 David J. Newman
David J. Newman- Newman Consulting llc, Wayne, PA, United States
The initial sources of marine-derived compounds that on a small scale exhibited interesting biological activities, were often confined to collection levels ranging from <100 grams to a kilogram. Then if significant interest was shown, collections on a larger scale were made of the nominal source organism. Due to many reasons, including but not limited to realization of the environmental harm that can occur, and in some cases, has occurred, over the last 15 plus years, the paradigm moved to semi-synthesis, total synthesis (sometimes of simpler versions) and the use of genomic techniques both to identify the “source” and then to utilize the data in due course. In this short review, examples of how ideas have changed will range from the early days with the arabinose-containing nucleosides, the sourcing and use of didemnins and their derivatives, work with bryostatins, ecteinascidins and halichondrins, and then demonstrate how molecules derived from dolastatins are now used as “warheads.” In most cases the varying methods used will be discussed, albeit in brief in some cases, with the aim of demonstrating how various techniques were used to optimize these compounds and their derivatives. The review finishes with comments as to the current and future involvement of microbes as sources of these and other agents.
Introduction
The aim of this short review is to discuss bioactive compounds from marine organisms, particularly focusing of the growing evidence that microorganisms associated with marine invertebrates, can be sources of the compounds isolated from these organisms. In a significant number of cases, such an involvement has been shown well after the initial reports, and in some cases, well after the compounds entered clinical trials, usually as putative anti-tumor compounds. These reports have led to the view that there is a significant probability that a majority of bioactive compounds are in fact produced by associated microbes, and not solely by the host invertebrate. This may well be the case with sponges, nudibranchs, tunicates, and some bryozoans, though as will be seen later under both the ecteinascidin and bryostatin headings, there is a strong probability that a mutually beneficial synergistic relationship exists between the host and a microbe producing a bioactive agent. The realization that such microbes, whether currently amenable to fermentation or as yet uncultured, are a major biosynthetic source of these bioactive agents, opens up the possibility of a larger scale production by use of suitable biotechnology approaches, aiding to solve the supply problem, and unravel their full potential.
The Arabinose-Containing Nucleosides
Arabinose containing nucleosides represent perhaps the simplest class of drug candidates derived from marine sources. These compounds, basically nucleosides containing arabinose in place of ribose or deoxyribose in nucleosides, were first described in the 1950s by Bergmann’s group and include spongothymidine (Figure 1; 1), spongouridine (Figure 1; 2), and spongosine (2-methoxyadenosine; Figure 1; 3) from the sponge Tethya crypta collected in the Caribbean (Bergmann and Feeney, 1950, 1951; Bergmann and Burke, 1955). Early reports of bioactivity of these compounds include the observation that the uracil requirement in auxotrophs of Escherichia coli could be met by feeding them either spongocytidine (which is deaminated to spongouridine) or by spongouridine (Pizer and Cohen, 1960) and the potential of Ara-A (Figure 1; 4) as a potential antitumor agent (Lee et al., 1960). The first publication demonstrating that Ara-C (Figure 1; 5) had significant biological activity, was the report by Evans et al. (1961) on the antitumor activity of the compound. While these initial arabinose substituted nucleosides have not themselves been developed as drugs, their discovery was likely source of inspiration for workers at the then Burroughs Welcome laboratories, that subsequently led to the other sugar modifications of nucleosides that produced a whole variety of antiviral drugs, previously reviewed by Suckling (1991).
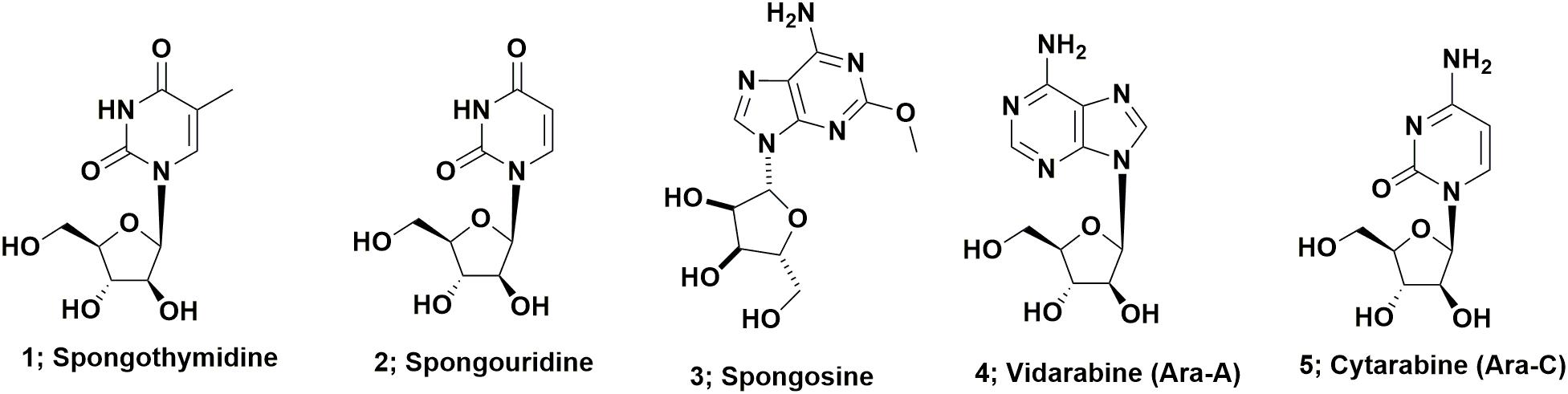
FIGURE 1. Nucleoside derivatives from marine sources.
Initial Synthetic Reports; Spongouridine and Ara-C (1956–1961)
If one searches the synthetic chemistry literature, Brown et al. (1956) described the synthesis of spongouridine (3-β-D-arabofuranosyluracil) from 3′,5′-di-O-acetyluridine via a series of reactions, including a cyclic phosphorylation step, that yielded “spongouridine” identical to the material reported by Bergmann and Burke (1955). Other methods of synthesis of arabino-nucleosides were then reported by Fox et al., 1957 in (spongothymidine) and by Walwick et al. (1959). It is of import that this latter paper described the first synthesis of what subsequently became known as Ara-C (Figure 1; 5) in addition to spongouridine and spongocytidine. Then Reist et al. (1961) further reported syntheses of the “spongo” nucleosides. Another interesting paper in the same time frame, though not in synthetic chemistry, but what was then known as microbial physiology, was the report by Pizer and Cohen (1960), that the requirement for uracil in auxotrophs of Escherichia coli could be met by feeding them either spongocytidine, which was deaminated to spongouridine, or by spongouridine itself, thus “a biological activity” was indirectly shown for such pentoses.
Production of Arabinose-Containing Nucleosides by Microorganisms
A patent report in 1969 by workers at Parke-Davis demonstrated fermentative production of Ara-A by the terrestrial microbe, Streptomyces antibioticus (Davis, 1969). This was followed 15 years later in 1984, by the report by Cimino et al. (1984) of the isolation of both Ara-A, which as mentioned above had previously been reported as a synthetic material by Lee et al. (1960) and spongouridine from the Mediterranean gorgonian coral, Eunicella cavolini. Finally in 2015, the Gerwick group at the Scripps Institute of Oceanography, reported the isolation from a fresh collection of the Caribbean sponge used by Bergmann 55 plus years earlier, of a Vibrio harveyi bacterial strain that when fermented, produced spongosine (Figure 1; 3) plus other nucleosides (Bertin et al., 2015). Thus the possibility exists that most, if not all of the nucleosides reported from this sponge species were produced by microbial endophytes of the sponge, is a reasonable assumption.
Didemnins and Associated Compounds
The depsipeptide, didemnin B (Figure 2; 6) was the first “direct from the sea” compound to go into clinical trials against cancer. It was one of a series of related compounds originally reported in 1981 that were isolated from the Caribbean tunicate Trididemnum solidum by Rinehart et al. (1981a,b). This was the first complex marine-derived compound requiring significant synthetic work in order to obtain enough material to perform preclinical and then clinical trials. Although none were initially suitable for the required scale, a variety of total syntheses were published relatively soon after these initial reports of activity.
