
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 15 December 2017
Sec. Marine Biogeochemistry
Volume 4 - 2017 | https://doi.org/10.3389/fmars.2017.00396
This article is part of the Research Topic Sensors for Autonomous Ocean Observations View all 7 articles
 Jennifer S. Clarke1,2*
Jennifer S. Clarke1,2* Matthew P. Humphreys1
Matthew P. Humphreys1 Eithne Tynan1
Eithne Tynan1 Vassilis Kitidis3Ian Brown3
Vassilis Kitidis3Ian Brown3 Matthew Mowlem4
Matthew Mowlem4 Eric P. Achterberg1,2
Eric P. Achterberg1,2The ocean is a major sink for anthropogenic carbon dioxide (CO2), with the CO2 uptake causing changes to ocean chemistry. To monitor these changes and provide a chemical background for biological and biogeochemical studies, high quality partial pressure of CO2 (pCO2) sensors are required, with suitable accuracy and precision for ocean measurements. Optodes have the potential to measure in situ pCO2 without the need for wet chemicals or bulky gas equilibration chambers that are typically used in pCO2 systems. However, optodes are still in an early developmental stage compared to more established equilibrator-based pCO2 systems. In this study, we performed a laboratory-based characterization of a time-domain dual lifetime referencing pCO2 optode system. The pCO2 optode spot was illuminated with low intensity light (0.2 mA, 0.72 mW) to minimize spot photobleaching. The spot was calibrated using an experimental gas calibration rig prior to deployment, with a determined response time (τ63) of 50 s at 25°C. The pCO2 optode was deployed as an autonomous shipboard underway system across the high latitude North Atlantic Ocean with a resolution of ca.10 measurements per hour. The optode data was validated with a secondary shipboard equilibrator-based infrared pCO2 instrument, and pCO2 calculated from discrete samples of dissolved inorganic carbon and total alkalinity. Further verification of the pCO2 optode data was achieved using complimentary variables such as nutrients and dissolved oxygen. The shipboard precision of the pCO2 sensor was 9.5 μatm determined both from repeat measurements of certified reference materials and from the standard deviation of seawater measurements while on station. Finally, the optode deployment data was used to evaluate the physical and biogeochemical controls on pCO2.
Atmospheric emissions of carbon dioxide (CO2) as a result of fossil fuel combustion and cement production averaged ca. 8.9 Gt C yr−1 (91% of total emissions) over the period between 2004 and 2013, while land use changes contributed an average of 0.9 Gt C yr−1 (9% of total emissions). The ocean is a significant sink for anthropogenic CO2, with an average global oceanic CO2 uptake of ca. 2.6 Gt yr−1 (26% of the total emissions) between 2004 and 2013 (Le Quéré et al., 2015). However, the ocean is not a uniform sink (DeVries, 2014). There are significant temporal and regional differences, which determine whether any given area acts as a CO2 sink or source (Takahashi et al., 2009). The high latitude North Atlantic, for example, is a globally significant sink for CO2, both storing anthropogenic CO2 and acting as a conduit to the rest of the worlds' oceans (Álvarez et al., 2003; Schuster et al., 2009). This is supported by data from the Bermuda Atlantic Time-series Study (BATS), which suggests that the North Atlantic mode waters formed at high latitudes have higher anthropogenic CO2 levels than those at lower latitudes (Gruber et al., 2002).
Air-sea CO2 flux calculated from cruise data is generally obtained from interpolated pCO2 measurements, with climatological wind speeds and spectrometric or chromatographically determined atmospheric pCO2 concentrations (Takahashi et al., 2009). Data at higher temporal resolution is required to allow a more robust statistical evaluations of CO2 fluxes over shorter time scales (Wanninkhof et al., 2013; Landschützer et al., 2014) and better constrain the mechanisms that drive the air sea fluxes (Schuster et al., 2013). This is of particular relevance to the North Atlantic sub-polar gyre, which forms an important CO2 sink with sparse measurements (Corbière et al., 2007). Previous data collected in this region, has been used to commence the evaluation of the controls on the carbon system in the north Atlantic, and to model biogeochemical dynamics in that region (Doney et al., 2009a).
The increasing seawater pCO2 has several implications for seawater biogeochemistry, including reductions in both ocean pH and calcium carbonate mineral saturation states (Ω). This may have effects on flora and fauna in the oceans that rely on aragonite and calcite production to build their skeletons and shells. To understand the potential implications of the increased CO2, and quantify future changes to the marine carbonate system, it is necessary to collect high quality, accurate, and precise pCO2 data. Autonomous in situ sensors allow widespread collection of this data. The ideal characteristics for in situ chemical sensors include small size, low power consumption and the possibility for high frequency measurements (Byrne, 2014) with a desired precision for autonomous seawater pCO2 sensors of 2 μatm (Pierrot et al., 2009). There are several in situ pCO2 systems currently available (Clarke et al., 2017) for measurement over the seawater range of 0–1,000 μatm with reported precisions of ca. 1 μatm (Battelle, 2011/2012; Sunburst Sensors LLC, 2014; Contros Systems and Solutions, 2015). However, these typically require gas equilibration, or have mechanical components that are not ideal for long term in situ deployment. Optodes are a small, simple alternative, comprising of an analyte sensitive sensor spot and electronic hardware with no mechanical parts. The suitability of fluorescent pCO2 sensor spots for marine measurements was previously demonstrated by Atamanchuk et al. (Atamanchuk, 2013; Atamanchuk et al., 2014), who applied the frequency domain-Dual Lifetime Referencing (f-DLR) method over the pCO2 range 0–10,000 μatm, and produced an eighth by third degree polynomial to convert the fluorescent response to pCO2 (Atamanchuk et al., 2014).
When using fluorescence methods, photobleaching of the dyes can be a significant limiting factor, causing irreversible changes to the dye such that it no longer fluoresces upon excitation. Photobleaching is the loss of fluorescence or color of a dye caused by light or illumination. This ultimately causes a reduced signal-to-noise ratio, and increased imprecision over time, until a response is no longer detectable. The photobleaching of the indicator and reference dyes is directly proportional to the light energy used in excitation, and therefore the duration of the excitation (Borisov et al., 2011). The time domain-Dual Lifetime Referencing (t-DLR) method used in this study applies short light emitting diode (LED) pulses, thereby reducing the overall irradiation time of the spot and reducing the photobleaching potential, theoretically prolonging the spot longevity.
In this paper, we present the calibration and initial deployment of the pCO2 optode spot using the t-DLR method. The performance of the optode as a shipboard underway measurement system is evaluated with respect to ocean deployment potential and validated with discrete samples, other pCO2 sensors, and in the context of other hydrographic variables and our existing understanding of the physical and biogeochemical processes that control pCO2 in our study region. We aim to demonstrate the suitability of t-DLR as an alternative low bleaching method for use with pCO2 optodes in the open ocean.
The cruise (08/06/2014 and 11/07/2014) covered the seasonally stratified sub-polar regions in the North Atlantic. The sub-polar region can be further subdivided into biogeographical provinces as defined by Longhurst et al. (1995), and refined by Lefèvre et al. (2004): Sub-Arctic (SARCT, 58–68°N, 24–10°W), Arctic (ARCT, 50–70°N, 60–10°W), and North Atlantic Drift (NADR, 44–58°N, 42–10°W) as shown in Figure 1. Further divisions can be made within the ARCT region, into the central Labrador Sea, and the four transects around the Greenland coastline (termed GL 1–4). The SARCT region comprised mostly the repeat hydrographic transect the Extended Ellett line (EEL), which is occupied by the National Oceanography Centre Southampton on an annual basis. The main warm water surface currents over the course of the cruise were the North Atlantic Current (NAC), and the Irminger current (IrC). The cooler polar surface water currents included the East Greenland Current (EGC), the west Greenland Current (WGC), and the Labrador Current (LabC). This paper focuses on the NADR and ARCT regions.
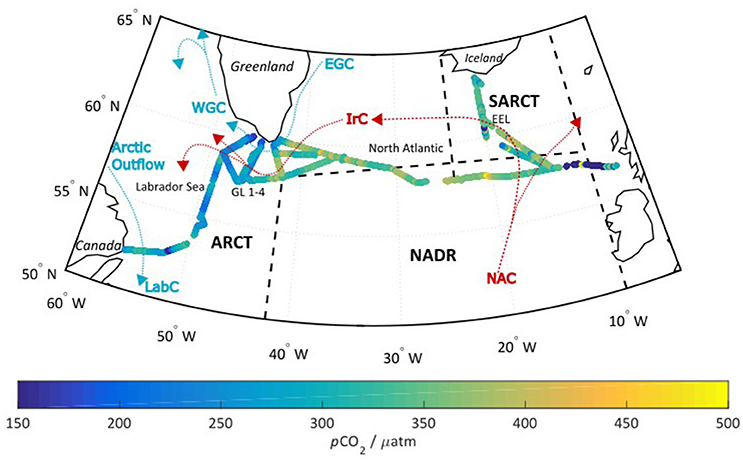
Figure 1. The cruise track overlaid with the optode measured pCO2 in μatm. The black dashed lines indicate the different biogeographical regions discussed in section Factors Controlling Surface Seawater pCO2 across the Subpolar North Atlantic, while the red and blue arrows indicate the respective “warm” and “cool” surface currents. SARCT = Sub-Arctic (58–68°N, 24–10°W), ARCT = Arctic (50–70°N, 60–10°W), and NADR = North Atlantic Drift (44–58°N, 42–10°W). Further divisions can be made within the ARCT region into the central Labrador Sea, and the four transects around the Greenland coastline (termed GL 1-4). The SARCT region comprises mostly the repeat hydrographic transect the Extended Ellett line (EEL). The main currents are the NAC, North Atlantic Current; IrC, Irminger Current; EGC, East Greenland Current; WGC, West Greenland Current; LabC, Central Labrador Sea water, with Arctic outflows referring to Arctic waters that reach the Labrador Sea via the Canadian archipelago.
