 Dewi Embong Bulan1,2
Dewi Embong Bulan1,2 Alisa Wilantho3
Alisa Wilantho3 Sissades Tongsima3
Sissades Tongsima3 Voranop Viyakarn1
Voranop Viyakarn1 Suchana Chavanich1*
Suchana Chavanich1* Naraporn Somboonna4,5*
Naraporn Somboonna4,5*- 1Reef Biology Research Group, Department of Marine Science, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
- 2Department of Aquatic Resources Management, Faculty of Fisheries and Marine Science, Mulawarman University, Samarinda, Indonesia
- 3Genome Technology Research Unit, National Center for Genetic Engineering and Biotechnology (BIOTEC), Pathum Thani, Thailand
- 4Department of Microbiology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
- 5Microbiome Research Unit for Probiotics in Food and Cosmetics, Chulalongkorn University, Bangkok, Thailand
Reef sites of Ko Samae San (S), Khao Ma Cho (K) and Ko Tao Mo (T) in the upper Gulf of Thailand have abundant corals and represent a hotspot of marine biodiversity. Coral reefs serve as major networks of food and energy, where bacteria, microbial eukaryotes (fungi) and small eukaryotes play significant roles as primary producers that convert inorganic compounds to organic compounds, degraders of toxic substances, and recyclers. These functions sustain food and energy supplies. Advances in metagenomics and next-generation sequencing can provide knowledge of diversity without limitations imposed by media and other conditions associated with laboratory cultures. Scientists have researched bacterial diversity of coral sites; however, a database for fungi and small eukaryotes from Thailand’s sites with abundant corals is lacking. The present study combined fungal ribosomal intergenic spacer analysis (F-RISA) and 18S rRNA gene sequencing to unveil the first culture-independent microbial and small eukaryotes from these sites at two times and across four species of coral (Porites lutea, Platygyra sinensis, Acropora humilis, and Acropora millepora), seawater and sediment. Results showed that the small eukaryotic communities on corals were distinct from communities in the surrounding seawater and sediment. The communities were relatively similar at the three sites and during the two periods of time. Pearson’s correlations indicated the community diversity were associated with water quality (e.g., dissolved oxygen concentrations and density of water).
Introduction
Coral reefs provide habitat where microbes and small eukaryotes (e.g., fungi, diatoms and copepods) live in potentially symbiotic relationships with corals, and these organisms play important roles in food and energy networks. These eukaryotic assemblages have been denoted “coral holobionts.” Similar to bacteria, microbial and small eukaryotes serve as primary producers and recyclers, for examples, microalgae Symbiodinium photosynthesizes (Mydlarz et al., 2010), copepods and fungi degrade toxic nitrate and nitrite compounds, and fungi fix nitrogen gas in seawater to forms that can be used by other organisms (Wegley et al., 2007). These activities support colonization, reproduction, growth, and resistance to stress (i.e., against coral bleaching, pathogen infection and pollution) for corals (Massana and Pedrós-Alió, 2008; Ainsworth et al., 2010; Mydlarz et al., 2010; Pootakham et al., 2018).
The upper Gulf of Thailand (GoT) harbored pristine reefs where corals remain abundant and healthy, including reefs at Ko Samae San (S), Khao Ma Cho (K) and Ko Tao Mo (T). These sites also harbor sone of the world’s most diverse assemblages (Phongsuwan et al., 2013; Ramírez et al., 2017). These sites are part of the Sea Natural History Museum of Thailand, thus they are protected from many anthropogenic activities. These fringing reefs include numerous colonies of Acropora, Platygyra, and Porites (Phongsuwan et al., 2013; Latypov, 2015). The microbes and small eukaryotes of these relatively pristine sites have not been studied in detail.
Our study provides the first data on microbial and small eukaryotic assemblages at the S, K, and T sites, using fungal ribosomal intergenic spacer analysis (F-RISA) and sequencing of the V9 region of 18S rRNA gene. F-RISA measures a DNA length that is characteristic for fungal species, so different communities yield different combinations of lengths (Ranjard et al., 2001; Gillevet et al., 2009). The next generation sequencing (NGS) provides more power to detect and identify natural microbial diversity, without limitations due to media or other conditions associated with laboratory cultures (Riesenfeld et al., 2004; Rusch et al., 2007; Somboonna et al., 2014; Mahé et al., 2015; Khitmoh et al., 2017). Our study primarily characterized and compared the eukaryotic assemblages of the prevalent coral species at the three sites (Porites lutea, Platygyra sinensis, Acropora humilis and Acropora millepora), and the surrounding niches (sediment and seawater) during the wet and dry seasons in 1 year (Figure 1A). Independent replicates were collected during all sampling events (3–10 replicates). Our study also correlated data on the relative abundances of representative genera of eukaryotes with parameters describing water quality. These initial data serve as a reference, and they may advance our understanding of similar reefs where data on microbial and small eukaryotes are inadequate.