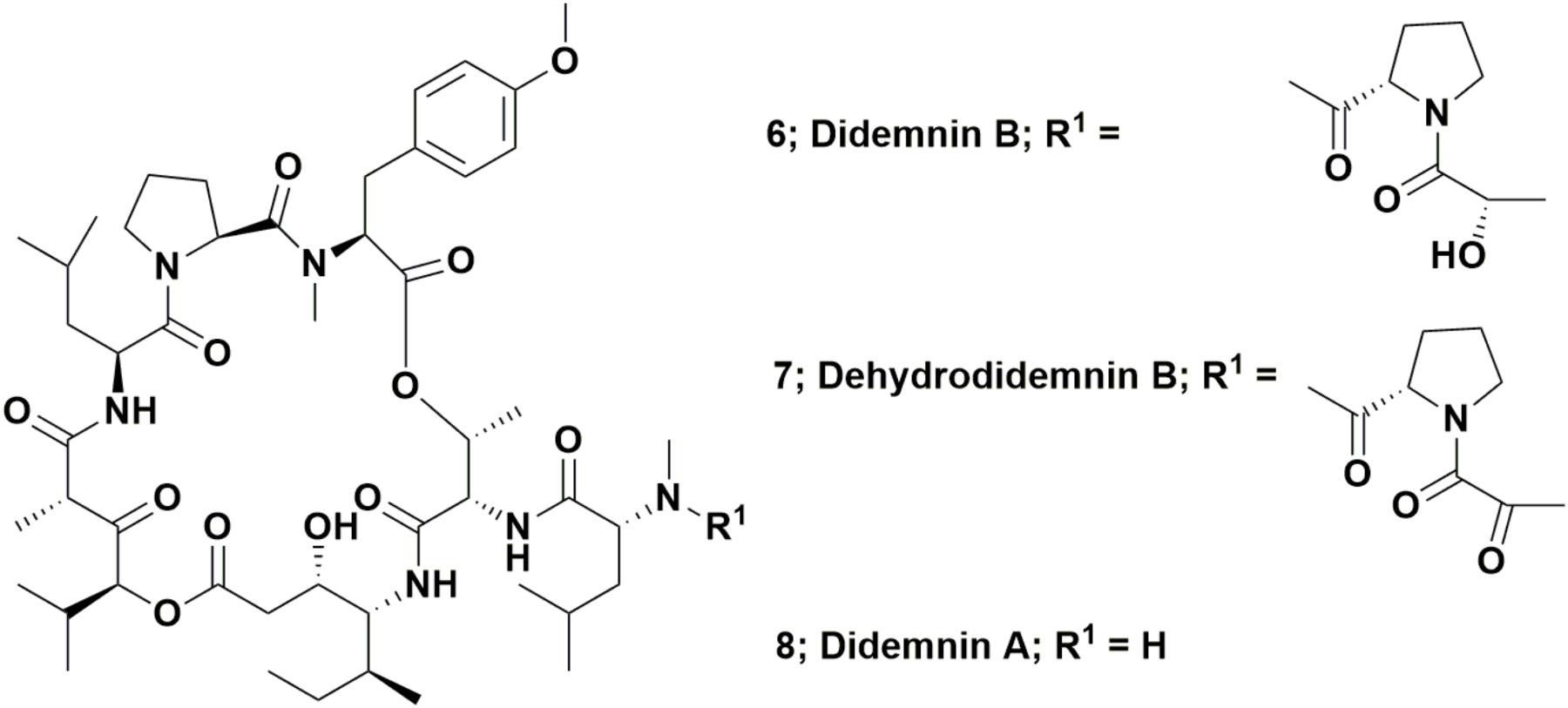
FIGURE 2. Didemnin B and derivatives.
The first synthesis was by the Rinehart et al. (1987). Three years later in 1990, the Joullie group at the University of Pennsylvania published their variation (Li et al., 1990). A paper in 1996 from the Rinehart group extended the initial biological information and gave the first significant details of the structure activity relationships amongst the didemnin series (Sakai et al., 1996). In addition to these earlier syntheses, this class of compounds was and is still of interest to synthetic chemists, as has been shown by the publications that were extensively reviewed by the Joullie group in 2002 (Vera and Joullie, 2002) and 2012 (Lee et al., 2012).
Clinical Fate of Didemnin B (and Future Potential?)
Didemnin B was pulled from clinical trials early in Phase II studies due to its significant toxicity, which with hindsight, might have been partially or totally due to the methodology used in those days [viz: single bolus dose at or close to the maximum tolerated level (MTD) in man]. On a more positive note for these agents, Potts et al. (2015) reported a potential pharmacogenomic method, using biomarkers from patients, that might be capable of identifying patients who might respond positively to treatment by this agent. To date, however, no further clinical work has been reported.
Dehydrodidemnin B (DDB or Aplidine)
Interestingly, the 1996 paper by Sakai et al. (1996) did not cover in any significant detail, a very simple modification of didemnin B, one where the pendant lactyl group was replaced by a pyruvyl group, yielding the molecule known as dehydrodidemnin B or aplidine (Figure 2; 7). There was mention “en passant,” in the paper, but only as a listing to a reference covering a 1991 patent (Rinehart and Lithgow-Bertelloni, 1991). That patent mentioned its isolation from the Mediterranean tunicate Aplidium albicans with the Spanish company PharmaMar being the ultimate assignee.
This compound entered preclinical trials under the aegis of PharmaMar (Rinehart was involved with this company from its start in 1988), with a synthesis of dehydrodidemnin B (DDB) reported in 1997 by a group from the University of Barcelona. Their strategy involved synthesis of didemnin A (Figure 2; 8), followed by production of DDB in an overall yield of 27% (Jou et al., 1997). Though not formally reported as such, this was probably the method used by PharmaMar for production of this agent for preclinical and clinical trials. The initial effects on mammalian cells of this agent were reported by Urdiales et al. (1996). In 2017, a review covering this compound (also known as Plitidepsin®) was published by Alonso-Álvarez et al. (2017) but in March of 2018, the European Medicines Agency (EMA; the EU equivalent to the US FDA) further denied approval of this agent as a treatment for multiple myeloma, by rejecting an appeal by PharmaMar of the 2017 EMA decision not to recommend approval (data from the PharmaMar web site/press release). As of the date of the submission of this review, it is currently in Phase II trials as a treatment for T-cell lymphoma.
Microbial Production of Didemnin B
In 2011, Japanese investigators reported that a free-living marine microbe isolated from Japanese waters, produced didemnin B when fermented (Tsukimoto et al., 2011). This was very surprising, as up until then, no involvement of microbes in the production of these agents had been mentioned, either in the scientific press or at scientific meetings. That this finding was not an isolated incident, was shown the following year, when investigators from Hong Kong, working with free-living microbes of the same genus, but isolated from the Red Sea in collaboration with microbiologists from Saudi Arabia, reported their findings. Following extensive cooperation with groups at the Scripps Institution of Oceanography and the University of California, San Diego, they demonstrated the complete previously proposed biosynthesis cycle for didemnin B, by very clever use of laser desorption mass spectroscopy on microscope plates containing the growing organism (Xu et al., 2012). What is quite interesting is that there have been no reports to date, on any work demonstrating that aplidine (DDB) is also produced by a microbe. Since the only difference is in the pendant prolyl-lactyl group, it is quite possible that such a finding may be reported in due course.
Although it took 30 years from the initial report by the Rinehart laboratory on the didemnins until the “actual” producer was identified, now that it is known, and the proposed biosynthetic pathways identified, the future of these depsipeptides as drug entities is not yet fully resolved. Whether or not didemnin B will again be a viable candidate (see heading above), perhaps produced by fermentative processes, rather than total synthesis, is not yet known. Also, from an ecological aspect, the following interesting questions still need to be answered. Does the tunicate also contain a microbe that produces the didemnin(s); or does it sequester the product from free-living microorganisms, or can the tunicate produce the other didemnin molecules with, or without, any other microbial or host involvement.
Bryostatins and Derivatives
These complex molecules are “temporal contemporaries” of the didemnins, with an indirect report of bryostatin 3 in 1970 (Pettit et al., 1970) and then a more thorough report in 1982 by the Pettit group at Arizona State University, of their isolation and identification of bryostatin 1 (Figure 3; 9) from the fouling bryozoan Bugula neritina (Pettit et al., 1982). Though more than 20 variations on the base structure have been reported over the subsequent years since that 1982 paper, only bryostatin 1 entered any clinical trial.
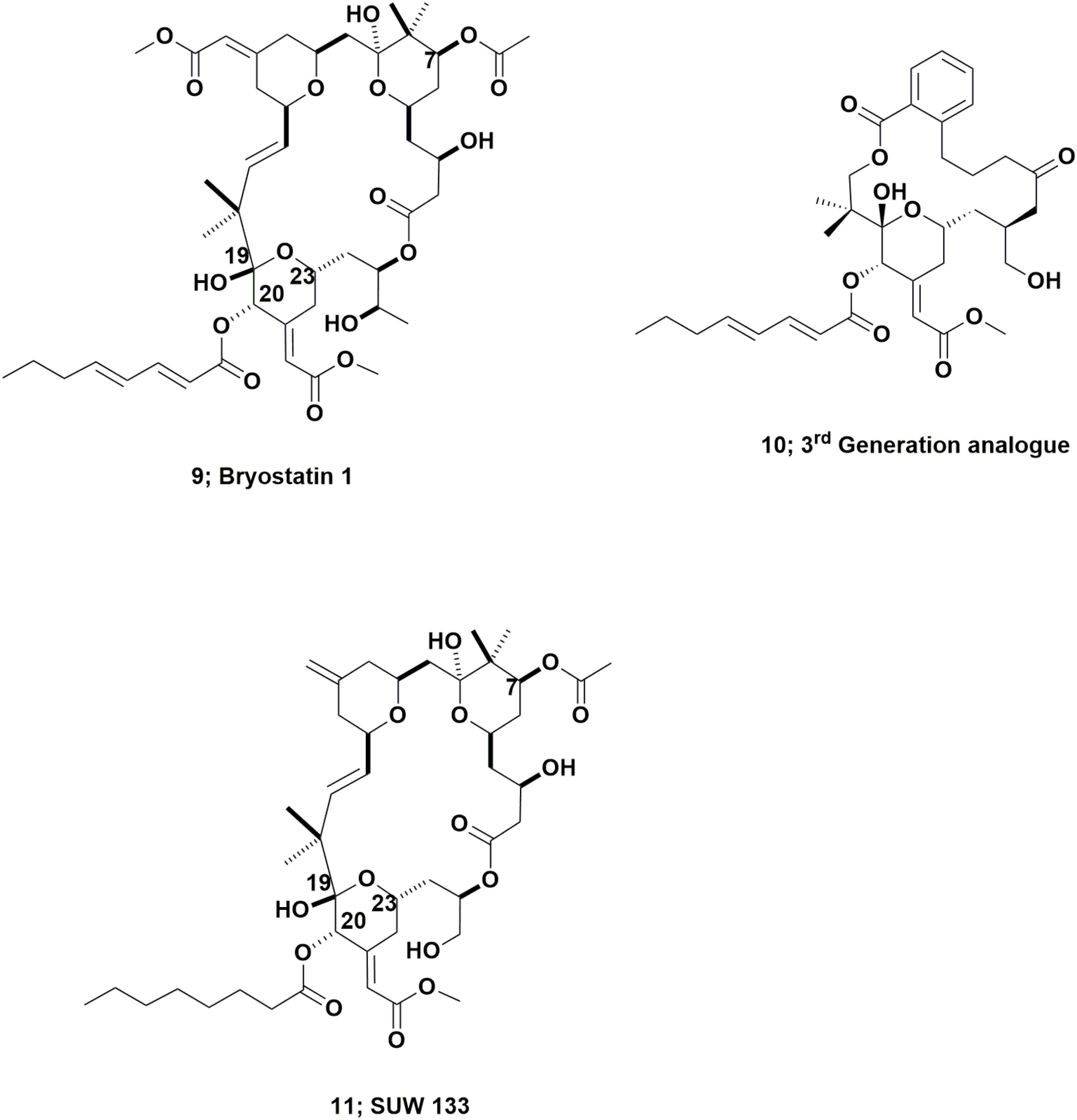
FIGURE 3. Bryostatin 1 and derivatives.
