 Sergio E. Morales
Sergio E. Morales Ambarish Biswas
Ambarish Biswas Gerhard J. Herndl
Gerhard J. Herndl Federico Baltar
Federico Baltar- 1Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
- 2Department of Limnology and Oceanography, Division of Bio-Oceanography, University of Vienna, Vienna, Austria
- 3Department of Marine Science, University of Otago, Dunedin, New Zealand
Fungal contributions to ecosystem processes are well documented for terrestrial systems yet oceans, which account for most of the Earth’s surface, have remained poorly explored with regards to organisms in this kingdom. Here, we demonstrate that, although in low relative abundance (i.e., fungal reads made up 1.4–2.9% of the metagenomes), fungi contribute to both phylogenetic and functional microbial diversity with a conserved fungal presence in global marine samples. Universally distributed taxa and functions implicate them in complex carbon and fatty acid metabolism, with depth stratification along pelagic zones. Functional differences in observed genes between epipelagic and mesopelagic waters indicate changes in UV protection, shift to carbohydrate limited diets, as well as alternative energy sources. Metagenomic data also provided evidence for a latitudinal gradient in fungal diversity linked to temperature shifts. Our results suggest that fungi contribute to multiple biogeochemical cycles in the pelagic ocean, and could be integral for ecosystem functioning through provision of key nutrients.
Introduction
Modern marine microbial ecology has focused on the prokaryotic component, with other organisms such as fungi being understudied in comparison to their terrestrial counterparts. Despite detection of fungi in many different marine environments via metabarcoding, from sub-seafloor sediments to open ocean waters (Nagahama et al., 2003; Pang and Mitchell, 2005; Bass et al., 2007; Zuccaro et al., 2008; Gao et al., 2010; Jebaraj et al., 2010; Edgcomb et al., 2011; Amend et al., 2012; Jones and Pang, 2012; Richards et al., 2012; Burgaud et al., 2013; Rama et al., 2014; Wang et al., 2014; Yarden, 2014; Gutiérrez et al., 2016; Taylor and Cunliffe, 2016), their diversity and functions in the ocean remains largely unknown. Recent evidence suggests that fungi interact with other members of marine ecosystems (O’Rorke et al., 2013; Yarden, 2014), and may play key roles in biogeochemical cycles in deep-sea sediments (Orsi W. et al., 2013; Orsi W.D. et al., 2013). However, concerning the pelagic fungi, little is known about their function, distribution, and the factors controlling both their diversity and function.
In contrast, we know that the biogeography of pelagic marine bacteria is governed by latitude and temperature (Fuhrman et al., 2008). We also know that dramatic shifts in prokaryotic community composition occur between epipelagic and mesopelagic zones (Giovannoni and Stingl, 2005), and that these zones are linked to concurrent shifts in functions (DeLong et al., 2006). Whether pelagic fungi share the same or related distribution patterns as marine bacteria is currently unknown. Studies addressing some of these questions have identified temperature and salinity (Booth and Kenkel, 1986), as well as location (Rama et al., 2014) as potential drivers of fungal community composition. Next generation sequencing (of fungal ITS region) showed changes in fungal communities linked to specific habitats (i.e., estuarine, coastal, and oceanic) associated to shifts in salinity, temperature, oxygen, and nutrients (Jeffries et al., 2016). In the only global-scale analysis of marine fungal community structure thus far available, depth, oxygen, and nitrate strongly correlated with fungal community composition (based on the nuclear ribosomal small subunit V9 region) (Tisthammer et al., 2016).
Far less is known about specific fungal functions in the ocean, the mechanisms that control the abundance and activity of these functions, and their relevance to the marine ecosystem. Recent evidence points to high activity of fungi in marine sediments linked to nutrients dynamics, with 5–20% of identified transcripts mapped to a fungal host (Orsi W.D. et al., 2013). Fungi were also found in sea ice where members of the fungal phylum Chytridiomycota were reported to be actively parasitizing light-stressed diatoms (Hassett and Gradinger, 2016). Also, a study conducted in the upwelling zone off Chile reported a main role for fungi in the extracellular enzymatic hydrolysis of organic polymers (Gutiérrez et al., 2011). Recent work taking advantage of next generation sequencing on fungal marker genes (Jeffries et al., 2016; Tisthammer et al., 2016), have started to shed some light on the distribution and roles of pelagic fungi, but direct links between taxonomy and function are still sparse. The application of metagenomics to marine pelagic fungal communities might provide unprecedented and relevant information about their ecology and function, although the lack of fungal gene database makes identifying fungal genes (particularly of novel clades and chytridiomycota) in the marine environment a difficult task.
In this study we take advantage of metagenomics to characterize the community structure and functional potential of pelagic fungi at both local and global scales. We aimed at identifying globally dominant taxonomic groups of marine fungi, conserved (core) functions, and patterns of biogeography and potential drivers of biogeochemical cycles. To do this, we used local data from the Munida Microbial Observatory Time-Series (MOTS) transect across the subtropical frontal zone in the Pacific east of New Zealand, where strong shifts were recently found in microbial community composition and function (Baltar et al., 2015, 2016; Morales et al., 2018), and from the TARA Oceans project (Armbrust and Palumbi, 2015). We hypothesized that pelagic fungi would be involved in multiple biogeochemical cycles in the pelagic realm as previously reported for the deep sediment biosphere [primarily linked to nitrogen, carbohydrate, amino acid, and lipid metabolism (Orsi W. et al., 2013)]. We also hypothesized, based on prior evidence for vertical changes in marine fungi (Gao et al., 2010; Wang et al., 2014), that fungal pelagic community composition and functions will shift in response to depth, as observed for marine bacteria (DeLong et al., 2006).
Materials and Methods
Sample Collection
Samples were collected at the MOTS1 (Supplementary Figure S1 and Supplementary Table S1). The sampling strategy and nucleic acid extraction methods used to collect samples for the MOTS transect are described elsewhere (Baltar et al., 2015, 2016) and samples used in the present study are listed with corresponding metadata in the SI Metadata. Briefly, surface (2 m) samples were collected on 5 l acid-washed plastic bottles and 0.5 and 0.6 l of seawater were filtered through a 0.22 μm polycarbonate filter. From the MOTS transect, four samples were utilized for metagenomic sequencing representative of biological duplicates for surface waters within the nutrient-poor subtropical waters (STW) and the cold, macronutrient rich (but iron-limited) sub-Antarctic waters (SAW). Two additional mesopelagic (500 m) samples from SAW were included for a total of six samples analyzed from MOTS. Total community DNA was extracted from each filter separately using a MoBio PowerSoilTM DNA Isolation Kit (MoBio, Solana Beach, CA, United States) as per manufacturer’s instructions, with final elution into 50 μl. DNA quality and quantity was assessed using a Nanodrop Spectrophotometer (Thermo Fisher Scientific). Metagenomes from the MOTS transect are available through the MG-RAST server with ID numbers 4662524.3–4662529.3. Samples of the Tara Oceans project (Armbrust and Palumbi, 2015) were also used in the present study, their accession numbers are listed in the SI Appendix. A total of 42 metagenomic libraries (paired end Illumina HiSeq 2000) were selected from the Tara Oceans project representing 14 stations across five oceanic regions (the Pacific, Atlantic, Indian, Southern, and Arctic Oceans) and covering multiple sampling depths (Supplementary Figure S1 and Supplementary Table S1). Samples were selected to provide broad representation of oceanic ecosystems across the globe, and major transitions across both surface and depth. Libraries were selected to ensure that for each site in our study data was available from at least two depths representative of the epipelagic and mesopelagic zones, and that all data was generated from the same fraction size (0.2 μm). Samples were collected during two time periods, with all samples within neighboring regions collected during the same period. Additional information is found in Supplementary Dataset.
Metagenomic Analysis
Metagenomes from the MOTS transect were generated using the Illumina Nextera XT library preparation kit. Samples were then run on a single MiSeq 2 × 250 base paired end run. Sequences were annotated using the MG-RAST server (Meyer et al., 2008). Sample counts ranged from 2,015,852 to 5,249,491 before quality control. After removal of low-quality reads, sequences were classified using the M5NR protein and Silva SSU ribosomal databases using default settings. Relative abundance matrices generated from either database were imported into JMP for principal component analysis. Bray Curtis distance matrices generated from either the M5NR protein or the SILVA SSU results where compared to each other using a Mantel correlation using the “vegdist” and “Mantel test” functions in the vegan and ade4 R packages (R Development Core Team, 2008). An additional protein classification using eggNOG was also carried out. A Bray Curtis distance matrix was generated based on the relative abundance matrix and used to compare sample clustering to the SILVA SSU results as above (Supplementary Figure S2). Matches to fungal taxa were extracted and used for plotting and multivariate analysis.
