 Anastasija Zaiko
Anastasija Zaiko Xavier Pochon
Xavier Pochon Eva Garcia-Vazquez
Eva Garcia-Vazquez Sergej Olenin
Sergej Olenin Susanna A. Wood
Susanna A. Wood- 1Coastal and Freshwater Group, Cawthron Institute, Nelson, New Zealand
- 2Marine Research Institute, Klaipeda University, Klaipeda, Lithuania
- 3Institute of Marine Science, University of Auckland, Auckland, New Zealand
- 4Department of Functional Biology, University of Oviedo, Oviedo, Spain
To enable successful management of marine bioinvasions, timely and robust scientific advice is required. This knowledge should inform managers and stakeholders on the magnitude of a pressure (rate of human-mediated introductions), the environmental state of an ecosystem (impacts of non-indigenous species), and the success of management response (prevention, eradication, mitigation). This advice often relies on baseline biodiversity information in the form of measureable parameters (metrics). This can be derived from conventional approaches such as visual surveys, but also by utilizing environmental DNA/RNA-based molecular techniques, which are increasingly being touted as promising tools for assessing biodiversity and detecting rare or invasive species. Depending on the stage of incursion, each approach has merits and limitations. In this review we assess the performance of biosecurity-relevant biodiversity parameters derived from eDNA/eRNA samples and discuss the results in relation to different stages of invasion and management applications. The overall performance of considered methods ranged between 42 and 90% based on defined criteria, with target-specific approaches scoring higher for respective biosecurity applications, followed by eDNA metabarcoding. Caveats are discussed along with avenues which may enhance these techniques and their successful uptake for marine biosecurity surveillance and management. To facilitate and encourage uptake of these techniques, there is a need for an international collaborative framework aimed at unifying molecular sampling and analysis methodologies. Improvement of quantitative capacity and cost-efficiency will also enhance their integration in biosecurity programmes.
Introduction
Translocation of marine organisms within and between biogeographic regions is an unavoidable consequence of modern society and has been enhanced with increasing globalization. When these organisms, referred to as non-indigenous species (NIS), arrive in a new location they can spread and become invasive (Occhipinti-Ambrogi and Galil, 2004; Katsanevakis et al., 2014), sometimes with unpredictable and undesirable effects on native communities and ecosystem functioning. This can ultimately compromise socio-economic values, and human health and wellbeing (Bax et al., 2003; Lodge and Shrader-Frechette, 2003; Molnar et al., 2008).
The importance of NIS introductions as a potential pressure on marine ecosystems is now recognized through established international organizations (e.g., International Maritime Organization [IMO], International Council for the Exploration of the Sea [ICES], Helsinki Commission [HELCOM], International Union for Conservation of Nature [IUCN]), and is addressed in a number of recent legislative initiatives worldwide (e.g., International Convention for the Control and Management of Ships' Ballast Water and Sediments [BWM], European Strategy on Invasive Alien Species and Marine Strategy Framework Directive [MSFD], New Zealand Craft Risk Management Standard [CRMS], and the United States Ballast Water Regulations). However, the range of possible responses to marine NIS introductions is limited and different response measures are applicable at different stages of the introduction and establishment process (Lodge et al., 2006; Olenin et al., 2011; Figure 1). Measures against NIS introductions are usually more efficient when applied at the pre-introduction stage rather than at the state change or impact stages (Lehtiniemi et al., 2015; Oesterwind et al., 2016).
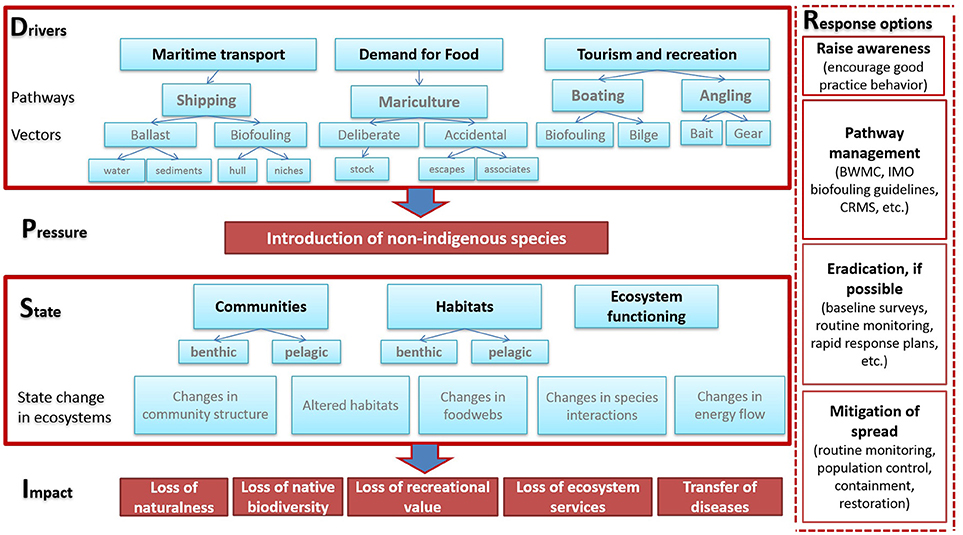
Figure 1. Available response and management measures to non-indigenous species introductions, separated across different levels of an ecosystem based management (driver-pressure-state-impact-response) framework (based on Oesterwind et al., 2016).
It is generally accepted that for successful management of environmental issues (including NIS), timely and robust scientific advice is required (Raymond et al., 2010; Olenin et al., 2011). Ideally, this advice should inform managers and stakeholders on some or all of the following: the magnitude of the pressure/s, environmental state in relation to the pressure, appropriate, and adequate management approach/s (including effort, timeframes, and location), and the success of management response. For example, the European MSFD suggests that baseline assessments of marine ecosystems and follow-up monitoring of environmental status should be determined by assessing: NIS diversity, the number of new incursions, and their impacts on communities, habitats and ecosystem functioning (EU-COM, 2008; Lehtiniemi et al., 2015).
Baseline biodiversity information is required to support NIS related decision-making processes (Olenin et al., 2014; Lehtiniemi et al., 2015; Ojaveer et al., 2016; Darling and Frederick, 2017). This information needs to be reliably derived either pre-border (on a pathway), at the border (incursion/establishment stage), post-border (expansion/secondary spread), and sometimes from controlled experimental environments (e.g., for impact assessment or development/approval of management method). The information that is generally required can be placed into the following categories: (i) general species lists; (ii) presence/absence of target NIS species; (iii) viability of detected taxa; (iv) quantitative data on the abundance of NIS.
To derive biodiversity information by conventional morphological analysis, samples need to be collected using specific or generalist sampling devices (e.g., nets, electrofishing, filtering large water volumes, sediment cores, SCUBA diving), then sorted and individually taxonomically identified usually under the microscope. This limits how many samples and replicates can be collected and analyzed. Therefore, surveillance of extensive geographic areas is often constrained by available resources and likely to be limited to rapid assessment surveys (Awad et al., 2014; Nall et al., 2015; Minchin et al., 2016).
Molecular methods are increasingly touted as promising tools for assessing biodiversity and enhancing environmental management (Ji et al., 2013; Wood et al., 2013; Aylagas et al., 2014; Kelly et al., 2014). In recent years, rapid technological advancements have led to a range of different molecular techniques being used in biosecurity applications (Nathan et al., 2014; Comtet et al., 2015; Zaiko et al., 2015a,b; Devloo-Devla et al., 2016; Wood et al., 2017). Environmental DNA/RNA can be obtained from a range of samples, such as soil, water, and feces (Ficetola et al., 2008; Andersen et al., 2012; Epp et al., 2012; Ibáñez de Aldecoa et al., 2017). These samples contain bulk nucleic acids originating from living organisms present in the sample as well as dead cells and extracellular DNA/RNA (Taberlet et al., 2012). This overcomes the need to isolate or identify individual specimens and circumvents many of the difficulties associated with conventional morphological identification including: morphological complexities, cryptic life stages, and globally declining taxonomic expertise (Wheeler et al., 2004; Costello et al., 2013; Kelly et al., 2014).
The aim of this manuscript is to review and assess the performance of eDNA/eRNA-based molecular approaches for deriving NIS-related biological information in the context of different stages of invasion and management applications (Figure 1). In this review we assessed a range of molecular methods currently used or considered for use in marine biosecurity applications. Hereafter, we refer to biosecurity, as protection against any risk posed to economy, health through “biological harm” caused by introducing or spreading non-indigenous species (and other harmful organisms such as diseases) in the wild (Armstrong and Ball, 2005). Our approach is based on the method-related criteria adopted from existing indicator evaluation frameworks (e.g., ICES, 2013; Krause-Jensen et al., 2015; Queirós et al., 2016). In these frameworks, criteria for selection of good indicators are suggested, in order to account for the various aspects of indicator utility, quality of underlying data, scientific robustness, and practicality. The importance of each criterion can be individually weighted, ranging from “essential” via “desirable” to “informative” and indicator compliance with criteria is assessed as “fully fitted,” “not fitted,” and sometimes–“partially fitted.” Scoring is usually conducted based on expert knowledge and considering published information. The derived weighted scores allow shortlisting of the most effective approaches and assist managers in making informed decisions when selecting the best fit-for-purpose suite of indicators. Here, this approach was adopted for ranking the molecular tools based on their suitability while deriving biosecurity-relevant biodiversity information. Because the review only focuses on the molecular methods and assesses their suitability for biosecurity applications, we do not directly compare nor contrast them to conventional biodiversity assessments approaches.
Molecular Target-Based and Inventory Methods Used in Biosecurity Applications
In this section, a brief overview of selected molecular techniques currently used in marine biosecurity studies is provided. It is not our intension to provide an in-depth account of the technical details of these methods and readers wanting more detailed explanations are referred to the multitude of internet resources which provide animations and diagrams (e.g., the DNA Learning Centre - http://www.dnalc.org). Table 1 provides a summary of how each technique can contribute to deriving biodiversity information relevant for biosecurity applications.
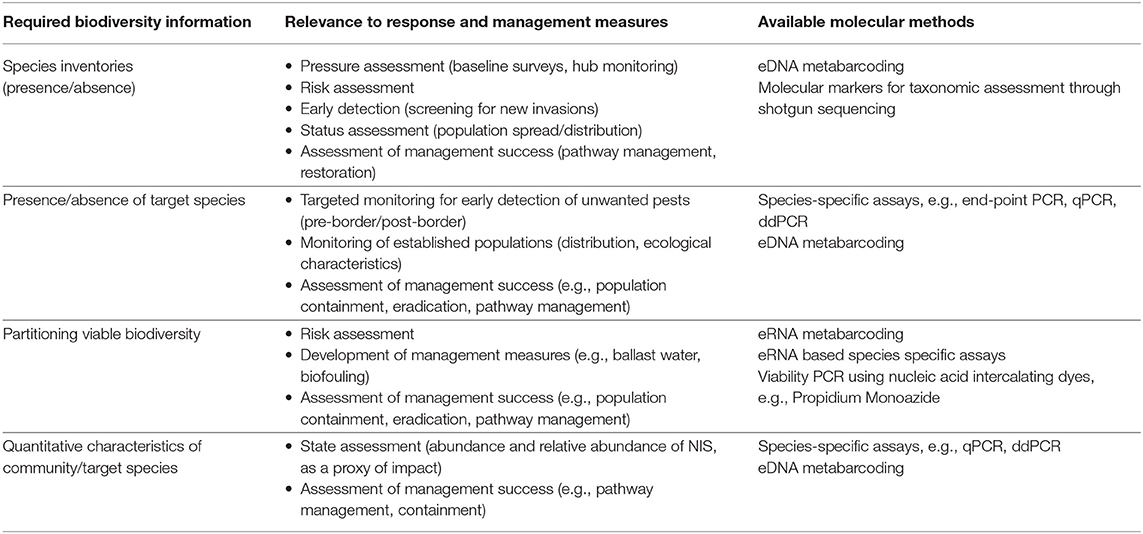
Table 1. Biodiversity information required for science-based biosecurity programmes, its relevance to specific management applications and available eDNA/eRNA methods for deriving this information.