Since the early 1990s, more than 80 clinical trials involving bryostatin 1, ranging from Phases I to II, have been either completed or stopped in man. A major reason for stopping most of the trials, aside from a lack of activity, or other problems found in some studies when combined with other cytotoxic agents, was the rapid onset of severe myalgia in many of the participants. Initially this was thought to be due to the dosing regimen, which as mentioned in the context of the early didemnin B trials referred to above, was a single bolus dose at the MTD. However, even using lower doses and/or spreading out the infusions with time, trials as an antitumor agent have ceased. A listing of the published clinical trials and results from 1993 to 2003 was published in a book chapter by Newman (2005) with an extension to 2010 again published as a book chapter in 2012 (Newman, 2012).
The two chapters just referred to, (Newman, 2005, 2012) in addition to a chapter published in 2016, (Newman and Cragg, 2016) cover the story of bryostatin 1 (and some of the other related compounds) from the aspect of obtaining enough material for clinical trials. These included very large-scale collections undertaken by the US National Cancer Institute (NCI, the largest Institute within the US Government’s National Institutes of Health), that yielded 18 g of cGMP bryostatin 1 from 13000 k of B. neritina collected in waters off California (Schaufelberger et al., 1991). This initial work was then followed by NCI-funded production using both in sea and on land aquaculture techniques as further large-scale collections were not feasible. The possibilities and problems associated with these techniques were later published by the NCI-funded scientist, Mendola, in the early 2000s (Mendola, 2000, 2003).
Chemical Syntheses of Bryostatins and Structural Variations
Though there have been significant numbers of synthetic strategies published over the last 25 plus years by many authors, of both bryostatins and their “truncated” variants, the best “compendium” of syntheses up through early 2010 was published by Hale and Manaviazar (2010). In spite of all the work over the last 25 plus years, no significant synthesis of bryostatin 1, the only bryostatin to enter clinical trials, had been published until 2017. In 2017, the Wender group at Stanford University published a route to the synthetic production of bryostatin 1 on a gram plus scale (Wender et al., 2017).
This work should be read in conjunction with two excellent reviews published by Wender et al. (2014, 2015), that demonstrated the ability, using modern synthetic techniques, to produce very active bryostatin variants, with an example being the third generation analog (Figure 3; 10). As a result of having readily available bryostatin 1, Wender and collaborators have now demonstrated that this bryostatin 1 and subtle variations such as SUW 133 (Figure 3; 11), can release latent HIV from cell depots in patients (Albert et al., 2017; Marsden et al., 2017).
Is an Uncultured Microbe the “True Bryostatin Producer?”
Anthoni et al. (1990) suggested from inspection of the structures of metabolites from bryozoans, that these might have been produced by a microbe within the organism. Haygood and Davidson (1997) published evidence demonstrating that the bryostatins found in B. neritina were possibly produced by an uncultured microbe. This microbe was identified in the organism and named as Candidatus Endobugula sertula (Haygood and Davidson, 1997). Later work by the same authors showed that there was a spatial (depth) differentiation, even in the same collection site, as to which bryostatins were found from the nominally same host organism (Davidson and Haygood, 1999). This was a very interesting finding, as the bryostatin 1 producers were below 9 m, whereas the major collection for NCI referred to earlier, combined B. neritina samples from shallow to deeper depths, with a majority above 9 m.
Work reported in 2001 from the Haygood group elaborated further on the biosynthetic mechanisms in the microbe/bryozoan complex (Davidson et al., 2001). Also of significant import, was the report by Lopanik et al. (2004) demonstrating that bryostatin was acting as a defense agent against predators, particularly at the larval stage, Further work from the Haygood group (California), the Lopanik group (Carolinas), both cooperating with the Sherman group (Minnesota/Michigan) identified the biosynthetic cluster in the microbe, with a publication in 2007 by Sudek et al. (2007). These studies were extended over the next 3 years, with further work reported by Lopanik et al. (2008) and by Buchholz et al. (2010). Taken together, the full biosynthetic scheme was identified by using information from the microbial genes, though the organism was not and still has not been cultivated.
The Current Status of Bryostatins
It should be emphasized that bryostatin 1 is still one of the most potent biological agents tested in antitumor clinical trials, and as mentioned earlier, though the material used in trials sponsored by the NCI, initially came by isolation from massive shallow-water collections off the coast of California, such efforts are no longer necessary. Since synthetic bryostatin 1 and derivatives are now available on a reasonable scale (Wender et al., 2014, 2015) with these new synthetic techniques, coupled to the ability to make modifications easily, then the biological activities of this class of compounds will definitely increase. As an example, recently there were reports of activity against HIV virus that has been “sequestered” in dendritic cells (Albert et al., 2017; Marsden et al., 2017). To finish this section, although the biosynthetic gene clusters have been identified, no work has yet been reported on cloning of these clusters in a surrogate host, and then coupled to production by fermentation. Thus, use of genetic manipulation to produce quantities of bryostatin (and biosynthetic precursors) is still to be demonstrated.
Ecteinascidin 743
In 1969, Sigel et al., reported that the Caribbean tunicate, Ecteinascidia turbinata exhibited cytotoxic activity against mammalian cells. This report was often missed as it was initially an oral presentation at a conference, followed by a paper (effectively the conference proceedings), published the following year as a book chapter (Sigel et al., 1970). In 1986, Holt, a graduate student in the Rinehart laboratory at the University of Illinois-Champaign, published the initial structural work on these agents from this source as part of his Ph.D. thesis, and named them as the ecteinascidins (Holt, 1986). Following up on this initial report, in 1990, two cooperating groups, the Rinehart laboratory and the Wright group in Florida, published back to back papers in the Journal of Organic Chemistry. Both providing details of the structure of the cytotoxic agent ET743 (Figure 4; 12), and closely related compounds (Rinehart et al., 1990; Wright et al., 1990).
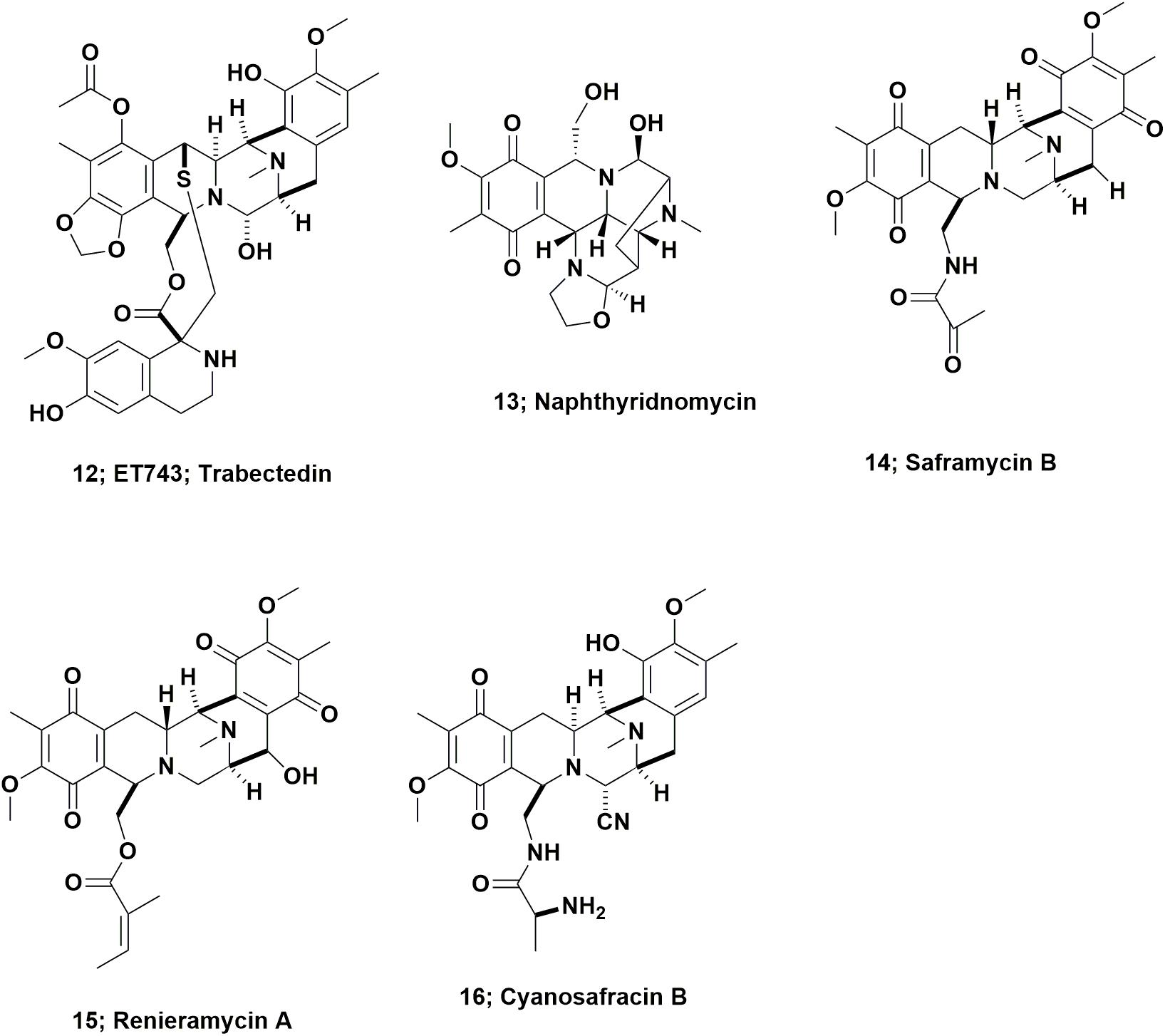
FIGURE 4. Ecteinascidin 743 and derivatives.