To allow for detection and comparison of functions specific to fungi, the paired end FASTQ files for all samples were merged with PEAR (Version 0.9.5) and reads that successfully merged to its pair with a minimum length of 120 nucleotides were retained. The resulting data were then subsampled at random to an equal depth of 1.472,710 reads. A local database was created for each of the following: NCBI RefSeq fungi (release 72, September 2015) and fuNOG (eggNOG v4.0). All subsampled libraries were screened against RefSeq fungi and fuNOG protein sequences using BLASTX (word size 3 and e-value 10). Only those reads matching both databases (with a minimum length of 40 amino acids and identity of 60%) were retained as putative fungal reads. Reads with matches to either bacterial (bactNOG) or archaeal (arcNOG) databases were removed. Taxonomy for putative fungal reads were assigned by using the best match with the database (fuNOG) and retrieving lineages for each match from NCBI taxonomy files (gi_taxid_prot.dmp, names.dmp, and nodes.dmp). An abundance matrix was created consisting of a unique taxa + function cluster (reads classified to closest function and host organisms) based on fuNOG and NCBI refseq genomes. A summary table containing a unique “function + species” was generated with the number of hits per sample. While this approach will decrease our sensitivity (increase our rate of false negatives), it ensures high specificity (low rate of false positives) to fungally derived reads. Data was ordinated using the “ordinate” functions on a Bray Curtis distance in the phyloseq (McMurdie and Holmes, 2013) R package. To correlate specific variables potentially linked to clustering of samples a distance matrix was created from a mapping file variable (i.e., salinity and depth) using the “distance_matrix_from_mapping.py” command and compared using a Mantel test using the “compare_distance_matrices.py “ command in MacQiime (Caporaso et al., 2010). To display changes in taxonomic groups across depth, reads were agglomerated based on phylum and plotted. This was repeated based on both genera and function, but only the top 50 most abundant groups were used to plot results. To determine the clustering of samples based on physicochemical parameters, metadata for the Tara ocean project metagenomes was trimmed to a basic file containing only those variables with values present for all samples. This file was imported into JMP and used for a principal component analysis (Supplementary Figure S3) with resulting factor analysis for the first three components presented in Supplementary Table S3. Results supported the clustering of samples based on epipelagic vs. mesopelagic samples. In order to determine the abundance and distribution of fungal functions in the absence of taxonomy, sequences were agglomerated based on function using the “tax_glom” function in the phyloseq R package. Samples were then grouped as either epipelagic or mesopelagic. The top 40 most abundant functions were determined for each group of samples separately. To identify those functions that were enriched in specific depths an exact test was performed using the edgeR R package. Identified functions where then trimmed to only those with a p value <0.05 and more than 2-fold difference in abundance between the two depths. After agglomerating results by function (as described above), alpha diversity (observed number of functions and Shannon diversity of detected functions) were calculated within the phyloseq R package (using either the “estimate_richness” or the “plot_richness” functions) for mesopelagic and epipelagic samples. To determine the role of temperature in driving the latitudinal pattern, diversity values were plotted against the mean temperature for each sample. To determine whether changes in functional patterns were associated with changes in taxonomic diversity results fungal phylogenetic alpha diversity was determined as described above for function and plotted by oceanic region. The data tables generated from these analyses were processed and analyzed with R (R Development Core Team, 2008) and JMP (SAS Institute, Cary, NC, United States).
Results and Discussion
Fungal Community Structuring Across a Regional Gradient
The “MOTS transect” has recently been used as a model system to study gradients at small scales leading to the identification of oceanic fronts as boundaries for microbial community changes (Baltar et al., 2015, 2016). To further explore this and to identify functional responses to these gradients, a metagenomic approach was applied. Surprisingly, a de novo assembly identified fungal contigs when blasted against available sequenced genomes (Supplementary Table S2). While abundant organisms like Pelagibacter ubique HTCC1062 and Synechococcus were detected (∼1% of reads mapped to each genome) and followed patterns consistent to those observed in 16S profile, fungal genomes recruited less sequences albeit at comparable numbers to other important but non dominant organisms (e.g., Nitrosopumilus). To confirm the presence of fungal sequences, metagenomes from surface waters representative of two major water masses (the nutrient-poor STW and the cold, macronutrient-rich but iron-limited SAW), along with a deep (500 m) sample from the SAW were analyzed with a focus on the fungal component. Results confirmed fungi as consistently detected across samples, with Ascomycota and Basidiomycota as the dominant fungal taxa independent of the database used [protein or SSU based assignment (Figure 1 and Supplementary Figure S2)]. Observed discrepancies between protein and SSU based results suggest a bias in protein-based assignments toward taxa with higher genomic representation in databases (Figure 1 and Supplementary Figure S2). In one of the samples (STW) unclassified fungi shared a large relative abundance (>50%) (Figure 1), which might be related to the lack of representation of the early diverging fungi, such as Chytridiomycota and Cryptomycota, in protein database. The fungal community was primarily structured by depth as previously observed for bacterioplankton (Baltar et al., 2015, 2016) and no difference in sample clustering was detected between SSU [SILVA] and protein [M5NR] analyses (Monte-Carlo test p = 0.97). Additional analysis using a different protein database [eggNOG (Powell et al., 2013)] confirmed these results (Monte-Carlo test p = 0.41; Supplementary Figure S2).
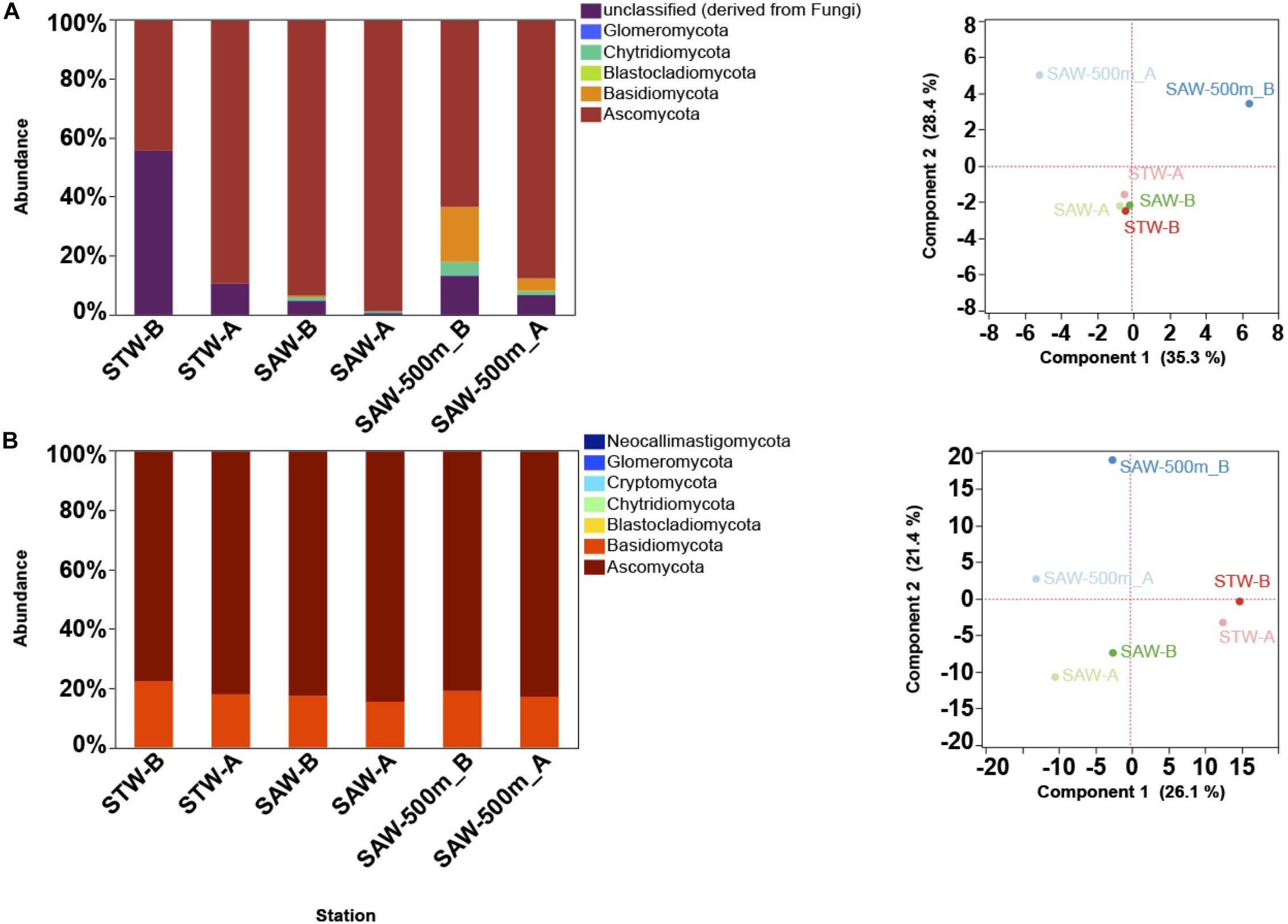
Figure 1. Taxonomic description of fungal community along the Munida transect based on MG-RAST results. Each Station represents a replicate sample taken at a different water mass (Surface samples were taken in nutrient-poor and salty subtropical waters (STW) and the cold, macronutrient-rich (but iron limited) subantarctic waters (SAW). A 500 m deep sample was also taken at the same SAW location. (A) Relative abundance (reads mapped) of fungal phyla based on SSU classification against SILVA database (left) and PCA plot (right) of sample clustering based on strain level taxa abundance. (B) Relative abundance (reads mapped) of fungal phyla based on M5NR protein database (left) and PCA plot (right) of sample clustering based on strain level taxa abundance.