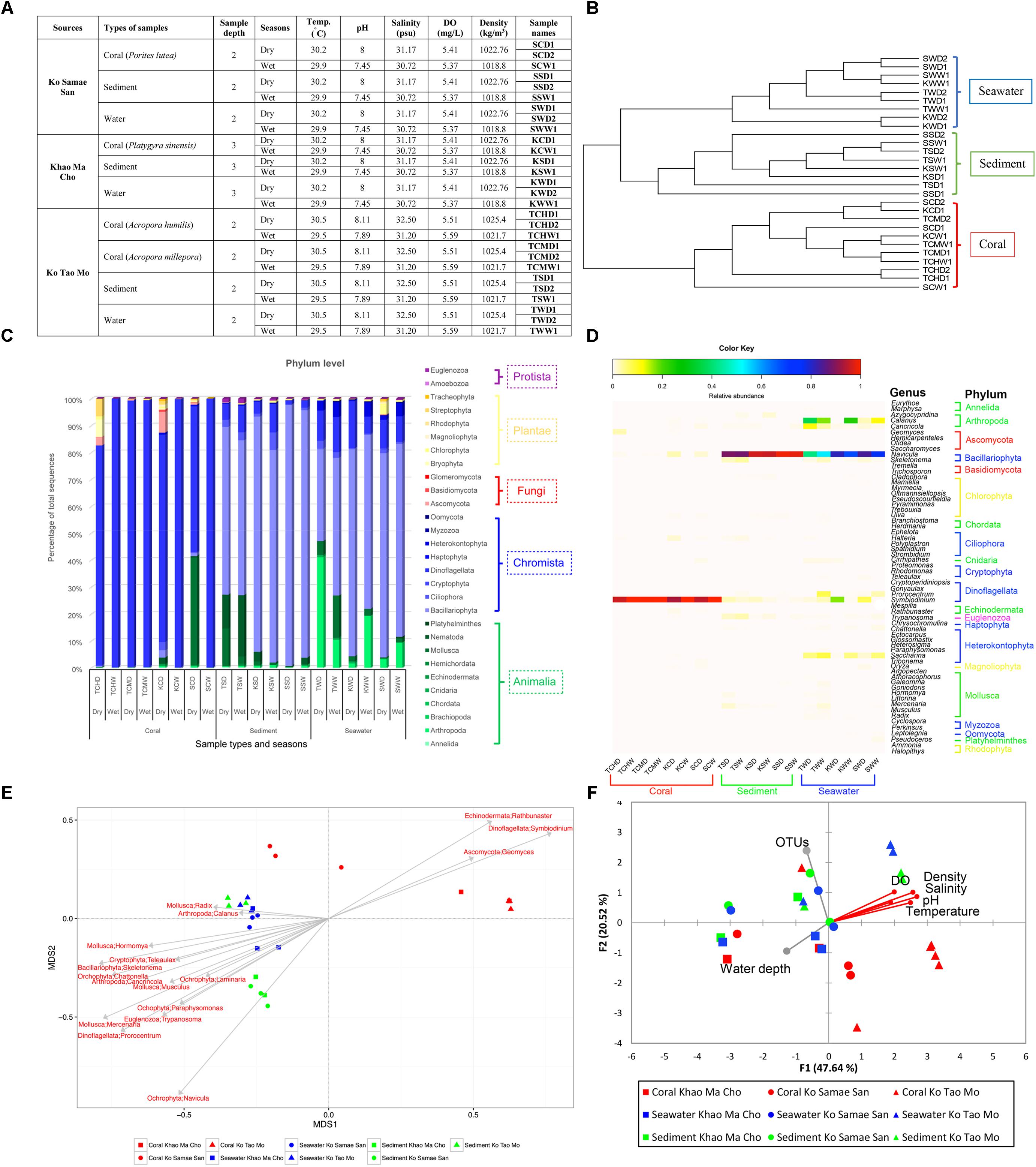
FIGURE 1. Samples collected (A), unweighted pair group with arithmetic mean (UPGMA) clustering to differentiate communities (B), relative abundances at the phylum (C) and genus (D) levels, and Pearson’s correlations for genera (E) and variables characterizing water quality (F). Unclassified phyla were not displayed in (C,D). In (D), organisms with relative abundances <1% were omitted. In (F), the vector direction and length represent the direction and strength of the correlation of that variable with the relative abundances of organisms. Red vectors represent significant correlations (p < 0.01), and gray vectors represent non-significant correlations (p ≥ 0.01).
