et al., 2017). Meanwhile, symbionts require the host to deliver oxygen, hydrogen sulfide and other intermediates to live an intracellular life and synthesize nutrients for their host (Flores et al., 2005).
Large, symbiont-hosting vesicomyid clams are capable of taking up water and hydrogen sulfide through their foot inserted into the sediment (Ohishi et al., 2016). Dense populations of symbiotic bacteria have been observed in bacteriocytes in the gill epithelium cells of the inner zone of gill filaments (Fiala-Médioni and Métivier, 1986). These endosymbionts are thioautotrophic bacteria capable of utilizing hydrogen sulfide as the main energy source (Ohishi et al., 2016). These clams have a highly reduced and functionally rudimentary digestive system (Van Dover, 2000) and rely heavily on the organic carbon and nutrients provided by the endosymbionts (Fiala-Médioni and Métivier, 1986; Barry et al., 1997; Ohishi et al., 2016).
The genomes of two species of thioautotrophic bacterial symbionts from two species of vesicomyid clams have been reported (Kuwahara et al., 2007; Newton et al., 2007). For example, both species of bacteria have complete pathways for metabolizing sulfur and for synthesizing essential amino acids, vitamins, and cofactors, but neither possesses transporter genes for exporting nutrients to their host or genes involved in the tricarboxylic acid (TCA) cycle (Kuwahara et al., 2007; Newton et al., 2007). These symbionts also lack the ftsZ gene encoding a circumferential ring to separate cells from one another during the cell division process, suggesting that they may have an unusual proliferation mechanism (Newton et al., 2007). How these symbiotic bacteria survive with an incomplete TCA cycle and how they provide nutrients to their host without transporters remain unknown (Kuwahara et al., 2007; Newton et al., 2007). Furthermore, the genetic adaptation of the host or host–symbiont interactions is largely unclear, particularly from the host’s perspective, due to the lack of genomic information of the host.
Genomes or transcriptomes of several deep-sea tube worms and mussels have recently been sequenced (Li et al., 2015, 2017; Sun et al., 2017). The results of both gene expression and positive selection provide a better understanding of the possible mechanisms of molecular adaptation behind developing and maintaining endosymbiosis as well as host–symbiont relationships in the horizontal mode of transmission (Sun et al., 2017). Transcriptomic information coupled with gene expression and positive selection analyses can provide a better understanding of the host–symbiont interactions in vesicomyid clams transmitting symbionts vertically. Therefore, in the present study, we sequenced the transcriptome of two vesicomyid clams, Phreagena okutanii (Kojima and Ohta, 1997) from a hydrothermal vent and Archivesica marissinica (Feng et al., 2018) [originally described as “Calyptogena” marissinica C. Chen et al. (2018)] from a methane seep (Chen et al., 2018), and compared their transcriptomes with those of shallow-water clam species. Further analysis of the gene expression pattern and genes under positive selection enhanced our understanding of host–symbiont interactions in vesicomyid clams.
Materials and Methods
Sample Collection, RNA Extraction, and Sequencing
A Phreagena okutanii individual was collected by the remotely operated vehicle (ROV) KAIKO with vehicle Mk-IV from a hydrothermal vent of the Sakai hydrothermal field (21°31.4749′ N, 126°59.021′ E) (Nakamura et al., 2015), Okinawa Trough at a depth of 1,550 m during the Japan Agency for Marine-Earth Science and Technology (JAMSTEC) R/V Kairei cruise KR15-17 in November 2015 (with Hiroyuki Yamamoto as the principal investigator). An Archivesica marissinica individual was collected by the ROV Haima from a methane seep at a depth of approximately 1,400 m on the northwestern slope of the South China Sea in March 2016 (see Figure 1 in a previous study (Liang et al., 2017) for the sampling location). The gills and mantle of P. okutanii and the gills, adductor muscle, foot and mantle of A. marissinica were dissected, separately fixed in RNAlater (Invitrogen, United States), and stored at −80°C until use. The tissues were ground in TRIzol reagent (Invitrogen, United States) to extract the total RNA following the manufacturer’s instructions. The tissues were lysed by adding 0.2 volume chloroform to the TRIzol mixture. After centrifuging the mixture to obtain the aqueous phase, the RNA was recovered by isopropanol precipitation. The RNA pellets were resolved in DEPC (diethyl pyrocarbonate) water and stored at −80°C until cDNA synthesis. For the total RNA of the gills of A. marissinica, the rRNA from the host and the symbionts were first removed with the Ribo-ZeroTM Magnetic Kit (Human/Mouse/Rat) (Epicenter, United States) and the Ribo-ZeroTM Magnetic Kit (Bacteria) (Epicenter, United States) following the manufacturer’s protocols. Then the remaining RNA was used to synthesize cDNA. The mRNA of other tissues was enriched by using Oligo-dT probes to construct cDNA libraries. The cDNA was synthesized by sequentially generating first-strand cDNA and second-stand cDNA using random primers reverse transcription. The synthesized cDNA was further used to generate sequencing library through end-repair, 3′ adenylated, ligation to the sequencing adaptors and PCR amplification for an Illumina HiSeq 4000 platform. The host species were determined based on morphological characteristics and cox1 sequences assembled using transcriptome sequencing data (Fujikura et al., 2012; Chen et al., 2018).
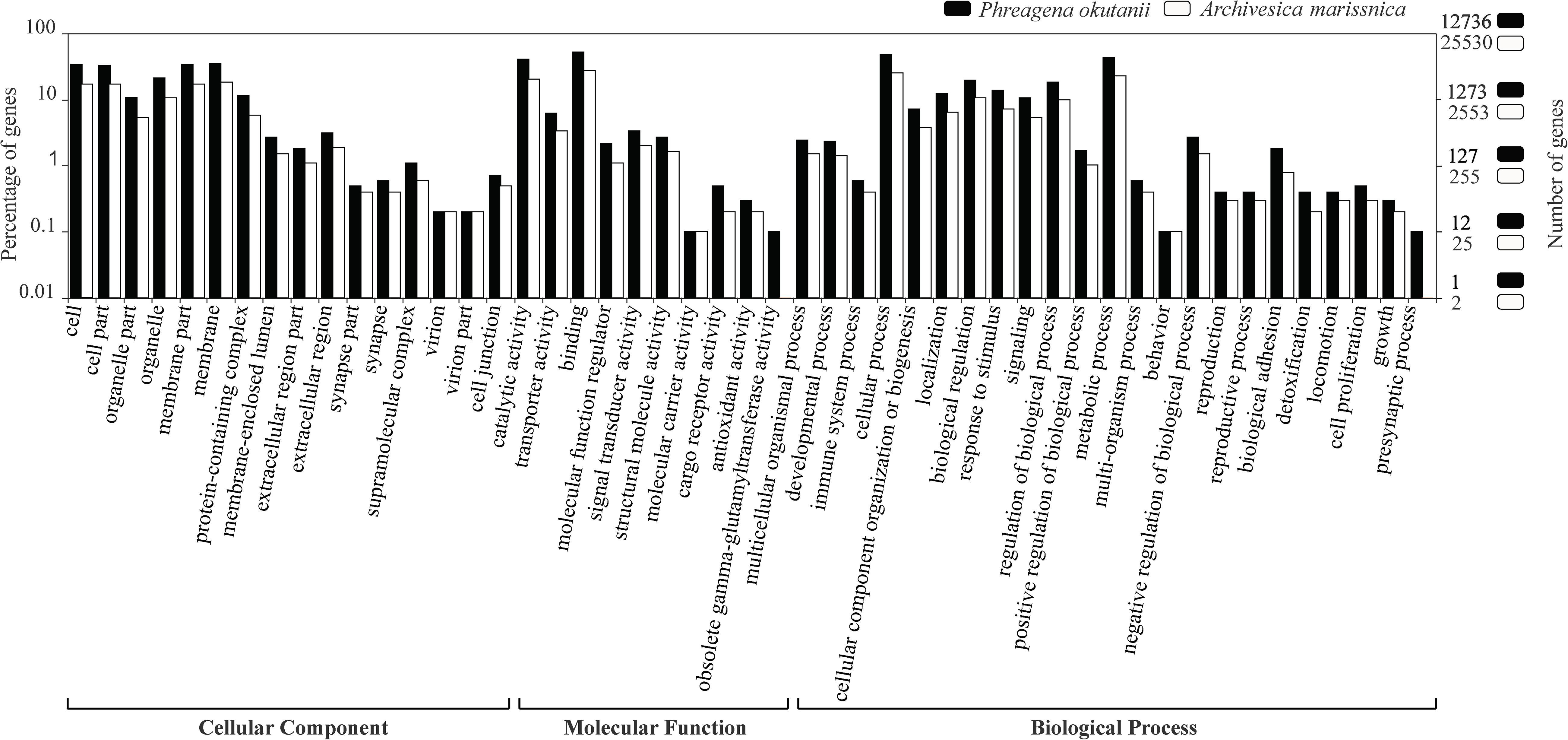
Figure 1. Gene ontology distribution of BLASTp hits from the predicted protein sequences of Phreagena okutanii (solid) and Archivesica marissinica (open). The left and right y-axis show the percentage and number of genes classified into different gene ontology items, respectively.