Global Functional and Taxonomic Fungal Community Structuring
Since patterns observed using MG-RAST (in MOTS samples) using a variety of databases where consistent, all further analyses were performed using eggNOG, or one of their taxonomically refined subsets (i.e., fuNOG) as described in Materials and Methods. To exclude the possibility for a regional anomaly, we used a global approach to determine the distribution and functional contribution of fungi to oceanic biomes. Forty two metagenomes from the Tara Oceans Expedition (Armbrust and Palumbi, 2015) were selected. These samples corresponded to six oceanic regions, and four different depth layers representative of the epipelagic and mesopelagic realms. To date the Tara Oceans Expedition has expanded our knowledge of prokaryotic, viral, and eukaryotic plankton communities in the ocean, but pelagic fungi remain fairly unexplored. This might be due to the prevalence of protist (>85% of total eukaryotic ribosomal diversity) with respect to other eukaryotes (such as fungi) in the supergroup-level classification of all Tara Oceans OTUs (De Vargas et al., 2015). The only specific mention to fungi found in that previous study basically highlighted the high phylogenetic diversity of the osmotrophic fungi Ascomycota (410 OTUs).
Metagenomes were analyzed by classifying each read (assigning both a function and a taxonomic placement) after subsampling to equal sequencing depth. Analysis of metagenomes confirmed the presence of fungi in all samples, and the observed dominance of both the Ascomycota (∼70%) and Basidiomycota independent of region or depth, as observed for the “MOTS transect” (Figure 2). Recent work presents evidence for the dominance of alternative fungal groups (chytrids) within certain ocean regions (Hassett and Gradinger, 2016; Jeffries et al., 2016). Although our results support the presence of these groups in the oceanic realm, the dominance of the Ascomycota and Basiodiomycota in our study may be due to our focus on proteins and classification based on sequenced genomes. At the time of analysis, 640 fungal genomes were available in the NCBI database with Ascomycota (439) and Basiodiomycota (139) being the most common, while all other fungal taxa being represented by only 62 genomes and poorly represented groups like the Chytrids only with four genomes. Cumulatively, a larger role is anticipated for other fungal taxa once low scoring hits (novel fungi) are accounted for in future studies (Richards et al., 2012, 2015; Hassett and Gradinger, 2016). Independent of classification, the ubiquitous presence of fungi in the global ocean suggests that fungi might play a major, hitherto largely unrecognized role in the oceanic biomes.
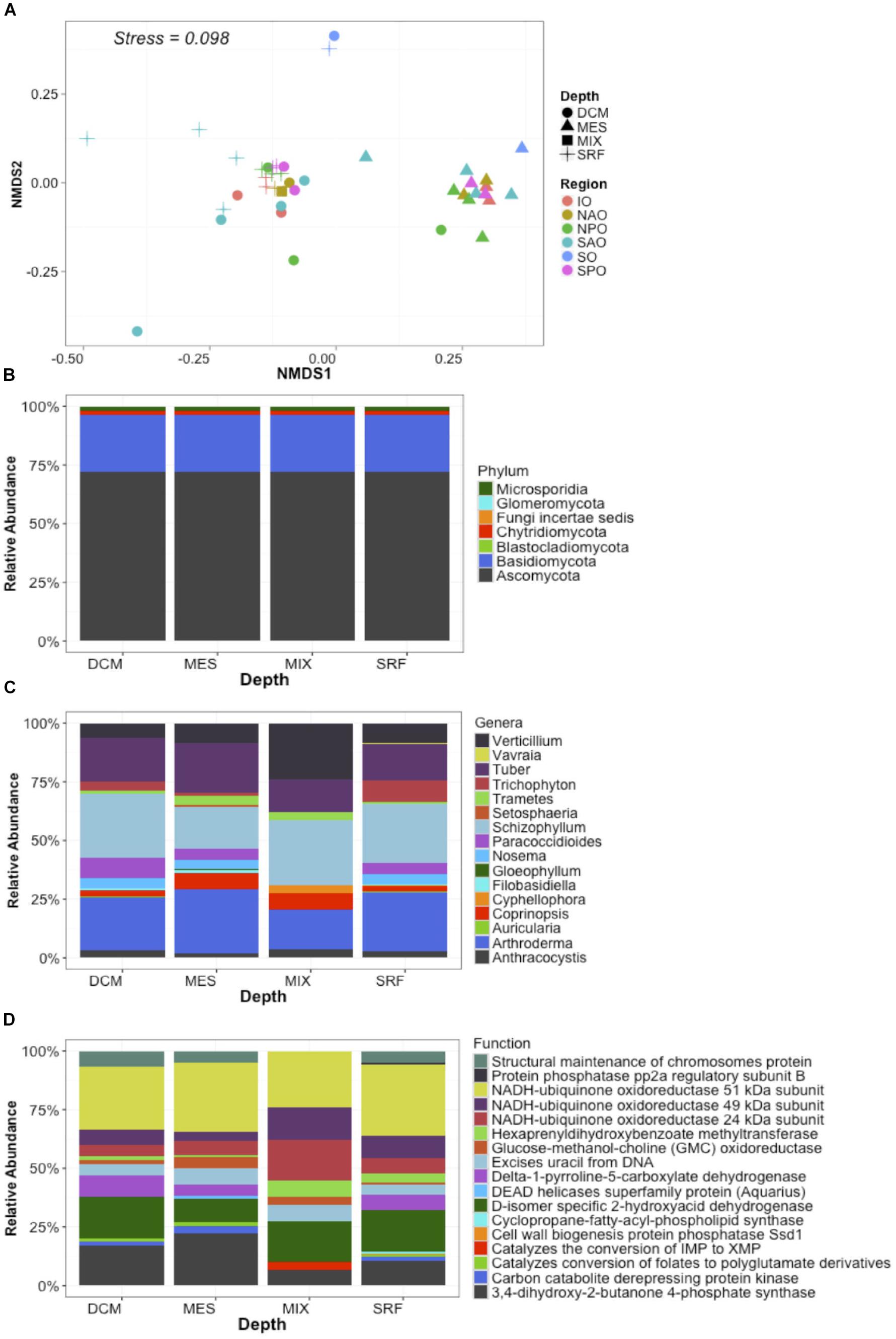
Figure 2. Global distribution of fungi in the ocean. Data represent abundance (% of mapped reads) of unique taxa + function clusters (reads classified to closest function and host organisms) based on eggNOG and NCBI refseq genomes. In this way data represents unique functions separated by each unique lowest taxonomic classification. (A) NMDS plots with samples color coded by ocean region and depth zone [Stressplot provided in Supplementary Figure S4]. (B) Relative abundance of fungal reads by Phyla based on all identified fungal reads. (C) Relative abundance of dominant genera based on the 50 most abundant unique taxa + function clusters. (D) Relative abundance of dominant functions based on the 50 most abundant unique taxa + function clusters. X-axis for B–D represent four depth layers analyzed (DCM, deep chlorophyll maximum; MES, mesopelagic; MIX, subsurface epipelagic mixed layer; SRF, surface).