Target-Based Tools: End-Point and Real-Time Polymerase Chain Reaction Amplification Methods
Despite being somewhat superseded by more sensitive methods (discussed below), traditional end-point Polymerase Chain Reaction (PCR) is still regularly used in marine biosecurity assessments. When applied to environmental samples, target-specific primers are used with the follow-up visualization of amplicons on an agarose gel, and (if needed) Sanger sequencing of the PCR amplicons. The sequences derived from these amplicons are then compared to global databases such as Barcode Of Life Data system (BOLD) or National Center for Biotechnology Information (NCBI) database to verify the identification of target organism or undertake follow-up phylogenetic analyses.
Examples of the use of species-specific PCR in marine biosecurity include development and application of a specific end-point PCR assays to detect NIS from environmental samples include: Atlantic wedge clam Rangia cuneata (Ardura et al., 2015), soft-shell clam Mya arenaria (Ardura and Zaiko, 2018), and the Australian tubeworm Ficopomatus enigmaticus (Muñoz-Colmenero et al., 2017).
Quantitative Polymerase Chain Reaction (qPCR, also known as real-time PCR) is an advancement on end-point PCR and is one of the most promising molecular tools for highly specific and sensitive detection of one or a few targets. It enables rapid turnaround and simultaneous analysis of multiple samples. Quantitative PCR assays rely on primers, or primers and a probe that have been designed to be specific for the target species. The amplification of this target can then be measured in real-time either through the use of intercalating dyes (Becker et al., 1996) or probe-based detection systems (Heid et al., 1996). In recent years, assays have been designed specifically for marine pests including: dinoflagellates Alexandrium spp. (Galluzzi et al., 2004; Vandersea et al., 2017), sea squirts Styela clava (Gillum et al., 2014), and Didemnum spp. (Simpson et al., 2017), Amur River clam Potamocorbula amurensis (Smith et al., 2012), and Mediterranean fan worm Sabella spallanzanii (Wood et al., 2017).
From a biosecurity perspective, a potentially useful extension of the qPCR application is viability assessment for applications where determining the presence of a living organism is essential (e.g., assessing the success of applied treatment or a pest management programme; Darling and Frederick, 2017; Pochon et al., 2017). Nucleic acid intercalating dyes added to the sample before extraction (e.g., propidium monoazide [PMA]), that only penetrate damaged lipid membranes (i.e., dead cells), bind and covalently crosslink with double-stranded nucleic acids, inhibiting their extraction, and amplification (Nogva et al., 2003; Nocker et al., 2006). This approach is being used to distinguish between viable and dead cells of bacteria (Schnetzinger et al., 2013; Desneux et al., 2015). The so-called viability PCR has not yet been used for biosecurity applications and its applicability to assess viability of multicellular organisms is unknown. Substantial loss of DNA signal in viability PCR (Nocker et al., 2006) may cause false negative results in targeted surveys, e.g., failing to detect extracellular DNA of living organisms. Therefore, this method should be considered for application in combination with conventional end-point or qPCR, implying additional costs and effort. Further research is required to assess the applicability of this approach for characterizing viable eukaryotic biodiversity in the context of marine biosecurity.
Alternatively, to infer viability of the target organism(s), PCR tools can be applied to eRNA samples. Ribonucleic acid is a crucial component for protein synthesis, usually single stranded in a cell and transcribed from DNA by enzymes, i.e., produced in biologically active (living) organisms. Compared to eDNA, eRNA degrades more rapidly in the marine environment (typically hours to days, Thomsen et al., 2012b; Sassoubre et al., 2016), and is therefore considered a better proxy for detecting living biota. On the other hand, susceptibility of RNA makes it difficult to work with. Collection of RNA samples requires dedicated sampling protocols, more careful preservation, and storage. There is also additional processing time and costs associated with isolation and reverse transcription of RNA (Laroche et al., 2016), making it more expensive and challenging and thus a less attractive molecule to work with.
A recent advancement in PCR methods is droplet digital PCR (ddPCR), where target DNA is randomly allocated into discrete droplets via microfluidics and each droplet is then thermally cycled and individually screened via fluorescence measurement for the presence of target DNA. Quantitation of DNA is very accurate using ddPCR and is calculated using Poisson statistics (Hindson et al., 2011; Pinheiro et al., 2012). This negates the need for the use of standard curves and enables extremely low-level detection. A recent comparative application of qPCR and ddPCR for detecting invasive aquatic species in the Laurentian Great Lakes (Nathan et al., 2014), suggested similar sensitivities between the two methods, with higher cost-efficiency demonstrated for ddPCR (when capital expenditure was not considered). An additional advantage provided by ddPCR platforms with two fluorescence filters, is that they enable duplex or with some developments and optimization greater multiplexing capability (Dobnik et al., 2016).
Inventory-Based Tools: Metabarcoding and Polymerase Chain Reaction Free High-Throughput Methods
The advent of high-throughput sequencing (HTS) has made it possible to produce enormous volumes of sequence data rapidly. Metabarcoding has become a well-established method for characterizing the biodiversity in different types of environmental samples (Chariton et al., 2010; Shokralla et al., 2012; Taberlet et al., 2012; Aylagas et al., 2014; de Vargas et al., 2015; Domaizon et al., 2017). It enables the identification of many taxa by matching short (typically 100–600 base pair) sequence reads obtained from HTS of PCR amplicons to reference sequences. Metabarcoding has proven to be very effective for characterizing marine communities and identifying potential pests (Pochon et al., 2013; Comtet et al., 2015; Zaiko et al., 2015c; Brown et al., 2016). However, there are some prerequisites required when applying metabarcoding for characterizing biotic assemblages and identifying potential NIS: (i) sufficient taxonomic resolution provided by the target gene, (ii) “universality” of the primers (i.e., capacity to amplify the target gene from a wide variety of taxa; see Geller et al., 2013), and (iii) availability of robust reference databases for reliable taxonomic assignments of obtained sequences (discussed in sections below).
Environmental RNA metabarcoding (targeting cDNA obtained through reverse-transcription PCR) is increasingly being used for characterizing viable assemblages associated with recent environmental change due to human marine activities (Pawlowski et al., 2014, 2016a; Dowle et al., 2015; Lejzerowicz et al., 2015; Pochon et al., 2015a; Visco et al., 2015; Laroche et al., 2016; Birrer et al., 2018), and is being considered for biosecurity applications (Pochon et al., 2017; Rey et al., 2018). However, there are uncertainties regarding potential biases associated with eRNA metabarcoding, resulting from e.g., overrepresentation of organisms with complex genomes and numerous copies of transcriptionally active marker genes (Gong et al., 2013), or a number of artifacts potentially occurring during RNA processing and PCR amplification (Laroche et al., 2017). Therefore, despite the potential of eRNA metabarcoding for differentiating living biodiversity, for example, in bilge (Pochon et al., 2017) or ballast water (Darling and Frederick, 2017; Rey et al., 2018), it remains challenging and requires further dedicated research and technological advancements to facilitate its uptake for biosecurity applications.
There are several emerging and rapidly advancing PCR-free methods, such as shotgun sequencing (Wang et al., 2013), mitochondrial enrichment (Zhou et al., 2013), and gene enrichment (Mertes et al., 2011; Dowle et al., 2016). These have limited application for routine biosecurity surveys to date, as they require considerable sequencing and computing effort or additional laboratory processing which increases the associated cost. These methods have an important advantage though, since they overcome PCR-inherent biases which result in the preferential amplification of certain DNA (or cDNA) templates leading to the overrepresentation of some taxa and potentially missing others.
Assessment of Biosecurity-Relevant Biodiversity Information Derived From Molecular Methods
Different criteria can be applied to select the most efficient and practical biological information or indicators for environmental management purposes (Mazik et al., 2010; Elliott, 2011; James et al., 2012; Queirós et al., 2016). Some criteria refer to intrinsic qualities of indicators, for example “scientific basis,” “responsiveness to pressure,” “social relevance.” However, there are also criteria associated with the methodology employed for deriving underlying data (i.e., biodiversity information) and quality of that data. In Table 1 we provide an overview of the relevance of biodiversity information derived from molecular approaches applied to environmental DNA/RNA, in the context of its utility for biosecurity management and response purposes.
Our assessment of molecular methods suitability for deriving biosecurity-relevant biodiversity information is based on the set of method-related criteria (Table 2) adopted from existing indicator evaluation frameworks (Elliott, 2011; ICES, 2013; Krause-Jensen et al., 2015). The relative importance of each criterion for informing adequate biosecurity response and management measures was assigned as either essential, desirable or informative, applying importance weighting factors 3, 2 and 1, respectively. Compliance of each considered method with selected criteria was assessed as either a criterion is fully met = 1.0, partially met = 0.5, or not met = 0.0. This assessment is based on evidence from our comprehensive literature review and expert knowledge. The performance score for each indicator against each criterion calculated as the product of these two values (compliance × weighting factor). For benchmarking the overall performance of the methods, the final scores were derived for each method/biosecurity application combination by summing the performance scores and then dividing the sum-product by a maximum possible score per method/application. The resulting performance value was expressed as percentage.

Table 2. Method-related criteria (adopted from ICES, 2013; Krause-Jensen et al., 2015 and other relevant literature sources3−10), scoring categories and importance weighting applied in this assessment.
Compliance Scores for Technical Feasibility, Precision, and Repeatability Criterion
End-point PCR (eDNA and eRNA) = 1
qPCR/ddPCR (eDNA and eRNA) = 1
Viability PCR using nucleic acid intercalating dyes = 0.5
Metabarcoding (eDNA and eRNA) = 0.5
Shotgun sequencing and mitochondrial enrichment (eDNA) = 0.5Shotgun sequencing and mitochondrial enrichment (eRNA) = 0
Gene enrichment (eDNA) = 0.5
Gene enrichment (eRNA) = 0
The aforementioned molecular methods (Table 1) are increasingly being applied in biosecurity research with multiple studies addressing species detection on pathways, in introduction hubs, and in ongoing monitoring programmes (Bott et al., 2010; Darling and Mahon, 2011; Mountfort et al., 2012; Comtet et al., 2015; Pochon et al., 2015a; Zaiko et al., 2015c, 2016; Montes et al., 2016). In general, molecular methods can be considered technically feasible for providing qualitative biodiversity information (species lists and presence/absence of target species).