The pCO2 sensor spots (SP-CD1-D5-rMy-US, PreSens GmbH, Germany) were 5 mm in diameter and consisted of a gas permeable, ion-impermeable membrane, secured to a support structure. They contained a pH sensitive indicator dye, a pH-insensitive reference dye and a pH buffer solution (Atamanchuk et al., 2014), however the exact composition of the pCO2 spots is proprietary information. Prior to use, the spots were preconditioned in a solution with a salinity close to that of the North Atlantic as per Atamanchuk et al. (2014). We used the same hardware, spot interrogation and data analysis (t-DLR) here as in our previous study on pH optodes (Clarke et al., 2015), briefly described below.
Custom-made hardware (Sensoptics, SGS 42000; Sensoptics Ltd.1, UK) and software (Sensoptics Photon Counter V1.2, Sensoptics, UK) were used to record the fluorescence decay curves. The sensor hardware and software is not commercially available however the chemical sensor spots are available through PreSens GmbH. The hardware is described in more detail below.
A blue LED (5 mm diameter, λ 470 nm, Farnell, UK) with excitation filters (SemRock single bandpass filter 475 nm and SemRock short pass edge filter 532 nm, USA), was pulsed at a low intensity (0.2 mA, 0.72 mW). The blue light was channeled through a fiber optic cable (600 μm diameter multimode optical fiber, 3.8 mm diameter, 2 m length, Thorlabs, UK) to the sensor spot. The fluorescence (red light, λ 630 nm) traveled back through the fiber optic cable to a dichroic beamsplitter (SemRock single edge BrightLine, 560 nm, USA). The red light passed through the dichroic beamsplitter (while blue light is reflected), and through three emission filters (SemRock single bandpass filter 609 nm, SemRock short pass edge filter 785 nm and SemRock long-pass edge filter 568 nm, USA) to a photon multiplier tube (PMT; Hamamatsu, Japan). The PMT and LEDs were controlled by a field-programmable gate array (FPGA, Xilinx, CA, USA).
This interrogation is as per Clarke et al. (2015). The blue light LED was pulsed, on for 5 μs (tex) causing the dyes to fluoresce, and off for 20.5 μs (tem), where the fluorescence decayed, with one averaged spectra (LED on and off) recorded every 0.5 s. Optode precision and signal-to-noise ratio were improved by measuring each sample for 180 s and integrating the fluorescence decay profiles over 10 s intervals.
The fluorescence emission (tex) when the LED was on, was considered a combination of fluorescence from the pH sensitive (ApH) and reference dye (Aref-ex). The integrated fluorescence intensity when the LED was off (tem) was considered to be derived entirely from the decay of the pH-inert reference dye emission (Aref-em), due to the significantly shorter decay lifetime of the pH sensitive dye. The ratio of intensity-integrated tex to intensity-integrated tem (R, Equation 1) was then related to the solution pCO2.
Artificial seawater solutions were made up with sodium chloride (35 g kg−1). A mixture of CO2 in nitrogen (N2) gas (1,000 ppm CO2, BOC Ltd., UK) was used to saturate the artificial seawater solutions with CO2 in the gas calibration rig (see Dissolved Gas Calibration Rig section). Solutions were degassed using N2 gas (BOC Ltd., UK). All chemicals were analytical grade (Sigma Aldrich, UK). All solutions were prepared and diluted with deionized water (MilliQ, <18.2 MΩ cm−1, Millipore, UK).
Certified Reference Material (CRM) (batches 135, 136, 142, 144) for dissolved inorganic carbon (DIC) and total alkalinity (TA) were obtained from Prof. A. Dickson at Scripps Institute of Oceanography (USA).
The photobleaching of the indicator and reference dyes is directly proportional to the light energy used during the excitation process (Borisov et al., 2011). Irradiation of the spot at higher light intensity than normal allowed evaluation of bleaching (Zhu et al., 2006) and the potential total number of measurements (at normal illumination intensity, 0.2 mA for 200 s) that could be undertaken prior to bleaching, in the absence of a long term bleaching experiment. The spot was initially measured with the LED powered normally, the spot was then illuminated with the LED powered with 2 mA (10 times the normal intensity) for 2 h continuously (no LED pulses). The response of the spot was then re-measured with the LED powered normally to check for any changes or drift. The exposure to enhanced LED illumination for 2 h was equivalent to 18,360 measurements.
The spot response time was determined by measuring the pCO2 of the seawater sample (~280 ppm) with the optode for 1 min before injection of a high pCO2 seawater solution (~800 ppm) and measurement for a further 2 min. This was performed at 25°C.
To determine if chlorophyll fluorescence in the ocean would interact with the optode measurements, cultures of defined chlorophyll concentrations were measured in six samples. The phytoplankton Emiliania huxleyi (coccolithophore, Strain RCC1228 from the Roscoff Collection) was cultured in artificial seawater at 16°C, under light conditions of 100 μE. The chlorophyll concentration in the stock solution was calculated based on a chlorophyll concentration per cell (Daniels et al., 2014), and the counted cell concentration of the stock solution. The culture was diluted with artificial seawater to 100 ml, with final chlorophyll concentrations of 0.1, 1, 2, 2.5, 5, 10 μg L−1. Samples were stored for a maximum of 2 h at 4°C in the dark between dilution and measurement. The pH and salinity of each sample was measured at 20°C, and CO2 gas (as above) bubbled through each sample in the dark until a constant pHT (8.022) was reached immediately prior to measurement. The experiments were all performed in the dark, in a water bath (20 ± 0.01°C; Grant TX150 ST26), and the optode inserted directly into the sample solutions. The solutions were measured three times consecutively before being discarded to avoid potential growth of the coccolithophores.
The DIC/TA CRM was used to check the precision and accuracy of the pCO2 optode, and calibrate the Apollo DIC analyzer (AS-C3, Apollo Sci-Tech, USA, ± 4 μmol kg−1), the VINDTA (3C; Marianda, Germany, ±3 μmol kg−1), and the bench top pH system (Orion VersaStar with a Fischer Semi-micro glass pH electrode, UK, ± 0.01 pH). Short- term precision was calculated as the standard deviation of 10 repeats CRM (Batch 144). Long-term precision was calculated from the pooled standard deviations of the CRMs (Batches 135 and 136, at 20°C) measured throughout the period of the research cruise.
The software program CO2SYS (v1.1 for Matlab) was used to determine pCO2 with carbon dissociation constants from Mehrbach et al. (1973), refitted by Dickson and Millero (1987), the sulfate dissociation constants from Dickson (1990) and boron-chlorinity ratio from Lee et al. (2010). pCO2 calculated from pH/DIC is referred to as pCO2-calcpH, while pCO2 calculated from DIC/TA is referred to as pCO2-calcTA.
A gas calibration rig developed by Sosna et al. (2007), originally designed for dissolved oxygen microelectrode calibrations, was applied to the calibration of the pCO2 optodes (Figure 2).
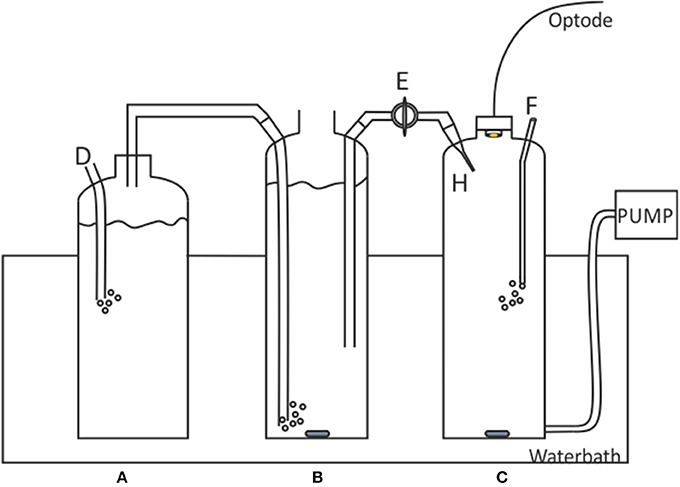
Figure 2. Schematic of the Sosna calibration rig. A, 1.5 L Gas humidifier; B, 2L saturated solution; C, 2L sample solution; D, CO2 gas tube to CO2 /N2 gas cylinder; E, Bridge with valve; F, nitrogen gas inlet; H, Saturated solution inlet capillary. Figure adapted from Sosna et al. (2007).
The artificial seawater filled Chamber B (2L) was saturated with CO2 using CO2/N2 gas humidified in chamber A. The artificial seawater in Chamber C was degassed using N2 gas. After ca. 20 min of gassing/degassing the solutions, the optode was slotted into place ensuring no headspace between the optode head and the port. The bridge was filled by opening valve E. A single pump cycle was then run to ensure there were no bubbles or air entering the measurement cell. Any bubbles found in the bridge or chamber C may cause dissolved CO2 to diffuse into the air bubble, thereby changing the pCO2SW concentration in chamber C. Chamber B was left open to the atmosphere, unsealed to prevent formation of a vacuum, with a lid resting on top to limit gas exchange. Sample aliquots of the saturated chamber and the degassed chamber were collected to verify the pCO2 levels (using DIC and pH measurements to calculate the pCO2, referred to as pCO2-calcpH).
A calibration curve was generated by sequentially adding small amounts of solution B (1.2 ml ± 0.05, 2 s pump time) to solution C. Each pump cycle pulled a small volume of solution C out of the chamber, which was replaced with the same volume of solution B (saturated solution). After each pump action, the solution was stirred for 3 min to ensure solution homogeneity before the optode measurements began. The concentration of CO2 at each step was calculated using Equation (2) from Sosna et al. (2007)
where ci is the concentration of solution C after the ith addition of CO2 saturated solution, Vex amount injected/removed with each pump, Vtot total size of chamber C (2,000 ml), csat concentration of CO2 in saturated solution (B).
