 Cornelia Jaspers1*†
Cornelia Jaspers1*† Nancy Weiland-Bräuer2*†
Nancy Weiland-Bräuer2*† Martin A. Fischer2
Martin A. Fischer2 Sven Künzel3
Sven Künzel3 Ruth A. Schmitz2
Ruth A. Schmitz2 Thorsten B. H. Reusch1
Thorsten B. H. Reusch1- 1GEOMAR Helmholtz Centre for Ocean Research Kiel, Marine Evolutionary Ecology, Kiel, Germany
- 2Institute for General Microbiology, Christian-Albrechts-Universität zu Kiel, Kiel, Germany
- 3Max Planck Institute for Evolutionary Biology, Plön, Germany
The translocation of non-indigenous species around the world, especially in marine systems, is a matter of concern for biodiversity conservation and ecosystem functioning. While specific physical and ecological traits are often recognized to influence the success in the establishment of non-indigenous species, the impact of the associated microbiota for the fitness and performance of basal marine metazoans remains vastly unknown. However, for the elucidation of the microbial impact on host fitness, first the diversity of the associated microbiota has to be characterized. In this study, we compared bacterial composition patterns of the invasive ctenophore Mnemiopsis leidyi in different native and invasive sub-populations along with the genetic structure of the host (polymorphic microsatellite markers). Amplicon sequencing of the 16S rRNA gene (V1-V2 hypervariable regions) revealed that M. leidyi as representative of the phylum Ctenophora, the sister group to all metazoans, harbored a distinct microbiota compared to the ambient seawater, which significantly differed across two major tissues, namely epidermis and gastrodermis. Moreover, we identified significant differences in bacterial community compositions between native and invasive sub-populations of M. leidyi. This might indicate, that the microbiota can be influenced by the genotypic background of the host. To detangle such interactions, laboratory controlled experiments in a common garden set up are needed.
Introduction
All multicellular organisms are intimately associated with microbes and thus jointly constitute metaorganisms, a fact that is attaining large public and scientific attention. For model organisms it has been shown that the microbiota contributes to host fitness and can be essential for host functioning (Bosch and McFall-Ngai, 2011; Rook et al., 2017; Jaspers et al., 2019a). Ctenophores, being a basal metazoan phylum (Dunn et al., 2008; Whelan et al., 2017), have so far not been considered in the metaorganism context (Bosch and McFall-Ngai, 2011). However, it is expected that a more detailed understanding on host-microbe interactions at the base of the metazoan tree can reveal important information about metaorganism evolution. For example, in other marine basal metazoans such as corals and jellyfish, bacteria and their exudates have a crucial impact on life history decision such as metamorphosis and settlement induction to the benthos (Neumann, 1979; Tebben et al., 2011). Additionally, bacterial exudates have been documented to trigger sexual reproduction in choanoflagellates (Woznica et al., 2017) as well as the evolution of multicellularity (Alegado et al., 2012).
The ctenophore Mnemiopsis leidyi is not only a basal metazoan species, but is becoming an ecological model organism for biological invasions (Jaspers et al., 2018) with large and widespread impacts on invaded ecosystems (e.g., Kideys, 2002; Tiselius and Møller, 2017). For western Eurasia, two distinct invasion events can be differentiated with a first introduction into the Black Sea with subsequently spreading throughout adjacent waters including the Mediterranean Sea, and a more recent introduction into north-western Europe (Reusch et al., 2010; Jaspers et al., 2018). Both introductions can be traced to different sub-population origins in the native habitat with the north European invasion originating from the north-east coast of the United States, while the southern invasion can be traced back to the Gulf of Mexico and south-east coast of the United States (Bolte et al., 2013; Bayha et al., 2015). Some earlier studies already characterized the associated microbiome of M. leidyi in invaded (Hao et al., 2015) and native (Daniels and Breitbart, 2012) sub-populations, but came to inconclusive results. Moreover, the study by Hammann et al. (2015) even claimed that M. leidyi epithelia in invaded habitats do not contain an associated microbiome (Hammann et al., 2015), which was, however, not confirmed by others (Dinasquet et al., 2012). Thus, the objectives of this study are to investigate (1) the effect of M. leidyi sub-population origin on the microbiota diversity, (2) the bacterial community differentiation between specific M. leidyi tissue compartments and 3) assess differences in the microbiome of native versus invasive sub-populations by using standardized sampling and sequencing methods.
Materials and Methods
Sampling of Mnemiopsis leidyi
Specimen of M. leidyi (8 to 17 individuals) were sampled during summer 2012 (approximately during temperature peak) in northern and southern areas of its native as well as invasive distribution range (see Jaspers et al., 2019b). In more detail, samples from native habitats originated from Mosquito Lagoon, Florida (17 ctenophores, 11 water samples), Atlantic Coast of south-east United States; Woods Hole (Cape Cod), Massachusetts, Atlantic Coast of north-east United States (10 ctenophores, nine water samples); whereas invasive M. leidyi were sampled in Marseille, France, north-west Mediterranean Sea (10 ctenophores, six water samples); and Helgoland, Germany, German Bight/North Sea (eight ctenophores, five water samples). Animals were caught by use of a dip net and animals were individually kept in sterile filtered seawater for up to 6 h before taxidermy of epi- and gastrodermis was conducted. All animals were ensured to have empty guts upon dissection and were washed thoroughly with sterile filtered seawater to remove non-associated microbes. Epi- and gastrodermis samples were attained by cutting approximately 1 × 1 cm of the respective tissue out of the animals on sterile petri dishes using a sterile scalpel. Excised samples comprising epithelial tissue as well as mucus were transferred into reaction tubes and homogenized by use of sterile pipet tips for later DNA extraction resulting in isolation of 1 mL v/v samples from the outer epithelia and 200 μL v/v samples from the gastric cavity. Ambient seawater was collected from the sampling sites (5 to 11 replicates per site) by bucket sampling and 1L was subsequently and successively filtered through 10 μm isopore (TCTP 04700) membranes and 0.22 μm polyethersulfone (GPWP 04700) membrane filters (Millipore Corporation, Bedford, MA, United States). All samples were immediately frozen at −80°C until processing.