In regard to documented evidence of feasibility, methods such as shotgun sequencing, gene enrichment, and viability PCR are currently less advanced comparing to more widely used metabarcoding and traditional PCR-based methods. We were unable to find any documented examples of eRNA-based PCR-free methods been applied for marine biosecurity, therefore the criterion was considered not met for these methods. There are also some uncertainties regarding the reproducibility and signal-to-noise ratio in data derived from metabarcoding (both eDNA and eRNA), as sensitivity and accuracy for individual species detection can be impeded by incompleteness of reference databases, marker resolution, and amplification biases, especially in complex samples from diverse communities (Briski et al., 2016; Hatzenbuhler et al., 2017; Leray and Knowlton, 2017). Extrinsic factors, that may affect precision and repeatability of these methods include heterogeneous distribution of eDNA/eRNA in the water, susceptibility to both field and laboratory cross-contamination, variability of nucleic acid degradation due to microbial activity and environmental conditions, and variation in eDNA/eRNA shedding rates (Goldberg et al., 2016).
Compliance Scores for Comprehensibility Criterion
End-point PCR (eDNA) = 1
End-point PCR (eRNA) = 0.5
qPCR/ddPCR (eDNA) = 1
qPCR/ddPCR (eRNA) = 0.5
Viability PCR using nucleic acid intercalating dyes = 0.5
Metabarcoding (eDNA and eRNA) = 0.5
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 0.5
Gene enrichment = 0.5
Generally, biodiversity information derived by target-based tools is more straight-forward, as positive eDNA signals are easy to interpret and communicate to stakeholders. This is much trickier for inventories, as there is a number of factors that should be considered for robust interpretation of eDNA-based biodiversity estimates, including resolution of the marker used, quality and completeness of reference databases, bioinformatics pipelines, etc. (Cristescu, 2014). There are also some difficulties interpreting detection or non-detection of rare taxa due to sequencing errors, preferential sequencing and aforementioned caveats (Darling and Frederick, 2017). The complex analyses applied to the molecular inventory data also makes communication of results to stakeholders more difficult than simple presence-absence data from targeted detections. Interpretation of viability assessment results from molecular analyses can also be complex and difficult to understand, given all potential uncertainties (see the above sections).
Compliance Scores for Quantitative Capacity Criterion
End-point PCR (eDNA and eRNA) = 0
qPCR/ddPCR (eDNA and eRNA) = 0.5 (Note: can be 1 for prokaryotes, provided an appropriate empirical calibration is undertaken)
Viability PCR using nucleic acid intercalating dyes = 0.5
Metabarcoding (eDNA and eRNA) = 0.5
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 0.5
Gene enrichment = 0.5
The ability to quantify the impacts of NIS using standardized methods has been identified as a priority in biosecurity research (Olenin et al., 2007, 2011; Lehtiniemi et al., 2015; Ojaveer et al., 2016). Although no universal framework has been agreed upon, most impact assessments utilize quantitative information on NIS populations: abundance, biomass, proportional contribution to relevant communities, distribution range (Kotta et al., 2001; Olenina et al., 2010; Zaiko et al., 2011; Ojaveer and Kotta, 2014). One of the key limitation of most molecular methods is that they usually cannot provide exact measures of abundance and/or biomass information (Yu et al., 2012), although some methods have higher quantitative capacity (e.g., qPCR, ddPCR, PCR-free approaches) than others (e.g., end-point PCR, HTS metabarcoding; Dowle et al., 2016).
Semi-quantitative biological information can be derived from molecular studies, for example, relative quantities of species in the community can be inferred from the percentage of sequence reads obtained through metabarcoding (Hajibabaei et al., 2011; Zhan et al., 2013; Evans et al., 2016; Pawlowski et al., 2016b). However, a number of studies have highlighted clear discrepancies between microscopically-derived absolute abundance data and metabarcoding-based abundance data (Medinger et al., 2010; Stoeck et al., 2014; Sun et al., 2015).
Although qPCR, ddPCR, and PCR-free methods enable quantification of targeted genes (e.g., Hindson et al., 2011), there are still a plethora of possible factors influencing biological biases when estimating abundances. These include intra- and inter-species variation due to the presence of different number of nuclei, gene copies, genome sizes, and/or biovolume (Martin-Laurent et al., 2001; Weber and Pawlowski, 2013), PCR and sequencing errors, primer biases, and differential amplification efficiencies (Farrelly et al., 1995; Suzuki and Giovannoni, 1996; Polz and Cavanaugh, 1998; Acinas et al., 2005). It is generally accepted that it is extremely challenging to determine absolute abundance, and some authors advocate for the exclusive use of presence-absence data when using molecular information generated from HTS or quantitative PCR (Chariton et al., 2015; Zaiko et al., 2016). Many conventional biotic indices require only relative abundance data (e.g., AZTI Marine Biotic Index [AMBI], Borja et al., 2000; Benthic Quality Index [BQI], Rosenberg et al., 2004; Infaunal Quality Index [IQI], Kennedy et al., 2011). Recent studies have used or developed metabarcoding-based indices and demonstrated the potential of using semi-quantitative HTS data for measuring diversity change of marine benthic assemblages along pollution gradients (Lejzerowicz et al., 2015; Pochon et al., 2015a; Pawlowski et al., 2016a; Keeley et al., 2018).
The intrinsic nature of eDNA may differ between multicellular eukaryotes and prokaryotes or micro-eukaryotes. For example, there is a higher probability of detecting intracellular DNA from microbes, as intact individuals are more readily isolated from small volumes of material. For larger eukaryotes, there is increased probability of detecting free-floating or extracellular DNA and DNA from non-living cells or unicellular/small propagules (gametes, eggs; Ibáñez de Aldecoa et al., 2017). Consequently, any quantitative biodiversity information derived from eDNA samples may need to be treated differentially for micro- and macro-communities in biosecurity applications, especially if only intact or viable organisms are of interest (e.g., for confirming compliance with the BWMC regulations; IMO, 2004).
Compliance Scores for Standardization Criterion
End-point PCR(eDNA and eRNA) = 0.5
qPCR/ddPCR (eDNA and eRNA) = 0.5
Viability PCR = 0
Metabarcoding (eDNA and eRNA) = 0
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 0
Gene enrichment = 0
While this criterion is being met at least to some extent for developed species-specific assays, further work is needed for international cross-validation to account for regional genetic variation (Bohmann et al., 2014; Ardura et al., 2016).
Despite recent attempts to develop and optimize standardized protocols for eDNA metabarcoding (Taberlet et al., 2012; Deiner et al., 2015; Elbrecht and Leese, 2015; Aylagas et al., 2016; Porter and Hajibabaei, 2018), they are often restricted to a portion of the biotic community (e.g., macrofauna), or only focus on a segment of the protocol (e.g., sampling, DNA extraction, library preparation, bioinformatics). A unified, internationally calibrated protocol for marine biosecurity applications is clearly needed. Currently, such a document does not exist and research laboratories employing metabarcoding for marine biosecurity applications often develop their own analytical workflows and in-house reference sequence databases, which impedes the transferability/comparability of results at international and regional scales.
Two widely used genes for characterizing entire eukaryotic communities are the 18S ribosomal RNA (18S rRNA) and the mitochondrial Cytochrome C Oxidase subunit I (COI). While it is relatively straightforward to design universal primers for the highly conserved 18S rRNA gene, it does not provide sufficient resolution to identify organisms at the species (or even genera) level within many groups (Pochon et al., 2013; Fletcher et al., 2017). Use of the COI gene enables finer resolution in taxonomic assignments, and primers which cover a short region of this gene are now widely used (Hajibabaei and McKenna, 2012; Leray and Knowlton, 2015; Wangensteen et al., 2017). The common drawback of applying these two markers for biosecurity assessment, however, is incompleteness of open-access reference databases (Ratnasingham and Hebert, 2013; Zaiko et al., 2016). Despite efforts to build standardized barcode reference libraries for many major animal and plant phyla (Ratnasingham and Hebert, 2010; Guillou et al., 2013; Quast et al., 2013) including recent pest-focused initiatives (Diaz et al., 2004; Dias et al., 2017), numerous gaps remain. Even among groups with well-developed reference databases greater than 20% of entries may be incorrectly identified or lack important data e.g., geographic location (Nilsson et al., 2006). Other marker genes suggested or considered for metabarcoding applications, e.g., 16S ribosomal RNA (Zhan et al., 2014) or taxon-specific markers proposed for meiofauna, protists and plants, e.g., chloroplast DNA or the ribosomal Internal Transcribed Spacer ITS region (Medinger et al., 2010; Yao et al., 2010; Freeland, 2016; Hume et al., 2018) are less frequently applied for determining marine biodiversity, and therefore are less likely to have substantial representation of relevant taxa in publicly available databases. The deficiency of reference sequence databases for international marine pests (Briski et al., 2016) limits effective uptake and standardized application of metabarcoding in marine biosecurity programmes (Bohmann et al., 2014; Zaiko et al., 2016).
Comparability of metabarcoding results can be further impeded by a broad array of bioinformatics pipelines currently applied and with continuously emerging novel analytical tools coupled with ever-developing computing and sequencing technologies (Coissac et al., 2012; Cristescu, 2014; Callahan et al., 2016; Anslan et al., 2017). Biodiversity information produced by different sequencing platforms (Zaiko et al., 2015a; Speranskaya et al., 2018) and/or algorithms used for filtering, de-noising, clustering, and taxonomic assignments are generally comparable at high-level overview of community composition, but can be divergent at genus or species levels (Plummer et al., 2015). When data is not easily reconcilable at lower taxonomic levels this can create challenges for biosecurity applications over extended spatio-temporal scales (e.g., long-term national hub monitoring or implementing international policies).
The need for uniformity and standardization (from protocols to databases and bioinformatics pipelines) has been already recognized by large consortia working in the field of molecular research, for example, 1000 Genomes (The 1000 Genomes Project Consortium, 2015) and CBOL Protist Working Group (Pawlowski et al., 2012). Similar initiatives should be also encouraged for biosecurity applications, through inter-calibration sequencing experiments and developing consistent analyses approaches (Muir et al., 2016).
Compliance Scores for Cost-Efficiency Criterion
End-point PCR (eDNA) = 1
End-point PCR (eRNA) = 0.5
qPCR/ddPCR (eDNA) = 1
qPCR/ddPCR (eRNA) = 0.5
Viability PCR = 1
Metabarcoding (eDNA and eRNA) = 0.5
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 0
Gene enrichment (eDNA and eRNA) = 0
Molecular methods can rapidly detect target species or be used to characterize comprehensive biodiversity from small sample sizes, and can be effectively applied to a wide range of environmental samples and spatio-temporal scales: from localized mesocosm experiments to temporally replicated site or nation-wide surveys (Shaw et al., 2016; Minamoto et al., 2017; Stat et al., 2017). Only small volumes of water or sediments are needed to obtain molecular traces of an organism (Thomsen et al., 2012a; Pochon et al., 2015a; Shaw et al., 2016). Thus, when compared to the traditional morphology-based biodiversity assessment, the estimated cost per sample when applying molecular methods is significantly less due to the constantly increasing high-throughput and multiplexing capacities (Ji et al., 2013; Shokralla et al., 2015). The actual cost however can vary significantly depending on the methodology (e.g., end-point PCR is generally cheaper than quantitative PCR, which is less expensive than HTS metabarcoding), the aim of the study, and the scale of the survey.
Sequencing services are becoming increasingly affordable for routine application, enabling larger numbers of samples to be processed and enormous volumes of sequencing data to be generated (Muir et al., 2016). Analysis of these data requires increased computational resources and analytical efforts. This affects the overall cost structure of biodiversity research projects requiring a larger budget allocated to the analysis component, compared to traditional research where most of the cost is spent on experimental work and data generation (Sboner et al., 2011). Such shift in focus should be accounted for when planning molecular-based studies, and allocating funding resources for biosecurity surveillance.