The Vex must be small such that none of the newly added saturated solution is removed during the pumping. The sensitivity of the pCO2 optode was determined by incrementally increasing the pumping time (total pumped volume) until a change was detected by the optode, ensuring the pumping time was below the maximum Vex (pump time of 5 s, 3.21 ml). The optode was able to detect changes at a 2 s pump time, equivalent to 1.2 ml of seawater at salinity 35 and 1.1 μatm. Therefore, when a bubble was observed at the end of the calibration run (or at any point during), the results were discarded. All work using the Sosna gas-calibration rig was carried out in a dark room in order to minimize optode spot dye photobleaching from excess ambient light, due to the clear glass of the calibration rig and the long run-times of the rig.
At the end of each experiment, further sample aliquots were taken to verify the final pCO2, calculated from measured pH and DIC, pCO2-calcpH. The uncertainty in the calculated start and end point pCO2-calcpH was ± 18 μatm.
The temperature during the measurements was maintained using a thermostatted water bath (Grant TX150 ST26, ± 0.01°C). Temperature of the solutions was verified prior to measurement with the optodes, using a thermometer (ATP DT-612 dual input K-type, ± 0.1°C). Temperature variations were achieved by changing the temperature of the water bath in which the Sosna rig was placed, and allowing 1–2 h for solutions to equilibrate prior to gassing or degassing and measurement. The partial pressure (or fugacity) of CO2 in seawater has a temperature dependence of δln(pCO2)/δT = 0.043 − 8.7·10−5 T (Takahashi et al., 2009), and therefore for a 0.1°C temperature change (the error of the thermometer), a pCO2 change of 1.6 μatm will occur, which is below the determined long term optode precision (±9.5 μatm, section Precision of pCO2 Optode Measurements). The uncertainty in the water bath temperature (±0.01°C) led to a ±0.17 μatm uncertainty in pCO2-calcpH, which is smaller than the best short-term laboratory precision of 0.8 μatm, and therefore the water bath induced temperature uncertainty was considered negligible.
The field data was collected in the period between 08/06/2014 and 11/07/2014 during cruise JR302 of the R.R.S. James Clark Ross, as part of the RAGNARoCC (Radiatively Active Gases from the North Atlantic Region and Climate Change) and OSNAP (Overturning in the Sub-polar North Atlantic Program) research programs. Underway sensor measurements and discrete surface water samples were obtained from the ship's underway seawater supply, with an intake at the hull at ca. 5 m depth. Where water column profile data are shown, they were discretely sampled water from rosette CTD casts along the cruise route. For more details on the cruise, please see King and Holliday (2015).
Continuous temperature, conductivity, and chlorophyll-a fluorescence were obtained using a shipboard thermosalinograph (TSG, SeaBird Electronics, Inc., SBE 45) connected to the underway supply. Discrete salinity samples were collected, analyzed with an AutoSal salinometer (Guildline), and used to calibrate the TSG and CTD conductivity sensors.
The pCO2 optode was operated near-continuously, stopping only near sea ice due to cessation of underway water intake, and for measurements of CRMs (DIC/TA CRM, batches 135 and 136, where pCO2 calculated from DIC and TA is referred to as pCO2-calcTA) to determine potential drift from the optode. The optode managed 5,438 successful measurements over the whole cruise. Seawater was transferred from the underway supply to a custom-built 15 ml chamber using a solenoid pump controlled by LabView software.
The optode was suspended in a water bath (Grant TX150-ST26, ±0.01°C) maintained at 20°C, with the tubing from the underway supply also placed in the water bath to enhance temperature equilibration. The tubing in the water bath was cleaned weekly while a CRM measurement was being undertaken.
The obtained data were converted to pCO2 using Equation (1), and as the measurement temperature remained at 20°C for the duration of the cruise, the pCO2-values were subsequently corrected to the underway in situ temperature using the software programme CO2SYS v1.1 (Van Heuven et al., 2011) and the same constants as listed in section Dissolved Gas Calibration Rig.
An underway equilibrator-based pCO2 instrument (PML-Dartcom Live pCO2, referred to as pCO2UWIR) was operated on the R.R.S. James Clark Ross during the entire cruise. The setup and data correction were as described in Ribas-Ribas et al. (2014) and Hardman-Mountford et al. (2008). Regular working standards (0, 250, 450 ppmv CO2) were used to reference the data every 5 min, which were calibrated against NOAA primary reference standards both pre- and post-deployment. The uncertainty in the infrared PML pCO2 measurement was ±1.5 μatm.
Discrete seawater samples were collected from CTD casts (total of 245) and three times daily from the underway supply. Seawater was collected according to the Guide for Best Practices (Dickson et al., 2007), into 250 ml borosilicate glass bottles that were filled and allowed to overflow with sample until a full bottle volume had been replaced. Each sample was poisoned with 50 μL of saturated mercuric chloride solution and a 2.5 ml air headspace was introduced. The bottles were then sealed with silicone grease (Apiezon L) and stored in a dark and cool environment until analysis.
Analysis of DIC and TA samples was performed on-board the ship. All samples were measured on a combined DIC and TA analyzer (VINDTA 3c; Marianda), which has a DIC precision of ±3.1 μmol kg−1 and a TA precision of ±4.1 μmol kg−1, determined from the repeated measurement of samples. DIC was measured coulometrically (Johnson et al., 1987) and TA measured with a potentiometric titration (Mintrop, 2004). The DIC was corrected using the CRM measurements. The TA was corrected using the CRM acid molality with the use of the calkulate functions (Matlab® v0.1.2, Humphreys, 2015). Data is available at doi: 10.5285/29ea7e19-d89c-3ab1-e053-6c86abc0284b.
Inorganic nutrient (nitrate, nitrite, phosphate, silicate) and dissolved oxygen (DO) samples were collected from the CTD rosette bottles and analyzed using a 7-channel Seal Analytical AA3 auto-analyzer with 2–10 CTD stations per run. The instrument was calibrated according to Seal Analytical Protocols, with application of additional CRMs from Hansell's Lab, University of Florida, USA and The General Environmental Technos Co., Ltd., (KANSO), (King and Holliday, 2015). Dissolved oxygen was sampled directly into pre-calibrated glass flasks using tygon tubing and fixed with manganese chloride and sodium hydroxide/sodium iodide solutions. The DO was determined during the cruise by Winkler titration according to Dickson (1994) and Holley and Hydes (1994).
The negative relationship between R and pCO2-calcpH (Figure 3) was expected based on the presence of the pH indicator immobilized within the foil, and the decrease of pH with an increase of pCO2. Clarke et al. (2015) demonstrated a corresponding positive sigmoidal relationship between R and pH.
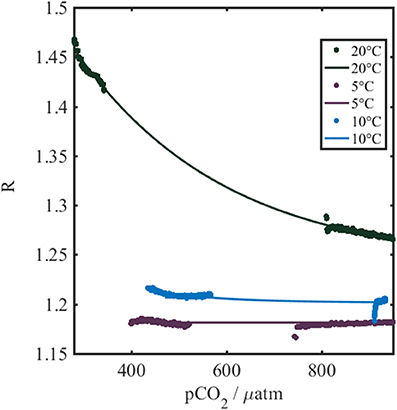
Figure 3. Temperature dependence of the sensor spot output (R) measured using the gas calibration rig described in section Dissolved Gas Calibration Rig. Colored dots represent the actual collected data, while the thin colored lines are the best fits to the data. The colors indicate the water temperature during the calibration. The pCO2 refers to pCO2-calcpH.
The optode shows a temperature dependence (Figure 3), which has also been reported for other optical chemical sensors by Clarke et al. (2015) and Fritzsche et al. (2017). The observed patterns in our study are consistent with the previous work and are in line with thermodynamic considerations dictating temperature influences on the indicator acid dissociation constant, as well as changes to the indicator fluorescence intensity and the permeability of the membrane. We see steeper gradients and greater sigmoidal inflection points at enhanced temperatures, increasing the sensitivity of the optodes with temperature as expected based on increased fluorescence intensity, and shifts in indicator acid dissociation constants to more acidic values (Clarke et al., 2015).
A three dimensional fit to calibration results yielded a first by second degree polynomial (SSE = 0.081, R2 = 0.99, Equation 3, Table 1), in agreement with previous optical pCO2 sensor work (Atamanchuk, 2013; Atamanchuk et al., 2014; Fritzsche et al., 2017).
Where B, C, D, and E are the slopes, A is the offset (as listed in Table 1), R is the ratio value from Equation (1) and T is the in situ temperature.
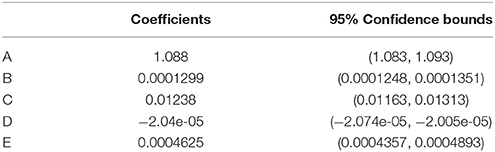
Table 1. Coefficients with 95% confidence bounds for Equation 3, determined from the data in Figure 3.
The importance of the temperature on the sensor response is indicated by the final squared term in Equation (3). Salinity was previously reported to have a limited influence on the pCO2 sensor response (Atamanchuk, 2013), particularly over the small salinity changes (typically 34 < S < 36) observed in the open ocean. It is unlikely this will change with a different interrogation method as the salinity influence is expected to be a physical effect on the membrane, such as osmotic pressure induced membrane swelling or shrinking. Deployment in environments with large salinity changes (e.g., estuarine) may have slightly larger influences on the final response.
Photobleaching is directly proportional to the light energy that the spot is exposed to (Borisov et al., 2011), therefore a higher intensity illumination was used to simulate a larger number of measurements. The residuals (mean R − Robs) showed no significant trend either before or after the high intensity illumination (Figure 4). The deviation from the mean was well within the 2σ limits confirming the spot was still performing well after illumination equivalent to 18,360 measurements. Previous work by Atamanchuk et al. (2014) demonstrated 10,000 measurements collected over 7 months, with no detected drift or bleaching. These two results combined suggest the optode spot would be suitable for long term in situ deployment with minimal effect from bleaching.