Nucleic Acid Isolation and 16S rRNA Gene Amplicon Sequencing
DNA was isolated from two different tissue types of M. leidyi, namely mucus and epidermis tissue samples from the outer epithelia (1 mL v/v) as well as tissue samples from the gastric cavity (pharynx: 200 μL v/v), and ambient seawater (from filters - 0.22 μm). DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega, Madison, United States) according the manufacturers protocol. PCR amplification of the V1–V2 hypervariable region of the 16S rRNA gene was performed with primers V2_A_Pyro_27F (5′-CGTATCGCCTCCCTCGCGCCATCAGTCAGAGTTTGATCCTGGCTCAG-3′) and V2_B_Pyro_27F (5′-CTATGCGCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3′). Amplicons were size checked and purified using MinElute Gel Extraction Kit (Qiagen, Hilden, Germany). Purified amplicon DNAs were quantified using Quant-iT PicoGreen kit (Thermo Fisher Scientific, Waltham, United States) and equal amounts of purified PCR products were pooled for subsequent sequencing using the Illumina MiSeq platform with library preparation according to manufacturer’s instructions at the Max-Planck Institute for Evolutionary Biology.
Data Processing and Bioinformatics
All steps of sequence processing were conducted with the program mothur v1.27.0 (Schloss et al., 2009). The Greengenes reference taxonomy (DeSantis et al., 2006), available on http://www.mothur.org/wiki/Greengenes-formatted_databases, was used with reference sequences trimmed to the V1-V2 hypervariable region of the 16S rRNA gene to improve accuracy of classification (Werner et al., 2012). The sequence depth of all 117 analyzed samples ranged from 23,622 to 675,908 single sequences. A random subset of 23,622 sequences per sample (corresponding to the smallest number of reads across samples) was generated to eliminate bias due to unequal sampling effort. The aligned dataset was used to compute a distance matrix (dist.seqs) for binning sequences into operational taxonomic units (OTU) by average neighbor clustering (cluster.split). 2,829 OTUs at a 97% similarity threshold (roughly corresponding to species level) distinction were considered. All downstream computations were performed in R v2.15.1 (R Core Team, 2012), following published protocols (Weiland-Bräuer et al., 2015). Briefly, the extent of change in OTU composition was explored by beta diversity analysis in terms of redundancy analysis (RDA) with Hellinger-transformed OTU abundances (Benjamini and Hochberg, 1995; Langfeldt et al., 2014; Stratil et al., 2014). In general, the RDA model was formulated as “OTU abundances F,” with factor F being either “provenance,” or “compartment.” Models were evaluated with the vegan function rda, followed by anova.cca, using 10,000 random permutations. Homoscedasticity was checked by using the vegan betadisper function, followed by anova.betadisper to assess significance. Upon determination of the significance (P < 0.05) of the overall model, pairwise factor-level combinations were tested for significance. OTUs significantly correlated with any axis in a significant RDA model were determined by using the envfit function with 105 permutations, followed by Benjamini-Hochberg correction.
Sequence data were submitted to the NCBI Sequence Read Archive under Accession No. PRJNA548849 and are additionally stored in a sequence data management system at the Institute of Clinical Molecular Biology (IKMB) at Kiel University, Germany.
Host Genotyping
Genomic DNA from M. leidyi epithelia tissue samples, isolated with the Wizard Genomic DNA Purification Kit (Promega, Madison, United States), were used for genotype analyses. Sampled M. leidyi representing different sub-populations were examined using 7 highly polymorphic microsatellite markers (Reusch et al., 2010). For genotyping, tissue was lysated in 100 μL low TE buffer (10 mM Tris, 0.1 mM EDTA) for 1 h at 56°C, while shaking and 1 μl extract used as template for the PCR reaction. Primer concentrations and PCR conditions followed published protocols (Reusch et al., 2010; Bolte et al., 2013; Jaspers et al., 2018). Amplified fragments were analyzed on an ABI 3130 genetic analyzer using Rox-350 (Applied Biosystems) as internal size standard. Allele sizes were scored using the software GENEMARKER v1.91 (SoftGenetics, LLC). Pairwise FST values were computed in ARLEQUIN v3.5.1.2 (Excoffier and Lischer, 2010) using non-parametric permutation procedures. All tests were performed with an initial alpha level of 0.05 and corrected following the Benjamini-Hochberg False Discovery Rate (B-H FDR) procedure (Benjamini and Yekutieli, 2001).
Results
Taxonomic Composition of Ctenophore Associated Bacteria Shows Difference to Ambient Seawater
The microbiota associated with different epithelia (epi- and gastrodermis) and sub-populations of M. leidyi as well as the bacterial communities of ambient water were analyzed by Illumina sequencing the hypervariable V1-V2 regions of the bacterial 16S rRNA gene. Sequence depths of 117 analyzed samples ranged between 23,622 to 675,908 single sequences. In total, 2,829 OTUs at a 97% similarity threshold (roughly corresponding to species level) were considered. Abundances of OTUs (97% sequence similarity) between individual samples are depicted at genus level in Figure 1. Consequently, main representative bacterial classes in the samples were Alphaproteobacteria (e.g., including genera Nautella, uncl. Pelagibacteraceae, uncl. Rhodobacteraceae, Octadecabacter), Gammaproteobacteria (e.g., including genera Marinomonas, Halomonas, Shewanella), Mollicutes (e.g., including genera uncl. Entoplasmatales, Mycoplasma), Cyanobacteria (e.g., including genus Synechococcus) and Bacteroidia (e.g., including genus Bacteroides). While OTU abundances between the different sample types were significant, in the following the different comparisons are considered in more detail.