Data Cleaning and de novo Assembly
Trimmomatic version 0.35 (Bolger et al., 2014) was used to remove adaptors and low-quality bases using the following settings: ILLUMINACLIP: Truseq3-PE-2.fa:2:30:10, LEADING: 3 TRAILING: 3, SLIDINGWINDOW: 4:15, and MINLEN: 36. Subsequently, the clean reads of all organs from each species were combined and used to de novo assemble the transcriptome of P. okutanii and that of A. marissinica, using Trinity version 2.4.0 (Haas et al., 2013) with the default settings.
Functional Annotation and Identification of Highly Expressed Genes
To assess the expression levels of the transcripts, mapped reads were normalized to transcripts per million (TPM) values using Salmon version 0.7.2 with quasi-mapping settings (Patro et al., 2017). In fact, TPM values were calculated twice. For the first calculation, all reads were mapped to the assembly sequences in order to obtain accurate and non-redundant (NR) transcripts. But for the second calculation, the reads of each tissue were separately mapped to the clean transcripts with an open reading frame to quantify the gene-level abundance of each gene in each tissue. In detail, to remove the assembly error, the transcripts with a TPM value below 0.1 were eliminated as in our previous studies (Lan et al., 2017, 2018). The isoforms with the highest TPM value were retained to predict genes. CD-HIT-EST version 4.6 (Li and Godzik, 2006) was further used to remove the redundant transcripts with a sequence similarity of 95%. The coding DNA sequences and proteins of the remaining NR transcripts were predicted using TransDecoder version 3.0.11. The translated protein sequences were then searched against the NCBI NR protein database using BLASTp version 2.2.31 with an E-value threshold of 1 × e–5 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the KEGG Automatic Annotation Server (KAAS) with the bi-directional best hit method (Moriya et al., 2007). The KEGG mapper was used to reconstruct the pathway with KEGG Orthology assignments. The gene ontology (GO) items of the sequences were determined with Blast2GO version 4.1.9 (Conesa et al., 2005) according to their NR hits and were further used to plot the GO distribution using WEGO version 2.0 (Ye et al., 2018).
To separate the gill mixed reads of the endosymbionts and the host, their gill mixed transcripts were first separated so that the endosymbiont transcripts could be used as references. In detail, MEGAN5 (Huson et al., 2016) was used to classify the mixed transcripts of the holobiont according to the annotation results from taxonomy NCBI databases. The mixed reads were then mapped to the isolated endosymbiont transcripts as well as the coding DNA sequences of Candidatus Vesicomyosocius okutanii (AP009247.1, Kuwahara et al., 2007) using Bowtie2 with sensitive end-to-end settings (Langmead and Salzberg, 2012). The complete genome of Candidatus Vesicomyosocius okutanii from P. okutanii (Kuwahara et al., 2007) was used as an additional reference for isolating the symbiont reads and examining the gene expression of the endosymbionts in A. marissinica. This is because the symbiont of A. marissinica is very closely related to Candidatus Vesicomyosocius okutanii. Species of the symbionts of A. marissinica were determined according to its 16S rRNA gene sequence similarity to Candidatus Vesicomyosocius okutanii from P. okutanii (AP009247.1), which is 94%. Finally, the mapped reads of A. marissinica were determined as the symbiont reads and used to analyze the gene expression levels of its symbionts. The unaligned reads were defined as host reads and used in the gene expression analysis of the host gill.
Since the gill is the organ in which endosymbionts are housed, it is the key organ involved in the host–symbiont interaction. Therefore, we compared the gene expression in the gill against those in other tissues. Genes with a significant fold change value (cutoff = 0.01) as well as a higher TPM value were determined as high expression by using GFOLD version 1.0.8 (Feng et al., 2012). TPM values were normalized for each gene and were then used to plot a heat map in Excel using conditional formatting. To test whether a biological pathway was overrepresented among the highly expressed genes, a hypergeometric test (Falcon and Gentleman, 2008) was performed to enrich the highly expressed genes with KEGG annotation from all the predicted genes with KEGG annotation. If the pathways had false discovery rate (FDR) corrected P-values smaller than 0.05, they were regarded as biological pathways enriched with highly expressed genes. The reads of the symbiont from A. marissinica were mapped to the coding DNA sequences of the symbiont Candidatus Vesicomyosocius okutanii (Kuwahara et al., 2007). The clusters of orthologous groups (COGs) of the protein sequences of Candidatus Vesicomyosocius okutanii were annotated using eggNOG-mapper version 4.5.1 and searched against the eggNOG database (Huerta-Cepas et al., 2016, 2017). The genes with a top 10% of the highest TPM value were regarded as highly expressed genes of the symbiont from A. marissinica.
References for Positive Selection Analysis
To identify the genes as well as their references for positive selection analysis, all available transcriptome and genome datasets of the clams within the order Veneroida were used. The raw reads of the transcriptome of Coelomactra antiquata (marine; Mactridae), Corbicula fluminea (freshwater; Cyrenidae), Eurhomalea rufa (marine; Veneridae), Glossus humanus (marine; Glossidae), Limecola balthica (marine; Tellinidae), Mactra chinensis (marine; Mactridae), Mercenaria mercenaria (marine; Veneridae), Meretrix meretrix (marine; Veneridae), Paphia textile (marine; Veneridae), Ruditapes philippinarum (marine; Veneridae), Saxidomus purpuratus (marine; Veneridae), Sinonovacula constricta (marine; Solecurtidae), and Tridacna maxima (marine; Tridacnidae) downloaded from the NCBI’s Sequence Read Archive (SRA) were trimmed and assembled (see Supplementary Table S1 for the SRA accession numbers). The redundant transcripts were removed, and all remaining transcripts were used to predict open reading frames and translated into protein sequences using the same pipeline as applied to deep-sea clams.
To identify the positively selected genes in the symbiont of endosymbionts of P. okutanii and A. marissinica, almost all genomic resources of sulfur-oxidizing Gammaproteobacteria were used in the positive selection analysis. The references included Allochromatium vinosum (NC_013851.1), the endosymbiont of Bathymodiolus septemdierum (AP013042.1), Candidatus Thioglobus autotrophicus (CP010552.1), Candidatus Thioglobus singularis (CP006911.1), Ectothiorhodospira sp. Strain BSL-9 (CP011994.1), Marichromatium purpuratum (CP007031.1), Thioalkalimicrobium aerophilum (CP007030.1), Thioalkalimicrobium cyclicum (CP002776.1), Thioalkalivibrio nitratireducens (NC_019902.2), Thioalkalivibrio paradoxus (CP007029.1), Thiobacillus denitrificans (NC_007404.1), Thioflavicoccus mobilis (NC_019940.1), Thiolapillus brandeum (AP012273.1), and Thiomicrospira crunogena (NC_007520.2). Their coding DNA sequences were downloaded from the NCBI GenBank and directly used in the subsequent positive selection analysis. The coding cDNA sequence of Candidatus Vesicomyosocius okutanii genome (AP009247.1) and de novo assembled coding DNA sequences of symbiont of A. marissinica were used in the positive selection analysis. Only single-copy orthologs were analyzed in the positive selection analysis of both host and symbiont.
Ortholog Identification and Phylogenetic Analysis
The best hits shared among P. okutanii, A. marissinica, and the other shallow-water clams were determined by all-versus-all BLASTp with a cutoff value of 1 × e–5. Afterwards, the default pipeline of OrthoMCL version 2.0.9 (Li et al., 2003) was used to identify the homologous gene clusters in the present study. Only one-to-one single-copy orthologous genes shared by all of these species were retained for subsequent phylogenetic analysis. The coding DNA sequence of these single-copy orthologous genes was concatenated and aligned using MUSCLE version 3.8.31 (Edgar, 2004) with default settings. The alignments of coding DNA sequence used amino acid alignment as a reference by ParaAT version 1.0 (Zhang et al., 2012). Poorly aligned regions and gaps were removed with Gblocks version 0.91b (Castresana, 2000) with default settings and the remaining alignment of each gene was concatenated and used for phylogenetic analysis. GTR + Γ + I, the best fit model predicted by jModelTest version 2.1.10 (Guindon and Gascuel, 2003; Darriba et al., 2012), was applied in the maximum-likelihood analysis using RaxML GUI version 1.5b1 (Stamatakis et al., 2008) with 100 rapid bootstraps.