From inspection of the structure of ET743 (Figure 4; 12) and related compounds, it was obvious that these compounds were from the same basic structure as reported for naphthyridinomycin (Figure 4; 13) and saframycin B (Figure 4; 14), both originally isolated from terrestrial microbes, and renieramycin A (Figure 4; 15), which was reported from an Eastern Pacific sponge source. Details of these base structures and other related compounds were discussed at length in the 2002 review by Scott and Williams (2002) and significantly extended in the 2015 review by Le et al. (2015). As a result, one now had multiple bioactive compounds that must have been produced by a similar set of biosynthetic clusters, though it was unknown at the time what the organism or organisms might be, but microbes were thought to be prime candidates.
ET743 subsequently became the first approved antitumor drug that came “direct from the sea” under the aegis of the Spanish company PharmaMar. To obtain enough material for preclinical and clinical trials up to Phase II, the methods used by PharmaMar for its production initially involved massive large-scale collections in the Caribbean and the Mediterranean seas. The methods then progressed to tunicate aquaculture in seas and in lakes, which together produced over 100 metric tons of the tunicate, yielding 100 g of ET743 for clinical trials up to Phase II (Cuevas and Francesch, 2009). The method used for later clinical trials, and then production as an approved drug, was partial synthesis from the marine derived bacterial product, cyanosafracin B (Figure 4; 16). The story leading to the production of ET743 has been presented by a number of investigators, but the publications of import are those from the PharmaMar team (Cuevas et al., 2000, 2012; Menchaca et al., 2003; Cuevas and Francesch, 2009).
Microbial Involvement in ET743 Biosynthesis
There were reports dating from 2003 that the uncultured bacterium, Candidatus Endoecteinacidia frumentenis (AY054370), was involved in the production of these molecules. This organism was found in E. turbinata samples that produced ET743, whether they were collected in the Caribbean or the Mediterranean seas (Moss et al., 2003; Parez-Matos et al., 2007). Using this information, in 2011, the Sherman group at the University of Michigan were able to identify the “contig” that encoded the NRPS biosynthetic enzymes involved in the ET743 complex. In addition, they identified the probable producer as the uncultured microbe Candidatus Endoecteinascidia frumentensis (Rath et al., 2011). In order to identify these gene cluster(s), the Sherman group used experimental “suggestions” made by Piel (2006), to look at the possibility of a microbe being involved in the actual production of an agent “in situ.” They then coupled these concepts to knowledge of the organization of the biosynthetic gene clusters (BCGs) of saframycin A (Li et al., 2008) and safracin B, (Velasco et al., 2005) as markers to derive their identification.
Four years later, the Sherman group confirmed their initial report, (Schofield et al., 2015) and demonstrated that the producing bacterium, Cand. E. frumentensis, probably represents a member of a new family of Gammaproteobacteria. In addition, it has a “streamlined genome” analogous to what has been reported from other symbiotic microbes (McCutcheon and Moran, 2012). In those symbionts, most of the genetic machinery is devoted to production of the complex of compounds, which in turn acts as a protective agent for the tunicate (Kehr and Dittmann, 2015). This is similar in principle to what was described for the putative bryostatin producers described above.
Halichondrin B and Eribulin
The “modified “half-halichondrin B” molecule now known as eribulin is the most complex molecule ever directly synthesized as a drug candidate and may never be equaled as a synthetic drug, excluding polypeptides for which syntheses are straightforward. The marine natural product halichondrin B (Figure 5; 17) was first reported by Hirata and Uemura (1986) from a 600 kg collection of the Japanese sponge Halichondria okadai, though a close relative, norhalichondrin A had been reported the previous year from the same sponge (Uemura et al., 1985). What should also be noted is that okadaic acid was also produced by the same sponge, and it is highly probable that some of this very potent material was co-isolated with the halichondrin.
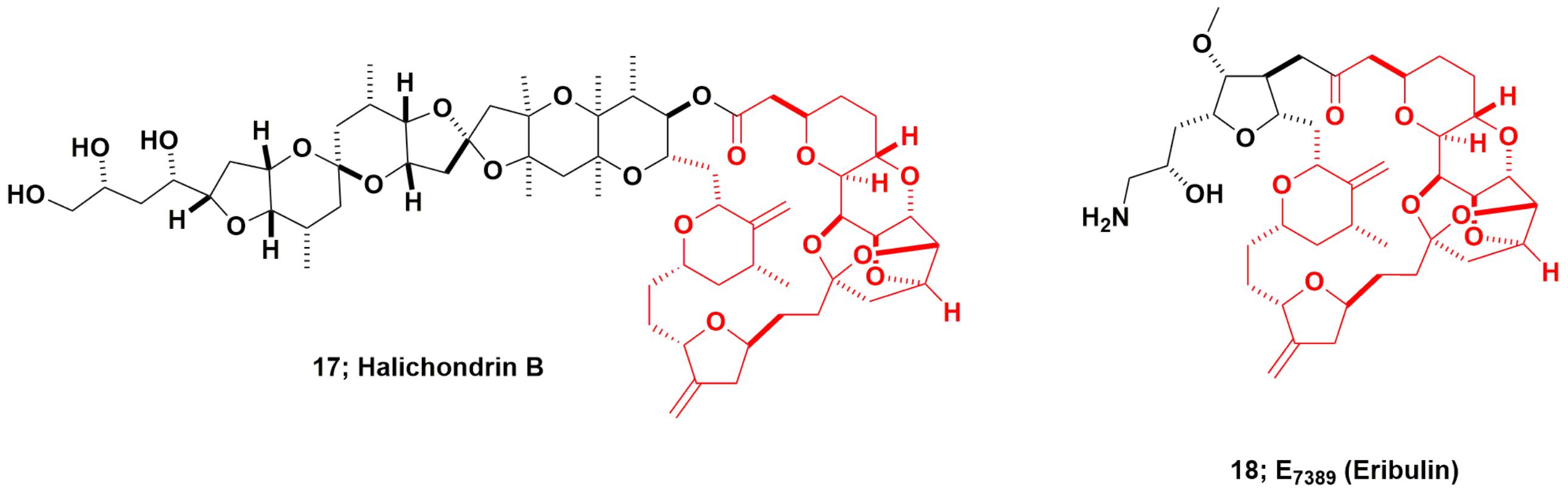
FIGURE 5. Halichondrin B and derivatives.
The Pettit group at Arizona State University, and the Blunt and Munro group at the University of Canterbury in New Zealand, later reported the same compound from different sponge genera collected in different parts of the Pacific Ocean. Using Pettit-supplied material, NCI reported its excellent anti-tubulin activity, (Bai et al., 1991) and subsequently, the compound was chosen for preclinical development in 1992 by the Developmental Therapeutics Program (DTP) at the NCI. The initial preclinical development was severely impeded due to the very limited amounts of compound available from natural sources, with very low milligram amounts being available.
Large-Scale Sponge Collections and Work-Up
In order to obtain enough material for initial preclinical work, NCI funded a 1 metric ton collection of a Lissodendoryx species from over 100 m depth off South Island, New Zealand. This multidisciplinary group, composed of chemists, marine biologists, marine engineers, researchers at the University of Canterbury together with New Zealand government agencies, produced 300+ milligrams of pure halichondrin B, minus any okadaic acid contamination. In order to develop a renewable resource for the future, during this isolation work, successful shallow water aquaculture of the sponge was also funded by NCI. Data from these studies demonstrated that halichondrin B could be produced by in sea aquaculture at 10 m depth yielding roughly 50% of the amount found in the deep-water samples (Munro et al., 1999).