Materials and Methods
Sampling
Samples were collected at S, K, and T sites during April-May (dry season) and September-October (wet season) in 2014. The coral reefs at these sites appeared healthy during both sampling events. Samples included 3–10 independent replicates for seawater, sediments, and prevalent coral species (P. lutea, P. sinensis, A. Humilis, and A. millepora). We collected coral colonies that were 5 cm in diameter, 5 L of seawater within 1 m above a colony, and 50 g of sediment within 1 m below a colony. All samples were placed in sterile bottles or bags, transported on ice, and analyzed within 7–14 days. Water quality was characterized by measuring temperature, pH, salinity, dissolved oxygen (DO) and density of water on-site (Figure 1A).
Metagenomic Extraction
Metagenomic DNA were extracted using Power Water DNA Isolation Kit for seawater and Power Soil DNA Isolation Kit for corals and sediments (MoBio, Carlsbad, CA, United States) according to the manufacturer’s instructions. Microorganisms in replicate 2.5 L aliquots of seawater were captured on a sterile 0.22-micron filter using a vacuum filtration system (Merck Millipore, Massachusetts, MA, United States). For coral and sediment samples, 1 g of ground coral and 1 g of sediment were used. Extractions were performed for individual replicates and checked for DNA quality and concentration by agarose gel electrophoresis and nanodrop spectrophotometry, respectively.
Fungal Ribosomal Intergenic Spacer Analysis for Subgrouping of Independent Sampled Replicates
Fungal ribosomal intergenic spacer analysis was performed according to established protocols, using universal primers 18S-2234C (5′-GTTTCCGTAGGTGAACCTGC-3′) and 28S-3126T (5′-ATATGCTTAAGTTCAGCGGGT-3′) (Ranjard et al., 2001; Gillevet et al., 2009). To prevent potential bias in PCR, for each replicate the minimum of two PCRs were performed and pooled. Each PCR comprised 1 × EmeraldAmp® GT PCR Master Mix (TaKaRa, Shiga, Japan), 0.3 μM forward primer, 0.3 μM reverse primer, and the metagenome. The thermocycling conditions were 95°C for 4 min, followed by 30–35 cycles of 94°C for 1 min, 55∘C for 1 min and 72∘C for 2 min, and ended by 72∘C for 10 min. Different communities or subgroups were identified by different banding patterns upon agarose gel electrophoresis.
18S rRNA Gene Library Construction and NGS
The 18S rRNA V9 gene libraries were constructed following Caporaso et al. (2012), using universal eukaryotic primers Illumina_Euk_1391F (5′-GTACACACCGCCCGTC-3′) and Illumina_EukBr (5′-TGATCCTTCTGCAGGTTCACCTAC-3′), appended with 5′ Illumina adapter and 3′ Golay barcode sequences. PCR comprised 1 × EmeraldAmp® GT PCR Master Mix (TaKaRa), 0.2 μM of each primer, and the metagenome. Triplicate PCRs were performed and pooled for each sample or subgroup to address potential bias. The thermocycling conditions were 94°C for 3 min, and 28–30 cycles of 94°C for 45 s, 50°C for 60 s and 72°C for 90 s, followed by 72°C for 10 min. The amplicons were purified using GenepHlowTM Gel Extraction Kit (Geneaid Biotech Ltd., New Taipei City, Taiwan), and quantified with Picogreen (Invitrogen, Eugene, Oregon, OR, United States). 200 ng of each sample or subgroup was pooled for NGS, along the sequencing primers and index sequence (Caporaso et al., 2012), using Miseq300 platform (Illumina, San Diego, CA, United States) housed at the Chulalongkorn Medical Research Center, Chulalongkorn University (Bangkok, Thailand).