Positive Selection Analysis
The identification of positively selected genes requires performing the likelihood ratio tests between the alternative branch-site model that allows the ratio ω [dN/dS: the non-synonymous substitution rate (dN) divided by the synonymous substitution rate (dS)] to vary in different lineages and the neutral branch-site model with a confined ratio ω. Both models rely on the phylogenetic relationship of the examined species (Yang and Dos Reis, 2011). The coding DNA sequences of single-copy orthologous genes shared by P. okutanii, A. marissinica, and other shallow-water veneroids were used for positive selection analysis using PosiGene (Sahm et al., 2017a) with the following settings: – target_species = Phreagena okutanii, Archivesica marissinica, – anchor species = Archivesica marissinica, – min_seq_num = 3, – min_omega = 0, – min_site_signifance = 0.9. Only the single-copy homologous gene in both P. okutanii and A. marissinica as well as other shallow-water species was considered for positive selection analysis, and shallow-water species with multiple copies of the gene were excluded from the list of references in the analysis of the given gene. The clade containing P. okutanii and A. marissinica was designated as the targeted lineage in the analysis using PosiGene (Sahm et al., 2017a). It has been reported that errors in the analysis are mainly caused by misalignment, which cannot be reduced by multiple test corrections (Sahm et al., 2017b). Therefore, the default pairwise similarity threshold of PosiGene (Sahm et al., 2017a) was applied to remove alignment errors, such as orthologs that were pseudo or poorly aligned, and nominal P-values were directly used to identify positively selected genes (Fletcher and Yang, 2010; Sahm et al., 2017b). If the genes had a P-value below 0.05 and amino acid sites with a bayes empirical bayes (BEB) probability above 0.9, they were regarded as the positively selected genes (Lan et al., 2017, 2018; Sun et al., 2017). For the positive selection analysis of symbionts, the settings of PosiGene was – target_species = symbiont Candidatus Vesicomyosocius okutanii, – anchor species = symbiont Candidatus Vesicomyosocius okutanii, – min_seq_num = 3, – min_omega = 0, – min_site_signifance = 0.9. Other settings and thresholds to identify positively selected genes was the same as that applied to the host.
Results
Transcriptome Sequencing, Assembly, and Annotation
The shells of P. okutanii and A. marissinica are shown in Supplementary Figure S1. The cox1 gene sequence information of P. okutanii and A. marissinica samples are shown in the Supplementary Table S1. The details of transcriptome sequencing data of both P. okutanii and A. marissinica are summarized in the Supplementary Table S2.
After removing the reads from the gill tissues that could be mapped to the genome of Candidatus Vesicomyosocius okutanii, 31,823,340 and 59,685,2585 reads were retained for the transcriptome assembly of P. okutanii and A. marissinica, respectively. After duplicate transcripts were removed, open reading frame prediction generated 25,402 transcripts with an N50 length of 1,545 bp for P. okutanii and 25,530 transcripts with an N50 length of 2,035 bp for A. marissinica (Supplementary Table S2). These transcripts were translated into proteins and compared with those of the shallow-water veneroid clams. Supplementary Table S1 summarizes all available transcriptome information of the clams.
Among the translated proteins of P. okutanii, 20,022 (∼78.8%) proteins matched those in the NR database, 12,736 (50.1%) matched the GO terms and 6,664 (26.2%) had hits in the KEGG pathway database. For A. marissinica, 20,141 (∼78.9%) predicted proteins had hits in the NCBI NR database, 13,259 (51.9%) matched the GO terms and 7,238 (28.4%) had hits in the KEGG pathway database (Supplementary Table S2). The functional annotation of P. okutanii and A. marissinica shared a similar GO distribution in terms of cellular component, molecular function, and biological process (Figure 1).
Gene Expression Profiling
For the highly expressed genes in the gills of P. okutanii and A. marissinica, genes with significant fold changes compared to other organs were listed in Supplementary Tables S3, S4, respectively. The TCA cycle was found to be incomplete in the symbionts from the transcriptome data of A. marissinica. While the host genes involved in TCA cycle, including cytoplasmic aconitate hydratase-like, 2-oxoglutarate dehydrogenase, isocitrate dehydrogenase (NADP) cytoplasmic-like and succinate ligase, were highly expressed in the gills of both P. okutanii and A. marissinica when compared with other organs (Figure 2 and Supplementary Figure S2). Among the highly expressed genes in the gills, many of them were the Rab gene families for vesicle trafficking and ones encoding lysozymes (Figure 3 and Supplementary Figure S3). For example, V-type proton ATPase, cathepsin B, cathepsin L, Rab 7 and lysozymes were highly expressed in the gills of both P. okutanii and A. marissinica (Figure 3 and Supplementary Figure S3). The Rab 5 and early endosome antigen 1 (EEA1) were highly expressed in the gill of A. marissinica (Figure 3). Rabenosyn-5 was highly expressed in the gill of P. okutanii (Supplementary Figure S3).
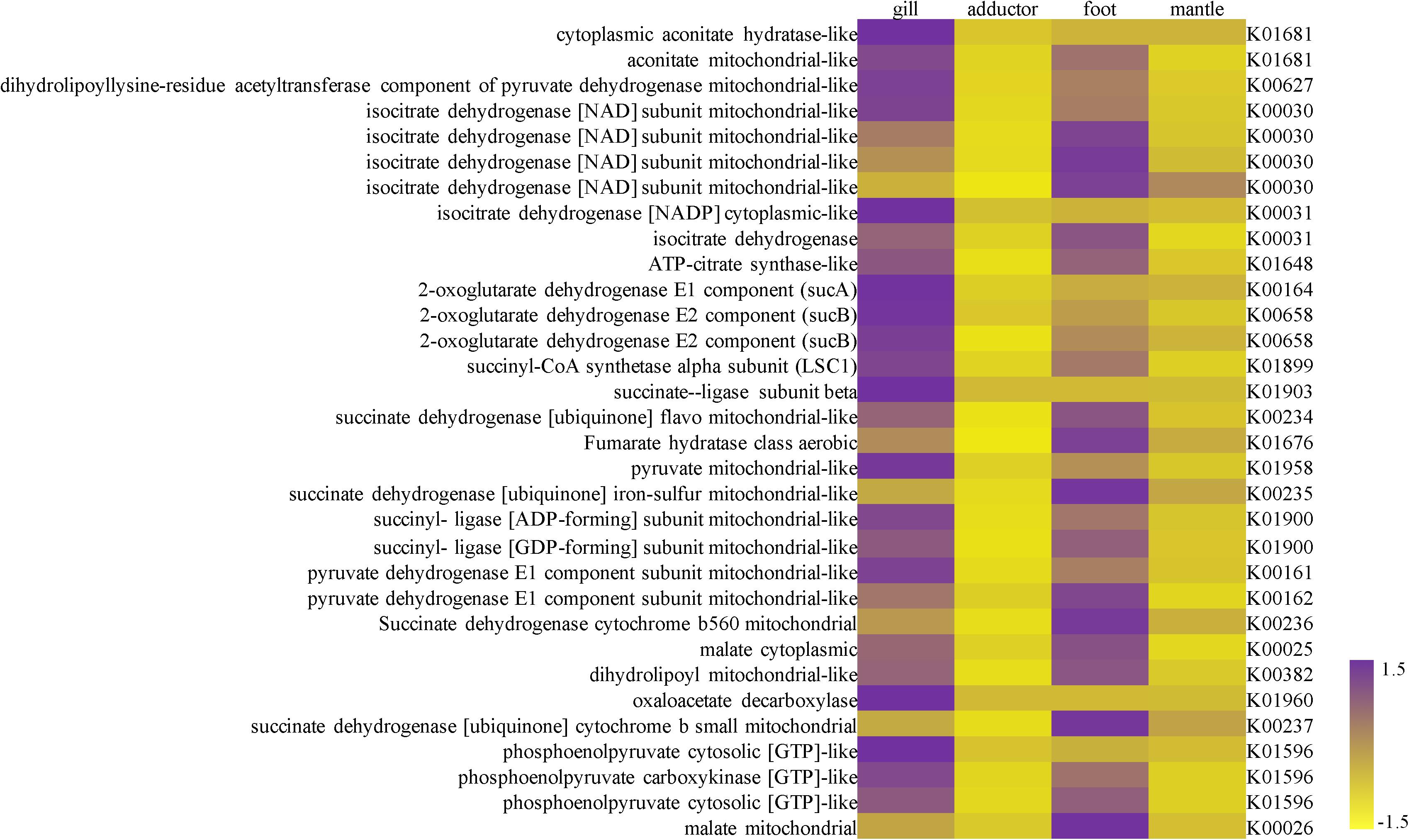
Figure 2. A heat map showing the expression level of genes in the TCA cycle in the gill, adductor muscle, foot, and mantle of Archivesica marissinica (left: NR annotation; right: KEGG annotation).