Chemical Synthesis of Halichondrin B
Kishi’s group at Harvard, funded by NCI some years before the 1991 report of tubulin activity (Bai et al., 1991) reported a total synthesis for halichondrin B in 1992, giving less than 1% overall yield (Aicher et al., 1992). In 1992, when approval was given by the then NCI Decision Network Committee to proceed with preclinical studies on halichondrin B (the author was the DTP scientist tasked in 1992 with procuring quantities of pure halichondrin B), NCI did not know that Kishi, in collaboration with scientists at the Eisai Research Institute (ERI) in Woburn, MA, United States, demonstrated that the right-hand (macrolide) half of the molecule (approx MW of 600), retained most of the potency of the much larger parent compound. This work was later published as part of US Patent 5436238 in 1995 (Kishi et al., 1995).
As reported much later, ERI, working very closely with Kishi’s group, then synthesized over 200 analogs of the truncated form of halichondrin B (Yu et al., 2012, 2013). Once this was known by DTP from presentations at the American Association for Cancer Research (AACR) by both Eisai (analog data) and DTP (pure halichondrin B data), this led to a collaboration in the1998–99 timeframe, using the pure New Zealand sourced material as a control. From these collaborative experiments, DTP and ERI scientists demonstrated that the modified truncated macrocyclic ketone, eribulin (E7389; Figure 5; 18), was a molecule that had greater in vivo stability, possessed comparable bioactivity to, and lower toxicity in vivo than halichondrin B. This work was not published as a single document but was presented as posters and oral presentations at AACR meetings, with a precis on the NCI/DTP web page at the URL https://dtp.cancer.gov/timeline/flash/success_stories/s4_halichondrinb.htm. A decision was then made in 2000 by NCI to further develop eribulin as a potential antitumor drug in conjunction with Eisai/ERI.
Between 2006 and 2011, reports that included biological activities, the probable site(s) of interaction (Dabydeen et al., 2006; Bai et al., 2011) and an initial review of the basic chemistry involved (Jackson et al., 2009) were published. However, the most important publications were three papers in 2013 from the Eisai process chemistry group, covering the methods used on a large scale under cGMP conditions (Chase et al., 2013; Austad et al., 2013a,b). These should be consulted for further information by interested readers, as they demonstrate the power of very clever synthetic chemistry to produce a viable antitumor agent.
Microbial Involvement in Halichondrin Biosynthesis
To date, no publications have surfaced that implicate any organism other than the host sponge in the production of this class of compounds in the marine environment. However, inspection of the basic structure(s) coupled to the presence of okadaic acid in the original sponge, suggests that dinoflagellates might be a possible source of compounds such as the halichondrins, but no hard evidence has been published. What is of interest, however, is that in the original paper by Hirata and Uemura (1986) they remarked upon the large number and variety of single-celled organisms found with the sponge.
Dolastatin Derivatives as “Warheads”
Monoclonal Antibodies-Natural Product Conjugates as Antitumor Drug Candidates
As found the case of dolastatin 10, when used as drug entities, some natural products, either “as is” or “slightly modified,” frequently resulted in toxicity due to their exquisite potencies, but usually low solubility in aqueous solutions. This was due to inadequate delivery to the required site of activity within the human body, coupled to less specific undesired interactions with other cellular processes. One potential method of overcoming (ameliorating) this problem, that has now come to the fore, is to utilize a combination of the “directional capability of monoclonal antibodies (mAbs)” and the innate activity of a very potent natural product. Early attempts coupled microbial toxins to mAbs, but today, relatively simple compounds (at least in comparison) based on natural products, or subtle chemical modifications thereof are used as the warhead.
Though not marine-sourced, the first ADC approved by the FDA in 2000 (Mylotarg®) used a modified calicheamicin (sourced from a terrestrial microbe) as the warhead. This was subsequently withdrawn in the United States, though still used in Japan. The next non-marine construct to be approved in 2013, was the anti-breast cancer ADC Kadcyla®, though it used the nominal plant-sourced compound, maytansine as the warhead. Maytansine is now known to be microbial in origin as it originates from an endophyte interaction in the roots of the plant (Kusari et al., 2014). In 2017, a new calicheamicin construct (Besponsa®) was approved by the FDA and Mylotarg® was reintroduced in the United States.
From the marine environment, dolastatin 10 (Figure 6; 19) which was originally isolated from the marine nudibranch Dolabella auricularia, entered up to Phase II clinical trials as the basic compound, but was dropped due a combination of toxicity and lack of efficacy. This agent (and other dolastatins) had to be synthesized due to the very low levels present in the nominal source, the nudibranch, D. auricularia. Today, however, it has been proven to be produced by a cyanobacterium of the genus Symploca that the sea hare consumes as part of its diet. Even though from a microbial source, the dolastatin-compounds required are much simpler to synthesize, as they are peptidic in nature, rather than attempt to modify cyanobacterium-sourced starting materials which would require significant purification before use.
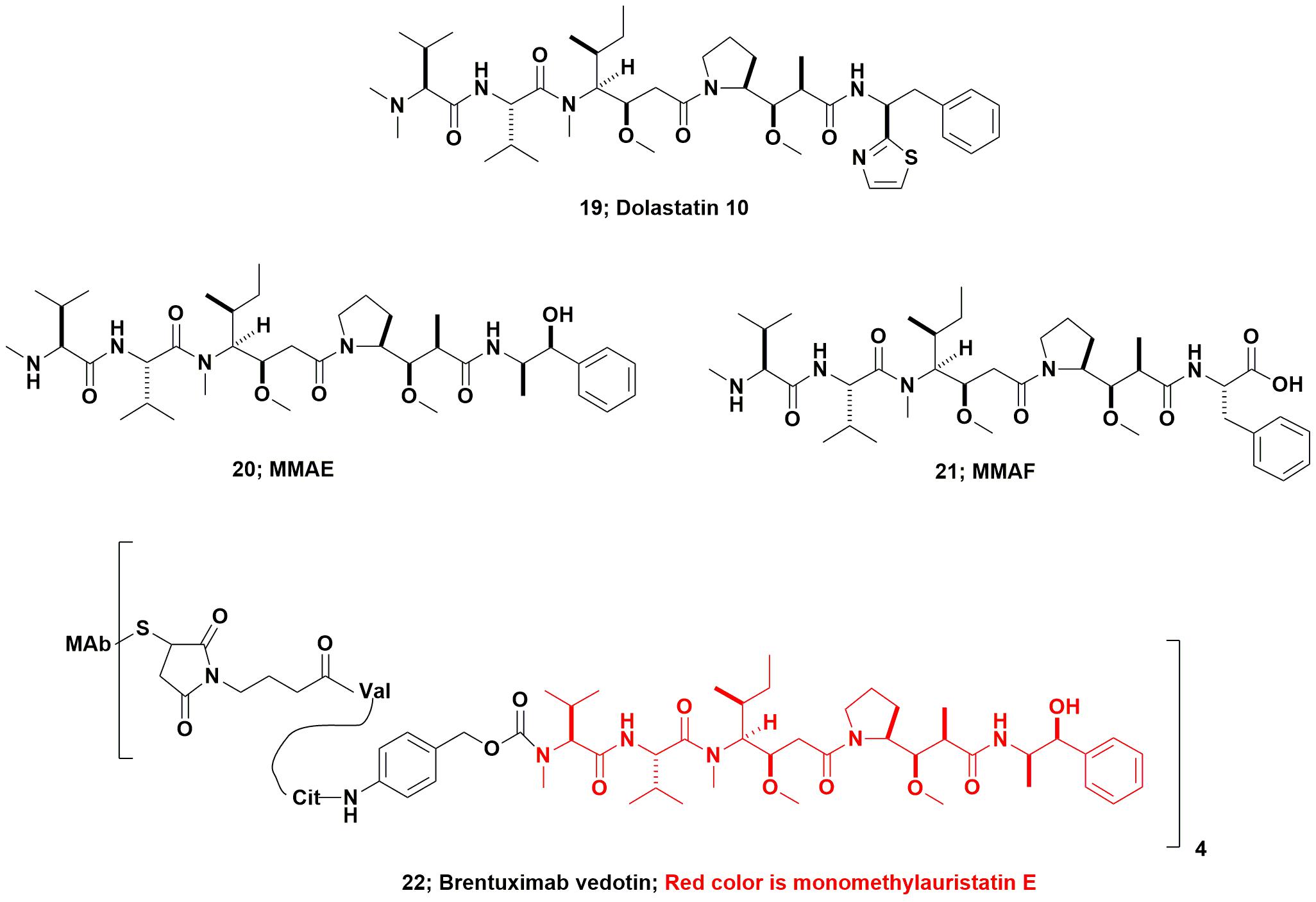
FIGURE 6. Dolastatin 10 and derivatives.
Using dolastatin 10’s basic structure, the Seattle Genetics group produced the linear peptides now known as monomethyl auristatin E (MMAE; Figure 6; 20), and monomethyl auristatin F (MMAF; Figure 6; 21) as warhead candidates. In 2011 the FDA approved the first marine-based ADC Adcetris® (Brentuximab vedotin, Figure 6; 22) and an excellent description of its development was published in 2012 by the Seattle Genetics inventors (Senter and Sievers, 2012). As a result, even compounds that had been effectively discarded as single agents many years ago, now have a new lease on life as ADC warheads. The relevant details and discussion of most of the then current marine-based warheads were given in a review by Newman and Cragg (2017). Further coverage of these and other “warhead sources,” was discussed the same year in a review by Beck et al. (2017).