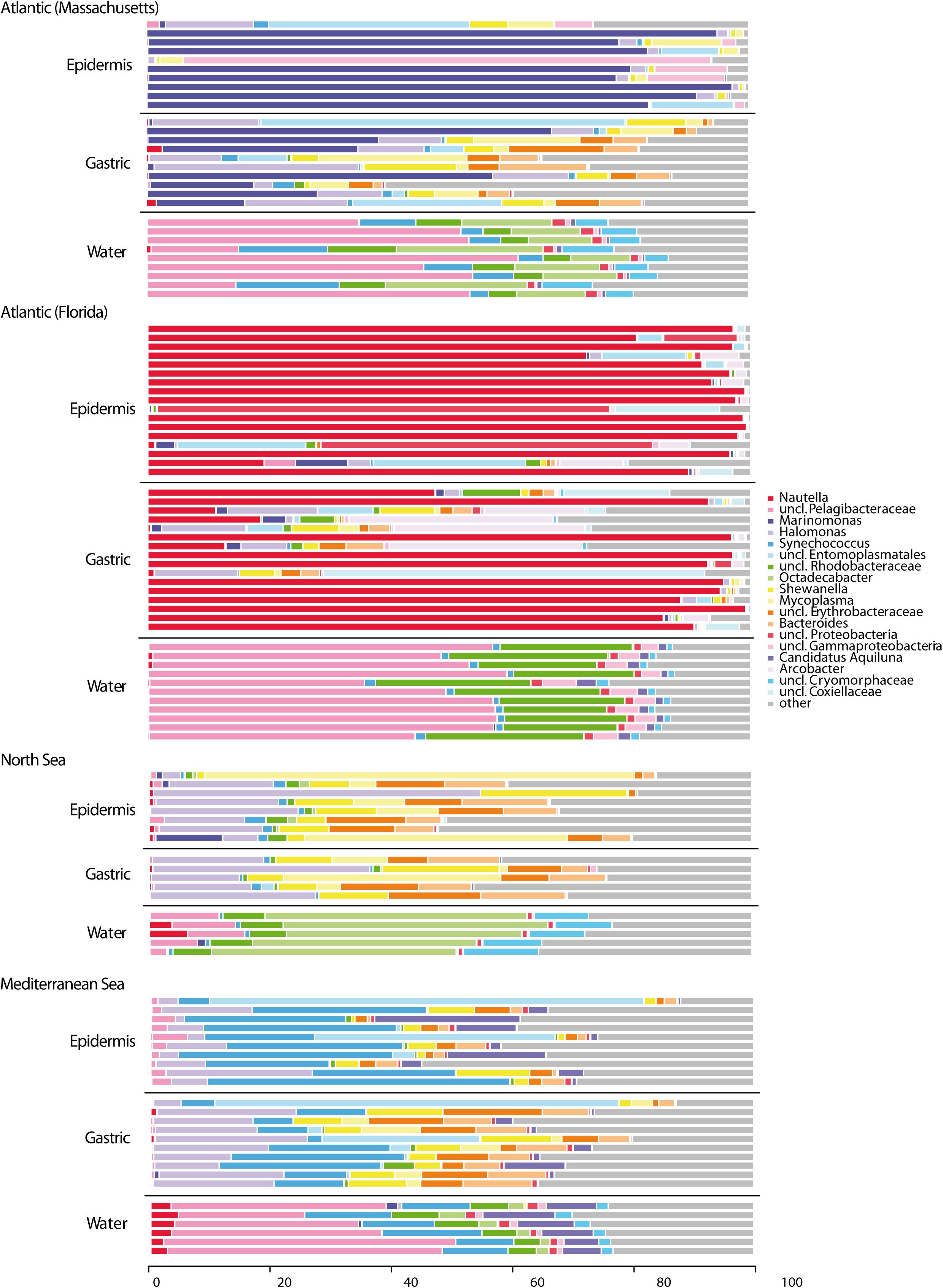
Figure 1. Bacterial composition patterns of individual Mnemiopsis leidyi samples. Microbial communities associated with different epithelia of several sub-populations (native and invasive) of M. leidyi were analyzed by sequencing the V1-V2 hypervariable regions of the bacterial 16S rRNA gene. Taxonomic assemblages inferred from non-chimeric operational taxonomic units (OTUs) using mothur at a 97% pairwise similarity cut-off; distribution patterns were calculated using R. OTU abundances were summarized at genus level. Bar plots are grouped according to the sample type characteristic “provenance”, splitted into “epidermis”, “gastric” and “water” each including at least five replicates. Taxa with a mean relative abundance of <1% across all samples are subsumed under “other”.
Significant Differences Between Associated Bacterial Communities of M. leidyi and Ambient Seawater
Beta diversity analysis demonstrated that the associated bacterial communities of the ctenophore sub-populations were significantly different from the community compositions present in the corresponding ambient water (Figure 2 and Supplementary Table S1). Water samples were mainly dominated by uncultured Pelagibacteraceae and uncultured Rhodobacteraceae of class Alphaproteobacteria, and Synechococcus of class Cyanobacteria, whereas the ctenophore tissues were mostly colonized by Halomonas, Shewanella, and Marinomonas of Gammaproteobacteria, and Mollicutes.