Bioinformatic Analyses
Raw sequences were quality screened following Mothur’s standard operating procedures for MiSeq (Schloss et al., 2009) that included removal of sequences that have (i) ambiguous bases, (ii) >1 mismatch base in the primer region, (iii) ≥1 mismatch base in the barcode, (iv) >10 homopolymers, (v) a minimum sequencing quality score of <35 over a 50-bp window, (vi) a read length of <350 bases, and (vii) chimera sequence (Huse et al., 2010; Edgar et al., 2011). Sequences were aligned against Silva database (Schloss et al., 2009). Operational taxonomic units (OTUs) were assigned using Naïve Bayesian method (Wang et al., 2007). Data for all samples (and subgroups) were normalized to an equal sequencing depth (78,068 sequences). Mothur was used to compute Good’s coverage to estimate a sequencing coverage, rarefaction curve, alpha diversity (species richness and evenness in a sample: Chao1 richness, Shannon diversity and inverse Simpson index), beta diversity (thetayc dissimilarity coefficients, unweighted pair group with arithmetic mean (UPGMA) clustering, and principal coordinates analysis (PCoA)), and AMOVA (Schloss et al., 2009). For sample with 2 subgroups, a student’s t-test was computed, and the subgroups were merged if p > 0.01. Correlations between the relative abundances of common genera and water quality variables were determined using Pearson’s correlation (Schloss et al., 2009) and XLSTAT-Ecology software1.
Availability of Supporting Data
Nucleic acid sequences were deposited in an open access Sequence Read Archive database of NCBI, accession number SRP095762.
Results
Fungal Ribosomal Intergenic Spacer Analysis for Subgrouping of Independent Sampling Replicates
The 3–10 replicate samples exhibited similar F- RISA banding patterns, suggesting limited diversity among replicates (no subgroup classification). Nonetheless, we analyzed bacterial ribosomal intergenic spacer analysis (B-RISA), and some samples showed two B-RISA patterns (e.g., SCD1 and SCD2 represent 2 B-RISA subgroups in Figure 1A and SCW1 represents one B-RISA subgroup). To ensure we did not miss any eukaryotic subgroup, we constructed and sequenced the 18S rRNA gene libraries based on the B-RISA subgroups.
18S rRNA Gene Sequencing
Libraries of 18S rRNA genes were constructed successfully for all subgroups, because the numbers of quality sequences per subgroup were high (avg. 218,885 sequences/subgroup). This sequencing depth was statistically sufficient, as indicated by Good’s coverage of 99.99–100% at genus-level OTUs (Supplementary Table S1A). These results are consistent with estimates of richness at the genus level derived from rarefaction curves that plateaued at this sequencing depth. The 18S rDNA data for fungi were analyzed separately, and they yielded Good’s coverage values of 87.33–97.91% (Supplementary Table S1B).
Shannon diversity indices indicated greater taxonomic richness in sediment and seawater (Supplementary Table S1A). These results highlighted the specificity of the community on corals. Eukaryotes in the Kingdoms Chromista (88%: Dinoflagellata 66% and Bacillariophyta 22%) and Animalia (mostly worms and molluscs) were more common than fungi (Supplementary Figures S1A,B). Among the four fungal phyla, Ascomycota was the most dominant (>90%), followed by Basidiomycota (5.609%), an unclassified phylum (3.741%) and Glomeromycota (0.003%; Supplementary Figure S1C).
Comparison of Communities From Species of Corals, Sites, and Times
In the UPGMA, the communities from corals clearly clustered separately from the communities from seawater and sediment across all sites and the two periods of time (Figure 1B). These results further highlight the unique community associated with corals, and AMOVA also identified a statistically significant difference between these communities (p < 0.001). However, the communities at the three sites and during the wet and dry sampled months were not statistically different (S vs. K vs. T, p = 0.082; months, p = 0.707). Moreover, pairwise comparisons between F-RISA subgroups supported the F-RISA results, because all corresponding subgroups were statistically similar (avg. p = 0.33; Supplementary Table S2). The corresponding subgroups were thereby merged.