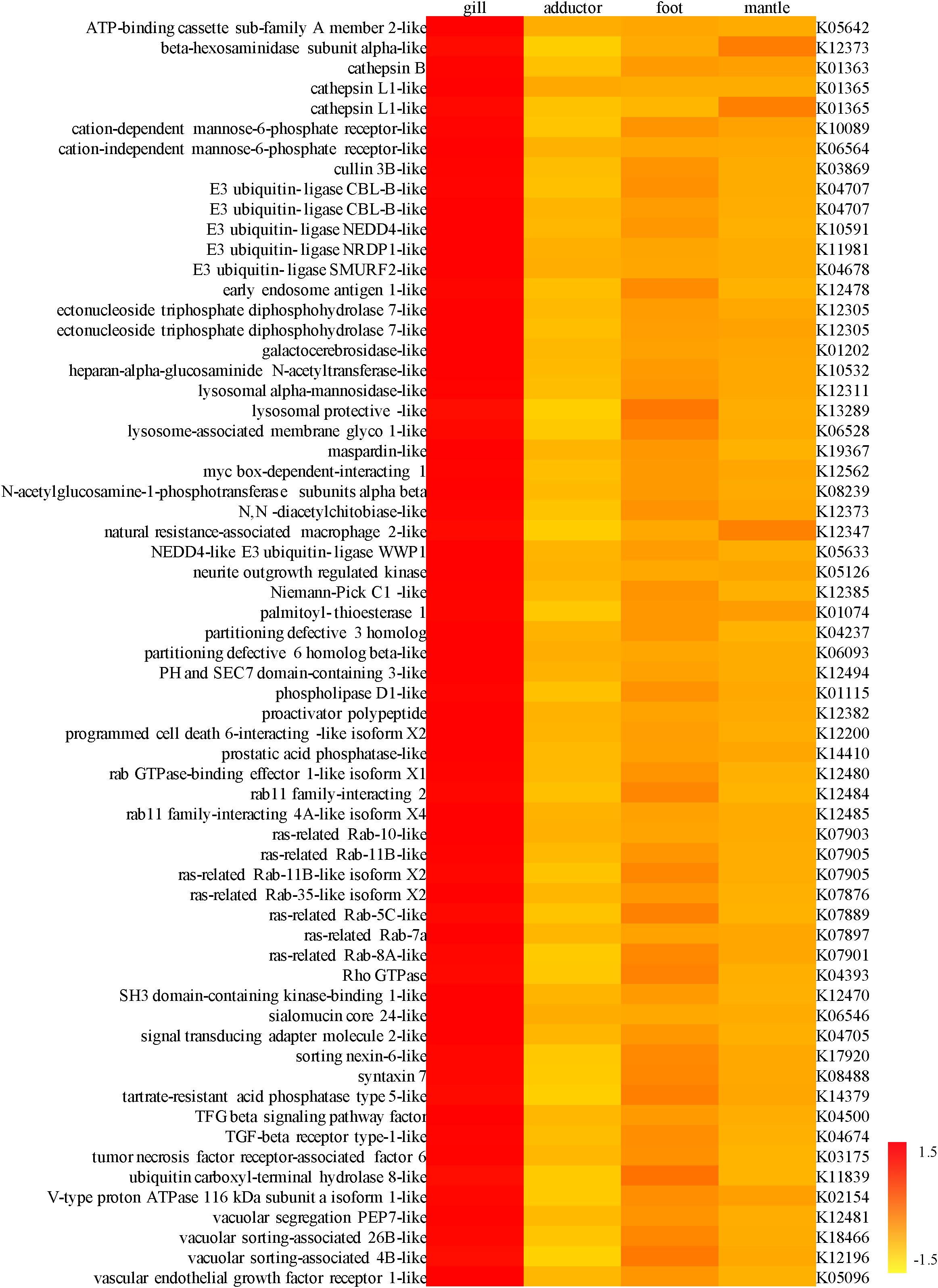
Figure 3. A heat map showing the expression level of genes involved in endocytosis and the lysosome pathway, especially the Rab families of gene and genes encoding lysozymes, in the gill, adductor muscle, foot, and mantle of Archivesica marissinica (left: NR annotation; right: KEGG annotation).
Among all the A. marissinica gill transcriptome reads, 6,548,372 were mapped successfully the coding DNA sequences of symbiont Candidatus Vesicomyosocius okutanii with an average mapping depth of 288. Around 97% Candidatus Vesicomyosocius okutanii coding DNA sequences were effectively covered by the A. marissinica symbiont reads. In the gills of A. marissinica, the top 10% of the highest expressed genes of the symbionts, including dsr, sqr, apr, sox and the gene encoding F0F1-ATPases, are involved in sulfur oxidation and ATP synthesis (Supplementary Table S5). Furthermore, these highly expressed genes were classified into a set of COG categories, such as energy production and conversion, amino acid metabolism and transport, carbohydrate metabolism and transport, translation, inorganic ion transport and metabolism, intracellular trafficking, and secretion (Supplementary Table S6). For the top 10% highest expressed genes of host included hemoglobin I, hemoglobin II, and ribosomal proteins (Supplementary Tables S7, S8).
Positive Selection
The proportions of amino acids in the protein sequences of deep-sea and shallow-water clams were similar, confirming that the subsequent positive selection analysis would not be biased due to amino acid usage (Supplementary Figure S4). A phylogenetic tree (Figure 4A) was constructed to provide the branch-site model with the phylogenetic relationship of the deep-sea and shallow-water veneroid clams. Among 4,648 orthologous genes that were used in the positive selection analysis, 86 were positively selected (Supplementary Table S9) and showed common amino acid substitutions between the deep-sea clams P. okutanii and A. marissinica. These amino acid substitutions occurred in the genes that are involved in vesicular transport, intracellular trafficking and lysis, including actin-related 2/3 protein complex (ARPC), V-type proton ATPase subunit C 1A-like (ATP6V1C1), cathepsin A (CTSA), CTSL, and cullin 3B-like (CUL3B) (Table 1 and Figure 4B). The amino acid alignments of these positively selected genes of host were provided in the Supplementary Material for future deep-sea studies (Supplementary Figure S5).
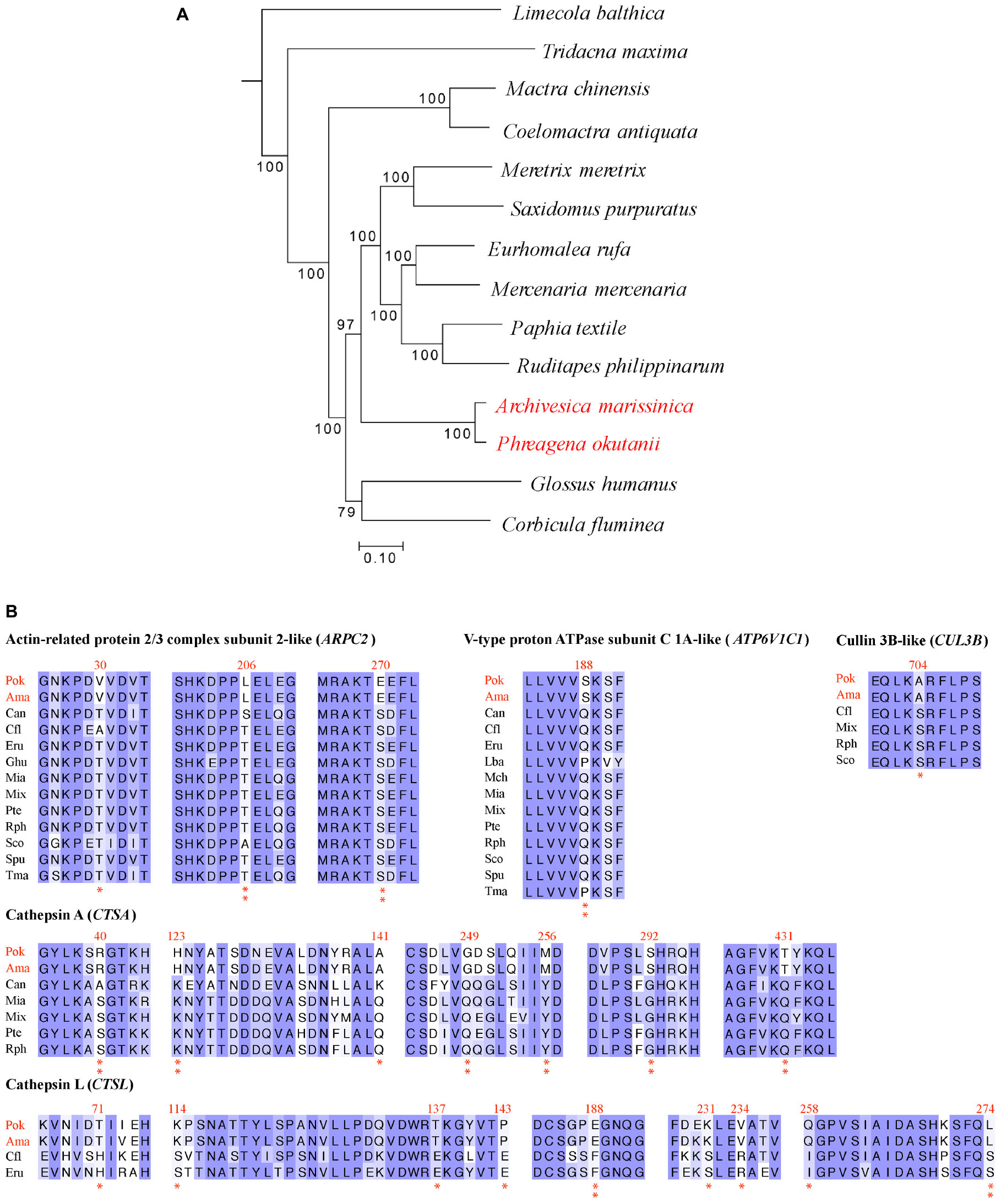
Figure 4. Positively selected genes identified in deep-sea Phreagena and Archivesica. (A) Phylogeny of Veneroida clams with transcriptomes. The number next to the nodes is the bootstrap value from maximum likelihood analysis. A total of 151,496 nt coding DNA sequences contributed to this phylogenetic tree. The outgroup is Sinonovacula constricta in the Solecurtidae family. (B) Partial alignments of the positively selected genes. The shallow-water species with multiple copies of homologs were excluded from the analysis of the given gene. The Arp2/3 complex, cathepsin A, cathepsin L, cullin 3B-like protein, and V-type proton ATPase subunit C 1A-like protein involved in the vesicle trafficking and lysis process have a clear sign of positive selection. Double asterisks denote the amino acids in both Phreagena okutanii and Archivesica marissinica with a BEB posterior probability exceeding 0.95 and a single asterisk indicates the sites with a posterior probability above 0.9 but below 0.95 (Pok: Phreagena okutanii; Ama: Archivesica marissinica; Can: Coelomactra antiquata; Cfl: Corbicula fluminea; Eru: Eurhomalea rufa; Ghu: Glossus humanus; Lba: Limecola balthica; Mch: Mactra chinensis; Mia: Mercenaria mercenaria; Mix: Meretrix meretrix; Pte: Paphia textile; Rph: Ruditapes philippinarum; Spu: Saxidomus purpuratus; Sco: Sinonovacula constricta; and Tma: Tridacna maxima).