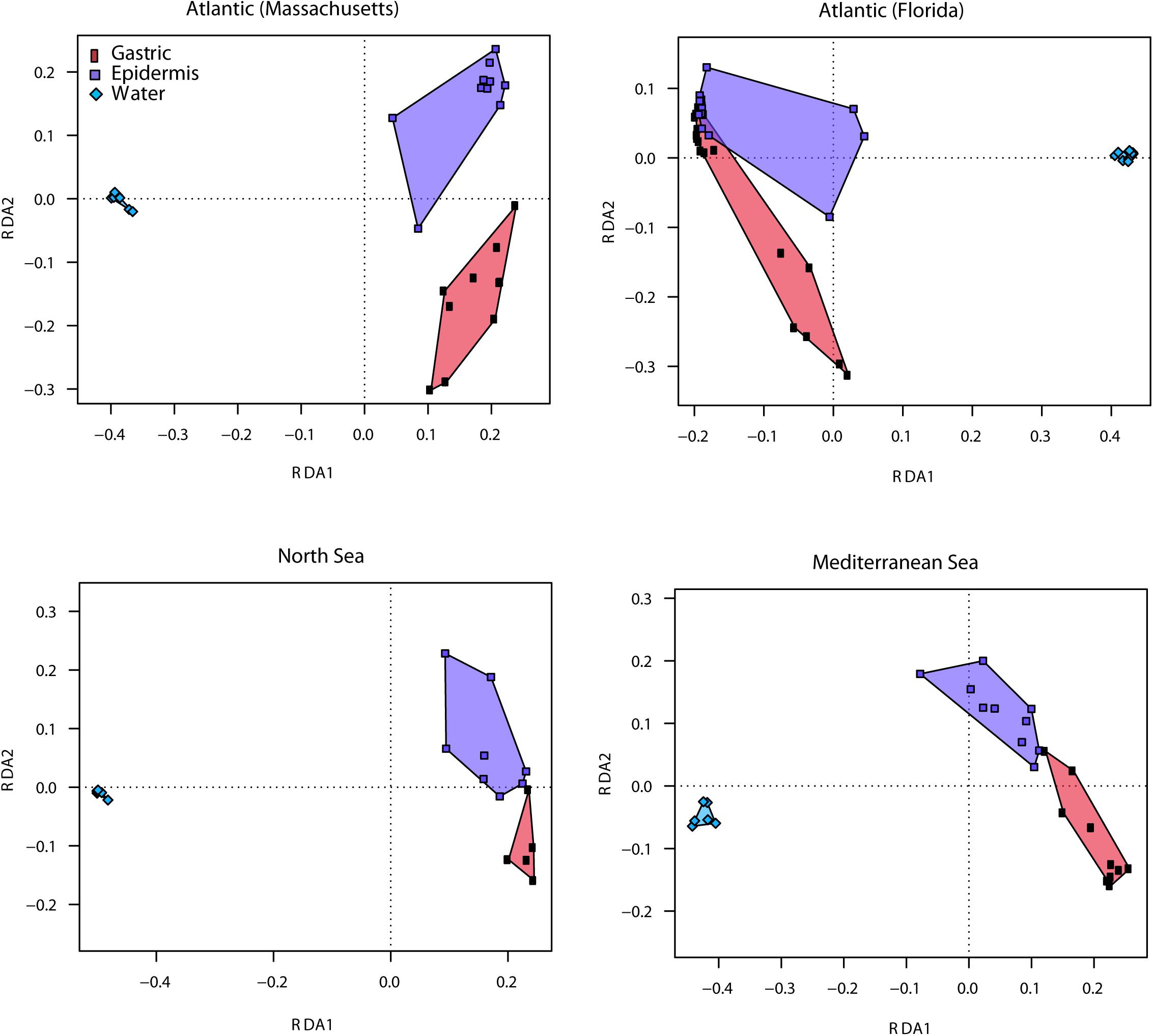
Figure 2. Redundancy analysis (RDA) models of Hellinger-transformed OTU abundances comparing Mnemiopsis leidyi associated bacteria of the gastric cavity and epidermis with the respective ambient water. Graphical representation (distance plots) of RDA model of Hellinger-transformed OTU abundances. Each point represents the whole microbiota community of an individual sample from gastric cavity, epidermis, and ambient seawater of the respective sub-population. Groups of related sample points are framed by polygons filled with a corresponding color to elucidate distribution and variability of sample groups in the ordination space.
Tissue Variation of Ctenophore-Associated Bacteria
Sequencing results based on the 16S rRNA gene amplicon sequencing revealed differences in the bacterial composition among two different tissues, the outer mucus layer and epidermis, and the epithelium in the gastric cavity (Figures 1, 2 and Supplementary Table S1). For all sub-populations, the tissues harbored a significantly different and specific microbiota (P = 0.001), except for the North Sea, where the microbiota composition differed only marginally (P = 0.059). Epidermis from native north-east animals (Atlantic, Massachusetts, United States) was dominated by Marinomonas and unclassified Pelagibacteraceae, whereas their gastric cavity was mainly colonized by Shewanella, Mycoplasma, and Bacteroides, unclassified Erythrobacteraceae and a multitude of low abundant OTUs. Both, epidermis and gastric cavity from native south-east animals (Atlantic, Florida, United States) were dominated by Nautella; epidermis additionally comprised unclassified Entoplasmatales, whereas gastric cavity was additionally colonized by unclassified Enterobacteriaceae, Bacteroides and Arcobacter. In comparison, epidermis and gastric samples of the invasive Mediterranean animals mostly comprised Synechococcus, while epidermis samples additionally contained unclassified Entoplasmatales and Candidatus Aquiluna, whereas gastric samples comprised Halomonas, Shewanella, and Mycoplasma, unclassified Erythrobacteraceae and Bacteroides. Main representatives in epidermis as well as gastric samples of invasive North Sea animals were Halomonas, Shewanella, and Mycoplasma, unclassified Enterobacteriaceae and Bacteroides. Epidermis and gastric cavity community patterns of those animals were almost similar, however, slight differences were observed in abundances of main representatives and presence of different low abundant taxa.