Compliance Scores for Early Warning Criterion
End-point PCR (eDNA and eRNA) = 1
qPCR/ddPCR (eDNA and eRNA) = 1
Viability PCR = 1
Metabarcoding (eDNA and eRNA) = 1
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 1
Gene enrichment (eDNA and eRNA) = 1
Surveillance programmes using eDNA-based tools may enable the detection of NIS arrival or population spread at an earlier stage compared to programmes relying exclusively on traditional survey methods (Pochon et al., 2015b; Brown et al., 2016; Xiong et al., 2016). A further benefit of molecular methods is the ability to detect cryptic marine species at early life stage, when visual identification is difficult or impossible.
Molecular surveillance can generally ensure higher sensitivity of detection due to legacy DNA (extracellular or non-living material). This may in some cases be an advantage as it provides information on potentially present, but not observable taxa (Zaiko et al., 2015c, 2016; Ardura et al., 2016). The down-side of legacy DNA is that it may lead to the detection of false positive signals, e.g., from non-viable organisms, untargeted sources, incidental contamination, or (in HTS metabarcoding) as a result of marker- or reference-related biases (Bohmann et al., 2014; Ficetola et al., 2015).
Eliminating false positives from biodiversity assessment remains a major challenge in eDNA-based studies (Bohmann et al., 2014). There are several pathways for minimizing the probability of false positives through e.g., stringent control of contamination, including the use of multiple blank controls at different steps of sample collection and processing (Jerde et al., 2011); and/or confirming species detection by HTS using complementary analyses such as multi-loci metabarcoding, and phylogenetic analyses or species-specific assays (Kelly et al., 2017; Wood et al., 2017). A range of statistical methods are also available for optimizing the quality and strategy of a survey and account for both imperfect detection and false positive signals (Ferguson et al., 2015; Ficetola et al., 2015; Lahoz-Monfort et al., 2015). It should be noted though, that appropriate fit-for-purpose sampling design and monitoring strategy is important for efficient implementation of molecular surveillance. Similarly as recommended for conventional approaches (Lehtiniemi et al., 2015), thorough consideration should be given to sampling time, locations, spatial coverage required, which will vary depending on the objectives of each survey or monitoring programme.
Compliance Scores for Low Impact (Non-Destructive) Criterion
End-point PCR (eDNA and eRNA) = 1
qPCR/ddPCR (eDNA and eRNA) = 1
Viability PCR = 1
Metabarcoding (eDNA and eRNA) = 1
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 1
Gene enrichment (eDNA and eRNA) = 1
Conventional biological surveillance methods vary in the magnitude of disturbance involved. High-impact methods such as electrofishing (Nielsen, 1998), may inflict significant injuries and mortality on non-target taxa. Medium-impact methods such as net sampling may interfere with a range of biota and induce some physiological stress and casual mortalities (e.g., Eleftheriou, 2013). Methods using underwater cameras and/or acoustic methodologies (Matsua et al., 2009; Doehring et al., 2011; Donaldson et al., 2014; Zhang et al., 2016) are considered least-to-non-destructive. The latter, however, have limited use for biosecurity applications as they cannot accurately identify most taxa, and are therefore not applicable for characterizing biodiversity and identifying the dispersive stages of potential pests.
The non-destructive nature of molecular surveillance methods has been highlighted in studies of endangered or protected species (Foote et al., 2012; Rees et al., 2014). Although NIS are not protected species, it is important to note that their efficient detection using traditional surveillance methods might require extracting substantial portions of accompanying native biota. This can be a particularly big issue in fragile ecosystems such as marine reserves or protected areas, like coral reefs and polar sanctuaries (Gutt, 2001; Leray and Knowlton, 2015).
Compliance Scores for Cross-Applicability Criterion
End-point PCR (eDNA and eRNA) = 0
qPCR/ddPCR (eDNA and eRNA) = 0
Viability PCR = 0
Metabarcoding (eDNA and eRNA) = 1
Shotgun sequencing and mitochondrial enrichment (eDNA and eRNA) = 1
Gene enrichment (eDNA and eRNA) = 1
Biodiversity information derived from molecular methods, even though intended for biosecurity applications, can be used to address many other questions in marine environmental research, from general biodiversity assessment and detection of spatio-temporal patterns to deducing environmental quality status for management purposes (Aylagas et al., 2014, 2016; Pochon et al., 2015a; Darling and Frederick, 2017; von Ammon et al., 2018). Target-specific methods like end-point PCR, qPCR, or viability PCR might not be widely applicable for other research and surveillance purposes. However, being more affordable for in-house implementation, they may be applicable in citizen science or local educational programmes (Ardura et al., 2015; Biggs et al., 2015).
Overall Results and Conclusions
As evidenced from the assessment above and resulting weighted scores (Table 3), the overall performance of the considered molecular methods for deriving biosecurity-relevant biodiversity information ranged from 42 to 90%. In general, target-specific tools like end-point PCR and qPCR/ddPCR scored higher for marine biosecurity applications, followed by eDNA metabarcoding which was especially applicable for deriving inventory information.
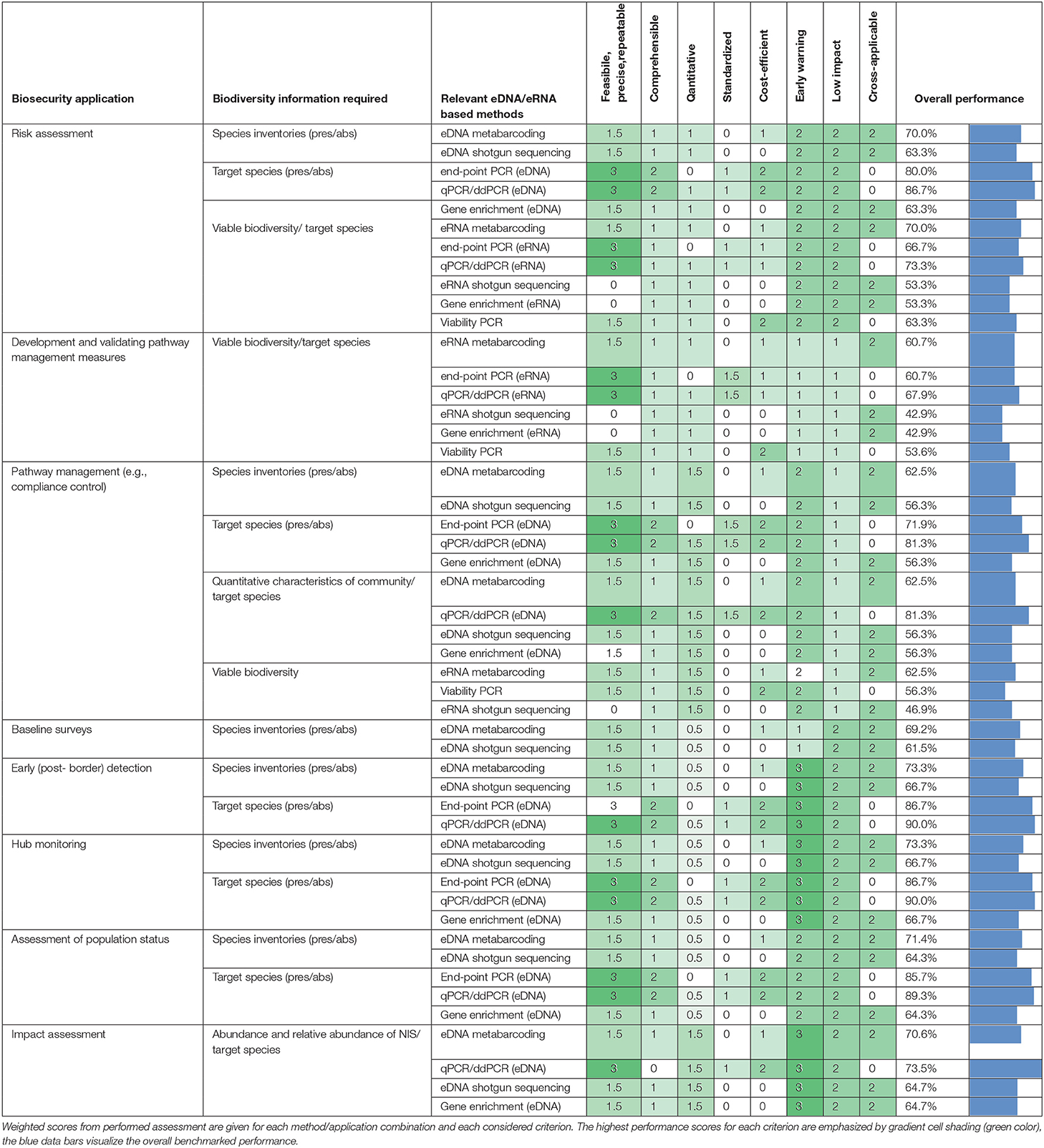
Table 3. Comparative performance of environmental DNA/RNA tools for deriving biodiversity information relevant for advising biosecurity response and management measures.
To facilitate and encourage effective uptake of molecular approaches, there is a need for an international collaborative framework aimed at unifying molecular sample processing and analysis methods for marine biosecurity applications. Given the complex range of potential biosecurity-related management questions, creating a single standardized protocol will be a challenging task. However, a universal/flexible protocol to ensure detection on different pathways, from varying habitats, and for a range of purposes could be developed through consultation with scientists and stakeholders working in this field globally.
Despite the current limitations of eDNA-based techniques, they have a great potential for deriving biodiversity information and complementing marine biosecurity programmes worldwide. For example, even in the lack of quantitativeness, biodiversity information derived from molecular analyses (e.g., community metabarcoding or target species detection with qPCR) often surpasses that from conventional approaches (e.g., microscopy and morphological assessment) in terms of taxonomic resolution, precision, and sensitivity (Zaiko et al., 2016; Fletcher et al., 2017). This information can be effectively used for identifying new arrivals at different temporal and geographic scales. Data on new arrivals can help assess introduction rates in relation to pathways and vectors of introduction, which is crucial for biosecurity management and, eventually, may be used to measure the effectiveness of legal and administrative instruments aimed at the prevention of new incursions.
Molecular techniques are advancing rapidly and it is likely that extensive scientific effort in this field will overcome many of the current caveats resulting in more robust and cost-efficient methods for use in a wide range of biosecurity applications. However, even with these advancements it is unlikely that all marine biosecurity questions will be answered by molecular methods alone. Comprehensive marine biosecurity programme should integrate complementary scientific approaches including traditional surveys, mathematical modeling, risk assessment frameworks, and molecular techniques. Effective collaboration and communication between experts working in different fields of marine biosecurity will be essential to successfully protect native marine biodiversity, marine ecosystems and associated environmental, economic, social, and cultural values.
Author Contributions
AZ, SW, and XP conceived the idea of the presented review. AZ developed the framework of the review and assessment, and drafted the manuscript with input from SW and XP. EG-V and SO critically revised the selected criteria and assessment, contributed to discussions, and revision of the initial draft. AZ, SW, and XP wrote the final version of the manuscript with input from all authors.
Funding
This contribution was supported by the BONUS Bio-C3 project (Art 185), funded jointly by the EU and the Research Council of Lithuania (Grant Agreement No. BONUS-1/2014) and the project BLUEPORTS from the Spanish Ministry of Economy and Competitiveness (Grant CGL2016-79209-R).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Drs. Aibin Zhan and Thierry Comtet for their time and valuable comments on the manuscript.