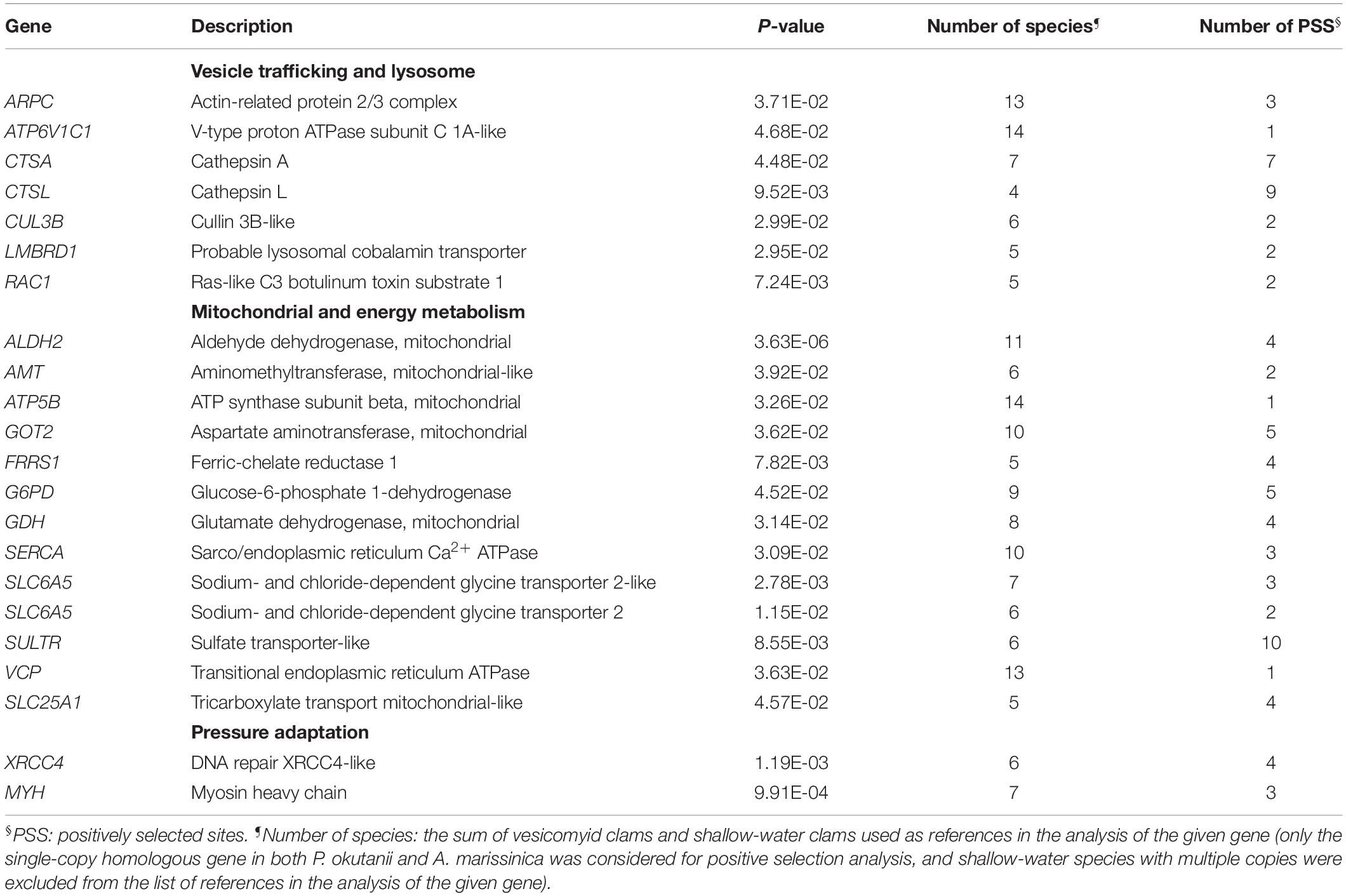
Table 1. Positively selected genes in Phreagena okutanii and Archivesica marissinica.
For symbiont, a total of 473 single-copy orthologs were applied in the positive selection analysis. Sixteen genes were identified under positive selection and listed in the Supplementary Table S10. Among them, pyruvate dehydrogenase that are involved in TCA cycle as well as NADH-quinone oxidoreductase subunit C and cytochrome-c oxidase cbb3-type subunit I that are involved in oxidative phosphorylation were all detected as having signals of positive selection.
Discussion
Previous studies (Kuwahara et al., 2007; Newton et al., 2007) have focused primarily on the symbiont of the vesicomyids that adopt the vertical transmission of symbiont, while the knowledge gap in the host genetics prevented us to gain a more comprehensive understanding of the mutual relationship between the symbiont and host. In the present study, we thoroughly analyzed the transcriptomes of both host and symbiont of the deep-sea vesicomyid clams to gain insights into the host–symbiont interaction.
Complementary Roles of the Two Partners in the Holobiont
Endosymbionts housed in deep-sea mussels and tubeworms have a transport system to take up various carbohydrates (Robidart et al., 2008; Ho et al., 2017; McCuaig et al., 2018). On the contrary, the endosymbionts of the deep-sea vesicomyid clams reportedly lacked the genes related to the systems for absorbing phosphoenolpyruvate-dependent sugar in the genomes (Kuwahara et al., 2007). This implies that the endosymbionts of the clams are unlikely to obtain sugar externally and appear to be more independent in the synthesis of nutrients, making them very different from the endosymbionts of mussels or tube worms that are horizontally transferred from the environment. Indeed, an essential enzyme for the carbon fixation step of the Calvin–Benson cycle, ribulose bisphosphate carboxylase, was in the top 10% of the highly expressed genes in the symbionts of A. marissinica (Supplementary Table S5), suggesting that these endosymbionts may rely strongly on the Calvin-Benson cycle to fix carbon for synthesizing nutrients.