Sub-Population Specific Community Patterns of M. leidyi
Population structure analyses using the polymorphism of seven microsatellite loci revealed that all tested sub-populations were significantly differentiated (FST range 0.03 to 0.33; P < 0.05 after correction for multiple testing, Table 1). The largest observed population differentiation was found between the southern native sub-population (Atlantic, Florida, United States) and the northern invasive sub-population (Helgoland, North Sea) with FST = 0.333 (P < 0.0001, see Table 1), while the smallest population difference was observed between native northern sub-populations (Atlantic, Massachusetts, United States) in comparison with their recently introduced counterparts in Northern Europe (Helgoland, North Sea) with FST = 0.032 (P < 0.05, Table 1). Based on the revealed genotyping of M. leidyi, bacterial community analysis of native northern (Atlantic, Massachusetts, United States) and southern (Atlantic, Florida, United States) as well as invasive (Mediterranean Sea and North Sea) M. leidyi sub-populations, bacterial community patterns where analyzed concerning ctenophore provenance (Figure 3 and Supplementary Table S1). RDA plots demonstrated that every single sub-population harbored a specific microbiota. Native sub-populations are significantly different and share just few OTUs. Invasive sub-populations shared a more similar microbiota and additionally were more similar to native northern sub-population animals (Atlantic, Massachusetts, United States), however, significantly different to native southern sub-population animals (Atlantic, Florida, United States) (Figure 3).
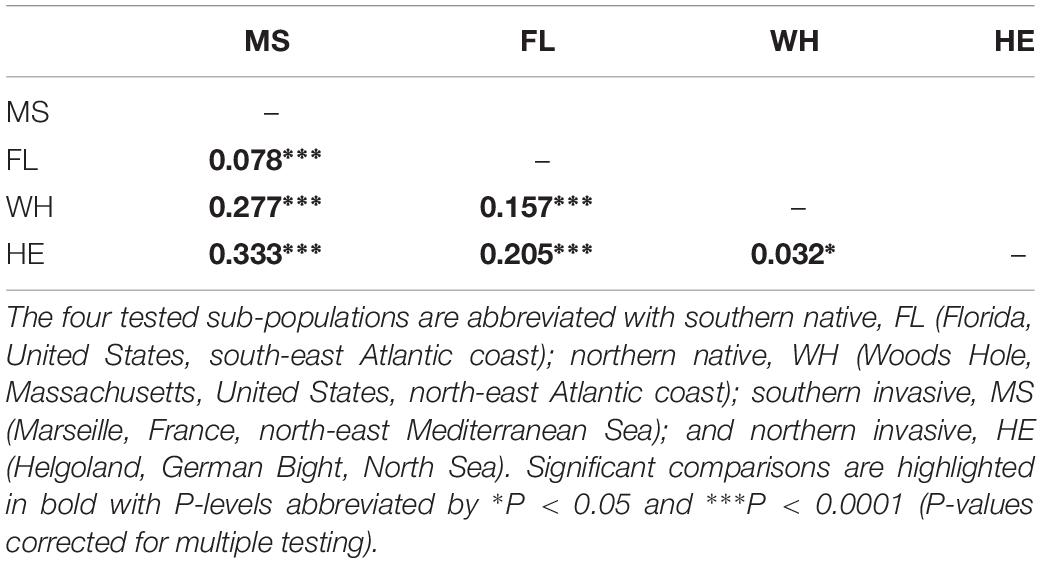
Table 1. Pairwise comparison of genetic differentiation (FST) between native and invasive Mnemiopsis leidyi sub-populations using highly polymorphic microsatellite markers.
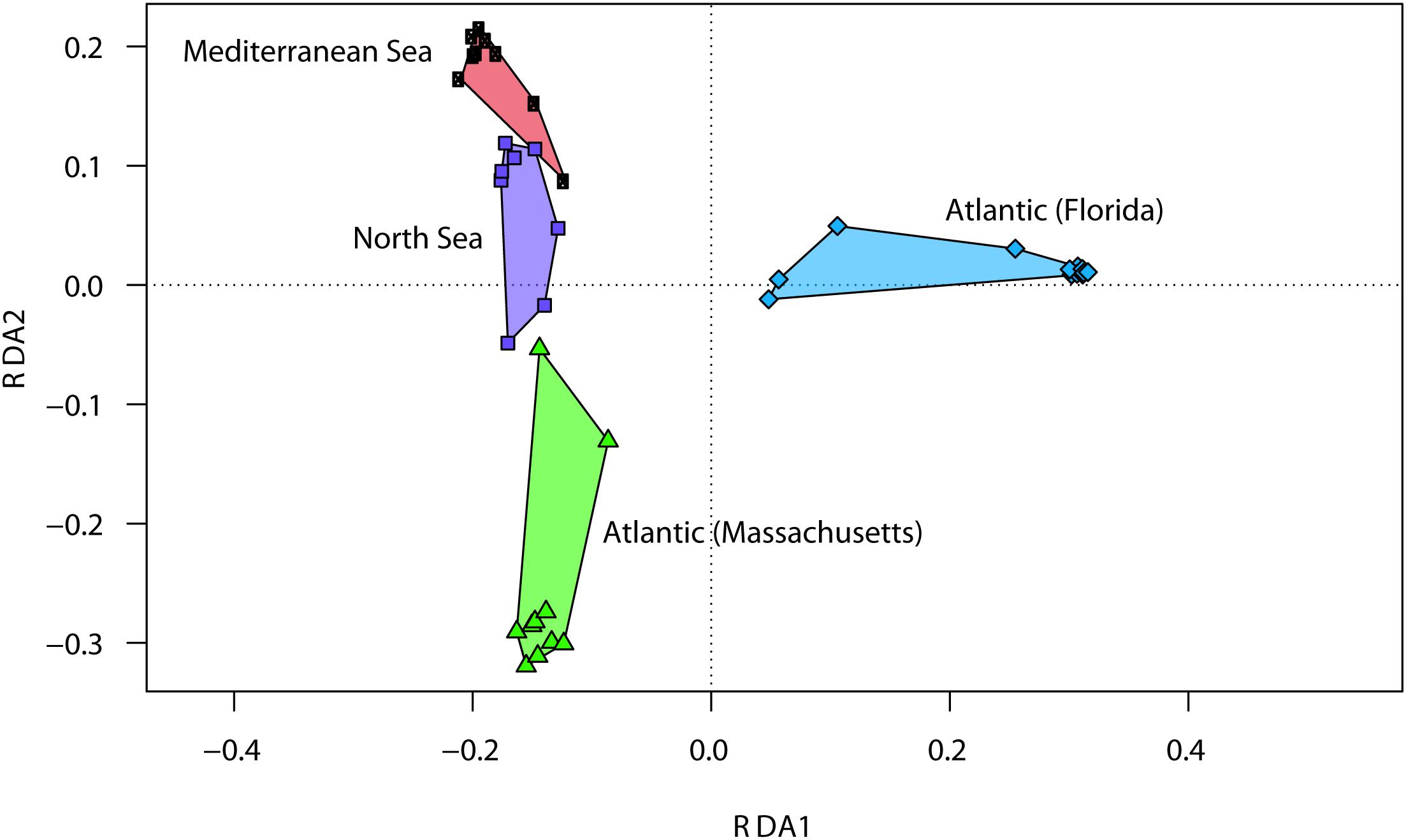
Figure 3. Redundancy analysis (RDA) models of Hellinger-transformed OTU abundances comparing Mnemiopsis leidyi associated bacteria of different sub-populations. Distance plots of RDA model of Hellinger-transformed OTU abundances. Each point represents the whole microbiota community of an individual sample associated with the respective native and invasive sub-population. Groups of related sample points are framed by polygons filled with a corresponding color.
Discussion
This is the first study comparing the microbiota of native and invasive sub-populations of the comb jelly M. leidyi using standardized high throughput amplicon sequencing methods based on the V1-V2 hypervariable region of the 16S rRNA gene. We demonstrated that the microbiota was significantly different from the surrounding seawater and specific to each analyzed sub-population. Contrary to the previous report of Hammann et al. (2015) describing that the outer surface of M. leidyi is free from associated microbes (Hammann et al., 2015), we observed a highly diverse microbiota on epithelial tissues. Additionally, and in line with Dinasquet et al. (2012), we detected different community patterns on the outer surface of the animals compared to the gastric cavity. Two published studies (Dinasquet et al., 2012; Hao et al., 2015) dealing with microbiota patterns of invasive M. leidyi observed that the core microbiome of M. leidyi is host-specific and main representatives were identified belonging to Gamma- and Alphaproteobacteria as well as Bacteroidetes with Marinomonas spp. (Gammaproteobacteria) as one abundant and most likely important colonizer. However, further work is needed to assess the functional roles of such ctenophore-associated microbes.
