 Daniel Petras1*
Daniel Petras1* Irina Koester2
Irina Koester2 Ricardo Da Silva1
Ricardo Da Silva1 Brandon M. Stephens2
Brandon M. Stephens2 Andreas F. Haas3
Andreas F. Haas3 Craig E. Nelson4
Craig E. Nelson4 Linda W. Kelly5
Linda W. Kelly5 Lihini I. Aluwihare2*
Lihini I. Aluwihare2* Pieter C. Dorrestein1*
Pieter C. Dorrestein1*- 1Collaborative Mass Spectrometry Innovation Center, University of California, San Diego, La Jolla, CA, United States
- 2Scripps Institution of Oceanography, University of California, San Diego, La Jolla, CA, United States
- 3NIOZ Royal Netherlands Institute for Sea Research and Utrecht University, Texel, Netherlands
- 4Center for Microbial Oceanography: Research and Education, Department of Oceanography and Sea Grant College Program, University of Hawai‘i at Mānoa, Honolulu, HI, United States
- 5Department of Biology, San Diego State University, San Diego, CA, United States
Dissolved organic matter (DOM) is arguably one of the most complex exometabolomes on earth, and is comprised of thousands of compounds, that together contribute more than 600 × 1015 g carbon. This reservoir is primarily the product of interactions between the upper ocean's microbial food web, yet abiotic processes that occur over millennia have also modified many of its molecules. The compounds within this reservoir play important roles in determining the rate and extent of element exchange between inorganic reservoirs and the marine biosphere, while also mediating microbe-microbe interactions. As such, there has been a widespread effort to characterize DOM using high-resolution analytical methods including nuclear magnetic resonance spectroscopy (NMR) and mass spectrometry (MS). To date, molecular information in DOM has been primarily obtained through calculated molecular formulas from exact mass. This approach has the advantage of being non-targeted, accessing the inherent complexity of DOM. Molecular structures are however still elusive and the most commonly used instruments are costly. More recently, tandem mass spectrometry has been employed to more precisely identify DOM components through comparison to library mass spectra. Here we describe a data acquisition and analysis workflow that expands the repertoire of high-resolution analytical approaches available to access the complexity of DOM molecules that are amenable to electrospray ionization (ESI) MS. We couple liquid chromatographic separation with tandem MS (LC-MS/MS) and a data analysis pipeline, that integrates peak extraction from extracted ion chromatograms (XIC), molecular formula calculation and molecular networking. This provides more precise structural characterization. Although only around 1% of detectable DOM compounds can be annotated through publicly available spectral libraries, community-wide participation in populating and annotating DOM datasets could rapidly increase the annotation rate and should be broadly encouraged. Our analysis also identifies shortcomings of the current data analysis workflow that need to be addressed by the community in the future. This work will lay the foundation for an integrative, non-targeted molecular analysis of DOM which, together with next generation sequencing, meta-proteomics and physical data, will pave the way to a more comprehensive understanding of the role of DOM in structuring marine ecosystems.
Introduction
On the surface of the ocean, unicellular photosynthetic organisms fix as much atmospheric CO2 into organic carbon as their terrestrial, multicellular counterparts, despite the standing biomass of marine primary producers being just 1% of the terrestrial biosphere (Siegenthaler and Sarmiento, 1993). This is the result of a fast and efficient recycling of biomass in this system. A significant fraction of the photosynthetic production is channeled through the dissolved organic matter (DOM) reservoir (Halewood et al., 2012; Stephens and Aluwihare, in review); and recycled by bacteria (Azam and Malfatti, 2007). The DOM reservoir comprises 600 Gt C, making it the largest reduced carbon (C) reservoir in the ocean and comparable in size to the atmospheric CO2 reservoir. In many areas of the ocean, DOM also represents the largest reservoir of nitrogen (N) and phosphorus (P), both essential nutrients for supporting marine food webs (Karl and Björkman, 2002; Sipler and Bronk, 2015). As such, DOM is a crucial intermediate in the cycling of carbon and nutrients in our oceans. Despite the central role of DOM in the ocean's elemental cycles, the identities of molecules underpinning this massive recycling effort and the microbial metabolisms that sustain it, remain poorly understood. The identification of molecules is however pivotal to determine accurate rates of remineralization and comprehensively understand the microbial community metabolism. This knowledge will better inform our predictions of how climate change may impact the size and composition of the DOM reservoir and thus, the future of atmospheric CO2 sequestration in the ocean.
From an analytical perspective DOM poses a special challenge. A single sample can be comprised of tens of thousands of individual molecules that together rarely exceed 1 mg C/L. The true chemical complexity of DOM is unknown because extraction methods capable of isolating this fraction from the much more abundant salts in seawater are not 100% efficient. The most widely used method is solid phase extraction (SPE) using the sorbent PPL, a proprietary functionalized, reversed phase, hydrophobic, styrene-divinylbenzene polymer (Dittmar et al., 2008). This resin typically isolates between 40 and 50% of DOC from the surface ocean with a molar C:N ratio of 20–30 (Dittmar et al., 2008; Arakawa et al., 2017) and contains various proportions of other elements including phosphorus (P) and sulfur (S) as detected primarily by mass spectrometry (Lechtenfeld et al., 2011; Herzsprung et al., 2016). This fraction is poorer in N and P than bulk DOM (Hopkinson and Vallino, 2005; Letscher and Moore, 2015) and is lacking in the more polar biomolecules such as dissolved proteins and carbohydrates (Hertkorn et al., 2013) consistent with the hydrophobic characteristics of the PPL resin. However, even this fraction shows marked and unique complexity, which is exemplified in Fourier transform ion cyclotron resolution (FT ICR) mass spectra of this PPL-DOM. In a general sense, FT ICR-MS has shown that PPL-DOM contains thousands of molecules with peaks located around every nominal mass unit between 200 and 1,000 m/z, with the most abundant masses concentrated between 300 and 400 m/z (D'Andrilli et al., 2010; Flerus et al., 2012; Hawkes et al., 2016; Zark et al., 2017). Although larger molecules are known to be present in DOM, various analyses confirm that most (at least 60%) of DOM is < 1 kDa (Guo et al., 1995; Dittmar and Kattner, 2003; Benner and Amon, 2015). Formula assignments to FT ICR-MS ions have provided further insight into elemental ratios and thus compound diversity (Dittmar and Paeng, 2009; Flerus et al., 2012; Hawkes et al., 2016; Herzsprung et al., 2016; Lucas et al., 2016; Osterholz et al., 2016), but molecular structures capable of identifying sources and cycling have remained elusive.
Recently, studies of DOM have focused on targeted molecules relevant for particular biogeochemical processes. These molecules have been identified in culture experiments and/or detected in field samples to highlight some of the important microbial interactions in the surface ocean (Amin et al., 2015; Johnson et al., 2016; Repeta et al., 2016; Heal et al., 2017; Kujawinski et al., 2017). Targeted metabolomic studies focus on individual, well-characterized substrates whose concentrations and cycling can be followed precisely in incubations and in field settings. Advantages of such an approach are clear – in general, targeted work is quantitative, precise, and the metabolic role of identified substrates may already be well established (Amin et al., 2015; Johnson et al., 2016; Repeta et al., 2016; Heal et al., 2017).
Still, thousands of unidentified molecules are present in DOM and uncovering their roles in elemental cycling and marine microbial ecology requires an unconstrained and non-targeted approach. Non-targeted studies aim to examine temporal and spatial variability of all detectable metabolites (specific to isolation method and analytical method). With the appropriate data analysis tools, this approach has the power to identify relevant metabolites and “metabolic interdependencies” at a faster pace (Sogin et al., 2017). Such an approach can also inform targeted approaches including those with stable isotope labeling studies and expression/transcription studies, for instance. Merits of both approaches are clear and they both have the ability to provide new insights and advance the study of DOM dynamics. The guiding scientific questions should determine the appropriate approach (Hawkes et al., 2016; Moran et al., 2016) and in the some cases, they can be combined (Kujawinski et al., 2017). The superior mass resolution of FT ICR-MS instruments approximates the compositional diversity of DOM most closely (Kujawinski et al., 2002; Reemtsma et al., 2008; Romano et al., 2014; Zark et al., 2017). Yet orbital iontrap MS instruments possess some advantages that, despite their slightly lower mass resolution, can augment the molecular level investigations of DOM. Besides the dependency of resolution and thus mass accuracy for molecular formula assignments, an interesting advantage of orbital ion traps, especially new generation high-field orbitraps, is the significantly higher scan speed (Makarov et al., 2009; Scheltema et al., 2014), also provided by high-end Time-of–Flight (TOF) mass analyzers. Fast scan speed is an essential requirement for the coupling with liquid chromatography. This enables structural annotations, which provide molecular level identification, through database comparison of tandem MS spectra and/or retention times (Vinaixa et al., 2016). Such an approach is common in metabolomic studies and has been extensively reviewed (Liesenfeld et al., 2013; Viant and Sommer, 2013; Gika et al., 2014; Rubert et al., 2015).