Fungal reads made up 1.4–2.9% (average of 2.3 ± 0.4%) of the metagenomes (Supplementary Table S3), with number of reads mapping to fungi significantly changing by depth (p < 0.01) but not oceanic region based on a Kruskal–Wallis test (Figure 3). This is likely to be a conservative estimate considering that only reads matching both fuNOG and RefSeq were retained, and only those sequences bearing no match to bacterial or archaeal databases were retained. This approach likely removes reads from novel fungal species, as well as non-coding, highly conserved homologs across kingdoms, or novel sequences. However, it ensures a high specificity to fungi. Clustering of samples was primarily associated with depth (Mantel Rho = 0.461; p < 0.001) and weakly with salinity (Mantel Rho = 0.200; p = 0.002). The identification of two major “clusters” of samples together with the low correlation with depth suggested the lack of a depth gradient, instead pointing to an abrupt transition between zones (epipelagic vs. mesopelagic). It also confirmed that this depth-related pattern is universal, not a local peculiarity, supporting previous findings (Wang et al., 2014; Zhang et al., 2014; Tisthammer et al., 2016). Also, a multivariate analysis of physicochemical parameters also clustered samples primarily according to depth, with two major clusters representing samples from either epipelagic vs. mesopelagic waters (Supplementary Figure S3). Depth was primarily linked to increases in nutrients (PO4, NO2/NO3) and a decrease in temperature (Supplementary Table S4). This also suggests an active link or adaptation of fungal assemblages to changes in the physicochemical conditions of their environment along the oceanic water column.
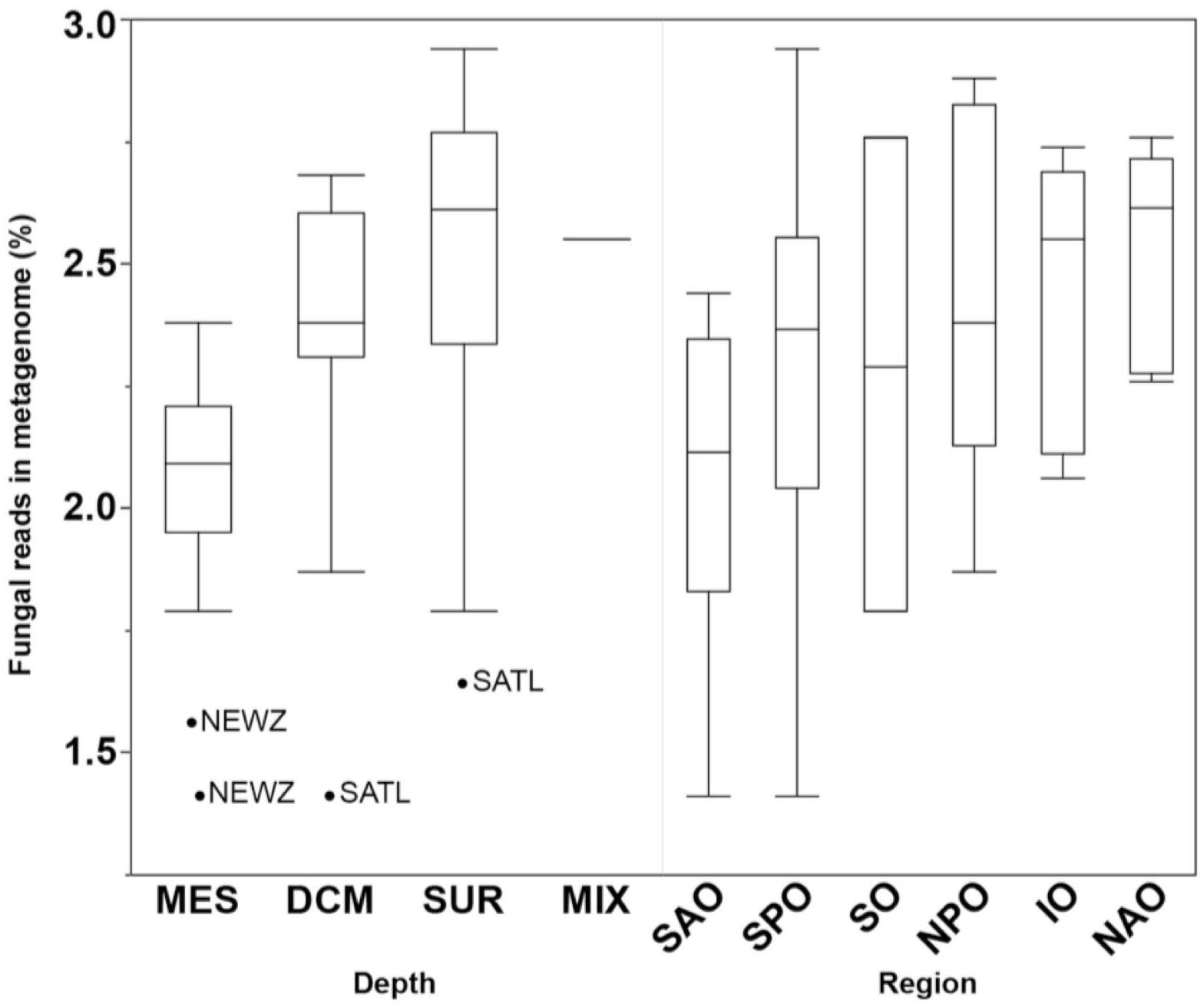
Figure 3. Boxplots comparing the abundance of all fungal reads grouped by depth (DCM, deep chlorophyll maximum; MES, mesopelagic; MIX, subsurface epipelagic mixed layer; SRF, surface) and oceanic region (IO, Indian Ocean; NAO, North Atlantic Ocean; NPO, North Pacific Ocean; SAO, South Atlantic; SO, Southern Ocean; SPO, South Pacific Ocean). Outliers shown as points with location indicated (NEWZ, New Zealand Coastal Province; SATL, South Atlantic Gyral Province).
Depth Partitioning of Putative Fungal Functions in the Ocean
To gain insight into the putative metabolic processes associated to fungi, the most common functions associated to highly abundant fungal reads were identified. These common functions were found across all depths and regions, and implicate them in vitamin, complex carbon, and fatty acid metabolism, potentially representing the core metabolism of oceanic fungi. These included the biosynthesis of B vitamins (pantoate-beta-alanine ligase), the biological activation of molybdenum (Mo cofactor biosynthesis protein), hemicellulose metabolism (xylose reductase and glycoside hydrolase family 43), degradation of extracellular fatty acids (peroxisomal fatty acid β-oxidation multifunctional protein) and processing of amino acids (phospho-2-dehydro-3-deoxyheptonate aldolase, 4-hydroxyphenylpyruvate dioxygenase (HPPD), aromatic L-amino acid decarboxylase) (Figure 4). Many of these functions have been previously described within cultured fungi. Stress and/or response to environmental stimuli was also identified as an important function. Genes involved in metabolic rearrangements (2-oxoglutarate dehydrogenase E1 component) and multidrug resistance involved in the resistance to Cycloheximide, which is an inhibitor of eukaryotic protein synthesis produced by bacteria suggests marine fungi are capable of responding to negative selective pressures of various forms (including through ecological interactions). Temperature as a key factor affecting oceanic fungi is supported by three genes linked to temperature changes (mitochondrial DnaJ chaperon, peptidyl-prolyl cis-trans isomerase, and the intermembrane space AAA protease IAP-1). Specifically, AAA protease IAP-1 serves the role of recognizing temperature shifts at the membrane with deletion of the gene in fungi leading to impaired growth in response to temperature changes (Klanner et al., 2001).