Our multivariate analyses indicated that native and invasive northern sub-populations are more similar compared with southern native and invasive sub-populations. For northern native sub-populations, Marinomonas spp. dominated the bacterial community. However, for invasive northern sub-populations, this species was only observed with a limited contribution to the microbiome of M. leidyi sampled during summer 2012. This is in contrast to previous findings, where during the years 2009/2010 Marinomonas spp. had been documented to be the most abundant bacterial species on M. leidyi from the invasive northern sub-population, i.e., Helgoland Road (Hao et al., 2015) and the Gulmar Fjord (Dinasquet et al., 2012). A change in the bacterial community composition with reduced contribution of Marinomonas spp. could be related to a significant change in winter conditions as observed during the early 2010s, where a series of cold winters had led to a retraction of the range expansion of M. leidyi in northern Europe with the extinction of M. leidyi in, e.g., the Gulmar Fjord (Jaspers et al., 2018). Hence, drastic physical conditions and extinction of local sub-populations could be reflected in a change of the microbiome, but deserve further investigation. It has been shown that the microbiome changes with season in southern native and northern invasive M. leidyi sub-populations (Daniels and Breitbart, 2012; Hao et al., 2015). In this study, we do not expect that seasonal differences impact the general conclusions of our study as all regions were sampled during the same seasonal time point.
In addition to the comparison of the microbiota on the comb jelly with the ambient water as well as between outer surface and gastric cavity, to the best of our knowledge, this is the first study focusing on the comparison between native and invasive M. leidyi. Therefore, our results are an essential first step to assess the impact of microbiota on the invasion process by describing basic diversity pattern.
A recent study investigating the microbiota of M. leidyi in comparison with other ctenophore species from the same environment documented significant difference between the microbiome of all studied ctenophore species (Hao et al., 2015). All investigated M. leidyi microbiomes were dominated by Proteobacteria, though variable through time (Hao et al., 2015). Our deep sequencing approach revealed that the native southern sub-population showed a large difference of the microbiota when compared to the invasive southern sub-population. Specifically, the native southern microbiome of M. leidyi was dominated by Nautella (Alphaproteobacteria), while this genus was absent from the corresponding invasive M. leidyi sub-population. In line with this, southern invasive and native M. leidyi sub-populations have also been differentiated for the longest time (Jaspers et al., 2018), which might explain the difference in their respective microbial community. Similar to our results, in a previous study, Alphaproteobacteria had been shown to be key bacterial community members of M. leidyi from the native southern range in the Gulf of Mexico during late spring and summer (Daniels and Breitbart, 2012). For the Mediterranean Sea, no previous microbiome characterization has been conducted. In our sequence data, we observed a large proportion of the Cyanobacteria species Synechococcus in epidermis as well as gastric samples. Even though this is a free living bacterium with a widespread distribution in marine and freshwater systems, Cyanobacteria have also been shown to contribute to the core microbiome of native southern M. leidyi during certain periods (Daniels and Breitbart, 2012) and might be more abundant during summer when they bloom in ocean surface waters (Wang et al., 2011).