In non-targeted LC-MS/MS experiments, tandem mass spectra are often acquired in data dependent acquisition (DDA), where the mass spectrometer decides in real time based on MS1 survey scans which ions to submit for subsequent MS/MS scans. This approach paired with high acquisitions speed (>1 Hz) of state of the art instruments results in thousands of spectra per LC-MS/MS run. For a reliable data analysis and reproducible interpretation of the results, bioinformatic workflows including comprehensive databases and statistical significance estimation are crucial (da Silva et al., 2015; Böcker, 2017; Scheubert et al., 2017; Weber et al., 2017) and have been very recently employed for marine metabolomic studies (Quinn et al., 2016; Hartmann et al., 2017; Kujawinski et al., 2017; Longnecker and Kujawinski, 2017). With these new bioinformatic tools and instrumental improvements in sensitivity, acquisition speed and resolution we anticipate that the techniques used for DOM characterization will further shift toward non-targeted analyses using high-resolution LC-MS/MS that provide inventories of molecular structures in complex environmental datasets. Therefore, the adaption and standardization of marine LC-MS/MS based metabolomics, an essential requirement for inter-dataset comparison, will be of high interest to the aquatic sciences community. Here we describe the implementation and assessment of an LC-MS/MS based workflow, capable of identifying untargeted metabolites in complex marine environmental samples. The addition of LC separation reduces sample complexity prior to ionization, which reduces ion suppression effects and improves quantification. The chromatographic separation also provides another dimension of information (retention time) for comparison across samples. ESI is a routinely applied ionization method for LC-MS based metabolomics, despite its selectivity toward polar compounds, as it is well suited to metabolite identification (Zhou et al., 2012). Identifying dissolved metabolites is a primary goal as it provides information that can be effectively compared with genomic and proteomic datasets and is essential for providing an integrative understanding of marine microbial communities. Untargeted metabolite identification is substantially improved by acquiring MS/MS information and coupling with a high throughput workflow used for the first time here to study DOM.
Experimental Concept
Our analytical workflow, shown in Figure 1, starts with a SPE enrichment using PPL resin (Dittmar et al., 2008). After extraction, we analyzed the compounds by ultra-high performance liquid chromatography (UHPLC) coupled through ESI to an orbital ion trap. The experimental design relies on high resolution MS1 experiments with orthogonal data acquisition in both positive and negative ionization modes. Subsequently, we also recorded positive high-resolution MS/MS information in data dependent acquisition (DDA) mode. The data analysis begins with MS1 features and relies on an initial creation and alignment of extracted ion chromatograms (feature extraction) using the software tool MZmine2 (Pluskal et al., 2010). The alignment of features was performed in two dimensions: first, between all samples, which resulted in a consensus feature table; and second, between positive and negative mode data to define consensus features of high confidence. After feature extraction from MS1, we calculated molecular formulas based on exact masses of individual and consensus features. In parallel, we performed clustering of identical MS/MS spectra and multiple spectra alignments using the Global Natural Product Social molecular networking (GNPS) (Wang et al., 2016). This analysis defines spectral proximity between all MS/MS spectra of a dataset and visualizes them in a spectral network (Watrous et al., 2012). For the molecular annotation we compared the MS/MS spectra to a spectral library including the GNPS community contributed spectral library as well as Massbank, ReSpect, HMDB, and NIST14 (Forsythe and Wishart, 2009; Horai et al., 2010; Sawada et al., 2012; Stein, 2014; Wang et al., 2016). The overall goal of such an analysis is to enable robust, untargeted comparison of multiple samples at the molecular level. Once features are defined, annotated and connected to sample metadata, molecular features can then be used for multivariate data reduction and visualization such as Principal Coordinate Analysis (PCoA) (Anderson and Willis, 2003) or multivariate component classification approaches such as Random Forest (Breiman, 2001). If spatial information is available, molecular intensities can be displayed on geographical coordinates to create extracted ion maps (Petras et al., 2017).
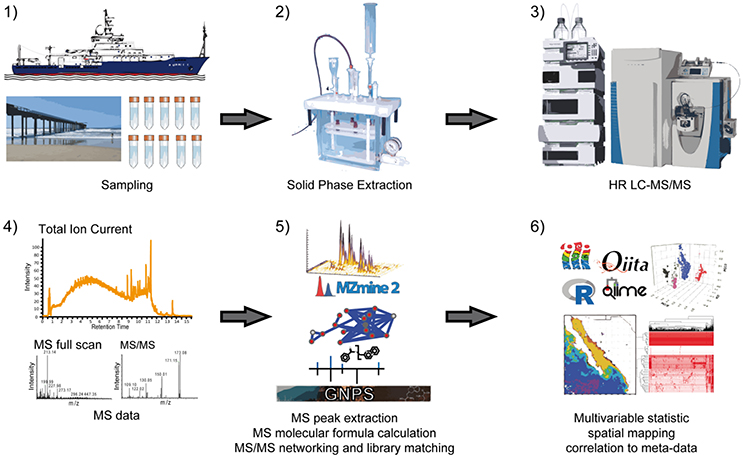
Figure 1. Overview of data acquisition and data analysis workflow. After sampling of sea water (1), dissolved organic matter is extracted by solid phase extraction (2) and samples are analyzed by non-targeted high-resolution liquid chromatography tandem mass spectrometry (HR LC-MS/MS) in data dependent acquisition mode (3). Following raw data acquisition (4), extracted ion chromatograms (XICs) are created and relative feature intensities are defined through integration of peak areas (5). For molecular annotation of features, molecular formulas based on exact masses of the XICs from negative and positive ionization experiments are calculated and spectrum library comparison of MS/MS spectra as well as spectral networking is performed (5). To facilitate the analysis of data, multivariable statistics such as Principal Coordinate Analysis (PCoA) can be performed in order to display sample-sample distance, and metabolite data can be interpreted in the context of oceanographic features and microbial community composition (6).
Results and Discussion
The overall goal of this study was to devise an LC-MS/MS data acquisition and analysis workflow that holds the potential to access the molecular level complexity of the marine DOM reservoir. The biggest barrier to any comprehensive molecular level survey of DOM composition is that a salt-free, concentrated sample is required, which means that DOM must be isolated from seawater (Dittmar et al., 2008). Most of the widely available methods for extracting DOM can only isolate ≤ 60% of the total dissolved organic carbon (DOC) in seawater. We chose to extract DOM from seawater using the modified styrene divinyl benzene polymer resin, PPL (Varian Bond Elut, Agilent Technologies, USA), as it is currently the most widely used method for extracting DOM from seawater (Dittmar et al., 2008). Before describing our LC-MS/MS protocol in detail we summarize the result of simple tests conducted to optimize the extraction protocol and to define a standard protocol for our future high throughput, open ocean, metabolic profiling. We tested five seawater replicates each of three different volumes (100, 500, and 1,000 mL) from a single, homogenized seawater sample from the Pacific Ocean (Scripps Pier, February 2018), that had been pre-filtered using a 0.2 μm cutoff, membrane filter (Acropak, Pall) to remove particles and large bacteria. Seawater was extracted with either 0.2 g (100, 500, and 1,000 mL) or 1.0 g (1,000 mL) of resin mass. We varied extraction volumes to determine the lower limit of extraction volume that was capable of providing representative compositional information.
Extraction Efficiency
We assessed extraction efficiency by measuring DOC concentrations in the bulk seawater sample and permeate of each replicate. Detailed results are shown in Figure S2 and Table S1. The input seawater had a concentration of approximately 70 μmol L−1. For the 0.2 g cartridges, the highest extraction efficiency calculated based on the permeate concentration was 54.4 ± 1% (mean ± SD) and was observed for the 100 mL samples, followed by 47.4 ± 3% and 42.3 ± 1% for 500 and 1,000 mL respectively. The 1.0 g cartridge extracted more DOC from 1,000 mL (53.1 ± 2.3%) than the 0.2 g cartridge, as would be expected. The data also suggested that the reactive sites on the 0.2 g bed mass cartridge became saturated at seawater volumes ≥500 mL. Extraction efficiencies calculated using the concentration of DOC that was eluted from the resin exhibited the same trend but were lower than those calculated from permeate concentrations. This is likely due to the fact that not all compounds extracted onto PPL resin could be eluted with methanol, but could also result from sample processing following the elution (e.g., drying). The approximate amount of injected carbon ranged from an average of 0.19 μmol for the 100 mL samples to 0.83 μmol for 500 mL to 1.48 μmol for the 1,000 mL samples extracted with 0.2 g PPL and 1.86 μmol per injection for the 1,000 mL samples extracted with 1 g PPL. The reported accuracy of injection concentrations is linked to the concentration of DOC in permeates of each SPE extraction and the dilution volume.
Global Analysis of Total Ion Currents and Extracted Ion Chromatograms
The simplest assessment of DOM MS data was performed by comparing overall intensities of total ion currents (TIC) (Figure 2). The featureless TIC, regardless of injected concentration, highlighted the overall complexity of the PPL-extracted DOM sample, and thus the depth of the analytical challenge faced by DOM analysts. As expected, the two 1,000 mL samples, which had a higher concentration, gave the highest response. The intensity of their TICs only differed in the early retention time range, where the 1,000 mL sample extracted onto 0.2 g of PPL resin showed a lower response. This difference indicated that polar compounds were less efficiently retained as the resin became saturated and is a response that is consistent with the hydrophobic nature of PPL.
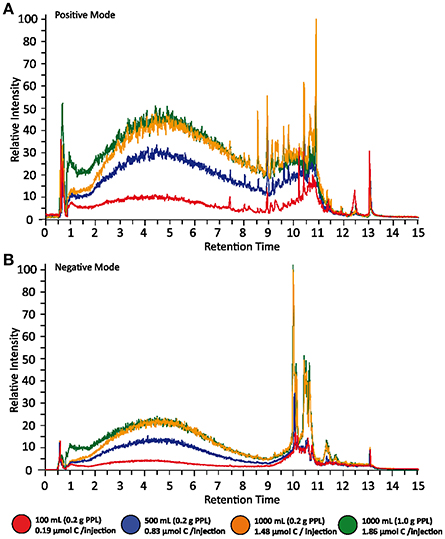
Figure 2. Total Ion Current (TIC) of different extraction volumes in positive (A) and negative (B) mode. Representative LC-MS chromatograms of five replicates of different water volumes are shown. Data was acquired in both positive and negative ionization mode. The relative TIC intensity is directly related to the abundance of analytes and shows a clear trend between the different amounts of total carbon injected. The signals of one liter samples extracted with different extraction cartridges (0.2 vs. 1.0 g bed mass) show the biggest difference in the lower retention time range, indicating that biases due to MS based extraction efficiencies mainly affects polar compounds.