Figures 1C,D exhibited phyla and genera that dominated and differentiated the communities. The genus Symbiodinium in phylum Dinoflagellata dominated samples of corals, whereas the genus Navicula in Bacillariophyta dominated in sediments and seawater. Fungi, although relatively rare, were 25–96 times more abundance in samples of corals (avg. 1.67%) than in sediments (0.017%) and seawater (0.066%; Figure 1C), and helped differentiate the communities on corals (phylum Ascomycota: genus Geomyces; Figure 1E). Additional genera specific to corals were Symbiodinium and Rathbunaster (Figure 1E).
To better understand the community associated with corals, genera that were reported to affect coral health were assessed. Symbiodinium, a well-known coral holobiont, was most abundant on all species of coral and more abundant in the dry (66%) months. In contrast, the relative abundance of Symbiodinium in sediment and seawater was only 1% (Figure 2). The presence and abundance of Geomyces on corals in the dry months raises some concern, because Geomyces can cause a skin disease called “white-nose syndrome” (Work and Meteyer, 2014). However, the corals we collected appeared healthy, so Symbiodinium and echinoderm Rathbunaster might limit the effects of Geomyces.
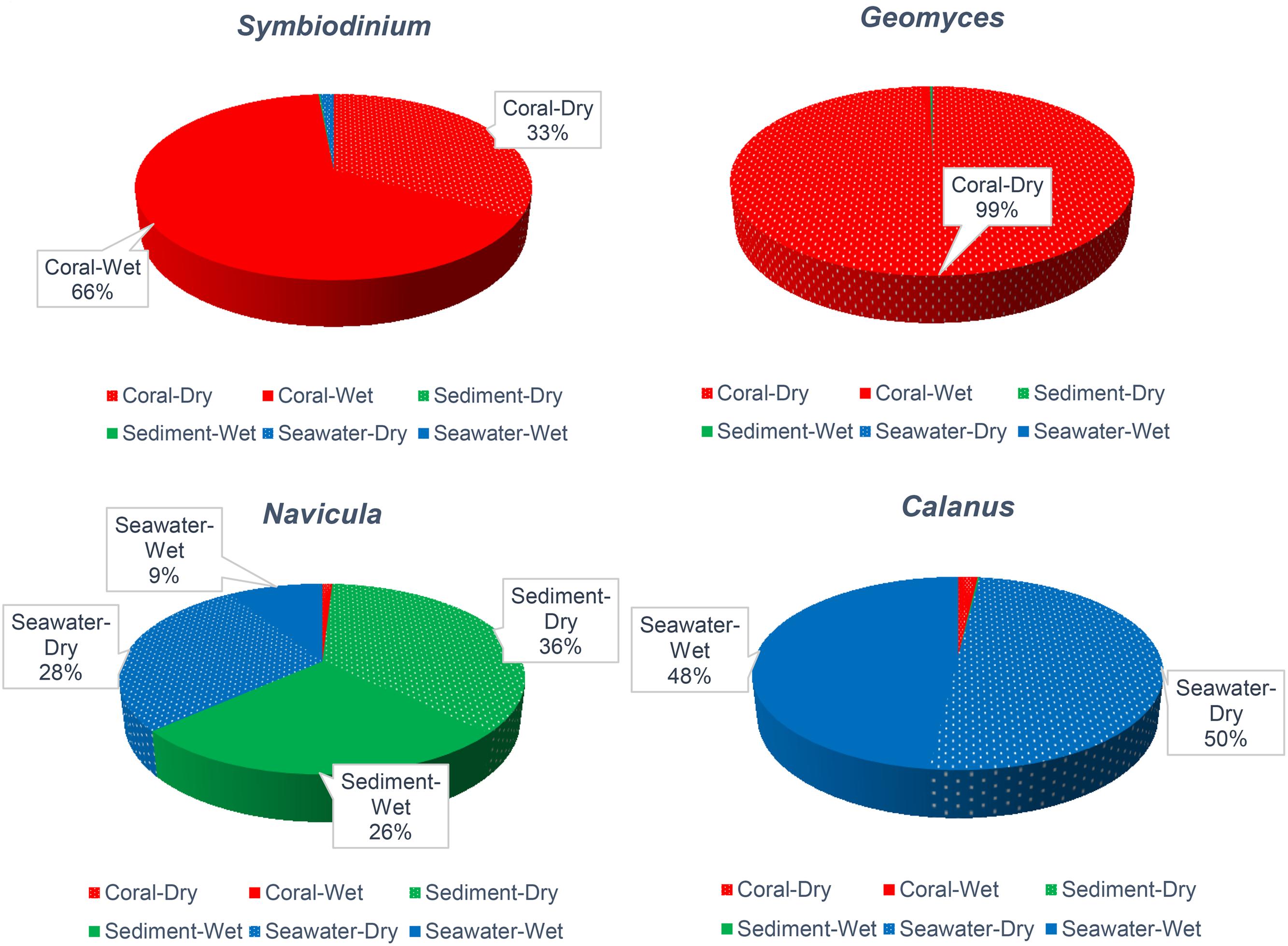
FIGURE 2. Relative abundance of Symbiodinium, Geomyces, Navicula, and Calanus across types of samples and the dry and wet months. Relative abundances <9% are not noted.