Weiland-Bräuer et al. (2015) showed significant differences in the microbiota associated to the polyp stage of the moon jellyfish Aurelia aurita among three source populations that were cultured for more than 10 years under similar artificial lab conditions. In the present study, we found the lowest host population differentiation between native north-east United States sub-population (Atlantic, Massachusetts, United States) and the invasive northern sub-population (Helgoland, North Sea). These host population structure results are in agreement with molecular results (Reusch et al., 2010). In detail, a recent investigation summarizing the range expansion of M. leidyi in western Eurasia has shown that the northern introduction of M. leidyi is very recent and dates back to 2005 (Jaspers et al., 2018). Hence, genetic background along with time since first introduction has an impact on microbiome evolution. We further showed that analyzed invasive sub-populations, in general, share a more similar microbiota, most likely due to a more similar environment they share, disclosing the impact of the environment. Moreover, we observed that the microbiota of those invasive M. leidyi sub-populations is more similar to the northern native sub-population from Massachusetts than to the southern native sub-population from Florida. We hypothesize that the observed microbiota reflects the invasion process of M. leidyi, since the southern invasive sub-population has been introduced from the southern native sub-population in the 1980’s, while the northern sub-population present in European waters has been introduced from the north-east United States coast during the early 2000’s as reviewed in Jaspers et al. (2018). Further common garden experiments have to be performed in the future to confirm this hypothesis.
In conclusion, significant variance in microbial community patterns and significant population differences between sub-populations suggest an impact of environment along with the genetic host background (host population affiliation) on the microbiota. Long-term experiments over several generations under controlled conditions are necessary to partition the influence of the environment from population affiliation and genotype of the animals. We found that M. leidyi harbors a diverse microbiota on its epithelia. The significant difference between water and the microbiota suggests that ctenophores might select the microbes in a host-specific way, thereby shaping the entire metaorganism. The direct interactions between microbes and host need to be studied in controlled environments to gain insights into the impact of the associated microbiota on the fitness and invasion success of the ctenophore M. leidyi.
Data Availability Statement
Sequence data were submitted to the NCBI Sequence Read Archive under Accession No. PRJNA548849 and are additionally stored in a sequence data management system at the Institute of Clinical Molecular Biology (IKMB) at Kiel University, Germany.
Author Contributions
CJ, NW-B, RS, and TR: conceptualization. CJ, NW-B, and SK: sample analyses. MF with support from NW-B: bioinformatic analyses. CJ with support from TR: genotyping and analyses. CJ and NW-B with support from MF, SK, RS, and TR: manuscript drafting. All authors read and approved the final version of the manuscript draft.
Funding
This study received financial support from the Collaborative Research Center (CRC) 1182 “Origin and Function of Metaorganisms” to TR and RS, funded through the German Research Foundation (DFG, Re1108/17-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sören Bolte for support and data collection, Katharina Bading for collection of animals in the native southern range, and Nicole Pinnow for experimental support. This manuscript has been released as a Pre-Print at https://www.biorxiv.org/content/10.1101/601419v1.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00635/full#supplementary-material
References
Alegado, R. A., Brown, L. W., Cao, S. G., Dermenjian, R. K., Zuzow, R., Fairclough, S. R., et al. (2012). A bacterial sulfonolipid triggers multicellular development in the closest living relatives of animals. eLife 1:e00013. doi: 10.7554/eLife.00013
Bayha, K. M., DeBoer, T. S., Harbison, G. R., Chang, M. H., Moseley, C., Mariani, C. L., et al. (2015). Worldwide phylogeography of the invasive ctenophore Mnemiopsis leidyi (Ctenophora) based on nuclear and mitochondrial DNA data. Biol. Invasions 17, 827–850. doi: 10.1007/s10530-014-0770-6
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188. doi: 10.1186/1471-2105-9-114
Bolte, S., Fuentes, V., Haslob, H., Huwer, B., Thibault-Botha, D., Angel, D., et al. (2013). Population genetics of the invasive ctenophore Mnemiopsis leidyi in Europe reveal source-sink dynamics and secondary dispersal to the Mediterranean Sea. Mar. Ecol. Prog. Ser. 485, 25–36. doi: 10.3354/meps10321
Bosch, T. C. G., and McFall-Ngai, M. J. (2011). Metaorganisms as the new frontier. Zoology 114, 185–190. doi: 10.1016/j.zool.2011.04.001
R Core Team (2012). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Daniels, C., and Breitbart, M. (2012). Bacterial communities associated with the ctenophores Mnemiopsis leidyi and Beroe ovata. FEMS Microbiol. Ecol. 82, 90–101. doi: 10.1111/j.1574-6941.2012.01409.x
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/aem.03006-05
Dinasquet, J., Granhag, L., and Riemann, L. (2012). Stimulated bacterioplankton growth and selection for certain bacterial taxa in the vicinity of the ctenophore Mnemiopsis leidyi. Front. Microbiol. 3:302. doi: 10.3389/fmicb.2012.00302
Dunn, C. W., Hejnol, A., Matus, D. Q., Pang, K., Browne, W. E., Smith, S. A., et al. (2008). Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749. doi: 10.1038/nature06614
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Hammann, S., Moss, A. G., and Zimmer, M. (2015). Sterile surfaces of Mnemiopsis leidyi (Ctenophora) in bacterial suspension - a key to invasion success? Open J. Mar. Sci. 5, 237–246. doi: 10.4236/ojms.2015.52019
Hao, W., Gerdts, G., Peplies, J., and Wichels, A. (2015). Bacterial communities associated with four ctenophore genera from the German Bight (North Sea). FEMS Microbiol. Ecol. 91, 1–11. doi: 10.1093/femsec/fiu006
Jaspers, C., Fraune, S., Arnold, A. E., Miller, D. J., Bosch, T. C. G., and Voolstra, C. R. (2019a). Resolving structure and function of metaorganisms through a holistic framework combining reductionist and integrative approaches. Zoology 133, 81–87. doi: 10.1016/j.zool.2019.02.007
Jaspers, C., Weiland-Bräuer, N., Fischer, M., Künzel, S., Schmitz, R. A., and Reusch, T. B. H. (2019b). Microbiota differences of the comb jelly Mnemiopsis leidyi in native and invasive sub-populations. bioRxiv 601419.