Of the six ADCs that are currently in Phase III trials against cancer, two, Polatuzumab vedotin and Enfortumab vedotin, use monomethyl auristatin E (MMAE). The third, Depatuxizumab mafodotin) uses monomethyl auristatin F (MMAF). The other three constructs at Phase III, use materials originally from terrestrial microbes. Of the 20 constructs currently in Phase II trials, six can be identified as using vedotins or variations thereof. Moving to those in Phase I and Phase I/II trials, one uses eribulin (Figure 5; 18) and others use a variety of agents based upon other variants of the dolastatin/vedotin structures. As they move into higher phases of clinical trials, more details should be available.
Closing Comments
In this short review, it has hopefully become obvious that the development of a pharmaceutical agent from marine sources is a very long process, involving marine biologists, natural product isolation chemists, microbiologists, pharmacologists, and very competent process chemists, all working in harmony. Although one may initially isolate a few milligrams of an agent from the marine source, currently, even with the advances in genomics and biotechnology, fermentative production on a large enough scale for drug development of the final desired product, is rare. However, as was shown above, in the case of Ecteinascidin 743, semi-synthesis from a marine-sourced bacterium is feasible. In some situations that involve cyanobacteria, however, direct fermentation of axenic cultures may provide reasonable quantities of material, allowing for significant progress to be achieved, but currently this has not proved to be the case for any drug candidate still in clinical trials.
From the perspective of utilization of genomic information, it is only relatively recently that the potential influence of “yet uncultured microbes” has been shown. Though not utilizing genetic data from complete genomes of yet uncultured microbes, the work by the Brady laboratory on recovery of soil metagenomic information and subsequent production in heterologous hosts should be noted at this stage (Chang and Brady, 2011; Katz et al., 2016).
From the aspect of working with the complete genome expressed from an as yet uncultured microbe, the seminal work from the Piel group demonstrating that the true source(s) of significant numbers of the cytotoxic agents discovered from Theonella swinhoei Y, was an uncultured bacterium, was a major advance in the field (Wilson et al., 2014). Further developments may lead to the ability to utilize genetic manipulation techniques to “recover” agents encoded in the genome of currently uncultured marine microbes using complete sequences as found by the Piel group. The aim then would be to have these genetic elements expressed in heterologous hosts, but to date, this has not been reported. However, the following papers, all from the Piel group (from 2013 to 2018) give an idea of where this field may be headed in the near future (Wilson and Piel, 2013; Freeman et al., 2016, 2017; Mori et al., 2018; Morinaka et al., 2018).
Finally three publications of interest that should be consulted, as they further demonstrate the potential for use of genomic information from as yet uncultured microbes, as distinct from genetic clusters from soil metagenomes, are a recent chapter by the Piel group, (Bhushan et al., 2017) the review by Blockley et al. (2017), on symbiotic microbes and their host interactions, and finally the review by Morita and Schmidt covering “parallel lives between host and symbiont microbes” (Morita and Schmidt, 2018).
Author Contributions
DN conceived and wrote the whole submission.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author is the sole proprietor of Newman Consulting llc, a single person operation in the United States. No funds from this entity were used in the production of this article.
References
Aicher, T. D., Buszek, K. R., Fang, F. G., Forsyth, C. J., Jung, S. H., Kishi, Y., et al. (1992). Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 114, 3162–3164. doi: 10.1021/ja00034a086
Albert, B. J., Niu, A., Ramani, R., Marshall, G. R., Wender, P. A., Williams, R. M., et al. (2017). Combinations of isoform-targeted histone deacetylase inhibitors and bryostatin analogues display remarkable potency to activate latent Hiv without global T-cell activation. Sci. Rep. 7:7456. doi: 10.1038/s41598-017-07814-4
Alonso-Álvarez, S., Pardal, E., Sánchez-Nieto, D., Navarro, M., Caballero, M. D., Mateos, M. V., et al. (2017). Plitidepsin: design, development, and potential place in therapy. Drug Des. Devel. Thera. 11, 253–264. doi: 10.2147/DDDT.S94165
Anthoni, U., Nielsen, P. H., Periera, M., and Christophersen, C. (1990). Bryozoan secondary metabolites: a chemotaxonomical challenge. Comp. Biochem. Physiol. 96B, 431–437. doi: 10.1016/0305-0491(90)90035-R
Austad, B. C., Benayoud, F., Calkins, T. L., Campagna, S., Chase, C. E., Choi, H.-W., et al. (2013a). Process development of Halaven®: Synthesis of the C14–C35 fragment via iterative Nozaki–Hiyama–Kishi reaction–Williamson ether cyclization. Synlett 24, 0327–0332.
Austad, B. C., Calkins, T. L., Chase, C. F., Fang, F. G., Horstmann, T. E., Hu, Y., et al. (2013b). Commercial manufacture of Halaven: Chemoselective transformations en route to structurally complex macrocyclic ketones. Synlett 24, 0333–0337.
Bai, R., Nguyen, T. L., Burnett, J. C., Atasoylu, O., Munro, M. H. G., Pettit, G. R., et al. (2011). Interactions of halichondrin B and eribulin with tubulin. J. Chem. Inf. Model. 51, 1393–1404. doi: 10.1021/ci200077t
Bai, R., Paull, K. D., Herald, C. L., Malspeis, L., Pettit, G. R., and Hamel, E. (1991). Halichondrin B and homohalichondrin B, Marine natural products binding in the vinca domain of tubulin: discovery of tubulinbased mechanism of action by analysis of differential cytotoxicity data. J. Biol. Chem. 266, 15882–15889.
Beck, A., Goetsch, L., Dumontet, C., and Corvaïa, N. (2017). Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Revs. Drug Discov. 16, 315–337. doi: 10.1038/nrd.2016.268
Bergmann, W., and Burke, D. C. (1955). Marine products. Xxxix. The nucleosides of sponges. Iii. Spongothymidine and spongouridine. J. Org. Chem. 20, 1501–1507. doi: 10.1021/jo01128a007
Bergmann, W., and Feeney, R. J. (1950). Isolation of a new thymine pentoside from sponges. J. Am. Chem. Soc. 72, 2809–2810. doi: 10.1021/ja01162a543
Bergmann, W., and Feeney, R. J. (1951). Marine products. Xxxii. The nucleosides of sponges. I. J. Org. Chem. 16, 981–987. doi: 10.1021/jo01146a023
Bertin, M. J., Schwartz, S. L., Lee, J., Korobeynikov, A., Dorrestein, P. C., Gerwick, L., et al. (2015). Spongosine production by a Vibrio harveyi strain associated with the sponge Tectitethya crypta. J. Nat. Prod. 78, 493–499. doi: 10.1021/np5009762
Bhushan, A., Peters, E. E., and Piel, J. (2017). “Entotheonella bacteria as source of sponge-derived natural products: Opportunities for biotechnological production,” in Blue Biotechnology Progress in Molecular and Subcellular Biology, Vol. 55, eds W. Müller, H. C. Schröder, and X. Wang (Berlin: Springer International Publishing), 291–314. doi: 10.1007/978-3-319-51284-6_9
Blockley, A., Elliott, D. R., Roberts, A. P., and Sweet, M. (2017). Symbiotic microbes from marine invertebrates: driving a new era of natural product drug discovery. Diversity 9:49. doi: 10.3390/d9040049
Brown, D. M., Todd, A. T., and Varadarajan, S. (1956). Nucleotides. Part Xxxvii. The structure of uridylic acids a and b, and a synthesis of spongouridine (3-beta-D-arabofuranosyl-uracil). J. Chem. Soc. 1956, 2388–2392. doi: 10.1039/JR9560002388
Buchholz, T. J., Rath, C. M., Lopanik, N. B., Gardner, N. P., Hakansson, K., and Sherman, D. H. (2010). Polyketide b-branching in bryostatin biosynthesis: Identification of surrogate acetyl-Acp donors for BryR, an Hmg-Acp synthase. Chem. Biol. 17, 1092–1100. doi: 10.1016/j.chembiol.2010.08.008
Chang, F.-Y., and Brady, S. F. (2011). Cloning and characterization of an environmental Dna-derived gene cluster that encodes the biosynthesis of the antitumor substance Be-54017. J. Am. Chem. Soc. 133, 9996–9999. doi: 10.1021/ja2022653
Chase, C. E., Fang, F. G., Lewis, B. M., Wilkie, G. D., Schnaderbeck, M. J., and Zhu, X. (2013). Process development of Halaven®: synthesis of the C1–C13 fragment from D-(–)-gulono-1,4-lactone. Synlett 24, 0323–0326. doi: 10.1055/s-0032-1317919
Cimino, G., De Rosa, S., and De Stefano, S. (1984). Antiviral agents from a gorgonian. Eunicella Eavolini. Exp. 40, 339–340.
Cuevas, C., and Francesch, A. (2009). Development of Yondelis (trabectedin, Et-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 26, 322–337. doi: 10.1039/b808331m
Cuevas, C., Francesch, A., Galmarini, C. M., Aviles, P., and Munt, S. (2012). “Ecteinascidin-743 (Yondelis(R)). Aplidin(R), and Irvalec(R),” in Anticancer Agents from Natural Products, 2nd Edn, eds G. M. Cragg, D. G. I. Kingston, and D. J. Newman (Boca Raton, FL: Taylor and Francis), 291–316.