The diatom Navicula was relatively abundant in sediment, and the free-living copepod Calanus was relatively abundant in seawater (Figure 2). These organisms may play important roles in the system. Navicula has been reported to play major role in cycling of nitrogen and phosphate (Kwon et al., 2013; Stock et al., 2014), and it is a food source for Calanus, which itself is a food for corals and other animals.
Correlation With Water Quality
Dissolved oxygen, density of water, salinity, pH, and temperature were correlated in the similar vector direction to the coral communities (p < 0.01; Figure 1F). The community diversity were not significantly correlated with numbers of OTUs and water depth. Given a sufficient sequencing depth, additional OTUs should not alter relative abundances substantially, and all samples were collected the similar depth (2–3 m) so the communities would not be expected to exhibit substantial variation.
Conclusion
This study provided initial data on the diversity of microbes and small eukaryotes on coral reefs in the upper GoT. The community on corals was consistently different from the communities in seawater and sediments, and all communities were similar at the three sites (S, K, and T) and in the two periods of sampling. Pearson’s correlations indicated that the relative abundances of various genera were correlated in different vector directions with several metrics characterizing water quality.
Author Contributions
DB did molecular biology experiments and performed the data analysis. AW helped the data analysis. ST and VV conceived the study. SC provided the samples and conceived the study. NS conceived the study, coordinated the experiments and data analysis, and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the 90th Anniversary of Chulalongkorn University Fund, and Chulalongkorn Academic Advancement into its 2nd Century Project, Thailand Research Fund (RSA6080087), Thailand Research Fund (RSA6180046), and EU-Horizon 2020 Project TASCMAR (634674).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledged the Plant Genetic Conservation Project under the Royal Initiative of Her Royal Highness Princess Maha Chakri Sirindhorn, and the Naval Special Warfare Command, Royal Thai Navy for their assistance in the field; and the National Center for Genetic Engineering and Biotechnology (BIOTEC), and the National Science and Technology Development Agency (NSTDA) for allowing us to compute the data on the server. The authors also thank P. Sodsai and W. Poomipak for sequencing advice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00436/full#supplementary-material
FIGURE S1 | Overall eukaryotic distribution by kingdoms (A) and phylum (B,C). In (C), only fungal phyla were displayed.
TABLE S1 | Estimates of sample coverage and diversity indices at the phylum and genus levels for all 18S rDNA profiles (A) and for fungal profiles (B).
TABLE S2 | Student’s t-test to evaluate statistically significant differences in the community structures of genus-level operational taxonomic unit (OTU) compositions between corresponding subgroups.
Footnotes
References
Ainsworth, T. D., Thurber, R. V., and Gates, R. D. (2010). The future of coral reefs: a microbial perspective. Trends Ecol. Evol. 25, 233–240. doi: 10.1016/j.tree.2009.11.001
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Gillevet, P. M., Sikaroodi, M., and Torzilli, A. P. (2009). Analyzing salt-marsh fungal diversity: comparing ARISA fingerprinting with clone sequencing and pyrosequencing. Fungal Ecol. 2, 160–167.
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Khitmoh, K., Wilantho, A., Wongsawaeng, D., Tongsima, S., and Somboonna, N. (2017). 16S rRNA gene sequencing assessment of the prokaryotic communities in the Southeast Andaman Sea, Thailand and potential environmental alerts. Chiang Mai J. Sci. 44, 15–28.