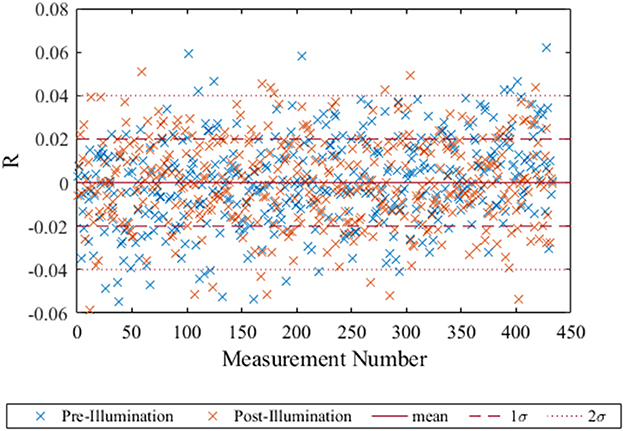
Figure 4. The residuals of R (mean–observed) for each of the post-bleach and pre-bleach measurements with the blue crosses representing the pre-bleach measurement and the orange the post-bleach measurement. The mean, 1σ and 2σ were calculated for the pre-bleach measurements and plotted here with red dashed and dotted line respectively. The straight red line indicates where measured values are the same as the mean.
The manufacturer of the pCO2 sensor spot specifies a dependency toward fluorescent compounds in the water. As such, in waters with enhanced fluorescent dissolved organic matter concentrations and chlorophyll (for example, during phytoplankton blooms), external fluorescent light may penetrate the spot to the photodetector. It is therefore of interest to evaluate the effect of fluorescing compounds in the sample matrix on the observed R-value (Equation 1). This was evaluated using seawater solutions with a known abundance of E. huxleyi, and therefore chlorophyll to allow a clear relationship with fewer interferences to be constructed.
The sensor response with increasing chlorophyll concentrations shows an approximately linear dependence (Figure 5, −4.4 μatm per μg L−1 chlorophyll or 0.0006 R per μg L−1 chlorophyll, where the error bars are the standard deviation over three repeat measurements each lasting 200 s). The large error bars are attributed to respiration processes of E. huxleyi under the low light conditions changing the desired pCO2. The observed decrease in pCO2 (increased R) with increasing chlorophyll would lead to underestimation of pCO2 in waters with enhanced phytoplankton abundance. At typical chlorophyll concentrations (<1.5 μg L−1) observed in remote surface ocean waters outside of bloom conditions (Yoder et al., 1993), the optode is unaffected by chlorophyll interferences. Additionally, the sensitivity to chlorophyll observed here is below the precision of the instrument, therefore this effect may not be relevant in real world deployments.
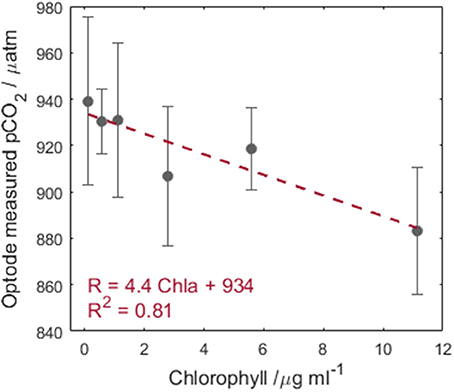
Figure 5. Chlorophyll concentration vs. optode pCO2 output. The error bars represent the standard deviation in the optode R across three repeat measurements. The red dashed line is a linear regression of the data, pCO2 = −4.4 Chla + 934 (n = 6, R2 = 0.81, p = 0.015).
As a caveat to this conclusion, chlorophyll is not the only fluorescent compound in the ocean and, this study was undertaken with only one phytoplankton species. Upon improvement of the optode precision, further testing will be required with fluorescent dissolved organic matter and external fluorescence in seawater to determine if additional compounds produce a stronger effect. Until such a time, it is recommended for deployments in known high chlorophyll regions, that water be filtered prior to pCO2 measurement to avoid potential chlorophyll interference. This brief investigation cannot be considered a robust assessment of the influence of chlorophyll on the optode performance, and we would not recommend using the linear regression to correct the data as such. Particularly as the sensitivity is below the determined precision of the optode.
Response time is here defined as the time for the R-value to reach 63% of the final stable response (τ63). A response time (τ63) of 50 s at 25°C was observed, which is comparable to a response time (τ63) of 88 s at 20°C reported by Atamanchuk et al. (2014). The shorter (50 s) response time was determined over a much smaller pCO2 range (280–800 μatm) compared to Atamanchuk (5,000–50,000 μatm), and at a higher temperature (25°C compared to 20°C). These response times are consistent with those for other pCO2 optode sensors [60–120 s, ~40 s, and up to 247 s (Oter et al., 2006; Chu and Lo, 2008; Borisov et al., 2011), respectively].
The short term optode precision was determined using repeated measurements of a DIC/TA CRM (CRM 135) in a water bath at 20°C. A short-term precision of 0.8 μatm was obtained as the standard deviation over 10 samples. This laboratory precision is significantly better than the desired 2 μatm precision (Pierrot et al., 2009), and is similar to 1 μatm achieved by Lefevre et al. (1993) with a spectrophotometric sensor, and 1.7 μatm reported by Ge et al. (2014).
The pCO2 optode data obtained at 6 min intervals over the course of cruise JR302 are presented in Figure 1. The cruise covered a wide range of oceanic environments from the polar waters of the ARCT region, with strong surface water pCO2 gradients along the Labrador Sea and Greenland transects, to the more homogenous Atlantic surface waters of the Irminger and Iceland Basins in the NADR and SARCT regions. Strong temperature gradients along the cruise track, would influence the optode response (section pCO2 Calibration of Optode), with decreasing temperatures increasing the response times due to a reduced diffusion rate of the analyte across the membranes (section Laboratory Metrology Assessment). The deployment of the pCO2 optode in a thermostatted water bath at 20°C kept the optode's response time constant, and allowed higher resolution measurements in colder ocean waters, but required an additional data correction to in situ oceanic temperatures. A wide range of pCO2-values (100 to 400 μatm) were observed across the Labrador Sea and Arctic influenced regions (Figures 1, 9). The pCO2 variability was lower in the Irminger and Iceland Basins (Table 3, NADR and Figure 9, North Atlantic region).
There is a lack of a suitable CRM for pCO2 optodes that facilitates accuracy and precision assessments in seawater, aside from purging seawater with a certified CO2 gas. To simplify the determination of longer term precision, DIC and TA CRMs from A. Dickson were used. The pCO2-calcTA of any single CRM bottle was assumed to be constant, although pCO2 is not certified, and can be considered to be within the calculated batch value ±10 μatm (Millero, 2007). Therefore we considered that using the CRM for the determination of precision is valid, however for drift and accuracy determination the lowest quotable value is ±10 μatm. The long-term precision [standard pooled deviation (NIST/SEMATECH, e-Handbook of Statistical Methods)] of the pCO2 optode measurements derived from CRM measurements, pCO2-calcTA, across the cruise period of 6 weeks was 9.5 μatm.
Further evaluation of the precision of the optode was achieved with repeat measurements of surface seawater whilst the ship was on station, similar to approaches reported by Bellerby et al. (2002). The ship remained on station for typically 2 h to complete the CTD cast and sampling, and stations were throughout the 24 h period at no specific time. The seawater was subject to the same transfer time from the underway intake to the optode, and the same filtration procedure, as the other pCO2 measurements conducted during the cruise. The surface seawater was expected to remain largely homogenous with near constant pCO2-values whilst the ship was stationary. Additionally, for our precision assessment approach no changes were required to the optode set up, such as switching the tubing from the underway supply to the CRM. This theoretically creates a more realistic approximation of the precision during the seawater pCO2 measurements. A pooled standard deviation of 23.9 μatm was obtained, while the mean standard deviation (averaged standard deviation across all samples) was 9.5 μatm. The precision recorded was variable between stations where the precision measurements were conducted, with later stations showing a larger standard deviation, perhaps indicating a stronger influence of currents with a resulting change in seawater pCO2 levels, or a biofilm build up on the sensor spot. This approach of evaluation of optode precision using repeated measurements may hence not provide the most reliable precision value because of pCO2 changes in the surface waters while on station, but provides an additional precision estimate. Previous deployments of pCO2 optodes in fjord environments had similar variability in the determined precision, but with lower values ranging from 2 to 10 μatm (Atamanchuk et al., 2015). The 9.5 μatm precision from the CRM measurements, and the average standard deviation from seawater measurements whilst at station was within the “weather” quality measurement as defined the global ocean acidification observing network measurement goals (Newton et al., 2015).
Figure 6 shows the residuals of pCO2 obtained from measurements of the CRM using the optode and the calculated CRM-value, pCO2-calcTA. The pCO2-calcTA-value obtained for each CRM measurement was not within the precision of the optode nor the error of the calculated value. The reason for this is not clear, although it could be caused by CO2 exchange of the CRM solution with the atmosphere, as the bottles were open for a short period of time (45 min) to allow the optode pCO2 measurements and calibration of the DIC analyzer. The optode drift determined from the measurements of the CRMs over the course of the cruise was −4.6 μatm (R2 = 0.24, p = 6.9 × 10−5). The lack of a CRM for pCO2 sensors that allows analysis of pCO2 directly in aqueous solutions is a recognized issue within the sensor community, particularly when reference materials for in situ sensors are increasingly desirable to improve drift assessment. There are ongoing efforts to further evaluate if DIC/TA CRMs can be used for this purpose and for the determination of the associated relative uncertainty. Due to the lack of certainty in pCO2 for each CRM bottle (the error in the calculated batch value is ±10 μatm) and the large difference between the overall pCO2-calcTA and the optode data, the observed drift of −4.6 μatm (33 days)−1 needs further validation to be considered credible. Therefore, the drift of the optode is evaluated relative to the pCO2 data obtained with other techniques (infrared PML pCO2 analyzer and the discretely collected DIC/TA samples).