Cuevas, C., Perez, M., MartiN, M. J., Chicharro, J. L., Fernandez-Rivas, C., Flores, M., et al. (2000). Synthesis of Ecteinascidin Et-743 and Phthalascidin Pt-650 from Cyanosafracin B. Org. Lett. 2, 2545–2548. doi: 10.1021/ol0062502
Dabydeen, D. A., Burnett, J. C., Bai, R., Verdier-Pinard, P., Hickford, S. J. H., Pettit, G. R., et al. (2006). Comparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulin. Mol. Pharmacol. 70, 1866–1875. doi: 10.1124/mol.106.026641
Davidson, S. K., Allen, S. W., Lim, G. E., Anderson, C. M., and Haygood, M. G. (2001). Evidence for the Biosynthesis of Bryostatins by the Bacterial Symbiont, “Candidatus Endobugula sertula” of the Bryozoan Bugula neritina. Appl. Environ. Microbiol. 67, 4531–4537. doi: 10.1128/AEM.67.10.4531-4537.2001
Davidson, S. K., and Haygood, M. G. (1999). Identification of sibling species of the bryozoan Bugula neritina that produce different anticancer bryostatins and harbor distinct strains of the bacterial symbiont “Candidatus Endobugula sertula”. Biol. Bull. 196, 273–280. doi: 10.2307/1542952
Evans, J. S., Musser, E. A., Mengel, G. D., Forsbled, K. R., and Hunter, J. H. (1961). Antitumor activity of 1-p-D-arabinofuranosylcytosine hydrochloride. Proc. Soc. Exp. Biol. Med. 106, 350–353. doi: 10.3181/00379727-106-26335
Fox, J. J., Yung, N., and Bendich, A. (1957). Pyrimidine nucleosides. II. The synthesis of 1-beta-D-arabinofuranosylthymine (“Spongothymidine”). J. Am. Chem. Soc. 79, 2775–2778. doi: 10.1021/ja01568a030
Freeman, M. F., Helf, M. J., Bhushan, A., Morinaka, B. L., and Piel, J. (2017). Seven enzymes create extraordinary molecular complexity in an uncultivated bacterium. Nat. Chem. 9, 387–395. doi: 10.1038/nchem.2666
Freeman, M. F., Vagstad, A. L., and Piel, J. (2016). Polytheonamide biosynthesis showcasing the metabolic potential of sponge-associated uncultivated ‘Entotheonella’ bacteria. Curr. Opin. Chem. Biol. 31, 8–14. doi: 10.1016/j.cbpa.2015.11.002
Hale, K. J., and Manaviazar, S. (2010). New approaches to the total synthesis of the Bryostatin antitumor macrolides. Chem. Asian J. 5, 704–754. doi: 10.1002/asia.200900634
Haygood, M. G., and Davidson, S. K. (1997). Small subunit ribosomal RNA genes and in situ hybridization of the bacterial symbionts in the larvae of the bryozoan Bugula neritina and proposal of “Candidatus Endobugula sertula”. Appl. Environ. Microbiol. 63, 4612–4616.
Hirata, Y., and Uemura, D. (1986). Halichondrins - antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 58, 701–710. doi: 10.1016/j.bmc.2008.10.093
Holt, T. G. (1986). The Isolation and Structural Characterization of the Ecteinascidins. Ph.D. thesis, University of Illinois at Urbana-Champaign, Champaign, IL.
Jackson, K. L., Henderson, J. A., and Phillips, A. J. (2009). The halichondrins and E7389. Chem. Rev. 109, 3044–3079. doi: 10.1021/cr900016w
Jou, G., Gonzalez, I., Albericio, F., Lloyd-Williams, P., and Giralt, E. (1997). Total synthesis of dehydrodidemnin B. Use of uronium and phosphonium salt coupling reagents in peptide synthesis in solution. J. Org. Chem. 62, 334–366. doi: 10.1021/jo961932h
Katz, M., Hover, B. M., and Brady, S. F. (2016). Culture-independent discovery of natural products from soil metagenomes. J. Ind. Microbiol. Biotechnol. 43, 129–141. doi: 10.1007/s10295-015-1706-6
Kehr, J.-C., and Dittmann, E. (2015). Protective tunicate endosymbiont with extreme genome reduction. Environ. Microbiol. 17, 3430–3432. doi: 10.1111/1462-2920.12941
Kishi, Y., Fang, F. G., Forsyth, C. J., Scola, P. M., and Yoon, S. K. (1995). Halichondrins and Related Compounds. U.S.Patent 5436238.
Kusari, S., Lamsho, M., Kusari, P., Gottfried, S., Zuhlke, S., Louven, K., et al. (2014). Endophytes are hidden producers of maytansine in Putterlickia roots. J. Nat. Prod. 77, 2577–2584. doi: 10.1021/np500219a
Le, V. H., Inai, M., Williams, R. M., and Kan, T. (2015). Ecteinascidins. A review of the chemistry, biology and clinical utility of potent tetrahydroisoquinoline antitumor antibiotics. Nat. Prod. Rep. 32, 328–347. doi: 10.1039/c4np00051j
Lee, J., Currano, J. N., Carroll, P. J., and Joullie, M. M. (2012). Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 29, 404–424. doi: 10.1039/c2np00065b
Lee, W. W., Benitez, A., Goodman, L., and Baker, B. R. (1960). Potential anticancer agents. Xl. Synthesis of the beta-anomer of 9-(D-arabinofuranosyl)adenine. J. Am. Chem. Soc. 82, 2648–2649. doi: 10.1021/ja01495a070
Li, L., Deng, W., Song, J., Ding, W., Zhao, Q.-F., Peng, C., et al. (2008). Characterization of the saframycin A gene cluster from Streptomyces lavendulae Nrrl 11002 revealing a nonribosomal peptide synthetase system for assembling the unusual tetrapeptidyl skeleton in an iterative manner. J. Bact. 190, 251–263. doi: 10.1128/JB.00826-07
Li, W.-R., Ewing, W. R., Harris, B. D., and Joullie, M. M. (1990). Total synthesis and structural investigations of didemnins A, B, and C. J. Am. Chem. Soc. 112, 7659–7672. doi: 10.1021/ja00177a030
Lopanik, N., Lindquist, N., and Targett, N. (2004). Potent cytotoxins produced by a microbial symbiont protect host larvae from predation. Oecologia 139, 131–139. doi: 10.1007/s00442-004-1487-5
Lopanik, N. B., Shields, J. A., Buchholz, T. J., Rath, C. M., Hothersall, J., Haygood, M. G., et al. (2008). In vivo and in vitro trans-acylation by BryP, the putative bryostatin pathway acyltransferase derived from an uncultured marine symbiont. Chem. Biol. 15, 1175–1186. doi: 10.1016/j.chembiol.2008.09.013
Marsden, M. D., Loy, B. A., Wu, X., Ramirez, C. M., Schrier, A. J., Murray, D., et al. (2017). In vivo activation of latent Hiv with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PLoS Pathog. 13:e1006575. doi: 10.1371/journal.ppat.1006575
McCutcheon, J. P., and Moran, N. A. (2012). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
Menchaca, R., Martınez, V., Rodrıguez, A., Rodrıguez, N., Flores, M., Gallego, P., et al. (2003). Synthesis of natural ecteinascidins (ET-729, ET-745, ET-759B, ET-736, ET-637, ET-594) from cyanosafracin B. J. Org. Chem. 68, 8859–8866. doi: 10.1021/jo034547i
Mendola, D. (2000). “Aquacultural production of bryostatin 1 and ecteinascidin 743,” in Drugs from the Sea, ed. N. Fusetani (Basel: Karger), 120–133.
Mendola, D. (2003). Aquaculture of three phyla of marine invertebrates to yield bioactive metabolites: process developments and economics. Biomol. Eng. 20, 441–458. doi: 10.1016/S1389-0344(03)00075-3
Mori, T., Cahn, J. K. B., Wilson, M. C., Meoded, R. A., Wiebach, V., Martinez, A. F. C., et al. (2018). Single-bacterial genomics validates rich and varied specialized metabolism of uncultivated Entotheonella sponge symbionts. Proc. Nat. Acad. Sci. U.S.A. 115, 1718–1723. doi: 10.1073/pnas.1715496115
Morinaka, B. I., Lakis, E., Verest, M., Helf, M. J., Scalvenzi, T., Vagstad, A. L., et al. (2018). Natural noncanonical protein splicing yields products with diverse beta-amino acid residues. Science 359, 779–782. doi: 10.1126/science.aao0157
Morita, M., and Schmidt, E. W. (2018). Parallel lives of symbionts and hosts: chemical mutualism in marine animals. Nat. Prod. Rep. 35, 357–378. doi: 10.1039/c7np00053g
Moss, C., Green, D. H., Perez, B., Velasco, A., Henriquez, R., and Mckenzie, J. D. (2003). Intracellular bacteria associated with the ascidian Ecteinascidia turbinata: phylogenic and in situ hybridization analysis. Mar. Biol. 143, 99–110. doi: 10.1007/s00227-003-1060-5
Munro, M. H. G., Blunt, J. W., Dumdei, E. J., Hickford, S. J. H., Lill, R. E., Shangxiao, L., et al. (1999). The discovery and development of marine compounds with pharmaceutical potential. J. Biotechnol. 70, 15–25. doi: 10.1016/S0168-1656(99)00052-8
Newman, D. J. (2005). “The bryostatins,” in Anticancer Agents from Natural Products, 1st Edn, eds G. M. Cragg, D. G. I. Kingston, and G. M. Cragg (Boca Raton, FL: Taylor and Francis), 137–150.