The overall number of features (defined as a single peak in an extracted ion chromatogram; XIC) in each sample was identified based on thresholds and deconvolution settings in the software tool used (MZmine2) and the strict requirement that features must appear in the XIC of 4 of 5 replicates. After blank subtraction, it was found that the number of features increased with injection concentration and that more features were identified in positive mode than negative mode. The overall sum of features observed in positive and negative mode across all groups was 13,987 and 7,328. The number of positive (negative) features (Figure S3) increased from 1,235 (465) in the lowest concentration samples (100 mL extraction volume) to 5,653 (2,715) of the medium concentrated samples (500 mL extraction volume) to 7,766 (3,408) and 8,167 (4,217) features in the highest concentrated samples (1,000 mL/0.2 g and 1,000 mL/1 g) respectively. These data highlight the concentration dependency of the mass spectrometer, which needs to be carefully controlled when performing comparative analyses. The feature tables are available in the Supporting Information of this article.
The two samples with the highest extraction efficiency (100 mL/0.2 g and 1,000 mL/1 g) also had the highest proportion of unique MS features in positive mode (45 and 41%, respectively, compared with 22 and 33% for the other two samples, absolute values shown in Table S2). The trend was similar for features identified in negative mode as well but all samples exhibited a higher proportion of unique features in negative mode. The observed similarity of the two samples with high extraction efficiencies may have been a coincidence, but is not unexpected given that polar compounds, for example, were extracted more efficiently if the PPL column was not saturated. Besides differences in sample composition, different injection concentrations affect signal/noise ratios and can result in concentration dependent changes in peak shape, which can lead to retention time misalignment between different sample groups and thus more unique features. More relaxed retention time tolerances or stricter filtering toward minimum repetition of features will likely alleviate this problem. If samples are likely to be low in DOC concentration then injecting a short dilution series will be useful.
Molecular Formula Assignment
From a global dataset perspective, the number of features with assigned formulas within a 5 ppm mass error tolerance increased with injected sample concentration. This finding is consistent with the hypothesis that low S/N ratios will be omitted from the molecular formula assignment calculation at lower injection concentrations. For instance, the number of features with assigned formulas within a 5 ppm mass error tolerance is slightly lower for the smaller extraction volumes (shown in Figure 3B). This can be explained by the assumption that some features were either noise or had a molecular composition which was not considered through our calculation parameters (e.g., contained elements other than C, H, O, N, and S, Na, Cl).
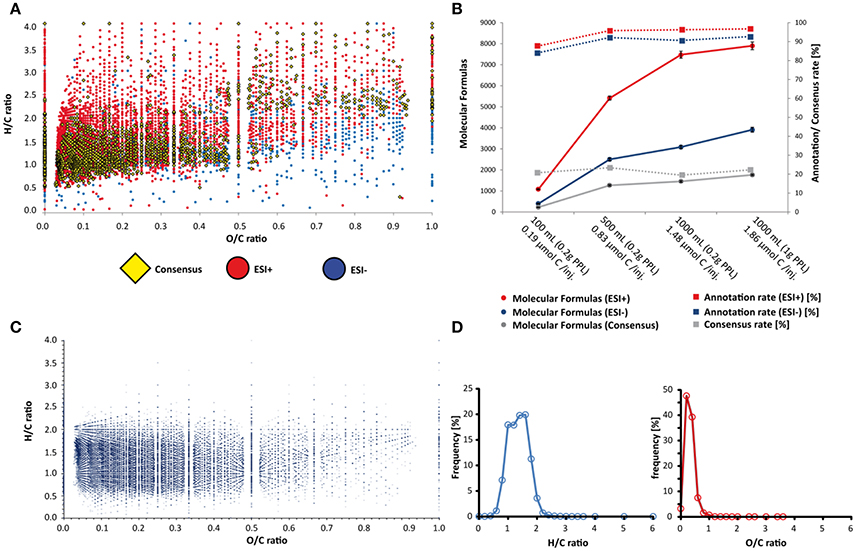
Figure 3. Visualization of molecular diversity by Van Krevelen diagram and feature distribution over sample and chemical space. In (A) the H/C vs. O/C ratios of computed molecular formulas from ESI+ and ESI– mode as well as from consensus features (matched XIC of at least two ion species/ adducts from both ESI+ and ESI– scans) are shown. In (B) total feature numbers and formula annotation rates are shown for different ionization modes and plotted against the different extraction volumes. In B, features are labeled based on extraction volumes and total injected carbon. (C,D) displays the Van Krevelen plot and frequency distributions of 233,692 molecular formulas from the supernatural chemical database (Banerjee et al., 2015).
To increase the confidence of molecular formula annotation, we made our data analysis more stringent through the alignment of XICs from both positive and negative ionization modes. This alignment groups different ion species from the same molecule e.g., adducts (M+H+, M+Na+, M-H+, and M+Cl−). If two or more matching ion species were aligned, a consensus molecular formula (the highest ranked common formula) was created. The overall number of consensus features of all volume groups was 3,060 and resulted in 2,600 molecular formulas (shown in the Supporting Information). To display the chemical space of the molecular formulas observed in the different groups, we created Van Krevelen diagrams, displaying the H/C vs. O/C ratios of the molecular formulas, differentiated between positive mode, negative mode and consensus formulas (Figure 3A).
In order to loosely categorize “likely” and “unlikely” H/C and O/C ratios in DOM, we also mapped out the distributions of previously characterized metabolite formulas in Van Krevelen space using structures < 500 Da from the Supernatural database (Banerjee et al., 2015), which includes molecular formulas from more than 230,000 natural products. The Supernatural Van Krevelen plot and density plots of H/C and O/C ratios are shown in Figures 3C,D. The highest density of molecules in the Supernatural database is constrained by H/C ratios between 0.5 and 2.5 and O/C ratios between 0 and 0.5. There is a striking resemblance between this area and the hotspot region of H/C vs. O/C ratios linked to consensus (or shared) features between ionization modes and between DOM sample groups (Figures S7, S8). As such, we have higher confidence in molecular formula annotations for DOM features that appear in these Van Krevelen regions. Many masses resulted in multiple possible molecular formulas within our mass tolerance constraints, and our analysis displays only the best match (highest ranked consensus formula for various adducts). Therefore, the elemental ratio information provided by our high confidence region is more appropriate for describing a statistically significant global sample composition. However, such an approach may not be suitable for confident annotation of particular ion species. Furthermore, post-production modification of metabolites in seawater will likely drive some H/C and O/C ratios away from those predicted by a metabolite database (e.g., loss of unsaturation, photo-oxidation; Arakawa et al., 2017), and so, while the comparison with the Supernatural database is useful for constraining our dataset, we cannot exclude the remaining molecular formulas without further scrutiny of the data.
Tandem Mass Spectrometry and Spectral Networking
Statistical comparison to known compounds from large scale libraries can help to increase the confidence of molecular formula assignments. The frequency of individual molecular formulas in the supernatural database provides an empirical basis to demonstrate that many molecules often share the same molecular formula, but not the same chemical structures. The frequency of molecular formulas in this database ranges from many unique to several 100 redundant formulas, with an average of 5.5 structures per molecular formula. Chromatographic retention times (e.g., Figures S4, S5) could be used to differentiate between individual molecules with identical formulas. However, absolute and relative retention times often vary between studies due to differences in LC gradients, HPLC column type, and instrument conditions. Thus, relying on MS1 features, even when molecular formula assignments are robust, is not a suitable characteristic for compound library searches, unless the standardized methods are used for library generation and sample analysis.
For this reason, we examined the efficacy of tandem MS in data driven acquisition (DDA) mode, to provide molecular level information for DOM in an untargeted context. Unlike MS1 feature detection and molecular formula assignment, MS/MS spectra do not depend on chromatographic reproducibility and allow comparison between different instrument platforms if the same fragmentation methods and similar fragmentation energies are used. Tandem mass spectra matching to library entries are considered level two annotations according to the 2007 metabolomics standards initiative (Sumner et al., 2007) and probably the most effective approach for high-throughput annotation of small molecules in complex samples. In our workflow, we acquired MS/MS spectra in top 5 DDA mode, in which up to five precursor ions are submitted for MS/MS after each MS1 survey scan. With a medium resolution (Rm/z 200 = 17.000), the iterative scan cycles result in several thousand spectra per sample. To reduce redundancy and computation time we clustered identical spectra to consensus spectra (Frank et al., 2008) and searched them against several spectral libraries, including GNPS (Wang et al., 2016), MassBank (Horai et al., 2010), ReSpect (Sawada et al., 2012) HMDB (Forsythe and Wishart, 2009), and NIST 2014 (Stein, 2014). Hereby a strict precursor mass tolerance of m/z 0.01 was used first, followed by a maximum allowed precursor delta mass of up to m/z 100, with the intention to annotate putative analogs. Besides dereplication of known compounds from the libraries, an additional multiple spectral alignment of all clustered spectra was performed. Thereby, all spectra are compared to each other using cosine similarity scoring (Stein and Scott, 1994) and spectrum-spectrum matches, with a cosine score higher or equal to 0.7 are connected as “nodes” in a network (Watrous et al., 2012; Wang et al., 2016). If nodes yielded a database hit during a spectrum library search, they can be labeled as specific compounds and nodes around the “hit” can be assumed to have similar chemical scaffolds. If the delta masses, fragmentation patterns and chemical formulas of the database hit are carefully interpreted, putative structures of previously unknown spectra can be proposed with a certain confidence.