The TCA cycle is a pathway not only to synthesize ATP but also to generate various intermediates to fulfill other important metabolic pathways, such as the biosynthesis of amino acids and lipids. In the transcriptome of the endosymbionts of A. marissinica, many genes implicated in glycolysis and the pentose phosphate pathway were among the top 10% of the highest expressed genes and these genes allow symbionts to produce a substantial amount of substrate for the TCA cycle (Supplementary Table S5). However, the symbiont transcriptome data of A. marissinica have incomplete TCA cycles as well as previously reported symbiont genomes of Phreagena okutanii and Calyptogena magnifica (Kuwahara et al., 2007; Newton et al., 2007). For example, the gene encoding the enzyme 2-oxoglutarate dehydrogenase is not found in the transcriptome data, but this enzyme likely promotes the production of succinyl-CoA, 2-oxoglutarate, and oxaloacetate. This finding is consistent with the results of the symbionts of P. okutanii (Kuwahara et al., 2007) and Calyptogena magnifica (Newton et al., 2007). However, this missing gene was highly expressed in the host gill of the P. okutanii and A. marissinica compared with other host organs (Figure 2 and Supplementary Figure S2). Replenishment of these intermediates of the TCA cycle or the existence of alternate cellular metabolism are very crucial for the symbiont, in order to synthesize all essential nutrients (Ponnudurai et al., 2017).
Sulfur Metabolism and Energy Conservation in the Endosymbiont of Archivesica marissinica
Vesicomyid clams usually insert their foot into sediments to obtain hydrogen sulfide and transport this chemical to the bacteriocytes via sulfide-binding proteins (Arp et al., 1984; Childress and Girguis, 2011). The endosymbionts rely on the energy generated from sulfur oxidation to synthesize organic matter for the host (Morton, 1986; Kuwahara et al., 2007; Newton et al., 2007). Indeed, the top 10% of the highest expressed genes in the endosymbionts of A. marissinica include the dsrABCL gene complex that catalyzes sulfide to sulfite, the aprA gene that mediates the oxidation of sulfite to sulfate, and the genes (cytochrome c oxidase and F-type ATPase) that are involved in the oxidative phosphorylation pathway (Supplementary Table S5). These results suggest that these sulfide oxidation pathways are essential for driving the metabolism of the endosymbionts. Since the genes encoding glutamine synthase and glutamate synthase can respectively synthesize glutamine and glutamate using ammonia, the high expression of these two genes in the endosymbionts of A. marissinica (Supplementary Table S5) suggests that these biosynthesis pathways of amino acids are essential in the endosymbionts. In conclusion, these results confirm the earlier findings in the endosymbionts of P. okutanii but more importantly, they demonstrate that the endosymbionts of A. marissinica could perform the same functions as those of P. okutanii.
Digestion of Symbiont
The symbionts without transporters for nutrients such as amino acids and cofactors cannot transport the nutrients to the host (Kuwahara et al., 2007; Newton et al., 2007). In fact, we found that the genes that encode EEA1-like protein, ras-related Rab-5C-like (Rab5) protein, ras-related Rab-7a (Rab7) protein, syntaxin 7, cullin 3B-like protein, lysosomal hydrolases and cathepsins responsible for active fusion and cell lysis were highly expressed in the host gill tissues of A. marissinica compared with other tissues without endosymbionts (Figure 3). Among them, Rabenosyn-5 (a Rab5 effector; Nielsen et al., 2000), Rab 7, V-type proton ATPase, lysosomal hydrolases and cathepsins were also highly expressed in the gills of P. okutanii (Supplementary Figure S3). Moreover, these two deep-sea clams share same positively selected genes, including ARPC, V-type proton ATPase subunit C 1A-like, cathepsin A, cathepsin L, and cullin 3B-like (Table 1 and Figure 4B), which may contribute to the molecular adaptation of deep-sea chemosynthetic environment. To better understand the potential functions of these genes, we propose a step-by-step schematic model to illustrate the maturation of symbiont-containing vesicles and the host digestion process (Figure 5). Briefly, EEA1 and Rab5 first promote the fusion between Rab5-positive vesicles and their maturation into Rab7-positive vesicles (Mills et al., 1998; Wennerberg et al., 2005); Rab7 enhances the fusion between mature symbiont-containing vesicles and promotes their further maturation for fusion with lysosome (Chen et al., 2003); Syntaxin 7 is required for the fusion between Rab7-positive vesicles and lysosomes (Mullock et al., 2000); the actin-related protein 2/3 complex then mediates actin polymerization during vesicle trafficking (Daugherty and Goode, 2008); cullin 3B-like protein regulates the vesicle trafficking during the maturation of symbiont-containing vesicles (Hubner and Peter, 2012; Huotari et al., 2012); and lastly V-type proton ATPase subunit C 1A-like protein, lysosomal hydrolases, and cathepsin proteins in the lysosomes that were all highly expressed support the host in carrying out the vigorous lysosome activity to digest the symbiont (Fiala-Médioni et al., 1994; Beyenbach and Wieczorek, 2006). Moreover, among the highly expressed genes in the gill, the actin-related protein 2/3 complex, cullin 3B-like protein, V-type proton ATPase subunit C 1A-like protein, cathepsin A, and cathepsin L were under positive selection pressure in both deep-sea vesicomyid clams compared with shallow-water veneroid clams (Table 1 and Figure 4B), implying that the hosts have developed sophisticated mechanisms to efficiently utilize the endosymbiont cells and thus control the bacterial population. However, the limitations of this study are clear. Due to the challenges and difficulties of deep-sea sampling, only one individual of each clam species were collected and used in the gene expression analysis. Lacking of replicates may cause potential individual bias in the gene expression analysis. The gene expression patterns may have been affected by the changes in hydrostatic pressure, temperature or other environmental factors during the period of sample collection, although the samples were dissected and fixed immediately upon arriving on board. Further validation will be conducted in the future, if deep-sea clams fixed in situ become available.
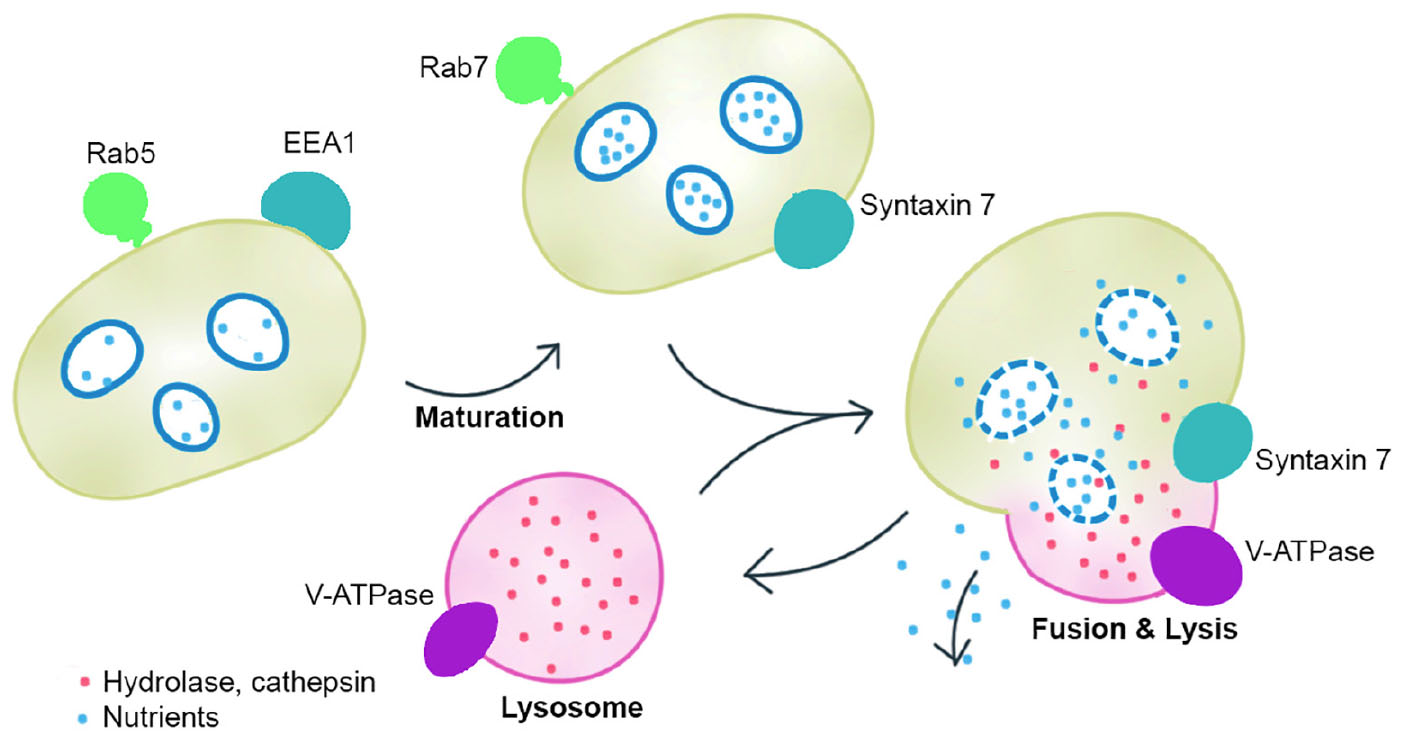
Figure 5. A working model showing the function of the highly expressed genes. Early endosome antigen 1 (EEA1) and Rab5 first promote the fusion between Rab5-positive vesicles and their maturation into Rab7-positive vesicles; Rab7 enhances the fusion between mature symbiont-containing vesicles and promotes their further maturation for fusion with lysosome; Syntaxin 7 is required for the fusion between Rab7-positive vesicles and lysosomes; and lastly V-type proton ATPase subunit C 1A-like protein, lysosomal hydrolases, and cathepsin proteins in the lysosomes support the host in carrying out the vigorous lysosome activity to digest the symbiont. The model indicates that the hosts have developed sophisticated mechanisms to efficiently digest the endosymbiont cells.