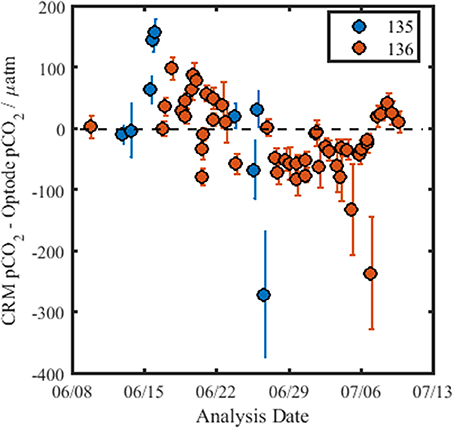
Figure 6. Residuals for pCO2 for the CRM measurements using the optode, over the course of the cruise. The black dashed line indicates no difference between the calculated pCO2-value of the CRM (pCO2-calcTA) and the pCO2-value obtained using the optode. The color of the data points denotes the CRM batch measured, and the error bars are calculated as the standard deviation of the repeat measurements of each CRM bottle (n ≥ 5).
The calculated residual pCO2-values between the different techniques (where residual is the pCO2 of the alternate method minus pCO2 of the optode) are presented in Figure 7. There is a strong positive relationship between the residuals of the surface water pCO2-calcTA from the CTD casts and the optode (Table 2, Row 2), but a smaller positive relationship between the optode and the infrared PML pCO2 analyzer (Table 2, Row 2). There is a positive trend between residuals from the surface water pCO2-calcTA from the CTD casts and the infrared PML pCO2 analyzer, (not shown in Figure 7, details in Row 2, Table 2), but with a smaller sample size (n = 7).
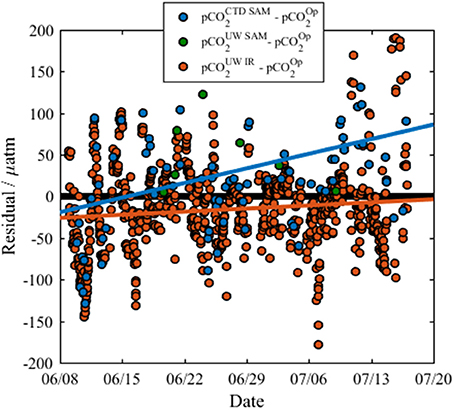
Figure 7. The residuals between the optode sensor data and other pCO2 data, where residual refers to the difference between the reference sample data and the optode as listed in the legend. pCO2op, optode measured pCO2; pCO2UWIR, Infrared PML pCO2 analyzer; pCO2UWSAM, pCO2-calcTA calculated from discrete underway DIC and TA samples; and pCO2CTDSurf, pCO2-calcTA calculated from discrete surface water DIC and TA samples collected from the CTD rosette. The residuals from the infrared PML pCO2 analyzer show a slight positive trend (orange line, residual = 0.53 × date − 390,000, R2 = 0.01, n = 698, p = 0.005) over the course of the cruise. The residuals from CTD surface samples show a stronger positive trend (blue line, residual = 2.5 × date − 1,800,000, R2 = 0.16, n = 71, p = < 0.001). The residuals from the underway samples show no significant trend due to the small sample size (n = 7). The solid black line indicates where there is zero difference between the data collected by the optode and data from a reference method.

Table 2. Row 1 details the Spearman's rank correlation coefficients between the different pCO2 measurements collected on the cruise where P = significance, N = number of samples, Coeff = the coefficient.
The consistently positive relationships may be attributed to a CO2 contamination of the seawater by organic matter respiration whilst flowing from the seawater intake point on the ship's hull to the pCO2 optode and infrared PML analyzer. However, the overall drift of the optode was considered negligible due to the good agreement with the infrared PML pCO2 analyzer, and the fact that both the small gradients in the residuals (m = 0.533 and m = 2.5) and the low drift determined from the CRM measurements [−4.6 μatm (33 days)−1] were within the shipboard precision of the optode.
Discrete surface water samples for DIC and TA analysis that were collected from the ship's underway supply and CTD rosette frame (<10 m) were used to calculate pCO2-calcTA, as a lower resolution validation of the sensor spot. The pCO2 optode (black dots, Figure 8) showed reasonable agreement with the pCO2-calcTA from the discretely collected samples (CTD rosette samples are displayed as triangles with the color indicating the sampling depth, and purple dots referring to the discrete underway samples; Figure 8). The infrared PML pCO2 analyzer was connected to the same underway seawater supply as the optode (orange dots in Figure 8).
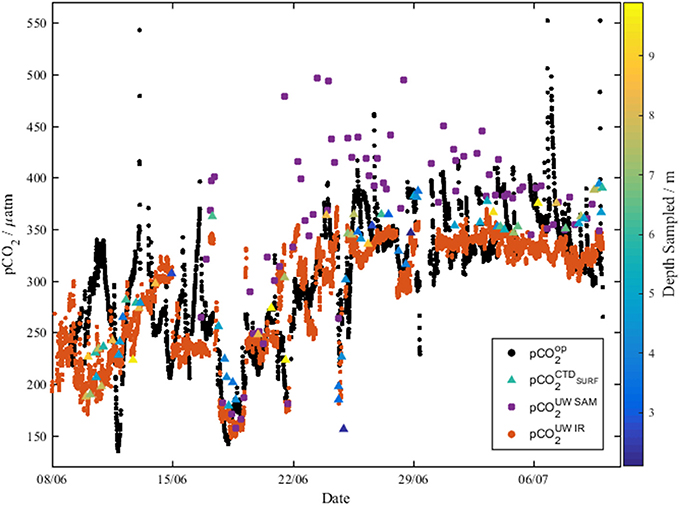
Figure 8. The underway optode pCO2 (black dots, pCO2op) compared to pCO2 measured with the infrared PML analyzer (orange dots, pCO2UWIR), and pCO2-calcTA calculated from both underway discrete DIC and TA measurements (purple dots, pCO2UWSAM), and CTD discrete DIC and TA measurements (triangles, colored according to the depth sampled, pCO2CTDSurf).
The pCO2-values measured by both the optode (135–369 μatm) and the infrared PML pCO2 analyzer (170–323 μatm) in the central Labrador Sea (8th−15th June) were consistent with summer values reported by DeGrandpre et al. (2006) of 220–350 μatm. Additionally, the pCO2 measured in the NADR by the pCO2 optode (228–408 μatm) and infrared PML pCO2 analyzer (301–389 μatm) was consistent with previous work of 200–360 μatm (Olsen et al., 2008). The discretely sampled TA in the central Labrador Sea showed homogeneity down to 200 m, and the concentrations of 2,280–2,310 μmol kg−1 were similar to the mean 2295 ± 4 μmol kg−1 reported by Azetsu-Scott et al. (2010). The DIC concentrations (2,090–2,125 μmol kg−1) across the central Labrador Sea were consistent with early summer values of 2,080–2,125 μmol kg−1 reported by DeGrandpre et al. (2006) and 2,060–2,085 μmol kg−1 reported for June by Körtzinger et al. (2008).
Over the period between 29th June and 11th July, the pCO2 measurements by the optode showed diurnal cycles, which were not apparent in the pCO2 results from the infrared PML pCO2 analyzer. The diurnal cycles measured by the optode is hypothesized to be caused by biofilm growth on the foil, which was not removed during flushing. The pCO2-calcTA determined from the discretely collected underway samples (purple dots, Figure 8) had significant offsets from the rest of the data from 22nd June. It is unclear why there is such a shift in the underway discrete samples.
The optode did not initially agree well with either the discrete samples (pCO2-calcTA) or the infrared PML pCO2 sensor, with large variations in pCO2 not present in data from the other samples. The offset does not have a clear trend or specific offset. Comparisons of the residuals to oxygen, and CTD do not reveal clear relationships. It is anticipated to be a combination of the increased chlorophyll concentrations in the water (ca. 2.5 μg L−1) observed during the first few days of the cruise, and temperature gradients between the optode measurement at 20°C and the in situ temperature being larger than corrected for. To correct for the former, filtration upstream of the optode is recommended. To correct for the latter, further deployments of the optode should have a temperature sensor in close proximity to the spot to allow accurate temperature corrections. Further work on temperature dependence within the calibration curve should allow for the optode to be deployed directly in the underway water, without the need for additional warming in the water bath. The temperature calibration should span the full range of temperatures and pCO2 to allow accurate in situ pCO2 measurement.
In addition to this, collaboration with the spot manufacturers may enable more suitable indicators to be immobilized in the membrane. Ideally, spots specifically designed to work in colder areas, with different CO2 ranges depending on the deployment location would be used, and would contribute to better precision values. Finally, future developments of the optode should include removal of the fiber optic cable to enable less light loss between the optode spot and the detector, and further development of the electrical hardware to prevent the disruption that occurred at the latter stages of the deployment.
Despite the issues, the Spearman Correlation Coefficient indicates overall agreement between the optode values and the infrared PML pCO2 analyzer (0.654, Table 2), the discrete underway samples (0.821, Table 2) and the discrete surface CTD samples (0.536, Table 2). Based on the good agreement of the optode data with both previous work and the other pCO2 data collected on board, the data was deemed suitable for further evaluation on the controls on surface water pCO2 in the region.
The high resolution optode data provided an opportunity to determine the factors that control surface water pCO2 across two different biogeographical regions. The CTD pCO2-calcTA dataset is much smaller in size and therefore leads to less statistically robust relationships. To determine the factors controlling surface seawater pCO2, Spearman's correlation coefficients were used to determine significant correlation (positive values) and anti-correlation (negative values) between optode measured pCO2 and other variables. From this we can determine the major controls on surface water pCO2 for each region. The dominant processes determining pCO2 in the surface ocean are photosynthesis which results in a reduction in pCO2 due to inorganic carbon uptake by phytoplankton, with organic matter remineralization yielding the opposite effect (Zeebe and Wolf-Gladrow, 2005), and an increase in surface water temperature which increases pCO2, with cooling having the opposite effect (Zeebe and Wolf-Gladrow, 2005).