References
Acinas, S., Sarma-Rupavtarm, R., Klepac-Ceraj, V., and Polz, M. (2005). PCR induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71, 8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005
Andersen, K., Bird, K. L., Rasmussen, M., Haile, J., Breuning-Mdsen, H., Kjaer, K. H., et al. (2012). Meta-barcoding of ‘dirt’ DNA from soil reflects vertebrate biodiversity. Mol. Ecol. 21, 1966–1979. doi: 10.1111/j.1365-294X.2011.05261.x
Anslan, S., Bahram, M., Hiiesalu, I., and Tedersoo, L. (2017). PipeCraft: flexible open-source toolkit for bioinformatics analysis of custom high-throughput amplicon sequencing data. Mol. Ecol. Resour. 17, 234–240. doi: 10.1111/1755-0998.12692
Ardura, A., and Zaiko, A. (2018). PCR-based assay for Mya arenaria detection from marine environmental samples and tracking its invasion in coastal ecosystems. J. Nat. Conserv. 43, 1–7. doi: 10.1016/j.jnc.2018.02.007
Ardura, A., Zaiko, A., Borrell, Y. J., Samuiloviene, A., and Garcia-Vazquez, E. (2016). Novel tools for early detection of a global aquatic invasive, the zebra mussel Dreissena polymorpha. Aquat. Conserv. Mar. Freshw. Ecosyst. 27, 165–176. doi: 10.1002/aqc.2655
Ardura, A., Zaiko, A., Martinez, J. L., Samuiloviene, A., Semenova, A., and Garcia-Vazquez, E. (2015). eDNA and specific primers for early detection of invasive species - A case study on the bivalve Rangia cuneata, currently spreading in Europe. Mar. Environ. Res. 112, 48–55. doi: 10.1016/j.marenvres.2015.09.013
Armstrong, K. F., and Ball, S. L. (2005). DNA barcodes for biosecurity: invasive species identification. Philos. Trans. R. Soc. B Biol. Sci. 360, 1813–1823. doi: 10.1098/rstb.2005.1713
Awad, A., Haag, F., Anil, A. C., and Abdulla, A. (2014). GEF-UNDP-IMO GloBallast Partnership Programme, IOI, CSIR_NIO and IUCN. Guidance on Port Biological Baseline Surveys, GloBallast Monograph 22. London: GEF-UNDP-IMO GloBallast Partnerships.
Aylagas, E., Borja, Á., Irigoien, X., and Rodríguez-Ezpeleta, N. (2016). Benchmarking DNA metabarcoding for biodiversity-based monitoring and assessment. Front. Mar. Sci. 3:96. doi: 10.3389/fmars.2016.00096
Aylagas, E., Borja, A., and Rodrigues-Ezpeleta, N. (2014). Environmental status assessment using DNA metabarcoding: towards a genetic based marine biotic index (gAMBI). PLoS ONE 9:e90529. doi: 10.1371/journal.pone.0090529
Bax, M., Williamson, A., Aguero, M., Gonzalez, E., and Geeves, W. (2003). Marine invasive alien species: a threat to global biodiversity. Mar. Pol. 27, 313–323. doi: 10.1016/S0308-597X(03)00041-1
Becker, A., Reith, A., Napiwotzki, J., and Kadenbach, B. (1996). A quantitative method of determining initial amounts of DNA by polymerase chain reaction cycle titration using digital imaging and a novel DNA stain. Anal. Biochem. 237, 204–207. doi: 10.1006/abio.1996.0230
Biggs, J., Ewald, N., Valentini, A., Gaboriaud, C., Dejean, T., Griffiths, R. A., et al. (2015). Using eDNA to develop a national citizen science-based monitoring programme for the great crested newt (Triturus cristatus). Biol. Conserv. 183, 19–28. doi: 10.1016/j.biocon.2014.11.029
Birrer, S. C., Dafforn, K. A., Simpson, S. L., Kelaher, B. P., Potts, J., Scanes, P., et al. (2018). Interactive effects of multiple stressors revealed by sequencing total (DNA) and active (RNA) components of experimental sediment microbial communities. Sci. Total Environ. 637–638, 1383–1394. doi: 10.1016/j.scitotenv.2018.05.065
Bohmann, K., Evans, A., Gilbert, M. T. P., Carvalho, G. R., Creer, S., Knapp, M., et al. (2014). Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 29, 358–367. doi: 10.1016/j.tree.2014.04.003
Borja, A., Franco, J., and Perez, V. (2000). A marine biotic index to establish the ecological quality of soft-bottom benthos within European Estuarine and coastal environments. Mar. Pollut. Bull. 40, 1100–1114. doi: 10.1016/S0025-326X(00)00061-8
Bott, N. J., Ophel-Keller, K. M., Sierp, M. T., Herdina Rowling, K. P., McKay, A. C., et al. (2010). Toward routine, DNA-based detection methods for marine pests. Biotechnol. Adv. 28, 706–714. doi: 10.1016/j.biotechadv.2010.05.018
Briski, E., Ghabooli, S., Bailey, S. A., and MacIsaac, H. J. (2016). Are genetic databases sufficiently populated to detect non-indigenous species? Biol. Invasions 18, 1911–1922. doi: 10.1007/s10530-016-1134-1
Brown, E. A., Chain, F. J. J., Zhan, A., MacIsaac, H. J., and Cristescu, M. E. (2016). Early detection of aquatic invaders using metabarcoding reveals a high number of non-indigenous species in Canadian ports. Divers. Distrib. 22, 1045–1059. doi: 10.1111/ddi.12465
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chariton, A. A., Court, L. N., Hartley, D. M., Colloff, M. J., and Hardy, C. M. (2010). Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Front. Ecol. Environ. 8, 233–238. doi: 10.1890/090115
Chariton, A. A., Stephenson, S., Morgan, M. J., Steven, A. D. L., Colloff, M. J., Court, L. N., et al. (2015). Metabarcoding of benthic eukaryote communities predicts the ecological condition of estuaries. Environ. Poll. 203, 165–174. doi: 10.1016/j.envpol.2015.03.047
Coissac, E., Riaz, T., and Puillandre, N. (2012). Bioinformatic challenges for DNA metabarcoding of plants and animals. Mol. Ecol. 21, 1834–1847. doi: 10.1111/j.1365-294X.2012.05550.x
Comtet, T., Sandionigi, A., Viard, F., and Casiraghi, M. (2015). DNA (meta)barcoding of biological invasions: a powerful tool to elucidate invasion processes and help managing aliens. Biol. Invasions 17, 905–822. doi: 10.1007/s10530-015-0854-y
Costello, M. J., May, R. M., and Stork, N. E. (2013). Can we name Earth's species before they go extinct? Science 339, 413–416. doi: 10.1126/science.1230318
Cristescu, M. E. (2014). From barcoding single individuals metabarcoding biological communities: towards an integrative approach to the study of global biodiversity. Trends Ecol. Evol. 29, 566–571. doi: 10.1016/j.tree.2014.08.001
Darling, J. A., and Frederick, R. M. (2017). Nucleic acids-based tools for ballast water surveillance, monitoring, and research. J. Sea Res. 133, 43–52. doi: 10.1016/j.seares.2017.02.005
Darling, J. A., and Mahon, A. R. (2011). From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ. Res. 111, 978–988. doi: 10.1016/j.envres.2011.02.001
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahe, F., Logares, R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. doi: 10.1126/science.1261605
Deiner, K., Walser, J.-C., Mächler, E., and Altermatt, F. (2015). Choice of capture and extraction methods affect detection of freshwater biodiversity from environmental DNA. Biol. Conserv. 183, 53–63. doi: 10.1016/j.biocon.2014.11.018
Desneux, J., Chemaly, M., and Pourcher, A.-M. (2015). Experimental design for the optimization of propidium monoazide treatment to quantify viable and non-viable bacteria in piggery effluents. BMC Microbiol. 15:164. doi: 10.1186/s12866-015-0505-6
Devloo-Devla, F., Miralles, L., Ardura, A., Borell, Y. J., Pejovic, I., Tsartsianidou, V., et al. (2016). Detection and characterisation of the biopollutant Xenostrobus securis (Lamarck 1819) Asturian population from DNA Barcoding and eBarcoding. Mar. Pollut. Bull. 105, 23–29. doi: 10.1016/j.marpolbul.2016.03.008
Dias, P. J., Fotedar, S., Munoz, J., Hewitt, M. J., Lukehurst, S., Hourston, M., et al. (2017). Establishment of a taxonomic and molecular reference collection to support the identification of species regulated by the Western Australian Prevention List for Introduced Marine Pests. Manag. Biol. Invasions 8, 215–225. doi: 10.3391/mbi.2017.8.2.09
Diaz, R. J., Solan, M., and Valente, R. M. (2004). A review of approaches for classifying benthic habitats and evaluating habitat quality. J. Environ. Manage. 73, 165–181. doi: 10.1016/j.jenvman.2004.06.004
Dobnik, D., Štebih, D., Blejec, A., Morisset, D., and Žel, J. (2016). Multiplex quantification of four DNA targets in one reaction with Bio-Rad droplet digital PCR system for GMO detection. Sci. Rep. 6:35451. doi: 10.1038/srep35451
Doehring, K., Young, R. G., Hay, J., and Quarterman, A. J. (2011). Suitability of Dual-frequency Identification Sonar (DIDSON) to monitor juvenile fish movement at floodgates. N. Z. J. Mar. Freshw. Res. 45, 413–422. doi: 10.1080/00288330.2011.571701
Domaizon, I., Winegardner, A., Capo, E., Gauthier, J., and Gregory-Eaves, I. (2017). DNA-based methods in paleolimnology: new opportunities for investigating long-term dynamics of lacustrine biodiversity. J. Paleolimnol. 58, 1–21. doi: 10.1007/s10933-017-9958-y
Donaldson, M. R., Hinch, S. G., Suski, C. D., Fisk, A. T., Heupel, M. R., and Cooke, S. J. (2014). Making connections in aquatic ecosystems with acoustic telemetry monitoring. Front. Ecol. Environ. 12, 565–573. doi: 10.1890/130283
Dowle, E., Pochon, X., Banks, J., Shearer, K., and Wood, S. A. (2016). Targeted gene enrichment and high throughput sequencing for environmental biomonitoring: a case study using freshwater macroinvertebrates. Mol. Ecol. Resour. 16, 1240–1254. doi: 10.1111/1755-0998.12488
Dowle, E., Pochon, X., Keeley, N., and Wood, S. A. (2015). Assessing the effects of salmon farming seabed wnrichment using bacterial community diversity and high-throughput sequencing. FEMS Microbiol. Ecol. 91:fiv089. doi: 10.1093/femsec/fiv089
Elbrecht, V., and Leese, F. (2015). Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass - sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10:e0130324. doi: 10.1371/journal.pone.0130324
Eleftheriou, A. (2013). Methods for the Study of Marine Benthos, 4th Edn. Oxford: John Wiley & Sons, Ltd.
Elliott, M. (2011). Marine science and management means tackling exogenic unmanaged pressures and endogenic managed pressures – a numbered guide. Mar. Pollut. Bull. 62, 651–655. doi: 10.1016/j.marpolbul.2010.11.033
Epp, L., Boessenkool, S., Bellemain, E. P., Haile, J., Esposito, A., Riaz, T., et al. (2012). New environmental metabarcodes for analysing soil DNA: potential for studying past and present ecosystems. Mol. Ecol. 21, 1821–1833. doi: 10.1111/j.1365-294X.2012.05537.x
EU-COM (2008). Directive 2008/56/EC of the European Parliament and of the Council establishing a framework for community action in the field of marine environmental policy (Marine Strategy Framework Directive). Official Journal of the European Union L164, 19e40.