The high expression level and positive selection of the genes related to vesicle trafficking and lysosomes suggest that the vesicomyid clams actively digest their symbionts through lysis to satisfy their nutritional demand. The symbionts actively generate and provide various nutrients to the hosts. Overall, the present study offers the first high-quality transcriptomes of deep-sea chemosymbiotic vesicomyid clams, which allow us to close the gap in our knowledge of their symbiotic relationships in terms of how the host contributes.
Data Availability Statement
All sequencing reads were submitted to the Sequence Read Archive of the National Center for Biotechnology Information (SRA accession number SRR7156763–4 for Phreagena okutanii and SRR7156765-8 for Archivesica marissinica). The non-redundant assembled transcripts of both Phreagena okutanii and Archivesica marissinica were deposited to the Transcriptome Sequencing Assembly database under BioProject PRJNA471131.
Ethics Statement
All animal experiments were approved by the Ethics Committee of the Hong Kong University of Science and Technology.
Author Contributions
P-YQ conceived the deep-sea symbiosis study and led the project. JS and CC collected the Phreagena okutanii clam. JT collected the Archivesica marissinica clam. CC confirmed the identity and taxonomy of the vesicomyid clams. TX extracted the RNA. YL performed the bioinformatics analyses and drafted the manuscript. WZ contributed to the bacterial analyses. J-WQ contributed to the manuscript writing. All authors contributed to the manuscript and approved it for submission and publication.
Funding
This work was supported by grants from the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC01) and from Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0409) as well as by a grant from China Ocean Mineral Resources Research and Development Association (DY135-E2-1-03) awarded to P-YQ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TY declared a shared affiliation, with no collaboration, with one of the authors, CC, to the handling Editor at the time of review.
Acknowledgments
The authors would like to thank Dr. Hiroyuki Yamamoto who served as the chief scientist of the R/V Kairei research cruise KR15-17, which was supported by the Council for Science, Technology, and Innovation (CSTI) of Japan as the Cross Ministerial Strategic Innovation Promotion Program (SIP), Next-generation Technology for Ocean Resource Exploration. The authors deeply appreciate the tireless support from the Captain and crew members of R/V Kairei, the technical team of the ROV KAIKO as well as all scientists on-board the research cruise. The authors also thank the crew of the ROV Haima and that of the R/V Haiyang-6 exploration vessel for sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00680/full#supplementary-material
Footnotes
References
Arp, A. J., Childress, J. J., and Fisher, C. R. Jr. (1984). Metabolic and blood gas transport characteristics of the hydrothermal vent bivalve Calyptogena magnifica. Physiol. Zool. 57, 648–662. doi: 10.1086/physzool.57.6.30155991
Barry, J. P., Kochevar, R. E., and Baxter, C. H. (1997). The influence of pore-water chemistry and physiology on the distribution of vesicomyid clams at cold seeps in monterey bay: implications for patterns of chemosynthetic community organization. Limnol. Oceanogr. 42, 318–328. doi: 10.4319/lo.1997.42.2.0318
Beyenbach, K. W., and Wieczorek, H. (2006). The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J. Exp. Biol. 209, 577–589. doi: 10.1242/jeb.02014
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bright, M., and Bulgheresi, S. (2010). A complex journey: transmission of microbial symbionts. Nat. Rev. Microbiol. 8, 218–230. doi: 10.1038/nrmicro2262
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, C., Okutani, T., Liang, Q., and Qiu, J. W. (2018). A noteworthy new species of the family vesicomyidae from the south china sea (bivalvia: glossoidea). Venus 76, 29–37. doi: 10.18941/venus.76.1-4_29
Chen, M. C., Cheng, Y. M., Sung, P. J., Kuo, C. E., and Fang, L. S. (2003). Molecular identification of Rab7 (ApRab7) in aiptasia pulchella and its exclusion from phagosomes harboring zooxanthellae. Biochem. Biophys. Res. Commun. 308, 586–595. doi: 10.1016/s0006-291x(03)01428-1
Childress, J. J., and Girguis, P. R. (2011). The metabolic demands of endosymbiotic chemoautotrophic metabolism on host physiological capacities. J. Exp. Biol. 214, 312–325. doi: 10.1242/jeb.049023
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Daugherty, K. M., and Goode, B. L. (2008). Functional surfaces on the p35/ARPC2 subunit of Arp2/3 complex required for cell growth, actin nucleation, and endocytosis. J. Biol. Chem. 283, 16950–16959. doi: 10.1074/jbc.M800783200
Decker, C., Olu, K., Arnaud-Haond, S., and Duperron, S. (2013). Physical proximity may promote lateral acquisition of bacterial symbionts in vesicomyid clams. PLoS One 8:e64830. doi: 10.1371/journal.pone.0064830
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Falcon, S., and Gentleman, R. (2008). “Hypergeometric testing used for gene set enrichment analysis,” in Bioconductor Case Studies, eds R. Hornik, K. Gentleman, and G. Parmigiani (New York, NY: Springer), 207–220. doi: 10.1007/978-0-387-77240-0_14
Feng, D., Qiu, J. W., Hu, Y., Peckmann, J., Guan, H., Tong, H., et al. (2018). Cold seep systems in the south china sea: an overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Feng, J., Meyer, C. A., Wang, Q., Liu, J. S., Shirley Liu, X., and Zhang, Y. (2012). GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28, 2782–2788. doi: 10.1093/bioinformatics/bts515
Fiala-Médioni, A., and Métivier, C. (1986). Ultrastructure of the gill of the hydrothermal vent bivalve calyptogena magnifica, with a discussion of its nutrition. Mar. Biol. 90, 215–222. doi: 10.1007/bf00569130
Fiala-Médioni, A., Michalski, J. C., Jollés, J., Alonso, C., and Montreuil, J. (1994). Lysosomic and lysozyme activities in the gill of bivalves from deep hydrothermal vents. C. R. Acad. Sci. 317, 239–244.
Fletcher, W., and Yang, Z. (2010). The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol. Biol. Evol. 27, 2257–2267. doi: 10.1093/molbev/msq115
Flores, J. F., Fisher, C. R., Carney, S. L., Green, B. N., Freytag, J. K., Schaeffer, S. W., et al. (2005). Sulfide binding is mediated by zinc ions discovered in the crystal structure of a hydrothermal vent tubeworm hemoglobin. Proc. Natl. Acad. Sci. U. S. A. 102, 2713–2718. doi: 10.1073/pnas.0407455102
Fujikura, K., Okutani, T., and Maruyama, T. (2012). Deep-Sea Life Biological Observations Using Research Submersibles, 2nd Edn, Kanagawa: Tokai University press.
Funkhouser, L. J., and Bordenstein, S. R. (2013). Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 11:e1001631. doi: 10.1371/journal.pbio.1001631
Guindon, S., and Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Ho, P. T., Park, E., Hong, S. G., Kim, E. H., Kim, K., Jang, S. J., et al. (2017). Geographical structure of endosymbiotic bacteria hosted by Bathymodiolus mussels at eastern pacific hydrothermal vents. BMC Evol. Biol. 17:121. doi: 10.1186/s12862-017-0966-3
Hubner, M., and Peter, M. (2012). Cullin-3 and the endocytic system: new functions of ubiquitination for endosome maturation. Cell. Logist. 2, 166–168. doi: 10.4161/cl.20372
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Huotari, J., Meyer-Schaller, N., Hubner, M., Stauffer, S., Katheder, N., Horvath, P., et al. (2012). Cullin-3 regulates late endosome maturation. Proc. Natl. Acad. Sci. U.S.A. 109, 823–828. doi: 10.1073/pnas.1118744109
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition – interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Ikuta, T., Igawa, K., Tame, A., Kuroiwa, T., Kuroiwa, H., Aoki, Y., et al. (2016). Surfing the vegetal pole in a small population: extracellular vertical transmission of an ‘intracellular’ deep-sea clam symbiont. R. Soc. Open Sci. 3:160130. doi: 10.1098/rsos.160130
Kojima, S., and Ohta, S. (1997). Calyptogena okutanii n. sp., a sibling species of calyptogena soyoae okutani, 1957 (Bivalvia: Vesicomyidae). Venus 56, 189–195. doi: 10.18941/venusjjm.56.3-189
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam. Calyptogena okutanii. Curr. Biol. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Lan, Y., Sun, J., Tian, R., Bartlett, D. H., Li, R., Wong, Y. H., et al. (2017). Molecular adaptation in the world’s deepest-living animal: insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 26, 3732–3743. doi: 10.1111/mec.14149
Lan, Y., Sun, J., Xu, T., Chen, C., Tian, R., Qiu, J. W., et al. (2018). De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genom. 19:394. doi: 10.1186/s12864-018-4720-z
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Levin, L. A. (2005). “Ecology of cold seep sediments: interactions of fauna with flow, chemistry and microbes,” in Oceanography and Marine Biology: An Annual Review, eds R. N. Gibson, R. J. A. Atkinson, and J. D. M. Gordon, (Boca Raton, FL: CRC Press), 11–56.