The spectral network of all samples acquired is shown in Figure 4A. Nodes that contained MS/MS spectra from blanks were subtracted from all samples. Depending on the origin of clustered spectra, nodes were labeled gray (100 mL, 0.2 g PPL), orange (500 mL, 0.2 g PPL), green (1,000 mL, 0.2 g PPL), and red (1,000 mL, 1 g PPL) if spectra came exclusively from this group. If a node contained spectra from more than one group, it was considered as shared feature and was labeled blue. The size of the nodes was increased proportionally to the number of samples which contained a particular MS/MS spectrum. Nodes with a library hit were shown with a diamond shape with a thick border if the precursor matched within 10 ppm, and as a thin line for hits observed with higher precursor delta masses (from analog search). Overall, we observed 6,272 nodes. Of these only 32 had an associated positive library hit. We also evaluated MS/MS information in the context of extraction volume and total injected carbon, by examining the total number of nodes contributing from each concentration/extraction group, as well as by tallying the number of unique nodes per group (Figure S6 as well as in a Table S2). The highest number of both total nodes and unique nodes were identified in the lowest concentration sample group (0.19 μmol injected C). The remaining samples had a smaller number of nodes and unique nodes, but the 1,000 mL/1 g PPL group had an elevated number of both types of nodes when compared with the other two concentration groups. This trend with respect to injection concentration was different from that observed for feature number and unique feature numbers as determined from MS1 (Figure S6, right). In that case, the number of both node types increased with injection concentration. In order to generate a metric for assessing the quality of the data resulting from differences in injection concentration, we investigated spectral annotation rates (Figure S6). Spectral annotation rates were low across all groups but the lowest annotation rate was found for the lowest injection concentration (100 mL extraction volume). Thus, while the number of nodes (total and unique) was highest for the lowest concentration injection, these nodes were annotated with lower efficiency. In addition, this sample group also showed a high number of self-looped nodes (i.e., many spectra were not connected to other nodes). The highest concentration injection groups (1.48 and 1.86 μmol injected C) had fewer self-looped nodes. Together, these observations suggest that the quality of MS/MS spectra generated from the lowest concentration group was so poor (low S/N) that even identical spectra could not be confidently identified. Thus, we propose the use of spectral annotation rate and number of unique nodes (Figure S6, left) as a quality control check, to determine whether certain samples in a dataset are likely to be poorly represented because they were injected at too low of a concentration.
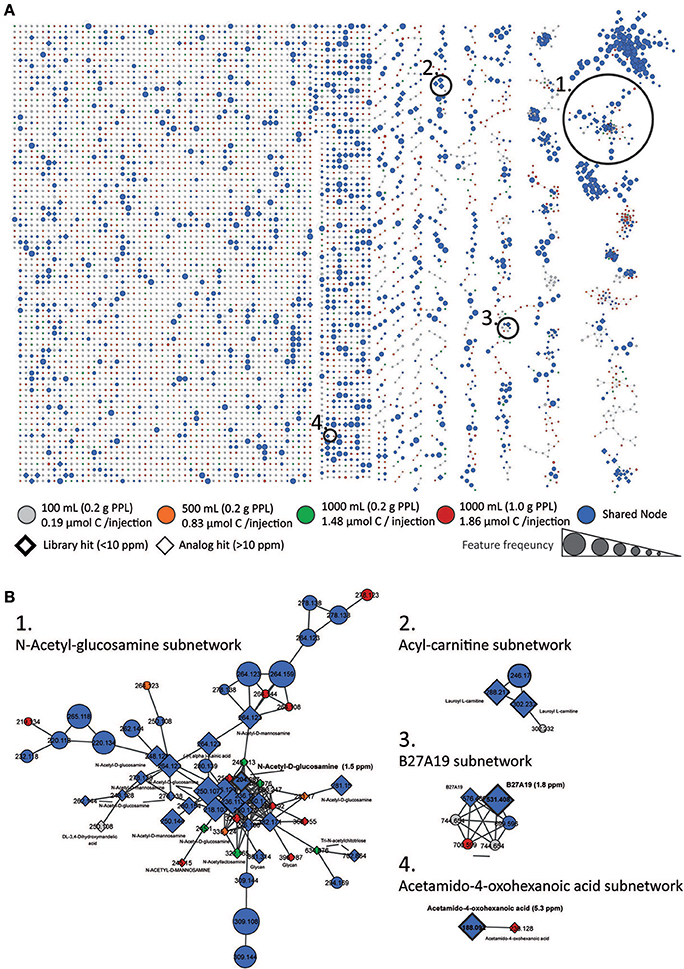
Figure 4. Molecular Network. Global spectral network from all sample groups is shown in (A). Nodes from blanks were subtracted. According to origin, nodes were labeled gray (100 mL, 0.2 g PPL, 0.19 μmol injected C), orange (500 mL, 0.2 g PPL, 0.83 μmol injected C), green (1,000 mL, 0.2 g PPL, 1.46 μmol injected C) and red (1,000 mL, 1 g PPL, 1.86 μmol injected C) if spectra came exclusively from this group. If a node was shared between two or more groups it was labeled blue. The size of the nodes represents the sample frequency, i.e. the number of samples which contained a particular MS/MS spectrum. Nodes with a library hit are displayed in diamond shape with a thick boarder line if the precursor matched within 10 ppm and with a thin line for hits with higher delta masses (from analog search). In (B) zoomed subnetworks of selected molecular families are shown.
The overall library annotation rate of the dataset was at 0.5%, and for the two 1,000 mL (1.48 and 1.86 μmol injected C) groups around 1%. The low annotation efficiency is likely a result of a combination of factors, all of which need to be addressed in future work.
One important reason for the low annotation efficiency is linked to the fact that more than one molecule is isolated prior to fragmentation in the collision cell, which results often in chimeric spectra (i.e., DOM MS/MS spectra are often a combination of fragments from multiple molecules with very similar masses that could not be separated by the unit resolution of the quadrupole, and naturally, yield lower matching scores to library MS/MS spectra). The general field of metabolomics has been grappling with the issue of chimeric spectra. Besides technical improvements in chromatographic or gas phase precursor separation (multi-dimensional chromatography or ion mobility), repetitive or large scale analysis of different samples could help to bypass this problem. Here, a possible solution could be an alignment of numerous chimeric spectra and searching for consensus fragments. However, the bioinformatic tools for the detection (Lawson et al., 2017) and separation of chimeric spectra are still in development. In addition, low annotation rates also result from the low number of available library spectra that match compounds in DOM. Nevertheless, the reported annotation rate is in a similar range as other non-targeted metabolomic datasets (Bouslimani et al., 2015; Petras et al., 2016; Floros et al., 2017) that are typically less complex than DOM.
This still poses a universal barrier to untargeted MS/MS analyses. This problem is independent from the data acquisition parameters and can be solved by expanding the chemical space in spectral libraries. Community driven databases such as GNPS (Wang et al., 2016) provide a good platform for researchers to contribute to the growth of spectral library knowledge, much like how nucleotide or protein databases were established in recent years (Bateman et al., 2004; Sayers et al., 2012).
Besides looking for perfect matches, spectral libraries were searched for spectral similarity/analog hits (the same way sequence libraries are used to search for homolog hits with BLAST Altschul et al., 1997). The analog search was set to allow precursors differences of up to m/z 100. Taking the resulting putative analogs into account, we could increase the overall annotation rate to 13.7% and to more than 20% for the two highest injection concentration groups. As a caution, we know that removing the stringent precursor mass filter can increase the rate of false positive hits. Therefore, analog hits should always be considered as putative annotations requiring subsequent statistical or ideally, manual validation. Assuming that the spectral networking approach connects similar spectra to subnetworks, then true library hits in a network should be surrounded by analog hits.
Following this logic, we could observe a subnetwork of amino-sugars (Figure 4B) containing one node with a library hit to N-acetyl-glucosamine, which is surrounded by several other analog hits to amino sugars or glycans. The inclusion of the library hit increases our confidence in the analog annotations in this subnetwork of an amino-sugar molecular family. The finding is in line with evidence from other studies showing that amino sugars are highly abundant in marine DOM (Ogawa et al., 2001; Aluwihare et al., 2005; Davis et al., 2009). In other subnetworks acetamido-oxohexanoic acid could be identified and a node connected to it was annotated as well as acetamido-oxohexanoic acid, but with a delta mass of m/z 48.036. Furthermore, we could identify subnetworks with several hits to the marine natural product B27A19 from a marine sponge library, and another molecular family of acyl-carnitines, both of which contained analog hits to the same compounds in the subnetworks. All spectrum library mirror plots of the above mentioned database hits are shown Figures S9–S11.
To further evaluate the effect of concentration on spectral quality, we examined the distribution of MS/MS library matches across the different injection concentrations. Acetamido-oxohexanoic acid, for example, was found in at least 4 of 5 replicates across all samples except for the lowest concentration injections. N-Acetyl-glucosamine and B27A19 were present in the three highest concentration samples as well, but only occurred in some of the replicates. The underlying reason could be the automatic triggering of MS/MS acquisition through data dependent acquisition (DDA). If a precursor ion is not among the most abundant ions in a survey scan then it will not be selected for subsequent MS/MS. By using a dynamic exclusion list for precursors that had already been submitted, we assumed that DDA would consider most of the ions, depending on the complexity of a given time point in the LC run, and the scan speed of the mass spectrometer. Nevertheless, for medium and low abundant compounds, the machine might not have had enough time to acquire MS/MS scans, and for some compounds, small shifts in chromatographic profiles may have changed the order of MS/MS selection, which triggered MS/MS acquisition in some but not all samples. Repeated measurements can alleviate this bias and increase annotation rates.