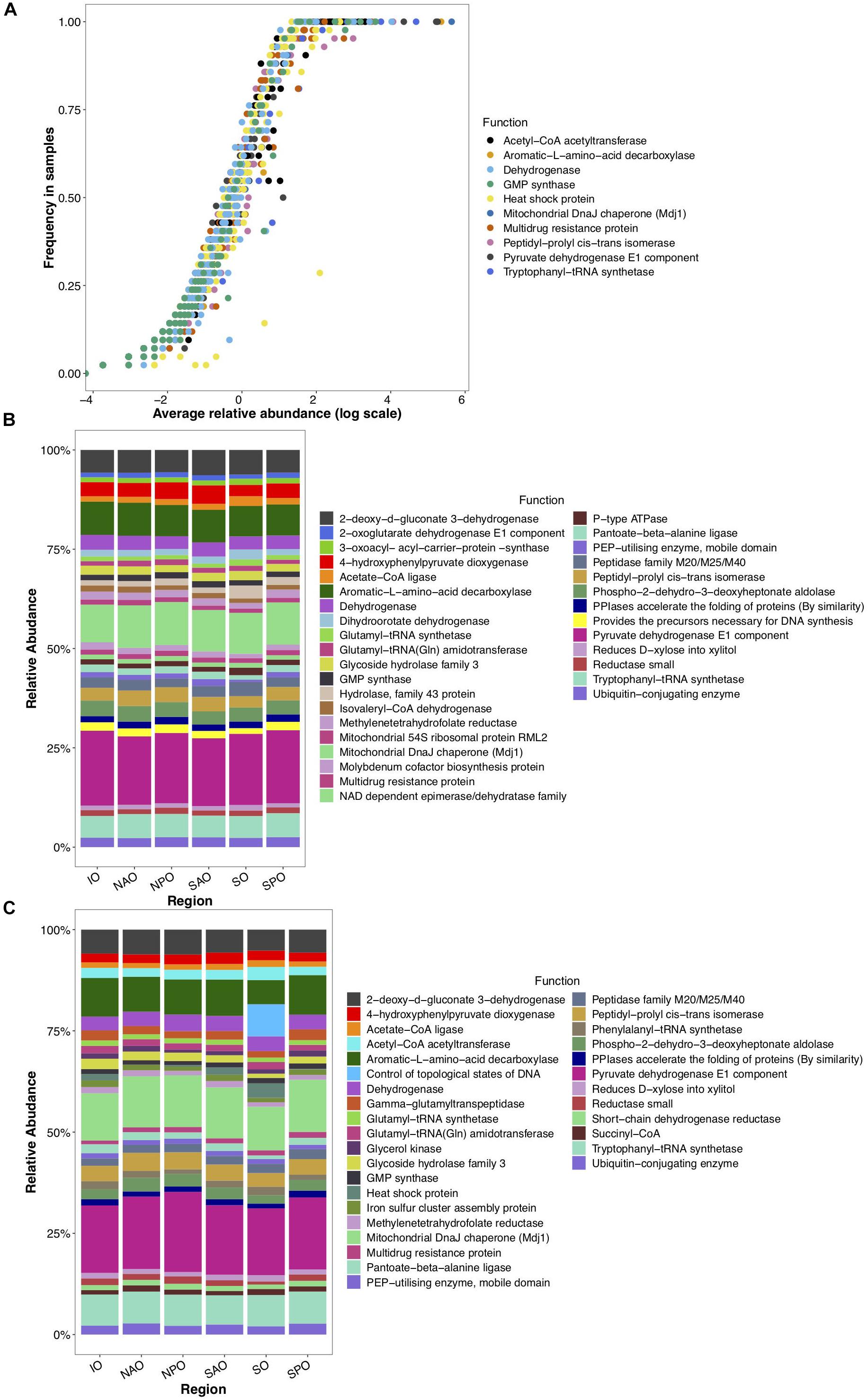
Figure 4. Globally abundant and differentially detected functions across depth and regions. Data represents abundance of unique functional clusters (reads classified to closest function based on classification to eggNOG). (A) Average relative abundance and frequency of top 10 most abundant functions in all samples. (B) Relative abundance of most abundant functions identified within epipelagic samples (representing 8.5% of all data linked to those samples) and clustered by oceanic region. (C) Relative abundance of most abundant functions identified within mesopelagic samples (representing 7.1% of all data linked to those samples) and clustered by oceanic region. X-axis for panels B and C represent oceanic regions (IO, Indian Ocean; NAO, North Atlantic Ocean; NPO, North Pacific Ocean; SAO, South Atlantic; SO, Southern Ocean; and SPO, South Pacific Ocean).
Depth was identified as a clear structuring factor of community composition in fungi as well as their encoded functions (Figures 2–4). Although the clustering of samples was associated with depth (Mantel Rho = 0.461; p < 0.001), as did physicochemical parameters (Supplementary Figure S3), the low correlation suggests no gradual decrease but rather a sharp transition from the surface to the deeper water layers. Consequently, we grouped samples based on pelagic zonation. Epipelagic (sunlit surface) waters exhibited significantly (>2-fold, p < 0.05; Figure 5) higher numbers of genes encoding the molybdenum cofactor biosynthesis protein, aspartate (homoserine dehydrogenase), purine (imidazole glycerol phosphate synthetase), serine and glycine (via serine hydroxymethyl transferase) amino acids synthesis than deep waters. Our analyses also suggested a preference in epipelagic waters for carbohydrate degradation (glycoside hydrolase, family 43) and the requirement for UV-light induced DNA repair (via DNA repair protein Msh2 and deoxyribodipyrimidine photolyase).

Figure 5. Significantly different (>2-fold change and p < 0.05) functions associated to either epipelagic (left) or mesopelagic (right) zones as identified through a Exact Binomial Test. logFC = log fold change.
In contrast, mesopelagic fungi were enriched in genes involved in the biosynthesis of different amino acids (primarily the aromatic amino acids phenylalanyl [phenylalanyl-tRNA synthetase], histidine [histidine biosynthesis trifunctional protein], threonine [threonine synthase], valine [dihydroxy-acid dehydratase], and particularly leucine [dihydroxy-acid dehydratase, 2-isopropylmalate synthase, 3-isopropylmalate dehydrogenase, and aconitate hydratase]). Other relatively enriched fungal genes in the mesopelagic were involved the response to environmental stimuli via signal transduction (histidine kinase, nucleotide-binding protein), to oxidative stress [UbiA prenyltransferase family (Kawai et al., 2015)], and to temperature (heat- shock proteins). Mesopelagic fungi were also enriched in genes related to vitamin B5 biosynthesis (dihydroxy-acid dehydratase). This is interesting as vitamin production is commonly assumed to be associated with phytoplankton (Croft et al., 2005; Bertrand et al., 2015), which are rare in mesopelagic waters. Thus, it would indicate that fungi might replace phytoplankton in supplying vitamins in deep waters.
Our analyses of the fungal gene repertoire also suggest a potential shift in available organic carbon sources for fungi with depth, with ketone body catabolism [3-oxoacid CoA transferase] potentially contributing to survival of fungi in the mesopelagic zone. Ketone bodies are formed by the breakdown of fatty acids or as byproducts of fermentation (Campbell and Farrell, 2014), and are generally found in marine sedimenting particles (Gagosian et al., 1982). Also, there was an enrichment of mesopelagic genes involved in fatty acid β-oxidation and break down, including the acyl-CoA dehydrogenase (initial step of fatty acid β-oxidation), electron transfer flavoprotein-ubiquinone oxidoreductase (links the oxidation of fatty acids to oxidative phosphorylation) and CoA-transferase family III (CaiB/BaiF) family (involved in the transport of fatty acids from the intermembraneous space in the mitochondria). Thus, in contrast to epipelagic fungi that seem to rely more on carbohydrates, mesopelagic fungi apparently experience a low carbohydrate affinity, which is in agreement with the decrease in carbohydrate concentration (Pakulski and Benner, 1994) and the increase in total fatty acids with depth (Wakeham et al., 2003).
Besides organic carbon limitation, fungi in the deep ocean also need to cope with energy limitation. Genes involved in alternative sources of carbon and energy, like the degradation of benzoate (carboxymuconolactone decarboxylase family) (Ornston, 1966; Giovannoni and Stingl, 2005), anaerobic glycolysis (L-lactate dehydrogenase, which transforms glucose to lactate under low oxygen concentrations), urea (urea active transporter for both urea and polyamine and argininosuccinate synthase) and sulfur metabolism (sulfite reductase and sulfate adenylyltransferase) were also relatively enriched in the mesopelagic.