The Arctic region (ARCT) comprised the Labrador Sea and the Greenland transects 1–4 (Figure 9), and was the best sampled region. Underway measurements of temperature, salinity and chlorophyll showed strong standard deviations, thereby indicating a pronounced spatial heterogeneity in the region (Table 3). The pCO2 in the ARCT region was positively correlated with temperature, salinity and inorganic nutrients, and negatively correlated with oxygen and chlorophyll (Table 4). These correlations suggest that both biological processes, water mass mixing, and changes to surface water temperature and salinity (e.g., sea ice melt) controlled surface water pCO2. Due to the high spatial heterogeneity, the ARCT region was subdivided into the Labrador Sea and the area around the four Greenland transects (highlighted in Figure 9) to evaluate the controls on pCO2 on smaller regional scales.
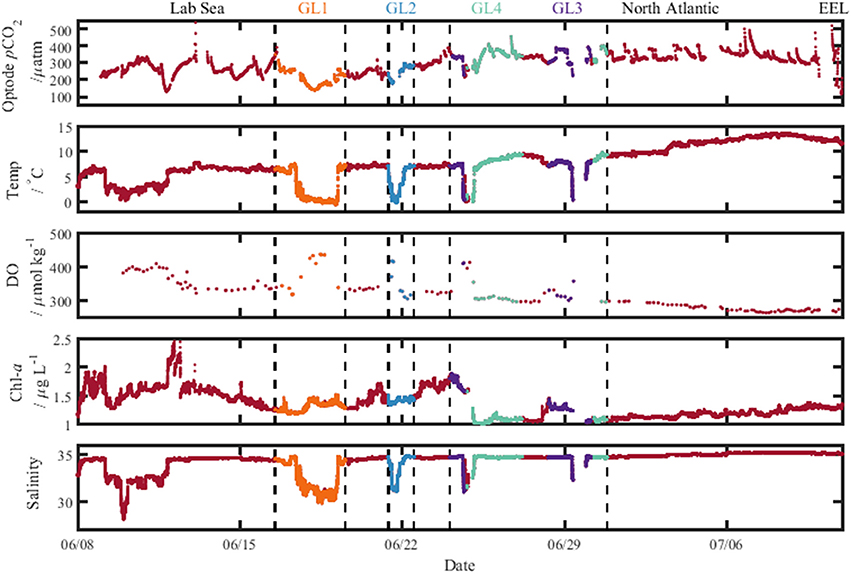
Figure 9. Plots of the measured variables from the ship's underway seawater supply along the cruise track, with pCO2 determined using the optode sensor. The different colors indicate data collected on the sections toward Greenland labeled at the top (GL1–4). Lab Sea, Labrador Sea; and North Atlantic, Irminger Basin; EEL, extended Ellett Line.
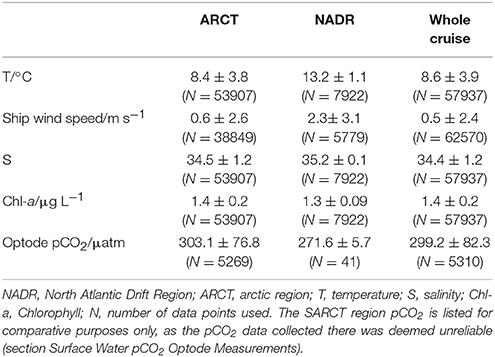
Table 3. Means ± standard deviations of underway measured variables for each biogeographical region (as defined in section Fieldwork Observations).
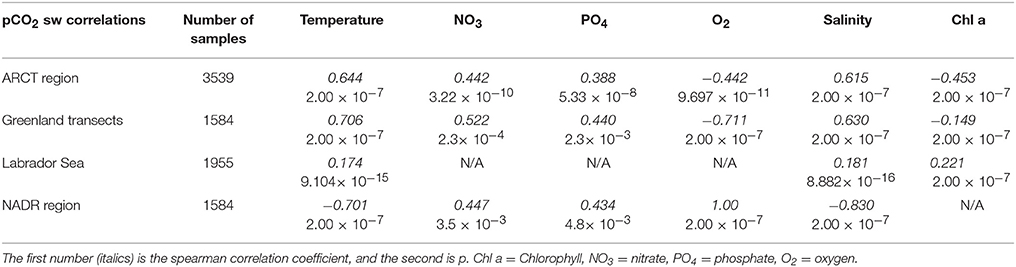
Table 4. Significant spearman correlation coefficients between pCO2 and the variable listed in the top of the table.
The Greenland transects showed similar correlation patterns between pCO2 and other variables as the larger ARCT region, with pCO2 positively correlated with temperature, salinity and inorganic nutrients, and negatively correlated to oxygen and chlorophyll. The Greenland transects (highlighted in separate colors in Figure 9), feature sharp drops in temperature and salinity, with increases in oxygen concentration and decreases in pCO2. This is a common occurrence upon sea ice melt (Fransson et al., 2009). Following rapid cooling and freshening processes during sea ice melt, seawaters readily re-equilibrate with the atmosphere with respect to oxygen, but become under saturated with respect to CO2, as CO2 has a longer equilibration timescale than oxygen [240 days compared to 12 days, respectively, on seasonal time scales under well-mixed tropospheric conditions considering only the mixed layer (Zeebe and Wolf-Gladrow, 2005)]. Melting sea ice combined with the slower equilibration time of CO2 hence explains the correlations to temperature, salinity and oxygen. The additional correlations between pCO2 and nutrients and chlorophyll are related to enhanced phytoplankton growth following freshening and cooling events (Tynan et al., 2016), which additionally enhances oxygen levels. The enhanced chlorophyll concentrations in the warmer sections of the Greenland transects (up to 2 μg L−1, Figure 9), compared to the cooler transects where sea ice was present (ca. 1.5 μg L−1, Figure 9), indicate an additional strong biological control on pCO2 in these areas situated further off shore of Greenland.
The surface water pCO2 in the Labrador Sea was controlled by a set of different processes to the larger ARCT region (Table 4), with only weak positive correlations between pCO2 and temperature, salinity and chlorophyll. Melting sea ice was present in the Labrador Sea toward the start of the cruise, with low temperatures, lower salinities, enhanced oxygen concentrations, and low pCO2 concentrations (Figure 9), as for the Greenland transects. Therefore, it is likely sea ice melt, as observed near the Greenland coast, and the associated temperature and salinity changes also determined pCO2 in the section close to the Canadian coast. The weak positive correlation between pCO2 and chlorophyll is likely related to a decaying spring phytoplankton bloom in the region, with enhanced chlorophyll concentrations still present (Figure 9), and respiration related to the consumption of the organic material driving up the pCO2 concentration (Körtzinger et al., 2008).
The pCO2 data for the NADR showed smaller standard deviations in the underway sampled variables, indicating a more pronounced spatial homogeneity (Table 3). The surface waters in this region are dominated by the warm salty North Atlantic Current (Figure 2). The NADR showed negative correlations of pCO2 to temperature and salinity, and this region features greater wind speeds compared to the ARCT region (Table 4), suggesting wind induced transfer of deeper waters to the surface ocean. The upward mixing of subsurface waters (such as the Subarctic Intermediate Water Holliday et al., 2006) results in fresher and cooler surface waters and a supply of CO2 and nutrients (Takahashi et al., 1985, 1993). This supports the observed positive correlations between pCO2 and nutrients. Additionally, oxygen solubility is increased in the colder and fresher waters, such that oxygen concentrations would have increased relative to CO2 due to sea-air gas exchange, resulting in a positive correlation between pCO2 and oxygen. Although the chlorophyll concentrations in the NADR are lower than in the ARCT region, they are elevated for the Irminger Basin region compared to winter values (Henson et al., 2006; Holliday et al., 2006). This could indicate a biological influence on the surface ocean carbonate system, despite the lack of a significant correlation between pCO2 and chlorophyll. In the sub-polar North Atlantic Ocean the spring blooms commence in the east and gradually moves to the west with the progression of the seasons (Olsen et al., 2008), and are additionally controlled by meteorological conditions (Henson et al., 2006). Hence, the lower chlorophyll concentration in NADR compared to the ARCT region could also be attributed to the fact the main spring bloom was in an initial phase, disrupted by the increased wind speed, such that chlorophyll concentrations were consequently low but nutrient drawdown had commenced. The combination of pCO2 upwelled from subsurface waters containing winter CO2 and surface photosynthesis and gas exchange increasing oxygen concentrations, produced the positive correlation between pCO2 and oxygen.
The high resolution dataset allowed for a detailed evaluation, and a distinction between control of surface water pCO2 concentration by temperature, salinity, sea ice melt and photosynthesis and respiration. In contrast, Doney et al. (2009b) reported only thermodynamic and biological controls on surface pCO2 from a biogeochemical model of the sub-polar North Atlantic, with no additional detail. This study demonstrates how high resolution precise pCO2 data can further our understanding of the temporal and spatial variability of the surface ocean carbonate system, and allow us to more fully assess potential implications resulting from the increasing anthropogenic CO2 emissions.
An experimental pCO2 optode was characterized in a laboratory before being deployed as a high resolution autonomous underway sensor. The spot has a proven working life of at least 1 month (5,438 measurements) during the deployment with a measurement resolution of 6 min, and laboratory experiments project the sensor to be capable of >18,000 measurements, based on photobleaching experiments. The short term precision measured in the laboratory (0.81 μatm) was significantly better than the desired 2 μatm for underway pCO2 measurements, while the shipboard determined precision was 9.5 μatm. A response time of 50 s at 25°C was found, comparable to other pCO2 sensors.
High resolution underway surface water pCO2 data was collected from both an experimental pCO2 optode, and an equilibrator based infrared pCO2 analyzer, along with lower resolution discrete DIC and TA sampling, pCO2-calcTA. The pCO2 obtained by the optode showed significant correlations with all of the reference measurements (equilibrator infrared pCO2 analyzer, and pCO2-calcTA from the lower resolution DIC and TA samples). The spot had a positive drift when compared to the infrared PML pCO2 analyzer, however this was less than the overall shipboard precision of the optode and therefore the drift was considered negligible. The optode pCO2 data was used to determine that the major controls on pCO2 across the subpolar North Atlantic were a combination of photosynthesis, respiration and water mass changes at the surfaces caused by sea ice melt and deeper water mass upwelling.