To test the initial assumption of intensity dependency, we plotted the sample frequency (number of samples contributing to one consensus spectrum) of all network nodes, shown in Figure 5. Only around 110 consensus spectra where found in all four groups (20 samples, grouped into five replicates of four different injection concentrations). The precursor intensity, plotted on the second y-axis, shows that these ions are among the most abundant compounds in DOM extracts (Figure 5). Looking at some of the chemical IDs, we observed that the fatty acid glycerin ester mono-laurine and the terpene dihydroactinidiolide, which shares structural similarity with the head group of beta-carotene, were present in all samples. In this context, it is very interesting that oxidation products of beta-carotene have been recently described as abundant in refractory DOM (Arakawa et al., 2017).
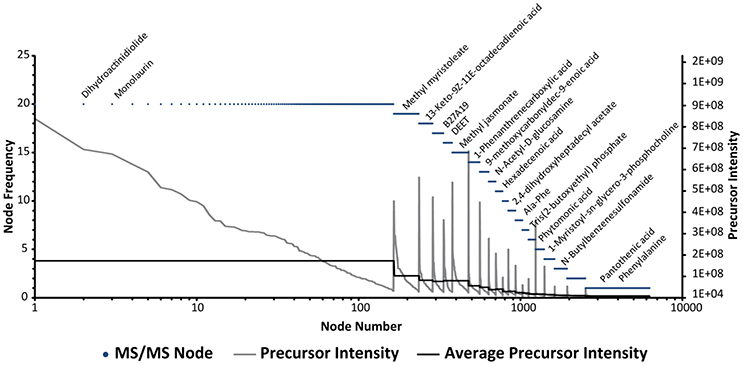
Figure 5. Network Node Frequency Plot. Frequency of MS/MS features in all 20 samples. Frequency is hereby defined as the number of samples contributing to one consensus spectrum. Each dot represents one network node. Library annotations of selected nodes are shown above. The order of nodes was sorted first by frequency and in a second level by precursor intensity which is drawn on the second y-axis. The average intensity of frequency bins is shown as a solid black line.
If we inspect the nodes that occur at lower frequencies, we can see that the average precursor intensity decreases accordingly, and that only around 10% of the nodes are shared by 10 or more samples (mainly from the 1,000 mL groups with 1.46 and 1.86 μmol injected C). Of the low abundance compounds, more than 50% of all nodes are only found in one sample. Pantothenic acid, a cofactor involved in fatty acid and secondary metabolite biosynthesis, provides an example of such a compound. Next to the above described problem of precursor selection, chimeric spectra, as noted previously, may be another reason for the high number of unique nodes. Chimeric spectra can result in an artificial diversification of MS/MS patters, which then appear as unique nodes. The repetitive analysis of the same sample can help to bypass this problem, for example through an alignment of chimeric spectra and search of consensus fragments. However, the bioinformatic tools for the detection (Lawson et al., 2017) and separation of chimeric spectra are still being developed in our data analysis workflow. Another missing piece of the software aided data analysis is the connection of MS1 and MS/MS features. Due to lower sensitivity of MS/MS and the overall higher numbers of MS1 features, our results indicate that for a comprehensive analysis of DOM, we need to merge MS1 feature tables and the networking database dereplication output. Currently, this is typically done either manually or with simple search functions in R or excel. The matching often results in difficulties, especially if precursor masses differ slightly from averaged MS1 masses from XICs or if the MS/MS event was triggered too far off the apex of the chromatographic peak. A more streamlined approach for large scale data analysis would likely involve implementing an MS/MS scan number assignment to XICs during MS1 feature extraction. Once established, this link could be used downstream during data analysis to link network nodes back to MS1 features and to reduce redundancy in the network, providing the most comprehensive molecular characterization of DOM. Most recently, such an approach has been described for MZmine2 (Olivon et al., 2017), but the software implementation and usage of the workflow is not yet straightforward. Recently, we tested a more user-friendly graphical software module directly implemented in MZmine2, which is now available within the latest version (MZmine2.28). We caution that the evaluation of this tool is still ongoing.
Conclusion
Our results show the successful implementation and assessment of non-targeted LC-MS/MS workflow for the analysis of DOM. We tested different sample volumes and sample volume to cartridge bed mass ratios. Both MS1 and MS/MS results indicate that the 1,000 mL sample groups with higher total carbon concentration, showed the most features and most database annotations. Given the general low variability of DOC concentrations in sea water (40–80 μmol L−1; Hansell, 2013), we suggest that 1,000 mL is an adequate and practical sample volume covering this DOC range. The 1 g cartridges showed slightly better results on MS1 level, but depending on the specific project may not warrant the considerably higher price. Furthermore, our results indicate that spectral annotation rate and unique node number are good predictors of when it is appropriate to compare compositional features in two samples. We recommend that these parameters are examined when comparing samples of significantly different concentrations. Concentrating samples with poor annotation rates and rerunning the series can alleviate this concern. For the global chemical characterization of DOM, our results show that through an alignment of chromatographic peak profiles and orthogonal ionization modes we could enhance the confidence in molecular formula annotations and reduce redundancy of different ion adducts. Through comparison to ~230,000 unique chemical structures from a natural product database we showed that the chemical space of our experimentally obtained molecular formulas falls in a similar range as those of known compounds. With the implementation of MS/MS networking and library dereplication specific structural annotations to observed ion species can be added. This allows for a more precise level of molecular annotation of DOM that will contribute toward advanced investigation of chemo-ecological relationships within marine ecosystems. We furthermore anticipate that, if data acquisition and data sharing is established and encouraged by funding agencies, the workflow presented here could be applied within multiple research groups in order to enable a global comparison of datasets at planetary scales.
Experimental Procedure
Sample Preparation and Solid Phase Extraction
Surface water from the Scripps Pier (La Jolla, USA) was collected with bucket and transported in a 20 L PTFE-carboy on February 2nd 2017 (10:05:39 (PST): temperature 14.58°C, chlorophyll a 0.47 μg/L; salinity 33.18 g/kg). The seawater sample was filtered through a membrane filter (0.2 μm pore size; Acropak, Pall) and adjusted to pH 2 with hydrochloric acid (38% p.a., LCMS trace metal grade, J.T. Baker, USA). The well mixed filtrate was poured into combusted glass bottles. Five replicates each of 100, 500, and 1,000 mL were extracted through 0.2 g bed mass PPL cartridges as well as five replicates of 1,000 mL through 1 g PPL cartridges.
Before use, the cartridges were rinsed and activated with one cartridge volume of methanol (LC-MS grade, Fisher Chemical, Belgium) and refilled with methanol for conditioning overnight (see Figure S1). Afterwards, the cartridges were rinsed with two cartridge volumes of water (LCMS grade, J.T. Baker, USA), two cartridge volumes of methanol and two cartridge volumes of water at pH 2 (acidified with HCl). For extraction, the filtered and acidified seawater [100 ml (0.2 g), 500 ml (0.2 g), 1,000 ml (0.2 g), 1,000 ml (1.0 g)] was gravity passed through each PPL cartridge with a flow rate below 5 ml min−1. Subsequently, remaining salt was removed with three cartridge volumes of pH 2 water. After drying with inert pure nitrogen gas, DOM was eluted with 3 ml (for 0.2 g PPL) and 6 ml (for 1.0 g PPL) of methanol into combusted glass vials. After taking an aliquot of extract for determining extraction efficiencies by direct concentration measurements (see below) the extracts were dried down with a vacuum centrifuge (Centrivap, labconco) and transferred in an amber glass GC vial. The extract was dried down again and stored at −20°C until later dissolved in 100 μL methanol for LC-MS/MS analysis. All glassware used during sampling and sample treatment was pre-combusted for 4 h at 450°C. All other materials were cleaned with acidified ultrapure water and rinsed with sample water before use. For verification of possible contamination, procedural blanks for each volume and cartridge size were performed by repeating the sample preparation with water (LC-MS grade, J.T. Baker, USA).