Fungal Global Biogeography
A biogeographical distribution pattern, particularly latitudinally, has been described for many oceanic organismal groups (Roy et al., 1998; Lambshead et al., 2002; Macpherson, 2002; Fuhrman et al., 2008). However, due to the limited information available on marine fungi, little is know about their global distribution in the ocean. When fungal taxonomic diversity was examined according to regions (Supplementary Figure S5), differences were found among oceanic regions, which could be a result of the environmental variability among regions. Changes in Shannon diversity index (variance) among different regions were more pronounced in the epipelagic than in the mesopelagic, which is consistent with the more dynamic nature (i.e., subjected to stronger physicochemical variability) of epipelagic relative to mesopelagic environments. In the epipelagic, the North Atlantic Ocean (NAO) and Indian Ocean (IO) showed the lowest Shannon index. A similar trend, although with smaller differences, was found in the mesopelagic, with NAO and IO presenting the lowest taxonomic diversities. In the epipelagic, higher variability (among and within regions) in fungal diversity was likely mediated by larger changes in physicochemical parameters, including temperature. Interestingly, the Shannon diversity was, in all oceanic regions, consistently higher (ANOVA p = < 0.0001; Supplementary Table S5) in the mesopelagic than in the epipelagic independent of whether data was analyzed using results based on taxonomic or functional assignment of reads (Figure 6 and Supplementary Figure S6). Significant increases in fungal diversity from surface to deep waters is consistent with the reported increase in bacterioplankton community diversity in the deep waters of the North Atlantic (Agogué et al., 2011) and the North Pacific and Atlantic (Hewson et al., 2006). This increased bacterioplankton diversity with depth is proposed to be linked to episodic inputs of organic matter from sunlit waters (Hewson et al., 2006), and to the generation of specific biogeochemical conditions on slowly sinking, or buoyant laterally advected suspended particles (Agogué et al., 2011), that would serve as important substrates for deep ocean microbes (Baltar et al., 2009; Bochdansky et al., 2010). Moreover, marine snow particles are colonized by marine fungi (Bochdansky et al., 2016), so they can also contribute to the diversity of fungi in the deep waters.
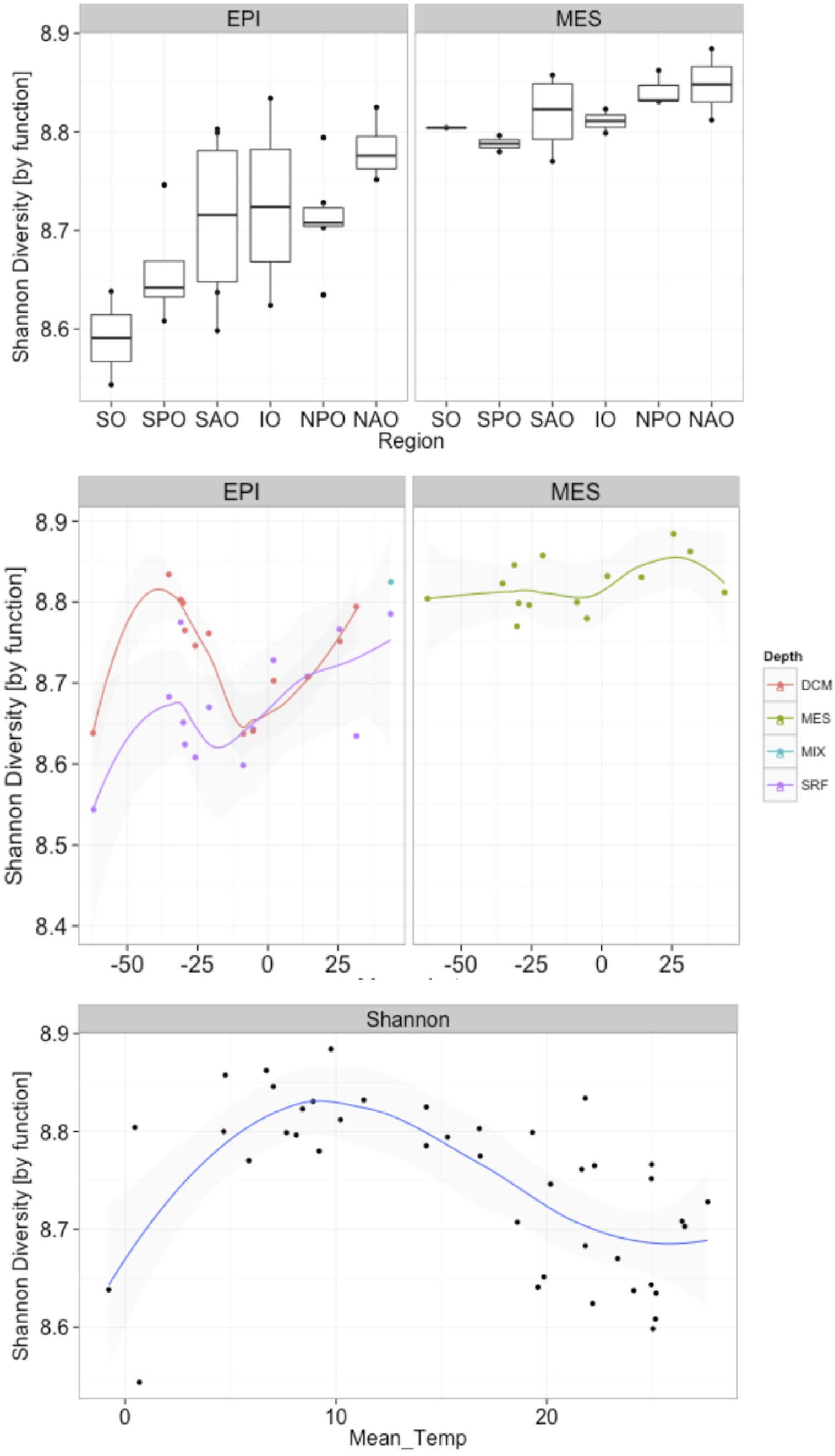
Figure 6. Changes in fungal functional diversity by oceanic region and pelagic zone (top), and in response to latitude (middle) and temperature (bottom). X-axes represent (top) oceanic regions (IO, Indian Ocean; NAO, North Atlantic Ocean; NPO, North Pacific Ocean; SAO, South Atlantic; SO, Southern Ocean; and SPO, South Pacific Ocean), (middle) latitude, and (bottom) mean temperature. Depths in middle panel represent four depth layers analyzed (DCM, deep chlorophyll maximum; MES, mesopelagic; EPI, epipelagic; MIX, subsurface epipelagic mixed layer; SRF, surface).
Our analyses, also revealed an increase in both fungal taxonomic and functional diversity with increasing distance from the equator (Figure 6 and Supplementary Figure S5). This trend of increasing fungal diversity with increasing distance from the equator was found also in mesopelagic waters, albeit the increase in diversity was not as pronounced in deep compared to epipelagic waters. In contrast to the general trend of increasing diversity with distance from the equator, a local decrease in diversity was observed in subpolar regions compared to the (sub)tropical and temperate regions (Figure 6 and Supplementary Figure S5). An observed decrease in diversity in subpolar regions (non-linear relationship with distance from equator) suggests a temperature-driven selection [which was clearer than a salinity-driven (Supplementary Figure S7)] with highest and lowest temperatures in epipelagic waters resulting in reduced diversity. This was also evident when correlating mean ocean epipelagic water temperatures to functional diversity. Diversity increased with increasing temperature until 8°C where the highest diversity was observed and decreased again toward higher surface water temperatures (Figure 6 and Supplementary Figure S5). Prior work has demonstrated a positive linear relationship between planktonic marine bacterial diversity and temperature (Fuhrman et al., 2008). A decrease in diversity with increasing latitude has been found for marine bacteria (Sul et al., 2013), phytoplankton (Barton et al., 2010), and zooplankton (Yasuhara et al., 2012). In contrast, other report suggested that surface bacterial diversity peaks in temperate latitudes across the world’s oceans during winter in each hemisphere (Ladau et al., 2013). Moreover, in a report mostly focused on polar biogeography of marine bacteria (but including low latitude sites as well), diversity for surface and deep communities were not correlated with latitude or temperature (Ghiglione et al., 2012). The latter authors also found that polar communities (from Arctic and Southern Ocean) were more similar to each other than to lower latitude pelagic communities, suggesting temperature as the main structuring factor of bacterioplankton communities. Although the conserved pattern of increasing richness and diversity from polar to tropical regions is generally recognized in both marine and terrestrial systems for most living organisms, our data suggest that fungi in the ocean do not follow this trend in biodiversity.