Evans, N. T., Olds, B. P., Renshaw, M. A., Turner, C. R., Li, Y., Jerde, C. L., et al. (2016). Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Mol. Ecol. Resour. 16, 29–41. doi: 10.1111/1755-0998.12433
Farrelly, V., Rainey, F., and Stackebrandt, E. (1995). Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 61, 2798–2801.
Ferguson, P. F. B., Conroy, M. J., and Hepinstall-Cymerman, J. (2015). Occupancy models for data with false positive and false negative errors and heterogeneity across sites and surveys. Methods Ecol. Evol. 6, 1395–1406. doi: 10.1111/2041-210X.12442
Ficetola, G. F., Miaud, C., Pompanon, F., and Taberlet, P. (2008). Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425. doi: 10.1098/rsbl.2008.0118
Ficetola, G. F., Pansu, J., Bonin, A., Coissac, E., Giguet-Covex, C., De Barba, M., et al. (2015). Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556. doi: 10.1111/1755-0998.12338
Fletcher, L. M., Zaiko, A., Atalah, J., Richter, I., Dufour, C. M., Pochon, X., et al. (2017). Bilge water as a vector for the spread of marine pests: a morphological, metabarcoding and experimental assessment. Biol. Invasions 19, 2851–2867. doi: 10.1007/s10530-017-1489-y
Foote, A. D., Thomsen, P. F., Sveegaard, S., Wahlberg, M., Kielgast, J., Kyhn, L. A., et al. (2012). Investigating the potential use of environmental DNA (eDNA) for genetic monitoring of marine mammals. PLoS ONE 7:e41781. doi: 10.1371/journal.pone.0041781
Forrest, B. M., and Hopkins, G. A. (2013). Population control to mitigate the spread of marine pests: insights from management of the Asian kelp Undaria pinnatifida and colonial ascidian Didemnum vexillum. Manage. Biol. Invas. 4, 317–326. doi: 10.3391/mbi.2013.4.4.06
Freeland, J. R. (2016). The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome 60, 358–374. doi: 10.1139/gen-2016-0100
Galluzzi, L., Penna, A., Bertozzini, E., Vila, M., Garces, E., and Magnani, M. (2004). Development of a real-time PCR assay for rapid detection and quantification of Alexandrium minutum (a Dinoflagellate). Appl. Environ. Microbiol. 70, 1199–1206. doi: 10.1128/AEM.70.2.1199-1206.2004
Geller, J., Meyer, C., Parker, M., and Hawk, H. (2013). Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. 13, 851–861. doi: 10.1111/1755-0998.12138
Gillum, J. E., Jimenez, L., White, D. J., Goldstien, S. J., and Gemmell, N. J. (2014). Development and application of a quantitative real-time PCR assay for the globally invasive tunicate Styela clava. Manage. Biol. Invas. 5, 133–142. doi: 10.3391/mbi.2014.5.2.06
Goldberg, C. S., Turner, C. R., Deiner, K., Klymus, K. E., Thosmsen, F. P., Murhpy, M. A., et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307. doi: 10.1111/2041-210X.12595
Gong, J., Dong, J., Liu, X., and Massana, R. (2013). Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 164, 369–379. doi: 10.1016/j.protis.2012.11.006
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The Protist Ribosomal Reference database (PR(2)): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Gutt, J. (2001). High latitude Antarctic benthos: a “coevolution” of nature conservation and ecosystem research? Ocean Polar Res. 23, 411–417. http://www.koreascience.or.kr/article/ArticleFullRecord.jsp?cn=HOGBB1_2001_v23n4_411
Hajibabaei, M., and McKenna, C. (2012). DNA mini-barcodes. Methods Mol. Biol. 858, 339–353. doi: 10.1007/978-1-61779-591-6_15
Hajibabaei, M., Shokralla, S., Zhou, X., Singer, G. A., and Baird, D. J. (2011). Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using river benthos. PLoS ONE 6:e17497. doi: 10.1371/journal.pone.0017497
Hatzenbuhler, C., Kelly, J. R., Martinson, J., Okum, S., and Piligrim, E. (2017). Sensitivity and accuracy of high-throughput metabarcoding methods for early detection of invasive species. Sci. Rep. 7:46393. doi: 10.1038/srep46393
Heid, C. A., Stevens, J., Livak, K. J., and Williams, P. M. (1996). Real time quantitative PCR. Genome Res. 6, 986–994. doi: 10.1101/gr.6.10.986
Hindson, B. J., Ness, K. D., Masquelier, D. A., Belgrader, P., Heredia, N. J., Makarewicz, A. J., et al. (2011). High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610. doi: 10.1021/ac202028g
Hume, B., Ziegler, M., Poulain, J., Pochon, X., Romac, S., Boissin, E., et al. (2018). An improved primer set and amplification protocol with increased specificity and sensitivity targeting the Symbiodinium ITS2 region. PeerJ 6:e4816. doi: 10.7717/peerj.4816
Ibáñez de Aldecoa, A. L., Zafra, O., and Gonzalez-Pastor, J. E. (2017). Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front. Microbiol. 8:1390. doi: 10.3389/fmicb.2017.01390
ICES (2013). Report of the Working Group on the Ecosystem Effects of Fishing Activities (WGECO). ICES, Copenhagen.
IMO (2004). International Convention for the Control and Management of Ships' Ballast Water and Sediments. London: International Maritime Organization.
James, C., Kershner, J., Samhouri, J., O'Neill, S., and Levin, P. (2012). A methodology for evaluating and ranking water quantity indicators in support of ecosystem-based management. Environ. Manage. 49, 703–719. doi: 10.1007/s00267-012-9808-7
Jerde, C. L., Mahon, A. R., Chadderton, W. L., and Lodge, D. M. (2011). “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 4, 150–157. doi: 10.1111/j.1755-263X.2010.00158.x
Ji, Y., Ashton, L., Pedley, S. M., Edwards, D. P., Tang, Y., Nakamura, A., et al. (2013). Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol. Lett. 16, 1245–1257. doi: 10.1111/ele.12162
Katsanevakis, S., Wallentinus, I., Zenetos, A., Leppakoski, E., Cinar, M. E., Ozturk, B., et al. (2014). Impacts of invasive alien marine species on ecosystem services and biodiversity: a pan-European review. Aquat. Invas. 9, 391–423. doi: 10.3391/ai.2014.9.4.01
Keeley, N. B., Wood, S. A., and Pochon, X. (2018). Development and preliminary validation of a multi-trophic metabarcoding biotic index for monitoring benthic organic enrichment. Ecol. Indic. 85, 1044–1057. doi: 10.1016/j.ecolind.2017.11.014
Kelly, R. P., Closek, C. J., O'Donnell, J. L., Kralj, J. E., Shelton, A. O., and Samhouri, J. F. (2017). Genetic and manual survey methods yield different and complementary views of an ecosystem. Front. Mar. Sci. 3:283. doi: 10.3389/fmars.2016.00283
Kelly, R. P., Port, J. A., Yamahara, K. M., Martone, R. G., Lowell, N., Thomsen, P. F., et al. (2014). Harnessing DNA to improve environmental management. Science 344, 1455–1456. doi: 10.1126/science.1251156
Kennedy, R., Wallace, A., and Keegan, B. F. (2011). Long-term trends in benthic habitat quality as determined by Multivariate AMBI and Infaunal Quality Index in relation to natural variability - A case study in Kinsale Harbour, south coast of Ireland. Mar. Pollut. Bull. 62, 1427–1436. doi: 10.1016/j.marpolbul.2011.04.030
Kotta, J., Orav, H., and Sandberg-Kilpi, E. (2001). Ecological consequence of the introduction of the polychaete Marenzelleria cf. viridis into a shallow-water biotope of the northern Baltic Sea. J. Sea Res. 46, 273–280. doi: 10.1016/S1385-1101(01)00088-0
Krause-Jensen, D., Bruhn, A., Carstensen, J., Queiros, A. M., Bruun, J., et al. (2015). Report on the Criteria for Good Indicators Selection. DEVOTES Deliverable 3.2 (Public Report).
Lahoz-Monfort, J. J., Guillera-Arroita, G., and Tingley, R. (2015). Statistical approaches to account for false-positive errors in environmental DNA samples. Mol. Ecol. Resour. 16, 673–685. doi: 10.1111/1755-0998.12486
Laroche, O., Wood, S. A., Tremblay, L. A., Ellis, J. I., Lejzerowicz, F., Pawlowski, J., et al. (2016). First evaluation of foraminiferal metabarcoding for monitoring environmental impact from an offshore oil drilling site. Mar. Environ. Res. 120, 225–235. doi: 10.1016/j.marenvres.2016.08.009
Laroche, O., Wood, S. A., Tremblay, L. A., Lear, G., Ellis, J. I., and Pochon, X. (2017). Metabarcoding monitoring analysis: the pros and cons of using co-extracted environmental DNA and RNA data to assess offshore oil production impacts on benthic communities. PeerJ 5:e3347. doi: 10.7717/peerj.3347
Lehtiniemi, M., Ojaveer, H., David, M., Galil, B., Gollasch, S., McKenzie, C., et al. (2015). Dose of truth - Monitoring marine non-indigenous species to serve legislative requirements. Mar. Pol. 54, 26–35. doi: 10.1016/j.marpol.2014.12.015
Lejzerowicz, F., Esling, P., Pillet, L., Wilding, T. A., Black, K. D., and Pawlowski, J. (2015). High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Sci. Rep. 5:13932. doi: 10.1038/srep13932
Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Acad. Sci. U.S.A. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Leray, M., and Knowlton, N. (2017). Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 5:3006. doi: 10.7717/peerj.3006
Lodge, D. M., and Shrader-Frechette, K. (2003). Nonindigenous species: ecological explanation, environmental ethics, and public policy. Conserv. Biol. 17, 31–37. doi: 10.1046/j.1523-1739.2003.02366.x
Lodge, D. M., Williams, S., MacIsaac, H. J., Hayes, K. R., Leung, B., Reichard, S., et al. (2006). Biological invasions: recommendations for U.S. policy and management. Ecol. Appl. 16, 2035–2054. doi: 10.1890/1051-0761(2006)016[2035:BIRFUP]2.0.CO;2
Magaletti, E., Garaventa, F., David, M., Castriota, L., Kraus, R., Luna, G. M., et al. (2017). Developing and testing an early warning system for non-indigenous species and ballast water management. J. Sea Res. 133, 100–111. doi: 10.1016/j.seares.2017.03.016
Martin-Laurent, F., Philippot, L., Hallet, S., Chaussod, R., Germon, J. C., Soulas, G., et al. (2001). DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl. Environ. Microbiol. 67, 2354–2359. doi: 10.1128/AEM.67.5.2354-2359.2001
Matsua, T., Mizuno, Y., and Cho, H. (2009). Monitoring of pipe clogging by mussels utilizing an optical fiber AE system. J. Acoustic Emission 27, 224–232. doi: 10.1007/s10921-011-0090-z
Mazik, K., Boyes, S., McManus, E., Ducrotoy, J.-P., Rogers, S., and Elliott, M. (2010). Healthy & Biologically Diverse Seas Evidence Group Technical Report Series: Evaluation and Gap Analysis of Current and Potential Indicators for Sediment Habitats. Technical Report Series, United Kingdom Marine Monitoring and Assessment Strategy (UKMMAS), JNCC, Peterborough.