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, Y., Kocot, K. M., Schander, C., Santos, S. R., Thornhill, D. J., and Halanych, K. M. (2015). Mitogenomics reveals phylogeny and repeated motifs in control regions of the deep-sea family siboglinidae (annelida). Mol. Phylogenet. Evol. 85, 221–229. doi: 10.1016/j.ympev.2015.02.008
Li, Y., Kocot, K. M., Whelan, N. V., Santos, S. R., Waits, D. S., Thornhill, D. J., et al. (2017). Phylogenomics of tubeworms (Siboglinidae, Annelida) and comparative performance of different reconstruction methods. Zool. Scripta. 46, 200–213. doi: 10.1111/zsc.12201
Liang, Q., Hu, Y., Feng, D., Peckmann, J., Chen, L., Yang, S., et al. (2017). Authigenic carbonates from newly discovered active cold seeps on the northwestern slope of the south china sea: constraints on fluid sources, formation environments, and seepage dynamics. Deep Sea Res. Part I 124, 31–41. doi: 10.1016/j.dsr.2017.04.015
McCuaig, B., Pena-Castillo, L., and Dufour, S. C. (2018). Metagenomic Analysis Suggests Broad Metabolic Potential in Extracellular Symbionts of the Bivalve Thyasira cf. Gouldi. BioRxiv. [Preprint]. doi: 10.1101/330373.
Mills, I. G., Jones, A. T., and Clague, M. J. (1998). Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr. Biol. 8, 881–884. doi: 10.1016/S0960-9822(07)00351-X
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Morton, B. (1986). The functional morphology of the organs of feeding and digestion of the hydrothermal vent bivalve calyptogena magnifica (Vesicomyidae). J. Zool. 208, 83–98. doi: 10.1111/j.1469-7998.1986.tb04711.x
Mullock, B. M., Smith, C. W., Ihrke, G., Bright, N. A., Lindsay, M., Parkinson, E. J., et al. (2000). Syntaxin 7 is localized to late endosome compartments, associates with vamp 8, and is required for late endosome–lysosome fusion. Mol. Biol. Cell 11, 3137–3153. doi: 10.1091/mbc.11.9.3137
Nakamura, K., Kawagucci, S., Kitada, K., Kumagai, H., Takai, K., and Okino, K. (2015). Water column imaging with multibeam echo-sounding in themid-okinawa trough: implications for distribution of deep-sea hydrothermal vent sites and the cause of acoustic water column anomaly. Geochem. J. 49, 579–596. doi: 10.2343/geochemj.2.0387
Newton, I. L., Girguis, P. R., and Cavanaugh, C. M. (2008). Comparative genomics of vesicomyid clam (Bivalvia: Mollusca) chemosynthetic symbionts. BMC Genom. 9:585. doi: 10.1186/1471-2164-9-585
Newton, I. L. G., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., Fisher, M. C., et al. (2007). The calyptogena magnifica chemoautotrophic symbiont genome. Science 315, 998–1000. doi: 10.1126/science.1138438
Nielsen, E., Christoforidis, S., Uttenweiler-Joseph, S., Miaczynska, M., Dewitte, F., Wilm, M., et al. (2000). Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J. Cell Biol. 151, 601–612. doi: 10.1083/jcb.151.3.601
Nussbaumer, A. D., Fisher, C. R., and Bright, M. (2006). Horizontal endosymbiont transmission in hydrothermal vent tubeworms. Nature 441, 345–348. doi: 10.1038/nature04793
Ohishi, K., Yamamoto, M., Tame, A., Kusaka, C., Nagai, Y., Sugimura, M., et al. (2016). Long-term cultivation of the deep-sea clam Calyptogena okutanii: changes in the abundance of chemoautotrophic symbiont, elemental sulfur, and mucus. Biol. Bull. 230, 257–267. doi: 10.1086/BBLv230n3p257
Ozawa, G., Shimamura, S., Takaki, Y., Takishita, K., Ikuta, T., Barry, J. P., et al. (2017). Ancient occasional host switching of maternally transmitted bacterial symbionts of chemosynthetic vesicomyid clams. Genome Biol. Evol. 9, 2226–2236. doi: 10.1093/gbe/evx166
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Ponnudurai, R., Kleiner, M., Sayavedra, L., Petersen, J. M., Moche, M., Otto, A., et al. (2017). Metabolic and physiological interdependencies in the bathymodiolus azoricus symbiosis. ISME J. 11, 463–477. doi: 10.1038/ismej.2016.124
Robidart, J. C., Bench, S. R., Feldman, R. A., Novoradovsky, A., Podell, S. B., Gaasterland, T., et al. (2008). Metabolic versatility of the riftia pachyptila endosymbiont revealed through metagenomics. Environ. Microbiol. 10, 727–737. doi: 10.1111/j.1462-2920.2007.01496.x
Sahm, A., Bens, M., Platzer, M., and Szafranski, K. (2017a). PosiGene: automated and easy-to-use pipeline for genome-wide detection of positively selected genes. Nucleic Acids Res. 45, e100. doi: 10.1093/nar/gkx179
Sahm, A., Bens, M., Szafranski, K., Holtze, S., Groth, M., Goerlach, M., et al. (2017b). Long-Lived Rodents Reveal Signatures of Positive Selection in Genes Associated with Lifespan and Eusociality. bioRxiv. [Preprint]. doi: 10.1101/191999.
Sibuet, M., and Olu, K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. Part II 45, 517–567. doi: 10.1016/S0967-0645(97)00074-X
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stewart, F. J., Young, C. R., and Cavanaugh, C. M. (2008). Lateral symbiont acquisition in a maternally transmitted chemosynthetic clam endosymbiosis. Mol. Biol. Evol. 25, 673–687. doi: 10.1093/molbev/msn010
Sun, J., Zhang, Y., Xu, T., Zhang, Y., Mu, H., Zhang, Y., et al. (2017). Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1:121. doi: 10.1038/s41559-017-0121
Van Dover, C. (2000). The Ecology of Deep-Sea Hydrothermal Vents. Princeton, NJ: Princeton University Press.
Wennerberg, K., Rossman, K. L., and Der, C. J. (2005). The ras superfamily at a glance. J. Cell Sci. 118, 843–846. doi: 10.1242/jcs.01660
Won, Y. J., Hallam, S. J., O’Mullan, G. D., Pan, I. L., Buck, K. R., and Vrijenhoek, R. C. (2003). Environmental acquisition of thiotrophic endosymbionts by deep-sea mussels of the genus Bathymodiolus. Appl. Environ. Microbiol. 69, 6785–6792. doi: 10.1128/aem.69.11.6785-6792.2003
Yang, Z., and Dos Reis, M. (2011). Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 28, 1217–1228. doi: 10.1093/molbev/msq303
Ye, J., Zhang, Y., Cui, H., Liu, J., Wu, Y., Cheng, Y., et al. (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46, W71–W75. doi: 10.1093/nar/gky400
Keywords: chemosynthesis, symbiosis, gene expression, Mollusca, vertical transmission
Citation: Lan Y, Sun J, Zhang W, Xu T, Zhang Y, Chen C, Feng D, Wang H, Tao J, Qiu J-W and Qian P-Y (2019) Host–Symbiont Interactions in Deep-Sea Chemosymbiotic Vesicomyid Clams: Insights From Transcriptome Sequencing. Front. Mar. Sci. 6:680. doi: 10.3389/fmars.2019.00680
Received: 25 July 2019; Accepted: 18 October 2019;
Published: 01 November 2019.
Edited by:
Sandie M. Degnan, The University of Queensland, AustraliaReviewed by:
Benedict Yuen, University of Vienna, AustriaAdam Michael Reitzel, The University of North Carolina at Charlotte, United States
Takao Yoshida, Japan Agency for Marine-Earth Science and Technology, Japan
Copyright © 2019 Lan, Sun, Zhang, Xu, Zhang, Chen, Feng, Wang, Tao, Qiu and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-Yuan Qian, Ym9xaWFucHlAdXN0Lmhr