Jaspers, C., Huwer, B., Antajan, E., Hosia, A., Hinrichsen, H.-H., Biastoch, A., et al. (2018). Ocean current connectivity propelling the secondary spread of a marine invasive comb jelly across western Eurasia. Global Ecol. Biogeogr. 27, 814–827. doi: 10.1111/geb.12742
Kideys, A. E. (2002). Fall and rise of the Black Sea ecosystem. Science 297, 1482–1484. doi: 10.1126/science.1073002
Langfeldt, D., Neulinger, S. C., Heuer, W., Staufenbiel, I., Kunzel, S., Baines, J. F., et al. (2014). Composition of microbial oral biofilms during maturation in young healthy adults. PLoS One 9:e87449. doi: 10.1371/journal.pone.0087449
Neumann, R. (1979). Bacterial induction of settlement and metamorphosis in the planula larvae of larvae of Cassiopea andromeda (Cnidaria, Scyphozoa, Rhizostomeae). Mar. Ecol. Prog. Ser. 1, 21–28. doi: 10.3354/meps001021
Reusch, T. B. H., Bolte, S., Sparwel, M., Moss, A. G., and Javidpour, J. (2010). Microsatellites reveal origin and genetic diversity of Eurasian invasions by one of the world’s most notorious marine invader, Mnemiopsis leidyi (Ctenophora). Mol. Ecol. 19, 2690–2699. doi: 10.1111/j.1365-294X.2010.04701.x
Rook, G., Bakhed, F., Levin, B. R., McFall-Ngai, M. J., and McLean, A. R. (2017). Evolution, human-microbe interactions, and life history plasticity. Lancet 390, 521–530. doi: 10.1016/S0140-6736(17)30566-4
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Stratil, S. B., Neulinger, S. C., Knecht, H., Friedrichs, A. K., and Wahl, M. (2014). Salinity affects compositional traits of epibacterial communitites on the brown macroalga Fucus vesiculosus. FEMS Microbiol. Ecol. 88, 272–279. doi: 10.1111/1574-6941.12292
Tebben, J., Tapiolas, D. M., Motti, C. A., Abrego, D., Negri, A. P., Blackall, L. L., et al. (2011). Induction of larval metamorphosis of the coral Acropora millepora by tetrabromopyrrole isolated from a Pseudoalteromonas bacterium. PLoS One 6:e19082. doi: 10.1371/journal.pone.0019082
Tiselius, P., and Møller, E. F. (2017). Community cascades in a marine pelagic food web controlled by the non-visual apex predator Mnemiopsis leidyi. J. Plankton Res. 39, 271–279.
Wang, K., Wommack, K. E., and Chen, F. (2011). Abundance and distribution of Synechococcus spp. and Cyanophages in the Chesapeake Bay. Appl. Environ. Microbiol. 77, 7459–7468. doi: 10.1128/AEM.00267-11
Weiland-Bräuer, N., Neulinger, S. C., Pinnow, N., Kunzel, S., Baines, J. F., and Schmitz, R. A. (2015). Composition of bacterial communities associated with Aurelia aurita changes with compartment, life stage, and population. Appl. Environ. Microbiol. 81, 6038–6052. doi: 10.1128/AEM.01601-15
Werner, J. J., Koren, O., Hugenholtz, P., DeSantis, T. Z., Walters, W. A., Caporaso, J. G., et al. (2012). Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 6, 94–103. doi: 10.1038/ismej.2011.82
Whelan, N. V., Kocot, K. M., Moroz, T. P., Mukherjee, K., Williams, P., Paulay, G., et al. (2017). Ctenophore relationships and their placement as the sister group to all other animals. Nat. Ecol. Evol. 1, 1737–1746. doi: 10.1038/s41559-017-0331-3
Keywords: ctenophore, metaorganism, bacteria, species translocations, invasive species
Citation: Jaspers C, Weiland-Bräuer N, Fischer MA, Künzel S, Schmitz RA and Reusch TBH (2019) Microbiota Differences of the Comb Jelly Mnemiopsis leidyi in Native and Invasive Sub-Populations. Front. Mar. Sci. 6:635. doi: 10.3389/fmars.2019.00635
Received: 03 June 2019; Accepted: 27 September 2019;
Published: 11 October 2019.
Edited by:
Sandie M. Degnan, The University of Queensland, AustraliaReviewed by:
Adam Michael Reitzel, The University of North Carolina at Charlotte, United StatesCynthia B. Silveira, San Diego State University, United States
Copyright © 2019 Jaspers, Weiland-Bräuer, Fischer, Künzel, Schmitz and Reusch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia Jaspers, Y2phc3BlcnNAZ2VvbWFyLmRl; Y29qYUBhcXVhLmR0dS5kaw==; Nancy Weiland-Bräuer, bndlaWxhbmRAaWZhbS51bmkta2llbC5kZQ==
†These authors have contributed equally to this work