The system used herein was designed as a benchtop system, therefore this paper is an evaluation of the performance of the sensor spot with the t-DLR technique rather than hardware suitability for in situ deployment. For an in situ deployment, the system requires a redesign as discussed above with further test deployments. Optodes are advantageous compared to more traditional technology due to their lack of moveable parts, and wet chemical requirements. Further development may see pCO2 optodes more widely deployed, similar to oxygen optodes. While the optode does not, currently, attain the desired precision as specified in the introduction, with the developments discussed above, it may be attained in future.
JC designed and performed the optode experiments, carried out the optode deployment, and led the writing of the manuscript. ET collected and analyzed the cruise samples. MH helped correct the discrete sample data and provided discussions around the data correlations. VK developed and corrected the PML pCO2 data, while IB deployed the instrument. MM and EA supervised the study and provided intellectual guidance throughout. All authors contributed to the interpretation of the results and writing of the manuscript.
The Analytical Chemistry Trust Fund, funded by the Royal Society for Chemistry and the UK Natural Environment Research Council (NE/I019638/1) funded the optode development and deployment. VK and IB and the PML pCO2 sensor development and deployment were funded by UK Natural Environment Research Council (NE/K00249X/1) and EU Horizons 2020 (AtlantOS, Grant No. 633211). The cruise was funded as part of the RAGNARoCC project, for the UK Natural Environment Research Council (NE/K002546/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the other scientists and crew on the JR302 cruise for sharing their data and expertise in this paper- in particular Hannah Donald, Carolyn Graves, Mark Stinchombe, Sinhue Torres and the JR302 PIs Penny Holliday and Brian King. In addition, we thank Alex Griffiths, Becky Garley, and Claudia Fry who helped with carbonate chemistry sampling and analysis on the cruise. The authors would like to thank Andrew Morris for the advice and help when setting up and using the Sosna rig. We are very grateful to Rosie Sheward and Lucie Munns for culturing the coccolithophores, and counting the stock solutions used in the paper.
1. ^Sensoptics is now a wholly owned subsidiary of Optasense (A QinetiQ company) www.optasense.com
Álvarez, M., Ríos, A. F., Pérez, F. F., Bryden, H. L., and Rosón, G. (2003). Transports and budgets of total inorganic carbon in the subpolar and temperate North Atlantic. Global Biogeochem. Cycles 17, 2-1–2-21. doi: 10.1029/2002GB001881
Atamanchuk, D. (2013). Development and Use of an Optical pCO2 Sensor in Marine Studies. Ph.D., Natural Science, University of Gothenburg.
Atamanchuk, D., Kononets, M., Thomas, P. J., Hovdenes, J., Tengberg, A., and Hall, P. O. J. (2015). Continuous long-term observations of the carbonate system dynamics in the water column of a temperate fjord. J. Mar. Syst. 148, 272–284. doi: 10.1016/j.jmarsys.2015.03.002
Atamanchuk, D., Tengberg, A., Thomas, P. J., Hovdenes, J., Apostolidis, A., Huber, C., et al. (2014). Performance of a lifetime-based optode for measuring partial pressure of carbon dioxide in natural waters. Limnol. Oceanogr. Methods 12, 63–73. doi: 10.4319/lom.2014.12.63
Azetsu-Scott, K., Clarke, A., Falkner, K., Hamilton, J., Jones, E. P., Lee, C., et al. (2010). Calcium carbonate saturation states in the waters of the Canadian Arctic Archipelago and the Labrador Sea. J. Geophys. Res. 115:C11021. doi: 10.1029/2009JC005917
Battelle (2011/2012). Battelle Designs Commercial Rugged Sensor System to Capture Our Oceans CO2 Data. Available online at: https://www.battelle.org/government-offerings/national-security/maritime-systems-technologies/sensors/pco2-monitoring-system (Retrieved 13:58 on November 22, 2012).
Bellerby, R. G. J., Olsen, A., Johannessen, T., and Croot, P. (2002). A high precision spectrophotometric method for on-line shipboard seawater pH measurements: the automated marine pH sensor (AMpS). Talanta 56, 61–69. doi: 10.1016/S0039-9140(01)00541-0
Borisov, S. M., Seifner, R., and Klimant, I. (2011). A novel planar optical sensor for simultaneous monitoring of oxygen, carbon dioxide, pH and temperature. Anal. Bioanal. Chem. 400, 2463–2474. doi: 10.1007/s00216-010-4617-4
Byrne, R. H. (2014). Measuring ocean acidification: new technology for a new era of ocean chemistry. Environ. Sci. Technol. 48, 5352–5360. doi: 10.1021/es405819p
Chu, C.-S., and Lo, Y.-L. (2008). Fiber-optic carbon dioxide sensor based on fluorinated xerogels doped with HPTS. Sens. Actuat. B 129, 120–125. doi: 10.1016/j.snb.2007.07.082
Clarke, J. S., Achterberg, E. P., Connelly, D. P., Schuster, U., and Mowlem, M. (2017). Developments in marine pCO2 measurement technology; towards sustained in situ observations. Trends Anal. Chem. 88, 53–61. doi: 10.1016/j.trac.2016.12.008
Clarke, J. S., Achterberg, E. P., Rérolle, V. M. C., Abi Kaed Bey, S., Floquet, C. F. A., and Mowlem, M. C. (2015). Characterisation and deployment of an immobilised pH sensor spot towards surface ocean pH measurements. Anal. Chim. Acta 897, 69–80. doi: 10.1016/j.aca.2015.09.026
Corbière, A., Metzl, N., Reverdin, G., Brunet, C., and Takahashi, T. (2007). Interannual and decadal variability of the oceanic carbon sink in the North Atlantic subpolar gyre. Tellus B 59, 168–178. doi: 10.1111/j.1600-0889.2006.00232.x
Daniels, C. J., Sheward, R. M., and Poulton, A. J. (2014). Biogeochemical implications of comparative growth rates of Emiliania huxleyi and Coccolithus species. Biogeosciences 11, 6915–6925. doi: 10.5194/bg-11-6915-2014
DeGrandpre, M. D., Körtzinger, A., Send, U., Wallace, D. W. R., and Bellerby, R. G. J. (2006). Uptake and sequestration of atmospheric CO2 in the Labrador Sea deep convection region. Geophys. Res. Lett. 33:L21SL03. doi: 10.1029/2006GL026881
DeVries, T. (2014). The oceanic anthropogenic CO2 sink: storage, air-sea fluxes, and transports over the industrial era. Global Biogeochem. Cycles 28, 631–647. doi: 10.1002/2013GB004739
Dickson, A. G. (1990). Standard potential of the reaction AgCl(s)+1/2H2(g) = Ag(s)+HCl(aq) and the standard acidity constant of the bisulfate ion in synthetic seawater from 273.15 K to 318.15 K. J. Chem. Thermodyn. 22, 113–127. doi: 10.1016/0021-9614(90)90074-Z
Dickson, A. G. (1994). Determination of Dissolved Oxygen in Seawater by Winkler Titration. Technical Report, WOCE Operations Manual.
Dickson, A. G., and Millero, F. J. (1987). A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep Sea Res. A Oceanogr. Res. Pap. 34, 1733–1743. doi: 10.1016/0198-0149(87)90021-5
Dickson, A. G., Sabine, C. L., and Christian, J. R. (eds.). (2007). Guide to Best Practices for Ocean CO2 Measurements. Sidney, BC: PICES Special Publication 3.
Doney, S. C., Lima, I., Feely, R. A., Glover, D. M., Lindsay, K., Mahowald, N., et al. (2009a). Mechanisms governing interannual variability in upper-ocean inorganic carbon system and air–sea CO2 fluxes: physical climate and atmospheric dust. Deep Sea Res. II Top. Stud. Oceanogr. 56, 640–655. doi: 10.1016/j.dsr2.2008.12.006
Doney, S. C., Tilbrook, B., Roy, S., Metzl, N., Le Quéré, C., Hood, M., et al. (2009b). Surface-ocean CO2 variability and vulnerability. Deep Sea Research II Top. Stud. Oceanogr. 56, 504–511. doi: 10.1016/j.dsr2.2008.12.016
Fransson, A., Chierici, M., and Nojiri, Y. (2009). New insights into the spatial variability of the surface water carbon dioxide in varying sea ice conditions in the Arctic Ocean. Cont. Shelf Res. 29, 1317–1328. doi: 10.1016/j.csr.2009.03.008
Fritzsche, E., Gruber, P., Schutting, S., Fischer, J. P., Strobl, M., Müller, J. D., et al. (2017). Highly sensitive poisoning-resistant optical carbon dioxide sensors for environmental monitoring. Anal. Methods 9, 55–65. doi: 10.1039/C6AY02949C
Ge, X., Kostov, Y., Henderson, R., Selock, N., and Rao, G. (2014). A low-cost fluorescent sensor for pCO2 measurements. Chemosensors 2, 108–120. doi: 10.3390/chemosensors2020108
Gruber, N., Keeling, C. D., and Bates, N. R. (2002). Interannual variability in the North Atlantic Ocean carbon sink. Science 298, 2374–2378. doi: 10.1126/science.1077077
Hardman-Mountford, N. J., Moore, G. F., Bakker, D. C. E., Watson, A. J., Schuster, U., Barciela, R., et al. (2008). An operational monitoring system to provide indicators of CO2-related variables in the ocean. ICES J. Mar. Sci. 65, 1498–1503. doi: 10.1093/icesjms/fsn110
Henson, S. A., Robinson, I., Allen, J. T., and Waniek, J. J. (2006). Effect of meteorological conditions on interannual variability in timing and magnitude of the spring bloom in the Irminger Basin, North Atlantic. Deep Sea Res. I Oceanogr. Res. Pap. 53, 1601–1615. doi: 10.1016/j.dsr.2006.07.009
Holley, S. E., and Hydes, D. J. (1994). Procedures for the Determination of Dissolved Oxygen in Seawater. Technical Report, James Rennell Centre for Ocean Circulation.