Newman, D. J. (2012). “The bryostatins,” in Anticancer Agents from Natural Products, 2nd Edn, eds G. M. Cragg, D. G. I. Kingston, and D. J. Newman (Boca Raton, FL: Taylor and Francis), 199–218.
Newman, D. J., and Cragg, G. M. (2016). “The travails of clinical trials with marine-sourced compounds,” in Marine Biomedicine, ed. B. J. Baker (Boca Raton, FL: Taylor and Francis), 439–462.
Newman, D. J., and Cragg, G. M. (2017). Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar. Drugs 15:99. doi: 10.3390/md15040099
Parez-Matos, A. E., Rosado, W., and Govind, N. S. (2007). Bacterial diversity associated with the Caribbean tunicate Ecteinascidia turbinata. Antonie Van Leeuwenhoek 92, 155–164. doi: 10.1007/s10482-007-9143-9
Pettit, G. R., Day, J. F., Hartwell, J. L., and Wood, H. B. (1970). Antineoplastic components of marine animals. Nature 227, 962–963. doi: 10.1038/227962a0
Pettit, G. R., Herald, C. L., Doubek, D. L., Herald, D. L., Arnold, E., and Clardy, J. (1982). Isolation and structure of Bryostatin 1. J. Am. Chem. Soc. 104, 6846–6848. doi: 10.1021/ja00388a092
Piel, J. (2006). Bacterial symbionts: prospects for the sustainable production of invertebrate-derived pharmaceuticals. Curr. Med. Chem. 13, 39–50. doi: 10.2174/092986706775197944
Pizer, L. I., and Cohen, S. S. (1960). Metabolism of pyrimidine arabinonucleosides and cyclonucleosides in Escherichia coli. J. Biol. Chem. 235, 2387–2392.
Potts, M. B., Mcmillan, E. A., Rosales, T. I., Kim, H. S., Ou, Y.-H., Toombs, J. E., et al. (2015). Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 11, 401–408. doi: 10.1038/nchembio.1797
Rath, C. M., Janto, B., Earl, J., Ahmed, A., Hu, F. Z., Hiller, L., et al. (2011). Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 6, 1244–1256. doi: 10.1021/cb200244t
Reist, E. J., Osiecki, J. H., Goodman, L., and Baker, B. R. (1961). Potential anticancer agents. Lxi. A novel synthesis of “spongo” nucleosides. J. Am. Chem. Soc. 83, 2208–2210. doi: 10.1021/ja01470a049
Rinehart, K., Holt, T. G., Fregeau, N. L., Stroh, J. G., Kiefer, P. A., Sun, F., et al. (1990). Ecteinascidins 729, 743, 745, 759A, 759B and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 55, 4512–4515. doi: 10.1021/jo00302a007
Rinehart, K. L. Jr., Gloer, J. B., and Cook, J. C. Jr. (1981a). Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J. Am. Chem. Soc. 103, 1857–1859. doi: 10.1021/ja00397a055
Rinehart, K. L. Jr., Gloer, J. B., Hughes, R. G. Jr., Renis, H. E., Mcgovren, J. P., Stringfellow, D. A., et al. (1981b). Didemnins:antiviral and antitumor depsipeptides from a Caribbean tunicate. Science 212, 933–935.
Rinehart, K. L. Jr., Kishore, V., Nagarajan, S., Lake, R. J., Gloer, J. B., Bozich, F. A., et al. (1987). Total synthesis of didemnins A, B, and C. J. Am. Chem. Soc. 109, 6846–6848. doi: 10.1021/ja00256a046
Rinehart, K. L., and Lithgow-Bertelloni, A. M. (1991). Novel antiviral and cytotoxic agent, dehydrodidemnin B. PCT. Int. Pat. Appl. 15:248086q.
Sakai, R., Rinehart, K. L. Jr., Kishore, V., Kundu, B., Faircloth, G., Gloer, J. B., et al. (1996)). Structure-activity relationships of the didemnins. J. Med. Chem. 39, 2819–2834.
Schaufelberger, D. E., Koleck, M. P., Beutler, J. B., Vatakis, A. M., Alvarado, A. B., Andrews, P., et al. (1991). The large-scale isolation of bryosttin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 54, 1265–1270. doi: 10.1021/np50077a004
Schofield, M. M., Jain, S., Porat, D., Dick, G. J., and Sherman, D. H. (2015). Identification and analysis of the bacterial endosymbiont specialized for production of the chemotherapeutic natural product ET-743. Environ. Microbiol. 17, 3964–3975. doi: 10.1111/1462-2920.12908
Scott, J. D., and Williams, R. M. (2002). Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 102, 1669–1730. doi: 10.1021/cr010212u
Senter, P. D., and Sievers, E. L. (2012). The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 30, 631–637. doi: 10.1038/nbt.2289
Sigel, M. M., Wellham, L. L., Lichter, W., Dudeck, L. E., Gargus, J. L., and Lucas, L. H. (1970). Food-drugs from the Sea: Proceedings 1969. Washington, DC: Marine Technology Society.
Sudek, S., Lopanik, N. B., Waggoner, L. E., Hildebrand, M., Anderson, C., Liu, H., et al. (2007). Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J. Nat. Prod. 70, 67–74. doi: 10.1021/np060361d
Tsukimoto, M., Nagaoka, M., Shishido, Y., Fujimoto, J., Nishisaka, F., Matsumoto, S., et al. (2011). Bacterial production of the tunicate-derived antitumor cyclic depsipeptide Didemnin B. J. Nat. Prod. 74, 2329–2331. doi: 10.1021/np200543z
Uemura, D., Takahashi, K., Yamamoto, T., Katayama, C., Tanaka, J., Okumura, Y., et al. (1985). Norhalichondrin A: an antitumor polyether macrolide from a marine sponge. J. Am. Chem. Soc. 107, 4796–4798. doi: 10.1021/ja00302a042
Urdiales, J. L., Morata, P., De Castro, I. N., and Sanchez-Jimknez, F. (1996). Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 102, 31–37. doi: 10.1016/0304-3835(96)04151-1
Velasco, A., Acebo, P., Gomez, A., Schleissner, C., Rodriguez, P., Aparicio, T., et al. (2005). Molecular characterization of the safracin biosynthetic pathway from Pseudomonas fluorescens A2-2: designing new cytoxic compounds. Mol. Microbiol. 56, 144–154. doi: 10.1111/j.1365-2958.2004.04433.x
Vera, M. D., and Joullie, M. M. (2002). Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 22, 102–145. doi: 10.1002/med.10003
Walwick, E. R., Roberts, W. K., and Dekker, C. A. (1959). Cyclisation during the phosphorylation of uridine and cytidine by polyphosphoric Acid : a new route to the 02,2’-cyclonucleosides. Proc. Chem. Soc 84, 415–423.
Wender, P. A., Donnelly, A. C., Loy, B. A., Near, K. E., and Staveness, D. (2014). “Rethinking the role of natural products: Function-oriented synthesis, bryostatin, and bryologs,” in Natural Products in Medicinal Chemistry, ed. S. Hanessian (Weinheim: Wiley-VCH), 475–543.
Wender, P. A., Hardman, C. T., Ho, S., Jeffreys, M. S., Maclaren, J. K., Quiroz, R. V., et al. (2017). Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science 358, 218–223. doi: 10.1126/science.aan7969
Wender, P. A., Quiroz, R. V., and Stevens, M. C. (2015). Function through synthesis-informed design. Acc. Chem. Res. 48, 752–760. doi: 10.1021/acs.accounts.5b00004
Wilson, M. C., Mori, T., Rückert, C., Uria, A. R., Helf, M. J., Takada, K., et al. (2014). An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506, 58–62. doi: 10.1038/nature12959
Wilson, M. C., and Piel, J. (2013). Metagenomic approaches for exploiting uncultivated bacteria as a resource for novel biosynthetic enzymology. Chem. Biol. 20, 636–647. doi: 10.1016/j.chembiol.2013.04.011
Wright, A. E., Forleo, D. A., Gunawardana, G. P., Gunasekera, S. P., Koehn, F. E., and Mcconnell, O. J. (1990). Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 55, 4508–4512. doi: 10.1021/jo00302a006
Xu, Y., Kersten, R. D., Nam, S.-J., Lu, L., Al-Suwailem, A. M., Zheng, H., et al. (2012). Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 134, 8625–8632. doi: 10.1021/ja301735a
Yu, M. J., Kishi, Y., and Littlefield, B. A. (2012). “Discovery of E7389, a fully synthetic macrocyclic ketone analog of Halichondrin B,” in Anticancer Agents from Natural Products, 2nd Edn, eds G. M. Cragg, D. G. I. Kingston, and D. J. Newman (Boca Raton, FL: Taylor and Francis), 317–345.
Keywords: arabinose-nucleosides, didemnins, ecteinascidin, bryostatins, halichondrins, dolastatins, synthetic routes
Citation: Newman DJ (2018) “From Large-Scale Collections to the Potential Use of Genomic Techniques for Supply of Drug Candidates”. Front. Mar. Sci. 5:401. doi: 10.3389/fmars.2018.00401
Received: 20 April 2018; Accepted: 10 October 2018;
Published: 29 October 2018.
Edited by:
Georgios Daletos, Heinrich-Heine-Universität Düsseldorf, GermanyReviewed by:
Antje Labes, Fachhochschule Flensburg, GermanyMarcelino T. Suzuki, Sorbonne Universités, France
Copyright © 2018 Newman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David J. Newman, ZGpuZXdtYW42NjRAdmVyaXpvbi5uZXQ=