Dissolved Organic Carbon (DOC) and Extraction Efficiencies
DOC concentrations were analyzed by high-temperature catalytic combustion using a TOC-VCPH/CPN Total Organic Carbon Analyzer equipped with an ASI-V autosampler (Shimadzu, Japan). Standard solutions ranging from 10 to 100 μmol C L−1 were used for calibration and Deep Atlantic Seawater reference material (DSR, D. A. Hansell, University of Miami, Florida, USA) as well as Deep Pacific Seawater (CCE P1604) and Scripps Pier Water reference material were measured to control for instrumental precision (1 μmol L−1) and accuracy (1.5 μmol L−1). Aliquots of the acidified filtrate (pre-extraction) were sampled for quantification of DOC. To calculate DOC concentrations of extracts, 250 μl of the methanol extracts were isolated based on weight and evaporated overnight at 50°C before re-dissolving in 15 ml ultrapure water at pH 2 for DOC analysis. Additionally, the last 40 mL of permeate from each SPE extraction was taken to determine the DOC concentration of the PPL flow through (post-extraction). Extraction efficiency was then calculated by subtracting this permeate DOC concentration from the pre-filtered DOC concentration in the seawater entering the SPE column.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
DOM samples were re-dissolved in 100 μL methanol and 1% formic acid of which 10 μL were injected into a ultra-high performance liquid chromatography (UPLC) system coupled to a Q-Exactive orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) in three independent runs, first in high resolution positive mode, then in high resolution positive DDA MS/MS mode and finally in UHR negative mode. For the chromatographic separation, a C18 core-shell column (Kinetex, 100 × 2 mm, 1.8 um particle size, 100 A pore size, Phenomenex, Torrance, USA) with a flowrate of 0.5 mL/min (Solvent A: H2O + 0.1% formic acid (FA), Solvent B: Acetonitrile (ACN) + 0.1% FA) was used. After injection, the samples were eluted during a linear gradient from 0 to 0.5 min, 5% B, 0.5 to 8 min 5 to 50% B, 8 to 10 min 50 to 99% B, followed by a 2 min washout phase at 99% B and a 3 min re-equilibration phase at 5% B. For positive mode measurements, the electrospray ionization (ESI) parameters were set to 52 L/min sheath gas flow, 14 L/min auxiliary gas flow, 0 L/min sweep gas flow and 400°C auxiliary gas temperature. The spray voltage was set to 3.5 kV and the inlet capillary to 320°C. 50 V S-lens level was applied. MS scan range was set to 150–1,500 m/z with a resolution at m/z 200 (Rm/z 200) of 140,000 with one micro-scan in positive mode. The maximum ion injection time was set to 100 ms with automated gain control (AGC) of 1.0E6. MS/MS spectra were recorded in data dependent acquisition (DDA) mode. Both MS1 survey scans (150–1,500 m/z) and up to 5 MS/MS scans of the most abundant ions per duty cycle were measured with Rm/z 200 of 17,500 with one micro-scan in positive mode. The maximum ion injection time was set to 100 ms with automated gain control (AGC) targets set to 1.0E6 for survey scans and 3.0E5 for MS/MS with minimum 10% C-trap filling. MS/MS precursor selection windows were set to m/z 1. Normalized collision energy was set to a stepwise increase from 20 to 30 to 40% with z = 1 as default charge state. MS/MS experiments were triggered at the apex of peaks within 2–15 s from their first occurrence. Dynamic exclusion was set to 5 s. Ions with unassigned charge states were excluded from DDA as well as isotope peaks. For negative mode measurements, the electrospray ionization (ESI) parameters were identical to the positive mode measurements besides the polarity switching and an adjusted spray voltage. MS scan range was also set to 150–1,500 m/z with R m/z 200 of 140,000, one micro-scan and maximum ion injection time of 100 ms with an AGC target of 1.0E6.
Raw Data Processing
Thermo.raw datasets were converted to.mzXML in centroid mode using MSConvert (Chambers et al., 2012). LC-MS/MS.raw and.mzXML data can be found on the Mass spectrometry Interactive Virtual Environment (http://massive.ucsd.edu/) with the accession number MSV000080562.
MS1 Feature Extraction
As the first step of data analysis MS1 feature extraction was performed with MZmine2 (Pluskal et al., 2010). For both positive and negative mode data, mass detection was performed with a signal threshold of 3.0e4 and 0.6 s minimum peak width. Extracted ion chromatograms were built with a minimum peak height of 9.0e4 and a relative mass tolerance of 5 ppm. Chromatographic deconvolution was performed with the baseline cutoff algorithm with a baseline level of 3.0e4 and a minimum peak height of 9.0e4. The maximum peak length was set to 2 min. For isotope peak grouping, mass and retention time tolerances were set to 5 ppm and 0.1 min respectively. For alignment of extracted ion chromatograms (XICs) between samples, the same mass and retention time tolerances as above were used. After alignment, only XIC which contained at least 2 isotope peaks and which occurred at least 4 times out of 5 replicates were further considered. The aligned peak list was further filtered with a duplicate peak filter in which duplicate XIC within 5 ppm mass windows and 0.1 min retention time windows. All peak areas of XICs as well as binary information of XIC (present/not present) can be found in the Data Sheet 2, positive-mode, negative-mode and consensus XICs.
Molecular Formula Calculation and Matching of Orthogonal Ionization Modes
Molecular formulas of MS1 features (XIC) < 500 m/z were calculated with an in-house R script applying the Rdisop Bioconductor package (https://bioconductor.org/packages/release/bioc/html/Rdisop.html), implementing the method developed by (Böcker et al., 2009). The generated formulas were filtered by the following rules: N rule, O/C ratio ≤ 1, H/C ratio ≤ 4 and >0, element counts: C ≤ 100, H ≤ 200, O ≤ 80, N ≤ 10, S ≤ 2 modified from (Cortés-Francisco et al., 2014) and our inspection of ~233.000 molecular formulas obtained from the supernatural structure database (Banerjee et al., 2015), and maximal mass error of 5 ppm. Resulting molecular formulas were scored by the method described by (Böcker et al., 2009) and the top1 hit was used for downstream statistical analysis. The code to perform the above formula calculations and filtering is available as a jupyter notebook at https://github.com/DorresteinLaboratory/adductMatchAndFormPrediction.
Following the filtering process presented above, molecular formulas were subsequently filtered by presence of matching ions detected in both positive and negative modes. For that, the most common ESI ion species (Huang et al., 1999) ([M+H]+, [M+Na]+ for positive mode and [M-H]+ and [M+Cl]− for negative mode) were searched in each ionization mode, matched, and tagged in the data. Calculated molecular formulas can be found in the Data Sheet 2.
Spectral Networking
MS/MS spectra were analyzed with GNPS (Wang et al., 2016). Therefore, the data was filtered by removing all MS/MS peaks within a 17 Da window of the precursor m/z and MS/MS spectra were filtered by choosing only the top 6 peaks in 50 Da windows. The data was then clustered with MS-Cluster (Frank et al., 2008), with a precursor mass tolerance of 0.01 Da and a MS/MS fragment ion tolerance of 0.01 Da. A spectral network was then created with a minimum spectral similarity of cosine 0.7 and more than 4 matched peaks. Only the top 10 edges connecting one node were kept in the network. Consensus spectra were searched against the GNPS spectral library as well as Massbank, ReSpect, HMDB, and NIST14 (Forsythe and Wishart, 2009; Horai et al., 2010; Sawada et al., 2012; Stein, 2014; Wang et al., 2016) with a precursor mass tolerance of 0.01 Da and a MS/MS fragment ion tolerance of 0.01 Da as well as in analog search mode with as maximum precursor delta mass of m/z 100. Library hits discussed in this article were inspected manually and mirror plots of spectrum library matches are shown in the Supplemental Information. Finally, spectral networks were the visualized in Cytoscape 3.4 (Shannon et al., 2003).
Data and Software Code Accessibility
All LC-MS/MS data can be found on the Mass spectrometry Interactive Virtual Environment (MassIVE) at https://massive.ucsd.edu/ with the accession number: MSV000080562
Molecular Networking Data and all results of the Spectra Library Comparison can be found at the Global Natural Product Social Molecular Networking (GNPS) website with the links:
http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e89fe14eddbb43b5baa2b3d65c257661,
http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=81b58b4ec67145b490cb0490f39165aa,
The code to perform ion species matching and molecular formula calculation is available at:
https://github.com/DorresteinLaboratory/adductMatchAndFormPrediction.