Medinger, R., Nolte, V., Pandey, R. V., Jost, S., Ottenwälder, B., Schlötterer, C., et al. (2010). Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol. Ecol. 19, 32–40. doi: 10.1111/j.1365-294X.2009.04478.x
Mertes, F., Elsharawy, A., Sauer, S., van Helvoort, J., van der Zaag, P., Franke, A., et al. (2011). Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genomics 10, 374–386. doi: 10.1093/bfgp/elr033
Minamoto, T., Fukuda, M., Katsuhara, K., Fujiwara, A., Hidaka, S., Yamamoto, S., et al. (2017). Environmental DNA reflects spatial and temporal jellyfish distribution. PLoS ONE 12:e0173073. doi: 10.1371/journal.pone.0173073
Minchin, D., Olenin, S., Liu, T.-K., Cheng, M., and Huang, S.-C. (2016). Rapid assessment of target species: Byssate bivalves in a large tropical port. Mar. Pollut. Bull. 112, 177–182. doi: 10.1016/j.marpolbul.2016.08.023
Molnar, J. L., Gamboa, R. L., Revenga, C., and Spalding, M. D. (2008). Assessing the global threat of invasive species to marine biodiversity. Front. Ecol. Environ. 6, 485–492. doi: 10.1890/070064
Montes, M., Rico, J. M., Garcia-Vazquez, E., and Borrell, Y. J. (2016). Morphological and molecular methods reveal the Asian alga Grateloupia imbricata (Halymeniaceae) occurs on Cantabrian Sea shore (Bay of Biscay). Phycologia 55, 365–370. doi: 10.2216/15-112.1
Mountfort, D., Smith, K. F., Kirs, M., Kuhajek, J., Adamson, J. E., and Wood, S. A. (2012). Development of single and multispecies detection methods for the surveillance and monitoring of marine pests in New Zealand. Aquat. Invas. 7, 125–128. doi: 10.3391/ai.2012.7.1.013
Muir, P., Li, S., Lou, S., Wang, D., Spakowicz, D. J., Salichos, L., et al. (2016). The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol. 17:53. doi: 10.1186/s13059-016-0917-0
Muñoz-Colmenero, M., Ardura, A., Clusa, L., Miralles, L., Gower, F., Zaiko, A., et al. (2017). New specific molecular marker detects Ficopomatus enigmaticus from water eDNA before positive results of conventional sampling. J. Nat. Conserv. 43, 173–178. doi: 10.1016/j.jnc.2017.12.004
Nall, C. R., Guerin, A. J., and Cook, E. J. (2015). Rapid assessment of marine non-native species in northern Scotland and a synthesis of existing Scottish records. Aquat. Invas. 10, 107–121. doi: 10.3391/ai.2015.10.1.11
Nathan, L. M., Simmons, M., Wegleitner, B. J., Jerde, C. L., and Mahon, A. R. (2014). Quantifying environmental DNA signals for aquatic invasive species across multiple detection platforms. Environ. Sci. Technol. 48:5034052. doi: 10.1021/es5034052
Nielsen, J. L. (1998). Scientific sampling effects: electrofishing California's endangered fish populations. Fisheries 23, 6–12. doi: 10.1577/1548-8446(1998)023<0006:SSEECE>2.0.CO;2
Nilsson, R. H., Ryberg, M., Kristiansson, E., Abarenkov, K., Larsson, K.-H., and Kõljalg, U. (2006). Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS ONE 1:e59. doi: 10.1371/journal.pone.0000059
Nocker, A., Cheung, C. Y., and Camper, A. K. (2006). Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 67, 310–320. doi: 10.1016/j.mimet.2006.04.015
Nogva, H. K., Drømtorp, S. M., Nissen, H., and Rudi, K. (2003). Ethidium monoazide for DNA-based differentiation of viable and dead bacteria by 5′-nuclease PCR. Biotechniques 34, 804–808.
Occhipinti-Ambrogi, A., and Galil, B. S. (2004). A uniform terminology on bioinvasions: a chimera or an operative tool? Mar. Pollut. Bull. 49, 688–694. doi: 10.1016/j.marpolbul.2004.08.011
Oesterwind, D., Rau, A., and Zaiko, A. (2016). Drivers and pressures – untangling the terms commonly used in marine science and policy. J. Environ. Manage. 181, 8–15. doi: 10.1016/j.jenvman.2016.05.058
Ojaveer, H., and Kotta, J. (2014). Ecosystem impacts of the widespread non-indigenous species in the Baltic Sea: literature survey evidences major limitations in knowledge. Biol. Invas. 19, 799–813. doi: 10.1007/s10750-014-2080-5
Ojaveer, H., Olenin, S., Narscius, A., Florin, A.-B., Ezhova, E., Gollasch, S., et al. (2016). Dynamics of biological invasions and pathways over time: a case study of a temperate coastal sea. Biol. Invas. 19, 799–813. doi: 10.1007/s10530-016-1316-x
Olenin, S., Elliott, M., Bysveen, I., Culverhouse, P. F., Daunys, D., Dubelaar, G. B. J., et al. (2011). Recommendations on methods for the detection and control of biological pollution in marine coastal waters. Mar. Pollut. Bull. 62, 2598–2604. doi: 10.1016/j.marpolbul.2011.08.011
Olenin, S., Minchin, D., and Daunys, D. (2007). Assessment of biopollution in aquatic ecosystems. Mar. Pollut. Bull. 55, 379–394. doi: 10.1016/j.marpolbul.2007.01.010
Olenin, S., Narscius, A., Minchin, D., David, M., Galil, B., Gollasch, S., et al. (2014). Making non-indigenous species information systems practical for management and useful for research: an aquatic perspective. Biol. Conserv. 173, 98–107. doi: 10.1016/j.biocon.2013.07.040
Olenina, I., Wasmund, N., Hajdu, S., Jurgensone, I., Gromisz, S., Kownacka, J., et al. (2010). Assessing impacts of invasive phytoplankton: the Baltic Sea case. Mar. Pollut. Bull. 60:046. doi: 10.1016/j.marpolbul.2010.06.046
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C., et al. (2012). CBOL Protist Working Group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 10:e1001419. doi: 10.1371/journal.pbio.1001419
Pawlowski, J., Esling, P., Lejzerowicz, F., Cedhagen, T., and Wilding, T. A. (2014). Environmental monitoring through protist next-generation sequencing metabarcoding: assessing the impact of fish farming on benthic foraminifera communities. Mol. Ecol. Resour. 14, 1129–1140. doi: 10.1111/1755-0998.12261
Pawlowski, J., Esling, P., Lejzerowicz, F., Cordier, T., Visco, J. A., Martins, C. I. M., et al. (2016a). Benthic monitoring of salmon farms in Norway using foraminiferal metabarcoding. Aquacult. Environ. Interact. 8, 371–386. doi: 10.3354/aei00182
Pawlowski, J., Lejzerowicz, F., Apotheloz-Perret-Gentil, L., Visco, J., and Esling, P. (2016b). Protist metabarcoding and environmental biomonitoring: time for change. Eur. J. Protistol. 55, 12–25. doi: 10.1016/j.ejop.2016.02.003
Pinheiro, L. B., Coleman, V. A., Hindson, C. M., Herrmann, J., Hindson, B. J., Bhat, S., et al. (2012). Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 84, 1003–1011. doi: 10.1021/ac202578x
Piola, R. F., Denny, C. M., Forrest, B. M., and Taylor, M. D. (2009). “Marine biosecurity: management options and response tools,” in Invasive Species Management: A Handbook of Principles and Techniques, eds P. A. Williams and M. N. Clout (New York, NY: Oxford University Press), 205–231.
Plummer, E., Twin, J., Bulach, D. M., Garland, S. M., and Tabrizi, S. N. (2015). A comparison of three bioinformatics pipelines for the analysis of preterm gut microbiota using 16S rRNA gene sequencing data. J. Proteomics Bioinform. 8, 283–291. doi: 10.4172/jpb.1000381
Pochon, X., Bott, N. J., Smith, K. F., and Wood, S. A. (2013). Evaluating detection limits of Next-Generation Sequencing for the surveillance and monitoring of international marine pests. PLoS ONE 8:e73935. doi: 10.1371/journal.pone.0073935
Pochon, X., Wood, S. A., Keeley, N. B., Lejzerowicz, F., Esling, P., Drew, J., et al. (2015a). Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding. Mar. Pollut. Bull. 100, 370–382. doi: 10.1016/j.marpolbul.2015.08.022
Pochon, X., Zaiko, A., Fletcher, L. M., Laroche, O., and Wood, S. A. (2017). Wanted dead or alive? Using metabarcoding of environmental DNA and RNA to distinguish living assemblages for biosecurity applications. PLoS ONE 12:e0187636. doi: 10.1371/journal.pone.0187636
Pochon, X., Zaiko, A., Hopkins, G. A., Banks, J. C., and Wood, S. A. (2015b). Early detection of eukaryotic communities from marine biofilm using high-throughput sequencing: an assessment of different sampling devices. Biofouling 31, 241–251. doi: 10.1080/08927014.2015.1028923
Polz, M., and Cavanaugh, C. (1998). Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64:3724.