Holliday, N. P., Waniek, J. J., Davidson, R., Wilson, D., Brown, L., Sanders, R., et al. (2006). Large-scale physical controls on phytoplankton growth in the Irminger Sea part I: hydrographic zones, mixing and stratification. J. Mar. Syst. 59, 201–218. doi: 10.1016/j.jmarsys.2005.10.004
Humphreys, M. P. (2015). Calkulate: Seawater Total Alkalinity from Open Cell Titration Data using a Modified Gran Plot Approach. National Oceanography Centre.
Johnson, K. M., Sieburth, J. M., Williams, P. J. L., and Brandstrom, L. (1987). Coulometric total carbon-dioxide analysis for marine studies - automation and calibration. Mar. Chem. 21, 117–133. doi: 10.1016/0304-4203(87)90033-8
King, B. A., and Holliday, N. P. (2015). RRS James Clark Ross Cruise 302, 06 Jun - 21 Jul 2014, The 2015 RAGNARRoC, OSNAP and Extended Ellett Line Cruise Report. National Oceanography Centre Southampton. 76.
Körtzinger, A., Send, U., Wallace, D. W. R., Karstensen, J., and DeGrandpre, M. (2008). Seasonal cycle of O2 and pCO2 in the central Labrador Sea: atmospheric, biological, and physical implications. Global Biogeochem. Cycles 22:GB1014. doi: 10.1029/2007GB003029
Landschützer, P., Gruber, N., Bakker, D. C. E., and Schuster, U. (2014). Recent variability of the global ocean carbon sink. Global Biogeochem. Cycles 28, 927–949. doi: 10.1002/2014GB004853
Le Quéré, C., Moriarty, R., Andrew, R. M., Peters, G. P., Ciais, P., Friedlingstein, P., et al. (2015). Global carbon budget 2014. Earth Syst. Sci. Data 7, 47–85. doi: 10.5194/essd-7-47-2015
Lee, K., Kim, T. W., Byrne, R. H., Millero, F. J., Feely, R. A., and Liu, Y. M. (2010). The universal ratio of boron to chlorinity for the North Pacific and North Atlantic oceans. Geochim. Cosmochim. Acta 74, 1801–1811. doi: 10.1016/j.gca.2009.12.027
Lefevre, N., Ciabrini, J. P., Michard, G., Brient, B., DuChaffaut, M., and Merlivat, L. (1993). A new optical sensor for PCO2 measurements in seawater. Mar. Chem. 42, 189–198. doi: 10.1016/0304-4203(93)90011-C
Lefèvre, N., Watson, A. J., Olsen, A., Ríos, A. F., Pérez, F. F., and Johannessen, T. (2004). A decrease in the sink for atmospheric CO2 in the North Atlantic. Geophys. Res. Lett. 31:L07306. doi: 10.1029/2003GL018957
Longhurst, A., Sathyendranath, S., Platt, T., and Caverhill, C. (1995). An estimate of global primary production in the ocean from satellite radiometer data. J. Res. 17, 1245–1271. doi: 10.1093/plankt/17.6.1245
Mehrbach, C., Culberso, C. H., Hawley, J. E., and Pytkowic, R. M. (1973). Measurement of apparent dissociation constants of carbonic acid in seawater at atmospheric pressure. Limnol. Oceanogr. 18, 897–907. doi: 10.4319/lo.1973.18.6.0897
Millero, F. J. (2007). The marine inorganic carbon cycle. Chem. Rev. 107, 308–341. doi: 10.1021/cr0503557
Mintrop, L. (2004). Versatile Instrument for the Determination of Titration Alkalinity, Manual for Versions 3S and 3C, Version 2.0. MARine ANalytics and DAta (MARIANDA), Kiel.
Newton, J. A., Feely, R. A., Jewett, E. B., Williamson, P., and Mathis, J. (2015). Global Ocean Acidification Observing Network: Requirements and Governance Plan. 2nd Edn. GOA-ON. Avaliable online at: http://www.goa-on.org/docs/GOA-ON_plan_print.pdf
Olsen, A., Brown, K. R., Chierici, M., Johannessen, T., and Neill, C. (2008). Sea-surface CO2 fugacity in the subpolar North Atlantic. Biogeosciences 5, 535–547. doi: 10.5194/bg-5-535-2008
Oter, O., Ertekin, K., Topkaya, D., and Alp, S. (2006). Emission-based optical carbon dioxide sensing with HPTS in green chemistry reagents: room-temperature ionic liquids. Anal. Bioanal. Chem. 386, 1225–1234. doi: 10.1007/s00216-006-0659-z
Pierrot, D., Neill, C., Sullivan, K., Castle, R., Wanninkhof, R., Lüger, H., et al. (2009). Recommendations for autonomous underway pCO2 measuring systems and data-reduction routines. Deep Sea Res. II Top. Stud. Oceanogr. 56, 512–522. doi: 10.1016/j.dsr2.2008.12.005
Ribas-Ribas, M., Rérolle, V. M. C., Bakker, D. C. E., Kitidis, V., Lee, G. A., Brown, I., et al. (2014). Intercomparison of carbonate chemistry measurements on a cruise in northwestern European shelf seas. Biogeosciences 11, 4339–4355. doi: 10.5194/bg-11-4339-2014
Schuster, U., McKinley, G. A., Bates, N., Chevallier, F., Doney, S. C., Fay, A. R., et al. (2013). An assessment of the Atlantic and Arctic sea–air CO2 fluxes, 1990–2009. Biogeosciences 10, 607–627. doi: 10.5194/bg-10-607-2013
Schuster, U., Watson, A. J., Bates, N. R., Corbiere, A., Gonzalez-Davila, M., Metzl, N., et al. (2009). Trends in North Atlantic sea-surface fCO2 from 1990 to 2006. Deep Sea Res. II Top. Stud. Oceanogr. 56, 620–629. doi: 10.1016/j.dsr2.2008.12.011
Sunburst Sensors LLC (2014). SAMI-CO2 Ocean CO2 Sensor. Avaliable online at: http://www.sunburstsensors.com/products/oceanographic-carbon-dioxide-sensor.html (Accessed October 29, 2014).
Sosna, M., Denuault, G., Pascal, R. W., Prien, R. D., and Mowlem, M. (2007). Development of a reliable microelectrode dissolved oxygen sensor. Sen. Actuators B Chem. 123, 344–351. doi: 10.1016/j.snb.2006.08.033
Takahashi, T., Olafsson, J., Broecker, W. S., Goddard, J., Chipman, D. W., and White, J. (1985). Seasonal variability of the carbon-nutrient chemistry in the ocean areas west and north of Iceland. Rit. Fiskideildar J. Mar. Res. Inst. Reykjavik 9, 20–36.
Takahashi, T., Olafsson, J., Goddard, J. G., Chipman, D. W., and Sutherland, S. C. (1993). Seasonal variation of CO2 and nutrients in the high-latitude surface oceans: a comparative study. Global Biogeochem. Cycles 7, 843–878. doi: 10.1029/93GB02263
Takahashi, T., Sutherland, S. C., Wanninkhof, R., Sweeney, C., Feely, R. A., Chipman, D. W., et al. (2009). Climatological mean and decadal change in surface ocean pCO2, and net sea–air CO2 flux over the global oceans. Deep Sea Res. II Top. Stud. Oceanogr. 56, 554–577. doi: 10.1016/j.dsr2.2008.12.009
Tynan, E., Clarke, J. S., Humphreys, M. P., Ribas-Ribas, M., Esposito, M., Rérolle, V. M. C., et al. (2016). Physical and biogeochemical controls on the variability in surface pH and calcium carbonate saturation states in the Atlantic sectors of the Arctic and Southern Oceans. Deep Sea Res. II 127, 7–27. doi: 10.1016/j.dsr2.2016.01.001
Van Heuven, S., Pierrot, D., Rae, J. W. B., Lewis, E., and Wallace, D. W. R. (2011). CO2SYS v 1.1, MATLAB Program Developed for CO2 System Calculations. ORNL/CDIAC-105b. Oak Ridge, TN: Carbon Dioxide Information Analysis Center; Oak Ridge National Laboratory; U.S. DoE.
Wanninkhof, R., Park, G. H., Takahashi, T., Sweeney, C., Feely, R., Nojiri, Y., et al. (2013). Global ocean carbon uptake: magnitude, variability and trends. Biogeosciences 10, 1983–2000. doi: 10.5194/bg-10-1983-2013
Yoder, J. A., McClain, C. R., Feldman, G. C., and Esaias, W. E. (1993). Annual cycles of phytoplankton chlorophyll concentrations in the global ocean: a satellite view. Global Biogeochem. Cycles 7, 181–193. doi: 10.1029/93GB02358
Zeebe, R. E., and Wolf-Gladrow, D. A. (2005). CO2 in Seawater: Equilibrium, Kinetics, Isotopes. Oxford: Elsevier.
Keywords: carbon dioxide, optode, seawater, North Atlantic, sensors, time domain dual lifetime referencing
Citation: Clarke JS, Humphreys MP, Tynan E, Kitidis V, Brown I, Mowlem M and Achterberg EP (2017) Characterization of a Time-Domain Dual Lifetime Referencing pCO2 Optode and Deployment as a High-Resolution Underway Sensor across the High Latitude North Atlantic Ocean. Front. Mar. Sci. 4:396. doi: 10.3389/fmars.2017.00396
Received: 23 June 2017; Accepted: 24 November 2017;
Published: 15 December 2017.
Edited by:
Marta Álvarez, Instituto Español de Oceanografía (IEO), SpainReviewed by:
Arvind Singh, Physical Research Laboratory, IndiaCopyright © 2017 Clarke, Humphreys, Tynan, Kitidis, Brown, Mowlem and Achterberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer S. Clarke, amVuc2NsYXJrZUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.