Author Contributions
DP, IK, LA, and PD designed the study. DP and IK collected the samples. DP and IK extracted and prepared the samples. DP acquired the mass spectrometry data. RD implemented the code for ion species matching and molecular formula calculation. DP and BS performed the data analysis. DP, IK, RD, BS, AH, CN, LK, and LA interpreted the results. DP, IK, LA, and PD wrote the manuscript. All authors read, discussed and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institute of Health with grant numbers P41 GM103484, S10RR029121, GM097509, the Deutsche Forschungsgemeinschaft with a postdoctoral research fellowship to DP with grant number PE 2600/1, Grants from the US National Science foundation OCE–1538567 to LWK, OCE–1538393 to CEN and OCE–1313747 to LA and a UCSD Frontiers of Innovation Scholars program grant to LA and PD to fund IK's participation in this work. We furthermore would like to thank Bryce Inman for assistance with graphic design of Figure S1. This paper is funded in part by a grant/cooperative agreement from the National Oceanic and Atmospheric Administration, Project R/WR-3, which is sponsored by the University of Hawai‘i Sea Grant College Program, SOEST, under Institutional Grant No. NA14OAR4170071 from NOAA Office of Sea Grant, Department of Commerce. The views expressed herein are those of the author(s) and do not necessarily reflect the views of NOAA or any of its subagencies. This is publication number 10277 of the School of Ocean and Earth Science and Technology of the University of Hawai‘i at Mānoa and publication UNIHI-SEAGRANT-JC-16-12 of the University of Hawai‘i Sea Grant College Program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2017.00405/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Aluwihare, L. I., Repeta, D. J., Pantoja, S., and Johnson, C. G. (2005). Two chemically distinct pools of organic nitrogen accumulate in the ocean. Science 308, 1007–1010. doi: 10.1126/science.1108925
Amin, S. A., Hmelo, L. R., van Tol, H., Durham, B., Carlson, L. T., Heal, K. R., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98. doi: 10.1038/nature14488
Anderson, M. J., and Willis, T. J. (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84, 511–525. doi: 10.1890/0012-9658(2003)084[0511:CAOPCA]2.0.CO;2
Arakawa, N., Aluwihare, L. I., Simpson, A. J., Soong, R., Stephens, B. M., and Lane-Coplen, D. (2017). Carotenoids are the likely precursor of a significant fraction of marine dissolved organic matter. Sci. Adv. 3:e1602976. doi: 10.1126/sciadv.1602976
Azam, F., and Malfatti, F. (2007). Microbial structuring of marine ecosystems, Nature reviews. Microbiology 5, 782. doi: 10.1038/nrmicro1747
Banerjee, P., Erehman, J., Gohlke, B. O., Wilhelm, T., Preissner, R., and Dunkel, M. (2015). Super Natural II—a database of natural products. Nucleic Acids Res. 43, D935–D939. doi: 10.1093/nar/gku886
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32, D138–D141. doi: 10.1093/nar/gkh121
Benner, R., and Amon, R. M. (2015). The size-reactivity continuum of major bioelements in the ocean. Ann. Rev. Mar. Sci. 7, 185–205. doi: 10.1146/annurev-marine-010213-135126
Böcker, S. (2017). Searching molecular structure databases using tandem MS data: are we there yet? Curr. Opin. Chem. Biol. 36, 1–6. doi: 10.1016/j.cbpa.2016.12.010
Böcker, S., Letzel, M. C., Lipták, Z., and Pervukhin, A. (2009). SIRIUS: decomposing isotope patterns for metabolite identification. Bioinformatics 25, 218–224. doi: 10.1093/bioinformatics/btn603
Bouslimani, A., Porto, C., Rath, C. M., Wang, M., Guo, Y., Gonzalez, A., et al. (2015). Molecular cartography of the human skin surface in 3D. Proc. Natl. Acad. Sci. U.S.A. 112, E2120–E2129. doi: 10.1073/pnas.1424409112
Chambers, M. C., Maclean, B., Burke, R., Amodei, D., Ruderman, D. L., Neumann, S., et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920. doi: 10.1038/nbt.2377
Cortés-Francisco, N., Harir, M., Lucio, M., Ribera, G., Martínez-Llad,ó, X., Rovira, M., et al. (2014). High-field FT-ICR mass spectrometry and NMR spectroscopy to characterize DOM removal through a nanofiltration pilot plant. Water Res. 67, 154–165. doi: 10.1016/j.watres.2014.08.046
D'Andrilli, J., Dittmar, T., Koch, B. P., Purcell, J. M., Marshall, A. G., and Cooper, W. T. (2010). Comprehensive characterization of marine dissolved organic matter by Fourier transform ion cyclotron resonance mass spectrometry with electrospray and atmospheric pressure photoionization. Rapid Commun. Mass Spectr. 24, 643–650. doi: 10.1002/rcm.4421
da Silva, R. R., Dorrestein, P. C., and Quinn, R. A. (2015). Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. 112, 12549–12550. doi: 10.1073/pnas.1516878112
Davis, J., Kaiser, K., and Benner, R. (2009). Amino acid and amino sugar yields and compositions as indicators of dissolved organic matter diagenesis. Organ. Geochem. 40, 343–352. doi: 10.1016/j.orggeochem.2008.12.003
Dittmar, T., and Kattner, G. (2003). Recalcitrant dissolved organic matter in the ocean: major contribution of small amphiphilics. Mar. Chem. 82, 115–123. doi: 10.1016/S0304-4203(03)00068-9
Dittmar, T., Koch, B., Hertkorn, N., and Kattner, G. (2008). A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater, Limnol. Oceanogr. Methods 6, 230–235. doi: 10.4319/lom.2008.6.230
Dittmar, T., and Paeng, J. (2009). A heat-induced molecular signature in marine dissolved organic matter. Nat. Geosci. 2, 175. doi: 10.1038/ngeo440
Flerus, R., Lechtenfeld, O., Koch, B. P., McCallister, S., Schmitt-Kopplin, P., Benner, R., et al. (2012). A molecular perspective on the ageing of marine dissolved organic matter. Biogeosciences 9, 1935. doi: 10.5194/bg-9-1935-2012
Floros, D. J., Petras, D., Kapono, C. A., Melnik, A. V., Ling, T. J., Knight, R., et al. (2017). Mass spectrometry based molecular 3D-cartography of plant metabolites. Front. Plant Sci. 8:429. doi: 10.3389/fpls.2017.00429
Forsythe, I. J., and Wishart, D. S. (2009). Exploring human metabolites using the human metabolome database. Curr. Protoc. Bioinformatics Chapter 14, Unit14 18. doi: 10.1002/0471250953.bi1408s25
Frank, A. M., Bandeira, N., Shen, Z., Tanner, S., Briggs, S. P., Smith, R. D., et al. (2008). Clustering millions of tandem mass spectra. J. Proteome Res. 7, 113–122. doi: 10.1021/pr070361e
Gika, H. G., Theodoridis, G. A., Plumb, R. S., and Wilson, I. D. (2014). Current practice of liquid chromatography–mass spectrometry in metabolomics and metabonomics. J. Pharm. Biomed. Anal. 87, 12–25. doi: 10.1016/j.jpba.2013.06.032
Guo, L., Santschi, P. H., and Warnken, K. W. (1995). Dynamics of dissolved organic carbon (DOC) in oceanic environments. Limnol. Oceanogr. 40, 1392–1403. doi: 10.4319/lo.1995.40.8.1392
Halewood, E. R., Carlson, C. A., Brzezinski, M. A., Reed, D. C., and Goodman, J. (2012). Annual cycle of organic matter partitioning and its availability to bacteria across the Santa Barbara Channel continental shelf. Aquatic Microbial Ecol. 67, 189–209. doi: 10.3354/ame01586
Hansell, D. A. (2013). Recalcitrant dissolved organic carbon fractions. Ann. Rev. Mar. Sci. 5, 421–445. doi: 10.1146/annurev-marine-120710-100757
Hartmann, A. C., Petras, D., Quinn, R. A., Protsyuk, I., Archer, F. I., Ransome, E., et al. (2017). Meta-mass shift chemical profiling of metabolomes from coral reefs. Proc. Natl. Acad. Sci. U.S.A. 114, 11685–11690. doi: 10.1073/pnas.1710248114
Hawkes, J. A., Dittmar, T., Patriarca, C., Tranvik, L., and Bergquist, J. (2016). Evaluation of the orbitrap mass spectrometer for the molecular fingerprinting analysis of natural dissolved organic matter. Anal. Chem. 88, 7698–7704. doi: 10.1021/acs.analchem.6b01624
Heal, K. R., Qin, W., Ribalet, F., Bertagnolli, A. D., Coyote-Maestas, W., Hmelo, L. R., et al. (2017). Two distinct pools of B12 analogs reveal community interdependencies in the ocean. Proc. Natl. Acad. Sci.U.S.A. 114, 364–369. doi: 10.1073/pnas.1608462114
Hertkorn, N., Harir, M., Koch, B. P., Michalke, B., and Schmitt-Kopplin, P. (2013). High-field NMR spectroscopy and FTICR mass spectrometry: powerful discovery tools for the molecular level characterization of marine dissolved organic matter. Biogeosciences 10, 1583–1624. doi: 10.5194/bg-10-1583-2013
Herzsprung, P., Hertkorn, N., von Tümpling, W., Harir, M., Friese, K., and Schmitt-Kopplin, P. (2016). Molecular formula assignment for dissolved organic matter (DOM) using high-field FT-ICR-MS: chemical perspective and validation of sulphur-rich organic components (CHOS) in pit lake samples. Anal. Bioanal. Chem. 408, 2461–2469. doi: 10.1007/s00216-016-9341-2
Hopkinson, C. S. Jr., and Vallino, J. J. (2005). Efficient export of carbon to the deep ocean through dissolved organic matter. Nature 433, 142. doi: 10.1038/nature03191
Horai, H., Arita, M., Kanaya, S., Nihei, Y., Ikeda, T., Suwa, K., et al. (2010). MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectr. 45, 703–714. doi: 10.1002/jms.1777
Huang, N., Siegel, M. M., Kruppa, G. H., and Laukien, F. H. (1999). Automation of a Fourier transform ion cyclotron resonance mass spectrometer for acquisition, analysis, and e-mailing of high-resolution exact-mass electrospray ionization mass spectral data. J. Am. Soc. Mass Spectr. 10, 1166–1173. doi: 10.1016/S1044-0305(99)00089-6
Johnson, W. M., Soule, M. C. K., and Kujawinski, E. B. (2016). Evidence for quorum sensing and differential metabolite production by a marine bacterium in response to DMSP. ISME J. 10, 2304. doi: 10.1038/ismej.2016.6
Karl, D., and Björkman, K. (2002). Dynamics of DOP, Biogeochemistry of Marine Dissolved Organic Matter. Amsterdam: Elsevier. 249–366.