Porter, T. M., and Hajibabaei, M. (2018). Scaling up: a guide to high-throughput genomic approaches for biodiversity analysis. Mol. Ecol. 27, 313–338. doi: 10.1111/mec.14478
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–596. doi: 10.1093/nar/gks1219
Queirós, A. M., Strong, J. A., Mazik, K., Carstensen, J., Bruun, J., Somerfield, P. J., et al. (2016). An objective framework to test the quality of candidate indicators of good environmental status. Front. Mar. Sci. 3:73. doi: 10.3389/fmars.2016.00073
Ratnasingham, S., and Hebert, P. D. N. (2010). BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. doi: 10.1111/j.1471-8286.2007.01678.x
Ratnasingham, S., and Hebert, P. D. N. (2013). A DNA-based registry for all animal species: the barcode index number (BIN) system. PLoS ONE 8:66213. doi: 10.1371/journal.pone.0066213
Raymond, C. M., Fazey, I. F., Reed, M. S., Stringer, L. C., Robinson, G. M., and Evely, A. C. (2010). Integrating local and scientific knowledge for environmental management. J. Environ. Manage. 91, 1766–1777. doi: 10.1016/j.jenvman.2010.03.023
Rees, H. C., Maddison, B. C., Middleditch, D. J., Patmore, J. R. M., and Gough, K. C. (2014). The detection of aquatic animal species using environmental DNA – a review of eDNA as a survey tool in ecology. J. Appl. Ecol. 51, 1450–1459. doi: 10.1111/1365-2664.12306
Rey, A., Basurko, O. C., and Rodríguez-Ezpeleta, N. (2018). The challenges and promises of genetic approaches for ballast water management. J. Sea Res. 133, 134–145. doi: 10.1016/j.seares.2017.06.001
Rosenberg, R., Blomquist, M., Nilsson, H. C., Cederwall, H., and Dimming, A. (2004). Marine quality assessment by use of benthic species-abundance distributions: a proposed new protocol within the European Union Water Framework Directive. Mar. Pollut. Bull. 49:12. doi: 10.1016/j.marpolbul.2004.05.013
Sassoubre, L. M., Yamahara, K. M., Gardner, L. D., Block, B. A., and Boehm, A. B. (2016). Quantification of environmental DNA (eDNA) shedding and decay rates for three marine fish. Environ. Sci. Technol. 50, 10456–10464. doi: 10.1021/acs.est.6b03114
Sboner, A., Mu, X. J., Greenbaum, D., Auerbach, P. K., and Gerstein, M. B. (2011). The real cost of sequencing: higher than you think! Genome Biol. 12:125. doi: 10.1186/gb-2011-12-8-125
Schnetzinger, F., Pan, Y., and Nocker, A. (2013). Use of propidium monoazide and increased amplicon length reduce false-positive signals in quantitative PCR for bioburden analysis. Appl. Microbiol. Biotechnol. 97, 2153–2162. doi: 10.1007/s00253-013-4711-6
Shaw, J. L. A., Clarke, L. J., Wedderburn, S. D., Barnes, T. C., Weyrich, L. A., and Cooper, A. (2016). Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biol. Conserv. 197, 131–138. doi: 10.1016/j.biocon.2016.03.010
Shokralla, S., Porter, T. M., Gibson, J. F., Dobosz, R., Janzen, D. H., Hallwachs, W., et al. (2015). Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Sci. Rep. 5:9687. doi: 10.1038/srep09687
Shokralla, S., Spall, J. L., Gibson, J. F., and Hajibabaei, M. (2012). Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 21, 1794–1805. doi: 10.1111/j.1365-294X.2012.05538.x
Simpson, T. J. S., Dias, P. J., Snow, M., Mu-oz, J., and Berry, T. (2017). Real-time PCR detection of Didemnum perlucidum (Monniot, 1983) and Didemnum vexillum (Kott, 2002) in an applied routine marine biosecurity context. Mol. Ecol. Resour. 17, 443–453. doi: 10.1111/1755-0998.12581
Smith, K. F., Wood, S. A., Mountfort, D., and Cary, S. C. (2012). Development of a real-time PCR assay for the detection of the invasive clam, Corbula amurensis, in environmental samples. J. Exp. Mar. Biol. Ecol. 412, 52–57. doi: 10.1016/j.jembe.2011.10.021
Speranskaya, A. S., Khafizov, K., Ayginin, A. A., Krinitsina, A. A., Omelchenko, D. O., Nilova, M. V., et al. (2018). Comparative analysis of Illumina and Ion Torrent high-throughput sequencing platforms for identification of plant components in herbal teas. Food Control 93, 315–324. doi: 10.1016/j.foodcont.2018.04.040
Staehr, P. A., Jakobsen, H. H., Hansen, J. L. S., Andersen, P., Storr-Paulsen, M., Christensen, J., et al. (2016). Trends in Records and Contribution of Non-Indigenous Species (NIS) to Biotic Communities in Danish Marine Waters. Scientific Report from DCE–Danish Centre for Environment and Energy No. 17, Aarhus University, p. 44.
Stat, M., Huggett, M. J., Bernasconi, R., DiBattista, J. D., Berry, T. E., Newman, S. J., et al. (2017). Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 7:12240. doi: 10.1038/s41598-017-12501-5
Stoeck, T., Breiner, H.-W., Filker, S., Ostermaier, V., Kammer-Lander, B., and Sonntag, B. (2014). A morphogenetic survey on ciliate plankton from a mountain lake pinpoints the necessity of lineage-specific barcode markers in microbial ecology. Environ. Microbiol. 16:12194. doi: 10.1111/1462-2920.12194
Sun, C., Zhao, Y., Li, H., Dong, Y., MacIsaac, H. J., and Zhan, A. (2015). Unreliable quantification of species abundance based on high-throughput sequencing data of zooplankton communities. Aquat. Biol. 24, 9–15. doi: 10.3354/ab00629
Suzuki, M., and Giovannoni, S. (1996). Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62, 625–630.
Taberlet, P., Coissac, E., Hajibabaei, M., and Rieseberg, L. H. (2012). Environmental DNA: a special issue on DNA metabarcoding. Mol. Ecol. 21, 1789–1793. doi: 10.1111/j.1365-294X.2012.05542.x
The 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Thomsen, P., Kielgast, J., Iversen, L., Wiuf, C., Rasmussen, M., Gilbert, M., et al. (2012a). Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 21, 2565–2573. doi: 10.1111/j.1365-294X.2011.05418.x
Thomsen, P. T., Kielgast, J., Iversen, L., Møller, P. R., Rasmussen, M., and Willerslev, E. (2012b). Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7:e41732. doi: 10.1371/journal.pone.0041732
Vandersea, M. W., Kibler, S. R., Sant, S. B. V., Tester, P. A., Sullivan, K., Eckert, G., et al. (2017). qPCR assays for Alexandrium fundyense and A. ostenfeldii (Dinophyceae) identified from Alaskan waters and a review of species-specific Alexandrium molecular assays. Phycologia 56, 303–320. doi: 10.2216/16-41.1
Visco, J. A., Apotheloz-Perret-Gentil, L., Cordonier, A., Esling, P., Pillet, L., and Pawlowski, J. (2015). Environmental monitoring: inferring the diatom index from next-generation sequencing data. Environ. Sci. Technol. 49, 7597–7605. doi: 10.1021/es506158m
von Ammon, U., Wood, S. A., Laroche, O., Zaiko, A., Tait, L., Lavery, S., et al. (2018). The impact of artificial surfaces on marine bacterial and eukaryotic biofouling assemblages: a high-throughput sequencing analysis. Mar. Environ. Res. 133, 57–66. doi: 10.1016/j.marenvres.2017.12.003
Wang, J., McLenachan, P., Biggs, P., Winder, L., Schoenfeld, B., Narayan, V., et al. (2013). Environmental bio-monitoring with high-throughput sequencing. Brief. Bioinformatics 14, 575–588. doi: 10.1093/bib/bbt032
Wangensteen, O. S., Palacín, C., Guardiola, M., and Turon, X. (2017). Metabarcoding littoral hard-bottom communities: unexpected diversity and database gaps revealed by two molecular markers. PeerJ Preprints 5:e3429v3421. doi: 10.7287/peerj.preprints.3429v1
Weber, A.-T., and Pawlowski, J. (2013). Can abundance of protists be inferred from sequence data: a case study of foraminifera. PLoS ONE 8:e56739. doi: 10.1371/journal.pone.0056739
Wheeler, Q. D., Raven, P. H., and Wilson, E. O. (2004). Taxonomy: impediment or expedient? Science 303:285. doi: 10.1126/science.303.5656.285
Williams, S. L., and Schroeder, S. L. (2004). Eradication of the invasive seaweed Caulerpa taxifolia by chlorine bleach. Mar. Ecol. Prog. Ser. 272, 69–76. doi: 10.3354/meps272069
Wood, S. A., Smith, K. F., Banks, J. C., Tremblay, L. A., Rhodes, L., Mountfort, D., et al. (2013). Molecular genetic tools for environmental monitoring of New Zealand's aquatic habitats, past, present and the future. N. Z. J. Mar. Freshwater Res. 47, 90–119. doi: 10.1080/00288330.2012.745885
Wood, S. A., Zaiko, A., Richter, I., Inglis, G. J., and Pochon, X. (2017). Development of a real-time polymerase chain reaction assay for the detection of the invasive Mediterranean fanworm, Sabella spallanzanii, in environmental samples. Environ. Sci. Poll. Res. 24, 17373–17382. doi: 10.1007/s11356-017-9357-y
Xiong, W., Li, H., and Zhan, A. (2016). Early detection of invasive species in marine ecosystems using high-throughput sequencing: technical challenges and possible solutions. Mar. Biol. 163:139. doi: 10.1007/s00227-016-2911-1
Yao, H., Song, J., Liu, C., Luo, K., Han, J., and Hansson, B. (2010). Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS ONE 5:e13102. doi: 10.1371/journal.pone.0013102
Yu, D. W., Ji, Y. Q., Emerson, B. C., Wang, X. Y., Ye, C. X., Yang, C. Y., et al. (2012). Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 3, 613–623. doi: 10.1111/j.2041-210X.2012.00198.x
Zaiko, A., Lehtiniemi, M., Narscius, A., and Olenin, S. (2011). Assessment of bioinvasion impacts on a regional scale: a comparative approach. Biol. Invasions 13, 1739–1765. doi: 10.1007/s10530-010-9928-z
Zaiko, A., Martinez, J. L., Ardura, A., Clusa, A., Borrell, Y. J., Samuiloviene, A., et al. (2015a). Detecting nuisance species using NGST: methodology shortcomings and possible application in ballast water monitoring. Mar. Environ. Res. 112, 1–9. doi: 10.1016/j.marenvres.2015.07.002
Zaiko, A., Martinez, J. L., Schmidt-Petersen, J., Ribicic, D., Samuiloviene, A., and Garcia-Vazquez, E. (2015b). Metabarcoding approach for the ballast water surveillance – an advantageous solution or an awkward challenge? Mar. Pollut. Bull. 92, 25–34. doi: 10.1016/j.marpolbul.2015.01.008
Zaiko, A., Samuiloviene, A., Ardura, A., and Garcia-Vazquez, E. (2015c). Metabarcoding approach for nonindigenous species surveillance in marine coastal waters. Mar. Pollut. Bull. 100, 53–59. doi: 10.1016/j.marpolbul.2015.09.030
Zaiko, A., Schimanski, K., Pochon, X., Hopkins, G. A., Goldstien, S., Floerl, O., et al. (2016). Metabarcoding improves detection of eukaryotes from early biofouling communities: implications for pest monitoring and pathway management. Biofouling 32, 671–684. doi: 10.1080/08927014.2016.1186165
Zhan, A., Bailey, S. A., Heath, D. D., and MacIsaac, H. J. (2014). Performance comparison of genetic markers for high-throughput sequencing-based biodiversity assessment in complex communities. Mol. Ecol. Resour. 14, 1049–1059. doi: 10.1111/1755-0998.12254
Zhan, A., Hulak, M., Sylvester, F., Huang, X., Adebayo, A. A., Abbott, C., et al. (2013). High sensitivity of 454 pyrosequencing for detection of rare species in aquatic communities. Methods Ecol. Evol. 4, 558–565. doi: 10.1111/2041-210X.12037
Zhang, D., Lee, D.-J., Zhang, M., Tippetts, B. J., and Lillywhite, K. D. (2016). Object recognition algorithm for the automatic identification and removal of invasive fish. Biosyst. Eng. 145, 65–75. doi: 10.1016/j.biosystemseng.2016.02.013
Keywords: non-indigenous species, early detection, metabarcoding, eRNA, species-specific assays
Citation: Zaiko A, Pochon X, Garcia-Vazquez E, Olenin S and Wood SA (2018) Advantages and Limitations of Environmental DNA/RNA Tools for Marine Biosecurity: Management and Surveillance of Non-indigenous Species. Front. Mar. Sci. 5:322. doi: 10.3389/fmars.2018.00322
Received: 24 May 2018; Accepted: 22 August 2018;
Published: 19 September 2018.
Edited by:
Stelios Katsanevakis, University of the Aegean, GreeceReviewed by:
Thierry Comtet, Station Biologique de Roscoff, FranceAibin Zhan, Research Center for Eco-environmental Sciences (CAS), China
Copyright © 2018 Zaiko, Pochon, Garcia-Vazquez, Olenin and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anastasija Zaiko, QW5hc3Rhc2lqYS5aYWlrb0BjYXd0aHJvbi5vcmcubno=