Conclusion
The role of fungi in the ocean remains largely enigmatic but recent advances have started to reveal considerable diversity in fungi. Genomic evidence presented here suggests that fungi might contribute to the production of vitamins, including B vitamins, known to be modulating micronutrients of ocean primary productivity. The data also point toward a thus far unrecognized role of marine fungi in carbon, sulfur, and nitrogen metabolism in the ocean as indicated by highly abundant fungal genes. Fungi and bacteria respond to latitudinal, or more likely temperature changes, suggesting that shifts in temperature caused by climate change could alter these relationships with unknown repercussion. Even without this added incentive, the phylogenetic and functional diversity of fungi in the ocean indicates that they might play a substantial, albeit hitherto largely ignored role in the ocean’s biogeochemical cycles.
Data Availability
The datasets generated for this study can be found in MG-RAST server, 4662524.3-4662529.3.
Author Contributions
SM and FB contributed equally in the design, analyses, and writing of this study. AB was involved in the analyses of the data. GH was involved in the writing and interpretation of the data.
Funding
FB was supported by a University of Otago Research Grant (UORG) and by a Rutherford Discovery Fellowship by the Royal Society of New Zealand. GH was supported by the European Research Council under the European Community’s Seventh Framework Program (FP7/2007-2013)/ERC Advanced Grant “MEDEA” agreement No. 268595 and by the Austrian Science Fund (FWF) project I486-B09.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Stephanie Roosa for sample processing and DNA extractions of samples from the MOTS Transect. We would also like to thank the skippers and crew of the RV Polaris II for their help during the sampling events, and the technical staff at the Portobello Marine Laboratory.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00131/full#supplementary-material
Footnotes
References
Agogué, H., Lamy, D., Neal, P. R., Sogin, M. L., and Herndl, G. J. (2011). Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol. Ecol. 20, 258–274. doi: 10.1111/j.1365-294X.2010.04932.x
Amend, A. S., Barshis, D. J., and Oliver, T. A. (2012). Coral-associated marine fungi form novel lineages and heterogeneous assemblages. ISME J. 6, 1291–1301. doi: 10.1038/ismej.2011.193
Armbrust, E. V., and Palumbi, S. R. (2015). Marine biology. Uncovering hidden worlds of ocean biodiversity. Science 348, 865–867. doi: 10.1126/science.aaa7378
Baltar, F., Arístegui, J., Gasol, J. M., and Sintes, E. (2009). Evidence of prokaryotic metabolism on suspended particulate organic matter in the dark waters of the subtropical North Atlantic. Limnol. Oceangr. 54, 182–193. doi: 10.4319/lo.2009.54.1.0182
Baltar, F., Currie, K., Stuck, E., Roosa, S., and Morales, S. E. (2016). Oceanic fronts: transition zones for bacterioplankton community composition. Environ. Microbiol. Rep. 8, 132–138. doi: 10.1111/1758-2229.12362
Baltar, F., Stuck, E., Morales, S., and Currie, K. (2015). Bacterioplankton carbon cycling along the subtropical frontal zone off New Zealand. Prog. Oceanogr. 135, 168–175. doi: 10.1016/j.pocean.2015.05.019
Barton, A. D., Dutkiewicz, S., Flierl, G., Bragg, J., and Follows, M. J. (2010). Patterns of diversity in marine phytoplankton. Science 327, 1509–1511. doi: 10.1126/science.1184961
Bass, D., Howe, A., Brown, N., Barton, H., Demidova, M., Michelle, H., et al. (2007). Yeast forms dominate fungal diversity in the deep oceans. Proc. R. Soc. B Biol. Sci. 274, 3069–3077. doi: 10.1098/rspb.2007.1067
Bertrand, E. M., McCrow, J. P., Moustafa, A., Zheng, H., McQuaid, J. B., Delmont, T. O., et al. (2015). Phytoplankton–bacterial interactions mediate micronutrient colimitation at the coastal Antarctic sea ice edge. Proc. Natl. Acad. Sci. U.S.A. 112, 9938–9943. doi: 10.1073/pnas.1501615112
Bochdansky, A. B., Clouse, M. A., and Herndl, G. J. (2016). Eukaryotic microbes, principally fungi and labyrinthulomycetes, dominate biomass on bathypelagic marine snow. ISME J. 11, 362–373. doi: 10.1038/ismej.2016.113
Bochdansky, A. B., van Aken, H. M., and Herndl, G. J. (2010). Role of macroscopic particles in deep-sea oxygen consumption. Proc. Natl. Acad. Sci. U.S.A. 107, 8287–8291. doi: 10.1073/pnas.0913744107
Booth, T., and Kenkel, N. (1986). “Ecological studies of lignicolous marine fungi: a distribution model based on ordination and classification,” in The Biology of Marine Fungi, ed. S. T. Moss (Cambridge: Cambridge University Press), 297–310.
Burgaud, G., Woehlke, S., Rédou, V., Orsi, W., Beaudoin, D., Barbier, G., et al. (2013). Deciphering the presence and activity of fungal communities in marine sediments using a model estuarine system. Aquat. Microb. Ecol. 70, 45–62. doi: 10.3354/ame01638
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Croft, M. T., Lawrence, A. D., Raux-Deery, E., Warren, M. J., and Smith, A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N. U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Science 311, 496–503. doi: 10.1126/science.1120250
De Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. doi: 10.1126/science.1261605
Edgcomb, V. P., Beaudoin, D., Gast, R., Biddle, J. F., and Teske, A. (2011). Marine subsurface eukaryotes: the fungal majority. Environ. Microbiol. 13, 172–183. doi: 10.1111/j.1462-2920.2010.02318.x
Fuhrman, J. A., Steele, J. A., Hewson, I., Schwalbach, M. S., Brown, M. V., Green, J. L., et al. (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 7774–7778. doi: 10.1073/pnas.0803070105
Gagosian, R. B., Smith, S. O., and Nigrelli, G. E. (1982). Vertical transport of steroid alcohols and ketones measured in a sediment trap experiment in the equatorial Atlantic-ocean. Geochim. Cosmochim. Acta 46, 1163–1172. doi: 10.1016/0016-7037(82)90002-3
Gao, Z., Johnson, Z. I., and Wang, G. (2010). Molecular characterization of the spatial diversity and novel lineages of mycoplankton in Hawaiian coastal waters. ISME J. 4, 111–120. doi: 10.1038/ismej.2009.87
Ghiglione, J.-F., Galand, P. E., Pommier, T., Pedrós-Alió, C., Maas, E. W., Bakker, K., et al. (2012). Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 109, 17633–17638. doi: 10.1073/pnas.1208160109
Giovannoni, S. J., and Stingl, U. (2005). Molecular diversity and ecology of microbial plankton. Nature 437, 343–348. doi: 10.1038/nature04158
Gutiérrez, M. H., Jara, A. M., and Pantoja, S. (2016). Fungal parasites infect marine diatoms in the upwelling ecosystem of the Humboldt current system off central Chile. Environ. Microbiol. 18, 1646–1653. doi: 10.1111/1462-2920.13257
Gutiérrez, M. H., Pantoja, S., Tejos, E., and Quinones, R. A. (2011). The role of fungi in processing marine organic matter in the upwelling ecosystem off Chile. Mar. Biol. 158, 205–219. doi: 10.1007/s00227-010-1552-z
Hassett, B. T., and Gradinger, R. (2016). Chytrids dominate arctic marine fungal communities. Environ. Microbiol. 18, 2001–2009. doi: 10.1111/1462-2920.13216
Hewson, I., Steele, J. A., Capone, D. G., and Fuhrman, J. A. (2006). Temporal and spatial scales of variation in bacterioplankton assemblages of oligotrophic surface waters. Mar. Ecol. Prog. Ser. 311, 67–77. doi: 10.3354/meps311067