Kujawinski, E. B., Freitas, M. A., Zang, X., Hatcher, P. G., Green-Church, K. B., and Jones, R. B. (2002). The application of electrospray ionization mass spectrometry (ESI MS) to the structural characterization of natural organic matter. Org. Geochem. 33, 171–180. doi: 10.1016/S0146-6380(01)00149-8
Kujawinski, E. B., Longnecker, K., Alexander, H., Dyhrman, S. T., Fiore, C. L., Haley, S. T., et al. (2017). Phosphorus availability regulates intracellular nucleotides in marine eukaryotic phytoplankton. Limnol. Oceanogr. Lett. 2, 119–129. doi: 10.1002/lol2.10043
Lawson, T. N., Weber, R. J., Jones, M. R., Chetwynd, A. J., Rodriguez-Blanco, G., Di Guida, R., et al. (2017). msPurity: automated evaluation of precursor ion purity for mass spectrometry-based fragmentation in metabolomics. Anal. Chem. 89, 2432–2439. doi: 10.1021/acs.analchem.6b04358
Lechtenfeld, O. J., Koch, B. P., Geibert, W., Ludwichowski, K. U., and Kattner, G. (2011). Inorganics in organics: quantification of organic phosphorus and sulfur and trace element speciation in natural organic matter using HPLC-ICPMS. Anal. Chem. 83, 8968–8974. doi: 10.1021/ac201765a
Letscher, R. T., and Moore, J. K. (2015). Preferential remineralization of dissolved organic phosphorus and non-Redfield DOM dynamics in the global ocean: impacts on marine productivity, nitrogen fixation, and carbon export. Global Biogeochem. Cycles 29, 325–340. doi: 10.1002/2014GB004904
Liesenfeld, D. B., Habermann, N., Owen, R. W., Scalbert, A., and Ulrich, C. M. (2013). Review of mass spectrometry–based metabolomics in cancer research. Cancer Epidemiol. Prev. Biomarkers 22, 2182–2201. doi: 10.1158/1055-9965.EPI-13-0584
Longnecker, K., and Kujawinski, E. B. (2017). Mining mass spectrometry data: using new computational tools to find novel organic compounds in complex environmental mixtures. Organ. Geochem. 110, 92–99. doi: 10.1016/j.orggeochem.2017.05.008
Lucas, J., Koester, I., Wichels, A., Niggemann, J., Dittmar, T., Callies, U., et al. (2016). Short-term dynamics of North Sea bacterioplankton-dissolved organic matter coherence on molecular level. Front. Microbiol. 7:321. doi: 10.3389/fmicb.2016.00321
Makarov, A., Denisov, E., and Lange, O. (2009). Performance evaluation of a high-field Orbitrap mass analyzer. J. Am. Soc. Mass Spectr. 20, 1391–1396. doi: 10.1016/j.jasms.2009.01.005
Moran, M. A., Kujawinski, E. B., Stubbins, A., Fatland, R., Aluwihare, L. I., Buchan, A., et al. (2016). Deciphering ocean carbon in a changing world. Proc. Natl. Acad. Sci. U.S.A. 113, 3143–3151. doi: 10.1073/pnas.1514645113
Ogawa, H., Amagai, Y., Koike, I., Kaiser, K., and Benner, R. (2001). Production of refractory dissolved organic matter by bacteria. Science 292, 917–920. doi: 10.1126/science.1057627
Olivon, F., Grelier, G., Roussi, F., Litaudon, M., and Touboul, D. (2017). MZmine 2 data-preprocessing to enhance Molecular Networking reliability. Anal. Chem. 89, 7836–7840. doi: 10.1021/acs.analchem.7b01563
Osterholz, H., Singer, G., Wemheuer, B., Daniel, R., Simon, M., Niggemann, J., et al. (2016). Deciphering associations between dissolved organic molecules and bacterial communities in a pelagic marine system. ISME J. 10, 1717–1730. doi: 10.1038/ismej.2015.231
Petras, D., Jarmusch, A. K., and Dorrestein, P. C. (2017). From single cells to our planet - Recent advances in using mass spectrometry for spatially resolved metabolomics. Curr. Opin. Chem. Biol. 36, 24–31. doi: 10.1016/j.cbpa.2016.12.018
Petras, D., Nothias, L. F., Quinn, R. A., Alexandrov, T., Bandeira, N., Bouslimani, A., et al. (2016). Mass spectrometry-based visualization of molecules associated with human habitats. Anal. Chem. 88, 10775–10784. doi: 10.1021/acs.analchem.6b03456
Pluskal, T., Castillo, S., Villar-Briones, A., and Oresic, M. (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 11:395. doi: 10.1186/1471-2105-11-395
Quinn, R. A., Vermeij, M. J., Hartmann, A. C., d'Auriac, I. G., Benler, S., Haas, A., et al. (2016). Metabolomics of reef benthic interactions reveals a bioactive lipid involved in coral defence. Proc. R. Soc. B 283, 20160469. doi: 10.1098/rspb.2016.0469
Reemtsma, T., These, A., Linscheid, M., Leenheer, J., and Spitzy, A. (2008). Molecular and structural characterization of dissolved organic matter from the deep ocean by FTICR-MS, including hydrophilic nitrogenous organic molecules. Environ. Sci. Technol. 42, 1430–1437. doi: 10.1021/es7021413
Repeta, D. J., Ferrón, S., Sosa, O. A., Johnson, C. G., Repeta, L. D., Acker, M., et al. (2016). Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat. Geosci. 9, 884–887. doi: 10.1038/ngeo2837
Romano, S., Dittmar, T., Bondarev, V., Weber, R. J., Viant, M. R., and Schulz-Vogt, H. N. (2014). Exo-metabolome of Pseudovibrio sp. FO-BEG1 analyzed by ultra-high resolution mass spectrometry and the effect of phosphate limitation, PLoS ONE 9:e96038. doi: 10.1371/journal.pone.0096038
Rubert, J., Zachariasova, M., and Hajslova, J. (2015). Advances in high-resolution mass spectrometry based on metabolomics studies for food–a review. Food Addit. Contam. A 32, 1685–1708. doi: 10.1080/19440049.2015.1084539
Sawada, Y., Nakabayashi, R., Yamada, Y., Suzuki, M., Sato, M., Sakata, A., et al. (2012). RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry 82, 38–45. doi: 10.1016/j.phytochem.2012.07.007
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2012). Database resources of the national center for biotechnology information, Nucleic Acids Res. 40, D13–D25. doi: 10.1093/nar/gkr1184
Scheltema, R. A., Hauschild, J. P., Lange, O., Hornburg, D., Denisov, E., Damoc, E., et al. (2014). The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteom. 13, 3698–3708. doi: 10.1074/mcp.M114.043489
Scheubert, K., Hufsky, F., Petras, D., Wang, M., Nothias, L. F., Düehrkop, K., et al. (2017). Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 8:1494. doi: 10.1038/s41467-017-01318-5
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Siegenthaler, U., and Sarmiento, J. (1993). Atmospheric carbon dioxide and the ocean, Nature 365, 119–125. doi: 10.1038/365119a0
Sipler, R., and Bronk, D. (2015). “Dynamics of dissolved organic nitrogen,” in Biogeochemistry of Marine Dissolved Organic Matter. 2nd Edn., eds D. A. Hansel and C. A. Carlson (Amsterdam: Elsevier), 127–232.
Sogin, E. M., Putnam, H. M., Nelson, C. E., Anderson, P., and Gates, R. D. (2017). Correspondence of coral holobiont metabolome with symbiotic bacteria, archaea and Symbiodinium communities. Environ. Microbiol. Rep. 9, 310–315. doi: 10.1111/1758-2229.12541
Stein, S. (2014). The NIST 14 Mass Spectral Library. Gaithersburg, MD: National Institute of Standards and Technology.
Stein, S. E., and Scott, D. R. (1994). Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectr. 5, 859–866. doi: 10.1016/1044-0305(94)87009-8
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221. doi: 10.1007/s11306-007-0082-2
Viant, M. R., and Sommer, U. (2013). Mass spectrometry based environmental metabolomics: a primer and review. Metabolomics 9, 144–158. doi: 10.1007/s11306-012-0412-x
Vinaixa, M., Schymanski, E. L., Neumann, S., Navarro, M., Salek, R. M., and Yanes, O. (2016). Mass spectral databases for LC/MS-and GC/MS-based metabolomics: state of the field and future prospects. Trends Anal. Chem. 78, 23–35. doi: 10.1016/j.trac.2015.09.005
Wang, M., Carver, J. J., Phelan, V. V., Sanchez, L. M., Garg, N., Peng, Y., et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837. doi: 10.1038/nbt.3597
Watrous, J., Roach, P., Alexandrov, T., Heath, B. S., Yang, J. Y., Kersten, R. D., et al. (2012). Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. U.S.A. 109, E1743–E1752. doi: 10.1073/pnas.1203689109
Weber, R. J., Lawson, T. N., Salek, R. M., Ebbels, T. M., Glen, R. C., Goodacre, R., et al. (2017). Computational tools and workflows in metabolomics: an international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 13, 12. doi: 10.1007/s11306-016-1147-x
Zark, M., Christoffers, J., and Dittmar, T. (2017). Molecular properties of deep-sea dissolved organic matter are predictable by the central limit theorem: evidence from tandem FT-ICR-MS. Mar. Chem. 191, 9–15. doi: 10.1016/j.marchem.2017.02.005
Keywords: mass spectrometry, LC-MS/MS, dissolved organic matter, non-targeted metabolomics, environmental metabolomics, marine microbial communities
Citation: Petras D, Koester I, Da Silva R, Stephens BM, Haas AF, Nelson CE, Kelly LW, Aluwihare LI and Dorrestein PC (2017) High-Resolution Liquid Chromatography Tandem Mass Spectrometry Enables Large Scale Molecular Characterization of Dissolved Organic Matter. Front. Mar. Sci. 4:405. doi: 10.3389/fmars.2017.00405
Received: 10 August 2017; Accepted: 28 November 2017;
Published: 12 December 2017.
Edited by:
Marta Álvarez, Instituto Español de Oceanografía (IEO), SpainReviewed by:
John Robert Helms, Morningside College, United StatesArvind Singh, Physical Research Laboratory, India
Helena Osterholz, University of Oldenburg, Germany
Copyright © 2017 Petras, Koester, Da Silva, Stephens, Haas, Nelson, Kelly, Aluwihare and Dorrestein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Petras, ZHBldHJhc0B1Y3NkLmVkdQ==
Lihini I. Aluwihare, bGFsdXdpaGFyZUB1Y3NkLmVkdQ==
Pieter C. Dorrestein, cGRvcnJlc3RlaW5AdWNzZC5lZHU=