Kwon, H. K., Oh, S. J., and Yang, H. S. (2013). Growth and uptake kinetics of nitrate and phosphate by benthic microalgae for phytoremediation of eutrophic coastal sediments. Bioresour. Technol. 129, 387–395. doi: 10.1016/j.biortech.2012.11.078
Latypov, Y. Y. (2015). Particular qualities of forming of reefs and coral communities in the Gulf of Thailand. Nat. Rev. Microbiol. 5, 1–8.
Mahé, F., Mayor, J., Bunge, J., Chi, J., Siemensmeyer, T., Stoeck, T., et al. (2015). Comparing high-throughput platforms for sequencing the V4 region of SSU-rDNA in environmental microbial eukaryotic diversity surveys. J. Eukaryot. Microbiol. 62, 338–345. doi: 10.1111/jeu.12187
Massana, R., and Pedrós-Alió, C. (2008). Unveiling new microbial eukaryotes in the surface ocean. Curr. Opin. Microbiol. 11, 213–218. doi: 10.1016/j.mib.2008.04.004
Mydlarz, L. D., McGinty, E. S., and Harvell, C. D. (2010). What are the physiological and immunological responses of coral to climate warming and disease? J. Exp. Biol. 213, 934–945. doi: 10.1242/jeb.037580
Phongsuwan, N., Chankong, A., Yamarunpatthana, C., Chansang, H., Boonprakob, R., Petchkumnerd, P., et al. (2013). Status and changing patterns on coral reefs in Thailand during the last two decades. Deep Sea Res. Pt. 2 96, 19–24.
Pootakham, W., Mhuantong, W., Putchin, L., Yocha, T., Sonthirod, C., Kongkachana, W., et al. (2018). Dynamics of coral-associated microbiomes during a thermal bleaching event. Microbiologyopen 23:e00604. doi: 10.1002/mbo3.604
Ramírez, F., Afán, I., Davis, L. S., and Chiaradia, A. (2017). Climate impacts on global hot spots of marine biodiversity. Sci. Adv. 3:e1601198. doi: 10.1126/sciadv.1601198
Ranjard, L., Poly, F., Lata, J. C., Mougel, C., Thioulouse, J., and Nazaret, S. (2001). Characterization of bacterial and fungal soil communities by automated ribosomal intergenic spacer analysis fingerprints: biological and methodological variability. Appl. Environ. Microbiol. 67, 4479–4487.
Riesenfeld, C. S., Schloss, P. D., and Handelsman, J. (2004). Metagenomics: genomic analysis of microbial communities. Ann. Rev. Genet. 38, 525–552.
Rusch, D. B., Halpern, A. L., Sutton, G., Heidelberg, K. B., Williamson, S., Yooseph, S., et al. (2007). The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 5:e77. doi: 10.1371/journal/pbio.0050077
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Somboonna, N., Wilantho, A., Assawamakin, A., Monanunsap, S., Sangsrakru, D., Tangphatsornruang, S., et al. (2014). Structural and functional diversity of free-living microorganisms in reef surface. Kra island, Thailand. BMC Genomics 15:607. doi: 10.1186/1471-2164-15-607
Stock, W., Heylen, K., Sabbe, K., Willems, A., and De Troch, M. (2014). Interactions between benthic copepods, bacteria and diatoms promote nitrogen retention in intertidal marine sediments. PLoS One 9:e111001. doi: 10.1371/journal.pone.0111001
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267.
Wegley, L., Edwards, R., Rodriguez-Brito, B., Liu, H., and Rohwer, F. (2007). Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ. Microbiol. 9, 2707–2719.
Keywords: diversity, fungi, microbiome, 18S rRNA gene, coral
Citation: Bulan DE, Wilantho A, Tongsima S, Viyakarn V, Chavanich S and Somboonna N (2018) Microbial and Small Eukaryotes Associated With Reefs in the Upper Gulf of Thailand. Front. Mar. Sci. 5:436. doi: 10.3389/fmars.2018.00436
Received: 01 April 2018; Accepted: 30 October 2018;
Published: 20 November 2018.
Edited by:
Jamal Ouazzani, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Aldo Cróquer, Simón Bolívar University, VenezuelaCharles Alan Jacoby, St. Johns River Water Management District, United States
Copyright © 2018 Bulan, Wilantho, Tongsima, Viyakarn, Chavanich and Somboonna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naraporn Somboonna, TmFyYXBvcm4uU0BjaHVsYS5hYy50aA== Suchana Chavanich, U3VjaGFuYS5DQGNodWxhLmFjLnRo