Jebaraj, C. S., Raghukumar, C., Behnke, A., and Stoeck, T. (2010). Fungal diversity in oxygen-depleted regions of the Arabian Sea revealed by targeted environmental sequencing combined with cultivation. FEMS Microbiol. Ecol. 71, 399–412. doi: 10.1111/j.1574-6941.2009.00804.x
Jeffries, T. C., Curlevski, N. J., Brown, M. V., Harrison, D. P., Doblin, M. A., Petrou, K., et al. (2016). Partitioning of fungal assemblages across different marine habitats. Environ. Microbiol. Rep. 8, 235–238. doi: 10.1111/1758-2229.12373
Jones, E. B. G., and Pang, K.-L. (2012). Tropical aquatic fungi. Biodivers. Conserv. 21, 2403–2423. doi: 10.1007/s10531-011-0198-6
Kawai, Y., Mercier, R., Wu, L. J., Domínguez-Cuevas, P., Oshima, T., and Errington, J. (2015). Cell growth of wall-free L-form bacteria is limited by oxidative damage. Curr. Biol. 25, 1613–1618. doi: 10.1016/j.cub.2015.04.031
Klanner, C., Prokisch, H., and Langer, T. (2001). MAP-1 and IAP-1, two novel AAA proteases with catalytic sites on opposite membrane surfaces in mitochondrial inner membrane of Neurospora crassa. Mol. Biol. Cell 12, 2858–2869. doi: 10.1091/mbc.12.9.2858
Ladau, J., Sharpton, T. J., Finucane, M. M., Jospin, G., Kembel, S. W., O’Dwyer, J., et al. (2013). Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 7, 1669–1677. doi: 10.1038/ismej.2013.37
Lambshead, P., Brown, C. J., Ferrero, T. J., Mitchell, N. J., Smith, C. R., Hawkins, L. E., et al. (2002). Latitudinal diversity patterns of deep-sea marine nematodes and organic fluxes: a test from the central equatorial Pacific. Mar. Ecol. Prog. Ser. 236, 129–135. doi: 10.3354/meps236129
Macpherson, E. (2002). Large-scale species-richness gradients in the Atlantic Ocean. Proc. Biol. Sci. 269, 1715–1720. doi: 10.1098/rspb.2002.2091
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Morales, S. E., Meyer, M., Currie, K., and Baltar, F. (2018). Are oceanic fronts ecotones? Seasonal changes along the subtropical front show fronts as bacterioplankton transition zones but not diversity hotspots. Environ. Microbiol. Rep. 10, 184–189. doi: 10.1111/1758-2229.12618
Nagahama, T., Hamamoto, M., Nakase, T., and Horikoshi, K. (2003). Rhodotorula benthica sp. nov. and Rhodotorula calyptogenae sp. nov., novel yeast species from animals collected from the deep-sea floor, and Rhodotorula lysiniphila sp. nov., which is related phylogenetically. Int. J. Syst. Evol. Microbiol. 53, 897–903. doi: 10.1099/ijs.0.02395-0
Ornston, L. N. (1966). The conversion of catechol and protocatechuate to beta-ketoadipate by Pseudomonas putida. IV. Regulation. J. Biol. Chem. 241, 3800–3810.
O’Rorke, R., Lavery, S. D., Wang, M., Nodder, S. D., and Jeffs, A. G. (2013). Determining the diet of larvae of the red rock lobster (Jasus edwardsii) using high-throughput DNA sequencing techniques. Mar. Biol. 161, 551–563. doi: 10.1007/s00227-013-2357-7
Orsi, W., Biddle, J. F., and Edgcomb, V. (2013). Deep sequencing of subseafloor eukaryotic rRNA reveals active Fungi across marine subsurface provinces. PLoS One 8:e56335. doi: 10.1371/journal.pone.0056335
Orsi, W. D., Edgcomb, V. P., Christman, G. D., and Biddle, J. F. (2013). Gene expression in the deep biosphere. Nature 499, 205–208. doi: 10.1038/nature12230
Pakulski, J. D., and Benner, R. (1994). Abundance and distribution of carbohydrates in the ocean. Limnol. Oceangr. 39, 930–940. doi: 10.4319/lo.1994.39.4.0930
Pang, K. L., and Mitchell, J. I. (2005). Molecular approaches for assessing fungal diversity in marine substrata. Bot. Mar. 48, 332–347. doi: 10.1515/bot.2005.046
Powell, S., Forslund, K., Szklarczyk, D., Trachana, K., Roth, A., Huerta-Cepas, J., et al. (2013). eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42, D231–D239. doi: 10.1093/nar/gkt1253
R Development Core Team (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rama, T., Norden, J., Davey, M. L., Mathiassen, G. H., Spatafora, J. W., and Kauserud, H. (2014). Fungi ahoy! Diversity on marine wooden substrata in the high North. Fungal Ecol. 8, 46–58. doi: 10.1016/j.funeco.2013.12.002
Richards, T. A., Jones, M. D. M., Leonard, G., and Bass, D. (2012). Marine fungi: their ecology and molecular diversity. Annu. Rev. Mar. Sci. 4, 495–522. doi: 10.1146/annurev-marine-120710-100802
Richards, T. A., Leonard, G., Mahé, F., Del Campo, J., Romac, S., Jones, M. D. M., et al. (2015). Molecular diversity and distribution of marine fungi across 130 European environmental samples. Proc. R. Soc. B Biol. Sci. 282:20152243. doi: 10.1098/rspb.2015.2243
Roy, K., Jablonski, D., Valentine, J. W., and Rosenberg, G. (1998). Marine latitudinal diversity gradients: tests of causal hypotheses. Proc. Natl. Acad. Sci. U.S.A. 95, 3699–3702. doi: 10.1073/pnas.95.7.3699
Sul, W. J., Oliver, T. A., Ducklow, H. W., Amaral-Zettler, L. A., and Sogin, M. L. (2013). Marine bacteria exhibit a bipolar distribution. Proc. Natl. Acad. Sci. U.S.A. 110, 2342–2347. doi: 10.1073/pnas.1212424110
Taylor, J. D., and Cunliffe, M. (2016). Multi-year assessment of coastal planktonic fungi reveals environmental drivers of diversity and abundance. ISME J. 10, 2118–2128. doi: 10.1038/ismej.2016.24
Tisthammer, K. H., Cobian, G. M., and Amend, A. S. (2016). ScienceDirect. Fungal Ecol. 19, 39–46. doi: 10.1016/j.funeco.2015.09.003
Wakeham, S. G., Pease, T. K., and Benner, R. (2003). Hydroxy fatty acids in marine dissolved organic matter as indicators of bacterial membrane material. Org. Geochem. 34, 857–868. doi: 10.1016/S0146-6380(02)00189-4
Wang, X., Singh, P., Gao, Z., Zhang, X., Johnson, Z. I., and Wang, G. (2014). Distribution and diversity of planktonic fungi in the West Pacific Warm Pool. PLoS One 9:e101523. doi: 10.1371/journal.pone.0101523
Yarden, O. (2014). Fungal association with sessile marine invertebrates. Front. Microbiol. 5:228. doi: 10.3389/fmicb.2014.00228
Yasuhara, M., Hunt, G., Dowsett, H. J., Robinson, M. M., and Stoll, D. K. (2012). Latitudinal species diversity gradient of marine zooplankton for the last three million years. Ecol. Lett. 15, 1174–1179. doi: 10.1111/j.1461-0248.2012.01828.x
Zhang, X.-Y., Tang, G.-L., Xu, X.-Y., Nong, X.-H., and Qi, S.-H. (2014). Insights into deep-sea sediment fungal communities from the East Indian Ocean using targeted environmental sequencing combined with traditional cultivation. PLoS One 9:e109118. doi: 10.1371/journal.pone.0109118
Keywords: fungi, metagenomes, Tara ocean project, biogeography, epipelagic and mesopelagic
Citation: Morales SE, Biswas A, Herndl GJ and Baltar F (2019) Global Structuring of Phylogenetic and Functional Diversity of Pelagic Fungi by Depth and Temperature. Front. Mar. Sci. 6:131. doi: 10.3389/fmars.2019.00131
Received: 03 December 2018; Accepted: 04 March 2019;
Published: 19 March 2019.
Edited by:
Toshi Nagata, The University of Tokyo, JapanReviewed by:
Maiko Kagami, Yokohama National University, JapanYantao Liang, Qingdao Institute of Bioenergy & Bioprocess Technology (CAS), China
Copyright © 2019 Morales, Biswas, Herndl and Baltar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio E. Morales, c2VyZ2lvLm1vcmFsZXNAb3RhZ28uYWMubno= Federico Baltar, ZmVkZXJpY28uYmFsdGFyQHVuaXZpZS5hYy5hdA==